
Abstract
Antibody-drug conjugates (ADCs) combine the high target specificity and favorable pharmacokinetics of monoclonal antibodies with the potent tumor-killing properties of cytotoxic agents, and have demonstrated convincing antitumor effect in both animal models and patients. However, the inherent complexity of ADCs with their multiple components often makes their development challenging. Pharmacokinetic and absorption, distribution, metabolism, and excretion (ADME) characterization of ADCs reflects the dynamic interactions between the biological system and ADC, and provides critical assessments in lead selection, optimization, and clinical development. A rational strategy integrating the mechanistic understanding of pharmacokinetic/pharmacodynamics and ADC disposition helps to inform target selection, drug selection, and linker design, and ultimately to maximize the therapeutic window. In this chapter, we give an overview of ADC PKPD and disposition, and discuss our current understanding of the major determinants, unique challenges, and lessons learned from current ADC landscape. The utility of pharmacokinetics–pharmacodynamics (PKPD) modeling is also discussed in the context of providing guidance to assist in the successful development of these complex molecules.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Pharmacokinetics (PK)
- Pharmacodynamics (PD)
- Disposition
- Monoclonal antibody (mAb)
- Antibody-drug conjugates (ADCs)
- Targeted therapy
- Optimization
1 Introduction
There is a growing need for evaluating and developing new therapeutic modalities in our battle against cancer. In the past few decades, the development of targeted therapies resulted in drugs with improved efficacy and safety (Gerber 2008). Monoclonal antibodies, as a class of highly targeted therapeutics, have been used for the treatment of a variety of cancers. Recently, antibody- drug conjugates (ADCs) have shown impressive potential in advancing cancer treatment to the next level (Sievers and Senter 2013). Conjugation of potent cytotoxic drugs to an antibody can increase potency of the antibody itself, which usually acts by blocking/activating signal transduction, antibody-dependent cellular cytotoxicity (ADCC), and/or complement-dependent cytotoxicity (CDC) (Waldmann 2003). By combining the high target specificity of monoclonal antibodies with potent tumor-killing properties of cytotoxic agents, ADCs have demonstrated convincing antitumor effect in both animal models and patients (Sievers and Senter 2013).
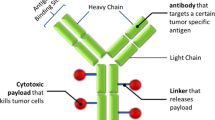
ADCs consist of a potent cytotoxic agent conjugated through a linker molecule to an antibody that can bind with high specificity to a target antigen. The expression of target antigen is selected for its overexpression in tumor compared to normal cells with the exception of B cell targets, such as CD22 and CD79 (Bander 2013). The antibody part of the ADC is designed to bind with high affinity and selectivity to its target antigen. Cytotoxic drugs that are attached to the antibody are usually potent antimitotic or DNA-modifying agents (Ducry and Stump 2010). Administration of these drugs by themselves may have minimal therapeutic index, but conjugation to an antibody renders an acceptable window through enhanced delivery to tumor, reduction of systemic exposure, and minimizing distribution and uptake of drug to nontarget tissues. The third component of an ADC is the linker that conjugates the cytotoxic agent with that of the antibody usually through the cysteine or lysine amino acid residues (Nolting 2013).
There are two ADCs that are currently approved for marketing. Adcetris (brentuximabvedotin) is an anti-CD30 antibody conjugated to an antimitotic agent monomethylauristatin E (MMAE) through a cathepsin cleavable linker and was approved for the treatment of patients with Hodgkin’s lymphoma after failure of autologous stem cell transplant (Cao et al. 2013). Kadcyla (ado-trastuzumabemtansine) consists of transtuzumab (targeted against HER2 antigen) conjugated to mertansine (DM1 ) and was approved for the treatment of HER2-positive metastatic breast cancer (Ballantyneand Dhillon 2013). In addition, more than 30 ADCs with different cytotoxic drug and linker combinations are in various stages of clinical development against a variety of targets (Mullard 2013). Furthermore, there is tremendous breakthrough in developing next-generation ADCs through antibody engineering, site-specific conjugation, and novel linker/cytotoxic drug combination (Flemming 2014).
However, the inherent complexity of ADCs with their multiple components often makes their development challenging. Pharmacokinetic and absorption, distribution, metabolism, and excretion (ADME) characterization of ADCs reflects the dynamic interactions between the biological system and ADC , and provides critical assessments in lead selection, optimization, and clinical development. A rational strategy integrating the mechanistic understanding of pharmacokinetics/pharmacodynamics and ADC disposition helps to inform target selection, drug selection, linker design, and ultimately helps to maximize the therapeutic window.
Target, antibody , linker, cytotoxic drug, and their interactions make distinct contributions to the mechanism of action for a given ADC , and the disposition of an ADC defines its therapeutic window (Sievers and Senter 2013). Two pharmacologically distinct components, the antibody and the cytotoxic small-molecule drug, necessitate the understanding of the behavior and fate of both components in vivo, and monitoring of their PK/disposition requires comprehensive analytical strategy.
