
Abstract
Iron is of great importance for many metabolic processes since the redox potential between its two valence states Fe2+ and Fe3+ lies within the range of physiological processes. Actually, iron is not a rare element, it is fourth in abundance in the earth crust, but it is not readily available for microorganisms. In the soil ferric oxide hydrates are formed at pH values around seven and the concentration of free Fe3+ is at best 10−17 mol/dm3 while about 10−6 mol/dm3 would be needed. In living organisms iron is usually strongly bound to peptidic substances such as transferrins. To increase the supply of soluble iron microorganisms other than those living in an acidic habitat may circumvent the problem by reduction of Fe3+ to Fe2+ (182), which seems to be of major importance for marine phytoplankton (151); see also amphiphilic marine bacteria (Sect.2.8) and Fe2+ binding ligands (Sect. 7) below. An important alternative is the production of Fe3+ chelating compounds, so-called siderophores. Siderophores are secondary metabolites with masses below 2,000 Da and a high affinity to Fe3+. Small iron-siderophore complexes can enter the cell via unspecific porins, larger ones need a transport system that recognizes the ferri-siderophore at the cell surface. In the cell, iron is released mostly by reduction to the less strongly bound Fe2+ state (137), and the free siderophore is re-exported (“shuttle mechanism”); for a modified shuttle system see pyoverdins (Sect. 2.1) and amonabactins (Sect. 2.7). Rarely the siderophore is degraded in the periplasmatic space as, e.g. enterobactin (Sect. 2.7). Alternatively Fe3+ is transferred at the cell surface from the ferri-siderophore to a trans-membrane transport system (“taxi mechanism”). A probably archaic and unspecific variety of the taxi mechanism comprises the reduction of Fe3+ at the cell surface (see ferrichrome A, Sect. 2.6 (99, 105)). The terms “shuttle” and “taxi mechanism” were coined by Raymond and Carrano (296).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Introduction
Iron is of great importance for many metabolic processes since the redox potential between its two valence states Fe2+ and Fe3+ lies within the range of physiological processes. Actually, iron is not a rare element, it is fourth in abundance in the earth crust, but it is not readily available for microorganisms. In the soil ferric oxide hydrates are formed at pH values around seven and the concentration of free Fe3+ is at best 10−17 mol/dm3 while about 10−6 mol/dm3 would be needed. In living organisms iron is usually strongly bound to peptidic substances such as transferrins. To increase the supply of soluble iron microorganisms other than those living in an acidic habitat may circumvent the problem by reduction of Fe3+ to Fe2+ (182), which seems to be of major importance for marine phytoplankton (151); see also amphiphilic marine bacteria (Sect. 2.8) and Fe2+ binding ligands (Sect. 7) below. An important alternative is the production of Fe3+ chelating compounds, so-called siderophores. Siderophores are secondary metabolites with masses below 2,000 Da and a high affinity to Fe3+. Small iron-siderophore complexes can enter the cell via unspecific porins, larger ones need a transport system that recognizes the ferri-siderophore at the cell surface. In the cell, iron is released mostly by reduction to the less strongly bound Fe2+ state (137), and the free siderophore is re-exported (“shuttle mechanism”); for a modified shuttle system see pyoverdins (Sect. 2.1) and amonabactins (Sect. 2.7). Rarely the siderophore is degraded in the periplasmatic space as, e.g. enterobactin (Sect. 2.7). Alternatively Fe3+ is transferred at the cell surface from the ferri-siderophore to a trans-membrane transport system (“taxi mechanism”). A probably archaic and unspecific variety of the taxi mechanism comprises the reduction of Fe3+ at the cell surface (see ferrichrome A, Sect. 2.6 (99, 105)). The terms “shuttle” and “taxi mechanism” were coined by Raymond and Carrano (296).
A microbial strain may produce more than one siderophore. There are variations in fatty acid chains of a lipophilic part or in the amino acids making up the backbone, as well as released intermediates of the biosynthetic chain. These variations belong all to the same structural pattern. However, there is also the possibility that so-called secondary siderophores are encountered. They constitute a different structural type, usually less complex in their constitution but also less efficient in binding Fe3+ than the primary ones. Secondary siderophores will be produced when the demand for iron is not so severe or in case there is a genetic defect impeding the production of the primary ones. Examples will be found throughout the review.
