
Abstract
The prevalence of type 2 diabetes mellitus and of the metabolic syndrome is rising worldwide and reaching epidemic proportions. These pathologies are associated with significant morbidity and mortality, in particular with an excess of cardiovascular deaths. Type 2 diabetes mellitus and the cluster of pathologies including insulin resistance, central obesity, high blood pressure, and hypertriglyceridemia that constitute the metabolic syndrome are associated with low levels of HDL cholesterol and the presence of dysfunctional HDLs. We here review the epidemiological evidence and the potential underlying mechanisms of this association. We first discuss the well-established association of type 2 diabetes mellitus and insulin resistance with alterations of lipid metabolism and how these alterations may lead to low levels of HDL cholesterol and the occurrence of dysfunctional HDLs. We then present and discuss the evidence showing that HDL modulates insulin sensitivity, insulin-independent glucose uptake, insulin secretion, and beta cell survival. A dysfunction in these actions could play a direct role in the pathogenesis of type 2 diabetes mellitus.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Type 2 Diabetes Mellitus and the Metabolic Syndrome
Diabetes mellitus (DM) is characterized by increased fasting and/or postprandial glucose levels. The disease had been known for many centuries (Laios et al. 2012). Its etiologyFootnote 1 however started to be unraveled in the late nineteenth century/early twentieth century. During this period, it was eventually discovered that a compound in pancreatic extracts (later identified to be insulin) could cure young diabetic patients (Banting et al. 1922). From the early work of H. P. Himsworth in 1936, a concept emerged that diabetes could be divided into what he termed “insulin sensitive” and “insulin insensitive” [the pioneering work of Himsworth is nicely portrayed in a historical perspective written by Gerald Reaven (Reaven 2005)]. But it is only once a reliable radioimmunoassay became available to measure circulating insulin levels (Bray 1996) that it was realized that some patients suffered from a disease associated with low to undetectable insulin levels and prone to ketoacidosis (type 1 DM [T1DM]), while others, the vast majority, had high circulating insulin levels despite increased glucose levels (type 2 DM [T2DM]) (Inzucchi 2012). Patients with T2DM are often overweight and obese, and their diabetes develops later in life. These patients are characterized as having insulin resistance (IR), i.e., a decreased response of insulin-sensitive tissues to the effects of the hormone, in particular its ability to induce glucose uptake. Insulin resistance is usually associated with compensatory hyperinsulinemia in an attempt to maintain normoglycemia. Insulin resistance is a hallmark of T2DM as well as of overweight and obesity. Insulin resistance precedes the development of T2DM, and it is once that the beta cell can no longer compensate to the insulin resistance that the full-blown disease with hyperglycemia develops (Tabak et al. 2009). Gerald Reaven, in his seminal Banting Lecture in 1988 (Reaven 1988), summarized his work that put insulin resistance not only at the center of diabetes research but proposed that insulin resistance may directly contribute to the increased risk of coronary artery disease associated with T2DM or obesity. In this chapter, he suggested that IR and hyperinsulinemia were, together with increased VLDL/triglyceride levels, low HDL cholesterol levels, and hypertension, directly associated with and responsible for the development of diabetes. He termed “syndromeFootnote 2 X” the clustering of these features. Syndrome X was also named “insulin resistance syndrome” but is now generally called “metabolic syndrome.” This syndrome corresponds to a cluster of increased abdominal obesity (a good proxy of insulin resistance), increased blood glucose levels, high blood pressure and elevated triglycerides, and low HDL cholesterol levels. Several definitions of the metabolic syndrome have been proposed with the National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP III) definition being the one most widely used. The World Health Organization (WHO) and the International Diabetes Federation (IDF) have suggested other definitions. Table 1 summarizes these various definitions. These current definitions, albeit arbitrary in the cutoffs that were chosen for the different variables included, are intended to provide a useful tool for clinicians to identify individuals at increased risk for the development of T2DM, atherosclerotic cardiovascular disease, and cardiovascular death. T2DM and the metabolic syndrome are characterized not only by insulin resistance but also by a dyslipidemia that includes high triglyceride and low HDL cholesterol levels. With the presence of small dense LDLs in T2DM, these lipid abnormalities substantially contribute to the increased cardiovascular morbidity and mortality associated with these conditions (Grundy 2012). Several epidemiological studies have clearly shown that these lipid abnormalities are present before the development of T2DM. Furthermore, longitudinal studies have provided evidence that these abnormalities represent an independent risk factor for T2DM (von Eckardstein et al. 2000). Hence, the concept has emerged that lipid abnormalities associated with IR and the metabolic syndrome are not only bystanders of the metabolic dysfunctions but contribute directly to the progression to type DM.
2 Low HDL Cholesterol Levels in Diabetes and Metabolic Syndrome
T2DM and the metabolic syndrome are characterized by the presence of an atherogenic dyslipidemia typically including low HDL cholesterol, hypertriglyceridemia, and increased small dense LDL blood concentrations. Individuals with low HDL cholesterol are often insulin resistant and at increased risk of developing T2DM (Li et al. 2014; von Eckardstein and Sibler 2011). T2DM, the metabolic syndrome, and IR are characterized by hyperinsulinemia and increased free fatty acids (FFA). Both insulin resistance and FFAs appear to contribute to the pathogenesis associated with decreased HDL levels (Rohrer et al. 2004).
