
Abstract
Chronic Kidney Disease (CKD) is a frequent comorbid condition and a major determinant of outcomes in patients with heart failure (HF). In acute and chronic HF, deterioration of kidney function usually occurs. The cause of this deterioration is much more complex than first thought and represents a combination of various pathophysiological pathways. Apart from low cardiac output (forward failure), tubuloglomerular feedback and increased intraabdominal pressure; increased venous congestion including renal vessels plays a major role. Regardless of the cause, the sustained deterioration of kidney function and increased systemic and renal congestion is related with increased morbidity and mortality in HF. Thus the therapeutic aim must be the relief of decongestion without compromising renal function. To target this issue we need novel diagnostic markers which shown the damage earlier and evidence based therapeutic options. To accomplish these issues active research is necessary. Based on these data in the present discussion, the mechanisms, outcomes, prognostic markers and the treatment options especially related with renal congestion in HF is summarised.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara Clinical Case ScenarioPeter Anderson is now again admitted for decompensated heart failure, for the 3 rd time in the last 12 months. Peter is a 67 years old obese (BMI 38) previous smoker with two myocardial infarctions 5 and 8 years ago. He also suffers from type 2 diabetes with poor glucose control making him incapable of limiting his fluid intake. He was discharged 7 weeks ago on high doses of oral furosemide (250+120 mg) on top of his regular heart failure medications (full-dose ramipril, metoprolol and 25 mg spironolactone).
Peter was admitted last evening. His echocardiogram showed a surprisingly preserved EF of 45%, but an enlarged vena cava without respiratory change indicating a CVP >20 mmHg. He has already received an IV furosemide infusion of 250 mg during the night but this achieved little diuresis. His creatinine has increased from 185 to now 320 µmol/L, potassium to 6.0 mmol/L, and he is also severely constipated. You are contacted as consultant for a discussion on which of the heart failure medications to reduce/discontinue, and on how much more furosemide he should receive.
Introduction
Cardiorenal interactions in heart failure have become increasingly recognised, and achieving adequate control of congestion with simultaneous preservation of renal function has become a goal of good patient management. Renal congestion and deterioration of kidney function is common in patients with heart failure and associated with increased risk of hospital readmission and both in-hospital and post-discharge mortality [1]. The exact pathophysiologic mechanisms, prognostic markers and treatment options regarding renal congestion and deterioration of kidney function in heart failure are not known despite increased research. In the present discussion, the mechanisms, outcomes, prognostic markers and the treatment options related with renal congestion and deterioration of kidney function in heart failure is summarised.
The Pathophysiology of Renal Congestion in Heart Failure
The pathophysiology of renal congestion and deterioration of kidney function in heart failure is very complex and multiple pathways are involved simultaneously. Below, these mechanisms are summarized.
Firstly, age, hypertension, and diabetes may act as unifying factors that associate heart failure with renal dysfunction and their coexistence can be considered to be partly due to common effects of the process of atherosclerosis on the heart and the kidney [2].
Secondly, arterial underfilling was one of the mechanisms involved in this process. Indeed, traditionally this mechanism was accepted as a main cause of deterioration of kidney function. Regarding the hypo perfusion (as also called forward failure) when mean aortic pressure is reduced; the renal perfusion pressure may also be lowered to ≤80 mmHg that is the threshold of kidney autoregulation. This threshold is important since below this threshold renal perfusion becomes directly pressure dependent [3]. Additionally, the thresholds and responses probably depend on intact endothelial function and responses, which are deranged in CKD and heart failure. At this point a high degree of neural and humoral activation occurs. The reduction in perfusion pressure is sensed by baroreceptors that increase catecholamine release from the sympathetic nervous system and adrenal glands. This increase in sympathetic activity, and the reduced cardiac output itself, elicit release of renin from granular cells in the juxtaglomerular apparatus of the nephron. Renin cleaves angiotensinogen to angiotensin I, which angiotensin-converting enzyme (ACE) converts to angiotensin II (AngII). AngII elicits positive feedback on the sympathetic nervous system, facilitating further catecholamine release. Both AngII and catecholamines induce glomerular arteriolar vasoconstriction, decreasing renal plasma flow. Yet AngII has a disproportionate vasoconstrictive effect on the efferent arteriole, preserving the glomerular filtration rate (GFR) despite reduced renal plasma flow [4]. Thus initially the filtration fraction and glomerular filtration rate was preserved but when this activation progress (if AngII levels and/or catecholamine levels are very high), it causes more preglomerular vasoconstriction leading to decrease in GFR [5]. This in turn activates proximal tubular sodium and water reabsorption leading to more congestion [6, 7].
Thirdly, the other determinant of kidney function in heart failure is the tubuloglomerular feedback. In the tubuloglomerular feedback, distal chloride delivery is sensed by the loop diuretic–sensitive sodium/potassium/2 chloride co transporter (NKCC2) in the macula densa at the end of Henle’s loop. The hairpin orientation of the loop of Henle allows for close proximity of the macula densa with the other elements of the juxtaglomerular apparatus, the afferent arteriolar smooth muscle cells and the renin-secreting granular cells at the glomerular vascular pole. When volume expansion or increased GFR results in increased chloride delivery to the macula densa, TGF mediates afferent arteriolar vasoconstriction and decreased renin release. This afferent vasoconstriction, with efferent arteriolar vasodilatation from the fall in AngII, decreases GFR. In heart failure, high AngII and catecholamine levels increase proximal tubular reabsorption of solute. This reduces distal chloride delivery and the opposite downstream events occur: the afferent arteriole vasodilates and renin release increases, leading to increased efferent arteriolar tone [4].
Fourthly, the role of increased intra abdominal pressure (IAP) must be mentioned. Abdominal congestion, i.e., splanchnic, venous, and interstitial congestion, manifests in a substantial number of patients with advanced congestive heart failure, yet is poorly defined, and current pathophysiological models unsatisfactorily explain the detrimental link between congestion and deterioration in renal function [8]. The normal IAP is usually <5–7 mmHg and a constant elevation of IAP >12 mmHg defines intra-abdominal hypertension [9, 10]. Renal blood flow is determined by the abdominal perfusion pressure, which is directly related to mean arterial pressure and inversely related to IAP [11]. From the 1940s and onwards, it was shown that abdominal compression and intra abdominal hypertension decreased renal plasma flow and GFR [12]. Those findings are consistent with reports of elevated plasma renin activity and aldosterone levels during elevated IAP [13]. There has been also a concern that some of the renal dysfunction with intra abdominal hypertension is due to hypotension and low cardiac output. Nevertheless, when cardiac output is corrected by volume expansion in intra abdominal hypertension dogs, renal blood flow and GFR were still <25 % of normal [14]. Compromised capacitance function of the splanchnic vasculature and deficient abdominal lymph flow resulting in interstitial oedema might both be implied in the occurrence of elevated cardiac filling pressures and deterioration of kidney function [15]. Additional data suggest that gut-derived hormones might influence sodium homeostasis, while the entrance of bowel toxins into the circulatory system- as a result of impaired intestinal barrier function secondary to congestion- might further depress cardiac as well as renal function. Those toxins are mainly produced by microorganisms in the gut lumen, and undergo important alterations in the case of advanced heart failure, especially when renal function is depressed [8]. In fact, until recently, little attention had been paid to the role of the intestine and its microbial flora in the pathogenesis of CKD-associated inflammation and oxidative stress. Almeida Duarte et al. demonstrated penetration of bacteria across the intestinal wall and their detection in the mesenteric lymph nodes in uremic rats [16]. In more recent studies, uremia induced loss of tight junction proteins (which play important role in barrier function of intestine) has been clearly shown [17]. Thus in bowel wall oedema and ischemia occurring in decompensated heart failure has been shown to increase intestinal permeability and result in endotoxemia, systemic inflammation, and even bacterial translocation. Therefore, when present, severe oedema and hypervolemia can further impair intestinal barrier function in CKD patients. Indeed, direct evidence comes from the fact that intestinal microflora changed in patients with CKD with the abundance of Brachybacterium, Cateni-bacterium, Enterobacteriaceae, Halomonadaceae, Moraxellaceae, Nesterenkonia, Polyangiaceae, Pseudomonadaceae, and Thiothrix families compared to healthy population [18].
Taken together all these evidence suggests that changes in the composition of the gut microbiome and disruption of its barrier structure/function may result in production and absorption of noxious by-products that can contribute to the uremic toxicity and inflammation which are further exaggerated in the presence of heart failure.
Lastly, as important as the decreased perfusion, the tubuloglomerular feedback and IAP; elevated central venous pressure (CVP) and renal congestion (the specific subject of this chapter) also plays a role in renal dysfunction in heart failure (as also called backward failure). Indeed, experimental evidence from classic experiments demonstrates that blood flow through the kidney is reduced more by an increase in venous pressure than by an equivalent decrease in arterial pressure, and that there is a steeply graded relationship between change in renal venous pressure and reduction in urine flow [19]. These changes occur independently of reduction in cardiac output and mean arterial pressure, which occur much later in the progression of congestive heart failure [20]. In normal people without heart failure, the transient hypervolemic state leads to increased renal fluid and salt excretion and loss of extracellular fluid from the body becomes greater than fluid intake, and this decreases both blood volume and cardiac output, returning the pressure back to normal. However, in patients with heart failure despite an increase in blood volume (hypervolemic state) the elevated right atrial and central venous pressure causes reducing driving force of fluid and salt excretion in kidney and a vicious cycle of sodium retention, volume expansion and heart failure exacerbation occurs [21]. Indeed in patients with various cardiovascular disease, elevated CVP has been shown to reduce renal perfusion pressure and possible be associated to increased mortality [22, 23]. It was even suggested that that venous congestion (both with increased CVP on admission and inadequate decrease of venous pressure with treatment) is the strongest hemodynamic determinant of renal dysfunction and persistent reduction of cardiac output may not have a primary role in the development of the renal dysfunction. Mullens et al. studied the importance of CVP in advanced decompensated heart failure. In 145 patients with acute decompensated heart failure, worsening renal function had a greater CVP on admission (18 ± 7 mmHg vs.12 ± 6 mmHg, p < 0.001). The development of deterioration of kidney function occurred less frequently in patients who achieved a CVP < 8 mmHg (p < 0.01). Furthermore, the ability of CVP to stratify risk for development of deterioration of kidney function was apparent across the spectrum of systemic blood pressure, pulmonary capillary wedge pressure, cardiac index, and estimated glomerular filtration rate. Besides, systemic blood pressures were similar between those with versus without deterioration of kidney function [24].
