
Abstract
Alcoholic liver disease (ALD) is the major liver disease in the developed world and characterized by hepatic iron overload in ca. 50 % of all patients. This iron overload is an independent factor of disease progression, hepatocellular carcinoma and it determines survival. Since simple phlebotomy does not allow the efficient removal of excess iron in ALD, a better understanding of the underlying mechanisms is urgently needed to identify novel targeted treatment strategies. This review summarizes the present knowledge on iron overload in patients with ALD.
Although multiple sides of the cellular and systemic iron homeostasis may be affected during alcohol consumption, most studies have focused on potential hepatic causes. However, it should not be overlooked that more than 90 % of the major iron pool, the hemoglobin-associated iron, is efficiently recycled within the human body and it is also strongly affected by alcohol. The few available studies suggest various molecular mechanisms that involve iron regulatory protein (IRP1), transferrin receptor 1 (TfR1), and the systemic iron master switch hepcidin, but not classical mutations of the HFE gene. Notably, reactive oxygen species (ROS), namely, hydrogen peroxide (H2O2), are powerful modulators of these iron-steering proteins. For instance, depending on the level, H2O2 may both strongly suppress and induce the expression of hepcidin that could partly explain the anemia and iron overload observed in these patients. More studies with appropriate ROS models such as the novel GOX/CAT system are required to unravel the mechanisms of iron overload in ALD to consequently identify molecular-targeted therapies in the future.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Alcoholic liver disease
- Alcohol
- Carcinogenesis
- Iron
- HFE
- Hemochromatosis
- Hepatocellular carcinoma
- Reactive oxygen species
6.1 Introduction
Carcinogenesis by alcohol is complex and still poorly understood for many reasons [1]. One major reason is due to the fact that it is rather the metabolites and side products of alcohol turnover rather than direct actions of alcohol itself (see Fig. 6.1). Indeed, alcohol is rapidly converted into more reactive, toxic, or even carcinogenic compounds such as acetaldehyde or reactive oxygen species (ROS) via nonenzymatic or enzymatic reactions. The production of these metabolites during alcohol consumption is complicated since it is dependent on many additional factors that include the genetic background or the tissue-specific induction of detoxifying enzymes [2]. The complete understanding of alcohol-mediated carcinogenesis is also hampered by the fact that side products such as ROS are very difficult to measure and appropriate models to study ROS and ROS-mediated carcinogenesis are still lacking [3]. Finally, commonly used animal models such as rodents significantly differ from humans with regard to alcohol sensitivity and metabolism and consequently with regard to cancer development. Although iron and its accumulation in the liver during alcohol consumption has been appreciated for a long time, a better understanding of the underlying molecular mechanisms has been only gained in the last decade with the discoveries of key regulatory molecules such as hepcidin and the HFE gene. This overview aims to summarize the actual knowledge of iron toxicity and accumulation in ALD, which is considered an important single factor for the development of liver cancer and overall survival.

Iron accumulation and ROS are important factors in the alcohol-mediated hepatocarcinogenesis
6.2 Health Statistics of ALD
Chronic alcohol consumption is one of the major risk factors worldwide, significantly affecting both mortality and years of life loss (YLL) [4]. Ca. 5 % of the Western world show risky alcohol consumption and some countries such as China reach a yearly regional increase of alcohol consumption of 400 % [5]. The liver is the major target organ of alcohol consumption, although alcohol may affect many other organ systems such as the heart, nervous system, pancreas, breast, and others. According to the recently published “burden of disease study 2010,” liver cirrhosis and liver cancer are ranked at position 12 and 16 in the actual death statistics [6]. In 2010 ca. one million people died from liver cirrhosis with one third directly attributable to alcohol. This is a considerable number when comparing with coronary heart disease with about ten million deaths and ranking as the leading cause of mortality in the global death statistics. In Central Europe, liver cirrhosis even ranks at the fourth position of YLL and HCC is now the most common fatal complication of patients with alcoholic liver cirrhosis. Moreover, it has the second fastest increase of all tumors worldwide after kidney tumors, and alcohol-associated HCC ranks on third position after HCC caused by viral hepatitis B and C infections. These epidemiological data explain the high interest in gaining a better understanding of molecular mechanisms of alcohol-driven hepatocarcinogenesis to identify novel target approaches for future therapies.
6.3 Prevalence of Iron Overload in ALD
A significant iron deposition in patients with chronic alcohol consumption has been appreciated for a long time [7] (see Fig. 6.2). Considerable confusion still exists today with genetic hemochromatosis, which is characterized by a typical and severe hepatocellular iron overload and shows other classical symptoms such as arthritis, skin pigmentation, and diabetes mellitus. The genetic nature of hereditary hemochromatosis was already clear when the first monographs on hemochromatosis in 1935 by Sheldon and the extensive review by Finch et al. in 1955 [8, 9] appeared. However, it was also noted [8] that patients with hemochromatosis showed a high prevalence of alcoholism of ca. 30 %. Although the introduction of percutaneous needle biopsy of the liver in the 1950s added a new tool for exact quantitation of hepatic iron deposition, it further led to considerable confusion since histological stainable iron deposits are common in cirrhotic livers [10, 11]. This is especially the case in alcoholic subjects and some investigators have even equated mild to moderate alcoholic cirrhosis with hemochromatosis. Notably, it is still not clear in what form the iron is actually deposited and in what form the iron is toxic (ferritin, hemosiderin, or labile iron pool [12, 13]). When analyzing the Finch data for differences between alcoholic and nonalcoholic patients, Powell noted that laboratory signs of hepatocellular damage showed a significant higher prevalence in the alcoholic group (serum transaminase elevation 35.5 % vs. 15.4 %). Interestingly, most large studies seem to indicate that mutations in the HFE gene [14] are not responsible for the iron accumulation [15–17] in ALD patients. Moreover, since hereditary hemochromatosis shows a very weak clinical penetration of about 10 %, it still remains open what exact role alcohol plays in patients with hemochromatosis and whether it is an important but underestimated disease modifier.

Histological section of a liver biopsy from a patient with ALD. Staining of iron-loaded cells by Prussian blue (black arrows = intracellular inclusions of iron in hepatocytes) in the liver of ALD patient. Asterisk = inflammatory foci, arrow head = macrovesicular steatosis (fat droplet with displaced nucleus). In this patient, a predominant hepatocellular iron deposition is seen while Kupffer cells are not stained for iron
Important studies in the 1980s focused on pathologic abnormalities of available serum iron parameters. For instance, Chapman et al. determined the hepatic iron content in 60 alcoholics with liver disease and in 15 patients with untreated idiopathic hemochromatosis [18]. A significantly higher mean liver iron concentration was found in alcoholics as compared to controls (156.4 vs. 53 μg per 100 mg dry weight). However, liver iron was much higher in the hemochromatosis group (2,094.5 μg). In addition, no relationship between liver iron concentration and the amount of alcohol consumed was noted. Interestingly, Chapman also recognized that the serum ferritin concentrations reflected well the iron overload in patients with hemochromatosis and mild alcoholic liver disease while no association was found with serum ferritin in patients with severe alcoholic liver disease. He also concluded that serum iron and transferrin saturation were of little value in assessing iron status in patients with ALD. In 1994, Bell studied 312 patients with different liver diseases and, after a careful assessment of fasting serum iron, transferrin, and ferritin levels, found that serum ferritin is more frequently elevated in ALD than in other liver diseases [19]. In addition, measurement of ferritin should be postponed until the patients are abstaining because he noted that serum ferritin decreased from 1,483 to 388 μg/L after 1.5–6 weeks of abstaining from alcohol. The author claimed that most patients with increased serum ferritin had normal transferrin saturation. Again, ferritin levels higher than 1,000 μg/L were observed in 100 % of patients with hemochromatosis but only in 11 % in patients with ALD. Only 7.7 % of patients with other liver diseases had such high ferritin concentrations. It should be noted, however, that 15.2 % of ALD patients have increased transferrin saturation. In a recent study from Japan, excess iron accumulation was found in 22 hepatic tissues with alcoholic liver diseases, but not in any normal hepatic tissues [20].