2 Analytical Strategy and Its Application in ADC Selection and Optimization
A typical ADC assay strategy evolves with the stage of development with the goal of selecting the right analyte to inform the safety and efficacy, and describe the exposure–response relationship (Kaur et al. 2013). Multiple analytes help to capture the many facets of the behavior of these complex molecules, such as the rate of drug loss from an ADC (i.e., linker stability ), the effect of conjugation on ADC clearance, and ultimately the exposure–response relationship. However, the desire to be comprehensive must be balanced by the practicality, the availability of the technology and reagents, and ultimately by the purpose of each study. In discovery stage, many optimization factors are being explored, such as different linkers , drug, drug to antibody ratio (DAR), site of conjugation, and a diverse array of assays are often used to understand the stability and disposition of ADCs, and its initial interactions with biological systems. At clinical stage, with confirmation of preclinical prediction and availability of human data, the assay strategy may be streamlined to increase efficiency and patient compliance (Dere et al. 2013). In general, a panel of assays is required to measure these disparate components: total antibody (conjugated and unconjugated antibody), conjugated antibody, conjugated drug, unconjugated antibody, and unconjugated (free) drug (Kaur et al. 2013). For example, in the development of T-DM1 , a comprehensive assay strategy consisting of three validated assays was developed. These validated assays included an enzyme-linked immunosorbent assay (ELISA) designed to measure total trastuzumab, an ELISA to measure conjugated trastuzumab, and a small-molecule LC–MS/MS assay to measure the amount of free DM1 catabolite. These assays are capable of quantifying DAR analytes in circulation to characterize the PK and stability of T-DM1 in nonclinical and clinical studies (Dere et al. 2013).
The total antibody (Tab) concentration captures both the conjugated and unconjugated forms of an ADC and is usually determined using an ELISA-based format. It provides the best assessment of the in vivo stability and integrity of the antibody over time and serves a key role in ADC optimization , particularly in evaluating the impact of conjugation and selecting a drug load.
Conjugated antibody concentrations in systemic circulation are usually determined using an ELISA assay format that measures a mixture of ADC species bearing at least one conjugated cytotoxic drug. Since the detection in this assay requires the presence of both, intact antibody and cytotoxic drug components of the ADC, conjugated antibody concentration is commonly used as an estimate of the active ADC concentration, and is the basis for most ADC PK analyses. The limitation of this assay is its inability to differentiate ADCs with varying numbers of conjugated cytotoxic drugs (DARs). Given ADC species with different DARs may have different potencies, the measured concentrations may not accurately reflect the associated pharmacologic activity (Stephan et al. 2008).
On the other hand, measuring conjugated drug provides a measure of the total amount of cytotoxic drug covalently bound to the antibody (Sanderson et al. 2005; Xu et al. 2013), and changes in conjugated drug concentration could reflect both elimination of ADC from systemic circulation and loss of cytotoxic drug from the antibody. Affinity capture LC-MS is a method of choice by which the ADC is specifically extracted from plasma, then analyzed using LC-MS/MS (Xu et al. 2013). The power of this method lies in its ability to provide direct measurement of average concentrations of drug associated with antibody. Xu et al. used affinity capture LC-MS to assess the site-specific loss of cytotoxic drug from a thiomab ADC, providing valuable insights into the impact of conjugation site on linker stability , a critical factor in ADC safety and efficacy (Xu et al. 2011). These examples illustrate the value of evolving analytical technologies in exploring the behavior of ADCs, which in turn can lead to improvements in ADC design and development .
Unconjugated cytotoxic drug concentrations are used to infer the systemic exposure to the cytotoxic drug released from the ADC and are often associated with loss of efficacy and increased toxicity. Assays for drug-containing products usually employ LC-MS or ELISA methods (Wang et al. 2005; Xie et al. 2004). LC-MS methods are highly specific for the measured analyte, while ELISA methods may be less specific and able to quantitate multiple analytes of similar structure. A critical consideration of assay selection ensures that the analyte(s) selected for measurement is relevant for efficacy or toxicity and able to provide meaningful PKPD relationship.
3 Disposition of an ADC and Its Implication on PKPD
The systemic PK profiles of an ADC provide only partial narratives of its fate. By design, an ADC is a prodrug. Its activity depends on the interaction with target, subsequent internalization of antigen-ADC complex and final releasing of active drug inside the cells. The full ADME properties of ADCs are crucial for the therapeutic window rendered by ADCs. Biologically, the disposition of ADCs is strongly influenced by the underlying antibody backbone conferring properties such as target-specific binding, neonatal Fc receptor (FcRn)-dependent recycling, and Fc (fragment, crystallizable) effector functions. Similarly, the ADME properties of ADCs possess similar attributes associated with unconjugated antibodies (Lobo et al. 2004; Deng et al. 2012; Boswell et al. 2011; Linand and Tibbitts 2012).
3.1 Absorption
All ADCs that are currently on the market or in clinical development are dosed intravenously and hence absorption is not considered when assessing PK properties of ADCs. However, for convenience intraperitoneal dosing of ADCs are sometimes used in early preclinical studies and in the future, there is always a possibility for the development of subcutaneous dosing strategies for ADC similar to that of biologics treatment (Sharkey et al. 2011, 2012). In both cases, the absorption properties of ADCs in general will be similar to that of any monoclonal antibody . Although the mechanism is still unclear, it is generally accepted that lymphatic drainage is the major route for absorption from the site of subcutaneous administration to systemic circulation (Zhao et al. 2013;Wang et al. 2008). In addition, it is also acknowledged that diffusion of the molecule across blood vessels can contribute to the absorption kinetics.
3.2 Distribution
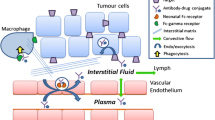
Distribution characteristics of ADC generally mimic that of monoclonal antibodies. Due to the cytotoxicity of conjugated drug, distribution and accumulation of an ADC to nontarget tissues, via either antigen specific or nonspecific processes, may have profound pharmacologic/toxic effects. Owing to the higher molecular weight, the initial distribution is limited to the vascular space, with the volume of distribution in the central compartment similar to the total plasma volume in any species (~40 mL/Kg) (Tabrizi et al. 2010; Mould and Green 2010). However, with time, distribution extends to tissue interstitial space. The extent of distribution into tissues is determined by a variety of factors including blood flow, tissue porosity, structure and heterogeneity, and target properties. The movement of the molecules into the tissues occurs by convection, transcytosis, and diffusion across the capillary and studies show that the partitioning of antibodies into tissues ranges between 5 and15 % of what is observed in circulation, with brain being an exception, having very low penetration of antibody because of the tough blood–brain barrier (Shahand and Betts 2013).
