
Abstract
Through an impressive range of dynamic interactions, proteins succeed to carry out the majority of functions in a cell. These temporally and spatially regulated interactions provide the means through which one single protein can perform diverse functions and modulate different cellular pathways. Understanding the identity and nature of these interactions is therefore critical for defining protein functions and their contribution to health and disease processes. Here, we provide an overview of workflows that incorporate immunoaffinity purifications and quantitative mass spectrometry (frequently abbreviated as IP-MS or AP-MS) for characterizing protein–protein interactions. We discuss experimental aspects that should be considered when optimizing the isolation of a protein complex. As the presence of nonspecific associations is a concern in these experiments, we discuss the common sources of nonspecific interactions and present label-free and metabolic labeling mass spectrometry-based methods that can help determine the specificity of interactions. The effective regulation of cellular pathways and the rapid reaction to various environmental stresses rely on the formation of stable, transient, and fast-exchanging protein–protein interactions. While determining the exact nature of an interaction remains challenging, we review cross-linking and metabolic labeling approaches that can help address this important aspect of characterizing protein interactions and macromolecular assemblies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Nonspecific Interaction
- Metabolic Label
- Transient Interaction
- Tandem Affinity Purification
- Immunoaffinity Purification
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
11.1 Overview
Proteins carry out the majority of functions in a cell, and their regulation at a core of health or disease processes. While cells contain a multitude of proteins, of various sizes and abundances, it is almost never the case for one protein to have only one function. Any given one protein usually has the remarkable ability to perform numerous functions. The main tactic through which proteins carry out these diverse functions is through formation of numerous interactions. These interactions can be dynamic, spatially and temporally defined, and stable or transient in nature. For example, one enzyme can have many substrates, and their regulation can have different downstream impacts on cellular pathways. Similarly, one protein can be part of multiple protein complexes that can have distinct functions. Given their fundamental contribution to cellular processes, the study of protein–protein interactions has become an essential part of biological discovery. In this chapter, we discuss one of the most commonly utilized approaches for studying protein interactions—immunoaffinity purification coupled with mass spectrometry analysis (IP-MS). We start by describing the types of optimizations that need to be considered when designing an IP-MS experiment to ensure efficient isolation and accurate characterization of protein complexes. Next, we discuss what controls should we performed and how mass spectrometry data can be used to distinguish specific versus background interactions. Within this context, we cover some of the most frequently implemented label-free and metabolic labeling approaches. Lastly, we describe some of the recent developments in capturing transient associations and measuring the relative stability of interactions. Application of cross-linking approaches for studying protein complex structures and transient interactions is also discussed.
11.2 Methods for Isolating Protein Complexes
Common workflows for characterization of protein complexes. Immunoaffinity purification (IP) of proteins is a powerful approach for characterizing proteins of interest, their direct and indirect interactions required for formation of complexes, as well as their posttranslational modifications (PTMs). This information provides critical insights into the functions of proteins in different pathways, as well as regulation of their functions by various mechanisms (e.g., inhibiting or activating PTMs) [1, 2]. A standard workflow for isolating protein complexes is illustrated in Fig. 11.1. This workflow starts with the selection of an appropriate cell line or tissue sample, effective lysis of the sample, isolation and elution of the target protein with its interactions, followed by mass spectrometry analysis and identification and quantification of the co-isolated proteins. Each step of this process can be modified and optimized based on the nature of the protein of interest, its subcellular localization and abundance, and the overall goal of the study. Several important considerations for these optimization experiments are detailed below.

Common workflow for immunoaffinity purification mass spectrometry experiments. Cells expressing the protein of interest are lysed and protein complexes are isolated by immunoaffinity purification. Eluted proteins are processed for MS analysis. MS spectra are analyzed to identify proteins within isolated complex(es) and bioinformatics tools are implemented to generate protein interaction networks
Optimizing conditions for immunoaffinity purifications. One of the first questions that needs to be addressed prior to IP is whether the endogenous protein or its tagged version would be a suitable candidate for the study. Endogenous proteins can be purified from tissue or cells at their biological levels, providing the best representation of their functional states. Isolation of an endogenous protein requires availability of antibody with high specificity and affinity to ensure efficient and clean purification. Endogenous protein isolation is extensively utilized in small- and large-scale studies [3–6]. For example, Li et al. isolated the nuclear DNA sensor IFI16 to define its localization-dependent antiviral functions [5]. Large-scale isolation of endogenous transcriptional and signaling proteins and their complexes was used by Malovannaya et al. to provide insight into the composition of human coregulator protein complexes [3]. However, antibody production can be costly and not all antibodies are commercially available or stored in a buffer compatible with IP conditions (e.g., large amounts of glycerol or storage in Tris buffer can interfere with coupling of an antibody to some resins), requiring additional purification steps. Therefore, these antibodies are routinely used in smaller-scale isolations for confirmatory studies. For example, Tsai et al. reported co-isolation of endogenous sirtuin 7 with B-WICH components and Pol I, supporting its role in regulation of Pol I transcription [4]. As an alternative to the use of antibodies for immunoaffinity purification studies, recent reports have proposed the use of small molecules, such as activity-based chemical probes or inhibitors, covalently linked to a resin for isolation of enzymes and their complexes [7–10]. For example, a large-scale study used histone deacetylase inhibitors to assess their affinity to different complexes [9]. Other approaches for isolation of endogenous targets include the use of nucleic acids and engineered binding proteins (reviewed by Ruigrok et al. [11]), which are actively incorporated in various biomedical studies [12, 13]. With their lower cost of production and higher stability than antibodies, these molecules have become valuable tools for isolating and characterizing protein complexes [14].
A more commonly utilized approach in studies of protein–protein interactions involves the tagging of the protein of interest, followed by its isolation using a tag-specific antibody. This method can be customized for the use of different tags (e.g., FLAG, EGFP, HA) and expression of the fusion protein from endogenous (genomic) or exogenous (e.g., tetracycline-inducible) promoters. For example, Quantitative BAC InteraCtomics (QUBIC) approach utilizes expression of tagged proteins from native promoters, followed by IP and quantitative MS analysis, which aids the identification of specific interacting partners [15]. The use of a fluorescent tag, such as green fluorescent protein (GFP), allows combining information regarding protein localization and interactions [16] and provides a complementary validation of protein–protein interactions, as shown for virus–host protein interactions [17, 18]. Tandem affinity purification (TAP) strategies, which use multiple isolation steps via different tags, are useful for achieving cleaner purifications, leading to the isolation of fewer nonspecific interactions, however, at the expense of weaker interacting partners [19–21]. A variation of TAP tagging can be used, where two proteins that are known to be present in a complex are tagged and purified simultaneously (bimolecular affinity purification), allowing for the specific isolation of a homogeneous population of protein complexes [22]. For all these approaches, one major concern that has to be addressed when using tags for protein IP is whether tagging alters protein function. To verify its functional state, the localization and function (e.g., enzymatic activity) of the tagged protein can be compared against endogenous control (e.g., [2]).