We have recently characterized a large cohort at the Heidelberg Salem Medical Center using biopsy, but also various newer noninvasive tests including transient elastography. Selected data [21] from a cohort of 235 patients with ALD, of whom 86 had underwent a liver biopsy, with detailed iron characterization regarding intensity and location (macrophages vs. hepatocytes), are shown in Table 6.1. It can be summarized that ca. 50 % of patients with ALD have significant pathological iron deposition. Interestingly, iron deposits are found both at the same rate in macrophages and hepatocytes. In addition, there is a mixture of patient groups with only macrophages, only hepatocytes, and mixed iron deposition. It should be clearly noted that this iron deposition depends on fibrosis stage, as has already been seen many decades before. However, with new noninvasive technologies to better quantitate the stage of fibrosis (e.g., transient elastography), more quantitative associations can be shown (see Table 6.2). It should be also noted that the Heidelberger alcohol cohort shows a rather representative fibrosis distribution with about 60 % without any fibrosis, 10 % with fibrosis stages F1 and F2, 10 % fibrosis stage F3, and 15 % fibrosis stage F4. According to our data, the mean histological iron score (0–3) is 1 in patients with advanced fibrosis stage F3 and F4. About 2/3 of these patients with advanced fibrosis stage have elevated ferritin levels higher 400 μg/mL and an increased transferrin saturation higher in 45 %. This is especially important to consider, since ferritin levels and transferrin saturation are commonly used to screen patients with hereditary hemochromatosis. In the subanalysis (Mueller et al. 2013, unpublished data), we could show that the serum ferritin levels were highly associated with GGT, transaminases levels, transferrin saturation, liver stiffness, and histological iron deposition in hepatocytes and to a lesser extent to iron in macrophages. Longitudinal analysis of serum ferritin levels during abstaining from alcohol showed that concentrations decreased by 50 % within 2.1 days. Importantly, a careful comparison between patients with alcoholic liver disease and patients with nonalcoholic liver disease (NALD) in 30 cases matched with regard to gender, age, and fibrosis stage showed significant differences between the two cohorts with regard to iron-related proteins. So ALD patients showed lower transferrin levels and lower erythrocyte counts and hemoglobin levels but significantly higher ferritin level [22].
Taken together, patients with ALD show a significantly increased histological iron deposition in about half of the population that increases with disease progression toward cirrhosis (see Fig. 6.3). This iron deposition is reflected by serum ferritin levels, but unfortunately overlayered by additional inflammatory conditions. In fact, according to our analysis, serum ferritin levels reflect hepatocyte iron deposition more closely than macrophage iron deposition in ALD. Moreover and often overlooked, a significant proportion of patients show elevated levels of transferrin saturation and ferritin levels that can be easily mistaken for hemochromatosis.

Natural course of ALD ultimately leading to liver cancer
6.4 Iron Toxicity and Carcinogenesis
Iron toxicity has been well known for many decades and usually attributed to Fenton chemistry, first described in 1896 by Fenton and referring to the reaction of reduced ferrous iron with H2O2 to ferric iron and hydroxyl radicals [23]. These radicals are highly and universally reactive to all cellular compounds including DNA, proteins, lipids, and carbohydrates. They cause oxidative damage to DNA, including base modifications and DNA strand breaks, and may change the structure of proteins and lipids, eventually leading to mutagenesis.
Although, the direct chemical nature of the iron toxicity in Fenton’s chemistry has been recently questioned by Saran et al. [24], it is generally assumed that at least Fenton-like reactions contribute to the toxicity of iron. For this reason, we will not further differentiate both reactions in this review. In vitro studies have shown that liver viability decreases in the presence of high amounts of iron and that the depletion of iron by iron chelators can protect cells from peroxide toxicity. Hepatic iron overload and the risk for liver cancer have also been extensively studied using in vivo experiments in the past. In patients with hereditary hemochromatosis due to HFE mutations, the hepatic iron overload ultimately determines the overall survival [25]. In a large transplant registry study, Ko et al. showed that iron alone increased the risk of HCC by a factor of 2.2 [26]. Turlin et al. showed in 1995 that the iron-associated risk of HCC was not dependent on whether the patient had cirrhosis or not [27]. In an interesting study from the NIH, Zacharski et al. could demonstrate that iron depletion by phlebotomy may have a protective effect on general carcinogenesis [28]. He studied prospectively 641 controlled subjects without iron depletion and 636 subjects in an iron reduction group. Iron reduction was achieved by monthly phlebotomy. Patients were followed up for an average for 4.5 years. Interestingly, the risk of new visceral malignancies was lower in the iron reduction group than controlled group (38 vs. 60), and among patients with new cancers, those in the iron reduction group had lower cancer-specific and all-cause mortality. Mean ferritin levels across all 6-monthly visits were lower among all patients who did not develop cancer. Notably, in a 1988 animal study, Hann et al. showed reduced tumor growth in iron-deficient mice [29], and Tsukamoto et al. in 1995 showed in rodents that a mixture of high fat diet and alcohol together with supplemented iron led to pronounced cirrhosis [30]. In a meta-analysis by D’Amico et al., it was concluded that liver iron is one among several independent prognostic factors of survival in liver cirrhosis [31]. Ganne-Carrie showed in 229 patients with ALD or hepatitis C virus-related cirrhosis that iron is the best parameter for predicting mortality in ALD patients (p = 0.007) followed over 57 months [32]. This study found no prognostic significance of hepatic iron for HCV.
Taken together, there seems to be enough evidence from human and animal studies to suggest that hepatic iron overload leads to hepatocellular damage. This in turn leads to increased fibrosis progression and risk for hepatocarcinogenesis, and ultimately iron seems to be an important prognostic factor for overall survival in patients with ALD. The most evident and generally accepted mechanisms of iron carcinogenesis include Fenton-like reactions that lead to the highly aggressive hydroxyl radicals.