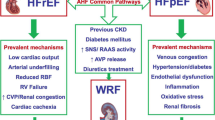
Thus as a combination, in decompensated heart failure defective renal perfusion pressure, tubuloglomerular feedback, elevated IAP and increased CVP and neurohumoral activation causes hypervolemia, renal congestion and renal dysfunction. The pathophysiologic mechanisms leading to renal congestion in heart failure are summarized in Fig. 9.1.

Mechanisms involved in renal congestion in heart failure
How Does Renal Congestion Lead to Worsening of Renal Function?
As suggested above venous congestion (including renal vein) is an important factor for kidney dysfunction. However, little is known regarding the mechanisms leading to deterioration of renal function in venous congestion. Some mechanisms such as reduced transglomerular pressure, increased interstitial pressure, interstitial fibrosis, tubular back leak probably play a role. Additionally, increased sympathetic renal nerve activity resulting in intrarenal arterial vasoconstriction and a fall in GFR play a role [25]. During renal venous hypertension there would be a rise in renal interstitial pressure that would affect the entire capillary bed and the tubules, possibly also involving local hypoxia. Compression of the tubules raises the luminal pressure, further attenuates the transglomerular pressure gradient, and lowers the GFR. It is important to appreciate that a rise in renal interstitial pressure due to venous congestion is physiologically different than that caused by elevations in arterial pressure which is associated with a natriuresis [26].
The inflammatory process is another mechanism which is thought to be involved in venous congestion mediated deterioration of kidney function [27, 28]. Inflammation can beget vascular dysfunction via endothelial activation and enhanced arterial stiffness. Second, inflammation may reduce myocardial contractility either through functional suppression of the contractile apparatus or through increased myocardial cell death. Third, inflammation may cause progressive renal dysfunction and fibrosis. Finally, inflammation may increase the permeability of the endothelium allowing extravasation of fluids into the alveolar space of the lungs and absorption of pro-inflammatory endotoxin from the bowel [27]. In heart failure and venous congestion activation of renin angiotensin system (RAS) and sympathetic system promotes inflammatory reaction. However, accumulating evidence suggests that volume overload and venous congestion independent of RAS and sympathetic system, are independently accepted as an additional source of inflammatory mediators [27]. Thus evidence is accumulating that inflammation is related with congestion. However, the exact mechanisms related to venous congestion and inflammation are not solved completely although some mechanisms are speculated. In volume overload state mesenteric venous congestion leads to bowel wall oedema with translocation of gram-negative bacteria through the endothelial cells of the intestinal villi. Lipopolysaccharide is then released into the circulation, and activating the inflammatory response [28]. The proof of this assumption comes from recent studies [29, 30]. Similarly, in a recent prospective study, endotoxin levels were higher in chronic kidney disease (CKD) patients with signs of fluid overload compared to CKD patients without fluid overload [31].
The other speculated mechanism is the activation of endothelial cells during congestion. Venous congestion itself may switch the synthetic and endocrine profile of the endothelium from quiescent toward an activated state that is pro-oxidant, proinflammatory, and vasoconstricting. Once “activated,” the endothelium can promote additional congestion through humoral, renal, and cardiac mechanisms, resulting in a deleterious positive feedback loop that leads, over time, more congestion [19].
Indeed it was clearly shown that during venous stretch endothelin-1 (ET-1), interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) can be secreted within hours of stretch exposure [32]. Besides, biomechanical signals such as stretch modulate endothelial production of reactive oxygen species [33]. Reactive oxygen species and cytokines may also trigger an inflammatory response through activation of nuclear factor (NF)-κB [34].
In summary, based on these reports, vascular stretch can activate endothelial pro-oxidant and proinflammatory programs. These results demonstrate that venous congestion and volume overload alone can promote an inflammatory state with elevations of inflammatory mediators in the circulation. Ultimately, the source of chronic inflammation in venous congestion syndrome is likely a combination of the several biologic mechanisms discussed (Fig. 9.2).

Mechanisms of increased inflammation during venous congestion
Venous Congestion: An Important Cause of Renal Dysfunction in Heart Failure
Various studies have shown that venous and renal congestion may play more important role in kidney dysfunction. In the ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) trial involved hospitalized decompensated heart failure patients in which kidney function did not correlate with cardiac index, pulmonary capillary wedge pressure, or systemic vascular resistance, but rather was associated with right atrial pressure [35]. In a retrospective analysis of 2,557 patients undergoing right heart catheterization, CVP was associated with low estimated GFR independently from cardiac index, and it predicted mortality [23]. Similarly, Guglin et al. described catheterization findings in 178 heart failure patients wherein low estimated GFR correlated with high CVP and low renal perfusion pressure but not the cardiac index or left ventricular ejection fraction [36]. Aranson et al. studied 475 patients with decompensated heart failure, of which 238 had right heart catheterization data available over the first 24 h. Net fluid loss was recorded in the first 24 h. Worsening renal function was defined as a >0.3 mg/dL increase in serum creatinine above baseline. The authors found that baseline right atrial pressure had a weak association with baseline renal function (r: −0.17, p: 0.009), but there was no association between baseline or change in right atrial pressure with the volume removed over the first 24 h, nor was there an association with the incidence of worsening renal function up to 14 days. The authors did, however, find a strong association between an increased volume of diuresis in the first 24 h of the hospitalization and a lower incidence of worsening renal function. The authors concluded that early net fluid loss is associated with worsening renal function [37]. Thus, as detailed below CVP may not be sensitive enough to detect subtle changes in volume status.
The role of haematocrit elevation (as a measure of extracellular fluid reduction) was also investigated in heart failure patients. In one study it was shown that 1,684 patients with heart failure were compared with respect to all cause mortality, cardiovascular mortality or heart failure depending on whether haemoconcentration (as a marker of decongestion) occurred or not. Haemoconcentration was defined as ≥3 % absolute increase in haematocrit. Haemoconcentration correlated with greater risk of in-hospital worsening renal function, but renal parameters generally returned to baseline within 4 weeks post-discharge. Patients with haemoconcentration were less likely to have clinical congestion at discharge and experienced greater in-hospital decreases in body weight and natriuretic peptide levels. They were also less likely to have dyspnoea, rales, and peripheral oedema at the time of discharge/day 7. After a median follow-up of 9.9 months and after adjustment for baseline clinical risk factors, every 5 % increase of in-hospital haematocrit change was associated with a decreased risk of all-cause death [hazard ratio (HR) 0.81, 95 % confidence interval (CI) 0.70–0.95]. Haematocrit change was also associated with decreased cardiovascular mortality or heart failure hospitalization at ≤100 day’s post-randomization (HR 0.73, 95% CI: 0.71–0.76). The authors concluded that haemoconcentration was associated with greater improvements in congestion and decreased mortality and heart failure re-hospitalization despite an increased risk of in-hospital worsening renal function in heart failure patients [38]. In the PROTECT trial authors also found haemoconcentration, defined as absolute in-hospital increases in haemoglobin, to correlate with favourable prognosis despite a decrease in renal function [39]. In the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness (ESCAPE) trial patients with haemoconcentration experienced greater net weight loss and substantially lower risk of mortality despite increased risk for worsening renal function [40]. On the other hand, Davila et al. demonstrated that in-hospital increases in haemoglobin associated with worsening renal function, but not mortality [41]. At this point one must mention that anaemia and iron deficiency may be important covariates in heart failure. As well known anaemia and iron deficiency is common in CKD. However, recent evidence suggests that these parameters are also important in heart failure. Indeed, patients with heart failure may be prone to the development of iron deficiency as a consequence of a depletion of iron stores or defective iron absorption and the reduced availability of iron recycled in the reticuloendothelial system [42, 43]. Indeed, in one prospective study it was shown that Treatment with ferric carboxymaltose for 24 weeks in patients who had chronic heart failure and iron deficiency with or without anaemia improved symptoms, functional capacity, and the quality of life. Importantly this benefit was seen in all patients with and without anaemia [44]. Thus apart from its role in showing extracellular fluid reduction, anaemia and iron deficiency may play a role in worse outcomes in heart failure patients with heart failure.
Is Renal Function Deterioration During Heart Failure Treatment Good or Bad?
Classically, it was accepted that deterioration in renal function was associated with increased mortality among patients with heart failure [45, 46]. A retrospective analysis of the ADHERE database suggests that serum creatinine >2.75 mg/dl is a significant risk factor for mortality in patients with heart failure [47]. However, recent findings challenged this concept and showed that deterioration in renal function had either no effect or beneficial impact [35, 48–52]. The cause of these contrasting findings is unknown but the effects of congestion may be responsible. To address this issue the independent effect of deterioration in renal function and presence of congestion during discharge in acute heart failure patients were investigated by Metra et al. Congestion was defined as the persistence of one or more signs or symptoms of fluid overload at discharge. The following symptoms and signs were prospectively considered: third heart sound, pulmonary rales, jugular venous stasis, hepatomegaly, and peripheral oedema. The outcome measures were post discharge mortality and acute heart failure readmission. There was no difference with respect to outcomes in patients with deterioration in renal function and no congestion and without deterioration in renal function and no congestion. However, the outcomes were worse in patients with congestion alone (without deterioration in renal function) and with congestion and deterioration in renal function. In the last patient group the hazard ratio for mortality was 2.44 (CI: 1.24–4.18). The authors concluded that deterioration in renal function alone, when detected using serial serum creatinine measurements, is not an independent determinant of outcomes in patients with acute heart failure. It has an additive prognostic value only when it occurs in patients with persistent signs of congestion [53]. Taken together, these data suggest that change in congestion status is perhaps a key variable underlying the impact of deterioration in renal function in this population.
Second selection bias may be another explanation for contrasting findings. For example, sicker patients who are more congested and have a longer hospital stay tend to have more creatinine measurements done and hence have a greater likelihood of showing a creatinine increase. Thus an increase in serum creatinine would be simply a marker of more severe heart failure rather than of progressive kidney disease [53]. Third, increases in serum creatinine levels may just be caused by renal haemodynamic abnormalities and diuretic therapy [5]. Indeed, low cardiac output and increased CVP and renal vein pressure may cause a reduction in the glomerular filtration pressure, progressive kidney disease and resistance to furosemide administration [23, 24]. Fourth, CVP a common surrogate for venous congestion was recently found to track poorly with fluid removal [37, 54]. The highly compliant nature of the venous system enables large changes in blood volume to be associated with small changes in pressure. Thus, even an effective treatment of volume overload may not be sufficient to produce a meaningful reduction in CVP and, in turn, reduce the risk of renal impairment. Besides, in most of the experimental studies showing relationship with venous pressure and deterioration in kidney function, the venous pressure was abruptly raised to extremely high values that are usually not seen even in patients with severe heart failure (e.g. 25–50 mmHg) [55]. For example, in the isolated perfused rat kidney model, GFR was not significantly altered until the imposed venous pressure reached 25 mmHg [56]. However, it is also possible that the kidneys may be more sensitive to elevated CVP in the setting of heart failure, such that GFR may fall with moderate CVP elevations [57]. Thus all these issues may be responsible for these divergent results. Indeed, today we still don’t know why some decompensated heart failure patients exhibit improvements in renal function with diuresis, whereas others display renal function deterioration, limiting attainment of euvolemia. Based on current data, one can speculate that deterioration in kidney function during heart failure is an adverse event given the fact that if the insult is progressive and long-lasting. However, transient deterioration in renal function may not pose worse prognosis.
