
Abstract
This article describes a split-marker approach, using two DNA fragments overlapping within the dominant selectable marker in fungi. This approach has been shown to increase homologous integration and thereby, facilitate targeted gene disruption in many fungi. Because the selectable marker gene is truncated in different fragments, the gene is not functional until homologous recombination takes place between two overlapping fragments. The truncated marker gene fragments flanking by homologous sequences of the target gene can be produced by two-round PCR without the need for cloning. The described method allows a faster and more efficient way of generating disruption strains and shall facilitate functional genomic analysis in filamentous fungi.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Genetic transformation and targeted gene disruption are essential for studying and understanding gene function in filamentous fungi. Targeted gene disruption in filamentous fungi can be troublesome because of low frequencies of homologous integration (Bird and Bradshaw 1997; Chung et al. 1999; Pratt and Aramayo 2002; Segers et al. 2001; Idnurm et al. 2003). In addition, integration of foreign genes in filamentous fungi often occurs via non-homologous integration, resulting in a large number of false-positive transformants. The frequency of targeted gene disruption can be improved by a split-marker disruption strategy by fusing the target DNA fragments with truncated but overlapping within the selectable marker gene (Fairhead et al. 1996; Fu et al. 2006). Only transformants harboring a functional dominant marker gene will grow on a medium containing the selection agent. Split-marker-based transformation decreases the occurrence of multiple and tandem integrations so as to decrease the overall numbers of transformants being screened. Split-marker approach has been shown to increase the frequency of targeted gene disruption and homologous integration as high as 100 % in Alternaria alternata and Cercospora spp. (Choquer et al. 2005; You et al. 2009; Lin and Chung 2010). Two truncated, overlapping marker gene fragments are joined with a gene of interest by fusion PCR without the need for cloning and the PCR products used directly for transformation. The methods described in this article could provide better tools to analyze gene functions in filamentous fungi.
2 Materials and Methods
2.1 Solution and Medium
2.1.1 Wash Solution
Dissolve 58.4 g NaCl and 1.47 g CaCl2 · H2O in water, bring the final volume to 1 L, and sterilize using an autoclave.
2.1.2 STC Solution
Dissolve 218.6 g sorbitol in 980 mL water, add 10 mL each of 1 M Tris–HCl (pH 7.5) and 1 M CaCl2 · H2O, and sterilize.
2.1.3 50 % PEG Solution
Dissolve 50 g polyethylene glycol (M.W. 3,350) in a pre-heated water (98 mL), add 1 mL each of 1 M Tris–HCl (pH 7.5) and 1 M CaCl2 · H2O, and filter sterilize. Discard after 4 months.
2.1.4 Enzyme Solution
Mix 0.2 mL β-glucuronidase (type H2, Sigma), 0.16 g β-d-glucanase, and 6 mg lyticase with 0.5 mL of 0.4 M Na2PO4 (pH 5.8), 0.4 mL of 1 M CaCl2 · H2O, and 1.4 g NaCl in water (final volume 20 mL), filter sterilize, and store at −20 °C (Chung et al. 2002).
2.1.5 Solution A
Dissolve 10 g Ca(NO3) · 4H2O in 100 mL water.
2.1.6 Solution B
Dissolve 2 g KH2PO4, 2.5 g MgSO4 · 7H2O and 1.5 g NaCl in 100 mL water (pH 5.3).
2.1.7 Regeneration Medium
Regeneration medium (RM) is prepared in both liquid and solid forms. In a 1-L bottle, add 10 mL each of Solution A and Solution B to 480 mL water. Add 15 g agar per liter for solid medium. In a 2-L bottle, dissolve 342.3 g sucrose and 10 g glucose in water (final volume 500 mL). Sterilize two solutions separately, mix, and dispense into sterile bottles.
2.2 Growth Conditions
Start culture by grinding fungal mycelium with 0.5 mL sterile water in a 1.5-mL centrifuge tube using a disposable mini pestle (Fisher Scientific) and adding the resulting suspension to 50 mL medium (potato dextrose broth or a synthetic medium). Incubate fungal culture on a rotary shaker at room temperature (ca. 25 °C) for 3–4 days. Blend the culture in a sterile blender cup (Fisher Scientific) for three or four 10 s pulses, add to 200 mL fresh medium, and incubate on shaker for an additional 16–18 h. Harvest fungal mycelium by low-speed centrifugation at 6,000 rpm for 10 min in an Allegra 21R centrifuge (Beckman Culter). Carefully remove supernatant using a disposable polyethylene transfer pipet. Resuspend fungal mycelium in 10 mL of wash solution, spin again, and discard supernatant.