Beyond these similarities, ADCs bring in a different level of complexity as a result of interactions between target antigen and ADCs. Factors such as binding affinity, tissue expression profile, target turnover/internalization rates, target density, and conjugation impact on ADC distribution and influence the therapeutic index of an ADC (Tabrizi et al. 2010). Unlike target for monoclonal antibodies which are developed for blocking specific function/pathway in tumors leading to tumor growth inhibition or killing, targets for ADCs do not need to be causal or implicated in tumor progression. The role of the target antigen for an ADC is to provide high specificity, either expressed only by the tumor or at levels significantly higher in tumor (Bander 2013; Silver et al. 1997). In some cases, low level of target antigen expression in normal tissues and their subsequent uptake of ADC may lead to decreased ADC delivery to tumor and/or increased delivery of cytotoxic drug to normal tissues; a phenomenon that could affect the therapeutic index of the ADC (Boswell et al. 2013). Employing a “predosing” or capping non-tumor expression site with an inactive antibody may be helpful to mitigate the toxicity (Boswell et al. 2013).
The level of target antigen expression and binding affinity of the target to the antibody are critical in determining the amount of ADC delivered to the target. Irrespective of how specific the target expression is, if the target expression levels and/or antibody binding affinity are low, ADCs delivered to tumor compared to normal tissues (nonspecific uptake) will be low resulting in a poor therapeutic ratio. Hence, in addition to being specific, the level of target expression (and binding affinity) should enable adequate accumulation of the drug at the site of interest (Bander 2013).
Most of cytotoxic agents in the ADC act intracellularly and hence internalization and rate of internalization of the target play an important role in determining the drug accumulation inside the tumor cells and its subsequent killing (Sievers and Senter 2013). The linker molecules are designed to be stable in circulation, but are cleaved in endosome/lysosome compartments to effectively release the cytotoxic agents inside the cell. In addition to a rapid internalization process, the recycling or replenishment of the target at the cell surface is important to sustain the delivery of ADCs into the tumor cells. Furthermore, shedding of target antigen from cell surface can play an important role in determining efficacy and toxicity of ADCs (Tolcher et al. 2003). If shed target antigens are present, ADC binding to these shed antigens will affect ADC distribution and can result in higher liver uptake (as these complexes are cleared by liver) and hence liver-related toxicities (Lovdal et al. 2000).
A few studies have assessed the effects of conjugation on the distribution of ADCs compared to unconjugated antibodies (Erickson and Lambert 2012). Distribution of DM1- and DM4- conjugated ADCs (lysine conjugation) in mice was similar to that of typical unconjugated IgG molecules (Xie et al. 2004 ; Erickson and Lambert 2012). However, auristatin-conjugated ADCs (cysteine conjugation) show modest differences in tissue distribution compared to unconjugated antibodies (Boswell et al. 2011). For example, MMAE-conjugated STEAP1 ADC showed a general trend towards increased hepatic uptake and reduced levels in other highly vascularized organs in rats. Similarly, CMD-193 antibody conjugated with calicheamicin showed increased hepatic uptake compared to the parental huS193 unconjugated antibody, in a phase I biodistribution and PK study in patients with advanced epithelial cancer (Scott et al. 2007). Conjugation likely increases the hydrophobicity of the antibody, which leads to enhanced uptake by the liver (Boswell et al. 2011). Efforts on antibody engineering fronts have also shown promise in altering distribution and mitigating the toxicity. Mutated antibody with deficiency in FcR binding with attenuated nonspecific uptake led to longer circulation time and enhanced efficacy (Sussman et al. 2011).
3.3 Metabolism and Elimination
Therapeutic antibodies are eliminated from the body predominantly via target-mediated and nonspecific uptake into cells followed by proteolytic degradation into inert small peptides and amino acids (Lobo et al. 2004). ADCs, bearing cytotoxic drugs, become active following release from, or degradation of, their associated antibodies. Understanding the mechanism of ADC metabolism and elucidating the identity of its active metabolites have practical implications on ADC optimization , clinical monitoring, and PKPD relationship. Metabolism of ADCs includes at least three processes: deconjugation as cytotoxic drug releasing from the ADC via enzymatic or chemical processes; proteolytic metabolism of the antibody , which can result in small peptides or amino acids that are still conjugated to the cytotoxic small-molecule drug (Tabrizi et al. 2006); and the metabolism of the cytotoxic drug (primarily the free drug) by the typical small-molecule phase I and phase II metabolizing enzymes (Lin and Lu 1997).