The choice of the affinity resin also has an impact on the success of the IP experiment, influencing the efficiency of the isolation and level of nonspecific interactions. Common choices include sepharose and agarose beads, as well as the steady growth in popularity magnetic beads [22–24]. The surface area of the bead determines not only its capacity for the number of antibody molecules that it can bind to, but also the nonspecific associations to the resin itself. Resins with various chemistries are available for antibody binding (i.e., antibody-binding proteins, primary amines reactive groups, cross-linking to affinity ligands) and can help reduce the amount of eluting immunoglobulin molecules, limiting the interference in MS analysis. In the “Determining specificity of interactions” section in this chapter, the impact of the resin choice on the amount of nonspecific interactions is discussed in detail.
The lysis of the selected cells or tissue is the first step of an IP experiment, and it can impact the preservation of protein–protein interactions. Therefore, the procedure selected for lysis and the composition of the lysis buffers require careful considerations. Mechanical disruption can be performed on wet or frozen samples. For example, cryogenic lysis was shown to provide an effective and reproducible disruption of cellular organelles and membranes, while helping to maintain protein complexes and PTMs [25, 26]. This method is appropriate for different cell types and was successfully applied in studies in bacteria, yeast, mammalian cells, and tissues, as well as following viral infection (as reviewed in [27]). If it is necessary to preserve intact intracellular structures, fractionation steps can be added to the protocol. For example, nuclear-cytoplasmic fractionation was used for assessment of localization-dependent protein–protein interactions of HDAC5 mutated at different phosphorylation sites that regulate its nuclear-cytoplasmic shuttling [1]. Importantly, the stringency of the lysis buffer in an IP experiment determines the nature of isolated interactions. Low salt concentrations and mild detergents may allow preservation of weak interactions, while isolation in a more stringent buffer will enrich for strongly bound interacting partners [28]. Miteva et al. compared the presence of distinct SIRT6 interactions under mild or stringent lysis conditions, which allowed determining their relative stability, as well as validation of specificity [6]. Addition of sonication step and RN/DNases to the lysis buffer can help remove interactions dependent on nucleic acid binding. It is also important to consider the compatibility of the lysis buffer detergents with the downstream sample analysis by MALDI or ESI/LC-MS/MS. For example, analysis of membrane-bound proteins can be hindered by the necessity to use harsh detergents that are detrimental for MS analysis, although it was shown that n-octylglucoside detergent is compatible with MALDI-MS [29]. Another solution is to use cleavable detergents that can be removed from the sample prior to the analysis [30, 31]. Of course, the composition of lysis buffer and duration of lysis has a profound impact on the level of observed nonspecific interacting partners, as discussed in detail in the “Determining specificity of interactions” section.
Following the isolation of the protein of interest, the elution conditions from the beads can also be optimized for different purposes, such as to reduce immunoglobulin contamination, to preserve native protein folding, or to assess the stability of interactions. Most commonly utilized elution buffers are sodium dodecyl sulfate- or lithium dodecyl sulfate-based, which denature the isolated proteins and are suitable for in-solution digest prior to MS analysis [32]. Basic (e.g., ammonium hydroxide and ethylenediaminetetraacetic acid) or acidic (e.g., trichloroacetic acid) elutions are also denaturing, but reduce the amount of background protein contamination [26]. For analysis of native proteins and their complexes, non-denaturing conditions can be used, such as competitive binders [33, 34].
Isolated protein complexes can be analyzed by bottom-up, middle-down, or top-down MS approaches [35–37]. For reduction of sample complexity, protein mixtures can be resolved by gel electrophoresis prior to digestion. Combination of different proteases can be used to improve sequence coverage and identification of PTMs [5]. Separation by liquid chromatography, performed either offline or online with the mass spectrometer, is also used to decrease the sample complexity and provide an in-depth analysis. Different types of peptide fragmentation, such as collision-induced dissociation (CID), electron transfer dissociation (ETD), and higher energy C-trap dissociation (HCD), have also significantly enhanced the current ability to characterize proteins (as reviewed in [38]). Targeted mass spectrometry-based approaches, such as selective reaction monitoring (SRM), further help the identification and quantification of low levels of proteins [39].
11.3 Determining Specificity of Interactions
The complex and dynamic nature of protein–protein interactions coexisting in a cell presents challenges for any IP study. During cell lysis, proteins lose their intracellular localizations, triggering an opportunity for numerous nonspecific interactions to occur. Additionally, nonspecific associations can occur with the resin, tags, or antibodies used for the study. In this section, we discuss some of the sources of nonspecific interactions and how these can be minimized when designing the IP workflow, as well as approaches used for determining the specificity of observed interactions following data analysis.
Sources of nonspecific interactions. The presence of background proteins in affinity-purified protein mixtures is determined by multiple factors (Fig. 11.2). Nonspecific interactions can include proteins that bind to resin (e.g., magnetic beads), to immunoglobulin molecules, to the tag, and to other isolated proteins. For instance, although polyclonal antibodies have higher affinities and provide more efficient isolations, they also tend to accumulate more nonspecific associations than monoclonal antibodies. As for the choice of the IP resin, it was observed that it can also differentially impact the level and type of nonspecific binders [24, 40]. Sepharose beads seemed to preferentially isolate nonspecific nucleic acid-binding factors, while magnetic beads are prone to association with cytoskeletal and structural proteins [24]. However, magnetic beads can be collected on a magnet, removing the need for a centrifugation step, which reduces sample loss and effectively removes flow-through. Additionally, magnetic beads were preferred for isolating organelles or larger structures or macromolecules, given their feature of surface binding [41, 42].