2.1 Insulin Resistance, Hypertriglyceridemia, and Decreased HDL Cholesterol Levels
Insulin has pleiotropic effects on lipid metabolism, in particular on the metabolism of HDLs and the TG-rich VLDLs. In rats, insulin can acutely decrease the hepatic production of ApoB100 (Chirieac et al. 2000), the apolipoprotein required for VLDL assembly, by favoring ApoB100 degradation. In the Hep2G liver cell line, insulin also decreases the levels of microsomal TG transfer protein that is essential for VLDL synthesis and secretion (Lin et al. 1995). Insulin could therefore negatively affect the hepatic production of VLDL. However, the situation is more complex than this because there are two VLDL subtypes that are differentially regulated by insulin, the larger and more buoyant VLDL1 particles and the smaller and denser VLDL2 particles. VLDL2 particles are intermediates in the synthesis of VLDL1 in the liver, but they can also be secreted directly by this organ. Additionally, VLDL2 can be generated in the circulation from VLDL1 through lipoprotein lipase activity (Taskinen 2003). Insulin does not appear to affect the hepatic production of VLDL2 (at least not in humans, see below) but seems to only inhibit hepatic VLDL1 production by inhibiting the conversion of VLDL2 into VLDL1 (Taskinen 2003). VLDL1 particles are seen by some authors as “liver-derived” chylomicron-like particles (Taskinen 2003). It makes sense therefore that insulin inhibits their production following meals in a situation where chylomicrons are produced by the intestine. In contrast, VLDL1 particle production could be favored in the fasting state. While insulin inhibits VLDL1 production, it favors the hepatic production of VLDL2 particles because insulin can increase the activity of the sterol regulatory element-binding protein (SREBP) 1c transcription factor that induces the expression of enzymes required for fatty acid and TG synthesis, such as fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC) (Horton et al. 2002). However, insulin was not found to modulate VLDL2 blood levels in humans (Malmstrom et al. 1997a, b). The physiological importance of the regulation of fatty acid and TG synthesis by insulin on VLDL production is therefore unclear. In type 2 diabetic individuals (i.e., in the presence of an insulin-resistant state), insulin is no longer able to inhibit VLDL1 production (Malmstrom et al. 1997a), and this can thus favor overall VLDL hepatic secretion. This presumably contributes to the hypertriglyceridemia seen in type 2 diabetic and metabolic syndrome patients (Brown and Goldstein 2008). In addition, hyperinsulinemia resulting from insulin resistance can enhance SREBP1c transcription and thereby microRNA33b (miR33b) that is encoded by an intron of the SREBP1c gene. Both SREBP1c and miR33b can drive VLDL production and favor hypertriglyceridemia by stimulating fatty acid and triglyceride synthesis (Rayner et al. 2011).
A high production of VLDLs can decrease the stability of HDL particles by increasing the transfer of TG from TG-rich particles to HDLs via the activity of the cholesteryl ester transfer protein (CETP) enzyme (Rashid et al. 2003). This is one possible mechanism linking high TG blood levels with decreased HDL cholesterol levels. Insulin participates in the clearance of TG from TG-rich lipoproteins by stimulating the secretion of lipoprotein lipase (LPL) in certain adipose tissues (e.g., subcutaneous depot) (Fried et al. 1993). Consequently, insulin resistance would lead to increased circulating TG-rich particles and augmented TG loading of HDLs. Additionally, reduced TG hydrolysis in VLDLs and chylomicrons will decrease the release of surface remnants which contribute to the maturation of HDL (Eisenberg 1984; Rashid et al. 2003). These two effects combined—decreased surface remnant production and enhanced CETP activity—favor the reduction in HDL cholesterol levels via the mechanism mentioned above (i.e., decreased HDL stability due TG enrichment of the particle).
Insulin has the potential to directly affect HDL production by the liver, either in a positive or in a negative manner. In the Hep2G liver cell line, insulin can stimulate the expression of apolipoprotein A-I (ApoA-I) (Murao et al. 1998), the most abundant apolipoprotein in HDLs (Eisenberg 1984; Rye and Barter 2012), raising the possibility that hepatic insulin resistance contributes to decreased production of HDLs. The relevance of this effect has however not been validated in vivo. In contrast, insulin may also negatively impact on hepatic HDL production by enhancing miR33 transcription. Indeed, miR33 suppresses the production of the adenosine triphosphate-binding cassette transporter A1 (ABCA1) at the posttranscriptional level and thereby compromises phospholipid and cholesterol efflux needed for HDL formation (Besler et al. 2012; Rader 2006; Rayner et al. 2011; Tang and Oram 2009).
2.2 FFAs and Decreased HDL Levels
In vitro, FFAs decrease the expression of SREBP1c by inhibiting the transcriptional activity of liver X receptor (LXR) (Ou et al. 2001; Yoshikawa et al. 2002). This represents a negative feedback loop ensuring that the production of VLDLs occurs when there is a need to deliver TGs to peripheral tissues (muscles, adipose tissue, etc.). Reduction in LXR activity, and consequently VLDL production, would be expected to lead to increased HDL cholesterol blood levels as a result of increased HDL stability. However, in the liver, unsaturated FFAs inhibit the expression of ABCA1 (Uehara et al. 2002). Moreover, free fatty acids stimulate the proteasomal degradation of ABCA1 (Wang and Oram 2005, 2007). So even though FFAs can lower the production of hepatic triglycerides, which will favor HDL stability, FFAs, at the same time, directly inhibit the generation of HDL particles.
2.3 HDL Metabolism, Subclasses, and Catabolism
Nascent HDLs are the precursors of the mature HDL3 and HDL2 particles. They are generated from lipid-free ApoA-I that acquires phospholipids and cholesterol extracellularly. ABCA1-mediated lipid efflux from cells is important for this initial lipidation. Further acquisition of lipids and subsequent LCAT-driven cholesterol esterification leads to the formation of mature HDL3 particles. These particles can absorb more phospholipids and free cholesterol by a mechanism involving ABCG1. Altogether, this transforms the HDL3 particles into larger and less dense HDL2 particles. Like ABCA1, this transporter is regulated by LXR and hence suppressed by unsaturated fatty acids at the transcriptional level (Uehara et al. 2007) so that the production of larger HDL2 is compromised. In addition, the conversion of HDL2 into HDL3 is enhanced in diabetic dyslipidemia by increased activities of CETP and hepatic lipase: First, the transfer rate of cholesteryl esters from HDL to VLDL in normal individuals depends on the concentration of CE acceptors, that is, VLDL particles, but not on CETP activity (Mann et al. 1991). However, in individuals with hypertriglyceridemia, cholesteryl ester transfer from HDL to VLDL can be augmented by increasing CETP plasma activity (Mann et al. 1991). Second, HL and endothelial lipase can convert HDL2 particles back to HDL3 particles that begin a new cycle of maturation or are further degraded by the liver or cleared through the kidneys (Borggreve et al. 2003; Ginsberg 1998; Rader 2006). HL activity is increased in T2DM (Tan et al. 1999) and contributes to the production of smaller and denser HDL particles (i.e., HDL2 to HDL3) that are then catabolized as indicated above.
3 Does HDL Play a Causal Role in the Pathogenesis of Diabetes?
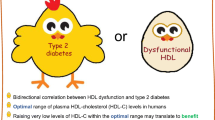
Several epidemiological studies have shown the association between low plasma concentrations of HDL-C and increased risks not only of CHD but also T2DM (Di et al. 2009; von Eckardstein et al. 2000).