Obviously siderophores can be potent virulence factors of pathogenic bacteria. Siderophores in many cases have elaborate structures providing recognition only by the receptor site of the producing species. This renders a pirating by competing microorganisms more difficult. The structural specificities of siderophores have been used for classification purposes of bacterial species (see especially pyoverdins, Sect. 2.1).
Whether a Fe3+ binding metabolite is actually involved in the iron transport has not always been established firmly. Criteria are the pronounced production under iron starvation and growth after feeding, or labeling studies (at best simultaneously of Fe3+ and of the ligand, see, e.g. parabactin, Sect. 3.2, and schizokinen, Sect. 4.1). Chelators whose function is uncertain will be included in this review with an explanatory remark. Incompletely characterized siderophores will be mentioned when at least some structural elements have been identified. However, the mere statement that color reactions for catecholates (8a) or hydroxamates (9a) were positive will not be sufficient. Not included will be the sideromycins, conjugates of siderophores with antibiotically active residues, produced mainly by Streptomyces spp., which use the iron transport paths for “Trojan Horse” strategies. For further references see (34, 97, 187).
Due to its high charge density, small ion radius, and low polarizability, Fe3+ is a hard Lewis acid and can bind strongly to hard Lewis bases such as oxide ions. It forms octahedral d5 high spin complexes providing six coordination sites, which can accommodate three bidentate ligands. The major ligands types are catecholates, hydroxamates, and α-hydroxy carboxylates; other ligands are encountered occasionally. Siderophores containing different ligand types are not uncommon. Three bidentate ligands are often connected by aliphatic segments keeping them in place for complexation. This results in an entropic advantage over three non-connected ligands. Siderophores containing only two binding sites form either (Fe3+)2Lig3 complexes or the remaining two octahedral loci accommodate some external ligand (see, e.g. pyochelin, Sect. 5). For (Fe3+)2Lig3 structures with three bridges or with one bridging ligand have been discussed. The latter variety has been proven for alcaligin (Sect. 3.3). Three bidentate ligands can be arranged around the Fe3+ nucleus in two ways forming a left-handed or a right-handed screw, designated as Λ or Δ. Three identical ligands can point all in the same direction (cis) or one of them is reversed (trans). The chiral arrangement of the Fe3+ complex can be determined by X-ray analysis or can be deduced from the sign of the broad CD extremum at ca. 500 nm correlated with the metal-to-ligand charge transfer band. A positive Δε indicates a Λ configuration.
Ga3+ complexes are frequently analyzed for two reasons. Ga3+ also forms octahedral structures and it has almost the same ion radius as Fe3+ (62 vs. 65 pm). In contrast to Fe3+ it is diamagnetic and its complexes are therefore amenable to NMR analysis. Also in contrast to Fe3+ it cannot be reduced and therefore it is used for uptake studies interested in the fate of the complex in the cell.
Siderophores can be classified by different criteria. In this review related structural types will be grouped together. Some arbitrariness cannot be avoided due to the occurrence of “mixed types”. Cross-references will then be given. Trivial names have either been given to the free ligands or to their iron complexes. In the latter case the free ligands are referred to as “desferri” or “deferri” (sometimes in a shortened form as “desferrioxamines” for “desferriferrioxamines”) or as “pro” (see Sect. 3.3). Occasionally the name applied originally to the iron complex was used later for the free ligand (e.g. ferribactins, Sect. 2.1). These variations should be kept in mind when literature search programs are used.
For earlier compilations of siderophores see (97, 255a, 276), specifically for fungal siderophores (157a, 300, 383), and for biosynthesis pathways (19, 63, 403). Reviews for specific classes of siderophores will be mentioned where applicable.