Dependence of interaction specificity on IP conditions. Optimized lysis and incubation conditions, such as stringency of lysis buffer and incubation time with beads and antibodies, allow retention of specific stable and transient interactions, while reducing the number of nonspecific associations. The latter include proteins that bind to beads, tag, immunoglobulin molecules, or to specific isolated proteins
Proteins that nonspecifically associate with isolated protein complexes are a significant source of background contamination that can be reduced by optimizing IP conditions (Fig. 11.2). For instance, the composition of the lysis buffer and the incubation period with the beads/resin and the antibody greatly influence nonspecific binding. More stringent buffers that contain higher concentrations of salts and detergents can be used to focus on the isolation of strong interactions, helping to reduce nonspecific interactions that tend to be weaker. On the other hand, very stringent buffers that are used to improve extraction of proteins from membranes and intracellular vesicles can lead to protein denaturation, which introduces additional surfaces for nonspecific binding (e.g., to heat shock proteins [43]). In addition, Cristea et al. demonstrated that the length of time used for the incubation of cell lysates with the beads affects the number and abundance of nonspecific interactions [16]. It was suggested to keep the incubation periods as short as possible, ranging from several minutes up to 1 h, depending on the abundance of the targeted protein and the affinity of the antibody. Therefore, optimization of lysis buffer composition and immunoaffinity purification period allows achieving cleaner isolations, while preserving specific interactions (Fig. 11.2). Additionally, nonspecific interactions can occur upon cell disruption and mixing of proteins from different intracellular compartments. This type of background can be avoided by performing a fractionation step prior to the IP workflow and the lysis/time optimizations mentioned above.
Designing appropriate control experiments and validating interactions. Suitable controls have to accompany every IP experiment because, even with optimized conditions, low levels of nonspecific binders will still be present in the final isolation. Control experiments have to be carefully designed to follow the same conditions as the IP of the protein of interest. For instance, when isolating a tagged protein, a suitable control would be a cell line that expresses only the tag. If the targeted protein is localized within a particular cellular compartment, the control cell line should be designed to afford localization of the tag alone within the same compartment (e.g., by addition of a nuclear localization signal for expression within the nucleus [18]). Similarly, isolation of an endogenous protein requires a control incubation done in parallel using beads coupled to immunoglobulin molecules, which will capture proteins that associate nonspecifically to the antibody. If isolations are performed at different stages of a biological process, such as cell cycle or viral infection, it is necessary to introduce a control experiment for each time point [17]. This will account for variations in the type and abundance of nonspecific interactions throughout the process. Altogether, these control isolations will help differentiate nonspecific associations that occur via the tag, the antibody, and the resin type.
In a collaborative effort, several proteomic laboratories have provided their data from numerous control isolations performed in different cell types and using various resins, tags, and antibodies to generate a repository freely available to the scientific community [40]. This resource, termed the CRAPome database, allows users to determine the frequency of appearance of a protein of interest in control IPs or to analyze their own datasets in comparison to controls available online and assess the presence of common background contaminants. This repository is continuously expanding with new data submitted and processed according to the established workflow.
Validation of isolated interactions is another critical step in all studies aimed at characterizing protein–protein interactions. Such experiments include reciprocal isolations, where an identified prey protein of interest is used as bait in a follow-up IP experiment to confirm the co-isolation of the initial targeted protein. However, it is necessary to keep in mind that reciprocal IPs might prove challenging in identifying a target protein that has low levels of expression. As a consequence, its signal in the prey IP might be suppressed by the presence of more abundant interactions. On the other hand, the prey IP might not be feasible if an antibody is not available for it, but the use of tagging can overcome this problem. Co-localization studies using confocal microscopy or simple binary approaches (e.g., yeast two-hybrid) are also successfully applied for validating interactions [4, 6, 44].
Label-free methods for determining interaction specificity. Qualitative and quantitative data derived from MS analysis of co-isolated proteins contain valuable information regarding the specificity of identified interactions. Therefore, several algorithms were developed for this purpose [49].
Various scoring systems, such as the socio-affinity index and purification enrichment score, were utilized for the analysis of interactions isolated from large-scale IP studies in yeast [45–48]. Their purpose was to decrease the number of identified false-positive interactions, while retaining abundant interactions that are frequently assigned as false-negatives. For example, V-ATPase, an abundant and common contaminant in numerous studies, can be selectively rescued if assigned as a possible specific interaction [45]. Computational approaches were also applied in studies of mammalian interactomes. Among them is the interaction reliability score that was derived for the study of transcription and RNA processing complexes to assign high-confidence interactions [49].
Quantitative data generated by MS analysis in the form of spectral counts (total number of spectra observed per protein) are becoming increasingly utilized for predicting the specificity of interactions [2, 50–52]. In this approach, the number of spectra observed for a particular protein in the bait versus control IP indicates whether this interaction is likely specific for the targeted protein. For further analysis, normalized spectrum abundance factor (NSAF) was introduced by Paoletti et al. to account for the number of amino acids that a protein contains, with larger proteins expected to generate a higher number of spectral counts in MS analysis [53]. When combined with the protein abundance factor (PAX) [54] that reflects total protein abundance in a cell, resulting NSAF/PAX ratio becomes a good indicator of the enrichment of a particular interaction among co-isolated proteins [4]. However, it should be kept in mind that PAX values are not yet derived for all cell types and can change drastically under different environmental conditions or under stress (e.g., during viral infection).
The SAINT (significance analysis of interactome) algorithm was developed by Nesvizhskii et al. to generate a probability model for distributions of false-positives and false-negatives in IP data and to assign confidence scores to identified interactions [52]. For example, SAINT was utilized in a large-scale interactome study of the insulin receptor/target of rapamycin pathway in Drosophila, helping the identification of interactions important in controlling cell growth upon stimulation with insulin [55]. More recently, the SAINT algorithm was further optimized to account for the large dynamic range of spectral counts that is frequently observed in human interactomes, such as in the global interaction network of all eleven human histone deacetylases [2]. This study led to the identification of numerous HDAC-containing protein complexes, as well as a previously unrecognized function for HDAC11 in mRNA splicing. The CRAPome database mentioned above also utilized the SAINT algorithm for analyzing the collection of control IPs derived from different cell types and performed in various laboratories [40]. Another spectral counting-based program, called CompPASS, uses several scoring systems to derive confidence scores for interactions found in multiple parallel nonreciprocal IPs, without the use of control IPs [51]. Algorithms that utilize other MS data, such as MS1 signals (MasterMap) and peak intensities (MiST), are also being developed with the goal of overcoming some of the limitations of spectral counting approaches, such as dependence of interaction abundances on bait and prey levels, efficiency of IP, and detection by MS [56, 57]. A more complete summary of available algorithms is reported in [27].
The label-free approaches mentioned above have several advantages over labeling approaches that will be discussed in the next section. They do not require expensive reagents, can be used for the analysis of tissue samples, and can be applied in both small- and large-scale studies. However, these approaches have certain limitations. For example, protein abundance has a significant impact on the assessment of specificity (i.e., cannot reliably quantify changes for proteins with low spectral counts). Additionally, as is the case for most methods, these approaches cannot fully segregate background proteins from specific interactions within the multitude of co-isolated proteins.