6.5 Mechanism of Iron Overload
Enormous progress has been made in the last two decades to better understand the molecular mechanisms of iron regulation and homeostasis both at the cellular and systemic level [33–35]. These milestones include the discovery of iron regulatory proteins (IRP) and their posttranscriptional regulation of cellular iron homeostasis in the late 1980s and early 1990s as well as identification of the systemic master switch peptide hepcidin about 10 years ago [36]. Despite this progress, numerous aspects of iron regulation still remain unexplained in humans. Apart from the diagnosis of rare and common variants of hereditary hemochromatosis, the progress has led to significant improvements in the diagnosis and therapy of iron overload. The discovery of the HFE gene that causes the majority of hereditary hemochromatosis forms has clearly helped to resolve the role of hemochromatosis in alcoholics [14]. Interestingly, most large studies seem to indicate that mutations in the HFE gene [14] are not responsible for the iron accumulation in patients with ALD [15–17]. These studies thus seem to rule out HFE as important genetic disease modifier. In contrast, as briefly discussed above, alcohol could be an important disease modifier in patients with hemochromatosis, but typically with weak clinical penetration. In addition, at the systemic level, ALD appears rather complex since alcohol affects not only the liver but also many other organ systems including the bone marrow and the immune system so that iron regulation obviously is altered at many different levels and locations throughout the body. In the following, a brief description of the cellular and systemic iron homeostasis is given and the status quo of molecular findings in patients with ALD is discussed.
6.6 Systemic Regulation
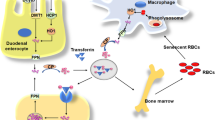
The majority of body iron, roughly 2 g in humans, is distributed in the oxygen carrier hemoglobin of red blood cells and developing erythroid cells (Fig. 6.4). Excess iron is usually stored in the liver, which normally contains about 1 g of iron, predominantly in the form of ferritin [34]. Ferritin is a protein composed of 24 subunits with a ferrihydrite core where 4500 iron atoms can be stored in cage-like structures. It consists of ferritin H chain (heavy or heart) and ferritin L chain (liver or light) subunits and can be regulated by IRPs [37]. Other significant amounts of iron are found in macrophages (0.6 g) and in the myoglobin of muscles (0.3 g). It is generally believed that mammals lose iron from regular sloughing of the mucosa and skin cells or during bleeding and do not possess any mechanisms for iron excretion from the body [33]. Therefore, the balance is maintained by the tight control of dietary iron absorption at the brush border of enterocytes in the proximal duodenum (Fig. 6.4). Dietary iron uptake involves the reduction of ferric iron to ferrous iron in the intestinal lumen by ferric reductases, such as duodenal cytochrome B (Dcytb), and subsequent transport of iron via the apical membrane of enterocytes by divalent metal transporters (such as DMT1 encoded by SCL11A2 (solute carrier family 11, member 2) gene). Iron can also be transported across the enteral membrane in the form of heme by unknown mechanisms and the iron is then released inside enterocytes via heme oxygenase 1 (HO-1). Cytosolic iron is exported by the basolateral iron transporter ferroportin (FP-1 or SLC40A1) from the enterocytes to the blood compartment and then bound to transferrin, the major abundant iron-carrying protein within serum. Before binding, ferrous iron is oxidized by ferroxidases (e.g., hephaestin/ceruloplasmin) to the ferric form. Iron is then distributed within the body and used in various pathways, but mainly utilized in the bone marrow for the synthesis of new heme.

Systemic iron homeostasis and utilization in the body. Black circles indicate potential target sites of iron regulation affected by chronic or acute alcohol consumption. Dietary iron is absorbed in the duodenum and bound to transferrin. Iron is then mainly delivered to the bone marrow for erythropoiesis while senescent erythrocytes are phagocytosed by macrophages. This efficient mechanism recycles >90 % of all iron for new heme synthesis. Excess iron is stored in hepatic ferritin or temporarily in macrophage ferritin. Regulation of iron metabolism by hepcidin and upstream factors are also shown
About 1–2 mg per day of iron is absorbed to keep a stable iron balance. Of note, iron undergoes an efficient recycling. Senescent erythrocytes that have a life span of 120 days are sequestered by macrophages and the iron is reused for new heme synthesis. This recycling machinery accounts for about 90 % of newly synthesized hemoglobin. It is important to note that the iron export pump, ferroportin, is not only found on enterocytes but also on macrophages and hepatocytes (Fig. 6.5). The ferroportin-mediated efflux of ferrous iron from enterocytes and macrophages into the serum is critical for systemic iron homeostasis and mainly controlled via the liver-secreted 25 AS peptide hormone, hepcidin. Mechanistically, hepcidin binds to ferroportin and promotes its phosphorylation, internalization, and lysosomal degradation [38, 39]. Hepcidin is primarily expressed in hepatocytes as a precursor pro-peptide, although other locations of secretion have been described, such as macrophages [40] (Fig. 6.5) and to a lesser extent cardiomyocytes. Hepcidin efficiently blocks ferroportin, which leads to accumulation of iron within macrophages and blocks the iron absorption via enterocytes (Fig. 6.4).

Interaction of systemic and cellular iron homeostasis at four major sites: enterocyte, hepatocyte, erythroblast, and macrophage
An important evolutionary conserved mechanism to induce hepcidin is an infection or inflammatory state. Via cytokines, namely, IL-6, but also microbial molecules (such as lipopolysaccharide), hepcidin is strongly induced leading to a rapid decrease of serum iron, which is thought to function as an antibacterial defense mechanism. More recently, another important inflammatory cofactor, H2O2, has been identified as potent inducer of hepcidin. In contrast, hepcidin levels seem to be suppressed in patients with genetic hemochromatosis, leading to increased uptake of iron via the duodenum and increased release of iron through macrophages. Cytokine-mediated induction of hepcidin is thought to be the reason for anemia of chronic disease [41], while the disruption of hepcidin is generally associated with the systemic iron overload (e.g., genetic hemochromatosis). Despite the progress and the discovery of various upstream regulators of hepcidin (such as C/EBPα; BMP6; SMAD 1, 5, 8, and 4; TMPRSS6; IL-6; CREBH; CHOP; and TLR4), an overall and conclusive understanding of the regulatory network with respect to the control of iron is not yet completely understood. Hitherto unexplained features of hepcidin regulation include:
-
1.
Expression of hepcidin in cells other than hepatocytes
-
2.
The nature of co-expressing ferroportin in hepatocytes and macrophages
-
3.
The experimental and clinical finding that hepcidin responds differentially to iron overload in vitro and in vivo
-
4.
The controversial findings of hypoxia-mediated regulation of hepcidin
-
5.
The controversial and conflicting findings of the response of hepcidin toward ROS
At least to the latter point we could recently identify a partial explanation [42]. Thus, the central ROS, H2O2 induces hepcidin in hepatocytes independent of IL-6 when exposed in continuous manner. Bolus treatments, however, which reflect an artificially high H2O2 exposure blocked hepcidin expression [43]. They are even toxic at H2O2 concentrations higher than 50 μmol and hepcidin suppression at such conditions was due to unspecific inhibition of the mRNA transcription machinery [43].