Nonspecific cleavage of the linker could happen in circulation resulting in the deconjugation of one or more molecule of the cytotoxic drug from the antibody . Once internalized into the cell (by target-mediated cellular uptake in target cells or nonspecific pinocytosis), depending on the type of linker, the linker can be cleaved by specific proteases (like cathepsin B involved in the cleavage of valine-citrulline (vc) dipeptide linkage used for MMAE or MMAF conjugates) or the proteolytic processing in endosomes/lysosomes (Sutherland et al. 2006 ; Chuand and Polson 2013). For maleimide-containing drug linkers which are conjugated to antibody cysteine residues to form thiosuccinimide bonds, there are two competing reactions: exchange with sulfhydryl-containing molecules with direct consequence of drug loss (Alley et al. 2009) versus hydrolysis of the thiosuccinimide ring resulting in stable bond (Xu et al. 2013 ; Lyon et al. 2012). Site of conjugation and solvent accessibility have direct influence on drug loss and subsequent efficacy (Shen et al. 2012), which led to the selection of LC, HC over FC variants, meanwhile, taking advantage of chemically induced hydrolysis, Lyon et al. showed the improved therapeutic window by making self-stabilizing maleimido-DPR (Lyon et al. 2012).
The other dominant mechanism that results in cytotoxic drug-containing catabolites is ADC catabolism, a process driven by either receptor-mediated endocytosis or fluid-phase pinocytosis with subsequent trafficking to the lysosome, followed by enzymatic degradation (Alley et al. 2009 ; Erickson et al. 2006 ; Okeley et al. 2010). Both deconjugation and proteolytic degradation of the antibody can occur simultaneously and the contribution of each process to the release of the cytotoxic drug (or drug-containing products) depends on multiple factors including target properties, linker stability , drug load per antibody, and conjugation site. For example, apart from the free DM1 (cytotoxic drug), MCC-DM1 (linker conjugated to drug) and lysine-MCC-DM1 were detected in circulation in both preclinical studies and in patients treated with T-DM1 (Shen et al. 2012b). Furthermore, for some ADCs, studies have shown other mechanisms of cytotoxic drug release or deconjugation like the transfer of linkerdrug from the antibody to circulating albumin in the case of MC-MMAF and VC-MMAE (Shen et al. 2012a). Nonspecific uptake and processing of ADCs may be the main source of ADC toxicity, and modulation of this process has potential benefit in expanding TI. For example, by mutating Fc residues that are in contact with FcγRIII, Sussman et al. have shown that S239°C variant does not bind FcγRIII, blocks localization to nontarget tissues and decreases off-target toxicity (Sussman et al. 2011 ; Jeffrey et al. 2013).
Once the cytotoxic drug is released from the antibody , they are subject to the metabolism and elimination processes associated with small-molecule drugs, including CYPs and drug transporters, and possesses the theoretical potential for drug–drug interactions (DDIs; Lu et al. 2013). In vitro studies in human liver microsome preparations suggest that DM1 is metabolized mainly by CYP3A4 and to a lesser extent by CYP3A5 (Shen et al. 2012b); Davis et al. 2012). The elimination of DM1-containing products and its metabolites was primarily through the biliary/fecal route in rats with 80 % recovery of the radio-labeled drug. Similar observations were made with VC-MMAE-conjugated ADCs, with ~80 % of the radio-labeled MMAE observed in feces and only ~6 % of MMAE was recovered from urine (Pastusovas et al. 2005).
It has been recognized that one of the key factors that modulates ADC disposition is heterogeneity of an ADC, the consequence of both its conjugation process during manufacturing and its deconjugation in vivo. Traditional conjugation through either cysteine and lysine residues on the monoclonal antibody results in a mixture of ADC species differing not only in the number of drugs attached to the antibody but also in the sites of drug linkage (Hamblett et al. 2004 ; Singhand and Erickson 2009). For instance, ADCs conjugated by cysteines are mixtures of conjugated antibodies with a DAR ranging from 0 to8, representing drugs conjugated to some or all of the cysteines that in unconjugated antibodies form the interchain disulfide bonds (Singhand and Erickson 2009). ADCs conjugated via lysine residues have the potential for even greater variability in the number of conjugated drugs and their locations (Wang et al. 2005 ; Hamblett et al. 2004). Heterogeneity from ADC mixtures potentially could diminish the therapeutic window with higher DAR species contributing to significant toxicity while lower DAR species compromise on efficacy. In addition, it presents a major challenge in describing the collective ADC PKPD profile with multiple species with different pharmacological potency and PK behavior. One particular concern has been the presence of higher DAR species. For the reasons that have been touched upon before, multiple site conjugation tends to increase hydrophobicity and decrease the overall stability on two fronts: faster loss of drug or deconjugation and accelerated antibody clearance (Sun et al. 2011).
Several studies helped assess the heterogeneity on PK, efficacy and toxicity, and guided the field toward more homogenous ADC production. McDonagh et al. engineered the cysteines associated with the interchain disulfides, resulting in an antibody with fewer cysteines for conjugation and potentially resulting in less heterogeneity (McDonagh et al. 2008). ThioMab-drug conjugates (TDC) with site-specifically engineered cysteines provided a defined DAR of 2 per mAb with minimized heterogeneity and increased therapeutic index (Junutula et al. 2008). Alternatively, biosynthetically incorporating nonnative amino acids into a given Ab scaffold provides homogeneous ADCs with precise control over the site and stoichiometry of drug conjugation (Tian et al. 2014). Minimizing heterogeneity and optimizing DARs also help to establish a more defined PKPD relationship.
4 Application of PK in ADC Optimization
Integrating information from multiple assays is critical in characterizing the in vivo behavior of these complex molecules, interpreting ADC pharmacologic effects and ultimately building exposure–response relationship. Accumulated knowledge on ADC PK helps to shed lights on mechanism of its stability , such as the effect of conjugation on ADC clearance, the rate of drug loss from an ADC (i.e., linker stability), and has profound impact on ADC optimization .
