
Abstract
Three main topics are described in this chapter, namely (i) physical and chemical structural of cellulosic materials, (ii) fire and flame retardancy finishing of cellulosic materials, and (iii) fire and flame retardancy finishing of cellulosic materials and its composite. The first subject describes in detail different cellulose sources and their chemical and physical structures. Furthermore, the types, structure, and chemical composition of different fibers (cotton, linen, jute, bamboo, hemp, and wood) and their blend have been described in detail. The second subject contains the uses of flame retardant fabrics, and describes the deference in the definition of retardant/resistant terms; in addition, the theory of combustion or burning process, and the mechanism of fire and flame retardant action are explained in detail. Also, different phosphorous flame retardant synergism, types of flame resistant finishes, and their classification based on durability and nature have been mentioned. By the end of this chapter, different Flammability Tests for Textile are described in detail. The last section of the chapter shows the finishing of cellulosic materials and their composites in detail.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
10.1 Physical and Chemical Structural of Cellulosic Materials
10.1.1 Sources for Cellulose
Cellulosic fibres are natural polymers of vegetable origin, like cotton, linen, jute, ramie, hessian and sisal. So, it would be useful to study the chemical and the physical structure of these natural polymers.
Cellulose is found in plants as micro-fibrils (2–20 nm diameter and 100–40,000 nm long). These form the structurally strong framework in the cell walls. Cellulose is mostly prepared from wood pulp. Cellulose is also produced in a highly hydrated form by some bacteria (for example, Acetobacter xylinum) [1].
10.1.2 Structural Unit
In the plant kingdom, cellulose is an important structure material [2]. It is the main construction material of the plant cell walls that are made out of it [3]. Native cellulose occurs in plant (such as cotton and ramie which contain a highly pure cellulose, as lignocelluloses) and in bast fibres (such as flax, hemp, jute and wood) where it occurs in combination with lignin, homocellulose [4].
Cellulose is a linear polymer of β-(1 → 4)-D-glucopyranose units often the so-called 4C1 chair conformation. This is illustrated in Fig. 10.1 where the ring oxygen is at the back, the 4-carbon is ‘up’ and the 1-carbon is ‘down’. Conversely, furanose rings can oscillate and have a more flexible structure than pyranose rings, which means that they are less likely to have a fixed interaction with a molecule of water as energy will be lost in this process. The fully equatorial conformation of β-linked glucopyranose residues stabilizes the chair structure, minimizing its flexibility (for example, relative to the slightly more flexible α-linked glucopyranose residues in amylose).

Chemical structure of cellulose
10.1.3 Molecular Structure
Cellulose is an insoluble molecule consisting of between 2,000–14,000 residues with some preparations being somewhat shorter. It forms crystals (cellulose Iα) where intra-molecular (O3-H → O5′ and O6 → H-O2′) and intra-strand (O6-H → O3′) hydrogen bonds holds the network flat allowing the more hydrophobic ribbon faces to stack. Weak C6-H → O2′ hydrogen bonds may also make some contribution to the crystal stability. Each residue is oriented 180° to the next with the chain synthesized two residues at a time. Although individual strand of cellulose are intrinsically no less hydrophilic, or no more hydrophobic, than some other soluble polysaccharides (such as amylose) this tendency to form crystals utilizing extensive hydrophobic interactions [5] in addition to intra- and intermolecular hydrogen bonding makes it completely insoluble in normal aqueous solutions (although it is soluble in more exotic solvents [6] such as aqueous N-methylmorpholine-N-oxide (NMNO, ~0.8 mol water/mol, then up to 30 % by weight cellulose at 100 °C [7]).
Moreover, these crystal regions are responsible for the limited solubility of cellulose and make it difficult for solvents and reagents to access areas within the cellulose fibres. It is thought that water molecules catalyse the formation of the natural cellulose crystals by helping to align the chains through hydrogen-bonded bridging, as shown in Fig. 10.2 [8].

Unit cell of the crystal lattice of cellulose in the cotton fibre
10.1.3.1 Molecular and Supramolecular Features of Cellulose
The chemical composition of cotton fibres has been reported in detail in several studies before [9, 10]. The cotton consists of an assembly of cellulose chains connected via inter-chain hydrogen bonding. Cellulose makes up 95 % of the cotton fibre after ginning. The secondary wall, which is almost entirely cellulose, consists of crystalline cellulose in multiple layers, called micro-fibrils.
Cellulose is a complex composite material, which structurally comprises three hierarchical levels: (i) the molecular level of the single molecule; (ii) the super-molecular level concerning the packing of the molecules in crystals called micro-fibrils; and (iii) the morphological level. Figure 10.3 illustrates the arrangement of micro-fibrils and interstitial voids in relation to the cell wall [7].

Plant cell walls are constructed from a combination of a variety of polysaccharides that can be generally grouped into cellulose, hemicelluloses, and pectic polysaccharides (Fig. 10.3), and whose relative proportions depend on the plant species, specific tissue, and growth stage. Cellulose, the most abundant structural polysaccharide in cell walls (comprising 15–50 % of the dry weight of plant biomass).
The macro-fibrils consist of numerous micro-fibrils of cellulose interspersed by micro-porosities containing non-cellulosic wall materials. Micro-fibrils consist of bundles of cellulose molecules, partly arranged into micelles. Micelles are crystalline because of regular spacing of glucose residues which are connected by β-1,4-glucosidic bonds [11].
10.1.3.2 Chemical Structure of Cellulose
Haworth has proposed that the structural formula of sugars be written in a roughly three-dimensional prescriptive manner, which when applied to cellulose yield the formula in Fig. 10.1. Furthermore, it is a polysaccharide made up of β-D (+) glucose residues. These molecules condensed and linked together linearly by means of 1-4, β-glucosidic bonds [13–15].
Each repeating anhydroglucose unit has three reactive hydroxyl groups, so they are polyhydroxyl alcohols [14, 16]. Two of these groups in the position 2 and 3 are secondary hydroxyl groups, whereas the last one in the position 6 is a primary hydroxyl group [17]. Cellulose molecules have the opportunity of forming many hydrogen bonds with its primary hydroxyl groups because it is more reactive than secondary hydroxyl group [3, 13]. In addition, the primary hydroxyl group is more acidity than the secondary [17].
Steric hindrance is an important consideration, particularly in the case of bulky reacting species; position 6 is least sterically hindered [3]. Whenever the distance between the various oxygen and hydrogen atoms in the cellulose molecule reaches 3 Å or less, they interact with each other to form intermolecular hydrogen bonding [14].
10.1.3.3 Physical Structure of Cellulose
10.1.3.3.1 Crystalline and Amorphous Regions
Cellulose has the capability of forming many hydrogen bonds along the length of the polymer chain with its three-hydroxyl groups. These bonds, combined with the other (principally the Van der Waals attraction), bind together, and it is ranging perfect geometrical packing of crystal lattice (crystalline region) to random condition (amorphous region) [13].
10.1.3.3.2 Cellulose Accessibility and Reactivity
Reactivity and accessibility have been used to describe the easiness with which cellulose can react. More properly, reactivity of cellulose is the ability of the chain molecules to react with other molecules, whereas accessibility of cellulose defines the ease by which the functional groups of the chain molecule can be by reached the reactant molecule [18, 19].
It has been proposed that the accessibility of cellulose depends mainly on the number and size of the pores in the cellulose structure; the size and type of solvent or reagent; the internal surface, as determined by the size of fibrils or fibril aggregates, that is accessible; and the structure of the cellulose molecules, which will determine which hydroxyl groups are accessible. Therefore, to increase cellulose accessibility, the pores must be opened, and both the fibril aggregates and the highly ordered regions must be altered [20].
10.1.4 Natural Fibres
Natural fibres are composed primarily of cellulose, hemicellulose, and lignin, with the balance being made up of pectins, water soluble compounds, waxes, and inorganic, non-flammable substances, which are generally referred to as ash. Examples of the chemical composition of some plant fibres are shown in Table 10.1 [21].
The decomposition of cellulose, between 260 and 350 °C, results in the formation of flammable volatiles and gases, non-combustible gases, tars, and some char [23–26]. A high content of cellulose tends to increase the flammability of the fibre. Hemicellulose decomposes between 200 and 260 °C but forms more non-combustible gases and less tar than cellulose.
Lignin contributes more to char formation than either cellulose or hemicellulose [27, 28]. Therefore, based only on chemical composition, coir, with a low cellulose (36–43 %) and high lignin (41–45 %) content, and deciduous wood fibre, with a low cellulose (38–49 %), high lignin (23–30 %), and hemicellulose (19–26 %) content, should have lower flammability than, for example, cotton, which has a very high cellulose (85–90 %) content. Lignin starts decomposing from about 160 °C and continues to decompose until about 400 °C. At the lower temperatures, relatively weak bonds break, whereas at higher temperatures, cleavage of bonds in the aromatic rings of the lignin takes place [29].
Manfredi et al. [30] showed the importance of lignin in the thermal decomposition of sisal, flax, and jute fibres, having lignin contents of 9.9, 2.0, and 11.8 %, respectively. They concluded that the lower lignin content in flax contributed to a higher decomposition temperature but resulted in a lower oxidation resistance, which would be provided by the aromatic structure of lignin [31].
Furthermore, there is another an important parameter is the fine structure of the fibre, it is also play a role in the flammability of the fibre [32, 33]. For cellulosic fibres in particular, higher levels of levoglucosan give higher levels of crystallinity during pyrolysis. A higher ignition temperature, however, also results from increased crystallinity as more energy is required to decompose the crystalline structure. For example, the activation energy of crystalline celluloses is about 200 kJ/mol whereas it is about 120 kJ/mol for amorphous cotton and ramie [33].
Increased orientation or degree of polymerization results in decreased pyrolysis. Basch and Lewin [32, 34] attributed the high thermal stability of ramie, when compared to cotton, to its very high orientation since the degree of polymerization and crystallinity of the two fibres are similar.
In addition, the higher the orientation, the lower the permeability of the fibre to oxygen [32]. A fibre with low crystallinity and a high degree of polymerization and orientation would, therefore, from a flammability aspect, be the best choice to use as reinforcement in a composite material.
10.1.4.1 Cotton Fibre
Cotton is a natural vegetable single elongated fibre, developed from an epidermal cell of the cotton seed, which grows in many countries of the world. The individual cotton fibres consist of a single long tubular cell. Its length is about 1,200–1,500 times than its width, where, its length is varies from 12 to 60 mm and a width of 15–24 μ depending upon its source [35].
The main producing countries of cotton fibre are the United States, Egypt and India. The highest quality cotton, which has very long and fine fibres, is growing in Egypt. It is generally recognized that, cotton is the most used textile fibre in textile industry. World textile fibre consumption in 1998 was approximately 45 million tons of this totals, cotton represented approximately 20 million tons [36]. Due to its versatile uses; most consumers prefer cotton personal care items to those containing synthetic fibre, where it has several attractive and useful properties, such as comfortable, soft hand, absorbent, good colour retention, machine washable and dry cleanable, good strength, well durability and easily sewing and handling (Fig. 10.4).

Amorphous and crystalline regions of cellulose [37]
10.1.4.1.1 Distinctive Features Morphology of Cotton
As it is noticeable from the Fig. 10.5 [27], cotton fibre consists of primary and secondary layers, while lumen is present in the centre. Primary layer holds up to 30 % cellulose and non-cellulosic materials. This cellulose is of lower molecular weight with the degree of polymerization (DP) between 2,000 and 6,000. Secondary wall is rich in cellulose of higher weight with DP of 14,000 [38].

Schematic structure of a mature cotton fibre, identifying its six parts [27]
More detailed the cotton fibre has the following composition:
-
(a)
Cellulose: 88–96 %, it is the main component of the cotton fibre and secondary wall possess highest percentage of the total cellulose.
-
(b)
Pectin’s: 0.9–1.2 %, these are made of ploygalacturonic acid, and its magnesium salts, methylester, xylose. They are present mainly in primary wall.
-
(c)
Proteins: 1.1–1.9 %, these are made of protoplasm rest in lumen and aspartic, glutamic acid and praline. The primary wall contains 0.2–0.3 % nitrogen.
-
(d)
Waxes: 0.3–1.00 %, they are composed of higher monovalent alcohol-tractional, palmitic, oleic acid, glycerine. Its melting point is 77 °C. They are found on surface of cotton and in primary wall.
-
(e)
Organic acids: 0.5–1.00 %, these are salts of citric and L-maleic acid.
-
(f)
Mineral salts: 0.7–1.6 %, these are hypochlorites, sulphates, phosphates, oxides of silicon, calcium, potassium, magnesium.
-
(g)
Sugar: 0.3 % made of glucose, galactose, fructose and pentose.
-
(h)
Toxine: 0.9 %, endotoxine, evolved from bacterial cells (0.017–100 g per bale of mass 218 kg).
-
(i)
Vitamins and pigment (flavones compound).
10.1.4.2 Linen (Flax)
A flax fibre is a biodegradable natural composite material which exhibits good specific mechanical properties. Consequently, this fibre is foreseen as a reinforcement material in polymeric based structural composites in replacement of the largely used E-glass fibres. 1.2 tons of short flax fibres, 80 % of which is used in the textile industries and 20 % in composite materials, can be produced.
The multilayer composite structure of the flax fibre is presented in Fig. 10.6 [39]. The fibres are located within the stems, between the bark and the xylem. Around twenty bundles can be seen on the section of a stem and each bundle contains between ten and forty fibres linked together by a pectic middle lamella. Each fibre is made of a thin external layer, called the primary cell wall, and a thick secondary cell wall, which is divided into three layers.

Structure of flax: from the stem to the fibre [39]
The cell walls are made of cellulose microfibrils laid in spirals around the fibre axis and embedded in a pectic matrix. In the secondary cell wall, the angle between the microfibrils and the longitudinal axis (called the “microfibril angle”) is about 10° [28, 40, 41]. In a flax bundle, the weight fraction of cellulose has been evaluated at 65–75 %, the one of non-cellulosic polymers (i.e. pectins, hemicelluloses and lignin) at 20–25 % and the one of water at 8–10 %. Considering the microstructural arrangement, the cellulose microfibrils act as the reinforcement of the pectic matrix and the interface between these two materials is mainly composed of hemicelluloses [39, 42].
10.1.4.3 Jute
Jute is a natural biodegradable fibre with advantages such as high tensile strength, excellent thermal conductivity, coolness, ventilation function [43, 44]. Jute fibre is a bast fibre obtained from the bark of jute plant containing three main categories of chemical compounds namely cellulose (58–63 %), hemicellulose (20–24 %) and lignin (12–15 %), and some other small quantities of constituents like fats, pectin, aqueous extract. Jute fibre is composed of small units of cellulose surrounded and cemented together by lignin and hemi-cellulose [45, 46]. The low cellulose content, coarseness, stiffness, low extensibility, low grip performance.
A bast fibre used for sacking, burlap, and twine as a backing material for tufted carpets. Jute is one of the most affordable natural fibres and is second only to cotton in amount produced and variety of uses of vegetable fibres. Jute fibres are composed primarily of the plant materials cellulose (major component of plant fibre) and lignin (major components of wood fibre).
10.1.4.3.1 Chemical Composition of Jute Fibre
• Cellulose | 65.2 % |
• Hemi-cellulose | 22.2 % |
• Lignin | 12.5 % |
• Water soluble matter | 1.50 % |
• Fat and wax | 0.60 % |
10.1.4.3.2 Defects in Jute
-
Rooty Jute: in these jute the lower parts of jute fires contain barks.
-
Specky jute: this defects occur because of insufficient washing which causes the outer barks to adhere in some places.
-
Croppy Jute: this is a defect where the top end of the fibre become rough and hard. It is usually caused by careless steeping.
-
Knotty jute: the jute fibres contain knots in places and it is caused by insect bite or punctures.
-
Dezed or Dead fibres: due to over retting in moist condition, the fibre becomes dull, lose strength and becomes inferior for spinning.
-
Runners: this is a defect where long and hard barky ribbon of fibres remains in jute fibre.
-
Hunka: defects caused by non-removal of dried up base and hard bark from the fibres.
-
Mossy jute: fibres from short plants that cannot be properly stripped and cleaned contain broken piece of jute sticks etc.
-
Flabby or Fluffy jute: due to careless stripping, fibre loses firmness and becomes flabby and hairy.
-
Heart damage: These defects occur when jute fibre contains excess moisture when baled. The centre of the bale becomes badly tendered and in some cases fibres are reduced to powder.
10.1.4.4 Bamboo
Bamboo is a naturally occurring composite material which grows abundantly in most of the tropical countries. It is considered a composite material because it consists of cellulose fibres imbedded in a lignin matrix. Cellulose fibres are aligned along the length of the bamboo providing maximum tensile flexural strength and rigidity in that direction [47].
It has been used widely for household products and extended to industrial applications due to advances in processing technology and increased market demand. The chemistry of bamboo is important in determining its utilization potential. Several studies have investigated the chemical composition of bamboo. But systematic and thorough research on a commercially important bamboo species is needed to determine utilization potential for the products such as medium density fibreboard (MDF). Most of previous studies provide either only general information of several bamboo species or focuses on only one aspect of one species.
The chemical composition of bamboo is similar to that of wood. The main constituents of bamboo culms are cellulose, hemi-cellulose and lignin, which amount to over 90 % of the total mass. The minor constituents of bamboo are resins, tannins, waxes and inorganic salts. Compared with wood, however, bamboo has higher alkaline extractives, ash and silica contents [48].
Bamboo contains other organic composition in addition to cellulose and lignin. It contains about 2–6 % starch, 2 % deoxidized saccharide, 2–4 % fat, and 0.8–6 % protein. The carbohydrate content of bamboo plays an important role in its durability and service life. Durability of bamboo against mold, fungal and borers attack is strongly associated with its chemical composition. Bamboo is known to be susceptible to fungal and insect attack [49].
10.1.4.5 Hemp
Hemp is a natural, cellulosic and bast fibre. Hemp fibre is collected from the hemp plant. The plant from which hemp fibre is collected that is called Cannabis sativa. This plant is generally grows in the bank of Capsicum sea. It is interesting that this type of plant is growth naturally or in some reason it is cultivated manually. The cultivation process is near flax cultivation. It is a natural fibre and it has a chemical composition which helps us to identify this fibre from the others.
Hemp is a cellulosic natural fibre and cellulose is a main component of hemp fibre. The chemical composition of hemp fibre is presented as follow [50].
• Cellulose | 77.77 % |
• Hemicellulose | 10 % |
• Lignin | 6.8 % |
• Pectin | 2.9 % |
• Fat and wax | 0.90 % |
• Water soluble | 1.73 % |
10.1.4.6 Wood
Wood fibre are natural composite structures in which cellulose fibrils are held together by lignin and hemicellulose. The major constituents of wood fibre are lignin, cellulose, hemicellulose, and extractives. Each of these components contributes to fibre properties, which ultimately impact product properties.
A model for wood structure of a typical softwood tracheid is described in Fig. 10.7. The middle lamella and primary cell wall of these fibres are often referred to as the compound middle lamella. The middle lamella contains a high proportion of amorphous material which holds neighbouring fibres together. The primary cell wall is approximately 0.03–1.0 µm thick, and also contains a high percentage of lignin. The secondary cell wall consists of three layers, labelled S1 through S3 from the outer to the inner layer. The S1 and S3 layers are thin, at 0.1–0.3 µm, while the middle layer (S2) is thick at 1–5 µm, and is said to be most responsible for the strength properties of individual fibres. The fibrils of secondary cell wall layers are wound helically around the fibre axis, while those of the primary wall are randomly oriented [51] .