2 Peptide Siderophores
In this group the ligands are incorporated in a peptide chain usually containing both d- (underlined in the structural formulas below) and l-configured amino acids. Frequently the two ends of the peptide chain are blocked by the formation of cyclic structures or otherwise. This prevents the degradation by proteolytic enzymes. Non-proteinogenic amino acids are encountered (homoserine, Hse; ornithine, Orn; 2,4-diaminobutyric acid, Dab; 2,3-dehydrobutyric acid, Dhb), lysine and ornithine may be incorporated in the chain by their ε/δ- rather than by their α-amino group, and amino acids may be modified to form ligand sites (3-hydroxy-aspartic acid, OHAsp; 3-hydroxy-histidine, OHHis; N 5-acyl-N 5-hydroxy-ornithine, acylOHOrn; N-hydroxy-cyclo-ornithine, i.e. 3-amino-1-hydroxy-piperidone-2, cOHOrn). Diaminobutyric acid frequently condenses with the preceding amino acid (Chart 1) giving a tetrahydropyrimidine ring (116). These condensation products are indicated below by a parenthesis as e.g. (Hse-Dab) in azoverdin (Sect. 2.2).


Condensation of a Dab residue with the preceding amino acid residue
2.1 Pyoverdins and Related Siderophores from Pseudomonas spp.
The most thoroughly investigated representatives are the pyoverdins, also spelled pyoverdines and occasionally named pseudobactins (353), produced by the fluorescent members of the genus Pseudomonas. For reviews see (44, 231); for a detailed study of the siderophores of this genus (37). Pyoverdins consist of three distinct structural parts, a chromophore (Chra) (Fig. 1, a), a peptide chain comprising six to twelve amino acids, and a dicarboxylic acid (succinic acid – Suc-, malic, glutamic, and 2-oxoglutaric acid) or monoamides (succinamide, malamide). For glutamic and 2-oxoglutaric acid the binding to the chromophore by their γ-carboxyl groups has been established by chemical degradation (124). For malic acid some not really convincing and partially contradictory NMR arguments have been advanced (319) for the binding by the carboxyl group neighboring the CH2 group. Recently mass spectrometric arguments were reported suggesting a binding via the other carboxyl group (45).


Chromophore types: (a) pyoverdin, (b) isopyoverdin, (c) ferribactin, (d) azotobactin
The about fifty pyoverdins for which structures have been proposed can be divided into three structural types exemplified by the three pyoverdins of Pseudomonas aeruginosa (234) (Fig. 2), viz. pyoverdins (a) with a C-terminal tri- or tetra-cyclopeptidic substructure (lactam formation between the C-terminal carboxyl group and an in-chain lysine or ornithine), e.g. ATCC 15692 (PAO1) (1), (b) with a C-terminal cOHOrn, e.g. ATCC 27853 (2), and (c) with a C-terminal free carboxyl group, e.g. Pa6 (R) (3). The free carboxyl group is probably the hydrolysis product of a depsipeptidic substructure (ester formation between the C-terminal carboxyl group and an in-chain serine or threonine). In several cases both the cyclic and the hydrolyzed open-chain form were found (e.g. (50)). Binding sites for Fe3+ are the catecholate part of the chromophore Chra and two units in the peptide chain, hydroxamate (acylOHOrn, cOHOrn) and/or α-hydroxycarboxylate (OHAsp, OHHis).


Pyoverdins from Pseudomonas aeruginosa representing the three structural types
Complete structural analysis requires mass spectral and NMR data as well as chemical degradation and analysis of the chirality of the constituent amino acids, determination of the mode of linkage of lysine (α- or ε-), the size of the cyclopeptide or cyclodepsipeptide ring, etc. (37). In some cases structures have been proposed based only on mass spectral data. Difficulties arising in this approach were discussed (44). To determine the three-dimensional structure an X-ray analysis (Plate 1) so far only of the Fe3+ complex of the pyoverdin B10 (4) was performed (353).


X-ray structure (stereo view) of ferri-pyoverdin B10 (ferri-4)
As an alternative strategy the investigation of the isomorphic Ga3+ complexes by NMR analysis was developed (241) for the pyoverdin GM-II (5) and extended to other pyoverdins, e.g. PL8 (16) (Plate 2) (H. Budzikiewicz, unpublished).


Calculated three-dimensional structure of the Ga3+-complex of pyoverdin PL8 (without side chain) Chra-Lys-acetylOHOrn-Ala-Gly-aThr-Ser-cOHOrn
In all cases the metal ion was found to lie at the surface of the complex. This facilitates its uptake and release. For the pyoverdins both Λ and Δ arrangements have been reported (37).