It has been observed that conjugation causes an increase in the clearance of the antibody for several ADCs (Boswell et al. 2011 ; Hamblett et al. 2004 ; Herbertson et al. 2009), which may result in more rapid delivery of cytotoxic drug-bearing ADC to the normal organs or tissues with potentially toxic consequences (Hamblett et al. 2004). Comparison of Tab PK of the ADC with the Tab PK of unconjugated antibody (administered as unconjugated antibody) provides information regarding the effect of conjugation on antibody clearance (Xie et al. 2004); (Sapra et al. 2005). There are several mechanisms being postulated ranging from the disruption of tertiary structure, weakening of disulfide bonds, to increase in hydrophobicity from conjugation (Boswell et al. 2011 ; Hamblett et al. 2004). Among them, the hydrophobicity charge seems to be plausible. Conjugation with higher DAR leading to faster clearance seems to corroborate this hypothesis. For example, cAC10-vc MMAE ADCs with high DARs were observed to have a faster Tab clearance than lower DAR ADCs (Hamblett et al. 2004). Adem et al. showed that high-drug-load species leads to thermal unfolding and aggregation and affects the physical stability of ADCs (Jeffrey et al. 2013).
Another key parameter in ADC optimization is the rate of drug loss from ADC. Loss of drug from the ADC can result in decreased efficacy and changes in the toxicity associated with ADC administration. Comparative assessment of Tab PK with either conjugated antibody PK or conjugated drug PK can provide qualitative guidance on the rate of drug loss from the ADC (Linand and Tibbitts 2012). When comparing Tab PK with conjugated antibody PK, it is typically observed that conjugated antibody concentrations decline more rapidly than Tab concentrations, and the degree of divergence of the curves is indicative of the rate of complete drug loss from the ADC (Tolcher et al. 2003; Tijink et al. 2006 ; Burris et al. 2011 ; Kantarjian et al. 2012). Better understanding of mechanism of drug loss from conjugated antibody helps assess the stability of linkers , conjugation sites, and impact of DARs. Illustrative cases for differences in linker stability between disulfide and thioether linkers, and the effect of conjugation site on thiomab-ADC linker stability have been also reported (Shen et al. 2012a ; Lewis Phillips et al. 2008). One example of different efficacy among anti-Her2 ThioMab Drug Conjugate (TDC) variants (light chain, heavy chain, and Fc site-specific) was evidenced by the differential rate of drug loss or deconjugation despite similar total antibody clearance (Shen et al. 2012a).
Conjugated drug measurements have provided insights when compared with total antibody profiles. Antibody-conjugated drug concentrations decline more rapidly than Tab concentrations because two processes drive the decrease in conjugated drug concentrations: loss of drug from the ADC and elimination of ADC, while Tab concentration changes are driven solely by elimination of ADC and unconjugated antibody. As such, the difference in the concentration decrease can be used to infer the rate of drug loss from the ADC (Kaur et al. 2013 ; Sanderson et al. 2005 ; Xie et al. 2004). Assessment and integration of ADC PK can be valuable not only in understanding a single ADC but also in evaluating multiple ADCs with different structural and pharmacologic characteristics; allowing improved design and development of these complex molecules.
5 PKPD Modeling for ADCs
As described earlier, ADC is a heterogeneous mixture of multiple species described by a variety of analytes. Due to this, heterogeneity and dynamics between different species quantitative characterization of ADC pharmacokinetics and pharmacodynamics becomes challenging. Mathematical modeling approaches can provide help in gaining a better understanding of the complex PK and PD behavior of ADCs. In addition, it can help in the integration of the dynamic changes in different ADC species and provide a way to assess the drivers of efficacy and toxicities.
A variety of PKPD mathematical models ranging from simple data-driven empirical models to semi-mechanistic and physiology-based models have been developed to characterize and describe ADC disposition and actions. For example, for T-DM1 , Jumbe et al. described the mouse drug-conjugated antibody pharmacokinetics using a simple two-compartment model with linear drug clearance (Jumbe et al. 2010). In this study, drug-conjugated antibody was assumed to be the driving force for efficacy and the simple model could describe reasonably well the disposition profile. A first-order growth model with tumor transit compartments was used for describing the tumor progression and tumor killing was modeled as nonlinear killing driven by the drug-conjugated antibody (T-DM1 ). In contrast, Bender et al. developed a mechanism-based multi compartment model that described the pharmacokinetic profile of different heterogeneous species representing T-DM1 ADC (Bender et al. 2012). Preclinical data for individual DAR species were utilized and each DAR species was explicitly modeled with a central and peripheral compartment for distribution. Deconjugation of the cytotoxic drug DM1 from the antibody was modeled with variable rate constants (differentiating deconjugation between higher and lower DAR species). Both in vivo and in vitro measurements were utilized to develop the model. Gibiansky et al. adapted the target-mediated drug disposition model to describe ADC PK and with number of approximations including rapid binding, quasi-steady-state and Michealis–Menten approximations, derived a reduced model that can still describe ADC PK with load-independent properties (Gibiansky and Gibiansky 2014).
Although most of the modeling work has been done with T-DM1 , some of the recent efforts have been in modeling other ADCs like that of brentuximab-vedotin which consists of a cleavable linker conjugating the cytotoxic drug with the cysteine residues of the antibody . Shah et al. developed a multiscale-mechanism-based model incorporating a variety of physiological process including ADC and drug payload PK at the cellular level, in circulation, and tumor tissue to characterize brentuximab-vedotin disposition and action and to predict the clinical response to the drug in cancer patients including progression-free survival rates and complete response rates (Fig. 7.1; Shah et al. 2012). Known mechanisms of ADC and payload disposition and tumor growth kinetics were used for the model development and the model utilized invitro, preclinical, and clinical measurements of ADC and payload and tumor-growth inhibition data in xenograft mouse models to develop the model.