2.3 Preparation of Protoplasts
Successful transformation of fungi requires competent protoplasts. Resuspend fungal mycelium in 20 mL of enzyme solution by pipetting up and down with a disposable polyethylene pipet and transfer to a 100-mL flask. Incubate the resulting suspension at 30 °C on a rotary shaker set at 100 rpm. Check protoplast release under microscope regularly. After 2 h digestion, passage the solution through Miracloth. Harvest protoplasts by low-speed centrifugation at 4,000 rpm in a F0850 Beckman rotor at 4 °C for 5 min. Discard supernatant. Wash protoplasts twice with 10 mL of STC solution. Collect protoplasts by centrifugation between washes. Discard supernatant. Gently resuspend protoplasts in 1 mL of STC and check concentration with a hemacytometer. Adjust the concentration to 107 protoplasts per mL in four parts of STC and one part of 50 % PEG solution (polyethylene glycol 3,350). Dispense protoplasts into a small volume (100 μL) and store them at −80 °C.
2.4 Generation of Split-Marker Fragments
The split-marker gene fragments flanked with a gene of interest are constructed using a fusing PCR approach (Fig. 15.1). This method completely eliminates tedious cloning procedures and allows quick generation of split-marker fragments for targeted gene disruption. Two truncated but overlapping gene fragments (WY/ and /YZ) are first amplified from a plasmid containing a suitable gene cassette. A bacterial phosphotransferase gene conferring resistance to hygromycin is often used a dominant selectable marker in filamentous fungi. Primers S1 and S2 are designed to amplify the WY/ fragment; primers S3 and S4 are used to amplify the /YZ fragment using a GoTag DNA polymerase (Promega) in a 50-μL solution using a standard PCR protocol. Two DNA fragments (0.5–1.5 kb) of a gene of interest are amplified separately by PCR with two gene-specific primers from fungal genomic DNA. The length of homologous sequences can be varied depending on the desired extent of deletion of target gene sequences. Using longer homologous sequences may increase the efficiency of homologous integration.

Schematic illustration of fusion PCR for generating overlapping truncation of a dominant selectable marker gene (WYZ) fused with homologous sequence of a gene of interest. PCR is used to amplify two overlapping fragments WY/ and /YZ with the primers S1 pairing with S2 and S3 pairing with S4, respectively. Primer P2 contains a tail sequence completely complementary to the sequence of S1 and primer P3 contains partial sequence completely complementary to the sequence of S4. The 5’ truncation of the target gene is amplified with the primers P1 and P2 and joined to the WY/ fragment. The 3’ truncation of the target gene is amplified with the primers P3 and P4 and joined to the /YZ fragment
As illustrated in Fig. 15.1, primers P1 and P2 are designed to amplify the 5’ region of the target gene; primers P3 and P4 are used to amplify the 3’ region of the gene. The tail sequence of the P2 primer is designed to be completely complementary to the sequence of S1 and the tail sequence of P3 is completely complementary to the sequence of S4. This is designed so that the marker gene fragment (WY/) is fused with the 5’ truncation of a gene of interest with the primers P1 and S2 and the /YZ fragment fused with the 3’ truncation with the primers P4 and S3 to form two chimeric DNA fragments. Note: It is not necessary to clean up PCR fragments prior to second-round amplification. The cycling profile for PCR amplification begins with a cycle of 95 °C for 3 min, immediately followed by 30 cycles of 95 °C for 30 s, 56 °C for 30 s, 72 °C for 1.5–3 min and completed by incubating at 72 °C for 10 min.