Markers for Worsening Renal Function During Heart Failure
Although the detailed description of markers used to detect renal damage during heart failure is beyond the scope of this chapter some important points must be remembered.
BUN and Creatinine
Blood Urea Nitrogen (BUN) is one of the most used and traditional markers for detecting renal function. A pooled analysis of the Prospective Randomized Evaluation of Cardiac Ectopy with Dobutamine or Nesiritide Therapy (PRECEDENT) trial conducted in 541 New York Heart Association (NYHA) Class III-IV, heart failure patients with systolic dysfunction assessed the prognostic importance of four different measures of renal function namely BUN, serum creatinine, BUN/creatinine ratio and estimated creatinine clearance. After 1-year follow-up, BUN was the only significant predictor of mortality, with an adjusted relative risk (RR) of 2.3 in patients in the upper compared with the lower quartiles (95 % CI 1.3–4.1; p: 0.005). BUN/creatinine ratio yielded similar prognostic information as BUN (adjusted RR = 2.3; 95 % CI 1.4–3.8; p: 0.007) for patients in the upper compared with the lower quartiles [58]. Consistently, a post hoc analysis of the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Chronic Heart Failure (ACTIV in congestive heart failure) trial showed that among 319 patients with reduced systolic function stratified into quartiles according to baseline BUN, those in the highest quartile (40 mg/dL) had the highest 60-day mortality compared with those in the lower quartile (14.3 vs. 0 % respectively, p: 0.001) and the highest rate of death or heart failure hospitalization (30.0 vs. 8.6 %, p: 0.001). After adjustment for covariates, BUN remained a significant predictor of both mortality and the composite endpoint of death or heart failure hospitalization at 60 days after hospital discharge. Serum creatinine and creatinine clearance did not predict mortality after covariate adjustment [59]. Other studies also show its relation with morbidity and mortality in patients with heart failure [58, 59]. In the randomized Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF), BUN had a stronger relationship with outcomes as compared with GFR, calculated on the basis of serum creatinine levels [60]. Thus there is growing evidence that urea is a stronger predictor of outcome than creatinine in patients with heart failure. The exact cause of this association is not known but BUN has been accepted as a mediator of neuro-humoral activation during heart failure. Additionally, drawbacks of serum creatinine may play a role. These include the dependence of serum creatinine to other variables, namely, age, gender and muscle mass, the insensitivity of serum creatinine in detecting early renal injury (relatively large amount of renal damage can occur without producing a change in GFR calculated on serum creatinine levels which start to increase only at advanced stages of renal dysfunction) and inability of serum creatinine to measure renal injury. For example changes in renal function may occur as a consequence of changes in volume status in the absence of any renal damage.
Cystatin C
It has been suggested that cystatin C might be superior to creatinine in terms of predicting prognosis in patients with heart failure [61–63]. Wen et al. explored early markers of renal impairment in experimental post-myocardial infarction heart failure and found that it is the high blood cystatin C levels, rather than serum creatinine and BUN that predict increased post-MI heart failure incidence [64]. In acute heart failure, some studies have demonstrated that cystatin C can be a good prognostic marker. Lassus et al. measured cystatin C on admission and at 48 h in 292 patients hospitalized for acute heart failure. Acute kidney injury defined by an increase in cystatin C 0.3 mg/l within 48 h. The increase of cystatin C occurred in 16 % of patients. This increase was associated with longer length of hospitalization (P: 0.01) and with a significantly higher in-hospital mortality (odds ratio 4.0 95 % CI 1.3–11.7, P: 0.01). At 90 days, the increase in cystatin C was an independent predictor of mortality (adjusted odds ratio 2.8 95 % CI 1.2–6.7, P: 0.02) [65]. However, given the fact that studies regarding the cystatin in heart failure are scarce, more studies are needed to explore the role of cystatin C in patients with heart failure.
Neutrophil Gelatinase-Associated Lipocalin (NGAL)
In patients with acute heart failure, neutrophil gelatinase-associated lipocalin (NGAL) can predict deterioration of kidney function more accurately and in an earlier stage than serum creatinine. In fact, NGAL level rises about 24 h before serum creatinine values. In 91 patients admitted for acute heart failure, deterioration of kidney function was observed in 38 % within 5 days of follow-up. Patients who developed deterioration of kidney function had significantly higher median admission serum NGAL levels (194 ng/ml vs. 128 ng/ml, P = 0.001) with an increase in risk of developing deterioration of kidney function [66]. Besides, it was suggested that levels of urinary NGAL may be more sensitive as makers of tubular damage than serum levels [67, 68].
Kidney Injury Molecule 1 (KIM-1)
In chronic heart failure, Kidney injury molecule 1 (KIM-1) demonstrated a correlation with plasma N-terminal pro-brain natriuretic peptide levels and, independently of GFR values; it was associated with an increased risk of death or heart failure hospitalizations [68]. KIM-1 is highly sensitive to acute tubular injury but in the setting of acute heart failure its role is still unsettled.
Uric Acid
Current evidence suggests that uric acid may be either a marker of poor prognosis [69] or an active player in the pathogenesis of heart failure [70]. A study of 112 NYHA class III or IV patients found that serum uric acid level is a strong predictor of poorer outcomes, defined as mortality and need for transplant [71]. Janakowska et al. had similar findings regarding elevated uric acid levels and poorer outcomes in 119 NYHA class I–III patients, suggesting uric acid levels correlate with mortality and morbidity even in mild heart failure [72]. The Framingham Offspring cohort showed that incidence rates of heart failure were approximately sixfold higher among those in the highest quartile of uric acid compared with those at the lowest quartile [73]. The relationship between uric acid and heart failure was thought to be associated with inflammation, renal congestion and oxidative stress. The mechanism of renal congestion is a special concern. In 50 patients with reduced left ventricular systolic function, uric acid correlated significantly with pulmonary artery pressure, pulmonary capillary wedge pressure, and with clinical signs of volume overload (rales, oedema, and paroxysmal nocturnal dyspnoea). It also inversely correlated with left ventricular ejection fraction, suggesting that uric acid may be a non-invasive indicator of elevated filling pressures [74]. These findings were further supported by Kittelson et al. who found that not only were higher uric acid levels were associated with increased pulmonary artery and wedge pressure but with increased right atrial pressures as well [75]. Moreover, when patients with heart failure were monitored longitudinally, uric acid correlated with clinical status of patients. Odds ratios of hyperuricemia were 1.67 (95 % CI, 1.21–2.32) for heart failure decompensation and 0.21 (95 % CI, 0.08–0.55) for compensation [76].
Natriuretic Peptides
It was stated that patients admitted with acute breathlessness due to heart failure and an elevated natriuretic peptide level (generally 600 pg/ml for B-type natriuretic peptide (BNP) or 6,000 pg/ml for N-terminal pro-BNP) have a high filling pressure secondary to volume overload [77]. High atrial natriuretic peptide (ANP) levels not only would be a marker of venous congestion, but there is also evidence that its beneficial natriuretic effects are attenuated in heart failure. High renal interstitial pressure due to venous congestion may impair preservation of GFR by loss of ANP’s effects modulating transforming growth factor β [78]. A recent study has shown that BNP levels correlate with capillary wedge pressure, it can also serve as an indirect marker for deterioration of kidney function during the treatment of acute decompensated heart failure [79].
Vasopressin
Excess vasopressin levels have long been recognized in patients with heart failure, particularly those with severe clinical manifestations and have the potential to exert deleterious effects on various physiological processes in heart failure [80]. However, measurement of circulating vasopressin levels has been challenging because it is released in a pulsatile pattern, unstable and is rapidly cleared from plasma. Arginine vasopressin is derived from a larger precursor peptide (preprovasopressin) along with copeptin, which is released from the posterior pituitary in an equimolar ratio to arginine vasopressin and is more stable in the circulation and closely reflects arginine vasopressin levels. Copeptin levels have been found to closely mirror the production of arginine vasopressin and have been proposed as a prognostic marker in acute illness [81]. Thus studies are needed whether vasopression and/or copeptin levels are promising in heart failure.
Bioimpedance
Bioimpedance technique was introduced to study changes in the volume in a tissue in 1940s. Bioimpedance is based on the principle that fluid (oedema) is a good conductor of electrical current and is associated with a low impedance value. Decreased bioimpedance reflects total body water excess, with total body water derived from these values by making certain electrophysical assumptions [82]. Bioimpedance vector analysis has been used in heart failure patients [83, 84]. More studies are needed regarding the efficiency of bioimpedance in heart failure patients.
Treatment of Renal Congestion in Heart Failure
There are various treatment options in renal congestion in heart failure (Table 9.1). The main aim is the decongestion of excess fluid without compromising renal function. The forthcoming section deals with the main treatment strategies regarding decongestion in heart failure.