Model for a pulpwood fibre. The middle lamella (ML), primary wall (P), outer (S1), middle (S2), and inner (S3) layers of the secondary wall, and the warty layer (W) are labeled accordingly
Table 10.2 presents major chemical compositions of some wood species. Each of these components contributes to fibre properties, which ultimately impact product properties.
10.1.5 Regenerated Fibres
The first man-made fibres which were developed and produced used polymers of natural origin, more precisely of cellulose which is a raw material available in large quantities in the vegetable world.
Cellulose is the natural polymer that makes up the living cells of all vegetation. It is the material at the centre of the carbon cycle, and the most abundant and renewable biopolymer on the planet.
Cotton linters, wood pulp, viscose rayon, Cupra-ammonium, Cellulose Acetate (secondary and triacetate) and polynosic are High Wet Modulus (HWM).
-
Cellulose is one of many polymers found in nature.
-
Wood, paper, and cotton all contain cellulose. Cellulose is an excellent fibre.
-
Cellulose is made of repeat units of the monomer glucose.
-
The three types of regenerated cellulosic fibres are rayon, acetate and triacetate which are derived from the cell walls of short cotton fibres called linters.
-
Paper for instance is almost pure cellulose.
10.1.5.1 Rayon (Viscose) Fibre
Originally, the word rayon was applied to any cellulose-based manufactured fibre, and therefore included the cellulose acetate fibres. However, the definition of rayon was clarified in 1951 and now includes textiles fibres and filaments composed of regenerated cellulose, excluding acetate. In Europe the fibres are now generally known as viscose, the term viscose rayon being used whenever confusion between the fibre and the cellulose xanthate solution (also called viscose—see below) is possible.
-
Rayon is a manufactured regenerated cellulosic fibre
-
It is the first manmade fibre
-
It has a serrated round shape with smooth surface
-
It loses 30–50 % of its strength when it is wet
-
Rayon is produced from naturally occurring polymers and therefore it is not a synthetic fibre, but a manufactured regenerated cellulosic fibre
-
The fibre is sold as artificial silk.
There are two principal varieties of rayon namely viscose and cupra ammonium rayon.
10.1.5.2 Cellulose Acetate
A manufactured fibre, in which, the fibre forming substance is cellulose acetate. Acetate is derived from cellulose by reacting purified cellulose from wood pulp with acetic acid and acetic anhydride in the presence of sulphuric acid.
The Acetate Fibre Characteristics
-
Luxurious feel and appearance
-
Wide range of colours and lustres
-
Excellent drapability and softness
-
Relatively fast drying
-
Shrink, moth and mildew resistant
-
Special dyes have been developed for acetate since it does not accept dyes ordinarily used for cotton and rayon.
Acetate fibres are the manufactured fibres in which the fibre-forming substance is cellulose acetate. The cellulose esters triacetate and acetate are formed through acetylation of cotton linters or wood pulp using acetic anhydride and an acid catalyst in acetic acid.
Acetate and triacetate fibres are very similar in appearance to the regular-tenacity viscose rayons. Acetates and triacetates are moderately stiff fibres and possess good resiliency on bending and deformation, particularly after heat treatment.
The abrasion resistance of acetate and triacetate is poor, and these fibres cannot be used in applications requiring high resistance to rubbing and abrasion; however, the resistance of these fibres to pilling is excellent. While acetate and triacetate are moderately absorbent, their absorbencies cannot compare with the pure cellulosic fibres. The hand of acetate fabrics is somewhat softer and more pliable than triacetate, which possesses a crisp firm hand. Fabrics of both fibres possess excellent draping characteristics. Fabrics of acetate and triacetate have a pleasing appearance and a high degree of lustre, but the lustre of these fabrics can be modified through addition of delusterants.
Both acetate and triacetate are susceptible to attack by a number of household chemicals. Acetate and triacetate are attacked by strong acids and bases and by oxidizing bleaches. Acetate has only fair sunlight resistance, whereas the sunlight resistance of triacetate is superior. Both fibres have good heat resistance below their melting points.
Acetate and triacetate cannot be dyed by dyes used for cellulosic fibres. These fibres can be satisfactorily dyed with disperse dyes at moderate to high temperatures to give even, bright shades. Acetate and triacetate dry quickly and may be tumbling dried or drip-dried.
10.2 Fire and Flame Retardancy Finishing of Cellulosic Materials
The flame retardant treatment is one of the oldest forms of textile finishing; hazards associated with ready combustibility of cellulosic materials were recognized as early as the 4th century Bc [53]. The concept of protecting textiles from burning dates back to 1821 in France when Gay Laussac treated hemp, jute and linen fabric with a mixture of ammonium phosphate, ammonium chloride and borax. The first successful, launder-resistant, flame retardant finish for fabric was based on the work of Perkin who precipitated stannic oxide within the fibre. This fabric was flame resistant but after glow was severe and persistent enough to completely consume the fabric [54].
10.2.1 Uses of Flame Retardant Fabrics
Flame retardant fabrics are needed for a variety of uses such as for [55]:
-
1.
Apparels and garments, sleepwear, nightwear, children’s wear, loose garments, sarees, shawls, kitchen wear, etc., where there are chances of accidental contact with flame.
-
2.
Uniforms for fire—fighting personnel.
-
3.
Dresses, boiler suits and protective clothing for work men in many industries like petroleum and petrochemicals, oils, paints and varnishes, mining, iron and steel, explosives, matches, organic chemicals and solvents, electricity generation and distribution, foundry, welding, petrol and diesel pumps, cooking gas storage and distribution etc.
-
4.
Home furnishing and decorations—curtains, drapes, upholstery, bedding, mattresses, wall coverings and trimmings etc.
-
5.
Carpets and rugs.
-
6.
Industrial fabrics-Brattice cloth for coal mines, carpet backing fabrics, barrier fabrics as overlay on foam and rubber cushions, underlay in motor vehicles, wall coverings, decorative fabrics etc.
-
7.
Hotels, restaurants, clubs, rest houses, dormitories, auditoriums, theatres, cinema halls, religious worship and congregation halls, marriage halls, hospitals, schools, colleges etc.
-
8.
Armed forces (Defence)-clothing for airmen and fire fighting crew, overalls, parachutes, awning, tarpaulins, canvas including skop (support kit overhead protection), claddings and shelters, etc. also for paramilitary, police etc.
-
9.
Exports-garments, curtains, wall coverings, bad covers, mattresses, quilts, airline furnishings, automobiles fabrics.
In addition to satisfying obligatory fire requirements and regulations, an efficient fire retardant has to have the following features: [56–58]
-
It must have thermal stability at the usual polymer processing temperature
-
The retardant should have compatibility with the polymer and no leaching should occur
-
The additive has to conserve its fire retardant properties when exposed to fire action
-
The retardant has to decrease the creation of toxic gases and smoke during combustion
-
The amount of required additive to achieve flame retardancy should be at a low level [usually not more than 10 % wfw] to reduce costs and its effect on the mechanical properties of the material
-
The retardant has to be easy to introduce into the polymer mass
-
The retardant has to be easily removed from the polymer during recycling
-
The additive must have no harmful environmental properties nor be harmful to health
-
The additive should be commercially available and cost efficient
-
The retardant must not provoke corrosion.
10.2.2 Flame Retardant Cellulosic Fabric
Cellulosic fabrics have low fire resistance [55]. They are composed of carbon and hydrogen (fuels) and oxygen (supporter or combustion) and burn very easily. The burning process of cellulosic materials is an oxidation process. This process may be accompanied by a flame or glow; most organic fibres undergo a glowing action after the flame has been extinguished. The glow may cause much damage as the flaming itself, since it can completely consume a textile. Flaming and glowing are distinctly different processes, taking place at different temperatures [54]. It is useful, however to define a number of terms before going into details of fire and flame retardant finishing of textile.
10.2.2.1 Fire-Proof Textile
This term is applied to textiles, which are essentially unaffected fire. This means that they do not support fire (flame or glow) and that there is little or no chemical or physical change when the textile is exposed to flames. Few textiles fall in this category. Carbon, asbestos, basalt and glass fibres are the one in common use today [54, 55, 59].
10.2.2.2 Flame Resistant Textile
Fabric that will not support a flame after the source of ignition has been removed is said to be flame resistance. A flame resistance fabric may continue to burn by the glow mechanism although a flame does not exist. Flame resistant fabrics can also char or melt [54, 60].
10.2.2.3 A Glow Resistant Textile
Fabric will not continue to burn by the glow mechanisms once the source of ignition has been removed. Glow-resistant textile may char or melt [55, 60].
10.2.2.4 Fire Resistant Textile
Fabric is not only flame resistant but also glow resistant. Thus, flaming and glowing cease once source of ignition has been removed. There will be a change in the physical and chemical characteristics, the textile may char or melt [55, 60].
10.2.3 Theory of Combustion
When solid materials are heated, physical and chemical changes occur at specific temperature depending on the chemical make-up of the solid. Thermoplastic polymers soften at the glass transition temperature (Tg), and subsequently melt (Tm) at some higher temperature (Tp), both thermoplastic and non-thermoplastic solids will chemically decompose (pyrolysis) into lower molecular weight fragments. Chemical change begins at (Tp) and continues through the temperature at which combustion occurs (Tc). These four temperatures are very important when considering the flame resistance of fibres. Another important factor in combustion is the limiting oxygen index (LOI). This is the amount of oxygen in the fuel mix needed to support combustion. The higher the number, the more difficult it is for combustion to occur. For non-thermoplastic fibres, (Tp) and or (Tc) are less than (Tg) and/or (Tm), however for thermoplastic fibres (Tp) and/or (Tc) is greater than (Tg) and or (Tm). Natural fibres are not thermoplastic, therefore when they are subjected to a heat source, pyrolysis and combustion temperatures are encountered before softening or melting temperatures are reached and eventually ignite. On other hand, low melting thermoplastic fibres will melt and drip away from the flame before pyrolysis and combustion temperature is reached. However if the melt doesn’t shrink away from the flame front, pyrolysis and combustion temperatures are eventually reached and ignition will occur [61, 62].
Natural fibres can be made flame retardant and like some synthetic fibres, nomex, Kevlar and PBI, offer protection to a wearer because they do not shrink away from a flame. Thermoplastic fibres may appear to offer protection because they pass the ignition test by shrinking away from the flame, however, in reality this exposes to wearer to direct heat and burns caused by contact of the molten mass with the body [62, 63].
10.2.3.1 Combustion or Burning Process in Textile
Combustion is defined as fast, self-accelerating exothermal redox process that is able to spread in the environment and accompanied by luminosity and the formation of flame [61, 64].
Combustion is a chain reaction that may be initiated and propagated by free radicals like the hydroxyl free radical. Hydroxyl radicals may be produced by the reaction of oxygen with macro alkyl radicals. Halogen radicals produced by the reaction of hydroxyl radicals with halides such as HX, may serve as terminators for the chain reaction [61] (Fig. 10.8).

Combustion exothermal redox process
Combustion is usually a gas phase phenomenon. Volatile combustible species oxidize exothermically in the gas phase. Afterglow or glowing combustion is a form of non-gas phase combustion. Here the substrate is oxidized in the condensed phase to form both solid and gaseous products. This usually takes place at temperatures well below the ignition temperature of the material. For instance, the carbon residue in a carbon rich material is oxidized in the solid phase.
Emman’s fire triangle is generally used to illustrate how combustion works and is shown in Fig. 10.9 [65]. For a sustained fire, three elements are needed [66]:

Emman’s fire triangle
-
Fuel—Volatile combustibles from carbon rich substance thermally degrading
-
Heat—Supplied by the exothermic oxidative destruction of fuel
-
Oxidizing agent—Oxygen provided by air
In the case of polymers, an external heat source is needed to ignite the substrate. The heat, to which the material is exposed, causes the high molecular weight polymer to thermally decompose [67]. On thermal decomposition, the polymer releases smaller volatile compounds that act as fuel to the fire. These combustible species mix with the oxygen in the air to form an ignitable mixture. Exothermic oxidation of the volatiles occurs and the material burns. Light and more heat are generated. The process becomes self-sustaining and functions with a feedback loop as can be seen in the fire triangle (Fig. 10.9).
The product of the fire differs for each combustible compound. In the case of polymers, the gases produced by combustion tend to be mainly carbon dioxide (CO2), carbon monoxide (CO) and water vapour (H2O). The solid residue is mostly carbon (C) and ash (oxidised metals) [65, 66].
A flame retardant system is a compound or compositions added to materials, which increases a given material’s resistance to combustion. The flame retardant system can either be added to the polymer during the polymers manufacturing step, during master batching of the polymer additives or during the production of the plastic artefacts.
An effective name retardant needs to hinder the supply of one or more of the elements required for sustained combustion [66]. The objective of flame retardants is to lower a polymers inherent fire risk by lowering the rates of combustion and flame spreading under fire conditions [68]. The use of flame retardants may prevent a small fire from becoming a major catastrophe [69]. In order for a flame retardant to be effective, it must interact and interfere with the degradation of the host polymer at the polymer’s degradation temperature [70]. The degradation temperatures for the most widely used polymers are between 200 and 400 °C [71]. Increased fire resistance can be achieved through several mechanisms, as can be seen in Fig. 10.10 [72]. The interference with the combustion process may take place in the gas or vapour (flame zone) or condensed or solid phases (polymer melt).

A simplified model for combustion and flame retardancy
A flame retardant is not designed to prevent the material from igniting, but to keep the flame spread rate to a minimum and prevent sustained burning. Flame retardants tend to retard the spread of flames by increasing the given polymer’s resistance to ignition. Ignition is unavoidable, because most substances will flame up if subjected to high enough levels of fire stress—thermal radiation [66].
A useful and complete description of the mechanisms of general retardant systems is provided in the review articles of Green [69, 73]. Different flame retardant systems can be identified, all of which function by different mechanisms. Flame retardants interfere with the thermal decomposition pathway of the polymeric material. Different compositions interact differently with different polymers and a flame retardants use is thus very specific to the particular substrate for which it was designed.
Some flame retardants which form acids during combustion—like the halogens operate through gas-phase free-radical inhibition. Other groups of retardants produce many non-combustible gases and dilute the amount of fuel or oxygen supplied to the fire. The formation of solid residues on the surface of the burning material is another way of reducing flame spread. Some reduce the rate of heat release during combustion by affecting the heat transfer pathway to the polymer substrate. Another group of flame retardants form a foaming char on the surface of the combusting material. These additives are called intumescent systems. A system can also operate through combinations of the above-mentioned mechanisms [69].
The following terms and definitions are used to describe the different mechanisms by which flame-retardants operate [74]:
-
Inert gas dilution: Large quantities of inert and non-combustible gasses are produced on thermal decomposition of the additive. The concentration of oxygen and combustible species are reduced and the fire dies.
-
Thermal quenching: The surface temperature of the polymer is reduced or kept low by the endothermic degradation of the additive. Due to the lower substrate temperature, less combustible products are produced and the thennal degradation is retarded.
-
Physical dilution: Large quantities of inorganic fillers (such as glass fibre) are added to the polymer matrix. The amount of flamable material (polymer) is thus reduced and the substrates fire resistance increased.
-
Chemical interaction: Some flame retardants thermally dissociates into radical species that then interferes with the gasphase combustion of the combustibles.
-
Protective char: On thermal decomposition, the additive forms an insulating char barrier on the surface of the polymer. This char reduces heat transfer for to the polymer, diffusion of oxygen to the area of decomposition and diffusion of combustibles to the flame zone, retarding the combustion.
An ideal flame retardant will have the following properties: [65, 66, 69, 73, 75]
-
It reduces flammability to the required standard
-
It is thermally stable at the processing temperatures
-
Have long term compatibility with the polymer matrix (i,e, does not “bloom”)
-
Maintains or improves the mechanical properties of the polymer
-
Represents no health hazard; and
-
Is cost effective.
The flame retardant must not decompose at temperatures below the processing temperatures of the polymer. Ideally, the flame retardants decomposition temperature should be several degrees higher than the polymer processing temperature.
Large-molecule flame-retardants in the form of polymeric and oligomeric compounds are gaining ground in the industry because they are more resistant to blooming and leach out and promise greater compatibility with host plastics than traditional flame retardant additives [76].
Figure 10.11 shows the mechanism of burning of polymers. The endothermic Step 1 produces low molecular weight pyrolysis products that combine with oxygen in exothermic Step 2 gas phase reaction to produce oxidation products. The heat generated in this step goes back (Step 3) to polymer to continue its decomposition.