We have seen that some of the metabolic perturbations (hyperglycemia, hypertriglyceridemia, etc.) associated with diabetes (and the metabolic syndrome) are mechanistically linked to decreased HDL cholesterol levels in blood. Traditionally, a reduced plasmatic level of HDL cholesterol has been interpreted to be the result of such metabolic perturbations, which are already present in the prediabetic state, and hence an innocent bystander of T2DM. However, several more recent studies suggest that HDLs help to maintain euglycemia via both insulin-dependent and insulin-independent pathways (Drew et al. 2012; von Eckardstein and Sibler 2011). The infusion of artificially reconstituted HDL (rHDL) was found to improve glycemia in patients with T2DM (Drew et al. 2009). In a post hoc analysis of data from diabetic participants in the ILLUMINATE trial, the addition of the CETP inhibitor torcetrapib to atorvastatin was found to increase HDL-C and to improve glycemic control (Barter et al. 2011). Several in vitro and animal experiments provided evidence that HDL improves the function and survival of beta cells as well as glucose-lowering metabolism in the muscle, liver, and adipose tissue (Drew et al. 2012; von Eckardstein and Sibler 2011; von Eckardstein and Widmann 2014). In mice, the absence of ApoA-I leads to hyperinsulinemia and hyperglycemia as well as altered responses to insulin tolerance tests. Vice versa, ApoA-I transgenic mice exhibit lower fasting glucose levels and improved glucose tolerance test compared with wild-type mice (Han et al. 2007; Lehti et al. 2013). These data point to a modulating role of HDLs in maintaining normal glucose homeostasis. Moreover, genetically increased HDL cholesterol and ApoA-I levels in mice prevented high-fat diet-induced glucose homeostasis impairment (Lehti et al. 2013).
Although it remains to be determined whether this is related to differences in skeletal muscle insulin resistance, adipose tissue mass, or differences in beta cell function, these data obtained in mice indicate that ApoA-I plays a modulatory role in the regulation of glucose homeostasis. This notion is also supported by studies in humans displaying altered HDL metabolism due to polymorphisms in the ApoA-I and ABCA1 genes that have been associated with increased risk of diabetes and impaired glucose uptake (Morcillo et al. 2005; Villarreal-Molina et al. 2008). Heterozygotes for rare mutations in the ABCA1 gene that profoundly decrease HDL-C levels were found to be less glucose tolerant than their unaffected relatives (Vergeer et al. 2010). However, it should also be noted that larger population studies did not find any association between heterozygosity for ABCA1 mutations and risk of diabetes (Schou et al. 2012).
3.1 Effects of HDL on Obesity
Augmented adipose tissue content is the hallmark of obesity. There is increasing evidence that HDL or ApoA-I regulate adipose tissue content. In humans, polymorphisms within the ApoA-I gene have been associated with increased risk of developing obesity and hypertension in a Brazilian cohort (Chen et al. 2009). More evidence for a potential role of HDL cholesterol on adipose tissue content regulation and obesity stems from genetic mouse models either lacking or overexpressing ApoA-I or treated with ApoA-I mimetic peptides. For example, ApoA-I-deficient mice have an increased fat content (Han et al. 2007). On the other hand, mice overexpressing ApoA-I or treated with the d-4F ApoA-I mimetic peptideFootnote 3 (Navab et al. 2005; Van Lenten et al. 2009) showed decreased white adipose tissue mass gain when fed a high-fat diet for 3 months (Ruan et al. 2011). ApoA-I transgenic and d-4F-treated mice had slightly improved insulin sensitivity when fed a high-fat diet compared to wild-type mice. This was associated with increased energy expenditure as measured in metabolic cages and related to increased expression of uncoupling protein 1 (UCP-1) in brown adipose tissue and AMPK phosphorylation (Ruan et al. 2011). Consistent with these results is the observation that administration of d-4F and l-4F in high-fat diet-fed mice reduced weight gain and improved insulin sensitivity when compared with age-matched vehicle-treated mice (Peterson et al. 2009). Taken together, these observations implicate a potential antiobesity effect of ApoA-I. In another study, ApoA-I gene transfer in the liver of mice increased adiponectin plasma levels and potently attenuated the rise of triglycerides and free fatty acids induced by lipopolysaccharide administration (Van Linthout et al. 2010). In vitro, incubation of 3 T3-L1 adipocytes with HDLs induced adiponectin expression in a phosphatidylinositol-3-kinase-dependent manner (Van Linthout et al. 2010). This set of experiments showed that ApoA-I and HDLs affect adipose tissue metabolism and adiponectin expression. ApoA-I and HDL have been shown to have anti-inflammatory properties in adipocytes similarly to what has been observed in macrophages and endothelial cells. It was found that both HDL and ApoA-I inhibit palmitate-mediated gene induction of pro-inflammatory chemotactic factor (e.g., monocyte chemotactic protein-1, serum amyloid A3) in cultured adipocytes (Umemoto et al. 2013). Furthermore, there was a reduction in chemotactic factor expression and macrophage accumulation in ApoA-I transgenic mice fed a high-fat diet compared to similarly treated control mice (Umemoto et al. 2013). Because macrophage accumulation and adipose tissue inflammation are strongly correlated with insulin resistance, these observations provide potential implications for the prevention and management of insulin resistance.
The molecular mechanisms involved in the antiobesity effects of ApoA-I are poorly understood. A recent suggestion is that ApoA-I and HDL might regulate autophagy in adipocytes. Decreased autophagy in pre-adipocytes has been associated with a preferential differentiation into “brown-like” adipocytes and improved energy expenditure, with reduced generation of white adipose tissue that serves mostly as an energy storage tissue (Wang and Peng 2012). Another potential mechanism is the direct capture of cholesterol from adipocytes by HDLs or ApoA-I (von Eckardstein and Sibler 2011; Zhang et al. 2010).