Iron transport through the cell membrane follows a modified shuttle mechanism. Evidence has been presented that the iron-free siderophore of P. aeruginosa PAO1 (1, Fig. 2 ) binds strongly to the receptor protein (67, 314a). This suggests two scenarios for the subsequent steps of iron transfer, an exchange of the ligands or a transfer or Fe3+ between them. By 3H- and 55Fe-labeling as well as fluorescence studies it was shown that an exchange between the approching ferri-pyoverdin and the bound iron-free pyoverdin occurs and that the former one enters the cell, i.e. that no Fe3+ exchange between the two ligands takes place (314b). Model studies with Aeromonas (Sect. 2.7) demonstrated there the iron-exchange variety. Binding of the iron-free siderophore to the receptor protein seems to be a common feature of the transport systems of P. aeruginosa and Escherichia coli (145a).
The peptide chains of the pyoverdins are responsible for the recognition of the ferri-siderophore at the cell surface of the producing species. It is usually highly strain specific. Cross-recognition between two strains is only observed when structurally closely related pyoverdins are produced (125, 233). An exception seems to be P. aeruginosa ATCC 15692, which besides its own ferripyoverdin (Fe-1), accepts several foreign ones (128a).
Without going into structural details, the pyoverdins stemming from the saprophytic group Pseudomonas aeruginosa/fluorescens/putida contain either two hydroxamic acid units or one hydroxamic and one α-hydroxycarboxylic acid, and those from the phytopathogens P. syringae etc. two α-hydroxycarboxylic acids. Structural differences of pyoverdins have been used recently to characterize species newly defined by breaking up the classical cluster of P. fluorescens/putida (e.g. (229, 231)). A listing of all pyoverdins from Pseudomonas spp. for which structural data have been published up to December 2009 is contained in the Appendix.
Pyoverdin-like siderophores with other chromophores have also been observed (see Fig. 1) (45). The 5,6-dihydropyoverdins (Chra without the 5,6-double bond) and the ferribactins (Chrc) are considered to be biogenetic precursors of the pyoverdins (318) (the term “ferribactin” was originally used for the Fe3+ complex (221) and later for the free ligand). An azotobactin chromophore (Chrd, see also below Sect. 2.2) is occasionally found in Pseudomonas isolates (e.g. (146)). Siderophores produced by a specific Pseudomonas strain but differing in the chromophore always have identical peptide chains.
Isopyoverdins contain the siderophore Fig. 1, Chrb with aspartic acid as the first amino acid. They have been encountered so far only in isolates from Pseudomonas putida strains, e.g. BTP1 (168) (6).
2.2 Azomonas and Azotobacter Siderophores
For a detailed discussion of this class of compounds see (37).
Azomonas macrozytogenes produces a siderophore with an isopyoverdin chromophore, azoverdin, but with a peptide chain 7 related to those of azotobactins, viz. (236).
The three-dimensional structure of the Ga3+ complex was determined by NMR techniques as outlined above. Also here the metal ion lies at the surface of the complex (377).
From Azotobacter vinelandii the structures of two siderophores were elucidated. They contain the chromophore Chrd (Fig. 1) and Hse units: azotobactin 87-I (8) (from the three Hse in this sequence two are l and one d configured) from the strain ATCC 12837 (314), and azotobactin D (9) (76) from the strain CCM 289.
Both of them are accompanied by compounds where the C-terminal Hse forms a γ-lactone ring (azotobactin 87-II and δ). An azotobactin O for which also a structure had been proposed (120) was shown later to be identical with azotobactin D (272). For secondary metabolites see protochelin and its constituents (Sect. 3.2).
Azotobacter chroococcum produces ornithine-containing hydroxamate siderophores with molecular masses 800 and 844 Da (difference of one carboxyl group?) of unknown structure (115a).
2.3 Anachelin
Cyanobacteria were probably the first organisms to perform oxygenic photosynthesis resulting eventually in the oxidation of environmental Fe2+ to Fe3+ with all its consequences. To cope with this problem the production of siderophores was initiated. Not much is known about the siderophores of cyanobacteria. Schizokinen (see below under citrate siderophores, Sect. 4.1) (326) found to be produced by several bacterial species may have been acquired by gene transfer; see however also the citrate siderophores synechobactins.