Polymers burning mechanism
Any compound or method inhibiting one or more steps in this burning process is potential flame retardant [62, 77].
Figure 10.11 shows the consecutive reactions during burning. When a solid fibre is ignited two successive chemical processes decomposition and combustion take place and these processes are linked together via a thermal fed bake. The pyrolysis Step 1 required heat (Q1), while combustion Step 2 generated heat (Q2). The contact area of ignition (A) is also important. A high value of (Q1) and low values of (Q2) and (A) would lead to flame resistance [62, 77].
10.2.3.2 Mechanism of Fire and Flame Retardant
10.2.3.2.1 Burning of Cellulose
In the development of better flame retardants it is helpful to know what chemical reaction occur when cellulose burns and how the reaction are affected when fabrics are treated with a flame retardant. The burning characteristics of cellulose depend to a considerable extent on the chemical and thermal properties of the anhydroglucose units, on the availability of oxygen, and on the nature of noncellulosic materials that may also be present. Fibre properties such as the ignition temperature, the rate and heat of combustion products, and moisture content also affect the burning characteristic of cellulose [59, 62, 77].
Cotton is a combustible material. Presence of oxygen and a high temperature (360–420 °C) can initiated combustion of cotton which can be either burning (flaming combustion) or smoldering (smolder combustion). The intrinsic ignition time, on index of auto ignition, for cotton at 500 °C is 15.6 s, which comes down at 600 °C to 6.7 s and further at 700 °C to only 3.1 s. Cotton has an ignition temperature of 400 °C and burns readily to attain a maximum temperature of 860 °C char formation and after glow. The sequence of steps in the combustion of cotton is ignition; propagation and afterglow temperature within burning cellulose are reported at 413 °C during flaming and 600 °C during after glow. Ignition is controlled by various factors such as heat transfer from the source of ignition to cotton, thermal decomposition of cotton and reactions of decomposition products with the environmental oxygen. Thermal decomposition of cotton release a large number of combustible gases which can sustain ignition at and above certain concentration levels with oxygen of the air. A burning fabric generating more heat requires less ignition energy to continue its burning [77]. So, the effect of heat on cellulose is varying important, it makes property changes. The rapid reaction of degree of polymerization has already been noted, and it has been suggested [78] that the chain breaks occur at strain points at the crystalline-amorphous boundaries. The number of hydroxyl groups decrease while carboxyl group increase [79]. Even very short heating times decrease the accessibility of cellulose to H2O and dye molecules and strongly increase its rate of acid hydrolysis. An increase in the crystalline lattice spacing has been shown while the chain orientation is reported to be unaffected [80].
It has found that there are two decomposition reaction occurred by controlled temperature. In the first reaction, which occurs between 200 and 280 °C water is lost and anhydrocellulose is formed [60].
This anhydrocellulose decomposes further at elevated temperature to various volatile products such as alcohols, aldehydes and alkanes, flammable gases, carbon monoxide, ethylene, methane and non-flammable gases, carbon dioxide, water vapour and char.
The second reaction, involving thermal scission of glucosidic linkages and levoglucosan formation predominates at temperature above 280 °C.
-
Gases: combustible (methane, ethane, carbon monoxide) and non—combustible (formaldehyde, carbon dioxide etc.)
-
Liquids: Water, Alcohols, Aldehydes, Ketones, Organic acids. [80]
Levoglucosan is the major product formed by the pyrolysis of cellulose it is the cyclic created when the β-1,4-glucosidic linkage is split, and a molecule of water is lost between the C(1) and C(6) hydroxyls of the anhydroglucose.
The burning of cellulose (Fig. 10.12) takes place in two ways: flaming and glowing both processes originate from either an open flame or a hot surface (above 300 °C). In the flaming process the thermal decomposition of cellulose results in gaseous, liquid, tarry and solid products [81–88].

Cellulose pyrolysis products
The competition between volatile and char formation confirms a three-stage process which depends on both temperature and the exact nature of the flame retardant present [82]. Figure 10.13 shows the overall scheme, which builds on previously published mechanisms [22, 89].

Pyrolysis of cellulose and char formation
-
Stage I:
shows the well-established competing mechanisms of char formation and volatilisation within the temperature range 300–400 °C
-
Stage II:
within the range 400–600 °C, shows a competition between char oxidation and conversion of aliphatic char to an aromatic form. Volatiles from Stage I are also oxidised within this range to yield similar products to those formed from char oxidation and aromatisation.
-
Stage III:
During the higher temperature regime of 600–800 °C, some char decomposition to acetylene occurs, while above 800 °C, this stage follows during which complete combustion of all remaining carbonaceous species to CO and CO2 takes place.
As the flammable gases burn, the liquids are volatilize, and some of these volatile fractions burn, where as other portions give a carbonized residue that does not readily burn. This process continues until only carbonaceous, begins. The residual carbonaceous matter glows, oxidizes, and continuous to glow until essentially all organic matter is consumed and only a fluffy ash is left. At flaming cellulose temperature degradation of cellulose precedes first the formation of levoglucosan by scission of the (1–4) glucosidic linkages of cellulose and subsequently by intermolecular rearrangement of the fragments [62, 81–85]. The levoglocosan is thought to undergo dehydration and polymerization to form char.
The rate controlling reaction in the thermal decomposition of cellulose involves the formation of levoglocosan, chemically 1,6-anhydroglucopyranose. Levoglucosan remains a major pyrolysis product in air pyrolysis.
Levoglucosan yields are strongly influenced by even traces of impurities present in or added to the cellulose. It has been shown that acidic impurities can either increase or decrease the percentage of levoglucosan in the tar, depending on their concentration; while basic impurities depress levoglucosan formation at all levels [84, 85].
Flammability of the cellulose seems to vary, within narrow limits, with levoglucosan formation. These factors explain the observed increase in flammability of cellulosic textile.
When treated with low concentration of flame-retardants. The continued flammability of cellulose in the presence of sufficient flame retardant to depress or eliminate levoglucosan is attributed to formation of other volatile compounds [80].
Crosslinking with formaldehyde influences both levoglucosan yields and flammability [88]. The levoglucosan content decreases with increase in amount of formaldehyde reacted with cotton. This decrease is explained by the lower tendency of the crystalline faces with substituted C2 and C6 to undergo the unzipping reaction [80–85]. It is possible that the crosslinks bring about an additional pyrolysis path, different from that leading to levoglucosan and necessitating higher activation energy. In the case of rayon, the levoglucosan yield increases with the extent of crosslinking [85].
10.2.3.2.2 Burning of Flame-Resistant Cellulose
There are a number of theories proposed and postulated to achieve flame retardant properties and explain the function of them in cellulosic fabrics with the aid of additives. Such an additive, to be a successful player in this field, might take one or more of following roles and categories [62, 90]. It must inhibit one or more steps of the burning process—ignition, propagation and afterglow. It must either dissipate heat internally or conduct the heat away from the flame front like a “heat sink”—thermal theory.
It must form a glassy film of molten material at the fabric surface, which will acts as a shield between the fabric and oxygen thereby terminating flame propagation—Coating theory. It must dilute the flammable gases or blanket the substrate with a so—called atmosphere—gas theory. It, being a Lewis acid or base or their precursor, catalyses dehydration mechanism at the flaming temperature—Chemical theory.
10.2.3.2.2.1 Coating Theory
According to the coating theory, an effective compound forms a glassy skin and stable foam on the fibre surface. Such a flame retardant coating protects the fabric from the air by serving as a barrier to the flame and by entrapping the volatile tars evolved during combustion [59, 90].
10.2.3.2.2.2 Gas Theory
The gas theory states that the flame retardant decomposes at burning temperatures to give gases which do not burn but which will dilute the flammable gases produced by the decomposition of the cellulose to a concentration below which they can ignite and burn [59].
10.2.3.2.2.3 Thermal Theory
Two thermal mechanisms have been proposed for the retardation of the burning of cellulose. The first one is proposed that the caloric input from a source is dissipated by an endothermic change of retardant, such as fusion and sublimation of the flame proof. Such endotherms prevent propagation of the flame. The second mechanism is proposed that the heat supplied from the source is conducted away from the fibres so rapidly that the fabric never reaches combustion temperature [59].
10.2.3.2.2.4 Hydrogen Bonding Theory
It has been suggested that a majority of flame retardant are strong hydrogen bonding agents. Therefore, when water (the bridging medium between cellulose hydroxyl groups), is lose at higher temperatures, the bridges may be maintained by the strong hydrogen bonding activity of the flame retardant, thus stabilizing cellulosic fragment by reducing their volatility and hence, their combustibility. Although this mechanism may be a factor, it seems weak. It is improbable that such bonds could continue to exist at 400–500 °C [59, 85].
10.2.3.2.2.5 Catalytic Dehydration of Cellulose to Carbon and Water
Ideally flame retardant would direct the decomposition of cellulose into carbon and water. Theoretically this can be accomplished through dehydration reactions, and several dehydrating compounds are known to be good flame retardants for cellulosic fibres. Basically, the path of decomposition is altered, so that the amount of flammable gases and tars so reduced that combustion will not continue once the source of flame is removed [59]. The most recent and widely accepted modification of the theory states that flame retardancy is brought about by the catalytic dehydration of the cellulose through the reaction of Lewis acid with the cellulose via a carbonium ion mechanism. The flame retardant (Lewis acid) may be either present or produced from its precursor at a temperature close to that of burning cellulose [84].
In considering the dehydration theory, two types of burning will be considered flaming and glowing. The tar from flame resistant cellulosic fibres contains little or no levoglucosan, whereas that from untreated cellulose contains levoglucosan as a major component. The higher the ratio of carbon monoxide to carbon dioxide or the ratio of chars to tar, the better the flame resistance [81, 83].
Parks et al. [91] suggested that prevention of formation of levoglucosan, the major component of tar from untreated cellulose, or altering the path of decomposition to yield more char would result in flame resistance.
The prevention of formation of levoglucosan alone appears to impart flame resistance. It seems that formation of large percentage of carbon is much more important for fire resistance than lack of levoglucosan formation. Lack of levoglucosan in the decomposition product is important in that it can influence the amount of carbon resulting from dehydration reaction [81, 85].
10.2.3.2.2.6 Chemical Theory
Effective chemical retardants may be considered to act in either the solid or vapour phase, or a combination of the two. Retardants, which act in the gas phase, exert their effect by functioning as free radical [90].
Inhibitors, which slow the oxidation processes, and decrease the heat retardant to the polymer surface. Those retardants, which act in the condensed phase, may operate by inhibiting the decomposition of cellulose so that the chain does not break down to form flammable gases, or much more commonly, they act to alter rather than inhibit the degradation reaction. The mode of decomposition is changed to favour the formation of non-volatile residue rather than flammable gases [61].
10.2.3.2.3 Mechanism of Retardant Action on Cellulosic Materials
Cellulose flame retardant may operate through one of two basic mechanisms, solid phase (condensed phase) or vapor phase flame retardant.
Halogens are known to be efficient flame retardant agents for cellulose and other polymers [92] and act mainly in vapor phase via free radical inhibition.
However, this radically couples with free radicals produced in the combustion process and terminates the reaction.
A numerous investigator has characterized the effect of which solid (condensed) phase active flame-retardant exerts on cellulose [75, 93, 94]. Figure 10.14 illustrate the depolymerisation of cellulose.

Proposed depolymerization of cellulose
The more effective flame-retardant finishes for cotton have based on organophosphorus compounds. These phosphorus compounds are relatively expensive, compared to many other types of organic compounds and contribute greatly to the high cost of flame retardant. Substitution of low cost nitrogen compounds for the phosphorus component in the flame retardants is one method of reducing cost. The use of nitrogen compounds was recognized many years ago when melamine and urea resins where incorporated into flame retardants systems based on chlorinated paraffin [60, 95].
Pyrolysis of brominated wood was examined in detail by chemical, spectroscopic, and thermal analysis and determined the effect of bromine upon the cellulose and the distribution of the bromine in the products of pyrolysis [55].
A numerous investigators have characterized the effect of which solid (condensed) phase active flame-retardants exerts on cellulose [93, 94]. Their results are consistent in showing that reduction in tar formation, coupled with increased char yield, are the most important effect of the retardant action.
The reaction of thermal degradation of the cellulose (pyrolysis) is primarily unzipping depolymerization, which produces levoglucosan and secondary products, almost all of which are volatile and flammable. The reactions of the other set appear to have much lower energies of activation and proceeds at essentially the same moderately slow rate at all pyrolysis temperatures. These reactions lead mainly to nonvolatile char and nonflammable gases, such as water and carbon dioxide. Obviously then, condensed phase flame retardant action on cellulose could results from the catalysis of the latter reaction set and/or inhibition of the former.
Flame retardant based on phosphorus, sulfur, boron, and other acidic forming materials act via dehydration to produce water and char at expense of flammable tars. Most commercial treatments for cellulose are based on phosphorus, and consequently the effect of the structure of phosphorus based materials on the mechanism and efficiency of flame retardancy has been widely studied. Acidic phosphates act via phosphorlation, presumably at the C-6 hydroxyl of the anhydroglucose unit [64]. Consequently, increases of esterification are likely to be more efficient in promoting flame retardancy.
10.2.4 Phosphorous Flame Retardant Synergism
10.2.4.1 Halogen—Phosphorus Synergism
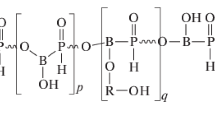
Halogen phosphorous synergism is often confused by analogy with the strong and well established halogen antimony synergism. Unlike antimony halogen synergism, phosphorous halogen synergism is not general. The postulated formation of phosphorous oxyhalides completely lacks experimental support. However, good additive results are often obtained with combinations of halogen- and phosphorous-based flame retardants [96]. An instance of bromine and phosphorous synergism is found in the structure of the following brominated phosphate ester:

10.2.4.2 Antimony—Phosphorus Synergism
There are a number of published formulations showing the attempted use of antimony oxide in combination with phosphorous and halogen flame-retardants. Results sometimes appear favourable, but quantitative studies show convincing evidence of an antagonism between antimony and phosphorous. In the most severe case, the one element cancels out the effect from the other, and in other cases, the effect is less than an additive. A detailed study of triaryl phosphate and antimony oxide in polyvinyl chloride (PVC) showed that this antagonism only occurred in a part of the composition range. The antagonistic effect probably is the result of the formation of antimony phosphates that are very stable and practically inert fillers [96].
10.2.4.3 Nitrogen—Phosphorus Synergism
As with many other multi-component products, flame retardant systems are regularly produced by simply mixing two or more ingredients together. The efficiency of flame retardants can be greatly influenced by synergy between the formulation components [96]. The properties of the final product or system depend on the proportion of the individual components used. Synergy is the phenomenon when a compound formulation (a combination of two or more components) leads to a desirable property that is better than the property obtained with the ratio of the individual compounds. However, such component combinations can unfortunately also lead to undesirable effects. A reduction in the desirable property due to the combination is called antagonism. Figure 10.15 shows possible responses for a two-component system [97].