3.2 Effects of HDL on Insulin Sensitivity and Glucose Utilization by Skeletal Muscle
There is increasing evidence that HDLs regulate glucose uptake in insulin-sensitive tissues. In a set of elegant experiments in patients with T2 DM, Drew et al. showed that reconstituted HDL (rHDL) infusion over 4 h induced larger decreases of plasma glucose levels than placebo. This effect was associated with increased insulin secretion (Drew et al. 2009). In the C2C12 myocyte cell line, ApoA-I increased glucose uptake in an insulin-independent manner and also potentiated insulin- and adiponectin-induced glucose uptake (Han et al. 2007). ApoA-I effects on glucose uptake were dependent on the endocytosis of the protein and subsequent phosphorylation of AMPK and acetyl-coenzyme A carboxylase (ACC). In agreement with these observations, ApoA-I also increased glucose uptake in isolated mouse soleus muscle. Furthermore, in ApoA-I-deficient mice, AMPK phosphorylation status was decreased in skeletal muscle as well as in liver (Han et al. 2007). These data suggest that in vivo, ApoA-I levels are able to regulate AMPK phosphorylation. In ApoA-I-/- mice, expression of liver enzymes regulating gluconeogenesis (e.g., phosphoenolpyruvate carboxykinase, glucose-6-phosphatase) was also increased (Han et al. 2007), suggesting that hepatic glucose output is increased in animals lacking the main HDL apolipoprotein.
HDLs were also found to directly enhance glucose oxidation by increasing glycolysis and mitochondrial respiration rate in C2C12 muscle cells (Lehti et al. 2013). ATP synthesis was blunted in mitochondria isolated from the gastrocnemius muscle of ApoA-I knockout mice. Endurance capacity of ApoA-I knockout mice was reduced upon exercise exhaustion testing. Conversely, mice transgenically overexpressing human ApoA-I exhibited increased lactate levels, reduced fat mass, and protection against age-induced decline of endurance capacity. Circulating levels of fibroblast growth factor 21, a factor potentially involved in mitochondrial respiratory chain activity and inhibition of white adipose lipolysis, were significantly reduced in ApoA-I transgenic mice (Lehti et al. 2013).
3.3 The Beneficial Effects of HDL in Pancreatic Beta Cells
HDLs also exert potentially antidiabetogenic functions on pancreatic beta cells by potently inhibiting stress-induced cell death and possibly enhancing glucose-stimulated insulin secretion.
3.3.1 Beneficial Effects of HDL on Insulin Secretion
In vivo and in vitro evidence suggests that HDLs support the insulin secretory capacity of beta cells (Drew et al. 2012). Infusion of reconstituted HDLs in type 2 diabetes patients increased their HOMA-B index, an indirect measurement of pancreatic beta cell function (Drew et al. 2009). Also, the increase of HDL cholesterol levels in healthy volunteers treated for 2 weeks with a CETP inhibitor was found to increase postprandial insulin and C-peptide plasma levels (Siebel et al. 2013). The plasma of the CETP inhibited volunteers but not the CETP inhibitor itself also showed an increased capacity to enhance glucose-stimulated insulin secretion from MIN6 cells.
In vitro data on the effects of HDL or ApoA-I on insulin secretion are however controversial. Fryirs and collaborators showed that stimulation of the Min6 insulinoma cell line for 1 h with lipid-free recombinant ApoA-I, ApoA-II, or discoidal reconstituted HDLs dose-dependently increased both basal and glucose-stimulated insulin secretion (Fryirs et al. 2010). By RNA interference, the authors showed that the stimulatory effect of lipid-free ApoA-I and reconstituted HDLs on insulin secretion depended on ABCA1 and ABCG1, respectively (Fryirs et al. 2010). By contrast, other experiments in Min6 cells as well as islets of mice and humans did not find conclusive evidence that HDL enhances insulin production or basal and glucose-stimulated insulin secretion (Abderrahmani et al. 2007; Roehrich et al. 2003; Rutti et al. 2009). Therefore and considering the supra-physiological concentrations of lipid-free ApoA-I used by Fryirs et al., it appears uncertain of whether HDLs have a direct effect on the insulin secretory capacity of beta cells.
However, there is good evidence that ABCA1 and ABCG1 modulate insulin secretion from pancreatic beta cells. Mice with a targeted knockout of ABCA1 in pancreatic beta cells and crossbred with hypercholesterolemic LDL-receptor knockout mice were found to be less glucose tolerant than LDL-receptor knockout only mice (Brunham et al. 2007). The beta cell-specific ABCA1 knockout mice also showed reduced insulin secretion in response to glucose administration. Islets isolated from these mice showed altered cholesterol homeostasis and impaired insulin secretion in vitro (Brunham et al. 2007). In vitro, beta cells lacking ABCA1 showed impaired depolarization-induced insulin granule release, disturbances in membrane micro-domain organization, and alteration in Golgi and insulin granule morphology. Acute cholesterol depletion rescued the exocytotic defect in beta cells lacking ABCA1, suggesting that elevated islet cholesterol accumulation directly impairs granule fusion and insulin secretion (Kruit et al. 2011). Posttranscriptional suppression of ABCA1 in beta cells by adenoviral overexpression of miR33a and miR145 also led to increased cholesterol levels and to decreased glucose-stimulated insulin secretion (Kang et al. 2013; Wijesekara et al. 2012). This compromised insulin secretion was again rescued by cholesterol depletion. Inhibition of miR33a expression in apolipoprotein E knockout islets and ABCA1 overexpression in beta cell-specific ABCA1 knockout islets rescued normal insulin secretion and reduced islet cholesterol (Wijesekara et al. 2012). ABCG1 knockout mice also show glucose intolerance due to reduced insulin secretion (Sturek et al. 2010). In contrast to ABCA1 knockout, this defect was rescued by the addition of cholesterol rather than by depletion of cholesterol (Sturek et al. 2010). These studies show that a delicate balance of cholesterol concentrations between different subcellular compartments must be achieved to allow optimal beta cell functionality. As the consequence of the nonredundant roles of ABCA1 and ABCG1 in beta cell activity, the combined inactivation of ABCA1 and ABCG1 increased intracellular cholesterol accumulation and induced inflammation in beta cells and aggravated the diabetic phenotype found in the single knockout animals (Kruit et al. 2012).
Taken together, these findings indicate that cholesterol homeostasis and its regulation by ABCA1 and ABCG1 are critical for the secretory function of beta cells. However, the human genetic data mentioned before do not unequivocally support the findings made in the genetic mouse models. On the one hand, decreased glucose-induced insulin secretion has been reported in ABCA1-deficient patients with Tangier disease or heterozygous carriers of ABCA1 mutations (Koseki et al. 2009; Vergeer et al. 2010). On the other hand, mutations in ABCA1 have not been associated with increased risk of diabetes (Schou et al. 2012).