Labeling methods for determining interaction specificity. Metabolic and chemical labeling approaches were introduced into MS analysis workflows to provide absolute or relative protein quantification. During the last decade, the application of these approaches within targeted or global studies has revolutionized the field of proteomics and its ability to contribute to critical biological discoveries. These labeling methods have certain limitations, such as their challenging application to tissue samples and variations in sample processing prior to chemical labeling. Nevertheless, in recent years, the application of these approaches was expanded to include their incorporation into IP workflows for the downstream analysis of interaction specificity.
Labeling with stable isotopes, such as 15 N, and later with heavy amino acids in cell culture (SILAC) were among the first metabolic labeling methods to be introduced within mass spectrometry-based workflows [58, 59]. For identification of interaction specificity using metabolic labeling, Chait and colleagues developed the I-DIRT (Isotopic Differentiation of Interactions as Random or Targeted) approach and applied it to the study of DNA polymerase ∈ complex in yeast [60]. In their workflow, cells expressing the affinity-tagged protein are grown in medium containing naturally occurring amino acids, termed isotopically light medium. In contrast, wild-type cells are grown in isotopically heavy medium that contains amino acids labeled with heavy isotopes (e.g., 13C). Proteins are immunoaffinity purified from a 1:1 mixture of these light and heavy cell lysates, and the specific interacting partners can be recognized as having only or predominantly light isotopic peaks (Fig. 11.3a). An “SRM-like” I-DIRT approach, in which the specificity of interaction for selected proteins of interest can be assessed using targeted MS/MS, may be utilized to analyze low abundance interactions [4]. To assess interaction specificity in studies of endogenous protein complexes, QUICK (quantitative immunoprecipitation combined with knockdown) strategy was developed [61–64]. In this workflow, light-labeled cell cultures are treated with RNAi against the protein of interest, while heavy-labeled cells serve as nontargeted controls. In subsequent MS analysis, light and heavy peptide intensities are compared to assign nonspecific (1:1 heavy to light rations) and specific (heavy isotopic peaks with higher intensity than light) interactions. QUICK can also be combined with cross-linking to stabilize protein complexes in cell extracts prior to IP, as demonstrated for VIPP1 complex functioning in chloroplast biogenesis [65]. Some of the disadvantages of the QUICK approach include SILAC-associated costs, arginine-to-proline conversions that produce difficulties in data interpretation, and uncontrollable alterations in protein expression due to RNAi knockdown. These concerns were addressed in an alternative QUICK method utilizing 15 N labeling and affinity modulation of protein–protein interactions [66]. SILAC approaches in the form of PAM (purification after mixing) SILAC and MAP (mixing after purification) SILAC were also used to assess the interaction specificity in several studies [67, 68]. These methods allow distinguishing between stable and transient interactions, which will be further discussed in the next section. Affinity purification of integral membrane proteins using nanodiscs, which circumvent the need for stringent detergents, was combined with SILAC in the analysis of interacting partners of bacterial channel, transporter, and integrase proteins [69]. In the study of phosphatase and tensin homologue (PTEN) interactions, IPs from two different cell lines using three different approaches (two tags and one endogenous) were combined in parallel affinity purification (PAP) SILAC approach to assign specific interactions with minimum number of false positives [70].

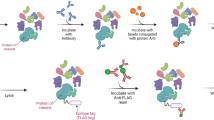
Determining specificity and relative stability of interactions using label-free and metabolic labeling approaches. (a) In the I-DIRT approach, wild-type cells grown in “heavy” medium and cells expressing the tagged protein of interest grown in “light” medium are mixed prior to IP. Isolated complexes are analyzed by MS and isotopic ratios for each protein are indicative of the specificity and stability of the interaction. (b) When label-free quantification (e.g., SAINT) is combined with a metabolic labeling approach (e.g., I-DIRT), the relative stability of interactions can be assessed. Specific transient interactions can be observed with high SAINT scores and low I-DIRT ratios
Chemical labeling was also successfully applied for the analysis of protein interactions and is typically done after IP, at either the protein or peptide level. In ICAT (isotope-coded affinity tag) approach, cysteine residues of intact proteins in bait and control experiments are labeled with heavy or light ICAT reagents, respectively [71, 72]. Therefore, specific interaction partners would produce peptide spectra with higher intensities for heavy isotope-containing peaks. Inherent to this method is the quantification of only cysteine-containing peptides. The iTRAQ multiplex labeling method [73], which tags peptides at N-terminal and lysine amines, was also applied to distinguish true interactions within co-isolated protein mixtures when comparing differentially labeled bait and control IPs, as shown for grb2 [74]. Isotope-coded protein labeling (ICPL) was also recently combined with IP-MS to analyze native β-tubulin complexes in bovine retinal tissue [75]. Besides being compatible with analysis of tissue samples, this method allows for simultaneous analysis of several control samples, which can account for nonspecific binders to the beads and immunoglobulin molecules. Currently, chemical labeling is not as widely applied as metabolic labeling to the analysis of interaction specificity. However, its compatibility with tissue samples and its multiplexing feature that allows direct comparison of multiple samples and diverse controls makes it a useful tool, expected to continue to aid the discovery of protein complex compositions relevant in biomedical research.
11.4 Cross-Linking Methods
In MS studies of protein complexes, cross-linking is used for covalent joining of two or more molecules. Chemical cross-linking reagents have various chemistries (e.g., amino-, sulfhydryl-, carboxyl-reactive), sizes that determine the distance between cross-linked peptides, and add-on features for easier detection and identification (e.g., reversible cross-linkers). In this section, the use of cross-linking in studies of protein complex structures is discussed, while its application to capturing transient protein interactions is described in the section on “Determining stability of interactions”.
Solving protein complex structures. Continuous advances in cross-linking methodologies, the improved sensitivity of MS instrumentation, and the development of automated algorithms for database searching have significantly expanded the use of cross-linking for characterization of protein complexes. Examples of elegant structures resolved using cross-linking methodologies include the yeast 19S proteasome lid, RNA Pol II, phage DNA packaging machinery, human protein phosphatase 2A (PP2A) complexes, INO80 nucleosome complex, TRiC/CCT chaperonin to name a few [76–80]. These cross-linking strategies are powerful at defining the exact points of contacts between proteins of interest. One caveat to keep in mind is that proteins can be part of multiple protein complexes that can have common components. For example, the histone deacetylases HDAC1 and HDAC2 are known to form the core of several distinct complexes, such as NuRD and Sin3A complexes [2]. Therefore, even after identifying the point of contact between two proteins, one may not know where the interaction takes place, i.e., which complex or which conformational state of the complex is represented. To partly address this issue, cross-linking is frequently combined with knowledge from X-ray crystallography studies. Nevertheless, as crystallography results usually reflect a more static or stable conformation of a protein, these issues should still be kept in mind.