Certainly of genuine origin is anachelin, a strange compound whose biosynthesis requires inter alia steps from the peptide and polyketide paths. It exists in an open (anachelin H, Fig. 3, (10)) and two cyclic forms arising from an interaction of the carbonyl group of the salicylic acid residue with one of the neighboring hydroxy groups (anachelin 1 and 2, Fig. 3, (11) and (12)) (22, 167). The relative and absolute stereochemistry of all chiral centers was established (22, 166) and confirmed by synthesis (121) (see Sect. 8.1). In solution anachelin forms a β-turn arrangement (122). Mass spectrometric analysis of the Fe3+ complex suggests a 1:1 ratio.


Anachelin H (10), anachelin 1 (11), anachelin 2 (12)
2.4 Actinomycetal Metabolites
Desferrimaduraferrin is a Fe3+ complexing metabolite of Actinomadura madurae (185). It consists of salicylic acid, β-Ala, Gly, l-Ser and N 5-hydroxy-N 2-methyl-l-Orn, with the latter incorporated in a heterocyclic system (Fig. 4, 13). From the same species the madurastatin group was obtained (136). The main representative A1 shows the sequence salicylic acid, d-azaridine carboxylic acid, l-Ala, β-Ala, N 5-hydroxy-N 2-methyl-Orn, l-cOHOrn (Fig. 4, 14). In A2 the azaridine ring is opened giving a Ser residue, A3 is an isomer of the open form with the salicylic acid bound to the hydroxy group of Ser. B1 and B2 are the precursors N-salicyloyl-azaridine carboxylic acid and N-salicyloyl-Ser. The madurastatin species A1 forms a 1:1 Fe3+ complex as shown by mass spectrometry.
Asterobactin from Nocardia asteroides (257) contains salicylic, 2,3-dihydroxypropionic, and 2-methyl-3-hydroxyundecanoic acid as well as derivatized Orn and Arg residues (Fig. 4, 15). It forms a Fe3+ complex. The stereochemistry of the various centers was not determined but l-configuration is proposed for Orn and Arg for biosynthetic reasons (general amino acid pool) and a negative [α] 25D of asterobactin. Whether the three compounds are involved in metal transport has not been investigated.


Desferrimaduraferrin (13), madurastatin A1 (14), asterobactin (15)
2.5 Bacterial Hydroxamate Siderophores
Exochelins (322, 323) are peptidic siderophores from Mycobacterium spp. (see also below mycobactins). Exochelin MS (16) from M. smegmatis comprises β-Ala and three N 5-OHOrn units, which are linked by their N5 atoms to acyl groups thus forming hydroxamic acids.
Exochelin MN (17) from M. neoaurum contains N 2-methyl-N 5-hydroxy-Orn linked by its N5 to β-Ala and by its carboxyl group to N2 of Orn, which in turn is bound amidically to cOHOrn; all amino acids are l configured.
The Fe3+ chelating properties of exochelin MN (17) were investigated in detail (pK a values, chelation constants, redox equilibria, etc.) (87). In one publication (128) siderophores from Mycobacterium tuberculosis otherwise referred to as carboxymycobactins (see below Sect. 2.8) were also named exochelins.
Vicibactin (18) (previously called hydroxamate K (61a)) from Rhizobium leguminosarum is a macrocyclic trilactone consisting of N 2-acetyl-N 5-hydroxy-d-Orn and (R)-3-hydroxybutyric acid (91).
Vicibactin 7101 from a mutant strain lacks the N-acetyl groups but shows comparable siderophore activity as demonstrated by 55Fe3+ uptake studies (91). The answer to the question why vicibactin is biosynthesized if vicibactin 7101 is as efficient in iron sequestering may be the greater stability of the acetylated compound (cf. fusarinines, Sect. 2.6). Vicibactin is identical with neurosporin produced by the fungus Neurospora crassa for which X-ray data of the Fe3+ complex are available. CD spectroscopy indicates a Λ-cis configuration both for crystals and for solution (108).
A hydroxamate siderophore from Salmonella typhimurium is described as containing isoleucine/leucine, phenylalanine and valine, but not serine and lysine. Further details are not given (290a). For other Salmonella siderophores see Sect. 2.7.