3.3.2 Effects of HDL on Pancreatic Beta Cell Survival and ER Stress
Several studies have shown that HDLs are very efficient in inhibiting apoptosis of beta cells induced by a variety of stimuli including inflammatory cytokines, free fatty acids (e.g., palmitate), thapsigargin, tunicamycin, or protein overexpression (Abderrahmani et al. 2007; Petremand et al. 2012; Roehrich et al. 2003; Rutti et al. 2009). Many of these stimuli induce endoplasmic reticulum (ER) stress. As ER stress has been proposed to be a driving parameter in beta cell dysfunction and death in the course of diabetes development (Eizirik et al. 2008; Oyadomari et al. 2002; Volchuk and Ron 2010), the capacity of HDLs to protect beta cells from ER stressors could be one mechanism underlying their potential ability to prevent T2DM.
Pancreatic beta cells are professional secretory cells. Half of the proteins produced by these cells are insulin. These cells have therefore developed an extensive ER network to fulfill their physiological function (Like and Chick 1970; Marsh et al. 2001). In response to the development of insulin resistance, triggered by obesity, for example, more insulin needs to be secreted by beta cells to compensate for insulin resistance and to achieve proper glycemic control. Such a sustained demand in insulin production can eventually cause ER dysfunction leading to chronic ER stress. This may promote beta cell dysfunction and ultimately beta cell death (Eizirik et al. 2008; Eizirik and Cnop 2010; Volchuk and Ron 2010). There is a wealth of evidence demonstrating that HDLs activate antiapoptotic responses in beta cells (Abderrahmani et al. 2007; Cnop et al. 2002; Petremand et al. 2009; Rutti et al. 2009). HDLs are particularly potent in inhibiting beta cell death induced by ER stressors (Petremand et al. 2009; Rutti et al. 2009). Recently, it was shown that HDLs maintain, despite the presence of ER stressors, a proper ER morphology and functionality in terms of the capacity of the ER to fold proteins and to allow protein trafficking out of the ER. Maintenance of the functionality of the ER was shown to be required for the beta cell protective activity of HDLs (Petremand et al. 2012). The ER stressors used included palmitate that is thought to mimic pro-diabetogenic conditions found in individuals with impaired glucose tolerance that are at risk of developing diabetes (Biden et al. 2004; Maedler et al. 2001; Shimabukuro et al. 1998). While this work identified a cellular mechanism mediating the beneficial effect of HDLs on beta cells against pro-diabetogenic factors, the underlying molecular mechanisms remain largely unresolved.
The agonist for the potentially antidiabetic activity of HDL on beta cell apoptosis has not yet been identified. The available information is mostly of negative nature. In beta cells, in contrast to endothelial cells, the SR-BI receptor is not required for HDL-mediated antiapoptotic responses (Petremand et al. 2012; Rutti et al. 2009). There is also no evidence that Akt, which plays a pivotal role in mediating the antiapoptotic activity of HDL in endothelial cells, participates in the protective function of HDLs in beta cells (Puyal et al. 2013). It appears therefore that the molecular mechanisms underlying the antiapoptotic property of HDLs in beta cells differ from those employed in endothelial cells. These mechanisms remain unknown (von Eckardstein and Widmann 2014).
Conceivably, individual components of HDLs may mediate their protective functions in beta cells. Actually, there is evidence that this could be the case. In primary islets, IL1β-induced beta cell apoptosis can be inhibited by either the delipidated HDL protein or the deproteinated lipid moieties of HDL as well as by ApoA-I and by S1P (Rutti et al. 2009). This is a good indication that proteins and lipids found in HDL particles carry pro-survival activities. However, pancreatic islets do not only contain beta cells (70–80 %) but also alpha cells (15–20 %), delta cells, PP cells, and immigrating blood-borne cells such as macrophages. Therefore, it is not clear whether ApoA-I and S1P exert their antiapoptotic effects on beta cells directly or indirectly, for example, by interfering with the release of proapoptotic signals from non-beta cells. To distinguish direct from indirect effects and to unravel the molecular basis for the antiapoptotic effects, it will be essential to substitute the complex model of pancreatic islets with purified beta cells or surrogate beta cell lines.
Conclusion
HDL possibly enhances glucose-stimulated insulin secretion (Drew et al. 2012; Fryirs et al. 2010; von Eckardstein and Sibler 2011) and reverses the proapoptotic effects of native and oxidized LDLs, IL1β, or thapsigargin (Abderrahmani et al. 2007; Petremand et al. 2012; Roehrich et al. 2003; Rutti et al. 2009). In addition, HDL and ApoA-I promote glucose uptake and activate AMP-kinase in primary human skeletal muscle cells and differentiated adipocytes by an insulin-independent way (Drew et al. 2009; Han et al. 2007; Zhang et al. 2011) and enhance oxidative metabolism in skeletal muscle through phosphorylation of acetyl-CoA carboxylase (Drew et al. 2009). Unfortunately, the identity of the agonists and their cognate receptors as well as the elicited signaling pathways and downstream targets of the antidiabetogenic HDL activities are yet poorly resolved. Understanding these pathways is a prerequisite for the development of drugs that stimulate or mimic the antidiabetic effects of HDLs and thereby help to lower the risk, for example, in overweight patients, to manifest T2DM.
Notes
- 1.
The study of causes or origins (of a disease).
- 2.
A group of symptoms that collectively indicate or characterize a disease.
- 3.
This peptide, albeit lacking any sequence homology to ApoA-I, forms a class A amphipatic helix similar to those found in ApoA-I. d-4F favors HDL formation and has anti-inflammatory and anti-atherogenic properties. The “d” in d-4F indicates that the peptide was synthesized with nonnatural d-amino acids (if synthesized with the natural l-amino acids, the peptide is called l-4F). d-amino peptides are very stable in biological fluids as they are not efficiently recognized by proteases (Chorev and Goodman 1995; Fischer 2003; Michod et al. 2009).