Loops Diuretics
Loop diuretics are commonly used in patients with heart failure. The detailed action of loop diuretic is beyond the scope of this chapter however some important issues must be remembered. First of all, in heart failure, the dose-response curve shifts downward and to the right and there is possibility that drug reaching the Henle’s loop decreases which necessitates a higher dose to achieve the same effect. Thus reaching a therapeutic threshold is an important concept [101]. At this point on may argue that higher loop diuretic dosing in heart failure is associated with worse clinical outcome [102]. However, these studies are criticized because patients with more severe disease and underlying renal insufficiency require higher diuretic doses [48]. This issue is examined in a recent trial which 183 patients with advanced heart failure stratified by baseline diuretic dose (furosemide < 80 or > 80 mg daily). Patients receiving high-dose diuretics (n = 113) had more markers of increased cardiovascular risk and were more likely to have had a recent history of clinical instability (33 % vs. 4 %). After adjusting for clinical stability, diuretic dose was no longer a significant predictor of increased risk [103]. Thus it is possible that a subgroup of patients with heart failure refractory to diuretics and thereby requiring higher doses of these drugs during heart failure decompensation are especially susceptible to the development of renal injury. In fact, these patients had significantly higher admission serum creatinine levels and included a higher percentage of patients requiring thiazide in addition to loop diuretics. Alternatively, patients with deterioration of kidney function required higher diuretic doses because of more advanced heart failure. Thus, the question of whether higher diuretic doses are responsible for deterioration of kidney function or are a marker of greater disease severity remains unanswered [104]. Secondly, in general loop diuretics are given as a single daily dose. However, a single furosemide bolus especially in heart failure, elicits a transient period of natriuresis followed by a longer period of increased renal tubular sodium avidity due to increase in systemic vascular resistance, plasma renin activity, and plasma levels of norepinephrine and arginine vasopressin [105, 106]. Thus there is an escape from the action of loop diuretics especially when given as a single dose. To overcome this effect, loop diuretic can be administered two or more times per day. Indeed, continuous loop diuretic infusion is an extrapolation of this concept in which the time interval between dosing is reduced to zero. In this scenario, there is no antinatriuretic rebound period and negative sodium balance is sustained [4]. However, it was shown that continuous dosing is not more effective than an intelligently prescribed bolus regimen as proven in the Diuretic Optimization Strategies Evaluation (DOSE) trial [48]. However, infusion may be preferred in patients with low systolic blood pressure. Thirdly, to overcome this escape the addition of a non-loop diuretic (i.e., thiazide or potassium-sparing diuretic) may be effective by decreasing the enhanced sodium absorption in the distal tubule above [107]. The excessive use of diuretics and sympathetic overactivity in heart failure promotes the activity of the RAS. Activation of the RAS leads to excessive salt and water retention and vasoconstriction of the venous beds, altering cardiac preload and afterload, which further worsens renal function. Thus addition of mineralocorticoid receptor antagonists, such as spironolactone and eplerenone, may attenuate the neurohumoral surge and prevent deterioration of kidney function. Previous small, nonrandomized, open-label trials have shown that these drugs at high doses overcome diuretic resistance in heart failure without a significant effect on serum creatinine [108, 109]. However, in an acute setting in which aggressive diuresis is needed, these drugs may also cause deterioration of kidney function. The balance is tight and careful attention to renal parameters should be given in such patients, with adjustment of doses of both drugs. Sometimes, it is prudent to withhold ACE inhibitors and angiotensin receptor blockers particularly in patients at high risk of developing renal injury, such as patients with advanced age and aggressive diuresis. Lastly, loop diuretics may also have independent actions of tubules irrespective of systemic and renal haemodynamic effects. In one interesting study in 30 patients with chronic systolic heart failure the effect of loop diuretic withdrawal and reinitiation on tubular dysfunction was evaluated. At baseline, subjects were withdrawn from their loop diuretics. After 72 h, their furosemide regimen was reinstated and patients were studied again 3 days later. Serum creatinine, atrial and B-type natriuretic peptide, KIM-1, urinary N-acetyl-beta-D- glucosaminidase (NAG), and serum as well as urinary NGAL were determined at various time points. Diuretic withdrawal resulted in increases in atrial and B-type natriuretic peptide (both p < 0.05). Serum creatinine was unaffected. Both urinary KIM-1 (p < 0.001) and NAG (p < 0.010) concentrations rose significantly, after diuretic withdrawal, whereas serum and urinary NGAL were not significantly affected. After reinitiation of furosemide, both urinary KIM-1 and NAG concentrations returned to baseline (both p < 0.05), but NGAL values were unaffected. The authors concluded that subclinical changes in volume status by diuretic withdrawal and reinstitution are associated with increases and decreases of markers of tubular dysfunction in stable heart failure and diuretic therapy may favourably affect renal and tubular function by decreasing congestion [110].
At this point one must also mention the importance of salt and fluid restriction in heart failure. Although salt and fluid restriction is the primary dietary therapy heart failure, this is mostly based on expert opinion and few controlled studies are available. Albert et al. demonstrated that fluid restriction (1,000 mL/day) in hyponatremic (serum sodium <137 mg/dl) patients improved quality of life at 60 days after discharge [111]. Hummel et al. showed that in heart failure patients with preserved ejection fraction sodium-restricted diet was associated with favourable changes in ventricular diastolic function, arterial elastance, and ventricular–arterial coupling [112].
Apart from direct effect on extracellular volume sodium restriction has been shown to improve endothelial function and arterial stiffness [113, 114]. Given the fact that studies are scarce, more randomised studies are needed to examine the effect of salt and fluid restriction in heart failure.
Diuretics vs. Ultrafiltration to Relief Renal Congestion
Diuretic therapy aimed at fluid withdrawal and relief of congestion which are the main currently available strategies for reducing venous congestion in decompensated heart failure. Ultrafiltration is another option for the same purpose. Although several guidelines state that ultrafiltration is reasonable for patients with refractory congestion not responding to medical therapy [115–117]; we do not know known which of the two is safer and more effective [118, 119]. Although the two treatments seem to work for decongestion some differences must be mentioned. Firstly, the amount of urine produced in response to IV diuretics is not predictable but fluid removal by ultrafiltration is completely controllable and adjustable. Secondly, a potential advantage of ultrafiltration over loop diuretics is that the ultrafiltrate is isotonic, whereas the urinary output with loop diuretics is hypotonic therefore ultrafiltration removes more sodium (and less potassium) than diuretics for an equivalent volume loss [120]. Thirdly, if fluid removal does not exceed the interstitial fluid mobilization rate of approximately 15 ml/min, then the intravascular volume can be preserved with ultrafiltration, potentially interrupting the vicious cycle of neurohormonal activation and renal impairment that can occur with loop diuretics [121]. Besides, adequacy of intravascular re-fill during ultrafiltration can be assessed by continuous monitoring of the haematocrit with sensors placed in the withdrawal line. When the haematocrit does not significantly change during ultrafiltration, regardless of the amount of fluid removed, this indicates a proportional shift of water from the extravascular to the intravascular space. An increase in haematocrit may indicate either that plasma re-fill rate is inadequate or that interstitial oedema has been eliminated. This hypothesis is supported by data demonstrating that patients receiving ultrafiltration have lower plasma renin, norepinephrine and aldosterone levels as long as 90 days after treatment compared with those receiving diuretics [122]. Fourthly, elimination of proinflammatory cytokines [26, 123] or sodium-retaining vasoconstrictive agents may occur during ultrafiltration which is potentially involved in improvement in urinary output or restoration of diuretic responsiveness during ultrafiltration [121, 124]. Lastly, ultrafiltration mediated neurohumoral regulation is more sustained. This is probably a reason why improvement in clinical signs and symptoms of volume overload and functional capacity were found to be persistent [122, 125] and rehospitalisations are lower in ultrafiltration compared in to diuretic therapy [126]. However, as stated earlier there are no strict recommendations regarding the preferential use of diuretics, ultrafiltration and or combination. In fact in the Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial it was found that the use of a stepped pharmacologic-therapy algorithm was superior to a strategy of ultrafiltration for the preservation of renal function at 96 h, with a similar amount of weight loss with the two approaches Ultrafiltration was associated with a higher rate of adverse events [49].
Peritoneal Dialysis
In peritoneal dialysis, there is a continuous slow ultrafiltration leading to a reduction in fluid overload. Lowering afterload and lowering CVP could be important physiological mechanisms by which peritoneal dialysis leads to clinical improvement in heart failure symptoms. Moreover, it provides some replacement of renal function, by removing metabolic waste products, so a possible decrease in renal function will be better tolerated. However, the role of peritoneal dialysis in heart failure and renal congestion is not examined satisfactorily. Additionally, there are no studies available comparing peritoneal dialysis, diuretic therapy and salt restriction head-to-head in congestive heart failure [127]. In some studies, there were improvements to echocardiographic or other cardiac parameters with ultrafiltration by peritoneal dialysis [128–130]. There are also reports that peritoneal dialysis can reduce the number of hospital admissions and improve quality of life in chronic heart failure patients, as supported by recent non-randomized and non-controlled prospective studies [131–133]. However, the number of studied patients is limited and these trials all lack a control group. Also most studied patients had advanced renal failure contributing to fluid overload, introducing a major confounding factor. Another potential concern is the elevation of intra abdominal pressure by peritoneal dialysis. Currently, neither the optimal dialysis schedule nor the optimal fluid status to be reached in heart failure patients is known. The available reports all describe different peritoneal dialysis intervals, types, fluids and dosages.
Vasopressin Type 2 Receptor Antagonists
Recently, vasopressin type 2 receptor (V2R) antagonists have shown promise for use in patients with heart failure by increasing free-water excretion and serum sodium level [93]. For example, the oral V2R antagonist tolvaptan caused an early and sustained reduction in body weight and improvement in serum sodium in the EVEREST trial although did not improve mortality or morbidity [85, 86]. In a recent experimental study, chronic tolvaptan administration in a rat hypertensive heart failure model was examined considering the functional and pathological effects on both the myocardium and kidney. The animals were chronically treated with low-dose or high-dose (HD) tolvaptan or vehicle from the left ventricular (LV) hypertrophic stage. Chronic tolvaptan treatment persistently increased urine volume but did not affect blood pressure. In the HD group, the animal survival significantly improved (log-rank test, P < 0.01). At the heart failure stage, the progression of LV dysfunction was prevented and lung congestion was suppressed. Activation of atrial natriuretic peptide, endothelin-1, AVP, and V1aR mRNA levels were significantly suppressed in the LV myocardium. Meanwhile, renal histopathologic damage including tubular fibrosis and glomerulosclerosis was ameliorated and renal function was improved in the HD group at the heart failure stage. Concomitantly, not only activation of aquaporin-2 but also those of V2R, V1aR, renin, and endothelin-1 in the kidney were significantly suppressed (all P < 0.05). V2R antagonists also caused redistribution of AQP2 from apical to intracellular domains [87]. Goldsmith et al. studied the effect of conivaptan on renal and hormonal effects compared with or in combination with loop diuretics in stable heart failure patients. There were no significant effects of conivaptan, furosemide, or the combination on any haemodynamic variable, neurohormonal level, renal blood flow, or glomerular filtration rate. Conivaptan and furosemide similarly increased urine volumes; the effect of the combination was significantly greater. Furosemide, but not conivaptan, increased urinary sodium excretion, and the combination was significantly greater than after furosemide alone. Without adversely affecting important haemodynamic variables, neurohormones, renal blood flow, or glomerular filtration rate, conivaptan significantly augmented both the diuretic and the natriuretic response to furosemide in patients with chronic heart failure [88].