2.6 Fungal l-Ornithine-Based Hydroxamate Siderophores
For other fungal siderophores see neurosporin above, pistilarin (a spermidine derivative, Sect. 3.2) and rhizoferrin (a citrate siderophore, Sect. 4.4); siderophores produced by marine fungi are treated in (147). The siderophores to be discussed here can be divided in three groups, the fusarinines, the ferrichromes, and the coprogens, all based on N 5-hydroxy-N 5-acyl-l-Orn. There exist some earlier reviews (204, 300, 383); for the early days see also (395). Lists of sidrophores and the producing fungi have been assembled (384, 385) to which the marine yeast Aureobasidium pullulans may be added (374a); see also (139). Chromatographic separation techniques were established (175, 192). For a number of siderophores and their Fe3+ complexes X-ray and other structural analyses are reported (366). In the text and the figures, the desferri ligands will be presented without adding the prefix “desferri” to their names.
Fusarinines (19) produced by several fungal genera comprise the acyl unit (Z)-5-hydroxy-3-methyl-pent-2-enoic acid (anhydromevalonic acid) (Fig. 5, a) bound to N 5-hydroxy-l-ornithine. They can be a linear monomer, dimer (fusarinine A) or trimer (fusarinine B) (the monomer can also be (E)-configured) (172). Fusarinine B is possibly identical with coprogen C (89).


Acyl residues encountered in fungal hydroxamate siderophores
The trimer by forming an ester bond between the two terminal functions results in a lactone ring (fusarinine C or fusigen) (88, 313). Since the fusarinines are rather labile it is not clear whether the open forms are genuine siderophores, precursors of fusigen or just hydrolysis products (204). The monomers (Z)- and (E)-fusarinine form in aqueous solution at neutral pH (Fe3+)Lig3 complexes, which are mixtures of Λ and Δ isomers (172).
The free α-amino groups of the ornithine units were also found in an acetylated form (90, 243). Since triacetylfusigen is resistant to hydrolysis, formation of the acetylated mono-, di-, and trimeric linear acetylfusarinines is assumed to be effected by enzymatic cleavage (103a, 243). X-ray and CD data of the Fe3+ complex of triacetylfusigen have been obtained (152). Depending on the solvent used for crystallization the crystals show Λ-cis or Δ-cis configuration, while in solution Δ-cis prevails.
The members of the ferrichrome group are cyclohexapeptides with the general structure [-(N 5-acyl-N 5-hydroxy-l-Orn)3-A-B-Gly-] where A and B can be Gly, Ala, or Ser (Table 1); the various acyl groups are depicted in Fig. 5. Exceptions are tetraglycylferrichrome, a cycloheptapeptide with four Gly units in sequence and three acetyl residues in the Orn part (ferrichrome with an additional Gly) (82), and des(diserylglycyl)ferrirhodin, a linear tripeptide containing only the three Orn units of ferrirhodin (169). One of the members of this group, ferricrocin was identified as an intra- and intercellular iron transporter for Aspergillus fumigatus (374).
Ferrichrome (as do also at least the members of the group for which structural data are available ((366) and references noted in Table 1) shows Λ-, synthetic enantio-ferrichrome based on d-Orn Δ-configuration (253). Uptake studies performed with Ustilago sphaerogena (103) using 59Fe3+ and [14C]-ferrichrome under optimal conditions (30°C, pH 7) showed rapid resorption of both labels during the first 30 min. The uptake of 59Fe3+ continued for further 30 min, then the level of radioactivity stayed constant, while the level of 14C dropped to a lower constant value. Desferri-[14C]-ferrichrome is not taken up or even bound to the cell surface. These findings are in agreement with shuttle mechanism and re-export of the ligand after detachment of iron. See also analogous experiments with parabactin (Sect. 3.2) and with schizokinen (Sect. 4.1). In contrast, ferrichrome A does not enter the cell. Fe3+ is rather reduced and given off at the cell surface and subsequently transported into the cell (99, 105). 55Fe uptake studies performed with Neurospora crassa showed the same incorporation rate for ferrichrome and tetraglycylferrichrome indicating that the peptide ring size is of minor importance for the acceptance by the transport system (82).