The property (P) evaluation of a two component system blend
Compounds containing nitrogen and phosphorus have been used for a long time as fire retardant for example (phosphoric acid/urea, tetrakis hydroxymethyl phosphonium chloride (THPC)/ammonia, urea or trimethylol melamine). It was established that incorporation of N—containing compounds in the finishing formulation with p—containing compounds increase the flame retardancy properties of the finished fabrics [90, 98].
In most of the above cases, the role of nitrogen was believed to influence the attachment of phosphorus to the cellulosic fabric [99].
It has reported that cellulose derivatives containing phosphorus are very much prone to dehydration reaction resulting in formation of char. A synergism enhancement in the efficiency of phosphorus—based flame retardants by compounds containing nitrogen atoms has been well documented. However, it is noteworthy that not all N-containing compounds are effective but only those containing nucleophilic nitrogen atoms, e.g. amines and amides are useful adjuncts [100].
Incorporation of a nitrogenous component to phosphorous—containing compounds has the advantage of minimizing acid degradation of cellulose by releasing phosphoric acid and also enhancing flame retardation [101].
Using the nitrogen compounds have many advantages, low toxicity, their solid state and in case of fire, the absence of dioxin and halogen acids as well as their low evolution of smoke. Their efficiency lies between that of halogen compounds and that of aluminium trihydrate and magnesium hydroxide. The metallic hydroxides split off water and are environmentally friendly, but their low activity requires high concentration which changes the mechanical properties of the matrix that they are applied to. Flame-retardants based on nitrogen are environmentally friendly because they do not add any new element to those already present in the polymers. The final and best argument is their moderate price [99]. Using a comparable level of flame retardant could be achieved using a smaller add—on of phosphorus/nitrogen mixture than with phosphorous or nitrogen alone. Synergistic effect occurs between phosphorous and nitrogen with respect to their ability of flame retardant cotton fabric [102].
It was reported that, using triazine formaldehyde and trimethylol melamine diammonium phosphate—treated cotton, the synergistic effect was found to be dependent on the type of nitrogen present. Reeves et al. [103] suggested that, amide and amine nitrogen generally increase flame retardancy whereas nitrile nitrogen can cause reduction [104].
Weil has reviewed mechanisms of nitrogen—phosphorous interaction on cellulose. Weil pointed out in his later study that bis(methoxy methyl) iron, which lacks secondary nitrogen, ceases to be non-synergistic and, indeed, non-additive at the higher phosphorus (2%P) levels used. In addition, the presence of triazine resin with its basic tertiary nitrogen shows no increase in flame retardant activity at 1%P and antagonism at 2%P. These resins were applied with the commercial phosphonamide flame retardant, pyrovatex CP [105].
Davies and Horrocks [105] using the ratio urea formaldehyde 1:1.6 which is typical cellulosic textile formulation, illustrate that the cured resin will have a low secondary nitrogen—content and so will not show any synergistic tendency. In fact based on Weil’s hypothesis, the observed antagonism of the urea—formaldehyde nitrogen should relate to its high tertiary nitrogen content. Nitrogen compounds such as amides and amines appear to catalyse the cellulose phosphate forming steps and are found to strongly synergize the action of phosphorus in cellulose systems.
Synergistic interaction between phosphorus and nitrogen was studied with various finishes, using the Limiting Oxygen Index (LOI) for evaluation. Clearly indicated the increasing of (LOI) value with the P% as the N% increased, or the increasing of (LOI) value with the N% as P% increased [99].
10.2.5 Types of Flame Resistant Finishes
Flame-retardants can be distanced into reactive and additive flame-retardants. Reactive flame-retardants are reactive components chemically built into a polymer molecule. Additive flame-retardants are incorporated into the polymer either prior to, during or (most frequently) following polymerization.
Classification of flame retardants
Flame-retardants based on different conditions were classified into four classes [59, 60, 106] these classes are (a) Nondurable flame resistant finishes, (b) Semidurable flame resistant finishes, (c) Durable flame resistant finishes and (d) Weather resistant flame retardants durability, nature of the compounds and methods of incorporation to the host polymer.
Flame-retardant chemicals based on nature of the compounds are divided into five families [65, 74, 107–110]. These are inorganic, halogenated, organo-phosphorus, halogenated phosphates and nitrogen-based flame-retardants.
10.2.5.1 Flame Retardants Based on Durability
10.2.5.1.1 Nondurable Flame Retardants
Nondurable flame retardants are suitable for use on cellulosic materials. The chemicals easily removed out of textiles when exposed to weather, leached, or laundered. It is should be treated again after each laundering. Even it is not laundered, it should be retreated again each 6 months because many of the nondurable type retardants crystallize on the surface of a fabric after having diffused from the inside of the fibre [54, 59, 60, 106].
The most important examples of this group are:
-
(a)
Borax (Sodium Borate Na2B4O7-10H2O) and Boric Acid
Fabrics treated with this retardant are flame resistant but have afterglow which can persist from about 30 s to several minutes [54, 60].
-
(b)
Borax (Sodium Borate Na2B4O7-10H2O) and Diammonium Phosphate (NH4)2 HPO4.
This flame retardant formulation applied to cotton fabrics by dissolving equal parts of borax and diammonium hydrogen phosphate in water [54, 60].
10.2.5.1.2 Semidurable Flame Retardants
Semidurable flame retardants can be defined as those that withstand one or more laundry cycles, with the upper limit being about 15 mild laundry cycles. The main advantage they have over the durable type, is lower cost [54, 111–115].
This class is exemplified by phosphorous compounds that are slowly dissolved and become resistant to leaching as well as to few mild launderings [112].
The most obvious means of attaining semidurable flame resistance is the application of insoluble salts. It must be taken into consideration that the flame retardant effect of the simple inorganic salts are based on their capability of decomposing in heat and releasing strong acid or alkali which is responsible for the reduction of flame propagation [113].
The most important examples of this group are:
-
(a)
Cellulose Phosphates
A solution of urea and diammonium phosphate was padded onto fabric; the fabric was then dried for about 7 min at 175 °C. By this technique, 10–15 % esterification is achieved easily, but cellulosic fibres are severely tendered during the process. The better tensile properties are obtained through very careful curing conditions and using formulation that would fix about 3 % phosphorus in the fabric [54, 111, 114, 115].
-
(b)
Cyanamide-Phosphoric Acid Process
A mixture of dimethylol dicyanamide and ammonium phosphate or phosphoric acid will polymerize to form an insoluble resin in cellulosic fibres, so, this modification cause less tendering of fabric. Other modifications involved replacement of urea with a mixture or diguandine thiosulphate with methylol melamine.
In a somewhat related process a pre-condensate of urea and ammonium phosphate could be insolubilized with CH2O to produce semidurable flame resistance. Semidurable flam resistant finishes closely resembling those made from urea and H3PO4 could be obtained by reacting ammonia with phosphorus oxychloride [54, 62, 111].
10.2.5.1.3 Weather-Resistant Flame Retardants
Weather resistant and flame resistant finishes are used extensively for many materials. Water—soluble flame resistant finishes are not weather resistant because rain and humid atmospheres readily remove them. The weather resistant and flame resistant finishes may be divided into two broad groups; (a) inorganic oxides, generally have good resistance to degradation by sunlight, however, they are poor flame resistant finishes and must be used in combination with some other material, such as chlorinated paraffin and (b) organo phosphorous compounds, generally degraded by ultraviolet light and must be protected by some additional shielding materials [62, 111, 116, 117].
10.2.5.1.4 Durable Flame Retardants
Durable finishes are durable to multiple launderings. These are more complex and difficult to apply. Although literature mentions a large number of treatments, only a small number has survived the test of time. These include phosphorus–based compounds, pre-condensates and fibre reactive compounds, requiring multiple process steps, heat treatments ammonia treatment [62, 118].
Some of the more important requirement for ideal durable retardant is the following:
-
Capability to impart fire resistance, that is, both flame and glow resistance
-
Applicability to any weight fabric and preferably from water solutions
-
Easy of application, preferably on existing finishing equipment without causing hazards
-
Adequate ability to withstand laundering and dry cleaning
-
Lack of physiological action
-
Production of tough char when exposed to a fire or high-energy radiation
-
Exertion of little or no adverse effect on the physical properties of the fabric
-
Resistance to bleaches, particularly hypochlorite type, Reasonable cost [62].
Durable flame retardant can be applied to the surface of fibres and/or penetrate inside the fibres. To be durable on the surface the retardant materials could be probably a polymer or bonded to the surface with a polymer. Best results from surface deposits are obtained by using thermoplastic polymers.
The retardant generally impart stiffness or adversely affect some other physical property to the cellulosic fabrics because the weight add-on required imparting flame resistance by surface deposition ranges upward from about 8 %.
Using another flame retardant inside the fibre can reduce the necessary amount of thermoplastic flame retardant. Flame retardant that is applied inside cellulosic fibres can be made impervious to laundering by either of three methods:
-
1.
Introduction of chemical substituents on the cellulose
-
2.
Crosslinking of cellulose
-
3.
In situ polymer formation
Use of flame retardants that crosslink cellulose molecules through primary valence bonds generally decreases tensile properties and increases resilience [112].
Through in situ polymerization technique, flame-retardants materials can be fixed in cellulosic fibres without adversely affecting strength. It is actually increases the breaking strength of fabrics without imparting a noticeable amount of stiffness or resilience [116, 117].
Therefore, one can conclude that a typical flame retardant formulation will contain:
-
Reactive Phosphorous Compound
N-methylol compound (which polymerize with the phosphorous compound and helps to bind the flame retardant to the fabric.
Considerable effort has been expended in recent years on developing flame retardant durable press (FR-DP) finishes for light weight fabrics. The efficiency of phosphorous flame retardant compounds has been enhanced by incorporating the N-methylol crosslinking agents through N-P synergism [112].
The most important examples of this group are:
-
(a)
Tetrakis(Hydroxymethyl)Phosphonium Derivatives
The bulk of today’s durable flame retardant for cellulose centres around the use of derivatives of tetrakis(hydroxymethyl)–phosphonium salts (THP). These derivatives can be applied by padding, drying, curing and oxidizing to yield serviceable flame-retardant fabrics. Add-ons are high and the handle of the fabric is stiffer so the finish is normally used for protective clothing applications [62, 123].
-
Tetrakis(Hydroxymethyl) Phosphonium Chloride (THPC)
THPC is the most important commercial derivative and it is prepared from phosphine, formaldehyde and hydrochloric acid at room temperature. It contains 11.5 % phosphorous and it is applied by a pad-dry-cure → oxidize → scour process.
The compound is highly reducing in character and the methylol groups condense with amines to form insoluble polymers. It is applied with urea, dried and cured. Control of pH and the oxidation state of the phosphorus is important in determining the flame retardant properties and the durability of the finish. HC1 releasing may cause tender to fabric during curing. The final step in finishing requires oxidation of P+3 to P+5 with hydrogen peroxide. This step must be controlled to prevent excessive tendering of the fabric. An alternative to the THPC is THPS. Sulphuric acid is used instead of HC1 and the corresponding phosphine sulphate is formed in place of the phosphine chloride [62, 123].
-
THPC-Urea Pre-condensate
Albright and Wilson replace heat curing with an ammonia gas curing at ambient temperature. This minimizes fabric tendering associated with heat and acids. A Pre-condensate of THPC with urea (1:1 mol ratio) is applied, dried and the fabric passed through an ammonia gas reactor. An exothermic reaction creates a polymeric structure within the voids of the cotton fibre [120, 123]. The ammonia cure gives a P:N ratio of 1:2. Weight percentages of the respective elements should be P, N >2 %. To enhance durability and light fastness of dyes, P+3 is oxidized to P+5 with hydrogen peroxide [116, 117].

-
Tetrakis(hydroxymethyl)phosphonium Hydroxide (THPOH)
From the forgoing discussion, THPC usually partly neutralized with amines, amides and/or alkali. Complete neutralization of THPC with sodium hydroxide yields a compound referred to as THPOH. The distinction between THPC used in a partially neutralized condition and THPOH is difficult to define. If the curing agent is basic as is ammonia, the distinction become meaningless [62, 111].

THPOH-ammonia has received a great deal of commercial attention. The major advantage over THPC is reduced fabric tendering and reduced stiffness. Fabrics padded with THPOH give off formaldehyde during drying [62].
-
(b)
Phosphate Esters with Bromine as Durable Flame Retardants [62, 111]
-
Bromoform-triallylphosphate ester polymer

-
Tris (2,3-dibromopropyl)phosphate

-
Bromine-contaning phsphonitrilates.

-
-
(c)
Azriridinyl Compounds [62, 111]
-
1.
APO—THPC
-
2.
APS—THPC
-
3.
APO
-
4.
APO—Diammonium phosphate
-
5.
APO—Thiourea.
-
1.
-
(d)
Phosphonates and Phosphine Oxides as Flame Retardants [62, 111]
-
1.
Phosphonomethyl cellulose.
-
2.
Allyl phosphonate
-
3.
Trichloro methyl phosphene and phosphene oxide.
-
4.
N-methylol amide of phosphene and phosphene oxide.
-
5.
Phosphopropionamides.
-
1.
-
(e)
N-Methyloldimethyl Phosphonopropioamide (PYROVATEX CP)



Pyrovatex CP provides a method of attaching phosphorus to cellulose making use of N-methylol reactivity with cellulose. It is applied with a methylolated melamine resin using a phosphoric acid catalyst by a pad-dry-cure process. The high nitrogen content of melamine provides synergistic activity to the phosphorus of the flame retardant [115, 123].

Fabric stiffening occurs when sufficient chemical is applied to give 2–3 % phosphorus on weight of fabric. In addition, the acid may cause high strength loss if left in the fabric after curing; therefore, it is desirable to wash the fabric after the curing step [62] (Fig. 10.16).

Chemical structure of APO and THPC
The finish tends to produce smoke in the curing oven. The smoke is composed of volatile fragments of the finish, which condense in the cooler reaches of the oven. The condensate may drip back onto the fabric causing unsightly spots [62, 116, 117, 123]. Figure 10.17 explain the proposed reaction between pyrovatex and melamine formaldehyde [55, 124].

-
(f)
-
1.
Inorganic durable flame retardants.
-
2.
Antimony trioxid—chlorinated parafines
-
3.
Antimony titanium complex.
-
1.
10.2.5.2 Flame Retardants Based on Nature
10.2.5.2.1 Inorganic Flame Retardants
The inorganic compounds are non-combustible materials and thus physically dilute the amount of combustible polymer in the solid phase. Rothon [125] discussed several inorganic flame retardant systems. Metal oxide residues are formed during the decomposition of the inorganic systems.
By the early 1970s the main inorganic flame-retardants used were Alumina trihydrate (ATH), zinc borate and antimony compounds [73, 75]. Although antimony was one of the most popular and frequently used compounds in flame retardant systems over the years, it was expensive and thus used sparingly [75].
Aluminum hydroxide (Al(OH)3), magnesium hydroxide [Mg(OH)2], ammonium polyphosphate and red phosphorus are the main flame-retardants in this group, which represents about 50 % by volume of the worldwide flame retardant production. In order to achieve necessary improvements in flame retardancy, metal hydroxides are employed alone or in combination with other flame-retardants. Antimony compounds (e.g. antimony trioxide) are used as synergistic co-additives in combination with halogen compounds to enhance fire characteristics such as smoke reduction or afterglow suppression. Ionic compounds have a very long history as flame-retardants for wool or cellulose based products.
Unfortunately, in practice very high dosage levels (50–60 %) of the inorganic fillers are needed in the polymer in order to be effective. The name-retarded material should also be processed at temperatures below 220 °C [109].
Alumina trihydrate (ATH) is the least effective in terms of dosage, but also the least expensive of all flame-retardants. Aluminum hydroxide (alumina/trihydrate) ATH decomposes when exposed to temperatures over 200 °C, which limits the polymers in which it can be incorporated. Magnesium hydroxide is stable at temperature above 300 °C and can be processed into several polymers. In the recent time, scientists have been using combination of the metal hydroxides in the concept of nanotechnology to improve the flame retardancy of polymer composites. This approach involves the dispensing of inorganic filler in a nanoscale as flame-retardants into a polymer matrix [126].
10.2.5.2.1.1 Antimony Compounds
For many years now, antimony based inorganic compounds have been used for flame retardants, for example Sb2O3, Sb2O5 and Na3SbO4 [109]. Unfortunately, antimony based compounds are very expensive. The basic Na3SbO4 is ideal for polymers which hydrolyse when processed in the presence of acidic additives. Currently antimony compounds are not used as flame retardant on their own, but are used as synergists with other flame retardants. The addition of antimony compounds to halogen systems can increase their efficiency considerably and thus reduces the total additive loading in the polymer. Antimony trioxide on its own has been reported to be a catalyst for carbon oxidation [127]. However, more recent studies showed that antimony trioxide has an inhibiting effect on char oxidation [128, 129] when used with other inorganic fillers. Unfortunately, a chemical explanation for this unexpected effect wasn’t given.
Antimony trioxide is one of the most widely used flame-retardants [73, 75, 109, 110]. Antimony trioxide is used as a synergist. It is utilized in plastics, rubber, textiles, paper and paints. Antimony oxides and antimonates must be converted to volatile species. This is accomplished by the reaction of halogen acids and antimony containing materials at fire temperatures to form antimony trihalide or antimony halide oxide which suppress flame propagation. Other antimony compounds include antimony pentoxide, available primarily as a stable colloid or as a re-dispersible powder. It is used in fibre and fabric. Sodium antimonate is recommended for formulations in which deep tone colours are required.
10.2.5.2.1.2 Boron Compounds
Boron compounds are also widely used as flame-retardants [109]. Compounds such as borax (sodium borate), zinc borate, barium metaborate (BaB2O4.XH2O) and ammonium fluoroborate (NH4BF) are well known retardant systems. Boron compounds act in both the condensed and vapour phase as fire suppressants. Most boron complexes are Lewis acids, which promote crosslinking of the polymeric material on thermal degradation and thus minimizing decomposition and volatile combustibles. Boron compounds can also react with the hydroxyl group in polymers containing such groups (e.g. cellulose) to form a glassy ester. This ester forms a char coating on the substrate surface and reduces solid-state carbon oxidation by protecting the underlying material. Moreover, it is common knowledge in commercial practice that the addition of zinc borate to metal hydroxide flame retardant systems can help to reduce the afterglow effect.
The most widely used in this class are boric acid (H3BO3) and sodium borate (borax; Na2B4O7.10H2). They are primarily used for cellulosic material such as cotton, paper, and roofing thatch [130]. They are effective in decreasing afterglow time and lengthening of ignition time; although their use is limited to products for which non-durable, flame retardancy is acceptable since both are very water-soluble.
Zinc borate is however water insoluble and is mostly used in plastics and rubber products. It is used as a complete or partial replacement for antimony oxide in PVC, nylon polyolefin, epoxy, etc. It functions as a flame retardant and smoke suppressant in condensed phase.
10.2.5.2.1.3 Silicon Compounds
There is a renewed interest in using silicon-based flame retardants as substitutes for the halogens or phosphorus [131]. Almost all forms of silicon have been explored: silicones, silica, organosilanes, silsesquioxanes, and silicates. The most common flame retardant based on silicon is in the form of polyorganosiloxane, in particular, polydimethylsiloxane (PDMS). The flammability of the block copolymers of various types of polycarbonate (PC) and poly(ether imide) with PDMS [132] show significant decreases. Silicon can also be incorporated into the branches of the polymer chains [133]. Under certain cases, the addition of silica can also affect the flammability properties of materials [134]. The formation of a silicon-based protective surface layer appears to be the flame-retardant mechanism for silicone and silica systems. Polycarbosilane, polysilastyrene, and polysilsesquioxane pre-ceramic polymers are shown in Fig. 10.18, are also used to blend with various thermoplastics, [135, 136].