2.5 Transformation
PCR-generated DNA fragments are directly transformed into fungal protoplasts without any additional cleanup. Transformation of fungal protoplasts was performed using CaCl2 and polyethylene glycol (Chung et al. 2002). Protoplasts frozen at −80 °C in 100 μL of STC: 50 % PEG (4:1, v/v) are placed in ice for at least 10 min. Mix split-marker DNA fragments (10 μL each) with 100 μL protoplasts in a sterile 15-mL centrifuge tube (Falcon). Leave at room temperature for 20 min. Add 1 mL of 50 % PEG gradually into the centrifuge tube, mix gently, and leave at room temperature for an additional 20 min. Add 3 mL liquid RM and place on a shaker set at 100 rpm at room temperature for 2–4 h. Add a selectable agent into each tube except the no selection control. Mix gently with molten solid RM (45 °C), pour into petri dish, and swirl gently. The plates are incubated at 28 °C. Examine daily for colony formation. Pick colonies and transfer to fresh medium. Successful disruption of a given gene in a wild-type strain can be identified quickly if the mutant strain shows any phenotypes, otherwise PCR verification is needed to confirm the disruption.
2.6 Homologous Integration
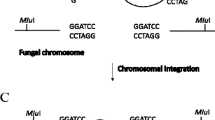
Because the dominant marker gene is split in separate fragments, the gene is not functional unless homologous recombination occurs between two overlapping fragments (Fig. 15.2). Fungal transformants will not grow on a medium containing the selection agent unless homologous recombination occurs between the overlapping regions of the dominant marker gene. The marker gene cassette fused with homologous flanking sequences is integrated into the target locus via double cross over recombination (Fig. 15.2). Successful integration of a marker gene fragment within a gene of interest can be validated by analytical PCR with the primers located just outside the targeted region (e.g., primers P5 and P6 in Fig. 15.1) and by Southern blot hybridization of fungal genomic DNA, digested with various endonucleases, to a gene-specific probe. For a given gene, six oligonucleotide primers are needed for generation of split-marker fragments and for verification of locus-specific integration.

Schematic illustration of targeted gene disruption by split-marker-based transformation. Two truncated, overlapping marker gene fragments flanked with the truncation of a target gene are directly transformed into protoplasts prepared from a wild-type fungal strain. Three cross-over events are required to generate functional marker gene and homologous integration. Only transformants containing a functional marker gene cassette will grow on a medium containing the selection agent. Employing split-marker-based gene disruption could enhance homologous recombination
Transformation of split-marker fragments in phytopathogenic fungi, Cercospora nicotianae, C. beticola, Elsinoë fawcettii, Colletotrichum acutatum, and A. alternata, has been shown to increase homologous integration frequency (You et al. 2007, 2009; Chen et al. 2007; Liao and Chung 2008; Weiland et al. 2010; Lin et al. 2010; Yang and Chung 2013). The split-marker approach could be useful for identifying disruptants with no obvious phenotypes because a high frequency of targeted gene disruption via homologous recombination can be achieved by screening less than 20–30 independent transformants. The target gene can be disrupted or completely replaced by the marker gene fragment, depending on the homologous DNA sequence within the gene of interest. Because the flanking fragments are generated by PCR, the deleted region can be precisely determined. The minimum flanking sequence required for efficient homologous integration varies among fungal species. However, we have observed that disruption frequency increases as the lengths of the flanking sequence on one end or both ends of the target gene increases (You et al. 2009). It is critical to have sufficient lengths of the flanking DNA sequence (>0.5 kb) when employing the split-marker approach for targeted gene disruption. The minimum overlapping sequence required for efficient recombination at the selectable marker gene remains uncertain. However, fungal disruptants have been successfully identified using two DNA fragments overlapping 200–450 bp at the selectable marker gene (Yang and Chung 2013).
3 Conclusion
Targeted gene disruption via homologous recombination has had a major impact on modern fungal biology. This split-marker-based transformation approach increases the frequency of recovering disruptants, presumably by increasing the frequency of homologous integration and/or by decreasing ectopic and tandem integration events in fungi. This approach was originally developed for rapid, gap repaired-mediated cloning in the budding yeast Saccharomyces cerevisiae (Fairhead et al. 1996). The split-marker fragments flanking by homologous sequences of target gene can be obtained by fusion PCR without the need for cloning, allowing a faster and more efficient method of generating disruption constructs. Efficient gene disruption strategies along with the other molecular techniques shall facilitate functional genomic analysis in filamentous fungi.