6.7 Cellular Iron Regulation
Developing erythrocytes, as well as most other cell types, require iron from plasma transferrin. Iron is loaded onto transferrin with a capacity of two atoms of ferric iron per molecule. Transferrin binds with a very high affinity to cell surface transferrin receptor 1 (TfR1) ubiquitously expressed and with much lower affinity to transferrin receptor 2 (TfR2) sharing 45 % homology to TfR1 [44, 45] (see Fig. 6.6). Homology aside, TfR2 exhibits different properties from TfR1. TfR2 is only expressed in liver and to a lesser extent in erythroid, spleen, lung, and muscle cells. Also, its mRNA levels are not directly regulated by cellular iron content. Its expression is higher in iron-replete conditions, suggesting a role as transferrin-iron sensor candidate rather than acting primary in iron uptake [46]. The TF-TfR1 complex is endocytosed via clathrin-coated pits. The release of ferrous iron into the intracellular compartment requires acidification of the endosome using a proton pump and the reduction of ferric iron to the ferrous form via Steap3. DMT1 transports the ferrous iron across the endosomal membrane into the cytosol where it can be stored in ferritin complexes in non-erythroid cells or incorporated into hemoglobin in erythroid cells [47] (see Fig. 6.6). Iron is then either reused for various synthesis pathways such as intracellular heme synthesis and iron cluster protein synthesis or transported into the major iron storage protein ferritin.

Cellular iron metabolism. Iron-loaded holo transferrin binds to its cell surface receptor (high affinity TfR1, low affinity TfR2) and the Tf-TfR complex is endocytosed. In the endosome, iron is released and reduced by Steap3 and transported across the endosomal membrane by DMT1. Internalized iron is directed to mitochondria for heme or ISC synthesis and excess iron is stored in ferritin. Cytosolic iron is exported by the iron transporter ferroportin to the blood compartment and then again bound to transferrin. Regulatory mechanisms by hepcidin or IRPs are shown in red
The expression of DMT1 and ferritin is coordinately posttranscriptionally regulated by binding of trans-acting iron-responsive proteins (IRP1 and IRP2) to iron-responsive elements (IRE) in the untranslated regions (UTRs) of their respective mRNAs [48–50]. In iron-starved cells IRPs bind with high affinity to cognate IREs (see Fig. 6.7a, b). IREs are evolutionary conserved hairpin structures of about 30 nucleotides with characteristic sequences. The effect of IRP binding to IREs is dependent on their position. TfR1 mRNA contains five IREs within its long 3′ UTR that stabilizes and protects the transcript from degradation leading to protein upregulation, but other mRNAs, for example, mRNAs encoding H- and L-ferritins, contain a single IRE in their 5′ UTRs where binding results in decreased protein translation by steric blockade. As a result increased TfR1 levels stimulated acquisition of iron from plasma transferrin to counteract iron deficiency. The inhibition of ferritin synthesis leads to decreased abundance of this protein as iron storage becomes obsolete under these conditions. Conversely, in cells with high iron content, both IRP1 and IRP2 become unavailable for IRE binding lowering TfR1mRNA degradation and ferritin mRNA translation. Thus, when iron supply exceeds cellular means, the IRE-IRP switch minimizes further iron uptake via TfR1 and favors the storage of excess iron in newly synthesized ferritin. Other IREs have been discovered in the genes of ALAS2, mitochondrial aconitase, ferroportin, HIF2α, β-APP, and α-synuclein, which in turn control iron storage and erythroid iron utilization, energy homeostasis, hypoxia responses, and neurological pathways, respectively.

Posttranscriptional regulation of iron-responsive element (IRE)-containing mRNAs either in the A) 5’UTR or B) 3′ UTR by iron regulatory proteins (IRPs). Binding of IRP in the 3′ UTR modulates TfR1 and DMT1 mRNA stability therefore influencing iron uptake and transport and renders translation of mRNAs with IREs in the 5′ UTR encoding H- and L-ferritins, ALAS2, m-aconitase, ferroportin, β-APP, and α-synuclein, which control iron storage, heme synthesis, energy homeostasis, iron efflux, and neurological functions, respectively
Although, the IRE-IRP network allows an autonomous independent control of iron homeostasis for individual cells, it can be overwritten by additional control mechanisms. For example, TfR1 expression is regulated transcriptionally by erythroid active element and its promoter in erythroid progenitor cells, which take up an enormous amount of iron for heme synthesis [51]. IRE-IRP independent regulation has been described in details elsewhere [52, 53]. An extensive phylogenetic analysis confirmed the IRE-containing mRNAs are exclusively found in metazoans [54]. The ferritin IRE motive may represent the exceptional prototype that was subsequently adapted during evolution by other genes in higher organisms. It should be also noted that IRP1 and IRP2 do not share sequence similarities with known RNA-binding proteins and do not contain any other established RNA-binding motives. IRPs belong to the family of iron sulfur cluster (ISC) isomerases, which are homologous to mitochondrial aconitase [48–50]. These enzymes catalyze the stereo-specific isomerization of citrate to isocitrate via the intermediate cis-aconitate during the citric acid cycle and contain a (4Fe-4S) cluster in its active side. ISC IRP1 assembles analogous to mitochondrial aconitase ISC. However, in contrast to m-aconitase, IRP1 only retains its ISC and its aconitase function in iron-repleted cells. Under iron deprivation, holo-IRP1 loses its labile cluster and is converted into apo-IRP1, which then binds to IRE-containing transcripts involved in iron uptake, storage, and transport. The ISC of IRP1 is also stabilized by hypoxia [55, 56]. Although IRP1 and IRP2 share 64 % sequence homology, they differ considerably in some aspects. IRP2 is neither assembled in (4Fe-4S) cluster nor retains aconitase activity. Consequently, IRP2 only exhibits an IRE-binding activity and does not have any enzymatic function. IRP2 is only regulated by its stability and is newly synthesized in response to low iron. Iron depletion or hypoxia leads to IRP2 stabilization [48, 50], whereas in iron-repleted cells, IRP2 becomes destabilized and undergoes rapid ubiquitination and degradation via the proteasome. More recently, an E3 ligase complex (SKP1-Cul1-FBXL5) was described to be responsible for iron-mediated IRP2 degradation [57, 58].
6.8 Redox Regulation of Iron Metabolism
Both IRP1 and IRP2 are sensitive to ROS and RNS (reactive nitrogen species) (reviewed in [59, 60]). This is evident through the redox regulation of IRP1 through its ISC. Exposure of cells to micromolar concentrations of ROS and RNS (especially NO) also leads to the destabilization of the IRP1-ISC complex with subsequent induction of IRE-binding activity via an incompletely characterized mechanism. This response can be antagonized by MPO-derived hypochlorite [61]. In vitro studies showed that ROS and RNS remove iron of the ISC and convert it to a nonfunctional cluster. IRP also responds to NO, which likewise induces IRE binding at the expense of its aconitase activity [62]. However, ROS and RNS may also modify potential cluster-destabilizing factors rather than the IRP1-ISC complex itself. Furthermore, some discrepancies are found in the literature about the regulation of IRP2 and NO. NO may either positively or negatively regulate IRP2, which could be explained by differential effects of the numerous NO species. Taken together, the redox regulation of IRE-IRP directly links the iron metabolism to inflammatory processes, hypoxia, and oxidative stress.
6.9 Non-hepatic Causes of Alcohol-Mediated Iron Overload
Although the liver is the major target organ of alcohol consumption, leading to the formation of steatosis in 90 %, inflammation (hepatitis) in 30 %, and cirrhosis in ca. 15 % (see also Fig. 6.3), various other target sites of iron homeostasis are affected (Fig. 6.4, black circles). Such potential sites include the duodenum, which is typically inflamed during chronic alcohol exposure. Chronic alcohol consumption may further affect the binding of iron to transferrin and the generation of newly formed erythrocytes via interference with vitamin B12 and folate metabolism, with secondary effects on iron utilization. Of course, overall iron metabolism can be affected in ALD patients simply by bleeding due to inflammatory conditions or ulcers in the upper gastrointestinal tract or due to complications of liver cirrhosis such as hypertensive gastropathy and esophageal varices. The stability of erythrocytes is also strongly affected by chronic alcohol consumption, leading to an earlier sequestration by macrophages as well as the release of iron from macrophages.