A simplified model representation of disposition and action of brentuximab-vedotin ADC and its payload described by Shah et al (2012). The integrated model takes into account the disposition of ADC and its payload in systemic circulation, tumor tissue, and inside cells and link intracellular payload concentration to tumor killing. ADC antibody- drug conjugates
6 Conclusions
As a hybrid between antibody therapeutics and small-molecule cytotoxic drugs, ADCs exhibit unique pharmacological and PK properties. Among them, the heterogeneity from ADC production and in vivo processing, the necessity to monitor multiple active ADC analytes, and less-understood catabolic and metabolic species, all of which demands meaningful PKPD relationships. As with other therapeutics, PKPD can aid in understanding exposure–response relationships, determining the optimal dose and dose regimen, predicting human PK, facilitating the translation of nonclinical data to clinical outcome, and allowing quantitative understanding of mechanistic pharmacology (Morgan et al. 2012). The growth of the interest in ADCs, the evolvement of powerful analytical tools, and generation of crucial mechanistic data indicate a promising future for ADC development .
References
Ballantyne A, Dhillon S (2013) Trastuzumabemtansine: first global approval. Drugs 73:755–765
Alley SC, Zhang X, Okeley NM, Anderson M, Law CL, Senter PD, Benjamin DR (2009) The pharmacologic basis for antibody-auristatin conjugate activity. J Pharmacol Exp Ther 330:932–938
Bander NH (2013) Antibody-drug conjugate target selection: critical factors. Methods Mol Biol 1045:29–40
Bender B, Leipold D, Liu L, Xu K, Shen BQ, Friberg LE, Tibbitts J (2012) A multicompartmental population PK model elucidating the complex disposition of trastuzumabemtansine (T-DM1): an antibody-drug conjugate for the treatment of HER2-positive cancer, Population Approach Group in Europe, 2012
Boswell CA, Ferl GZ, Mundo EE, Bumbaca D, Schweiger MG, Theil FP, Fielder PJ, Khawli LA (2011) Effects of anti-VEGF on predicted antibody biodistribution: roles of vascular volume, interstitial volume, and blood flow. PLoS ONE 6:e17874
Boswell CA, Mundo EE, Firestein R, Zhang C, Mao W, Gill H, Young C, Ljumanovic N, Stainton S, Ulufatu S, Fourie A, Kozak KR, Fuji R, Polakis P, Khawli LA, Lin K (2013) An integrated approach to identify normal tissue expression of targets for antibody-drug conjugates: case study of TENB2. Br J Pharmacol 168:445–457
Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle NR, Kozak KR, Fourie A, Chuh J, Koppada N, Saad O, Gill H, Shen BQ, Rubinfeld B, Tibbitts J, Kaur S, Theil FP, Fielder PJ, Khawli LA, Lin K (2011) Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjugate Chem 22:1994–2004
Burris HA 3rd, Tibbitts J, Holden SN, Sliwkowski MX, Lewis Phillips GD (2011) Trastuzumabemtansine (T-DM1): a novel agent for targeting HER2 + breast cancer. Clin Breast Cancer 11:275–282
Cao H, Yamamoto K, Yang LX, Weber R (2013) Brentuximabvedotin: first-line agent for advanced Hodgkin lymphoma. Anticancer Res 33:3879–3885
Chuand YW, Polson A (2013) Antibody-drug conjugates for the treatment of B-cell non-Hodgkin’s lymphoma and leukemia. Future Oncol 9:355–368
Davis JA, Rock DA, Wienkers LC, Pearson JT (2012) In vitro characterization of the drug-drug interaction potential of catabolites of antibody-maytansinoid conjugates. Drug Metab Dispos 40:1927–1934
Deng R, Jin F, Prabhu S, Iyer S (2012) Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opin Drug Metab Toxicol 8:141–160
Dere R, Yi JH, Lei C, Saad OM, Huang C, Li Y, Baudys J, Kaur S (2013) PK assays for antibody-drug conjugates: case study with ado-trastuzumabemtansine. Bioanalysis 5:1025–1040
Erickson HK, Lambert JM (2012) ADME of antibody-maytansinoid conjugates. AAPS J 14:799–805
Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, Lutz RJ, Goldmacher VS, Blattler WA (2006) Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res 66:4426–4433
Flemming A (2014) Antibody engineering: fine-tuning antibody-drug conjugates. Nat Rev Drug Discov 13:178
Gerber DE (2008) Targeted therapies: a new generation of cancer treatments. Am Fam Physician 77:311–319
Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, Meyer DL, Francisco JA (2004) Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 10:7063–7070
Herbertson RA, Tebbutt NC, Lee FT, MacFarlane DJ, Chappell B, Micallef N, Lee ST, Saunder T, Hopkins W, Smyth FE, Wyld DK, Bellen J, Sonnichsen DS, Brechbiel MW, Murone C, Scott AM (2009) Phase I biodistribution and pharmacokinetic study of Lewis Y-targeting immunoconjugate CMD –193 in patients with advanced epithelial cancers. Clin Cancer Res 15:6709–6715
Jeffrey SC, Burke PJ, Lyon RP, Meyer DW, Sussman D, Anderson M, Hunter JH, Leiske CI, Miyamoto JB, Nicholas ND, Okeley NM, Sanderson RJ, Stone IJ, Zeng W, Gregson SJ, Masterson L, Tiberghien AC, Howard PW, Thurston DE, Law CL, Senter PD (2013) A potent anti-CD70 antibody-drug conjugate combining a dimericpyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjug Chem 24:1256–1263