Adenosine Receptor Blockers
Adenosine concentration is increased in patients with heart failure. In the kidney, adenosine is released by the juxta-glomerular cells in response to increase in sodium load in the distal tubule, sensed by the macula densa cells. Adenosine binds to adenosine 1 receptors located in the proximal tubule and afferent arterioles of the glomerulus. This leads to a reduction in intracellular cyclic adenosine mono-phosphate and an increase the activity of basolateral Na+/HCO3− symporter in the proximal tubule and to constriction of the afferent glomerular arteriole. Thus, adenosine release following an increase of sodium load to the distal tubule, as during intensive diuretic treatment for acute heart failure, leads to sodium retention and reduces GFR. Thus adenosine release may be a major mechanism of renal dysfunction after high dose furosemide treatment [134]. In the PROTECT trial (A Placebo-controlled Randomized study of the selective A1 adenosine receptor antagonist rolofylline for patients hospitalized with acute heart failure and volume overload to assess treatment effect on congestion and renal function), 2,033 patients admitted for acute heart failure were enrolled and randomized 2:1 to the type 1A adenosine antagonist rolofylline or placebo. Worsening renal function, defined as an increase from baseline 0.3 mg/dl of serum creatinine at day 7 from enrolment persisting at day 14 was, for the first time, included as a component of the primary end-point and as an essential component, together with dialysis or haemofiltration, of a secondary end-point. However, PROTECT failed to show any beneficial effect of rolofylline on worsening renal function. Actually, the proportion of patients who developed deterioration of kidney function was numerically greater with rolofylline compared with placebo, whereas rolofylline, likely through its mild diuretic effects, had a favourable effect on dyspnoea as well as short-term mortality [89]. Rolofylline was also associated with higher rates of seizures and stroke, compared with placebo, and this has further inhibited any development of the drug by the sponsoring company [90].
Dopamine
Dopamine, when administered at low doses, may selectively improve renal blood flow in both the large conductance and small resistance renal blood vessels through its action on dopaminergic receptors and this was attended by an improvement in diuresis [91, 92]. In the recent Dopamine in Acute Decompensated Heart Failure (DAD-HF) trial, 60 consecutive patients hospitalized for acute heart failure were randomized to high doses of furosemide or low doses of furosemide plus low doses of dopamine. Deterioration of kidney function and hypokalemia were more frequent in the high doses arm (P = 0.042 and P = 0.003, respectively), although the 60 days outcomes were similar in both groups. The study had some limitations, related to its small sample size, which do not allow drawing conclusions regarding the effects on outcomes, as well as with regards of the relatively high average systolic blood pressure on admission [135]. In the double-blind, placebo-controlled ROSE trial 360 hospitalized patients with acute heart failure and renal dysfunction (eGFR 15–60 mL/min/1.73 m2), were randomized to placebo, low-dose dopamine or recombinant BNP (nesiritide; see below). Compared with placebo, low-dose dopamine or nesiritide had no effect on decongestion, renal function, or clinical outcomes [95]. No studies have shown favourable effects of dopamine infusion on major outcomes, defined as mortality, rehospitalisations and prevention of long-term renal damage [94]. Thus, there is no evidence to recommend dopamine administration for the protection of renal function in patients with fluid overload and need of diuretic treatment.
Natriuretic Peptides
Nesiritide, a recombinant form of endogenous human BNP has been shown to rapidly reduce cardiac filling pressure, increase cardiac output, promote diuresis and suppress RAS and release of norepinephrine [96]. In one study, nesiritide, has a borderline effect on dyspnoea without worsening renal function [97]. In order to assess the effects of nesiritide on symptoms and outcomes of the patients with acute heart failure, the Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF), was designed [136]. This study included 7,141 patients with heart failure, randomized to placebo or nesiritide. The trial confirmed that nesiritide administration was associated with an improvement in dyspnoea, compared with placebo, although not meeting the prespecified criteria for statistical significance and no effects on outcomes were found. Thus, ASCEND- heart failure showed the safety of nesiritide administration although with only mild effects on symptoms and no effects on outcomes. Also in the ROSE trial mentioned above, nesiritide did not improve short-term endpoints compared to placebo [95]. Meta-analyses of previous randomized trials had raised concerns regarding untoward effects on renal function and mortality of nesiritide infusion [137]. Recently, developed alternatively spliced BNPs (ASBNP and ASBNP.1) lacked the hypotensive side effects of nesiritide but increased the glomerular filtration rate, suppressed plasma renin and angiotensin, while inducing natriuresis and diuresis [98].
Novel Therapies
Although it seems paradoxical, combining hypertonic saline with furosemide was thought to prevent the rebound sodium reabsorption and promote effective dieresis. Paterna et al. demonstrated that a combination of high-dose furosemide with bolus hypertonic saline infusion in patients with NYHA class IV heart failure improved diuresis, shortened hospital stay, decreased BNP levels, and reduced readmissions compared with IV diuretic therapy alone [99]. In another trial namely RELAX-AHF (Efficacy and Safety of Relaxin for the Treatment of Acute heart failure), will provide definitive information of the impact of relaxin on congestion, renal dysfunction, and outcomes in acute decompensated heart failure [138]. Relaxin acts via the nitric oxide pathways and endothelin B receptors to produce systemic and renal vasodilatation. In a preliminary phase II trial (pre RELAX), relaxin was associated with relief of dyspnoea and a tendency to greater weight loss with smaller doses of diuretics and nitrates [100]. The FAIR-HF study has recently shown that among 459 patients with stable chronic heart failure, those receiving intravenous ferric carboxymaltose were more likely to report an improvement in their quality of life after 24 weeks of follow-up than those receiving placebo. It is not known whether intravenous iron application may positively affect symptom burden in patients with iron deficiency presenting with renal failure and acute decompensated heart failure [44].
Conclusions
We still do not understand the complex interactions between the failing heart and the kidneys. Apart from the classical underflow hypothesis, renal congestion is becoming realised as a major contributing factor for worse outcomes in heart failure patients. The management of renal congestion in heart failure remains an important but unresolved clinical challenge, owing to the lack of consistent data from randomized studies in this field, rendering it difficult to outline concise evidence based treatment guidelines. Pathophysiologic mechanisms on how renal congestion leads to worse outcomes are not completely understood. Additionally, renal congestion is not directly measurable at this moment although MRI and other imaging may allow detection of renal congestion in the future.
Renal congestion is part of a complex of process. The mechanisms may vary from patient to patient, and physicians should try to individualize the management of heart failure, with focus on preventing renal injury and decreasing renal congestion. Various treatment strategies alone or in combination can be used for this purpose. Novel therapeutic options such as BNP analogues, sympathetic denervation, and combination therapy with hypertonic saline with furosemide show some promise and may enter the clinic in the future.
References
Aronson D. Cardiorenal syndrome in acute decompensated heart failure. Expert Rev Cardiovasc Ther. 2012;10(2):177–89.
Giamouzis G, Butler J, Triposkiadis F. Renal function in advanced heart failure. Congest Heart Fail. 2011;17(4):180–8.
Ljungman S, Laragh JH, Cody RJ. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs. 1990;39 Suppl 4:10–21; discussion 22–4.
Sambandam KK. Effective use of loop diuretics in heart failure exacerbation: a nephrologist’s view. Am J Med Sci. 2014;347(2):139–45.
Ruggenenti P, Remuzzi G. Worsening kidney function in decompensated heart failure: treat the heart, don’t mind the kidney. Eur Heart J. 2011;32(20):2476–8.
Schrier RW. Role of diminished renal function in cardiovascular mortality – marker or pathogenetic factor? J Am Coll Cardiol. 2006;47(1):1–8.
Ichikawa I, Pfeffer JM, Pfeffer MA, Hostetter TH, Brenner BM. Role of angiotensin II in the altered renal function of congestive heart failure. Circ Res. 1984;55(5):669–75.
Giamouzis G, Kalogeropoulos AP, Butler J, Karayannis G, Georgiopoulou VV, Skoularigis J, Triposkiadis F. Epidemiology and importance of renal dysfunction in heart failure patients. Curr Heart Fail Rep. 2013;10(4):411–20.
Sugrue M. Abdominal compartment syndrome. Curr Opin Crit Care. 2005;11(4):333–8.
Lambert DM, Marceau S, Forse RA. Intraabdominal pressure in the morbidly obese. Obes Surg. 2005;15(9):1225–32.
Cheatham ML, White MW, Sagraves SG, Johnson JL, Block EF. Abdominal perfusion pressure: a superior parameter in the assessment of intra-abdominal hypertension. J Trauma. 2000;49(4):621–6; discussion 626–7.
Bradley SE, Bradley GP. The effect of increased intra-abdominal pressure on renal function in man. J Clin Invest. 1947;26(5):1010–22.
Bloomfield GL, Blocher CR, Fakhry IF, Sica DA, Sugerman HJ. Elevated intra-abdominal pressure increases plasma renin activity and aldosterone levels. J Trauma. 1997;42(6):997–1004.
Harman PK, Kron IL, McLachlan HD, Freedlender AE, Nolan SP. Elevated intra-abdominal pressure and renal function. Ann Surg. 1982;196(5):594–7.
Verbrugge FH, Dupont M, Steels P, Grieten L, Malbrain M, Tang WH, Mullens M. Abdominal contributions to cardio-renal dysfunction in congestive heart failure. J Am Coll Cardiol. 2013;62(6):485–95.
de Almeida Duarte JB, de Aguilar-Nascimento JE, Nascimento M, Nochi Jr RJ. Bacterial translocation in experimental uremia. Urol Res. 2004;32(4):266–70.
Vaziri ND, Yuan J, Rahimi A, Ni Z, Said H, Subramanian VS. Disintegration of colonic epithelial tight junction in uremia: a likely cause of CKD-associated inflammation. Nephrol Dial Transplant. 2012;27(7):2686–93.
Vaziri ND. CKD impairs barrier function and alters microbial flora of the intestine: a major link to inflammation and uremic toxicity. Curr Opin Nephrol Hypertens. 2012;21(6):587–92.
Ganda A, Onat D, Demmer RT, Wan E, Vittorio TJ, Sabbah HN, Colombo PC. Venous congestion and endothelial cell activation in acute decompensated heart failure. Curr Heart Fail Rep. 2010;7(2):66–74.
Blake WD, Wegria R, Keating RP, Ward HP. Effect of increased renal venous pressure on renal function. Am J Physiol. 1949;157(1):1–13.
Guazzi M, Gatto P, Giusti G, Pizzamiglio F, Previtali I, Vignati C, Arena R. Pathophysiology of cardiorenal syndrome in decompensated heart failure: role of lung-right heart-kidney interaction. Int J Cardiol. 2013;169(6):379–84.
Damman K, Navis G, Smilde TD, Voors AA, van der Bij W, van Veldhuisen DJ, Hillege HL. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9(9):872–8.
Damman K, van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53(7):582–8.
Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, Young JB, Tang WH. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53(7):589–96.
Dilley JR, Corradi A, Arendshorst WJ. Glomerular ultrafiltration dynamics during increased renal venous pressure. Am J Physiol. 1983;244(6):F650–8.
Ross EA. Congestive renal failure: the pathophysiology and treatment of renal venous hypertension. J Card Fail. 2012;18(12):930–8.