6.9.1 Alcohol and Hematopoietic System
Alcoholics are often found to have an abnormal hemogram because of direct damaging effects on hematopoietic cells and pathologic consequences on hematopoiesis. Until recently, the frequent coexisting formation of infections in liver diseases and malnutrition have been thought to be primary the cause of these changes. However, nowadays it is appreciated that alcohol alone is capable of producing several types of hematologic abnormalities such as diseases of red blood cells, white blood cells, and platelets. Red blood cells are pathologically affected by alcohol in nearly every stage of their life cycle [63]. Alcohol impairs red blood cell production in the bone marrow, which also has dramatic effects on erythrocyte maturation, delivery, and life span. Alcohol is also a potent folic acid antagonist. It blocks its absorption in the jejunum and decreases serum folate levels. Folic acid deficiency may be a key hematologic abnormality in ALD and the resulting anemia is almost invariably secondary to alcoholism and may therefore be the most common type of anemia seen in hospitals. Chronic alcohol abuse also leads to altered vitamin B6 (pyridoxine, pyridoxal, and pyridoxamine) metabolism often resulting in deficiency of the coenzyme pyridoxal phosphate, which is important for heme biosynthesis as described in a study by Hines et al. [64]. Lumeng and Li reported an alternative mechanism: acetaldehyde reduces the synthesis of intracellular pyridoxal phosphate by increasing membrane-bound phosphatases resulting in accelerated degradation of pyridoxal phosphate within erythrocytes [65]. Erythrocyte abnormalities are also due to alcohol and alcohol-mediated changes of the iron metabolism. Thus, a sideroblastic hypochromic anemia was found in alcoholic patients in whom the red blood cells appear similar to that seen in iron deficiencies. Further effects on red blood cell survival are seen as an effect of alcohol by producing several types of the hemolytic syndrome [66]. Chronic alcohol ingestion is also associated with a diminished granulocyte precursor pool in the bone marrow and functional disturbances that may lead to neutrocytopenia, brought into connection with the high susceptibility to infections of alcoholics [67]. Also dysfunctional monocyte-macrophages and lymphocytes may undoubtedly contribute to the predisposition to infections seen in ALD patients. Platelets are also affected by alcohol withdrawal, as thrombocytopenia has been frequently observed in ALD patients. Here, dietary folic acid deficiency is questionable as the underlying reason, but a direct effect of alcohol on platelet production and reduced survival may be the case [68]. All these changes and hematopoietic disorders seem to be reversible and return to normal levels after alcohol withdrawal, although, for example, an enlarged erythrocyte volume (MCV) can be detected still years after abstaining from alcohol.
6.9.2 Alcohol and Hemolysis
An increased sequestration and degradation of erythrocytes is observed in the spleen mainly due to alcohol-induced alterations to red blood cells. Typically observed morphological changes related to membrane lipids due to peroxidation of fatty acids [69] include stomatocytes, knizocytes (also called bridge cells or target cells), acanthoid cells, and irregularly spiculated cells [70]. Hemolysis often ultimately leads to an enlarged spleen (splenomegaly) rather than cirrhosis. Therefore, ALD patients often have elevated levels of heme oxygenases (HO-1) and indirect bilirubin (called hyperbilirubinemia), as a breakdown product from heme catabolism (unpublished observations). Disruption of the hepato-protective glutathione metabolism [71–73] may be another important reason contributing to hemolysis. A loss of reduced glutathione could sensitize cells for oxidative damage by iron overload or infections often coexisting in ALD patients.
6.9.3 Alcohol and Nutrition
Excessive and chronic alcohol consumption frequently causes malnutrition and vitamin deficiency. This malnutrition is often due to poor dietary intake because energy supply is to 50 % replaced by alcohol, therefore leading to inadequate calorie intake or periods without any food intake [74]. The status of malnutrition often correlates with the severity of ALD. Furthermore, gastrointestinal disturbances, e.g., maldigestion and malabsorption of nutrients, have also been observed [75]. These disturbances include dysgeusia (disorder of the sense of taste), anorexia, nausea, and early satiety [76]. Intestinal malabsorption and pancreatic dysfunction are seen in alcoholic patients with cirrhosis and in alcoholic patients with minimal and low liver disease. The substances that have been shown to be a malabsorbed are d-xylose, thiamine, folic acid, and lipid [77]. Deficiencies in nutrients like zinc, magnesium, and vitamin A, among many others, are also very common in these patients [78]. Therefore, additional sufficient nutritional support is an important therapeutic strategy [79].
6.10 Hepatic Causes of Iron Overload in ALD
The complete picture of the molecular changes in iron metabolism during chronic alcohol consumption is not yet completely understood (Fig. 6.8). There are various studies that focused on the central systemic master switch hepcidin. Both acute and chronic alcohol exposure seem to suppress hepcidin expression in the liver and in sera from patients with ALD and also pro-hepcidin levels are reduced. Ohtake and coworkers demonstrated that serum hepcidin is decreased in patients with ALD [80]. Most notably, Harrison-Findik et al. showed in their elegant study using an alcohol mouse model that the expression of hepatic hepcidin is rapidly suppressed upon exposure to alcohol [81]. 4-Methylpyrazole, a competitive inhibitor of alcohol-metabolizing enzymes (e.g., alcohol dehydrogenases), abolished this effect. However, ethanol did not alter the expression of TfR1 and ferritin or the activation of iron regulatory mRNA-binding proteins IRP1 and IRP2. Mice maintained on 10–20 % ethanol for 7 days displayed a downregulation of liver hepcidin without changes in liver triglycerides or histology. This was accompanied by elevated DMT1 and ferroportin expression in the duodenum [81]. Ethanol downregulated hepcidin promoter activity and the DNA-binding activity of the CCAT/NH-binding protein α (C/EBPα) but not β [82]. The same author showed later that the effect of alcohol and hepcidin was independent of Kupffer cell activation and TNFα signaling [83]. Conversely, there are observational studies showing that the hepatic expression of TfR1 is increased under conditions of alcohol consumption. In 2002 Suzuki et al. showed that TfR1 is increased in patients with ALD and that abstinence from drinking decreased TfR1 [20]. Furthermore, ethanol increased transferrin-bound iron uptake into hepatocytes, possibly due to ethanol-induced TfR1 expression and partly mediated by activation of IRP1 [84, 85]. TfR1 upregulation may be directly related to translational affects by ROS and not due to IRP1 [86].