Cross-linking was also integrated with other biochemical or mass spectrometry tools to help define protein structures and interactions within macromolecular assemblies. For example, a combination of cross-linking with hydrogen/deuterium exchange was used to decipher inter-subunit interactions critical in the assembly of HIV-1 capsid protein, which complemented studies performed using X-ray crystallography and cryo-electron microscopy [81]. In another study, the folding of the immune receptor NKR-P1C was resolved using cross-linking, molecular modeling, and ion mobility mass spectrometry [82].
One common limitation of cross-linking when combined with MS analysis is the possible low abundance of resulting cross-linked peptides, which can make spectra interpretation challenging. To solve this problem for the analysis of the 12-subunit Pol II complex structure, Chen et al. utilized a strong cation exchange chromatography to enrich for cross-linked peptides which have multiple charges [76]. For isotopic labeling, Zelter et al. digested cross-linked peptides in the presence of H2 18O, which were then identified by their characteristic isotopic peak distribution in MS spectra [83]. Several strategies were proposed that utilize modifications of the cross-linker itself to provide easier enrichment, detection, and identification. These include affinity tags, reporter tags, isotopic and fluorescence labeling, and cleavable cross-linking, all of which can be used in combination [84]. For example, Chowdhury et al. combined an alkyne enrichment tag and NO2 detection tag when constructing a CLIP (click-enabled linker for interacting proteins) cross-linking reagent [85]. For the study of the 20S proteasome complex in yeast cells, Kao et al. designed a disuccinimidyl sulfoxide (DSSO) cross-linker that is cleavable by collision-induced dissociation and can be identified at the MS3 level [86].
The expansion in types of cross-linking strategies and applications places demands on the developments of streamlined procedures for MS data interpretation. To make this process more automated, Herzog et al. utilized specialized xQuest search engine [79, 87]. In this workflow, isotopic pairs of cross-linked peptide ions were matched against a database of candidate peptides, upon which their sequences were assigned. This algorithm was used to solve the structures of human PP2A, INO80 nucleosome, TRiC/CCT eukaryotic chaperonin, and other complexes [79, 80, 88, 89]. However, this process has a laborious scoring procedure that requires manual verification of the cross-linked peptide spectra. In addition, reliable identification of isotopically labeled cross-linked peptides in this method can suffer from incomplete labeling. To overcome this limitation, Goodlett and coworkers developed an alternative cross-linking strategy that uses Popitam search engine [90] and can identify unlabeled cross-linkers [91]. In their workflow, cross-linked peptides are considered as complementary pairs of peptides modified by an unknown mass. The spectra are interpreted by matching to theoretical spectra of single linear peptides, and further analyzed against the masses of precursor tryptic peptides and manually validated. SEQUEST [92] searching was also further optimized for the identification of cross-linked peptides from a database containing all possible products of cross-linking, which allowed matching complex spectra of cross-linked peptide pairs more efficiently [93]. For automatic validation of database search results, Walzthoeni et al. introduced the xProphet software that uses a target-decoy strategy to estimate false discovery rates in large datasets derived from cross-linking studies [94]. Many other database processing algorithms and bioinformatics tools are being continuously developed and released to address challenges associated with deciphering complex cross-linked peptide spectra [95–100]. Overall, further improvements in cross-linking strategies for easier detection and identification, as well as in the software for analysis of MS spectra of cross-linked peptides are required. However, the combination of cross-linking strategies with crystallography studies, computational modeling, and other quantitative mass spectrometry methods provides powerful approaches in proteomics that will continue to shed light on the structures of heterogeneous protein complexes.
11.5 Studying Transient and Fast-Exchanging Interactions
Coupling of efficient IP strategies with highly sensitive MS analysis leads to identification of numerous interactions, direct and indirect, transient and stable, which provide valuable information about protein function. Several methods can be used to distinguish between direct and indirect interactions, such as yeast two-hybrid and protein arrays [101]. These methods do not utilize mass spectrometry analysis and are not discussed in this review. The identification of transient and fast-exchanging interactions, such as enzyme-substrate interactions, presents challenges in mass-spectrometry proteomic workflows. These interactions can be either lost during the IP process or can be falsely assigned as nonspecific in metabolic labeling experiments. Therefore, methods are continuously being developed to help the capture and identification of these interactions in MS-based experiments.
Determining interaction stability using metabolic labeling. As mentioned in the earlier section, time-controlled PAM SILAC and MAP SILAC approaches have also been used for identification of specific interactions that are dynamic in nature [102]. In PAM SILAC approach, samples are mixed prior to purification, which allows for transient interactions to exchange quickly between light and heavy forms, resulting in equivalent levels of heavy and light ions. However, with decreased incubation time during purification, the level of heavy ions will increase. Moreover, if mixing of heavy and light-labeled samples is done after purification (MAP SILAC), same interactions will have predominant heavy ions because there will be no “light” labeled proteins present in the isolated sample. The MAP SILAC approach also allows for identification of fast-exchanging interactions, which would require short incubation times in order to be accurately assigned as specific when using the PAM SILAC approach. This approach was applied in studies of 26S proteasome and COP9 signalosome complexes [67, 68].
A combination of spectral counting (SAINT) and metabolic labeling (I-DIRT) approaches was utilized by Joshi et al. to measure relative interaction stabilities within HDAC-containing protein complexes (Fig. 11.3) [2]. In their workflow, unlabeled bait samples were analyzed against control IPs to generate a list of interactions using SAINT scores, with scores >0.8 reflective of likely specific interactions. In parallel experiments, metabolic labeling of cells expressing tagged HDACs was performed, and an I-DIRT approach was used as described above. By integrating SAINT and I-DIRT scores for each isolated protein, stability profiles could be assigned to specific interacting partners. Proteins with >0.8 SAINT scores and ~0.5 I-DIRT scores corresponded to fast-exchanging proteins, while proteins with >0.8 SAINT scores and I-DIRT scores closer to 1.0 indicated stable interactions. For example, HDAC5 and HDAC7 interact transiently with the NCoR complex due to their nucleo-cytoplasmic shuttling, while HDAC3 is a stable component of this complex. Similarly, HDAC1 was shown to be an integral component of several chromatin remodeling complexes, while transiently associating with transcription factors and DNA-binding proteins. Therefore, this approach allows confident identification of novel specific interacting partners and assignment to transient or stable associations.