Jumbe NL, Xin Y, Leipold DD, Crocker L, Dugger D, Mai E, Sliwkowski MX, Fielder PJ, Tibbitts J (2010) Modeling the efficacy of trastuzumab-DM1, an antibody drug conjugate, in mice. J Pharmacokinet Pharmacodyn 37:221–242
Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, Gorrell J, Katta V, Kim A, Mc Dorman K, Flagella K, Venook R, Ross S, Spencer SD, Lee Wong W, Lowman HB, Vandlen R, Sliwkowski MX, Scheller RH, Polakis P, Mallet W (2008) Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 26:925–932
Lin K, Tibbitts J (2012) Pharmacokinetic considerations for antibody drug conjugates. Pharm Res 29:2354–2366
Kantarjian H, Thomas D, Jorgensen J, Jabbour E, Kebriaei P, Rytting M, York S, Ravandi F, Kwari M, Faderl S, Rios MB, Cortes J, Fayad L, Tarnai R, Wang SA, Champlin R, Advani A, O'Brien S (2012) Inotuzumabozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol 13:403–411
Kaur S, Xu K, Saad OM, Dere RC, Carrasco-Triguero M (2013) Bioanalytical assay strategies for the development of antibody-drug conjugate biotherapeutics. Bioanalysis 5:201–226
Ducry L, Stump B (2010) Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem 21:5–13
Gibiansky L, Gibiansky E (2014) Target-mediated drug disposition model and its approximations for antibody-drug conjugates. J Pharmacokinet Pharmacodyn 41:35–47
Lewis Phillips GD Li G Dugger DL Crocker LM Parsons KL Mai E Blattler WA Lambert JM Chari RV Lutz RJ Wong WL Jacobson FS Koeppen H Schwall RH Kenkare-Mitra SR Spencer SD Sliwkowski MX (2008) Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res 68:9280–9290
Lin JH, Lu AY (1997) Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol Rev 49:403–449
Lobo ED, Hansen RJ, Balthasar JP (2004) Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 93:2645–2668
Lovdal T, Andersen E, Brech A, Berg T (2000) Fc receptor mediated endocytosis of small soluble immunoglobulin G immune complexes in Kupffer and endothelial cells from rat liver. J Cell Sci 113(Pt 18):3255–3266
Lu D, Sahasranaman S, Zhang Y, Girish S (2013) Strategies to address drug interaction potential for antibody-drug conjugates in clinical development. Bioanalysis 5:1115–1130
Lyon RP, Meyer DL, Setter JR, Senter PD (2012) Conjugation of anticancer drugs through endogenous monoclonal antibody cysteine residues. Methods Enzymol 502:123–138
Mc Donagh CF, Kim KM, Turcott E, Brown LL, Westendorf L, Feist T, Sussman D, Stone I, Anderson M, Miyamoto J, Lyon R, Alley SC, Gerber HP, Carter PJ (2008) Engineered anti-CD70 antibody-drug conjugate with increased therapeutic index. Mol Cancer Ther 7:2913–2923
Morgan P, Van Der Graaf PH, Arrowsmith J, Feltner DE, Drummond KS, Wegner CD, Street SD (2012) Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today 17:419–424
Mould DR, Green B (2010) Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. Bio Drugs 24:23–39
Mullard A (2013) Maturing antibody-drug conjugate pipeline hits 30. Nat Rev Drug Discov 12:329–332
Nolting B (2013) Linker technologies for antibody-drug conjugates. Methods Mol Biol 1045:71–100
Okeley NM, Miyamoto JB, Zhang X, Sanderson RJ, Benjamin DR, Sievers EL, Senter PD, Alley SC (2010) Intracellular activation of SGN –35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res 16:888–897
Pastuskovas CV, Maruoka EM, Shen BQ, Koeppen H, Doronina SO, Senter PD, Zioncheck TF (2005) Tissue distribution, metabolism, and excretion of the antibody-drug conjugate Herceptin-monomethylauristatin E in rats. AACR Meeting Abstracts. 2005:1195-d-1196
Singh R, Erickson HK (2009) Antibody-cytotoxic agent conjugates: preparation and characterization. Methods Mol Biol 525:445–467, xiv
Sanderson RJ, Hering MA, James SF, Sun MM, Doronina SO, Siadak AW, Senter PD, Wahl AF (2005) In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatinimmunoconjugate. Clin Cancer Res 11:843–852
Sapra P, Stein R, Pickett J, Qu Z, Govindan SV, Cardillo TM, Hansen HJ, Horak ID, Griffiths GL, Goldenberg DM (2005) Anti-CD74 antibody-doxorubicin conjugate, IMMU –110, in a human multiple myeloma xenograft and in monkeys. Clin Cancer Res 11:5257–5264
Scott AM, Tebbutt N, Lee FT, Cavicchiolo T, Liu Z, Gill S, Poon AM, Hopkins W, Smyth FE, Murone C, Mac Gregor D, Papenfuss AT, Chappell B, Saunder TH, Brechbiel MW, Davis ID, Murphy R, Chong G, Hoffman EW, Old LJ (2007) A phase I biodistribution and pharmacokinetic trial of humanized monoclonal antibody Hu3s193 in patients with advanced epithelial cancers that express the Lewis-Y antigen. Clin Cancer Res 13:3286–3292
Shah DK, Haddish-Berhane N, Betts A (2012) Bench to bedside translation of antibody drug conjugates using a multiscale mechanistic PK/PD model: a case study with brentuximab-vedotin. J Pharmacokinet Pharmacodyn 39:643–659
Shah DK, Betts AM (2013) Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs 5:297–305