Present understanding of systemic and cellular changes of iron homeostasis in ALD ultimately leading to cancer
Notably, we could not find differences in the expression of various iron-related mRNAs in a preliminary microarray study comparing iron overload and normal ALD patients (Mueller, S. et al., unpublished). We also noted a high variability in the expression of these mRNAs, implicating some caution in the interpretation of liver samples from ALD patients. It seems that ALD lesions are quite heterogeneously scattered throughout the liver. Therefore, it remains an open question whether the effects observed in an acute alcohol-exposure model in rodents are directly related to alcohol and hepcidin. Additional indirect conditions on the perfusion of the splanchnic system could also be involved. Two recent publications have tried to further elucidate the mechanisms of alcohol-mediated hepcidin expression [43, 87]. Both studies showed that bolus H2O2 treatments, a major ROS insult, could drastically suppress hepcidin expression. However, we recently found that this effect is rather due to the artificial exposure to high H2O2 levels (>50 μM) than peroxide itself [42]. If H2O2 is released continuously at low concentrations (between 0.3 and 6 μM) to hepatoma and primary liver cells, as they are typically released by inflammatory cells or during cellular metabolism, a strong hepcidin upregulation rather than suppression was observed. H2O2 synergistically stimulates hepcidin promoter activity in combination with recombinant IL-6 or BMP-6 in a manner that requires the functional STAT3-responsive element. The H2O2-mediated hepcidin induction requires STAT3 phosphorylation and is effectively blocked by siRNA-mediated STAT3 silencing, overexpression of SOCS3 (suppressor of cytokine signaling 3), and antioxidants such as N-acetylcysteine [42].
In summary, numerous rodent and human studies suggest the involvement of key iron molecules in the iron overload of ALD patients. These molecules may include hepcidin, TfR1, IRP1, and ROS. However, no definite conclusions can be made due to methodological challenges and complex interactions among these molecules.
6.11 Therapy of ALD in the Context of Iron Overload
Based on our preliminary knowledge of underlying mechanisms of iron disturbances in patients with ALD, no treatment options are readily available. The idea of iron depletion by phlebotomy has been controversially discussed in patients with HCV that equally develop iron overload; however, phlebotomy is highly propagated in certain countries, such as Japan. There has been one trial initiated in France in 2012 and titled, “phlebotomy in risk of HCC in patients with compensated alcoholic cirrhosis” (Tirrox). Unfortunately, this study has stopped recruiting patients due to delayed recruitment compared to that anticipated (REF: ClinicalTrials.gov NCT01342705). For the moment it can be regularly perceived that iron overload is an important factor in the progression of ALD, also in combination with other liver diseases, such as HCV. It is an important challenge for the future to clearly identify the underlying molecular mechanisms and to develop novel targeted therapeutic strategies and to improve the early detection of hepatic iron overload in alcoholic liver disease (e.g., measurement of liver iron by susceptometry or hepcidin-ELISA for diagnostic purposes). Future studies will continue with the goal of preventing fibrosis progression and to prevent HCC development, thereby improving overall survival by targeted therapeutic approaches that may include a pharmacological intervention leading to stimulation of hepcidin.
6.12 Summary
Presently, the pathological progression regarding iron metabolism in patients with chronic alcohol consumption is not completely and conclusively understood. First findings on human subjects and animal models indicate that the systemic iron master switch hepcidin is suppressed. Hepcidin suppression likely contributes to the hepatic iron overload observed in these patients. On the other side, it seems not to be clear whether ROS play an important role in mediating this hepcidin suppression. Although, ROS are assuredly involved in alcohol metabolism, specific levels have never really been detected inside hepatic cells. Additionally, the suppression of hepcidin has only been shown in bolus treatment experiments that do not recapitulate the physiological condition. Contrary, low steady state levels of peroxide are able to induce hepcidin. Unfortunately, the clinical characterization of iron status in patients with ALD is complicated and heterogeneous. It is well known that these patients have low hemoglobin levels, which cannot be solely attributed to bleeding events. One mechanistic explanation includes an ROS-mediated induction of hepcidin. Furthermore, patients with ALD display increased transferrin saturation and ferritin levels, which are not only observed in cirrhotic patients and are clearly related to iron deposition found in macrophages and also in hepatocytes. This means that iron accumulation in the liver of ALD patients is not solely due to inflammatory events mediated by macrophages. The incomplete characterization of the iron physiology itself further aggravates the studies on ALD patients. For instance, it is not clear why in vivo and in vitro findings differ with regard to hepcidin responses toward iron exposure. Under in vivo conditions, iron loading leads to induction of hepcidin. The contrary is the case if liver cells in vitro were exposed to iron, which leads to subsequent downregulation of hepcidin. Therefore, it appears that the complicated network of ferroportin expression and potential systemic and paracrine effects of hepcidin are not yet fully understood. Presently, it should be well accepted that ROS are involved with and are responsible for iron disturbance in ALD. Additionally, low ROS levels lead to IRP1 and TfR1 upregulation that cause iron uptake and hepcidin upregulation that further results in iron retention in hepatocytes. Higher ROS levels that may destroy and damage hepatocytes can locally dramatically reduce and suppress hepcidin levels, what then could result in an “overflow” of iron and lead to further iron accumulation. It is quite conceivable that the presence of increased ROS (oxidative stress) and iron would dramatically increase Fenton-like reactions mentioned above, which then can be a highly cancerogenic environment. In summary, potential mechanisms of iron overload in ALD include:
-
1.
Suppression of hepcidin via impaired redox signaling (role of NADPH oxidases) and overproduction of ROS
-
2.
Impaired iron recycling (hemolysis)
-
3.
Ineffective erythropoiesis
-
4.
Retinoid signaling
-
5.
Other iron proteins, e.g., TfR1 and IRP1
-
6.