Detecting transient interactions using cross-linking. Several cross-linking methods were incorporated into IP-MS workflows to study stable and transient interactions in cell culture. The main requirement for a cross-linking reagent to be used for such analysis is its cell permeability. One of the most widely utilized reagents in these studies is formaldehyde. For example, TAP of formaldehyde cross-linked SCF ubiquitin ligase complex under denaturing conditions was utilized by Tagwerker et al. to preserve and characterize novel ubiquitination targets, as well as identify transient or weak interacting partners [103]. For a more quantitative analysis, Guerrero et al. combined TAP, formaldehyde cross-linking, and SILAC approaches to characterize 26S proteasome interactions in yeast [104]. Zero distance cross-linking using photo-inducible amino acids [105] introduced into growing mammalian cells allowed identification of a direct interaction between endoplasmic reticulum stress protein MANF and GRFP78 that regulates stress-induced cell death [106]. One of the disadvantages of irreversible cross-linking is that, following the immunoaffinity purification of a protein complex, there is a low accessibility for trypsin at the core of the isolated complex, hindering the identification of selected proteins by MS. To resolve this issue, as well as other challenges connected to irreversible cross-linking, numerous studies employ reversible cross-linking. For example, reversion of formaldehyde cross-links was used in the SPINE method for detection of interacting partners of Strep-tagged membrane proteins in bacteria [107]. Another reversible cross-linking methodology—ReCLIP—utilizes thiol-cleavable cross-links and was used in the study of p120-catenin and E-cadherin complex [108, 109].
To capture transient interactions and address specificity of interactions within the same experiment, a transient I-DIRT approach was reported. This approach used cross-linking combined with isotopic labeling in yeast culture and was applied to the study of NuA3 multi-subunit complex [110]. In this workflow, cells expressing the tagged protein are grown in light media, while wild-type cells are grown in heavy media. Upon mixing cross-linked heavy and light cell cultures and purifying the target protein, MS analysis is performed and used to assign stable specific (~100 % light peptides), nonspecific (1:1 light:heavy peptide ratio), and transient (intermediate ratios) interacting partners [111].
Recent years have also seen significant developments in cross-linking reagents. To overcome the challenge of identifying cross-linked peptides in the mixed spectra generated from a complex mixture of cross-linked proteins with various intermediate products, Bruce laboratory developed Protein Interaction Reporter (PIR) technology [112]. In their methodology, the cross-linking reagent is designed to contain two labile bonds that can be cleaved during MS/MS analysis. Upon cleavage, a reporter ion is released to mark the presence of a cross-linked peptide, while cleavage of the second labile bond generates single peptides from the cross-linked pair for further fragmentation and sequencing. This technology was utilized in the study of the Potato leafroll virus capsid structure and in defining interactions of bacterial chaperones and membrane proteins [113–115].
Cross-linking with formaldehyde has also been utilized in mouse models, where the reagent was introduced via transcardiac perfusion in a time-controlled manner [116]. This method was applied in combination with isotopic labeling with iTRAQ to assign interaction specificity in studies of cellular prion protein (PrPc), oxidative stress sensor DJ-1, and amyloid precursor protein interactomes [116–119]. In addition to identifying specific interactions of PrPc, Watts et al. also suggested that information derived from the MS/MS analysis of cross-linked proteins could be used to distinguish direct and indirect interactions [118]. For instance, proteins that have high sequence coverage and share similar domain structures were most likely to represent direct interacting partners.
The task of identifying transient, stable, direct, and indirect interactions is not trivial. However, metabolic labeling and cross-linking approaches incorporated into IP-MS workflows discussed above have significantly aided these studies. There is no doubt that studies of protein–protein interactions and resulting macromolecular complexes will continue to expand our understanding of critical biological processes. Further methodological developments are needed. Approaches that specifically capture one moment in a cellular process, temporally and spatially defined, are continuously being developed and improved. While studies in cell systems provide simple models with extraordinary specificity and insight into concrete cellular pathways, expansion of interaction studies to animal models allows for in vivo validation and a systems view of the changes caused by perturbations in a single protein functions. As protein interactions are at the core of cellular, tissue, and organ functions, their study will continue to shed light onto fundamental questions in both basic science and clinical research.
References
Greco TM et al (2011) Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol Cell Proteomics 10(2):M110 004317
Joshi P et al (2013) The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol 9:672
Malovannaya A et al (2011) Analysis of the human endogenous coregulator complexome. Cell 145(5):787–799
Tsai YC et al (2012) Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol Cell Proteomics 11(5):60–76
Li T et al (2012) Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A 109(26):10558–10563
Miteva YV, Cristea IM (2014) A proteomic perspective of SIRT6 phosphorylation and interactions, and their dependence on its catalytic activity. Mol Cell Proteomics 13(1):168–183
Adam GC, Sorensen EJ, Cravatt BF (2002) Chemical strategies for functional proteomics. Mol Cell Proteomics 1(10):781–790
Salisbury CM, Cravatt BF (2007) Activity-based probes for proteomic profiling of histone deacetylase complexes. Proc Natl Acad Sci U S A 104(4):1171–1176
Bantscheff M et al (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol 29(3):255–265
Cen Y et al (2011) Mechanism-based affinity capture of sirtuins. Org Biomol Chem 9(4):987–993
Ruigrok VJ et al (2011) Alternative affinity tools: more attractive than antibodies? Biochem J 436(1):1–13
Gronwall C, Stahl S (2009) Engineered affinity proteins—generation and applications. J Biotechnol 140(3–4):254–269
Brody E et al (2012) Life’s simple measures: unlocking the proteome. J Mol Biol 422(5):595–606
Wiens M et al (2011) Isolation of the silicatein-alpha interactor silintaphin-2 by a novel solid-phase pull-down assay. Biochemistry 50(12):1981–1990
Hubner NC, Mann M (2011) Extracting gene function from protein-protein interactions using Quantitative BAC InteraCtomics (QUBIC). Methods 53(4):453–459
Cristea IM et al (2005) Fluorescent proteins as proteomic probes. Mol Cell Proteomics 4(12):1933–1941
Cristea IM et al (2006) Tracking and elucidating alphavirus-host protein interactions. J Biol Chem 281(40):30269–30278
Li T, Chen J, Cristea IM (2013) Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14(5):591–599