Colombo PC, Ganda A, Lin J, Onat D, Harxhi A, Iyasere JE, Uriel N, Cotter G. Inflammatory activation: cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail Rev. 2012;17(2):177–90.
Anker SD, Egerer KR, Volk HD, Kox WJ, Poole-Wilson PA, Coats AJ. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am J Cardiol. 1997;79(10):1426–30.
Peschel T, Schönauer M, Thiele H, Anker SD, Schuler G, Niebauer J. Invasive assessment of bacterial endotoxin and inflammatory cytokines in patients with acute heart failure. Eur J Heart Fail. 2003;5(5):609–14.
Niebauer J, Volk HD, Kemp M, Dominguez M, Schumann RR, Rauchhaus M, Poole-Wilson PA, Coats AJ, Anker SD. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999;353(9167):1838–42.
Gonçalves S, Pecoits-Filho R, Perreto S, Barberato SH, Stinghen AE, Lima EG, Fuerbringer R, Sauthier SM, Riella MC. Associations between renal function, volume status and endotoxaemia in chronic kidney disease patients. Nephrol Dial Transplant. 2006;21(10):2788–94.
Colombo PC, Banchs JE, Celaj S, Talreja A, Lachmann J, Malla S, DuBois NB, Ashton AW, Latif F, Jorde UP, Ware JA, LeJemtel TH. Endothelial cell activation in patients with decompensated heart failure. Circulation. 2005;111(1):58–62.
Harrison DG, Widder J, Grumbach I, Chen W, Weber M, Searles C. Endothelial mechanotransduction, nitric oxide and vascular inflammation. J Intern Med. 2006;259(4):351–63.
Canty Jr TG, Boyle Jr EM, Farr A, Morgan EN, Verrier ED, Pohlman TH. Oxidative stress induces NF-kappaB nuclear translocation without degradation of IkappaBalpha. Circulation. 1999;100(19 Suppl):II361–4.
Nohria A, Hasselblad V, Stebbins A, Pauly DF, Fonarow GC, Shah M, Yancy CW, Califf RM, Stevenson LW, Hill JA. Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol. 2008;51(13):1268–74.
Guglin M, Rivero A, Matar F, Garcia M. Renal dysfunction in heart failure is due to congestion but not low output. Clin Cardiol. 2011;34(2):113–6.
Aronson KD, Abassi Z, Allon E, Burger AJ. Fluid loss, venous congestion, and worsening renal function in acute decompensated heart failure. Eur J Heart Fail. 2013;15(6):637–43.
Greene SJ, Gheorghiade M, Vaduganathan M, Ambrosy AP, Mentz RJ, Subacius H, Maggioni AP, Nodari S, Konstam MA, Butler J, Filippatos G. EVEREST Trial investigators. Haemoconcentration, renal function, and post-discharge outcomes among patients hospitalized for heart failure with reduced ejection fraction: insights from the EVEREST trial. Eur J Heart Fail. 2013;15(12):1401–11.
van der Meer P, Postmus D, Ponikowski P, Cleland JG, O’Connor CM, Cotter G, Metra M, Davison BA, Givertz MM, Mansoor GA, Teerlink JR, Massie BM, Hillege HL, Voors AA. The predictive value of short term changes in hemoglobin concentration in patients presenting with acute decompensated heart failure. J Am Coll Cardiol. 2013;61(19):1973–81.
Testani JM, Chen J, McCauley BD, Kimmel SE, Shannon RP. Potential effects of aggressive decongestion during the treatment of decompensated heart failure on renal function and survival. Circulation. 2010;122(3):265–72.
Davila C, Reyentovich A, Katz SD. Clinical correlates of hemoconcentration during hospitalization for acute decompensated heart failure. J Card Fail. 2011;17(12):1018–22.
Opasich C, Cazzola M, Scelsi L, De Feo S, Bosimini E, Lagioia R, Febo O, Ferrari R, Fucili A, Moratti R, Tramarin R, Tavazzi L. Blunted erythropoietin production and defective iron supply for erythropoiesis as major causes of anaemia in patients with chronic heart failure. Eur Heart J. 2005;26(21):2232–7.
Nanas JN, Matsouka C, Karageorgopoulos D, Leonti A, Tsolakis E, Drakos SG, Tsagalou EP, Maroulidis GD, Alexopoulos GP, Kanakakis JE, Anastasiou-Nana MI. Etiology of anemia in patients with advanced heart failure. J Am Coll Cardiol. 2006;48(12):2485–9.
Anker SD, Comin Colet J, Filippatos G, Willenheimer R, Dickstein K, Drexler H, Lüscher TF, Bart B, Banasiak W, Niegowska J, Kirwan BA, Mori C, von Eisenhart RB, Pocock SJ, Poole-Wilson PA, Ponikowski P, FAIR-HF Trial Investigators. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med. 2009;361(25):2436–48.
Krumholz HM, Chen YT, Vaccarino V, Wang Y, Radford MJ, Bradford WD, Horwitz RI. Correlates and impact on outcomes of worsening renal function in patients or > 65 years of age with heart failure. Am J Cardiol. 2000;85(9):1110–3.
Blair JE, Pang PS, Schrier RW, Metra M, Traver B, Cook T, Campia U, Ambrosy A, Burnett Jr JC, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Konstam MA, Gheorghiade M. Changes in renal function during hospitalization and soon after discharge in patients admitted for worsening heart failure in the placebo group of the EVEREST trial. Eur Heart J. 2011;32(20):2563–72.
Adams Jr KF, Fonarow GC, Emerman CL, LeJemtel TH, Costanzo MR, Abraham WT, Berkowitz RL, Galvao M, Horton DP, ADHERE Scientific Advisory Committee and Investigators. Characteristics and outcomes of patients hospitalized for HF in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated HF National Registry (ADHERE). Am Heart J. 2005;149(2):209–16.
Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, LeWinter MM, Deswal A, Rouleau JL, Ofili EO, Anstrom KJ, Hernandez AF, McNulty SE, Velazquez EJ, Kfoury AG, Chen HH, Givertz MM, Semigran MJ, Bart BA, Mascette AM, Braunwald E, O’Connor CM. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364(9):797–805.
Bart BA, Goldsmith SR, Lee KL, Givertz MM, O’Connor CM, Bull DA, Redfield MM, Deswal A, Rouleau JL, LeWinter MM, Ofili EO, Stevenson LW, Semigran MJ, Felker GM, Chen HH, Hernandez AF, Anstrom KJ, McNulty SE, Velazquez EJ, Ibarra JC, Mascette AM, Braunwald E. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med. 2012;367(24):2296–304.
Dupont M, Mullens W, Finucan M, Taylor DO, Starling RC, Tang WH. Determinants of dynamic changes in serum creatinine in acute decompensated heart failure: the importance of blood pressure reduction during treatment. Eur J Heart Fail. 2013;15(4):433–40.
Testani JM, McCauley BD, Chen J, Coca SG, Cappola TP, Kimmel SE. Clinical characteristics and outcomes of patients with improvement in renal function during the treatment of decompensated heart failure. J Card Fail. 2011;17(12):993–1000.
Testani JM, McCauley BD, Kimmel SE, Shannon RP. Characteristics of patients with improvement or worsening in renal function during treatment of acute decompensated heart failure. Am J Cardiol. 2010;106(12):1763–9.
Metra M, Davison B, Bettari L, Sun H, Edwards C, Lazzarini V, Piovanelli B, Carubelli V, Bugatti S, Lombardi C, Cotter G, Dei Cas L. Is worsening renal function an ominous prognostic sign in patients with acute heart failure? The role of congestion and its interaction with renal function. Circ Heart Fail. 2012;5(1):54–62.
Testani JM, Damman K. Venous congestion and renal function in heart failure it’s complicated. Eur J Heart Fail. 2013;15(6):599–601.
Doty JM, Saggi BH, Sugerman HJ, Blocher CR, Pin R, Fakhry I, Gehr TW, Sica DA. Effect of increased renal venous pressure on renal function. J Trauma. 1999;47(6):1000–3.
Firth JD, Raine AE, Ledingham JG. Raised venous pressure: a direct cause of renal sodium retention in oedema? Lancet. 1988;1(8593):1033–5.
Bishara B, Abu-Saleh N, Awad H, Ghrayeb N, Goltsman I, Aronson D, Khamaysi I, Assady S, Armaly Z, Haddad S, Haddad E, Abassi Z. Phosphodiesterase 5 inhibition protects against increased intra-abdominal pressure-induced renal dysfunction in experimental congestive heart failure. Eur J Heart Fail. 2012;14(10):1104–11.
Aronson D, Mittleman MA, Burger AJ. Elevated blood urea nitrogen level as a predictor of mortality in patients admitted for decompensated heart failure. Am J Med. 2004;116(7):466–73.
Filippatos G, Rossi J, Lloyd-Jones DM, Stough WG, Ouyang J, Shin DD, O’Connor C, Adams KF, Orlandi C, Gheorghiade M. Prognostic value of blood urea nitrogen in patients hospitalized with worsening heart failure: insights from the Acute and chronic therapeutic impact of a vasopressin antagonist in chronic heart failure (ACTIV in CHF) study. J Card Fail. 2007;13(5):360–4.
Klein L, Massie BM, Leimberger JD, O’Connor CM, Piña IL, Adams Jr KF, Califf RM, Gheorghiade M, OPTIME-CHF Investigators. Admission or changes in renal function during hospitalization for worsening heart failure predict postdischarge survival: results from the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF). Circ Heart Fail. 2008;1(1):25–33.
Pérez-Calvo JI, Ruiz-Ruiz FJ, Carrasco-Sánchez FJ, Morales-Rull JL, Manzano-Fernández S, Galisteo-Almeda L, Pascual-Figal D. Prognostic value of serum cystatin C and N-terminal pro b-type natriuretic peptide in patients with acute heart failure. Eur J Intern Med. 2012;23(7):599–603.
Manzano-Fernández S, Boronat-Garcia M, Albaladejo-Otón MD, Pastor P, Garrido IP, Pastor-Pérez FJ, Martínez-Hernández P, Valdés M, Pascual-Figal DA. Complementary prognostic value of cystatin C, N-terminal pro-B-type natriuretic Peptide and cardiac troponin T in patients with acute heart failure. Am J Cardiol. 2009;103(12):1753–9.
Lassus J, Harjola VP, Sund R, Siirilä-Waris K, Melin J, Peuhkurinen K, Pulkki K, Nieminen MS, FINN-AKVA Study group. Prognostic value of cystatin C in acute heart failure in relation to other markers of renal function and NT-proBNP. Eur Heart J. 2007;28(15):1841–7.