References
Abderrahmani A, Niederhauser G, Favre D, Abdelli S, Ferdaoussi M, Yang JY, Regazzi R, Widmann C, Waeber G (2007) Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic b cells. Diabetologia 50:1304–1314
Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA (1922) Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J 12:141–146
Barter PJ, Rye KA, Tardif JC, Waters DD, Boekholdt SM, Breazna A, Kastelein JJ (2011) Effect of torcetrapib on glucose, insulin, and hemoglobin A1c in subjects in the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial. Circulation 124:555–562
Besler C, Luscher TF, Landmesser U (2012) Molecular mechanisms of vascular effects of High-density lipoprotein: alterations in cardiovascular disease. EMBO Mol Med 4:251–268
Biden TJ, Robinson D, Cordery D, Hughes WE, Busch AK (2004) Chronic effects of fatty acids on pancreatic b-cell function: new insights from functional genomics. Diabetes 53(Suppl 1):S159–S165
Borggreve SE, De VR, Dullaart RP (2003) Alterations in high-density lipoprotein metabolism and reverse cholesterol transport in insulin resistance and type 2 diabetes mellitus: role of lipolytic enzymes, lecithin:cholesterol acyltransferase and lipid transfer proteins. Eur J Clin Invest 33:1051–1069
Bray GA (1996) Methods and obesity research: the radioimmunoassay of insulin. Obes Res 4:579–582
Brown MS, Goldstein JL (2008) Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 7:95–96
Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, Marsh BJ, Rodrigues B, Johnson JD, Parks JS, Verchere CB, Hayden MR (2007) b-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med 13:340–347
Chen ES, Mazzotti DR, Furuya TK, Cendoroglo MS, Ramos LR, Araujo LQ, Burbano RR, de Arruda Cardoso SM (2009) Apolipoprotein A1 gene polymorphisms as risk factors for hypertension and obesity. Clin Exp Med 9:319–325
Chirieac DV, Chirieac LR, Corsetti JP, Cianci J, Sparks CE, Sparks JD (2000) Glucose-stimulated insulin secretion suppresses hepatic triglyceride-rich lipoprotein and apoB production. Am J Physiol Endocrinol Metab 279:E1003–E1011
Chorev M, Goodman M (1995) Recent developments in retro peptides and proteins – an ongoing topochemical exploration. Trends Biotechnol 13:438–445
Cnop M, Hannaert JC, Grupping AY, Pipeleers DG (2002) Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology 143:3449–3453
Di AE, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J (2009) Major lipids, apolipoproteins, and risk of vascular disease. JAMA 302:1993–2000
Drew BG, Duffy SJ, Formosa MF, Natoli AK, Henstridge DC, Penfold SA, Thomas WG, Mukhamedova N, de Court FJM, Yap FY, Kaye DM, van Hall G, Febbraio MA, Kemp BE, Sviridov D, Steinberg GR, Kingwell BA (2009) High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 119:2103–2111
Drew BG, Rye KA, Duffy SJ, Barter P, Kingwell BA (2012) The emerging role of HDL in glucose metabolism. Nat Rev Endocrinol 8:237–245
Eisenberg S (1984) High density lipoprotein metabolism. J Lipid Res 25:1017–1058
Eizirik DL, Cnop M (2010) ER stress in pancreatic beta cells: the thin red line between adaptation and failure. Sci Signal 3:e7
Eizirik DL, Cardozo AK, Cnop M (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29:42–61
Fischer PM (2003) The design, synthesis and application of stereochemical and directional peptide isomers: a critical review. Curr Protein Pept Sci 4:339–356
Fried SK, Russell CD, Grauso NL, Brolin RE (1993) Lipoprotein lipase regulation by insulin and glucocorticoid in subcutaneous and omental adipose tissues of obese women and men. J Clin Invest 92:2191–2198
Fryirs MA, Barter PJ, Appavoo M, Tuch BE, Tabet F, Heather AK, Rye KA (2010) Effects of high-density lipoproteins on pancreatic beta-cell insulin secretion. Arterioscler Thromb Vasc Biol 30:1642–1648
Ginsberg HN (1998) Lipoprotein physiology. Endocrinol Metab Clin North Am 27:503–519
Grundy SM (2012) Pre-diabetes, metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol 59:635–643
Han R, Lai R, Ding Q, Wang Z, Luo X, Zhang Y, Cui G, He J, Liu W, Chen Y (2007) Apolipoprotein A-I stimulates AMP-activated protein kinase and improves glucose metabolism. Diabetologia 50:1960–1968
Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125–1131
Inzucchi SE (2012) Clinical practice. Diagnosis of diabetes. N Engl J Med 367:542–550
Kang MH, Zhang LH, Wijesekara N, de Haan W, Butland S, Bhattacharjee A, Hayden MR (2013) Regulation of ABCA1 protein expression and function in hepatic and pancreatic islet cells by miR-145. Arterioscler Thromb Vasc Biol 33:2724–2732
Koseki M, Matsuyama A, Nakatani K, Inagaki M, Nakaoka H, Kawase R, Yuasa-Kawase M, Tsubakio-Yamamoto K, Masuda D, Sandoval JC, Ohama T, Nakagawa-Toyama Y, Matsuura F, Nishida M, Ishigami M, Hirano K, Sakane N, Kumon Y, Suehiro T, Nakamura T, Shimomura I, Yamashita S (2009) Impaired insulin secretion in four Tangier disease patients with ABCA1 mutations. J Atheroscler Thromb 16:292–296
Kruit JK, Wijesekara N, Fox JE, Dai XQ, Brunham LR, Searle GJ, Morgan GP, Costin AJ, Tang R, Bhattacharjee A, Johnson JD, Light PE, Marsh BJ, Macdonald PE, Verchere CB, Hayden MR (2011) Islet cholesterol accumulation due to loss of ABCA1 leads to impaired exocytosis of insulin granules. Diabetes 60:3186–3196
Kruit JK, Wijesekara N, Westwell-Roper C, Vanmierlo T, de Haan W, Bhattacharjee A, Tang R, Wellington CL, LutJohann D, Johnson JD, Brunham LR, Verchere CB, Hayden MR (2012) Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired beta-cell function. Diabetes 61:659–664
Laios K, Karamanou M, Saridaki Z, Androutsos G (2012) Aretaeus of Cappadocia and the first description of diabetes. Hormones (Athens) 11:109–113