Li Y (2010) Commonly used tag combinations for tandem affinity purification. Biotechnol Appl Biochem 55(2):73–83
Gavin AC, Maeda K, Kuhner S (2011) Recent advances in charting protein-protein interaction: mass spectrometry-based approaches. Curr Opin Biotechnol 22(1):42–49
Rees JS et al (2011) In vivo analysis of proteomes and interactomes using parallel affinity capture (iPAC) coupled to mass spectrometry. Mol Cell Proteomics 10(6):M110 002386
Maine GN et al (2009) Bimolecular affinity purification (BAP): tandem affinity purification using two protein baits. Cold Spring Harb Protoc 2009(11):pdb prot5318
Ayyar BV et al (2012) Affinity chromatography as a tool for antibody purification. Methods 56(2):116–129
Trinkle-Mulcahy L et al (2008) Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J Cell Biol 183(2):223–239
Archambault V et al (2003) Genetic and biochemical evaluation of the importance of Cdc6 in regulating mitotic exit. Mol Biol Cell 14(11):4592–4604
Cristea IM, Chait BT (2011) Affinity purification of protein complexes. Cold Spring Harb Protoc 2011(5):pdb prot5611
Miteva YV, Budayeva HG, Cristea IM (2013) Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal Chem 85(2):749–768
Conlon FL et al (2012) Immunoisolation of protein complexes from Xenopus. Methods Mol Biol 917:369–390
Cadene M, Chait BT (2000) A robust, detergent-friendly method for mass spectrometric analysis of integral membrane proteins. Anal Chem 72(22):5655–5658
Norris JL, Porter NA, Caprioli RM (2003) Mass spectrometry of intracellular and membrane proteins using cleavable detergents. Anal Chem 75(23):6642–6647
Ye X et al (2009) Optimization of protein solubilization for the analysis of the CD14 human monocyte membrane proteome using LC-MS/MS. J Proteomics 73(1):112–122
Wisniewski JR et al (2009) Universal sample preparation method for proteome analysis. Nat Methods 6(5):359–362
Darie CC et al (2011) Identifying transient protein-protein interactions in EphB2 signaling by blue native PAGE and mass spectrometry. Proteomics 11(23):4514–4528
Dubois F et al (2009) Differential 14-3-3 affinity capture reveals new downstream targets of phosphatidylinositol 3-kinase signaling. Mol Cell Proteomics 8(11):2487–2499
Chait BT (2011) Mass spectrometry in the postgenomic era. Annu Rev Biochem 80:239–246
Tipton JD et al (2011) Analysis of intact protein isoforms by mass spectrometry. J Biol Chem 286(29):25451–25458
Aebersold R, Mann M (2003) Mass spectrometry-based proteomics. Nature 422(6928):198–207
Yates JR, Ruse CI, Nakorchevsky A (2009) Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng 11:49–79
Picotti P, Aebersold R (2012) Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods 9(6):555–566
Mellacheruvu D et al (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10(8):730–736
Alber F et al (2007) Determining the architectures of macromolecular assemblies. Nature 450(7170):683–694
Selimi F et al (2009) Proteomic studies of a single CNS synapse type: the parallel fiber/purkinje cell synapse. PLoS Biol 7(4):e83
Hendrick JP, Hartl FU (1993) Molecular chaperone functions of heat-shock proteins. Annu Rev Biochem 62:349–384
Bard-Chapeau EA et al (2013) EVI1 oncoprotein interacts with a large and complex network of proteins and integrates signals through protein phosphorylation. Proc Natl Acad Sci U S A 110(31):E2885–E2894
Gavin AC et al (2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440(7084):631–636
Krogan NJ et al (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440(7084):637–643
Collins SR et al (2007) Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol Cell Proteomics 6(3):439–450
Babu M et al (2012) Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature 489(7417):585–589
Jeronimo C et al (2007) Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell 27(2):262–274
Sardiu ME et al (2008) Probabilistic assembly of human protein interaction networks from label-free quantitative proteomics. Proc Natl Acad Sci U S A 105(5):1454–1459
Sowa ME et al (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138(2):389–403
Choi H et al (2011) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods 8(1):70–73
Paoletti AC et al (2006) Quantitative proteomic analysis of distinct mammalian mediator complexes using normalized spectral abundance factors. Proc Natl Acad Sci U S A 103(50): 18928–18933
Wang M et al (2012) PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteomics 11(8):492–500
Glatter T et al (2011) Modularity and hormone sensitivity of the Drosophila melanogaster insulin receptor/target of rapamycin interaction proteome. Mol Syst Biol 7:547
Rinner O et al (2007) An integrated mass spectrometric and computational framework for the analysis of protein interaction networks. Nat Biotechnol 25(3):345–352
Jager S et al (2012) Global landscape of HIV-human protein complexes. Nature 481(7381): 365–370
Oda Y et al (1999) Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci U S A 96(12):6591–6596
Ong SE et al (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1(5):376–386
Tackett AJ et al (2005) I-DIRT, a general method for distinguishing between specific and nonspecific protein interactions. J Proteome Res 4(5):1752–1756
Selbach M, Mann M (2006) Protein interaction screening by quantitative immunoprecipitation combined with knockdown (QUICK). Nat Methods 3(12):981–983
Ge F et al (2010) Identification of novel 14-3-3zeta interacting proteins by quantitative immunoprecipitation combined with knockdown (QUICK). J Proteome Res 9(11): 5848–5858
Meixner A et al (2011) A QUICK screen for Lrrk2 interaction partners—leucine-rich repeat kinase 2 is involved in actin cytoskeleton dynamics. Mol Cell Proteomics 10(1):M110 001172
Zheng P et al (2012) QUICK identification and SPR validation of signal transducers and activators of transcription 3 (Stat3) interacting proteins. J Proteomics 75(3):1055–1066
Heide H et al (2009) Application of quantitative immunoprecipitation combined with knockdown and cross-linking to Chlamydomonas reveals the presence of vesicle-inducing protein in plastids 1 in a common complex with chloroplast HSP90C. Proteomics 9(11):3079–3089
Schmollinger S et al (2012) A protocol for the identification of protein-protein interactions based on 15 N metabolic labeling, immunoprecipitation, quantitative mass spectrometry and affinity modulation. J Vis Exp 67(2):4083
Wang X, Huang L (2008) Identifying dynamic interactors of protein complexes by quantitative mass spectrometry. Mol Cell Proteomics 7(1):46–57
Fang L et al (2008) Characterization of the human COP9 signalosome complex using affinity purification and mass spectrometry. J Proteome Res 7(11):4914–4925
Zhang XX et al (2012) Nanodiscs and SILAC-based mass spectrometry to identify a membrane protein interactome. J Proteome Res 11(2):1454–1459
Gunaratne J et al (2011) Protein interactions of phosphatase and tensin homologue (PTEN) and its cancer-associated G20E mutant compared by using stable isotope labeling by amino acids in cell culture-based parallel affinity purification. J Biol Chem 286(20):18093–18103
Gygi SP et al (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 17(10):994–999
Ranish JA, Brand M, Aebersold R (2007) Using stable isotope tagging and mass spectrometry to characterize protein complexes and to detect changes in their composition. Methods Mol Biol 359:17–35