The third group comprises the coprogen family. Their characteristic element is a diketopiperazine ring formed by the head-to-head condensation of two N 5-acyl-N 5-hydroxy-l-Orn units. Rhodotorulic acid (20) first isolated from the yeast Rhodotorula pilimanae and subsequently found to be produced by many yeasts (9a) contains two acetyl groups (Fig. 6) (9), and dimerum acid (21) from Fusarium dimerum (89) and other fungi (172) two (E)-anhydromevalonyl residues (Fig. 6). An acetyl-dimerum acid of unknown structure has been encountered (157b). In the coprogens a third variously substituted (E)-fusarinine unit is added by means of an ester bond (Table 2) (300). Rhodotorulic and dimerum acid form (Fe3+)2Lig3 complexes, but also a mixed 1:1:1 complex of Fe3+ with dimerum acid and (Z)-fusarinine was observed. Various coprogens were shown to yield 1:1 complexes with Fe3+ (60, 153, 172). The CD-spectra of the coprogen and neocoprogen I/II Fe3+ complexes demonstrate Δ-configuration for the solutions and for the crystals of neocoprogen I (153). Palmitoylcoprogen (Table 2 last entry) from Trichoderma spp. is retained in the fungal mycelium and may therefore be considered as a candidate for an iron uptake taxi mechanism (5).


Relationships between the structure of the siderophores and the iron transport were investigated for the fungus Neurospora crassa (160, 160a). Apparently two different receptors exist for ferrichromes and for coprogenes. For the recognition and the binding to the cell surface the iron configuration and the nature of the acyl chains is of importance. However, the transport system seems to be the same for both siderophore types dependent on the peptide part of the molecules.
2.7 Catecholate Siderophores
For other catecholate siderophores see di-/tri-aminoalkane (Sect. 3.2) and citric acid (Sect. 4.3) derivatives below; for a review see (38).
2,3-Dihydroxybenzoic acid is produced by a series of microorganisms, viz. Aerobacter aerogenes (291), Azotobacter vinelandii (70, 273), Bacillus subtilis (282), Escherichia coli (261, 291), Klebsiella oxytoca (196), Micrococcus denitrificans (347), Nocardia asteroides (112), Rhizobium sp. (74), and Salmonella typhimurium (290), 3,4-dihydroxybenzoic acid by a mutant of Aerobacter aerogenes (291), Azomonas macrocytogenes (380), Bacillus anthracis (123), Escherichia coli (291), Magnetospirillum magneticum (54), and Mycobacterium smegmatis (291). Both dihydroxybenzoic acids can act as siderophores.
Condensation products of DHB (which usually is found also in the fermentation broth) with amino acids were reported, viz. with glycine from Bacillus subtilis (164) named subsequently itoic acid (282); with serine from Escherichia coli (261) and Klebsiella oxytoca (196); with threonine from Klebsiella oxytoca (196) and Rhizobium spp. (275, 327); with arginine from Pseudomonas stutzeri (62); with glycine and threonine from Rhizobium sp. (240); with threonine and lysine as well as with leucine and lysine from Azospirillum lipoferum (312, 320). In most cases the isolate (sometimes designated as being a siderophore) was hydrolyzed and the constituents were determined by paper chromatography. The relative amounts of the constituents, the chiralities of the amino acids and the molecular mass of the isolate have not been determined. Hence it is not known whether condensation products of the enterobactin type exist.
Ideally suited for Fe3+ complexation – exemplified by the extremely high complexing constant of 1049 (originally estimated as 1052) (210) – is enterobactin (enterochelin) first isolated from Salmonella typhimurium (286) and Escherichia coli as well as from Aerobacter aerogenes (261) and recently from Enterobacter cloacae (368). It is a cyclic trilactone of N-2,3-dihydroxybenzoyl-l-serine (DHB-Ser) (Fig. 7, 22). Syntheses have been reported (71, 321). DHB-Ser by itself can act as a siderophore. In the culture medium degradation products of enterobactin also were found, and are open-chain compounds comprising two or three constitutional units. Iron release in the cell is effected by degradation of enterobactin. Ferri-enterobactin shows a Δ-cis configuration, with the synthetic ferri-enantio-enterobactin based on d-Ser Λ-cis-configuration (256).