Wen Z, Cai M, Mai Z, Chen Y, Geng D, Wang J. Protection of renal impairment by angiotensin II type 1 receptor blocker in rats with post- infarction heart failure. Ren Fail. 2013;35(5):766–75.
Lassus JP, Nieminen MS, Peuhkurinen K, Pulkki K, Siirilä-Waris K, Sund R, Harjola VP, FINN-AKVA study group. Markers of renal function and acute kidney injury in acute heart failure: definitions and impact on outcomes of the cardiorenal syndrome. Eur Heart J. 2010;31(22):2791–8.
Aghel A, Shrestha K, Mullens W, Borowski A, Tang WH. Serum neutrophil gelatinase-associated lipocalin (NGAL) in predicting worsening renal function in acute decompensated heart failure. J Card Fail. 2010;16(1):49–54.
Damman K, van Veldhuisen DJ, Navis G, Voors AA, Hillege HL. Urinary neutrophil gelatinase associated lipocalin (NGAL), a marker of tubular damage, is increased in patients with chronic heart failure. Eur J Heart Fail. 2008;10(10):997–1000.
Damman K, Van Veldhuisen DJ, Navis G, Vaidya VS, Smilde TD, Westenbrink BD, Bonventre JV, Voors AA, Hillege HL. Tubular damage in chronic systolic heart failure is associated with reduced survival independent of glomerular filtration rate. Heart. 2010;96(16):1297–302.
Culleton BF, Larson MG, Kannel WB, Levy D. Serum uric acid and risk for cardiovascular disease and death: the Framingham Heart Study. Ann Intern Med. 1999;131(1):7–13.
Duan X, Ling F. Is uric acid itself a player or a bystander in the pathophysiology of chronic heart failure? Med Hypotheses. 2008;70(3):578–81.
Anker SD, Doehner W, Rauchhaus M, Sharma R, Francis D, Knosalla C, Davos CH, Cicoira M, Shamim W, Kemp M, Segal R, Osterziel KJ, Leyva F, Hetzer R, Ponikowski P, Coats AJ. Uric acid and survival in chronic heart failure: validation and application in metabolic, functional, and hemodynamic staging. Circulation. 2003;107(15):1991–7.
Jankowska EA, Ponikowska B, Majda J, Zymlinski R, Trzaska M, Reczuch K, Borodulin-Nadzieja L, Banasiak W, Ponikowski P. Hyperuricaemia predicts poor outcome in patients with mild to moderate chronic heart failure. Int J Cardiol. 2007;115(2):151–5.
Krishnan E. Hyperuricemia and incident heart failure. Circ Heart Fail. 2009;2(6):556–62.
Amin A, Vakilian F, Maleki M. Serum uric acid levels correlate with filling pressures in systolic heart failure. Congest Heart Fail. 2011;17(2):80–4.
Kittleson MM, St John ME, Bead V, Champion HC, Kasper EK, Russell SD, Wittstein IS, Hare JM. Increased levels of uric acid predict haemodynamic compromise in patients with heart failure independently of B-type natriuretic peptide levels. Heart. 2007;93(3):365–7.
Misra D, Zhu Y, Zhang Y, Choi HK. The independent impact of congestive heart failure status and diuretic use on serum uric acid among men with a high cardiovascular risk profile: a prospective longitudinal study. Semin Arthritis Rheum. 2011;41(3):471–6.
Maisel A, Mueller C, Adams Jr K, Anker SD, Aspromonte N, Cleland JG, Cohen-Solal A, Dahlstrom U, DeMaria A, Di Somma S, Filippatos GS, Fonarow GC, Jourdain P, Komajda M, Liu PP, McDonagh T, McDonald K, Mebazaa A, Nieminen MS, Peacock WF, Tubaro M, Valle R, Vanderhyden M, Yancy CW, Zannad F, Braunwald E. State of the art: using natriuretic peptide levels in clinical practice. Eur J Heart Fail. 2008;10(9):824–39.
Morsing P, Stenberg A, Casellas D, Mimran A, Müller-Suur C, Thorup C, Holm L, Persson AE. Renal interstitial pressure and tubuloglomerular feedback control in rats during infusion of atrial natriuretic peptide (ANP). Acta Physiol Scand. 1992;146(3):393–8.
Pfister R, Müller-Ehmsen J, Hagemeister J, Hellmich M, Erdmann E, Schneider CA. NT-pro-BNP predicts worsening renal function in patients with chronic systolic heart failure. Intern Med J. 2011;41(6):467–72.
Goldsmith SR. The role of vasopressin in congestive heart failure. Cleve Clin J Med. 2006;73 Suppl 3:S19–23.
Ronco C, Cicoira M, McCullough PA. Cardiorenal syndrome type 1: pathophysiological crosstalk leading to combined heart and kidney dysfunction in the setting of acutely decompensated heart failure. J Am Coll Cardiol. 2012;60(12):1031–42.
O’Brien C, Young AJ, Sawka MN. Bioelectrical impedance to estimate changes in hydration status. Int J Sports Med. 2002;23(5):361–6.
Parrinello G, Paterna S, Di Pasquale P, Torres D, Fatta A, Mezzero M, Scaglione R, Licata G. The usefulness of bioelectrical impedance analysis in differentiating dyspnea due to decompensated heart failure. J Card Fail. 2008;14(8):676–86.
Saunders CE. The use of transthoracic electrical bioimpedance in assessing thoracic fluid status in emergency department patients. Am J Emerg Med. 1988;6(4):337–40.
Konstam MA, Gheorghiade M, Burnett Jr JC, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, Zimmer C, Orlandi C, Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319–31.
Gheorghiade M, Konstam MA, Burnett Jr JC, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, Zimmer C, Orlandi C, Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials. JAMA. 2007;297(12):1332–43.
Morooka H, Iwanaga Y, Tamaki Y, Takase T, Akahoshi Y, Nakano Y, Fujiki H, Miyazaki S. Chronic administration of oral vasopressin type 2 receptor antagonist tolvaptan exerts both myocardial and renal protective effects in rats with hypertensive heart failure. Circ Heart Fail. 2012;5(4):484–92.
Goldsmith SR, Gilbertson DT, Mackedanz SA, Swan SK. Renal effects of conivaptan, furosemide, and the combination in patients with chronic heart failure. J Card Fail. 2011;17(12):982–9.
Metra M, O’Connor CM, Davison BA, Cleland JG, Ponikowski P, Teerlink JR, Voors AA, Givertz MM, Mansoor GA, Bloomfield DM, Jia G, DeLucca P, Massie B, Dittrich H, Cotter G. Early dyspnoea relief in acute heart failure: prevalence, association with mortality, and effect of rolofylline in the PROTECT Study. Eur Heart J. 2011;32(12):1519–34.
Massie BM, O’Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, Weatherley BD, Cleland JG, Givertz MM, Voors A, DeLucca P, Mansoor GA, Salerno CM, Bloomfield DM, Dittrich HC, PROTECT Investigators and Committees. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N Engl J Med. 2010;363(15):1419–28.
Maskin CS, Ocken S, Chadwick B, LeJemtel TH. Comparative systemic and renal effects of dopamine and angiotensin-converting enzyme inhibition with enalaprilat in patients with heart failure. Circulation. 1985;72(4):846–52.
Elkayam U, Ng TM, Hatamizadeh P, Janmohamed M, Mehra A. Renal vasodilatory action of dopamine in patients with heart failure: magnitude of effect and site of action. Circulation. 2008;117(2):200–5.
Schrier RW, Gross P, Gheorghiade M, Berl T, Verbalis JG, Czerwiec FS, Orlandi C, SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099–112.
Kellum JA, Decker J. Use of dopamine in acute renal failure: a meta-analysis. Crit Care Med. 2001;29(8):1526–31.
Chen HH, Anstrom KJ, Givertz MM, Stevenson LW, Semigran MJ, Goldsmith SR, Bart BA, Bull DA, Stehlik J, LeWinter MM, Konstam MA, Huggins GS, Rouleau JL, O’Meara E, Tang WH, Starling RC, Butler J, Deswal A, Felker GM, O’Connor CM, Bonita RE, Margulies KB, Cappola TP, Ofili EO, Mann DL, Dávila-Román VG, McNulty SE, Borlaug BA, Velazquez EJ, Lee KL, Shah MR, Hernandez AF, Braunwald E, Redfield MM, NHLBI Heart Failure Clinical Research Network. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA. 2013;310(23):2533–43.
Cheng JW. Nesiritide: review of clinical pharmacology and role in HF management. Heart Dis. 2002;4(3):199–203.
O’Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, McMurray JJ, Nieminen MS, Reist CJ, Rouleau JL, Swedberg K, Adams Jr KF, Anker SD, Atar D, Battler A, Botero R, Bohidar NR, Butler J, Clausell N, Corbalán R, Costanzo MR, Dahlstrom U, Deckelbaum LI, Diaz R, Dunlap ME, Ezekowitz JA, Feldman D, Felker GM, Fonarow GC, Gennevois D, Gottlieb SS, Hill JA, Hollander JE, Howlett JG, Hudson MP, Kociol RD, Krum H, Laucevicius A, Levy WC, Méndez GF, Metra M, Mittal S, Oh BH, Pereira NL, Ponikowski P, Tang WH, Tanomsup S, Teerlink JR, Triposkiadis F, Troughton RW, Voors AA, Whellan DJ, Zannad F, Califf RM. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365(1):32–43.
Pan S, Chen HH, Dickey DM, Boerrigter G, Lee C, Kleppe LS, Hall JL, Lerman A, Redfield MM, Potter LR, Burnett Jr JC, Simari RD. Biodesign of a renal-protective peptide based on alternative splicing of B-type natriuretic peptide. Proc Natl Acad Sci U S A. 2009;106(27):11282–7.
Paterna S, Di Pasquale P, Parrinello G, Fornaciari E, Di Gaudio F, Fasullo S, Giammanco M, Sarullo FM, Licata G. Changes in brain natriuretic peptide levels and bioelectrical impedance measurements after treatment with high-dose furosemide and hypertonic saline solution versus high-dose furosemide alone in refractory congestive heart failure: a double-blind study. J Am Coll Cardiol. 2005;45(12):1997–2003.
Teerlink JR, Metra M, Felker GM, Ponikowski P, Voors AA, Weatherley BD, Marmor A, Katz A, Grzybowski J, Unemori E, Teichman SL, Cotter G. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet. 2009;373(9673):1429–39.
Felker GM. Diuretic management in heart failure. Congest Heart Fail. 2010;16 Suppl 1:S68–72.
Hasselblad V, Gattis Stough W, Shah MR, Lokhnygina Y, O’Connor CM, Califf RM, Adams Jr KF. Relation between dose of loop diuretics and outcomes in a heart failure population: results of the ESCAPE trial. Eur J Heart Fail. 2007;9(10):1064–9.