References
Bird D, Bradshaw R (1997) Gene targeting is locus dependent in filamentous fungus Aspergillus nidulans. Mol Gen Genet 255:219–225
Chen H, Lee MH, Daub ME, Chung KR (2007) Molecular analysis of the cercosporin biosynthetic gene cluster in Cercospora nicotianae. Mol Microbiol 64:755–770
Choquer M, Dekkers K, Ueng PP, Daub ME, Chung KR (2005) The CTB1 gene encoding a fungal polyketide synthase is required for cercosporin biosynthesis and fungal virulence of Cercospora nicotianae. Mol Plant Microbe Interact 18:468–476
Chung KR, Jenns AE, Ehrenshaft M, Daub ME (1999) A novel gene required for cercosporin toxin resistance in the fungus, Cercospora nicotianae. Mol Gen Genet 262:382–389
Chung KR, Shilts T, Li W, Timmer LW (2002) Engineering a genetic transformation system for Colletotrichum acutatum, the causal fungus of lime anthracnose and postbloom fruit drop. FEMS Microbiol Lett 213:33–39
Fairhead C, Llorente B, Denis F, Soler M, Dujon B (1996) New vectors for combinatorial deletions in yeast chromosomes and for gap-repair cloning using “split-marker” recombination. Yeast 12:1439–1457
Fu J, Hettler E, Wickes BL (2006) Split marker transformation increases homologous integration frequency in Cryptococcus neoformans. Fungal Genet Biol 43:200–212
Idnurm A, Warnecke DC, Heinz E, Howlett BJ (2003) Characterisation of neutral trehalase and UDP-glucose:sterol glucosyltransferase genes from the plant pathogenic fungus Leptosphaeria maculans. Physiol Mol Plant Pathol 62:305–313
Liao HL, Chung KR (2008) Genetic dissection defines the roles of elsinochrome phytotoxin for fungal pathogenesis and conidiation of the citrus pathogen Elsinoë fawcettii. Mol Plant Microbe Interact 21:469–479
Lin CH, Chung KR (2010) Specialized and shared functions of the histidine kinase—and HOG1 MAP kinase–mediated signaling pathways in Alternaria alternata, the filamentous fungal pathogen of citrus. Fungal Genet Biol 47:818–827
Lin CH, Yang SL, Wang N, Chung KR (2010) The FUS3 MAPK signaling pathway of the citrus pathogen Alternaria alternata acts independently and cooperatively with the fungal redox-responsive AP1 regulator for diverse developmental, physiological and pathogenic functions. Fungal Genet Biol 47:381–391
Pratt R, Aramayo R (2002) Improving the efficiency of gene replacements in Neurospora crassa: a first step towards a large-scale functional genomics project. Fungal Genet Biol 37:56–71
Segers GC, Bradshaw N, Archer D, Blissett K, Oliver RP (2001) Alcohol oxidase is a novel pathogenicity factor for Cladosporium fulvum but aldehyde dehydrogenase is dispensable. Mol Plant Microbe Interact 14:367–377
Weiland JJ, Chung KR, Suttle JC (2010) The role of cercosporin in the virulence of Cercospora spp. to plant hosts. In: Lartey RT, Weiland JJ, Panella L, Crous PW, Windels CE (eds) Cercospora leaf spot of sugar beet and related species. APS Press, St. Paul, pp 109–117
Yang SL, Chung KR (2013) Similar and distinct roles of NADPH oxidases components in the tangerine pathotype of Alternaria alternata. Mol Plant Pathol 14:543–556
You BJ, Choquer M, Chung KR (2007) The Colletotrichum acutatum gene encoding a putative pH-responsive transcription regulator is a key virulence determinant during fungal pathogenesis on citrus. Mol Plant Microbe Interact 20:1149–1160
You BJ, Lee MH, Chung KR (2009) Gene-specific disruption in the filamentous fungus Cercospora nicotianae using a split-marker approach. Arch Microbiol 191:615–622
Acknowledgments
The author would like to thank current and former Chung lab members S.L. Yang, L.H. Chen, H.C. Tsai, C.H. Lin, and B.J. You for their contributions to this work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Chung, KR., Lee, MH. (2015). Split-Marker-Mediated Transformation and Targeted Gene Disruption in Filamentous Fungi. In: van den Berg, M., Maruthachalam, K. (eds) Genetic Transformation Systems in Fungi, Volume 2. Fungal Biology. Springer, Cham. https://doi.org/10.1007/978-3-319-10503-1_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-10503-1_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-10502-4
Online ISBN: 978-3-319-10503-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)