Lehti M, Donelan E, Abplanalp W, Al-Massadi O, Habegger KM, Weber J, Ress C, Mansfeld J, Somvanshi S, Trivedi C, Keuper M, Ograjsek T, Striese C, Cucuruz S, Pfluger PT, Krishna R, Gordon SM, Silva RA, Luquet S, Castel J, Martinez S, D'Alessio D, Davidson WS, Hofmann SM (2013) High-density lipoprotein maintains skeletal muscle function by modulating cellular respiration in mice. Circulation 128:2364–2371
Li N, Fu J, Koonen DP, Kuivenhoven JA, Snieder H, Hofker MH (2014) Are hypertriglyceridemia and low HDL causal factors in the development of insulin resistance? Atherosclerosis 233:130–138
Like AA, Chick WL (1970) Studies in the diabetic mutant mouse. II. Electron microscopy of pancreatic islets. Diabetologia 6:216–242
Lin MC, Gordon D, Wetterau JR (1995) Microsomal triglyceride transfer protein (MTP) regulation in HepG2 cells: insulin negatively regulates MTP gene expression. J Lipid Res 36:1073–1081
Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY (2001) Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 50:69–76
Malmstrom R, Packard CJ, Caslake M, Bedford D, Stewart P, Yki-Jarvinen H, Shepherd J, Taskinen MR (1997a) Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM. Diabetologia 40:454–462
Malmstrom R, Packard CJ, Watson TD, Rannikko S, Caslake M, Bedford D, Stewart P, Yki-Jarvinen H, Shepherd J, Taskinen MR (1997b) Metabolic basis of hypotriglyceridemic effects of insulin in normal men. Arterioscler Thromb Vasc Biol 17:1454–1464
Mann CJ, Yen FT, Grant AM, Bihain BE (1991) Mechanism of plasma cholesteryl ester transfer in hypertriglyceridemia. J Clin Invest 88:2059–2066
Marsh BJ, Mastronarde DN, Buttle KF, Howell KE, McIntosh JR (2001) Organellar relationships in the Golgi region of the pancreatic beta cell line, HIT-T15, visualized by high resolution electron tomography. Proc Natl Acad Sci USA 98:2399–2406
Michod D, Annibaldi A, Schaefer S, Dapples C, Rochat B, Widmann C (2009) Effect of RasGAP N2 fragment-derived peptide on tumor growth in mice. J Natl Cancer Inst 101:828–832
Morcillo S, Cardona F, Rojo-Martinez G, Esteva I, Ruiz-de-Adana MS, Tinahones F, Gomez-Zumaquero JM, Soriguer F (2005) Association between MspI polymorphism of the APO AI gene and type 2 diabetes mellitus. Diabet Med 22:782–788
Murao K, Wada Y, Nakamura T, Taylor AH, Mooradian AD, Wong NC (1998) Effects of glucose and insulin on rat apolipoprotein A-I gene expression. J Biol Chem 273:18959–18965
Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Yu N, Ansell BJ, Datta G, Garber DW, Fogelman AM (2005) Apolipoprotein A-I mimetic peptides. Arterioscler Thromb Vasc Biol 25:1325–1331
Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, Goldstein JL, Brown MS (2001) Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci USA 98:6027–6032
Oyadomari S, Araki E, Mori M (2002) Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 7:335–345
Peterson SJ, Kim DH, Li M, Positano V, Vanella L, Rodella LF, Piccolomini F, Puri N, Gastaldelli A, Kusmic C, L'Abbate A, Abraham NG (2009) The L-4F mimetic peptide prevents insulin resistance through increased levels of HO-1, pAMPK, and pAKT in obese mice. J Lipid Res 50:1293–1304
Petremand J, Bulat N, Butty AC, Poussin C, Rutti S, Au K, Ghosh S, Mooser V, Thorens B, Yang J-Y, Widmann C, Waeber G (2009) Involvement of 4E-BP1 in the protection induced by HDLs on pancreatic beta cells. Mol Endocrinol 23:1572–1586
Petremand J, Puyal J, Chatton JY, Duprez J, Allagnat F, Frias M, James RW, Waeber G, Jonas JC, Widmann C (2012) HDLs protect pancreatic beta-cells against ER stress by restoring protein folding and trafficking. Diabetes 61:1100–1111
Puyal J, Petremand J, Dubuis G, Rummel C, Widmann C (2013) HDLs protect the MIN6 insulinoma cell line against tunicamycin-induced apoptosis without inhibiting ER stress and without restoring ER functionality. Mol Cell Endocrinol 381:291–301
Rader DJ (2006) Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest 116:3090–3100
Rashid S, Watanabe T, Sakaue T, Lewis GF (2003) Mechanisms of HDL lowering in insulin resistant, hypertriglyceridemic states: the combined effect of HDL triglyceride enrichment and elevated hepatic lipase activity. Clin Biochem 36:421–429
Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ (2011) Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 478:404–407
Reaven GM (1988) Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37:1595–1607
Reaven GM (2005) Why Syndrome X? From Harold Himsworth to the insulin resistance syndrome. Cell Metab 1:9–14
Roehrich ME, Mooser V, Lenain V, Herz J, Nimpf J, Azhar S, Bideau M, Capponi A, Nicod P, Haefliger JA, Waeber G (2003) Insulin-secreting b-cell dysfunction induced by human lipoproteins. J Biol Chem 278:18368–18375
Rohrer L, Hersberger M, von Eckardstein A (2004) High density lipoproteins in the intersection of diabetes mellitus, inflammation and cardiovascular disease. Curr Opin Lipidol 15:269–278
Ruan X, Li Z, Zhang Y, Yang L, Pan Y, Wang Z, Feng GS, Chen Y (2011) Apolipoprotein A-I possesses an anti-obesity effect associated with increase of energy expenditure and up-regulation of UCP1 in brown fat. J Cell Mol Med 15:763–772
Rutti S, Ehses JA, Sibler RA, Prazak R, Rohrer L, Georgopoulos S, Meier DT, Niclauss N, Berney T, Donath MY, von Eckardstein A (2009) Low and high-density lipoproteins modulate function, apoptosis and proliferation of primary human and murine pancreatic beta cells. Endocrinology 150:4521–4530
Rye KA, Barter PJ (2012) Predictive value of different HDL particles for the protection against or risk of coronary heart disease. Biochim Biophys Acta 1821:473–480
Schou J, Tybjaerg-Hansen A, Moller HJ, Nordestgaard BG, Frikke-Schmidt R (2012) ABC transporter genes and risk of type 2 diabetes: a study of 40,000 individuals from the general population. Diabetes Care 35:2600–2606