Some pre-ceramic polymers
Nanoparticles of ZnO and amino functionalized silica have been used in order to functional finished cotton fabrics to produce technical textile with high performance, regarding the environmental aspect [137]. Cotton cellulose had been activated via grafting copolymerization in order to facilitate the reaction between cellulose and nanoparticles. A novel flame retardant system consisted of diethyl phosphite in combination with the ZnO and amino functionalized silica nanoparticles had been investigated. The performed treatment helps to form more non-flammable char residue and increases char formation after heating in addition to the improvement of the water repellency property. Nano ZnO was investigated as a novel flame retardant for cotton and fabrics.
In addition, the presence of phosphorus deposited on the diethyl phosphite treated samples is the most effective parameter in the char forming and decreasing the flammability of the treated fabrics [137].
Jia and Levalois-Grutzmacher [138], found that with argon plasma treatment, it is possible to polymerize a silicon-based acrylate monomer, in this case 3-(acryloyloxy)propyltrimethoxysilane (APTMS). However, APTMS is found to confer little or no flame retardant properties on cotton textiles. When introduced as a physical mixture with diethyl (acryloxyethyl) phosphoramidate (DEAEPN), the presence of Si-O bonds in APTMS does not contribute towards any synergistic effects with phosphorus and nitrogen in DEAEPN on the flame retardancy of cotton textiles.
10.2.5.2.1.4 Clays
Recently, there is a great interest in the flammability properties of polymer-clay (layered-silicate) nano-composites [139, 140].
Clays can be used as flame retardant systems. Experiments showed that the heat release rate—the most important parameter for predicting fire hazard—is reduced by 63 % in a nylon-6-clay nano-composite containing 5 % and of a modified montmorillonite clay [76]. Nano-composites used are clays. According to Oilman Kashiwagi, [76] the clay additive does not degrade the overall material properties and caused no increase in carbon monoxide or soot levels during combustion.
10.2.5.2.1.5 Phosphorus Compounds
Red phosphorus is effective as a flame retardant in polyurethane foams, polyamides and phenolic applications. Ammonium polyphosphate is mainly applied in intumescent coatings and paints. Intumescent systems are materials that puff up to produce foams [107]. Owing to these characteristics, they are used to protect materials such as wood, plastics and steel from high temperature. Other inorganic flame-retardants as ammonium sulfamate (NH4SONH2) and ammonium bromide (NH4Br) are mainly for cellulose-based products and in front forest fighting.
10.2.5.2.1.6 Other Metal Compounds
The effect of the metal cation on the intumescent behaviour and char formation was studied. Metals, representative of the main groups in the periodic table are listed below:
-
Alkali metals—sodium [Na]
-
Alkali earth metals—magnesium [Mg] and calcium [Ca]
-
Main group metals—aluminium [Al] and antimony [Sb]
-
Transition metals—titanium [Ti], vanadium [V], zirconium [Zr], zinc [Zn]. iron [Fe] and copper [Cu]
Potassium is known to improve char formation in cellulose (Tang and Neill 1964) and in polyamide 6 (nylon 6) when used as synergist with silica compounds [76].
Potassium nitrate can also be used as a flame retardant in nylon 6 as it improves charring of the polyamide [141]. In addition, potassium bitartrate showed intumescence and char formation on its own [72]. It has also been found poly(sodium styrene sulphonate) is a flame retardant providing intumescent char formation [142].
Copper Cu+2 complexes can act to improve char performance [133]. Copper is also added to halogen flame retardants as a smoke suppressant [69]. This is most likely due to the condensed phase reactions such as charring caused by the copper.
Ebdon et al. [143] found that vanadyl acetylacetonate (VO(CH3COCHOCH3)2) and VOCb increased the LOI of styrene/4-vinyl pyridine- and methylmethacrylate/4-vinyl pyridine copolymers by forming an intumescent char on the surface. The addition of Zn and Ni had little or no effect on char formation.
Other very popular flame-retardants are metal hydroxides such as aluminum trihydrate (ATH) and magnesium hydroxide [69, 72, 73, 144, 145]. Magnesium oxide and magnesium carbonate (Mg2(CO3)3(OH2)) are also used [65, 66, 72, 73, 75, 109, 146]. In some instances, calcium compounds such as calcium carbonate and calcium hydroxide, calcium sulphate and calcium oxide are also used [65, 66, 72, 75, 109].
Zinc compounds were also used. They mainly act as a source of boron (zinc borate), which helps with smoke suppression and to stabilise carbon chars [65, 66, 69, 72, 73, 75, 109, 147] or as a source of phosphate (ZnPO4)—the catalyst for intumescence [148].
To isolate the specific effects of the metals on intumescence, carbon char yield and thermal degradation, a common organic compound (backbone) had to be used. Acetylacetone, i.e. 2,4-pentanedione was selected for this purpose. It forms very stable complexes with most of the metals. The chemical properties of acetylactone are determined by the keto-enol tautomerism shown in Fig. 10.19. At moderate temperatures, it is generally accepted that the enol form predominates in the gas and liquid phases.

2,4-Pentanedione (acetylacetone) and its stable isomers
Acetylacetone is renowned for forming stable metal acetylacetonate complexes [149]. The metal reacts with the acetylacetonate in the enol form-to-form bonds between the two oxygen atoms and the metal ion. The inner complexes formed between the metal ions and the β-diketones are unusual since they possess properties usually associated with pure compounds, and not organometallic complexes.
Gluconic acid is a good chelating agent for most metals. Metal complexes of gluconic acid were used, e.g. sodium gluconate showed appreciable intumescence, high carbon char yield and good char properties. Calcium gluconate monohydrate, magnesium gluconate hydrate, iron (II) gluconate hydrate and copper (II) gluconate were used as commercial products. Ammonium was also used as a cation with this acid in order to be clear on the influence of the metals on intumescence and char properties.
Molybdenum compounds are also used as flame-retardants [109], and it was one of the first elements to be used as flame retardant additives for cellulose. Molybdic oxide, molybdenum trioxide, zinc molybdate and ammonia octamolybdate are used. Molybdenum trioxide acts as a condensed phase retardant by increasing char yields. Molybdenum compounds have been used as flame-retardants for cellulosic materials and in other polymers as smoke suppressants. Titanium and Zirconium compounds are used for textiles, especially wool [107].
10.2.5.2.2 Halogenated Organic Flame Retardants
The halogenated flame retardants are divided into three classes, namely aromatic, aliphatic and cycloaliphatic. Among the halogens, the compounds bromine and chlorine are significant as flame retardants. Fluorine compounds though expensive are ineffective because the C-F bond is too strong. Iodine compounds though effective are expensive and too unstable to be useful [74].
The thermal stability of halogenated flame retardants vary in the order of; aromatic brominated flame retardants greater than the aliphatic chlorinated flame retardant which in turn are more stable than aliphatic brominated flame retardants.
The aromatic bromated flame retardants can be used in thermoplastics at fairly high temperatures without stabilizers but at very high temperatures, they must be used with stabilizers like tin compounds [74].
10.2.5.2.2.1 Brominated Flame Retardants
Bromine-based flame-retardants are highly brominated organic compounds with a relative molecular mass ranging from 200 to that of large molecule polymers. They usually contain 50–85 % (by weight) of bromine [150]. Tetrabromo bisphenol A (TBBPA) and decabromo diphenyl ether (DeBDE) are the two brominated flame-retardants that have the highest usage by volume today.
TBBPA is an example of a flame retardant that can be used as an additive as well as a reactive. It is used in the production of flame retarded epoxy resins used in printed circuit boards. DeBDE is used in high impact polystyrene, which is in the production of television cabinets.
Other uses of DeBDE are in ABS, engineering thermoplastics, polyolefins, thermosets PVC and elastomers and textiles. Hexabromocyclo dodecane (HBCD) is a major cycloaliphatic flame retardant and its primary used is in polystyrene foam and textiles.
10.2.5.2.2.2 Chlorinated Flame Retardants
Chlorine-containing flame-retardants are divided into three classes: aliphatic, cycloaliphatic and aromatics. Chlorinated parafins are the most widely used aliphatic chlorine containing flame-retardants. The have applications in plastics, fabrics, paints, and coatings. Bis (hexachlorocyclo-pentadieno) cyclooctane is a flame-retardant having good thermal stability for chlorinated cycloaliphatic, with thermal stability comparable with brominated aromatics. It is used in several polymers especially polyamides and polyolyfins for wire and cable applications [74].
10.2.5.2.3 Organophosphorus Flame Retardants
The predominant phosphorus flame retardants in use in plastics and textiles are phosphorus, phosphorus-nitrogen, phosphorus-halogen and phosphate esters with or without halogen. Phosphorus-containing flame retardants are the most important material that impart durable flame resistance to cellulose [110, 151]. The major groups of additive, among organo-phosphorus compounds are phosphate esters, polyols, phosphonium derivatives and phosphonates.
The phosphate esters include trialkyl derivatives such as triethyl or triocytyl phosphate, triaryl derivatives such as triphenyl phosphate and aryl-alkyl derivatives such as 2-ethyl hexyl-diphenyl phosphate. Phosphonium salts are used to improve the flame retardancy for cellulosic products. The esters formed by reaction of the three functional groups of phosphoric acid with alcohols or phenols are excellent plasticizers. Plasticizers are mixed into polymers to increase flexibility and workability [152, 153]. Aryl phosphatem plasticizers are used in PVC based products, used as lubricants for industrial air compressors and gas turbines.
Other uses of aryl phopsphates are as pigments, dispersants and peroxide carriers, and additives in adhesives and wood preservatives.
10.2.5.2.4 Halogenated Phosphates
These are flame retardants that contain phosphorus and bromine or chlorine. They normally combine the flame retardancy properties of phosphorus and those of the halogens (bromine and chlorine).
Among this group, is tris (1-chloro-2-propyl) phosphate (TCPP) used in polyurethane foam. The other is the tris (2-chloroethyl) phosphate used in the manufacture of polyester resins, polyacrylates, polyurethanes and cellulose derivatives. In the bromine—phosphorus group, the commonest is tris (2,3-dibromopropyl) phosphate which has been withdrawn from use in many countries due to carcinogenic properties in animals [108].
10.2.5.2.5 Nitrogen—Based Flame Retardants
Nitrogen-based flame retardants are used primarily in nitrogen containing polymers such as polyurethanes and polyamides. They are also utilized in PVC and polyolefins and in the formulation of intumescent paint systems [154].
Melamine, melamine cyanurate, other melamine salts are currently the most used group of nitrogen—containing flame retardants. Melamine, melamine cyanurate are used for polypropylene, polyethylene, polyamindes epoxy and polyurethane.
10.2.6 Flammability Tests for Textile
To fully evaluate the fire behaviour of different polymers, it is necessary to develop standard tests for assessing the flammability and other combustion-related properties of polymers. Most countries have standards and codes for the classification of materials with respect to their combustion behaviour, but the experimental setup used in existing standard tests varies considerably, according to the nature, shape, and size of the polymeric materials to be tested [136]. The fire tests most commonly used are the ASTM E 84 Steiner Tunnel [155], and the ASTM E 622 National Bureau of Standards (NBS) smoke chamber [156]. However, these tests can be used only as guides and suffer from problems with precision and reproducibility.
The majority of fire tests used now are concerned with the determination of the following fire properties of materials [136, 157].
-
Ease of ignition—how readily a material ignites
-
Flame spread—how rapidly fire spreads across a surface
-
Fire endurance—how rapidly fire penetrates a wall or barrier
-
Rate of heat release—how much heat is released and how quickly
-
Ease of extinction—how rapidly or how easily the flame chemistry leads to extinction
-
Smoke evolution—amount, evolution rate, and composition of smoke released during stages of a fire
-
Toxic gas evolution—amount, evolution rate, and composition of gases released during stages of a fire.
Among them, the oxygen concentration test is a very important ignition test, from which the limiting oxygen index (LOI) [158] can be obtained. The LOI is defined as the minimum oxygen concentration at ambient temperature needed in an inert gaseous medium for the material to achieve sustained burning after ignition. The precision and reproducibility of the results are two reasons for the wide acceptance of this method.
10.2.6.1 Vertical Spread Method of Flame Test: AATCC 1934–1969 [54, 60, 111, 159]
The vertical flame test is the most severe test. The criteria evolutions are char length, after flame removal and after vanishing of glow. Fabric specimen dimensions are 1.5 × 72 in., conditioned at 70 °F and 65 % relative humidity. The specimen mounted in a holder kept in a cabinet, is subjected to standard flame (gas burner) at its lower edge, under controlled condition. The fabric specimen is suspended in a vertical position and flame is applied along the fabric width. The rate of flame spread is recorded and the flaming time, after glow time and char lengths are measured.
10.2.6.2 Horizontal Spread of Flame Test Method 5906–CCC–T–1916 [60, 160]
Several tests have been designed for measuring the spread of flame on horizontally placed fabric specimens. In federal specification method 5,906 specimen strips of 12.5 × 4.5 in. oven-dry fabric are used. Ignition is affected in one end of the sample, and the rate of burning is measured. In method 5,900 the ignition is effected in the centre of the horizontal specimen and the largest dimension of the charred area is reported. A Germen method similar to 5,906 was also announced. Specimens of 35.5 × 10 cm, is clamped of two matching rectangular frames and held horizontally in a certain conditioned at 20 °C and 65 % humidity, is ignited for 18 s by a propane gas burner of specified dimensions applied to the edge of the specimen the time required for the lower of the flame front to travel across the specified length is measured.
10.2.6.3 The 45° Angle Test: AATCC 1933–1962 [60, 161]
The test is designed to indicate the textiles, mainly used for apparel and applicable for easily flammable fabrics, which ignite easily and, once ignited, burned with sufficient intensity and rapidity to hazardous when worn. The test was first developed by AATCC following the adoption of flammable fabrics act in 1954. Oven-dry specimens of 2 × 6 in. are used in the test. The specimen is held at 45° in a rack placed in ventilated chamber containing an automatic timing device and a standardized ignition medium. The flame applied is of 5/8 in. length. The distance of fuel nozzle from the fabric surface is 5/16 in. The distance from the central point of flame impingement on the face of specimen to stop-cord made of mercerized cotton sewing thread 5 in. The time of ignition is adjusted 1 ± 0.05 s. The time of flame spread, including ignition from the impingement point to the stop-cord (5 in.) is measured. Flaming times higher than 3.5 s for textile without a raised fibre surface or 7 s for textiles with raised fibre surfaces are considered of “normal flammability”. Textile of intermediate flammability with raised fibre surfaces have a time of flame spread between 4 and 7 s. Lower times of flame spread characterize dangerously flammable textiles. The 45° test has recently been accepted in a modified form in Japan.
10.2.6.4 Ease of Ignition [111]
Oven-dry specimens of 7 × 7 in. are used. The specimen is placed horizontally and a micro burner is applied to one edge. The time needed to ignite the specimen is measured.
10.2.6.5 The Limiting Oxygen Index (LOI) Test ASTM 2863 [60, 106, 158]
This test, suggested in 1966 for testing the flammability of polymers. It has been recently adapted to the testing of fabrics. The (LOI) is defined as minimal volume fraction of oxygen in a rising gaseous atmosphere that will sustain candle like burning of the sample of solid polymer or fabric. A gas metring system and a Pyrex glass chimney (as a flame holder) is 15 in. high and 3.5 in. diameter.
The samples tested are in the form of 2.5 × 6 in. strips placed into a hinged U-shaped holder, while the free end of holder is held with clamps. The holder is mounted vertically in the chimney on the chimney axis. A series of known mixtures of O2 and N2 in varying ratios are passed upward through the chimney at a velocity of 3–11 cm/s for 1–2 min each the sample is ignited with a laboratory gas burner at its top end during the flow of mixture, the oxygen concentration below which the flame will not spread downward the entire length of samples found, and its volume fraction in the gas mixture is designated as LOI. Figure 10.20 illustrate the scheme of LOI tester.