Mielniczuk LM, Tsang SW, Desai AS, Nohria A, Lewis EF, Fang JC, Baughman KL, Stevenson LW, Givertz MM. The association between high-dose diuretics and clinical stability in ambulatory chronic heart failure patients. J Card Fail. 2008;14(5):388–93.
Butler J, Forman DE, Abraham WT, Gottlieb SS, Loh E, Massie BM, O’Connor CM, Rich MW, Stevenson LW, Wang Y, Young JB, Krumholz HM. Relationship between heart failure treatment and development of worsening renal function among hospitalized patients. Am Heart J. 2004;147(2):331–8.
Wilcox CS, Guzman NJ, Mitch WE, Kelly RA, Maroni BJ, Souney PF, Rayment CM, Braun L, Colucci R, Loon NR. Na+, K+, and BP homeostasis in man during furosemide: effects of prazosin and captopril. Kidney Int. 1987;31(1):135–41.
Francis GS, Siegel RM, Goldsmith SR, Olivari MT, Levine TB, Cohn JN. Acute vasoconstrictor response to intravenous furosemide in patients with chronic congestive heart failure. Activation of the neurohumoral axis. Ann Intern Med. 1985;103(1):1–6.
Dormans TP, Gerlag PG. Combination of high-dose furosemide and hydrochlorothiazide in the treatment of refractory congestive heart failure. Eur Heart J. 1996;17(12):1867–74.
van Vliet AA, Donker AJ, Nauta JJ, Verheugt FW. Spironolactone in congestive heart failure refractory to high-dose loop diuretic and low-dose angiotensin-converting enzyme inhibitor. Am J Cardiol. 1993;71(3):21A–8.
Hensen J, Abraham WT, Dürr JA, Schrier RW. Aldosterone in congestive heart failure: analysis of determinants and role in sodium retention. Am J Nephrol. 1991;11(6):441–6.
Damman K, Ng Kam Chuen MJ, MacFadyen RJ, Lip GY, Gaze D, Collinson PO, Hillege HL, van Oeveren W, Voors AA, van Veldhuisen DJ. Volume status and diuretic therapy in systolic heart failure and the detection of early abnormalities in renal and tubular function. J Am Coll Cardiol. 2011;57(22):2233–41.
Albert NM, Nutter B, Forney J, Slifcak E, Tang WH. A randomized controlled pilot study of outcomes of strict allowance of fluid therapy in hyponatremic heart failure (SALT-HF). J Card Fail. 2013;19(1):1–9.
Hummel SL, Seymour EM, Brook RD, Sheth SS, Ghosh E, Zhu S, Weder AB, Kovács SJ, Kolias TJ. Low-sodium DASH diet improves diastolic function and ventricular-arterial coupling in hypertensive heart failure with preserved ejection fraction. Circ Heart Fail. 2013;6(6):1165–71.
Gates PE, Tanaka H, Hiatt WR, Seals DR. Dietary sodium restriction rapidly improves large elastic artery compliance in older adults with systolic hypertension. Hypertension. 2004;44(1):35–41.
Jablonski KL, Racine ML, Geolfos CJ, Gates PE, Chonchol M, McQueen SMB, Seals DR. Dietary sodium restriction reverses vascular endothelial dysfunction in middle-aged/older adults with moderately elevated systolic blood pressure. J Am Coll Cardiol. 2013;61(3):335–43.
Jessup M, Abraham WT, Casey DE, Feldman AM, Francis GS, Ganiats TG, Konstam MA, Mancini DM, Rahko PS, Silver MA, Stevenson LW, Yancy CW. 2009 focused update: ACCF/AHA guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119(14):1977–2016.
Howlett JG, McKelvie RS, Costigan J, Ducharme A, Estrella-Holder E, Ezekowitz JA, Giannetti N, Haddad H, Heckman GA, Herd AM, Isaac D, Kouz S, Leblanc K, Liu P, Mann E, Moe GW, O’Meara E, Rajda M, Siu S, Stolee P, Swiggum E, Zeiroth S, Canadian Cardiovascular Society. The 2010 Canadian Cardiovascular Society guidelines for the diagnosis and management of heart failure update: Heart failure in ethnic minority populations, heart failure and pregnancy, disease management, and quality improvement/assurance programs. Can J Cardiol. 2010;26(4):185–202.
Dickstein K, Cohen-Solal A, Filippatos G, McMurray JJ, Ponikowski P, Poole-Wilson PA, Strömberg A, van Veldhuisen DJ, Atar D, Hoes AW, Keren A, Mebazaa A, Nieminen M, Priori SG, Swedberg K, ESC Committee for Practice Guidelines (CPG). ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J. 2008;29(19):2388–442.
Costanzo MR, Guglin ME, Saltzberg MT, Jessup ML, Bart BA, Teerlink JR, Jaski BE, Fang JC, Feller ED, Haas GJ, Anderson AS, Schollmeyer MP, Sobotka PA, UNLOAD Trial Investigators. Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J Am Coll Cardiol. 2007;49(6):675–83.
Shin JT, Dec GW. Ultrafiltration should not replace diuretics for the initial treatment of acute decompensated heart failure. Circ Heart Fail. 2009;2(5):505–11.
Felker GM, Mentz RJ. Diuretics and ultrafiltration in acute decompensated heart failure. J Am Coll Cardiol. 2012;59(24):2145–53.
Marenzi G, Grazi S, Giraldi F, Lauri G, Perego G, Guazzi M, Salvioni A, Guazzi MD. Interrelation of humoral factors, hemodynamics, and fluid and salt metabolism in congestive heart failure: effects of extracorporeal ultrafiltration. Am J Med. 1993;94(1):49–56.
Agostoni P, Marenzi G, Lauri G, Perego G, Schianni M, Sganzerla P, Guazzi MD. Sustained improvement in functional capacity after removal of body fluid with isolated ultrafiltration in chronic cardiac insufficiency: failure of furosemide to provide the same result. Am J Med. 1994;96(3):191–9.
Bock JS, Gottlieb SS. Cardiorenal syndrome: new perspectives. Circulation. 2010;121(23):2592–600.
Bart BA, Boyle A, Bank AJ, Anand I, Olivari MT, Kraemer M, Mackedanz S, Sobotka PA, Schollmeyer M, Goldsmith SR. Ultrafiltration versus usual care for hospitalized patients with heart failure: the Relief for Acutely Fluid-Overloaded Patients with Decompensated Congestive Heart Failure (RAPID-CHF) trial. J Am Coll Cardiol. 2005;46(11):2043–6.
Costanzo MR, Saltzberg M, O’Sullivan J, Sobotka P. Early ultrafiltration in patients with decompensated heart failure and diuretic resistance. J Am Coll Cardiol. 2005;46(11):2047–51.
Costanzo MR, Saltzberg MT, Jessup M, Teerlink JR, Sobotka PA, Ultrafiltration Versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Heart Failure (UNLOAD) Investigators. Ultrafiltration is associated with fewer rehospitalizations than continuous diuretic infusion in patients with decompensated heart failure: results from UNLOAD. J Card Fail. 2010;16(4):277–84.
Broekman KE, Sinkeler SJ, Waanders F, Bartels GL, Navis G, Janssen WM. Volume control in treatment-resistant congestive heart failure: role for peritoneal dialysis. Heart Fail Rev. 2014. [Epub ahead of print].
Nakayama M, Nakano H. Novel therapeutic option for refractory heart failure in elderly patients with chronic kidney disease by incremental peritoneal dialysis. J Cardiol. 2010;55(1):49–54.
Sotirakopoulos NG, Kalogiannidou IM, Tersi ME, Mavromatidis KS. Peritoneal dialysis for patients suffering from severe heart failure. Clin Nephrol. 2011;76(2):124–9.
Sánchez JE, Ortega T, Rodríguez C, Díaz-Molina B, Martín M, Garcia-Cueto C, Vidau P, Gago E, Ortega F. Efficacy of peritoneal ultrafiltration in the treatment of refractory congestive heart failure. Nephrol Dial Transplant. 2010;25(2):605–10.
Cnossen TT, Kooman JP, Konings CJ, Uszko-Lencer NH, Leunissen KM, van der Sande FM. Peritoneal dialysis in patients with primary cardiac failure complicated by renal failure. Blood Purif. 2010;30(2):146–52.
Núñez J, González M, Miñana G, Garcia-Ramón R, Sanchis J, Bodí V, Núñez E, Puchades MJ, Palau P, Merlos P, Llàcer A, Miguel A. Continuous ambulatory peritoneal dialysis as a therapeutic alternative in patients with advanced congestive heart failure. Eur J Heart Fail. 2012;14(5):540–8.
Koch M, Haastert B, Kohnle M, Rump LC, Kelm M, Trapp R, Aker S. Peritoneal dialysis relieves clinical symptoms and is well tolerated in patients with refractory heart failure and chronic kidney disease. Eur J Heart Fail. 2012;14(5):530–9.
Carubelli V, Metra M, Lombardi C, Bettari L, Bugatti S, Lazzarini V, Dei Cas L. Renal dysfunction in acute heart failure: epidemiology, mechanisms and assessment. Heart Fail Rev. 2012;17(2):271–82.
Giamouzis G, Butler J, Starling RC, Karayannis G, Nastas J, Parisis C, Rovithis D, Economou D, Savvatis K, Kirlidis T, Tsaknakis T, Skoularigis J, Westermann D, Tschöpe C, Triposkiadis F. Impact of dopamine infusion on renal function in hospitalized heart failure patients: results of the Dopamine in Acute Decompensated Heart Failure (DAD-HF) Trial. J Card Fail. 2010;16(12):922–30.
Hernandez AF, O’Connor CM, Starling RC, Reist CJ, Armstrong PW, Dickstein K, Lorenz TJ, Gibler WB, Hasselblad V, Komajda M, Massie B, McMurray JJ, Nieminen M, Rouleau JL, Swedberg K, Califf RM. Rationale and design of the Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure Trial (ASCEND-HF). Am Heart J. 2009;157(2):271–7.
Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293(15):1900–5.
Ponikowski P, Metra M, Teerlink JR, Unemori E, Felker GM, Voors AA, Filippatos G, Greenberg B, Teichman SL, Severin T, Mueller-Velten G, Cotter G, Davison BA. Design of the RELAXin in acute heart failure study. Am Heart J. 2012;163(2):149–55.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Afsar, B., Kanbay, M. (2015). Renal Congestion in Heart Failure. In: Goldsmith, D., Covic, A., Spaak, J. (eds) Cardio-Renal Clinical Challenges. Springer, Cham. https://doi.org/10.1007/978-3-319-09162-4_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-09162-4_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09161-7
Online ISBN: 978-3-319-09162-4
eBook Packages: MedicineMedicine (R0)