Shimabukuro M, Zhou YT, Levi M, Unger RH (1998) Fatty acid-induced b cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA 95:2498–2502
Siebel AL, Natoli AK, Yap FY, Carey AL, Reddy-Luthmoodoo M, Sviridov D, Weber CI, Meneses-Lorente G, Maugeais C, Forbes JM, Kingwell BA (2013) Effects of high-density lipoprotein elevation with cholesteryl ester transfer protein inhibition on insulin secretion. Circ Res 113:167–175
Sturek JM, Castle JD, Trace AP, Page LC, Castle AM, Evans-Molina C, Parks JS, Mirmira RG, Hedrick CC (2010) An intracellular role for ABCG1-mediated cholesterol transport in the regulated secretory pathway of mouse pancreatic beta cells. J Clin Invest 120:2575–2589
Tabak AG, Jokela M, Akbaraly TN, Brunner EJ, Kivimaki M, Witte DR (2009) Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet 373:2215–2221
Tan KC, Shiu SW, Chu BY (1999) Roles of hepatic lipase and cholesteryl ester transfer protein in determining low density lipoprotein subfraction distribution in Chinese patients with non-insulin-dependent diabetes mellitus. Atherosclerosis 145:273–278
Tang C, Oram JF (2009) The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim Biophys Acta 1791:563–572
Taskinen MR (2003) Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 46:733–749
Uehara Y, Engel T, Li Z, Goepfert C, Rust S, Zhou X, Langer C, Schachtrup C, Wiekowski J, Lorkowski S, Assmann G, von Eckardstein A (2002) Polyunsaturated fatty acids and acetoacetate downregulate the expression of the ATP-binding cassette transporter A1. Diabetes 51:2922–2928
Uehara Y, Miura S, von Eckardstein A, Abe S, Fujii A, Matsuo Y, Rust S, Lorkowski S, Assmann G, Yamada T, Saku K (2007) Unsaturated fatty acids suppress the expression of the ATP-binding cassette transporter G1 (ABCG1) and ABCA1 genes via an LXR/RXR responsive element. Atherosclerosis 191:11–21
Umemoto T, Han CY, Mitra P, Averill MM, Tang C, Goodspeed L, Omer M, Subramanian S, Wang S, Den Hartigh LJ, Wei H, Kim EJ, Kim J, O'Brien KD, Chait A (2013) Apolipoprotein AI and high-density lipoprotein have anti-inflammatory effects on adipocytes via cholesterol transporters: ATP-binding cassette A-1, ATP-binding cassette G-1, and scavenger receptor B-1. Circ Res 112:1345–1354
Van Lenten BJ, Wagner AC, Anantharamaiah GM, Navab M, Reddy ST, Buga GM, Fogelman AM (2009) Apolipoprotein A-I mimetic peptides. Curr Atheroscler Rep 11:52–57
Van Linthout S, Foryst-Ludwig A, Spillmann F, Peng J, Feng Y, Meloni M, Van Craeyveld E, Kintscher U, Schultheiss HP, De GB, Tschope C (2010) Impact of HDL on adipose tissue metabolism and adiponectin expression. Atherosclerosis 210:438–444
Vergeer M, Brunham LR, Koetsveld J, Kruit JK, Verchere CB, Kastelein JJ, Hayden MR, Stroes ES (2010) Carriers of loss-of-function mutations in ABCA1 display pancreatic beta-cell dysfunction. Diabetes Care 33:869–874
Villarreal-Molina MT, Flores-Dorantes MT, Arellano-Campos O, Villalobos-Comparan M, Rodriguez-Cruz M, Miliar-Garcia A, Huertas-Vazquez A, Menjivar M, Romero-Hidalgo S, Wacher NH, Tusie-Luna MT, Cruz M, Aguilar-Salinas CA, Canizales-Quinteros S (2008) Association of the ATP-binding cassette transporter A1 R230C variant with early-onset type 2 diabetes in a Mexican population. Diabetes 57:509–513
Volchuk A, Ron D (2010) The endoplasmic reticulum stress response in the pancreatic beta-cell. Diabetes Obes Metab 12(Suppl 2):48–57
von Eckardstein A, Sibler RA (2011) Possible contributions of lipoproteins and cholesterol to the pathogenesis of diabetes mellitus type 2. Curr Opin Lipidol 22:26–32
von Eckardstein A, Widmann C (2014) HDL, beta cells and diabetes. Cardiovasc Res 103(3):384–394
von Eckardstein A, Schulte H, Assmann G (2000) Risk for diabetes mellitus in middle-aged Caucasian male participants of the PROCAM study: implications for the definition of impaired fasting glucose by the American Diabetes Association. J Clin Endocrinol Metab 85:3101–3108
Wang Y, Oram JF (2005) Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a phospholipase D2 pathway. J Biol Chem 280:35896–35903
Wang Y, Oram JF (2007) Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a protein kinase Cd pathway. J Lipid Res 48:1062–1068
Wang S, Peng D (2012) Regulation of adipocyte autophagy – the potential anti-obesity mechanism of high density lipoprotein and ApolipoproteinA-I. Lipids Health Dis 11:131
Wijesekara N, Zhang LH, Kang MH, Abraham T, Bhattacharjee A, Warnock GL, Verchere CB, Hayden MR (2012) miR-33a modulates ABCA1 expression, cholesterol accumulation, and insulin secretion in pancreatic islets. Diabetes 61:653–658
Yoshikawa T, Shimano H, Yahagi N, Ide T, Amemiya-Kudo M, Matsuzaka T, Nakakuki M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Takahashi A, Sone H, Osuga JJ, Gotoda T, Ishibashi S, Yamada N (2002) Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J Biol Chem 277:1705–1711
Zhang Y, McGillicuddy FC, Hinkle CC, O'Neill S, Glick JM, Rothblat GH, Reilly MP (2010) Adipocyte modulation of high-density lipoprotein cholesterol. Circulation 121:1347–1355
Zhang Q, Zhang Y, Feng H, Guo R, Jin L, Wan R, Wang L, Chen C, Li S (2011) High density lipoprotein (HDL) promotes glucose uptake in adipocytes and glycogen synthesis in muscle cells. PLoS ONE 6:e23556
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this chapter
Cite this chapter
Vollenweider, P., von Eckardstein, A., Widmann, C. (2015). HDLs, Diabetes, and Metabolic Syndrome. In: von Eckardstein, A., Kardassis, D. (eds) High Density Lipoproteins. Handbook of Experimental Pharmacology, vol 224. Springer, Cham. https://doi.org/10.1007/978-3-319-09665-0_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-09665-0_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09664-3
Online ISBN: 978-3-319-09665-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)