Scheme of the limiting oxygen index (LOI) tester. A metering, B test gas, C pyrex cylinder, D glass beads, E sample holder, F test fabric
10.2.6.6 The Metal Cylinder (Carpet Flammability Test) [60, 111]
Which consists of placing a steal cylinder heated to 800 °C on a carpet for 12 s and measuring the time of flaming and the charred area.
10.2.6.7 Cigarette Test [162]
This test for household furnish like mattresses. Smouldering cigarettes (non–filter from natural tobacco) are applied at four types of locations smooth surface, tape edge, quilted location under two sheets on the mattress. Combustion distance and time for flaming and smouldering as well as char length are measured.
10.2.6.8 Pill Test DDD–C–95 [60, 163]
This test for carpets and rugs, a methylamine tablet is placed in the centre of a 6 × 6 in. carpet specimen and light with match. The tablet burns for about 2 min and the diameter of the char area is measured.
10.2.6.9 Smoke Test [156, 164]
A test specimen (7.6 × 7.6 cm) is vertically mounted and max. 2.5 cm is exposed to radiant heat and/or a gas flame in chamber (91.4 × 61 × 91.4 cm). A photometric system is used to measure smoke density.
10.2.6.10 Colorimeter Test (NBS Cone Colorimeter) Consider by ASTM E 84 and E 119 [155, 165]
The Cone Calorimeter test is probably the most widely used test method in use today to calculate the rate of heat released by a burning material. Ignitability polymer formulations can also be tested in the calorimeter. It is the bench-scale test method used most commonly for the majority of polymer fire testing and was developed in 1982 by Babraukas at the National Institute of Standards and Technology (NIST). A 100 mm by 100 mm sample (up to 50 mm thick) is placed horizontally in the apparatus and exposed to a radiant heat flux of 10–100 kW/m2 with a spark ignition system. The effluent is monitored and temperature, gas flow rate and oxygen concentration measured. The amount of smoke produced along with carbon dioxide and carbon monoxide concentrations can be determined with some instruments. Flame retarded coatings can also be evaluated with the cone calorimeter,
Flame retarded coatings are commonly tested under fire tests developed for materials used in the construction of buildings. Examples of such tests are the American Society for Testing and Materials (ASTM E 84 [155] and E 119 tests [165] ). The Steiner Tunnel is the basis for the ASTM E 84 test. It is possibly the oldest fire test for flame retarded materials in use in North America and is used to test the surface burning characteristics of building materials. The test was developed by Albert J. Steiner of Underwriters Laboratory Inc. and Simon Ingberg and was first described in 1943. A 0.514 m wide and 7.32 m long sample is placed in the ceiling of a 7.62 m (25 ft) long tunnel with a 88 kW burner at the one side of the tunnel and an air draft forced through the tunnel at a velocity of 1.22 m/s. The flame spread is recorded as a function of time. The tunnel is also fitted with a photometer to determine smoke. The test results for some plastics are however questionable. Due to its size, large samples were needed for testing. From the 25 ft Steiner tunnel test several smaller tunnel tests were developed e.g. the 18, 8 and 2 ft tunnel tests [166].
10.3 Fire and Flame Retardancy Finishing of Cellulosic Materials and Its Composite
10.3.1 Fibres and Fabrics
10.3.1.1 Cellulose (Cotton and Rayon) [167]
There are important historical and economic reasons for the fact that, the science and technology of flame-retardants for cellulose fibres are more advanced than for other fibres and polymers. Until perhaps 25 years ago, an overwhelming proportion of all textiles used was made of cellulosic fibres.
Thermal degradation of cellulose, which added flame-retardants, has led to a partial elucidation of the mechanism of fire-retardant action by Lewis acids. The principal role of acid or acid-forming flame-retardants in cellulose is to enhance dehydration and char formation in the condensed phase, suppressing the formation of combustible volatiles in the thermal degradation process.
Phosphorus acids are particularly efficient in catalysing cellulose dehydration, and phosphorus-containing flame-retardants have been studied most extensively. In the case of natural cellulosic fibres, modification with flame-retardant compounds has been accomplished almost exclusively by finishing of fabrics. In the case of regenerated cellulose fibres, the most important approach to modification has been the incorporation of flame-retardant additives in the spinning fluid.
Flame-retardant finishes for cotton fabrics have been studied and reviewed before in a large number of other publications [4, 31, 54, 60, 100, 124, 137, 168–173]. Milestones of the evolution from simple processes involving deposition of inorganic salts (readily removed by water) to sophisticated modifications of the cellulose molecule and/or in situ polymerization of appropriate monomers are summarized in Table 10.3.
Flame-retardant finishes based on chlorinated paraffins and antimony oxide in conjunction with resin binders were developed for tent fabrics. Large amounts of finish (up to 60 % solids applied based on fabric weight) were used, impairing the flexibility and air permeability of the treated fabrics. However, the finish succeeded in combining flame resistance, water and mildew resistance for a critically important application.
Attempts to obtain durable flame resistance in cotton by chemical modification of the cellulose molecule were made in the late 1940s. Work on phosphorylation of cellulose in the presence of large amounts of urea or other basic compounds to prevent acid degradation and depolymerization of the polymer [167] led to a process, which was used, on a commercial scale for a time. However, the phosphate ester linkage introduced into the cellulose is not sufficiently stable to hydrolysis to withstand repeated laundering and the presence of ion exchange sites in the modified cotton leads to replacement of NH: by metal cations with loss of flame resistance.
Reaction of cotton with titanium oxychloride and antimony oxychloride [167] enjoyed a brief period of intense interest. The development of “modern,” truly wash-resistant, or durable flame-retardant finishes for cotton fabrics began on the reactions of two organophosphorus molecules, APO and THPC (Fig. 10.16) with cellulose, with coreactants, and with each other.
Finishes based on THPC chemistry have been produced commercially for a decade, and remain important. Finishes based on APO chemistry have been abandoned primarily because of toxicity and/or suspected carcinogenic properties of the reagent (APO) and its precursor (ethylene imine).
10.3.1.2 Other Cellulosic Fabrics
In principle, approaches used for finishing of cotton fabrics are applicable to other cellulosics (rayon, linen, jute, etc.). In fact, the amounts of flame-retardant finish needed to attain a specified level of flame resistance. Commercial production of flame-resistant 100 % cellulosic fabrics by finishing has been limited to cotton, and some specific rayon fabrics [167].
10.3.1.2.1 Regenerated Cellulose (Rayon) Fibres
Flame-resistant rayon fibre can be used to manufacture fabrics, which are self-extinguishing in a vertical test. It has been made commercially by incorporating an alkoxyphosphazene in the spinning fluid (Fig. 10.21). The commercial fibre employs the n-propoxy compound (R = n-C3H7-) as the additive.

Chemical structure of alkoxyphosphazene
Other approaches in which the rayon fibre is modified by graft copolymerization reaction with a flame-retardant monomer prior to yarn manufacture have been suggested, but have not reached commercial status to date [167].
These fibres usually have flame retardant additives incorporated into the spinning dopes during their manufacture, which therefore yield durability and reduced levels of environmental hazard with respect to the removal of the need for a chemical flame retardant finishing process (see Table 4.8). Additives like phosphorus-based and so are similar to the majority of these fibre finishes in terms of their mechanisms of activity (condensed phase), performance and cost-effectiveness [89].
This fibre not only has removed the need for phosphorus, but also chars to form a carbonaceous and silica-containing mixed residue, which offers continued fire barrier properties above the usual 500 °C where carbon chars will quickly oxidise in air [89].
10.3.1.2.2 Cellulose Acetate and Triacetate
Cellulose acetate and triacetate fibres, or cellulose ester fibres which made by acetylation of natural cellulose are thermoplastic. They melt at relatively low temperature [174], they ignite, and they can propagate the flame even though they drip while they continue to burn. The only method used to attain fire resistance in cellulose acetate and triacetate fibres is the incorporation of a flame-retardant additive (specifically, 2,3-trisdibromopropyl phosphate) into the spinning solution before extrusion. The bromine-containing product (flame-resistant acetate) obtained is somewhat more resistant to ignition than the unmodified acetate. If a non-thermoplastic component is present in the system, it acts as a wick for the molten acetate or triacetate polymer and burning is sustained. In this case, the amount of flame-retardant additive commonly used in spinning (about 10 % based on polymer weight) is not sufficient to impart flame resistance and self-extinguishing behaviour [167].
10.3.2 Flame Retardant Cellulosic Blends
Blends are textile materials made from yarns containing two or more different fibres whether synthetic or natural. Blend fabrics have attained great commercial importance and have afforded opportunities for optimal utilization of fibre properties in textile products.
In practice, a number of technical limitations including dictates limitations: [175]
-
1.
Compatibility of fibres during spinning or fabric formation; fibres must be available with similar dimensions and be processible simultaneously with other types on the same equipment.
-
2.
Compatibility of fibre and textile properties during chemical finishing; for instance, flame retardant cotton treatments must not adversely influence the characteristics of the other fibres present in the blend during their chemical application.
-
3.
Additivity and, preferably synergy, should exist in the flame retardant blend; it is well known that with some flame retardant blends, antagonism can occur and the properties of the blend may be significantly worse than either of the components alone [175].
Flammability and flame resistance of blends have received attention since 1968, and it was soon established that modification of these multicomponent substrates with flame-retardants poses special problems [167, 176]. The large number of variables in the substrate (specific fibres, fibre content or percentage, melting vs. non-melting behavior, etc.) further increases the complexity of these problems.
The most important blend fabrics are those made from polyester and cotton, generally (but not necessarily) containing 50 or 65 % polyester. Modification of these fabrics with flame-retardants can be approached by (1) blending modified (flame-resistant) polyester fibre with cotton; (2) finishing the polyester/cotton blend fabric with flame-retardants; (3) a combination of (1) and (2) [167].
All these approaches have been explored, generally utilizing flame-retardant compounds, which showed effectiveness on polyester and on cotton. The use of modified polyester does not provide an adequate level of flame resistance since melt drip is inhibited by the presence of cotton.
Modifications of the polyester specifically designed for approach (1) have not been reported, and research and development efforts have been directed primarily towards approaches (2) and (3)-namely, finishing.
10.3.3 Flammability of Natural Fibre Composites
As with composites reinforced with glass or other synthetic fibres, natural fibre reinforced composites (NFRC) comprise a polymer matrix that is reinforced with natural fibres. A wide range of fibres, mainly plant, and polymer matrices utilized have been reported in Table 10.4 [31, 177, 178].
The flammability properties of the composite are different from the component materials and factors such as the structure of the composite, adhesion between the fibres and the matrix, the type of matrix polymer, and the type of fibre all play roles in determining these properties [31, 179].
Natural fibres are non-thermoplastic and their decomposition (pyrolysis) temperature is less than their glass transition and/or melting temperatures [21, 23]. Unlike protein fibres, plant fibres has poor fire resistance; for example, cotton has a LOI value of 18–20, whereas wool has a LOI value of 25 [180].
The thermal degradation of plant fibres involves a number of processes including the desorption of adsorbed water, cross-linking of cellulose chains with the evolution of water to form dehydrocellulose, decomposition of the dehydrocellulose to yield char and volatiles, formation of levoglucosan, and decomposition of the levoglucosan to yield flammable and nonflammable volatiles and gases, tar, and char [23, 32, 181].
Manfredi et al. [30] found that, the thermal degradation of sisal and jute was similar, with the main peak at 340 °C, whereas the flax started to degrade at higher temperatures, with the main peak at 345 °C. Slight variations in the flammability of different plant fibres can be attributed in part to the chemical composition of the fibres.
10.3.4 Wood and Paper
The importance of wood and wood-based products (insulation board, hardboard, and particle board) in our environment is obvious. Similarly, enormous quantities of paper and paper like products are consumed daily, frequently in situations where fire safety considerations are important [167].
Wood and paper products consist essentially of cellulose. Thus, flame-retardant compounds that inhibit flammability of cellulose are, in principle, effective for wood, paper, and cellulosic textiles. It is clear, however, that flammability and performance requirements, durability of flame resistance, critical side effects, and economics are different for these products.
When wood is progressively heated at raised temperatures, changes begin to occur in its structure, accelerated by further increase in temperature [182]. The three polymeric components in the wood begin to thermally decompose to a mixture of volatile gases, tar (levoglucosan) and carbonaceous char. The decomposition is often regarded as the superposition of the individual constituent’s decomposition mechanisms: hemicellulose decomposes first [180–350 °C] followed by cellulose [275–350 °C], and lignin [250–500 °C] [182, 183]. The thermal stability of lignin is considered to be due to its heavily cross-linked structure and high molecular weight [182, 184].
The subject of flame-retardants for cellulose must be divided into its logical components; namely, wood and wood-based products, paper and paper-based products.
10.3.4.1 Wood and Wood Based Products
Modification of wood and wood-based products with flame-retardants aims at preventing ignition and at reducing the rate of flame spread if ignition occurs.
Fire retardants are typically either coated onto the surface of the wood, or impregnated into the wood structure using a vacuum-pressure technique, although other technologies, such as plasma treatments [185], are being investigated.
The structure of wood being as similar to that of a sponge, with cell cavities and cell walls when impregnating.
To protect the structure from fire, these walls must coat with fire retardant. First, the vacuum removes the air from the cavities to create space for the fire retardant solution, which is then forced deep into the wood under high pressure.
Painted, sprayed or dipped into a solution of fire retardant are the ways to apply a protective surface coating to wood. Superficial treatments, such as paints, are often thought to be attractive for the ease with which they can be applied, and for the comparatively small amount of material required for fire protection. However, the associated re-application requirements and surface damage possibilities are considerable problems for the end-user.
Fire retardants exert their influence on the combustion of wood by numerous approaches. All the treatments are aim to delay the time to ignition of wood, reduce the rate of heat release during combustion [186] and reduce the surface spread of flames. The modes of action (how they work) are described briefly.
Browne [187] provided an in-depth literature survey on these concepts and summarised them into four basic theories. These are chemical, coating, thermal and gas theories; however, these strategies are by no means mutually exclusive because two or more of them may be, and probably are, operative in a given case.
Chemical mechanisms are exhibited by most commercial fire retardants for wood where the pyrolysis of wood is directed towards the production of increased char and water, and fewer volatiles. Coating theories suggest that the fire retardant coats the wood fibres to provide a ‘blanket’ of protection, and prevent the escape of flammable vapours and access of oxygen. Fire retardants displaying thermal action can operate in one of three ways: provide thermal insulation to the wood, absorb the surrounding heat by endothermal reactions, or increase the thermal conductivity of wood in order to dissipate the heat from the wood surface. Gas theories suggest that gases released by the fire retardant either dilute the flammable gases produced by wood to prevent the formation of a flammable mixture, or inhibit chemical reactions in the flame so that combustion is incomplete and less heat is available to sustain burning. Examples of substances used for each of the mechanisms described are outlined in Fig. 10.22 [182].