Sharkey RM, Karacay H, Govindan SV, Goldenberg DM (2011) Combination radioimmunotherapy and chemoimmunotherapy involving different or the same targets improves therapy of human pancreatic carcinoma xenograft models. Mol Cancer Ther 10:1072–1081
Sharkey RM, Govindan SV, Cardillo TM, Goldenberg DM (2012) Epratuzumab-SN –38: a new antibody-drug conjugate for the therapy of hematologic malignancies. Mol Cancer Ther 11:224–234
Shen BQ, Bumbaca D, Saad O, Yue Q, Pastuskovas CV, Khojasteh SC, Tibbitts J, Kaur S, Wang B, Chu YW, Lorusso PM, Girish S (2012a) Catabolic fate and pharmacokinetic characterization of trastuzumabemtansine (T-DM1): an emphasis on preclinical and clinical catabolism. Curr Drug Metab 13:901–910
Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR (2012b) Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 30:184–189
Sievers EL, Senter PD (2013) Antibody-drug conjugates in cancer therapy. Ann Rev Med 64:15–29
Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C (1997) Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res 3:81–85
Stephan JP, Chan P, Lee C, Nelson C, Elliott JM, Bechtel C, Raab H, Xie D, Akutagawa J, Baudys J, Saad O, Prabhu S, Wong WL, Vandlen R, Jacobson F, Ebens A (2008) Anti-CD22-MCC-DM1 and MC-MMAF conjugates: impact of assay format on pharmacokinetic parameters determination. Bioconjug Chem 19:1673–1683
Sun X, Widdison W, Mayo M, Wilhelm S, Leece B, Chari R, Singh R, Erickson H (2011) Design of antibody-maytansinoid conjugates allows for efficient detoxification via liver metabolism. Bioconjug Chem 22:728–735
Sussman D, Torrey L, Westendorf L, Zhang X, Okeley NM, Alley SC, Lyon R, Meyer D, Miyamoto JB, Benjam DR (2011) Engineered cysteine antibodies: Improved antibody-drug conjugate vehicles. In: Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, 2011
Sutherland MS, Sanderson RJ, Gordon KA, Andreyka J, Cerveny CG, Yu C, Lewis TS, Meyer DL, Zabinski RF, Doronina SO, Senter PD, Law CL, Wahl AF (2006) Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J Biol Chem 281:10540–10547
Tabrizi M, Bornstein GG, Suria H (2010) Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J 12:33–43
Tabrizi MA, Tseng CM, Roskos LK (2006) Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today 11:81–88
Tian F, Lu Y, Manibusan A, Sellers A, Tran H, Sun Y, Phuong T, Barnett R, Hehli B, Song F, De Guzman MJ, Ensari S, Pinkstaff JK, Sullivan LM, Biroc SL, Cho H, Schultz PG, Di Joseph J, Dougher M, Ma D, Dushin R, Leal M, Tchistiakova L, Feyfant E, Gerber HP, Sapra P (2014) A general approach to site-specific antibody drug conjugates. Proc Natl Acad Sci U S A 111:1766–1771
Tijink BM, Buter J, de Bree R, Giaccone G, Lang MS, Staab A, Leemans CR, van Dongen GA (2006) A phase I dose escalation study with anti-CD44v6 bivatuzumabmertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin Cancer Res 12:6064–6072
Tolcher AW, Ochoa L, Hammond LA, Patnaik A, Edwards T, Takimoto C, Smith L, de Bono J, Schwartz G, Mays T, Jonak ZL, Johnson R, DeWitte M, Martino H, Audette C, Maes K, Chari RV, Lambert JM, Rowinsky EK (2003) Cantuzumabmertansine, a maytansinoidimmunoconjugate directed to the CanAg antigen: a phase I, pharmacokinetic, and biologic correlative study. J Clin Oncol 21:211–222
Waldmann TA (2003) Immunotherapy: past, present and future. Nat Med 9:269–277
Wang L, Amphlett G, Blattler WA, Lambert JM, Zhang W (2005) Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci 14:2436–2446
Wang W, Wang EQ, Balthasar JP (2008) Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 84:548–558
Xie H, Audette C, Hoffee M, Lambert JM, Blattler WA (2004) Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumabmertansine (huC242-DM1), and its two components in mice. J Pharmacol Exp Ther 308:1073–1082
Xu K, Liu L, Saad OM, Baudys J, Williams L, Leipold D, Shen B, Raab H, Junutula JR, Kim A, Kaur S (2011) Characterization of intact antibody-drug conjugates from plasma/serum in vivo by affinity capture capillary liquid chromatography-mass spectrometry. Anal Biochem 412:56–66
Xu K, Liu L, Dere R, Mai E, Erickson R, Hendricks A, Lin K, Junutula JR, Kaur S (2013) Characterization of the drug-to-antibody ratio distribution for antibody-drug conjugates in plasma/serum. Bioanalysis 5:1057–1071
Zhao L, Ji P, Li Z, Roy P, Sahajwalla CG (2013) The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model. J Clin Pharmacol 53:314–325
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
Sukumaran, S., Lin., K. (2015). Pharmacokinetics/Pharmacodynamics and Disposition of Antibody-Drug Conjugates. In: Wang, J., Shen, WC., Zaro, J. (eds) Antibody-Drug Conjugates. AAPS Advances in the Pharmaceutical Sciences Series, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-319-13081-1_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-13081-1_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-13080-4
Online ISBN: 978-3-319-13081-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)