Interference of molecules with iron chelating and antioxidant property modifying iron absorption in the small intestine [88]
Abbreviations
- ALAS:
-
Aminolevulinic acid synthase
- ALD:
-
Alcoholic liver disease
- DMT1:
-
Divalent metal transporter 1
- FP:
-
Ferroportin
- GGT:
-
Gamma-glutamyltransferase
- HCC:
-
Hepatocellular carcinoma
- HCV:
-
Hepatitis C virus
- HFE:
-
Gene that is mutated in hereditary hemochromatosis
- HH:
-
Hereditary hemochromatosis
- HO-1:
-
Heme oxygenase 1
- IRE:
-
Iron-responsive element
- IRP:
-
Iron regulatory protein
- ISC:
-
Iron sulfur cluster
- MPO:
-
Myeloperoxidase
- NO:
-
Nitrogen oxide
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- SOCS3:
-
Suppressor of cytokine signaling 3
- STAT3:
-
Signal transducer and activator of transcription 3
- TfR:
-
Transferrin receptor
- TNF:
-
α Tumor necrosis factor α
- YLL:
-
Years of life loss
References
Seitz HK, Stickel F (2007) Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer 7:599–612
Seitz HK, Mueller S (2011) Ethanol metabolism and its consequences. In: Anzenbacher P, Zanger U (eds) Metabolism of Drugs and Xenobiotics. Weinheim, Wiley
Mueller S, Millonig G, Seitz HK, Waite GN (2012) Chemiluminescence detection of hydrogen peroxide. In: Schipper H, Pantopoulos K (eds) Principles of Free Radical Biomedicine. Nova Publishers, New York
Mueller S, Millonig G, Seitz HK (2009) Alcoholic liver disease and hepatitis C: a frequently underestimated combination. World J Gastroenterol 15:3462–3471
Cochrane J, Chen H, Conigrave KM, Hao W (2003) Alcohol use in China. Alcohol Alcohol 38:537–542
Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL et al (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2095–2128
Powell LW (1975) The role of alcoholism in hepatic storage disease. In: Seixas FA, Williams K, Seggleston S (eds) Medical consequences of alcoholism. New York Academy of Sciences, New York
Finch SC, Finch CA (1955) Idiopathic hemochromatosis, an iron storage disease. A iron metabolism in hemochromatosis. Medicine (Baltimore) 34:381–430
Sheldon JH (1935) Haemochromatosis. Oxford University Press, London
Bell ET (1955) The relation of portal cirrhosis to hemochromatosis and to diabetes mellitus. Diabetes 4:435–446
Conrad ME, Berman A Jr, Crosby WH (1962) Iron kinetics in laennec’s cirrhosis. Gastroenterology 43:385–390
Tavill AS, Qadri AM (2004) Alcohol and iron. Semin Liver Dis 24:317–325
Shoden A, Gabrio BW, Finch CA (1953) The relationship between ferritin and hemosiderin in rabbits and man. J Biol Chem 204:823–830
Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Wolff RK et al (1996) A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 13:399–408
Pascoe A, Kerlin P, Steadman C, Clouston A, Jones D, Powell L, Jazwinska E, Lynch S, Strong R (1999) Spur cell anaemia and hepatic iron stores in patients with alcoholic liver disease undergoing orthotopic liver transplantation. Gut 45:301–305
Sohda T, Takeyama Y, Irie M, Kamimura S, Shijo H (1999) Putative hemochromatosis gene mutations and alcoholic liver disease with iron overload in Japan. Alcohol Clin Exp Res 23:21S–23S
Dostalikova-Cimburova M, Kratka K, Stransky J, Putova I, Cieslarova B, Horak J (2012) Iron overload and HFE gene mutations in Czech patients with chronic liver diseases. Dis Markers 32:65–72
Chapman RW, Morgan MY, Laulicht M, Hoffbrand AV, Sherlock S (1982) Hepatic iron stores and markers of iron overload in alcoholics and patients with idiopathic hemochromatosis. Dig Dis Sci 27:909–916
Bell H, Skinningsrud A, Raknerud N, Try K (1994) Serum ferritin and transferrin saturation in patients with chronic alcoholic and non-alcoholic liver diseases. J Intern Med 236:315–322
Suzuki Y, Saito H, Suzuki M, Hosoki Y, Sakurai S, Fujimoto Y, Kohgo Y (2002) Up-regulation of transferrin receptor expression in hepatocytes by habitual alcohol drinking is implicated in hepatic iron overload in alcoholic liver disease. Alcohol Clin Exp Res 26:26S–31S
Mueller S (2013) Non-invasive assessment of patients with alcoholic liver disease. Clin Liver Dis 2:68–71
Mueller S (2013) The role of iron in alcoholic liver disease. Alcohol Alcohol 48:6
Fenton HJ (1894) Oxidation of tartaric acid in presence of iron. J Chem Soc Trans 65:899–910
Saran M, Michel C, Stettmaier K, Bors W (2000) Arguments against the significance of the Fenton reaction contributing to signal pathways under in vivo conditions. Free Radic Res 33:567–579
Niederau C, Fischer R, Purschel A, Stremmel W, Haussinger D, Strohmeyer G (1996) Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 110:1107–1119
Ko C, Siddaiah N, Berger J, Gish R, Brandhagen D, Sterling RK, Cotler SJ, Fontana RJ, McCashland TM, Han SH, Gordon FD, Schilsky ML, Kowdley KV (2007) Prevalence of hepatic iron overload and association with hepatocellular cancer in end-stage liver disease: results from the National Hemochromatosis Transplant Registry. Liver Int 27:1394–1401
Turlin B, Juguet F, Moirand R, Le Quilleuc D, Loreal O, Campion JP, Launois B, Ramee MP, Brissot P, Deugnier Y (1995) Increased liver iron stores in patients with hepatocellular carcinoma developed on a noncirrhotic liver. Hepatology 22:446–450
Zacharski LR, Chow BK, Howes PS, Shamayeva G, Baron JA, Dalman RL, Malenka DJ, Ozaki CK, Lavori PW (2008) Decreased cancer risk after iron reduction in patients with peripheral arterial disease: results from a randomized trial. J Natl Cancer Inst 100:996–1002
Hann HW, Stahlhut MW, Blumberg BS (1988) Iron nutrition and tumor growth: decreased tumor growth in iron-deficient mice. Cancer Res 48:4168–4170
Tsukamoto H, Horne W, Kamimura S, Niemela O, Parkkila S, Yla-Herttuala S, Brittenham GM (1995) Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest 96:620–630
D’Amico G, Garcia-Tsao G, Pagliaro L (2006) Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol 44:217–231
Ganne-Carrie N, Christidis C, Chastang C, Ziol M, Chapel F, Imbert-Bismut F, Trinchet JC, Guettier C, Beaugrand M (2000) Liver iron is predictive of death in alcoholic cirrhosis: a multivariate study of 229 consecutive patients with alcoholic and/or hepatitis C virus cirrhosis: a prospective follow up study. Gut 46:277–282
Wang J, Pantopoulos K (2011) Regulation of cellular iron metabolism. Biochem J 434:365–381
Andrews NC (1999) Disorders of iron metabolism. N Engl J Med 341:1986–1995
Ganz T (2007) Molecular control of iron transport. J Am Soc Nephrol 18:394–400
Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S (2001) Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A 98:8780–8785
Theil EC (2003) Ferritin: at the crossroads of iron and oxygen metabolism. J Nutr 133:1549S–1553S
Nemeth E, Ganz T (2009) The role of hepcidin in iron metabolism. Acta Haematol 122:78–86
Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of mammalian iron metabolism. Cell 142:24–38
Sow FB, Florence WC, Satoskar AR, Schlesinger LS, Zwilling BS, Lafuse WP (2007) Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J Leukoc Biol 82:934–945
Weiss G, Goodnough LT (2005) Anemia of chronic disease. N Engl J Med 352:1011–1023
Millonig G, Ganzleben I, Peccerella T, Casanovas G, Brodziak-Jarosz L, Breitkopf-Heinlein K, Dick TP, Seitz HK, Muckenthaler MU, Mueller S (2012) Sustained submicromolar H2O2 levels induce hepcidin via signal transducer and activator of transcription 3 (STAT3). J Biol Chem 287:37472–37482
Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA (2008) Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 48:1420–1429
Ponka P, Beaumont C, Richardson DR (1998) Function and regulation of transferrin and ferritin. Semin Hematol 35:35–54
Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, Koeffler HP (1999) Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem 274:20826–20832
Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP (2001) Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 98:2714–2719
Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, Sheftel AD, Ponka P (2010) Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A 107:10775–10782
Rouault TA (2006) The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol 2:406–414
Recalcati S, Minotti G, Cairo G (2010) Iron regulatory proteins: from molecular mechanisms to drug development. Antioxid Redox Signal 13:1593–1616
Wallander ML, Leibold EA, Eisenstein RS (2006) Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim Biophys Acta 1763:668–689
Lok CN, Ponka P (2000) Identification of an erythroid active element in the transferrin receptor gene. J Biol Chem 275:24185–24190
Torti FM, Torti SV (2002) Regulation of ferritin genes and protein. Blood 99:3505–3516
Ponka P, Lok CN (1999) The transferrin receptor: role in health and disease. Int J Biochem Cell Biol 31:1111–1137
Piccinelli P, Samuelsson T (2007) Evolution of the iron-responsive element. RNA 13:952–966
Deck KM, Vasanthakumar A, Anderson SA, Goforth JB, Kennedy MC, Antholine WE, Eisenstein RS (2009) Evidence that phosphorylation of iron regulatory protein 1 at Serine 138 destabilizes the [4Fe-4S] cluster in cytosolic aconitase by enhancing 4Fe-3Fe cycling. J Biol Chem 284:12701–12709
Sanchez M, Galy B, Muckenthaler M, Hentze MW (2007) Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat Struct Mol Biol 14:420–426
Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, Sangfelt O, Spruck C, Leibold EA, Wohlschlegel JA (2009) Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326:718–721
Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, Grishin NV, Bruick RK (2009) An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326:722–726
Fillebeen C, Pantopoulos K (2002) Redox control of iron regulatory proteins. Redox Rep 7:15–22
Mueller S (2005) Iron regulatory protein 1 as a sensor of reactive oxygen species. Biofactors 24:171–181
Mutze S, Hebling U, Stremmel W, Wang J, Arnhold J, Pantopoulos K, Mueller S (2003) Myeloperoxidase-derived hypochlorous acid antagonizes the oxidative stress-mediated activation of iron regulatory protein 1. J Biol Chem 278:40542–40549
Watts RN, Hawkins C, Ponka P, Richardson DR (2006) Nitrogen monoxide (NO)-mediated iron release from cells is linked to NO-induced glutathione efflux via multidrug resistance-associated protein 1. Proc Natl Acad Sci U S A 103:7670–7675
Hillman RS (1975) Alcohol and hematopoiesis. In: Seixas FA, Williams K, Seggleston S (eds) Medical consequences of alcoholism. New York Academy of Sciences, New York
Hines JD (1975) Hematologic abnormalities involving vitamin B6 and folate metabolism in alcoholic subjects. In: Seixas FA, Williams K, Seggleston S (eds) Medical consequences of alcoholism. New York Academy of Sciences, New York
Lumeng L, Li TK (1974) Vitamin B6 metabolism in chronic alcohol abuse. J Clin Invest 53:698
Herbert V, Tisman G (1975) Hematologic effects of alcohol. In: Seixas FA, Williams K, Seggleston S (eds) Medical consequences of alcoholism. New York Academy of Sciences, New York
Scharf R, Aul C (1988) Alcohol-induced disorders of the hematopoietic system. Z Gastroenterol 26(Suppl 3):75–83
Eichner ER (1973) The hematologic disorders of alcoholism. Am J Med 54:621–630
Lindenbaum J (1987) Hematologic complications of alcohol abuse. Semin Liver Dis 7:169–181
Niemela O, Parkkila S (2004) Alcoholic macrocytosis—is there a role for acetaldehyde and adducts? Addict Biol 9:3–10
Pitcher CS, Williams R (1963) Reduced red cell survival in jaundice and its relation to abnormal glutathione metabolism. Clin Sci 24:239–252
Krasnow SE, Walsh JR, Zimmerman HJ, Heller P (1957) Megaloblastic anemia in alcoholic cirrhosis. AMA Arch Intern Med 100:870–880
Maturu P, Reddy VD, Padmavathi P, Varadacharyulu N (2012) Ethanol induced adaptive changes in blood for the pathological and toxicological effects of chronic ethanol consumption in humans. Exp Toxicol Pathol 64:697–703
McClain CJ, Barve SS, Barve A, Marsano L (2011) Alcoholic liver disease and malnutrition. Alcohol Clin Exp Res 35:815–820
Mezey E (1975) Intestinal function in chronic alcoholism. In: Seixas FA, Williams K, Seggleston S (eds) Medical consequences of alcoholism. New York Academy of Sciences, New York
Madden AM, Bradbury W, Morgan MY (1997) Taste perception in cirrhosis: its relationship to circulating micronutrients and food preferences. Hepatology 26:40–48
Mezey E (1982) Liver disease and protein needs. Annu Rev Nutr 2:21–50
Hanje AJ, Fortune B, Song M, Hill D, McClain C (2006) The use of selected nutrition supplements and complementary and alternative medicine in liver disease. Nutr Clin Pract 21:255–272
Stickel F, Hoehn B, Schuppan D, Seitz HK (2003) Review article: Nutritional therapy in alcoholic liver disease. Aliment Pharmacol Ther 18:357–373
Ohtake T, Saito H, Hosoki Y, Inoue M, Miyoshi S, Suzuki Y, Fujimoto Y, Kohgo Y (2007) Hepcidin is down-regulated in alcohol loading. Alcohol Clin Exp Res 31:S2–S8
Harrison-Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, Clemens D, Fein E, Andriopoulos B, Pantopoulos K, Gollan J (2006) Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem 281:22974–22982
Harrison-Findik DD, Klein E, Crist C, Evans J, Timchenko N, Gollan J (2007) Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology 46:1979–1985
Harrison-Findik DD, Klein E, Evans J, Gollan J (2009) Regulation of liver hepcidin expression by alcohol in vivo does not involve Kupffer cell activation or TNF-alpha signaling. Am J Physiol Gastrointest Liver Physiol 296:G112–G118
Shindo M, Torimoto Y, Saito H, Motomura W, Ikuta K, Sato K, Fujimoto Y, Kohgo Y (2006) Functional role of DMT1 in transferrin-independent iron uptake by human hepatocyte and hepatocellular carcinoma cell, HLF. Hepatol Res 35:152–162
Suzuki M, Fujimoto Y, Suzuki Y, Hosoki Y, Saito H, Nakayama K, Ohtake T, Kohgo Y (2004) Induction of transferrin receptor by ethanol in rat primary hepatocyte culture. Alcohol Clin Exp Res 28:98S–105S
Andriopoulos B, Hegedusch S, Mangin J, Hd R, Hebling U, Wang J, Pantopoulos K, Mueller S (2007) Sustained hydrogen peroxide induces iron uptake by transferrin receptor-1 independent of the iron regulatory protein/iron-responsive element network. J Biol Chem 282:20301–20308
Nishina S, Hino K, Korenaga M, Vecchi C, Pietrangelo A, Mizukami Y, Furutani T, Sakai A, Okuda M, Hidaka I, Okita K, Sakaida I (2008) Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology 134:226–238
Ren Y, Deng F, Zhu H, Wan W, Ye J, Luo B (2011) Effect of epigallocatechin-3-gallate on iron overload in mice with alcoholic liver disease. Mol Biol Rep 38:879–886
Acknowledgment
This work was supported by the Dietmar-Hopp-Stiftung and the Manfred-Lautenschläger-Stiftung.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this paper
Cite this paper
Mueller, S., Rausch, V. (2015). The Role of Iron in Alcohol-Mediated Hepatocarcinogenesis. In: Vasiliou, V., Zakhari, S., Seitz, H., Hoek, J. (eds) Biological Basis of Alcohol-Induced Cancer. Advances in Experimental Medicine and Biology, vol 815. Springer, Cham. https://doi.org/10.1007/978-3-319-09614-8_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-09614-8_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09613-1
Online ISBN: 978-3-319-09614-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)