Examples of fire retardants acting by different mechanisms [182]
It can be approached either by impregnation of the substrate with flame-retardants, or by surface coatings. The former method has a long history, dating back to the first century B.c., when alum and vinegar solutions were used as fire-retardant treatments for wood. Although the flame-retardant chemicals are relatively inexpensive, the processing cost is high and the treatment increases the cost of the wood or wood products by 50–100 %.
The chemicals and treatment processes used commercially have been developed primarily from empirical knowledge and pragmatic observations over the years. Recent research on the manner in which flame-retardant compounds alter pyrolysis and combustion processes in wood cellulose now provides a conceptual framework for established approaches and for new developments as well [182].
It is now generally accepted that flame-retardant chemicals that are effective for cellulose (specifically wood) alter the course of thermal degradation reactions in the solid phase, increasing the amount of char, water, and carbon dioxide formed at the expense of combustible degradation products (organic volatiles-tars and gases).
Volatile degradation products from treated wood provide less energy on combustion than the same weight of volatiles from untreated wood, while the char yield is significantly increased. Thermal data showing the effect of additives in α-cellulose are shown in Table 10.5 [182], and are indicative of the approaches used for imparting flame resistance to wood, involving primarily inorganic salts.
These inorganic salts are generally used in mixtures containing several compounds (e.g., 10 parts diammonium phosphate +60 parts ammonium sulfate +10 parts borax +20 parts boric acid = “Minalith formulation”). Aqueous solutions containing 12–15 % concentrations of the flame-retardant salts are used for pressure impregnation of lumber or plywood, aiming for a dry-salt retention of 2.5–3 lb/ft3 for plywood or 2 in. lumber (and decreasing amounts for thicker lumber).
Pulp also treated before sheet formation, or the wet pressed mat may be treated before drying. Hardboards have also been treated by pressure impregnation after hot pressing. Related processes have been disclosed for particle board [54, 60].
Approaches for chemical modification of wood with organic and/or reactive flame-retardant compounds [182] are include four terms: (1) impregnation from emulsions of organic phosphates in combination with oil-borne preservatives, (2) impregnation from solvent solution of organic compounds of phosphorus and halogen, (3) impregnation with (unsaturated) organophosphorus monomers polymerized in situ by radiation and (4) bromination of lignin to produce bromolignin as the effective flame retardant.
Wood and wood-based products properly treated with fire-retardant formulations have decreased rates of surface flame spread (ASTM Test for surface burning characteristics of building materials E-84-13A [155]) and are self-extinguishing (flaming and glowing) when the external source of heat is removed.
In modifying wood by chemical treatment, the influence on many properties must be considered, including the effect of added flame-retardants on strength, durability, hygroscopicity, corrosiveness, painting, gluing, and machining characteristics. Reduced performance in some of these characteristics (in addition to the cost of fire-retardant treatment) has limited the use of wood-based products treated with fire retardants.
Many of the chemicals used as fire retardants are water-soluble inorganic salts, easily leached from the wood: therefore, the treatments are primarily limited to interior uses. Furthermore, some chemicals used in fire-retardant formulations are hygroscopic, and as water is absorbed, droplets may come to the surface and drop from the treated substrate, with consequent loss of fire retardant. Many of the inorganic salts used as fire retardants are corrosive to certain metals and alloys. Formulations can be balanced and neutralized, and commercial corrosion inhibitors added so that this does not pose a problem. However, if treated wood is exposed for long periods at high relative humidity, moisture and chemicals may be exuded on the metals, and produce various forms of electrolytic corrosion, which the inhibitors may not be able to control.
10.3.4.2 Fire Retardant of Wood-Plastic Composites
It has been noted that addition of lignocellulosic fibres to polypropylene reduces the heat release rate peak essentially. Characteristics such as heat released in combustion and mass loss rate are also reduced. However, criteria such as time to ignition and production of smoke deteriorate in comparison with pure polypropylene [188, 189]. The flammability of WPCs depends on several factors: type of raw material, structure, density, thermal conductivity, humidity, and the nature of composite [31, 190].
Fire retardants can be introduced into composites by two methods [58]:
-
1.
Mass treatment: The addition of fire retardants to the mass during the production process
-
2.
Surface protection: The addition of fire retardants onto the surface of the composite in the final stage of production [21, 191].
The flame retardancy of WPCs can be enhanced by several methods: [21, 58, 186]
-
1.
Insertion of lignocellulosic natural fibres together with flame retardants before the production process by impregnation in an autoclave
-
2.
Addition of fire retardants in liquid or solid form during the production of the composites
-
3.
Use of non-flammable polymers and resins
-
4.
Additior1 of nanoparticles to the composites
-
5.
Insulation of the composites to prevent penetration of heat flux (intumescent coatings and fire barriers).
The above methods can be used together or separately. The current ecological situation dictates the new tendency of use halogen-free fire retardants.
Commonly used fire retardant compounds for wood-plastic composites are magnesium hydroxide, boric acid, ammonium phosphates and borates, ammonium sulphate and chloride, zinc chloride and borate, phosphoric acid, dicyanodiamide, sodium borate and antimony oxide. These additives are generally introduced in a powder form and the quantity used is generally within the range 5–10 % of dry mass.
The dimensions of the fire retardant particles affect their effectiveness and the amount added, especially for the fire retardation of polymers [21, 192, 193]. Commonly used inert fillers for composite materials are silica, calcium carbonate, talc, glass fibre and other. They improve the fire retardancy and decrease smoke yield by mechanism of fuel dilution in the solid phase [194].
10.3.4.3 Paper and Paper Products
Flame-retardants recommended for paper and paper products have closely paralleled those suggested for other forms of cellulose, particularly wood. The most commonly used materials are ammonium sulfate and ammonium phosphates, with or without boric acid, but a large number of more sophisticated retardants have been suggested for various specialty applications [167].
There are several modes of adding flame-retardants to paper products, depending on three parameter: (a) the solubility of the compounds, (b) the paper product involved, and (c) the cost considerations.
References
Nishiyama, Y.: Structure and properties of the cellulose microfibril. J. Wood Sci 55(4), 241–249 (2009)
Agoudjil, N., et al.: Design and properties of biopolymer–silica hybrid materials: the example of pectin-based biodegradable hydrogels. Pure Appl. Chem. 84(12), 2521 (2012)
Alongi, J., et al.: Hybrid phosphorus-doped silica architectures derived from a multistep sol-gel process for improving thermal stability and flame retardancy of cotton fabrics. Polym. Degrad. Stab. 97(8), 1334–1344 (2012)
Alongi, J., et al.: Thermal and fire stability of cotton fabrics coated with hybrid phosphorus-doped silica films. J. Therm. Anal. Calorim. 110(3), 1207–1216 (2012)
Lindman, B., Karlström, G., Stigsson, L.: On the mechanism of dissolution of cellulose. J. Mol. Liq. 156(1), 76–81 (2010)
Medronho, B., Lindman, B.: Competing forces during cellulose dissolution: from solvents to mechanisms. Curr. Opin. Colloid Interface Sci. 19(1), 32–40 (2014)
Möller, M., Popescu, C.: Chapter 9.6: Natural fibres, in sustainable solutions for modern economies. In: Höfer, R. (ed.) The Royal Society of Chemistry, pp. 364–389. Academic Press, UK (2009)
Klemm, D., Schmauder, H.-P., Heinze, T.: Cellulose In: Baets, S.D., Vandamme, E.J., Steinbüchel, A. (eds.) Biopolymers: Biology, Chemistry, Biotechnology, Applications, Polysaccharide II pp. 275–319. Wiley-VCH, Weinheim (2002)
Wakelyn, P.J. et al.: Cotton fibers. In: Lewin, M., Pearce E.M. (eds.) Handbook of Fiber Chemistry, pp. 577–724. Marcel Dekker, Inc., New York (1998)
DeLanghe, E.A.L.: Cotton physiology. In: Mauney, J.R., Stewart, J.M. (eds.) The Cotton Foundation, pp. 325–349. Lint Development, Memphis (1986)
TutorVista: Polysaccharides Cellulose (2011). http://www.tutorvista.com/biology/polysaccharides-cellulose
Moran-Mirabal, J.M.: Advanced-microscopy techniques for the characterization of cellulose structure and cellulose-cellulase interactions. In: Ven, T.V.D., Godbout, L. (eds.) Cellulose—Fundamental Aspects (2013)
Berendjchi, A., Khajavi, R., Yazdanshenas, M.: Fabrication of superhydrophobic and antibacterial surface on cotton fabric by doped silica-based sols with nanoparticles of copper. Nanoscale Res. Lett. 6(1), 594 (2011)
Cheng, T., et al.: Fast response photochromic textiles from hybrid silica surface coating. Fibers Polym. 9(3), 301–306 (2008)
de Menezes, E.W., et al.: Ionic silica based hybrid material containing the pyridinium group used as an adsorbent for textile dye. J. Colloid Interface Sci. 378(1), 10–20 (2012)
Li, Y., Lo, L.Y., Hu, J.Y.: USA Patent (2007)
Liu, X., et al.: Photochromic textiles from hybrid silica coating with improved photostability. Res. J. Text. Appar 14(2), 1–8 (2010)
Toskas, G., et al.: Chitosan(PEO)/silica hybrid nanofibers as a potential biomaterial for bone regeneration. Carbohydr. Polym. 94(2), 713–722 (2013)
Hassabo, A.G.: Synthesis and Deposition of Functional Nano-Materials on Natural Fibres 2011, RWTH Aachen University, Aachen, p. 154 (2011)
Krässig, H.A.: Cellulose—structure, accessibility and reactivity. In: Huglin, M.B. (eds.) Polymer Monographs, pp. 1–375. Gordon and Breach Science Publishers, Yverdon, Switzerland (1993)
Kozlowski, R., Wladyka-Przybylak, M.: Uses of natural fibre reinforced plastics. In: Wallenberger, F.T., Weston, N.E. (eds.) Natural Fibres, Plastics and Composites, pp. 249–274. Kluwer Academic, Dordrecht, Boston (2004)
Kandola, B.K., et al.: Flame-retardant treatments of cellulose and their influence on the mechanism of cellulose pyrolysis. J. Macromol. Sci. C 36(4), 721–794 (1996)
Horrocks, A.R.: An introduction to the burning behaviour of cellulosic fibres. J. Soc. Dyers Colour. 99(7–8), 191–197 (1983)
Russell, L.J., et al.: Combining fire retardant and preservative systems for timber products in exposed applications—state of the art review. Australian Forest and Wood Products R & D Corporation, Victoria (2007)
Shafizadeh, F.: The chemistry of pyrolysis and combustion. In: Rowell, R. (ed.) The Chemistry of Solid Wood, pp. 489–529. American Chemical Society, Washington (1984)
LeVan, S.L., Winandy, J.E.: Effects of fire retardant treatments on wood strength: a review. Wood Fiber Sci. 22(1), 113–131 (1990)
Cottoninc: Cotton Nonwoven Technical Guide, 2011. http://www.cottoninc.com/Cotton-Nonwoven-Technical-Guide/
Mukherjee, P.S., Satyanarayana, K.G.: An empirical evaluation of structure-property relationships in natural fibres and their fracture behaviour. J. Mater. Sci. 21(12), 4162–4168 (1986)
Ferdous, D., et al.: Pyrolysis of lignins: experimental and kinetics studies. Energ. Fuels 16(6), 1405–1412 (2002)
Manfredi, L.B., et al.: Thermal degradation and fire resistance of unsaturated polyester, modified acrylic resins and their composites with natural fibres. Polym. Degrad. Stab. 91(2), 255–261 (2006)
Chapple, S. Anandjiwala, R.: Flammability of natural fiber-reinforced composites and strategies for fire retardancy: a review. J. Thermoplast. Compos. Mater. 23, 871–893 (2010)
Lewin, M., Basch, A.: Structure, pyrolysis and flammability of cellulose. In: Lewin, M., Atlas, S.M., Pearce, E.M. (eds.) Flame-retardant Polymeric Materials, pp. 1–41. Plenum Press, New York (1978)
Lewin, M.: Unsolved problems and unanswered questions in flame retardance of polymers. Polym. Degrad. Stab. 88(1), 13–19 (2005)
Basch, A., Lewin, M.: The influence of fine structure on the pyrolysis of cellulose. I. Vacuum pyrolysis. J. Polym. Sci. Polym. Chem. Ed. 11(12), 3071–3093 (1973)
Festucci-Buselli, R.A., Otoni, W.C., Joshi, C.P.: Structure, organization, and functions of cellulose synthase complexes in higher plants. Braz. J. Plant Physiol. 19(1), 1–13 (2007)
Shaw, L.H.: Cotton’s importance in the textile industry: the role of the ICAC. In: At a Symposium Lima, Peru. pp. 1–8 (1998)
van Zyl, W.H., et al.: Next-generation cellulosic ethanol technologies and their contribution to a sustainable Africa. Interface Focus 1, 196–211 (2011)
Kampalanonwat, P., Supaphol, P.: Preparation and adsorption behavior of aminated electrospun polyacrylonitrile nanofiber mats for heavy metal ion removal. Acs Appl. Mater. Interfaces 2(12), 3619–3627 (2010)
Karine, C., et al.: Morphology and mechanical behavior of a natural composite: the flax fiber. In: 16th International Conference on Composite Materials. Kyoto, Japan (2007)
Bos, H.L., Donald, A.M.: In situ ESEM study of the deformation of elementary flax fibres. J. Mater. Sci. 34(13), 3029–3034 (1999)
Wang, H.H., et al.: An improved fibril angle measurement method for wood fibres. Wood Sci. Technol. 34(6), 493–503 (2001)
Charlet, K., et al.: Characteristics of Hermès flax fibres as a function of their location in the stem and properties of the derived unidirectional composites. Compos. Appl. Sci. Manuf. 38(8), 1912–1921 (2007)
Ghosh, P., Samanta, A.K., Basu, G.: Effect of selective chemical treatments of jute fibre on textile-related properties and processibility. Indian J. Fibre Text. Res. 29(1), 85–99 (2004)
Wang, W.-M., Cai, Z.-S., Yu, J.-Y.: Study on the chemical modification process of jute fiber. J. Eng. Fibers Fabr. 3(2), 1–11 (2008)
Hattopadhyay, D.P.: Introduction, chemistry and preparatory processes of jute. Colourage 45(5), 23 (1998)
Pan, N.C., Day, A., Mahalanabis, K.K.: Chemical composition of jute and its estimation. Man Made Text. India 42, 467–473 (1999)
Lakkad, S.C., Patel, J.M.: Mechanical properties of bamboo, a natural composite. Fibre Sci. Technol. 14(4), 319–322 (1981)
Chen, Y.D., Qin, W.L.: The chemical composition of ten bamboo species. In: Rao, A.N., et al. (ed.) Recent Research on Bamboo. Proceedings of the International Bamboo Workshop, Hangzhou, China (1985)
Chaowana, P.: Bamboo: an alternative raw material for wood and wood-based composites. J. Mater. Sci. Res. 2(2), 90–102 (2013)
Cronier, D., Monties, B., Chabbert, B.: Structure and chemical composition of bast fibers isolated from developing hemp stem. J. Agric. Food Chem. 53(21), 8279–8289 (2005)
Wardrop, A.B.: Morphological factors involved in the pulping and beating of wood fibers. Sven. Papperstid 66, 231–247 (1963)
Sjostrom, E.: Wood Chemistry. Fundamentals and Applications, 2nd edn. Academic press, San Diego (1993)
Grayson, M.: Encyclopedia of Textiles Fibers and Nonwoven Fabrics, pp. 99–114, 188–211. Wiley Interscience, New York (1997)
Lewin, M., Atlas, S.A., Pearce, E.M.: Flame-Retardant Polymeric Materials, vol. 1, Plenum Press, New York (1978)
Mohamed, A.L.: Acrylamide Derivatives as Additives to Multifinishing Formulations of Cotton Fabrics, p. 378. Cairo, Egypt, Helwan University (2005)
Moon, S., Jo, B., Farris, R.J.: Flame resistance and foaming properties of NBR compounds with halogen-free flame retardants. Polym. Compos. 30(12), 1732–1742 (2009)
Zhang, S., Horrocks, A.R.: A review of flame retardant polypropylene fibres. Prog. Polym. Sci. 28(11), 1517–1538 (2003)
Nikolaeva, M., Kärki, T.: A review of fire retardant processes and chemistry, with discussion of the case of wood-plastic composites. Baltic For. 17(2), 314–326 (2011)
Norber, T., Blkales, M., Segal, L.: Cellulose and Cellulose Derivatives, vol. 5. Wiley Interscience, New York, (1976)
Lewin, M., Atlas, S.A., Pearce, E.M.: Flame-Retardant Polymeric Materials, vol. 2. Plenum Press, New York (1978)
Carraher, C.E.: Polymer Chemistry, 4th Edn. Marcel Dekker, New York (1996)
Tomasino, C.: Chemistry and Technology of Fabric Preparation and Finishing, p. 268. North Carolina State University, Raleigh, North Carolina (1992)
Hebeish, A., et al.: Modification of partially carboxymethylated cotton via crafting with acrylic acid and styrene using gamma radiation. J. Appl. Polym. Sci. 32(8), 6237–6257 (1986)
Kroschwitz, J.I.: Polymers: fibers and textile: a compendium. In: Encyclopedia Reprint Series, pp. 119–141, 433–482. Wiley, New York (1990)
Wolf, R., Kaul, B.L.: Plastics, additives. In: Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA (2000)
Sutker, B.J.: Flame retardants. In: Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim (2000)
Hirschler, M.M.: Chemical aspects of thermal decomposition of polymeric materials, In:Grand, A.F., Wilkie, C. (eds.) Fire Retardancy of Polymeric Materials, pp. 27–79. Marcel Dekker, New York, (2000)
Benbow, J.: Minerals in fire protection. Ind. Miner. 240, 61–73 (1987)
Green, J.: Mechanisms for flame retardancy and smoke suppression—a review. J. Fire Sci. 14(6), 426–442 (1996)
Schmidt, W.G.: Flame retardant additives in plastics and recent patents. Plast. Inst. (London), Trans. J. 33(108), 247–255
Pearce, E.M.: Flame retardants for polymer systems. Pure Appl. Chem. 58(6), 925–930 (1986)
Focke, W.W., Strydom, C.A., Bartie, N.: Thermal analysis of commercial inorganic flame retardants. S. Afr. Inst. Chem. Eng. 41–51 (1997)
Green, J.: 25 years of flame retarding plastics. J. Fire Sci. 15(1), 52–67 (1997)
Pettigrew, A.: Flame retardants, halogenated. In: Kirk-Othmer Encyclopedia of Chemical Technology, pp. 954–976. Wiley, New York (2000)