Ross PL et al (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3(12):1154–1169
Zieske LR (2006) A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J Exp Bot 57(7):1501–1508
Vogt A et al (2013) Isotope coded protein labeling coupled immunoprecipitation (ICPL-IP): a novel approach for quantitative protein complex analysis from native tissue. Mol Cell Proteomics 12(5):1395–1406
Chen ZA et al (2010) Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J 29(4):717–726
Sharon M et al (2006) Structural organization of the 19S proteasome lid: insights from MS of intact complexes. PLoS Biol 4(8):e267
Fu CY et al (2010) A docking model based on mass spectrometric and biochemical data describes phage packaging motor incorporation. Mol Cell Proteomics 9(8):1764–1773
Herzog F et al (2012) Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337(6100):1348–1352
Leitner A et al (2012) The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure 20(5):814–825
Lanman J et al (2003) Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J Mol Biol 325(4):759–772
Rozbesky D et al (2013) Structural model of lymphocyte receptor NKR-P1C revealed by mass spectrometry and molecular modeling. Anal Chem 85(3):1597–1604
Zelter A et al (2010) Isotope signatures allow identification of chemically cross-linked peptides by mass spectrometry: a novel method to determine inter-residue distances in protein structures through cross-linking. J Proteome Res 9(7):3583–3589
Paramelle D et al (2013) Chemical cross-linkers for protein structure studies by mass spectrometry. Proteomics 13(3–4):438–456
Chowdhury SM et al (2009) Identification of cross-linked peptides after click-based enrichment using sequential collision-induced dissociation and electron transfer dissociation tandem mass spectrometry. Anal Chem 81(13):5524–5532
Kao A et al (2011) Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol Cell Proteomics 10(1):M110 002212
Rinner O et al (2008) Identification of cross-linked peptides from large sequence databases. Nat Methods 5(4):315–318
Tosi A et al (2013) Structure and subunit topology of the INO80 chromatin remodeler and its nucleosome complex. Cell 154(6):1207–1219
Jennebach S et al (2012) Crosslinking-MS analysis reveals RNA polymerase I domain architecture and basis of rRNA cleavage. Nucleic Acids Res 40(12):5591–5601
Hernandez P et al (2003) Popitam: towards new heuristic strategies to improve protein identification from tandem mass spectrometry data. Proteomics 3(6):870–878
Singh P et al (2008) Characterization of protein cross-links via mass spectrometry and an open-modification search strategy. Anal Chem 80(22):8799–8806
Eng JK, McCormack AL, Yates JR (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5(11):976–989
McIlwain S et al (2010) Detecting cross-linked peptides by searching against a database of cross-linked peptide pairs. J Proteome Res 9(5):2488–2495
Walzthoeni T et al (2012) False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat Methods 9(9):901–903
Panchaud A et al (2010) xComb: a cross-linked peptide database approach to protein-protein interaction analysis. J Proteome Res 9(5):2508–2515
Maiolica A et al (2007) Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol Cell Proteomics 6(12):2200–2211
Chu F et al (2010) Finding chimeras: a bioinformatics strategy for identification of cross-linked peptides. Mol Cell Proteomics 9(1):25–31
Lee YJ et al (2007) Shotgun cross-linking analysis for studying quaternary and tertiary protein structures. J Proteome Res 6(10):3908–3917
Nadeau OW et al (2008) CrossSearch, a user-friendly search engine for detecting chemically cross-linked peptides in conjugated proteins. Mol Cell Proteomics 7(4):739–749
Yang B et al (2012) Identification of cross-linked peptides from complex samples. Nat Methods 9(9):904–906
Braun P (2012) Interactome mapping for analysis of complex phenotypes: insights from benchmarking binary interaction assays. Proteomics 12(10):1499–1518
Kaake RM, Wang X, Huang L (2010) Profiling of protein interaction networks of protein complexes using affinity purification and quantitative mass spectrometry. Mol Cell Proteomics 9(8):1650–1665
Tagwerker C et al (2006) A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivo cross-linking. Mol Cell Proteomics 5(4):737–748
Guerrero C et al (2006) An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol Cell Proteomics 5(2):366–378
Suchanek M, Radzikowska A, Thiele C (2005) Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells. Nat Methods 2(4):261–267
Glembotski CC et al (2012) Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J Biol Chem 287(31):25893–25904
Muller VS et al (2011) Membrane-SPINE: an improved method to identify protein-protein interaction partners of membrane proteins in vivo. Proteomics 11(10):2124–2128
Smith AL et al (2012) Association of Rho-associated protein kinase 1 with E-cadherin complexes is mediated by p120-catenin. Mol Biol Cell 23(1):99–110
Smith AL et al (2011) ReCLIP (reversible cross-link immuno-precipitation): an efficient method for interrogation of labile protein complexes. PLoS One 6(1):e16206
Smart SK et al (2009) Mapping the local protein interactome of the NuA3 histone acetyltransferase. Protein Sci 18(9):1987–1997
Byrum S et al (2012) Analysis of stable and transient protein-protein interactions. Methods Mol Biol 833:143–152
Tang X, Bruce JE (2010) A new cross-linking strategy: protein interaction reporter (PIR) technology for protein-protein interaction studies. Mol Biosyst 6(6):939–947
Chavez JD et al (2012) Cross-linking measurements of the Potato leafroll virus reveal protein interaction topologies required for virion stability, aphid transmission, and virus-plant interactions. J Proteome Res 11(5):2968–2981
Zheng C et al (2011) Cross-linking measurements of in vivo protein complex topologies. Mol Cell Proteomics 10(10):M110 006841
Zhang H et al (2009) Identification of protein-protein interactions and topologies in living cells with chemical cross-linking and mass spectrometry. Mol Cell Proteomics 8(3):409–420
Schmitt-Ulms G et al (2004) Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat Biotechnol 22(6):724–731
Bai Y et al (2008) The in vivo brain interactome of the amyloid precursor protein. Mol Cell Proteomics 7(1):15–34
Watts JC et al (2009) Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog 5(10):e1000608
Knobbe CB et al (2011) Choice of biological source material supersedes oxidative stress in its influence on DJ-1 in vivo interactions with Hsp90. J Proteome Res 10(10):4388–4404
Acknowledgments
We are grateful for funding from NIH grants DP1DA026192, R21AI102187, and R21 HD073044-01A1, an HFSPO award RGY0079/2009-C to IMC, and an NSF graduate fellowship to HGB.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Budayeva, H.G., Cristea, I.M. (2014). A Mass Spectrometry View of Stable and Transient Protein Interactions. In: Woods, A., Darie, C. (eds) Advancements of Mass Spectrometry in Biomedical Research. Advances in Experimental Medicine and Biology, vol 806. Springer, Cham. https://doi.org/10.1007/978-3-319-06068-2_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-06068-2_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-06067-5
Online ISBN: 978-3-319-06068-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)