Gann, R.G.: Flame retardants, overview. In: Kirk-Othmer Encyclopedia of Chemical Technology. Wiley, New York (2000)
Gilman, J.W., et al.: Characterisation of flame retarded polymer combustion char by solid state 13C and 29Si NMR and EPR. Polym. Prepr. 38(1), 802–803 (1997)
Oulton, D.P.: Fire-retardant textiles. In: Carr, C.M. (ed.) Chemistry of the Textiles Industry, pp. 102–124. Springer, Netherlands (1995)
Rusznák, I., Tánczos, I.: Influence of short-term thermal pretreatment on the rate of acidolysis of cellulose. J. Polym. Sci. Polym. Symp. 42(3), 1475–1480 (1973)
Chatterjee, P.K.: Characterization of fibers by dynamic thermoacoustical analysis. J. Macromol. Sci. A Chem. 8(1), 191–209 (1974)
Horrocks, A.R., Anand, S.C., Sanderson, D.: Complex char formation in flame retarded fibre-intumescent combinations: 1. Scanning electron microscopic studies. Polymer 37(15), 3197–3206 (1996)
Zhu, P., et al.: A study of pyrolysis and pyrolysis products of flame-retardant cotton fabrics by DSC, TGA, and PY–GC–MS. J. Anal. Appl. Pyrol. 71(2), 645–655 (2004)
Price, D., et al.: Influence of flame retardants on the mechanism of pyrolysis of cotton (cellulose) fabrics in air. J. Anal. Appl. Pyrol. 40–41, 511–524 (1997)
Lecoeur, E., et al.: Flame retardant formulations for cotton. Polym. Degrad. Stab. 74(3), 487–492 (2001)
Kandola, B.K., Horrocks, S., Horrocks, A.R.: Evidence of interaction in flame-retardant fibre-intumescent combinations by thermal analytical techniques. Thermochim. Acta 294(1), 113–125 (1997)
Radlein, D., Piskorz, J., Scott, D.S.: Fast pyrolysis of natural polysaccharides as a potential industrial process. J. Anal. Appl. Pyrol. 19, 41–63 (1991)
Madorsky, S.L., Hart, V.E., Straus, S.: Thermal degradation of cellulosic materials. J. Res. Nat. Bur. Stan. 60(4), 343 (1958)
Schuyten, H.A., Weaver, J.W., Reid, J.D.: Advances in Chemistry, vol. 9. Academic Press, New York (1954)
Rodrig, H., Basch, A., Lewin, M.: Crosslinking and pyrolytic behavior of natural and man-made cellulosic fibers. J. Polym. Sci. Polym. Chem. Ed. 13(8), 1921–1932 (1975)
Horrocks, A.R., Price, D.: Fire Retardant Materials (2001) Abington Hall, Abington, Cambridge CB1 6AH, England: Woodhead Publishing Limited
Troitzsch, J.H., Overview of Flame Retardants. Chimica Oggi/chemistry Today 16(January/February) (1998)
Parks, W.G., et al.: Mechanism of pyrolytic decomposition of cellulose. In: 127th Meetting of American Chemical Society (1955)
Jolles, Z.E.: Bromine and its compounds. Ernest Benn Ltd., London (1966)
Barker, R.H., Drews, M.J.: Development of Flame Retardants for Polyester/Cotton Blend. NBS—GRC—ETIP, pp. 59–77 (1976)
Kuryla, W.C., Papa, A.J.: Flame Retardant Science and Technology of Polymeric Materials. Marcel Dekker, New York (1979)
Nevell, T.P., Zeronian, S.H.: Cellulose chemistry and its applications. Ellis Horwood, John Wiley & Sons, Ltd, Chichester (1985)
Weil, E.D.: Synergists, adjuvants, and antagonists in flame-retardant systems. In: Wilkie, C.A., Grand, A.F. (eds.) Fire Retardancy of Polymeric Materials. New York, Marcel Dekker Inc. (2000)
Cornell, J.A.: Experiments with Mixtures: Designs, Models, and the Analysis of Mixture Data, 3rd Edn. Wiley, New York (2002)
Khanna, Y.P., Pearce, E.: Synergism and flame retardancy. Lewin, M., Atlas, S.M., Pearce E.M. (eds.) In: Flame—Retardant Polymeric Materials, pp. 43–61. Springer, New York (1978)
Horacek, H., Grabner, R.: Advantages of flame retardants based on nitrogen compounds. Polym. Degrad. Stab. 54(2–3), 205–215 (1996)
Waly, A.I., et al.: Special finishing of cotton to impart flame-retardancy, easy care finishing and antimicrobial properties. Res. J. Text. Apparel 13(3), 10–26 (2009)
Barker, R.H., Hendrix, J.E.: Flame Retardancy of Polymeric Materials. In: Kuryla, W.C., Papa, A.V. (eds.) pp. 39–78. Marcel Dekker, New York (1979)
Little, R.W.: Flame Proofing Textile Fabrics. Reinhold Publishing Corporation, New York (1947)
Reeves, W.A., et al.: Some chemical and physical factors influencing flame retardancy. Text. Res. J. 40(3), 223–231 (1970)
Weil, E.D.: Flame Retardancy of Polymeric Materials. In: Kuryla, W.C., Papa, A.V. (eds.) pp. 185–241. Marcel Dekker, New York (1979)
Davies, D., Horrocks, A.R.: Nitrogen–phosphorous antagonism in flame-retardant cotton. J. Appl. Polym. Sci. 31(6), 1655–1662 (1986)
Carty, P., Byrne, M.S.: The Chemical and Mechanical Finishing of Textile Materials, 2nd Edn. UNN Commercial Enterprises, Ltd (1987)
Troitzsch, J.H.: International Plastics Flammability Handbook: Principles Regulations, Testing and Approval, 2nd edn. Hanser publishers, Munch Germany (1990)
Green, J.: A review of phosphorus-containing flame retardants. J. Fire Sci. 10(6), 470–487 (1992)
Touval, T.: Antimony and other inorganic flame retardants. In: Krik-Othmer (ed.) Encyclopaedia of Chemical Technology. pp. 936–954, Wiley, New York (1993)
Weil, E.D.: Phosphorus Flame Retardants. In: Kirk-Othmer Encyclopedia of Chemical Technology, p. 559–570. Wiley, New York (2000)
Hebeish, A., Guthrie, J.T.: The Chemistry and Technology of Cellulose Copolymers. Springer, Berlin (1981)
Reeves, W.A., Marquette, Y.B.: Lightweight, durable-press cotton and polyester/cotton with ignition resistance. Text. Res. J. 49(3), 163–169 (1979)
Nair, G.P.: Flammability in Textiles and Routs to Flame Retardant Textiles- V Colourge 47(12), 31–34 (2000)
Nair, G.P.: Flammability in Textiles and Routs to Flame Retardant Textiles - XIII Colourge 48(9), 39–43 (2001)
Nair, G.P.: Flammability in Textiles and Routs to Flame Retardant Textiles - XII Colourge 48(8), 43–47 (2001)
Bullock, J.B., Welch, C.M., Guthrie, J.D.: The weathering characteristics of lightweight flame-retardant finishes for cotton fabrics. Text. Res. J. 34(8), 691–700 (1964)
Bullock, J.B., Welch, C.M.: Weathering durability of cotton fabrics treated with APO-THPC flame retardant. Text. Res. J. 36(5), 441–451 (1966)
Nair, G.P.: Flammability in Textiles and Routs to Flame Retardant Textiles - X Colourge 48(6), 37–41 (2001)
Holme, I.: Int. Dyer 184(6), 17 (1999)
Barber, A.J.: Int. Dyer 186(2), 29 (2001)
Nair, G.P.: Flammability in Textiles and Routs to Flame Retardant Textiles - XVII Colourage 49(3), 49–52 (2002)
Nair, G.P.: Flammability in Textiles and Routs to Flame Retardant Textiles - XVIII Colourage 49(4), 29–34 (2002)
Stowell, J.K., Yang, C.Q.: A durable low-formaldehyde flame retardant finish for cotton fabrics. AATCC Rev. 3(2), 17–20 (2003)
Waly, A.I., et al.: Processes of dyeing, finishing and flame retardancy of cellulosic textiles in the presence of reactive tertiary amines. Res. J. Text. Apparel 16(3), 66–84 (2012)
Rothon, R.N.: Particulate-filled polymer composites. The University of Michigan: Longman Scientific and Technical,—Science (1995)
Ekebafe, L.O.: Improving the flammability of polymeric materials: a review. J. Chem. Soc. Niger. 34(1), 128–137 (2009)
Day, J.E.: Preparation and catalytic oxidation of pure amorphous carbon. Ind. Eng. Chem. 28(2), 234–238 (1936)
Delfosse, L., et al.: Combustion of ethylene-vinyl acetate copolymer filled with aluminium and magnesium hydroxides. Polym. Degrad. Stab. 23(4), 337–347 (1989)
Baillet, C., Delfosse, L.: The combustion of polyolefins filled with metallic hydroxides and antimony trioxide. Polym. Degrad. Stab. 30(1), 89–99 (1990)
Eboatu, A.N., Rades, J., Shehu, D.: Flame retardant treatment of some roofing thatch. Niger. J. Renew. Energ. 3(1–2), 91–94 (1992)
Iji, M., Serizawa, S.: Silicone derivatives as new flame retardants for aromatic thermoplastics used in electronic devices. Polym. Adv. Technol. 9(10–11), 593–600 (1998)
Kambour, R.P., Klopfer, H.J., Smith, S.A.: Limiting oxygen indices of silicone block polymer. J. Appl. Polym. Sci. 26(3), 847–859 (1981)
Zaikov, G.E., Lomakin, S.M.: New aspects of ecologically friendly polymer flame retardant systems. Polym. Degrad. Stab. 54(2–3), 223–233 (1996)
Allen, N.S., et al.: Entrapment of stabilisers in silica: I. Controlled release of additives during polypropylene degradation. Polym. Degrad. Stab. 56(2), 125–139 (1997)
Chao, T.C.: et al.: In: 43rd International SAMPE Symposium. Anaheim (1998)
National Technical Information Service (NTIS) (2004) Fire-Safe Polymers and Polymer Composites. September 2004, Final Report, Office of Aviation Research, Springfield, Virginia, Washington
Mohamed, A.L., El-Sheikh, M.A., Waly, A.I.: Enhancement of flame retardancy and water repellency properties of cotton fabrics using silanol based nano composites. Carbohydr. Polym. 102, 727–737 (2014)
Low, J.E., Levalois-Grützmacher, J.: Investigation of synergistic effects of phosphorus, nitrogen and silicon in the flame retardancy of cellulose-based cotton textiles processed by plasma-induced graft polymerization. In: ISPC 21–21st International Symposium on Plasma Chemistry. Cairns Convention Centre, Australia (2013)
Giannelis, E.P.: Polymer layered silicate nanocomposites. Adv. Mater. 8(1), 29–35 (1996)
Gilman, J.W., et al.: Flammability properties of polymer—layered-silicate nanocomposites. polypropylene and polystyrene nanocomposites. Chem. Mater. 12(7), 1866–1873 (2000)
Levchik, S.V., et al.: Mechanistic study of combustion performance and thermal decomposition behaviour of nylon 6 with added halogen-free fire retardants. Polym. Degrad. Stab. 54(2–3), 217–222 (1996)
Wilkie, C.A.: Poly(sodium styrenesulfonate): a self intumescent material. In: 10th Conference Recent Advances in Flame Retardancy of Polymeric Materials. Stamford, Connecticut, USA (1999)
Ebdon, J.R., et al.: The effects of some transition-metal compounds on the flame retardance of poly(styrene-co-4-vinyl pyridine) and poly(methyl methacrylate-co-4-vinyl pyridine). Polym. Degrad. Stab. 60(2–3), 401–407 (1998)
Rothon, R.N., Hornsby, P.R.: Flame retardant effects of magnesium hydroxide. Polym. Degrad. Stab. 54(2–3), 383–385 (1996)
Brown, N.: New Aluminium hydroxides for printed circuit boards and public transport application. In: 10th Conference Recent Advances in Flame Retardancy of Polymeric Materials. Stamford, Connecticut, USA (1999)
Reyes, J.D., et al.: Flame Retardants for Enhanced Propeties in Polyolefins. In: 10th Conference Recent Advances in Flame Retardancy of Polymeric Materials. Stamford, Connecticut, USA (1999)
Shen, K.K., Ferm, J.: Maximizing fire test performances of polyolefins with zinc borate. In 10th Conference Recent Advances in Flame Retardancy of Polymeric Materials. Stamford, Connecticut, USA (1999)
Kuma, K., et al.: Flame retardant and flame retardant resin composition formulated with the same. In: US Patent. USA (1999)
Siegel, H., Eggersdorfer, M.: Ketones. In: Gerhartz, W. (eds.) Ullmann’s Encyclopedia of Industrial Chemistry, pp. 77–95. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (2000)
Birnbaum, L.S., Staskal, D.F.: Brominated flame retardants: cause for concern? Environ. Health Perspect. 112(1), 9–17 (2004)
Lomakin, S.M., Zaikev, G.E.: Polymer Flame Retardancy. VSP Publishers, Netherlands (2003)
Fried, J.R.: Polymer Science and Technology, 2nd edn. Prentice-Hall, New Delhi (2003)
Bouton, T.C.: N-Containing flame retardants: an alternative handout at OECD workshop. In: Brominated Flame Retardants, Flame Retardant, pp. 22–25. Neuchatel Switzerland (1997)
Grabner, R.S.: N-Containing flame retardants: an alternative handout at OECD workshop. In: Brominated Flame Retardants, Flame Retardants, pp. 22–25. Neuchatel Switzerland (1993)
ASTM Standard Test Method C33 (ASTM E84-13A).: Standard Test Method for Surface Burning Characteristics of Building Materials, in Institution. ASTM International, West Conshohocken, PA (2001)
ASTM Standard Test Method C33 (ASTM E–662).: Standard Test Methods for Optical Density of Smoke Generated by Solid Materials, in Institution. ASTM International, West Conshohocken, PA (2011)
Nelson, G.L.: Materials and tests for hazard prevention. In: Nelson G.L. (ed.) Fire and Polymers II, American Chemical Society, pp. 1–26. Washington, D.C (1995)
ASTM Standard Test Method C33 (ASTM D–2863–00).: Standard Test Method for Measuring the Minimum Oxygen Concentration to Support Candle-Like Combustion of Plastics (Oxygen Index), In Institution. ASTM International, West Conshohocken, PA (2011)
AATCC Test Method (1934–1969).: Vertical burning test method in fire resistance of textile fabrics. Technical Manual Method of the American Association of Textile Chemists and Colorists, USA. p. 201–202 (1972)
5906–CCC–T–191b.: Horizontal Spread of Flame Test Method. U.S. Dept. of Commerce, Federal Specification, USA (1951)
AATCC Test Method (1932–1962): Flammability of Clothing Textiles, pp. 203–206. Technical Manual Method of the American Association of Textile Chemists and Colorists, USA (1972)
Pads, S.F.F.O.M.A.M.: Test manual for flammability of mattresses and mattress pads resistance of a mattress or mattress pad when exposed to a lighted cigarette. Consum. Prod. Saf. Comm. Eng. Sci. (1991)
Federal Specification DDD-C-95.: Pill test, carpets and rugs, wool, nylon, acrylic, modacrylic (1972)
Standard Test Method: Smoke Test. National Bureau of Standards, NBS (1973)
ASTM Standard Test Method C33 (ASTM E119-00a): Standard test methods for fire tests of building construction and material. In: Institution. ASTM International, West Conshohocken, PA (2001)
Bishop, D.M., Bottomley, D., Zobel, F.G.R.: Fire-retardant paints. J. Oil Colour Chem. Assoc. 66(12), 373–396 (1983)
Tesoro, G.C.: Chemical modification of polymers with flame-retardant compounds. J. Polym. Sci. Macromol. Rev. 13(1), 283–353 (1978)
Waly, A., et al.: Especial finishing of cotton to impart flame-retardancy and easy care finishing. In: 3rd International Conference of Textile Research Division, NRC; Textile Processing: State of the Art and Future Developments, pp. 515–528. Cairo, Egypt (2006)
Waly, A., et al.: Process of single—bath dyeing, finishing and flam—retarding of cellulosic textiles in presence of reactive tertiary amines. In: 3rd International Conference of Textile Research Division, NRC; Textile Processing: State of the Art and Future Developments, pp. 529–543. Cairo, Egypt (2006)
Gaan, S., Sun, G.: Effect of phosphorus flame retardants on thermo-oxidative decomposition of cotton. Polym. Degrad. Stab. 92(6), 968–974 (2007)
Gaan, S., Sun, G.: Effect of phosphorus and nitrogen on flame retardant cellulose: a study of phosphorus compounds. J. Anal. Appl. Pyrol. 78(2), 371–377 (2007)
Waly, A., et al.: Flame retarding, easy care finishing and dyeing of cellulosic textiles in one bath. Egypt. J Text. Polym. Sci Technol. 12(2), 101–131 (2008)
Chruściel, J.J., Leśniak, E.: Modification of thermoplastics with reactive silanes and siloxanes in thermoplastic elastomers. In: El-Sonbati, P.A. (ed.) Tech: Europe University Campus STeP Ri, Slavka Krautzeka 83/A: Rijeka, Croatia (2012)
Mark, H.F., Atlas, S.M., Cernia, E.: Man-Made Fibers Science and Technology, vol. 2, 3. Wiley, New York (1986)
Heidari, S., Kallonen, R.: Hybrid fibres in fire protection. Fire Mater. 17(1), 21–24 (1993)
Tesoro, G.C., Meiser, C.H.: Some effects of chemical composition on the flammability behavior of textiles. Text. Res. J. 40(5), 430–436 (1970)
Puglia, D., Biagiotti, J., Kenny, J.M.: A review on natural fibre-based composites—Part II. J. Nat. Fibers 1(3), 23–65 (2005)
Blicblau, A.S., Coutts, R.S.P., Sims, A.: Novel composites utilizing raw wool and polyester resin. J. Mater. Sci. Lett. 16(17), 1417–1419 (1997)
Kandola, B.K., Kandare, E.: Composites having improved fire resistance. In: Horrocks, A.R., Price, D. (eds.) Advances in Fire Retardant Materials, Woodhead Publishinge, Cambridge (2008)
Mark, H.F., et al.: Combustion of polymers and its retardation. In: Lewin, M., Atlas, S.M., Pearce, E.M. (eds.) Flame-retardant Polymeric Materials, pp. 1–17. Plenum Press, New York (1975)
Biagiotti, J., Puglia, D., Kenny, J.M.: A review on natural fibre-based composites-Part I. J. Nat. Fibers 1(2), 37–68 (2004)
Lowden, L., Hull, T.: Flammability behaviour of wood and a review of the methods for its reduction. Fire Sci. Rev. 2(1), 1–19 (2013)
Kim, H.-S., et al.: Thermal properties of bio-flour-filled polyolefin composites with different compatibilizing agent type and content. Thermochim. Acta 451(1–2), 181–188 (2006)
Yang, H., et al.: In-depth investigation of biomass pyrolysis based on three major components: hemicellulose. Cellul. Lignin Energ. Fuels 20(1), 388–393 (2005)
Pabeliña, K.G., Lumban, C.O., Ramos, H.J.: Plasma impregnation of wood with fire retardants. Nucl. Instrum. Methods Phys. Res. Sect. B 272, 365–369 (2012)
Hakkarainen, T., et al.: Innovative Eco-Efficient High Fire Performance Wood Products For Demanding Applications. SP Tratek, Sweden; KTH Biotechnology: VTT, pp. 1–47. Finland, Sweden (2005)
Browne, F.L.: Theories of the Combustion of Wood and its Control—A Survey of the Literature. Forest Products Laboratory, Madison (1958)
Wang, G.-A., et al.: Characterizations of a new flame-retardant polymer. Polym. Degrad. Stab. 91(12), 3344–3353 (2006)
Borysiak, S., Paukszta, D., Helwig, M.: Flammability of wood–polypropylene composites. Polym. Degrad. Stab. 91(12), 3339–3343 (2006)
Kozłowski, R., Władyka-Przybylak, M.: Flammability and fire resistance of composites reinforced by natural fibers. Polym. Adv. Technol. 19(6), 446–453 (2008)
Bourbigot, S., Duquesne, S.: Fire retardant polymers: recent developments and opportunities. J. Mater. Chem. 17(22), 2283–2300 (2007)
Sain, M., et al.: Flame retardant and mechanical properties of natural fibre–PP composites containing magnesium hydroxide. Polym. Degrad. Stab. 83(2), 363–367 (2004)
Stark, N.M., et al.: Evaluation of various fire retardants for use in wood flour–polyethylene composites. Polym. Degrad. Stab. 95(9), 1903–1910 (2010)
Mouritz, A.P., Gibson, A.G.: Fire Properties of Polymer Composite Materials. Solid Mechanics and Its Applications, vol. 143. Springer (2006)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Mohamed, A.L., Hassabo, A.G. (2015). Flame Retardant of Cellulosic Materials and Their Composites. In: Visakh, P., Arao, Y. (eds) Flame Retardants. Engineering Materials. Springer, Cham. https://doi.org/10.1007/978-3-319-03467-6_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-03467-6_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-03466-9
Online ISBN: 978-3-319-03467-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)