Enterobactin (22), salmochelin S4 (23), corynebactin (24)
Escherichia coli and Salmonella enterica produce a derivative of enterobactin, salmochelin S4, where two of the aromatic rings are β-C-glucosylated in the 5-position (Fig. 7, 23). Also glycosylated degradation products or precursors (monomer: salmochelin SX, dimers: S1 and S5, linear trimer: S2) could be isolated (31, 135, 247). Salmochelin S4 is identical with pacifarin, a compound active against salmonellosis (378), and SX with pacifarinic acid, glucosylated DHB-serine (247).
From Corynebacterium glutamicum the siderophore corynebactin was obtained (41). It differs from enterobactin in being composed of three DHB-Gly-l-Thr units (Fig. 7, 24). Later the same siderophore was found to be produced also by Bacillus subtilis and named bacillibactin (223). Its complexation constant is ~1048 (84). The monomeric unit DHB-Gly-Thr was isolated from Bacillus licheniformis (357a).
Azospirillum brasilense under iron starvation produces spirilobactin. Hydrolysis yields DHB, ornithine, and serine of unknown chirality in a ratio of 1:1:1. The molecular mass was not determined and hence it is not known whether spirilobactin forms a (cyclic) trimer. Iron uptake was studied with the 59Fe3+ complex (10).
Erwinia chrysanthemi (278) and Serratia marcenscens (101) produce N 2-DHB-d-Lys-l-Ser named chrysobactin. The structure was confirmed by synthesis. At physiological pH values 2 or 3 chrysobactin residues are associated with Fe3+ (280). From Chryseomonas luteola in addition to chrysobactin a derivative (chrysomonin) was isolated where C-6 of the DHB unit is substituted with the N-atom of a pyridinium cation. Chrysomonin could be synthesized from chrysobactin (1a).
Amonabactins (25) were found to be excreted by Aeromonas hydrophila (355, 356) and by Pseudomonas stutzeri (398). They are based on the peptides Lys-Lys-Phe and Lys-Lys-Trp; N6 of the first l-Lys residues is derivatized by DHB or by a DHB-Gly residue, and that of the second l-Lys by a DHB group (Table 3). At high pH values and excess ligand a (Fe3+)2Lig3 complex is formed, while at neutral pH a 1:1 ratio prevails with H2O molecules satisfying the remaining coordination sites. The 2:3 complex is preferentially Δ-configured, and the 1:1 complex is achiral (357). Model uptake studies with Aeromonas were performed with 55Fe3+ and a 14C-labeled artificial synthetic siderophore. They demonstrate a modified shuttle mechanism. An iron-free siderophore molecule is strongly bound to the receptor protein and Fe3+ exchange occurs between an approaching ferri-siderophore and the bound one, which then is transported into the cell (337); cf. the pyoverdins (Sect. 2.1).
Alterobactin A is a cyclodepsipeptide from Alteromonas luteoviolacea, with N 8-DHB-(4S),8-diamino-(3R)-hydroxy-octanolyl-d-Ser-Gly-l-Arg-l-threo-3-hydroxy-Asp-Gly-l-threo-3-hydroxy-Asp having an ester bond between the C-terminal carboxyl group and Ser. It is accompanied by its hydrolysis product alterobactin B (Fig. 8, 26, 27) (298). Alterobactin A forms a 1:1 complex with Fe3+ with an unexpectedly high complexing constant (between 1049 and 1053), higher than that of enterobactin above, despite the fact that two complexing sites are α-hydroxy acids which bind Fe3+ less efficiently than DHB units (148). A synthesis has been reported (83) (see Sect. 8.2).


Alterobactins (26, 27), pseudoalterobactins (28, 29)
Related structures are the pseudoalterobactins A and B from Pseudoalteromonas sp. (Fig. 8, 28, 29) (183), one of the rare examples of bacterial metabolites containing an aromatic sulfonic acid (40). Chiralities of the constituents were not determined.
Heterobactin A and B (30) are produced by Rhodococcus erythropolis (59). They are based on the sequence Orn-Gly-cOHOrn. The N5-amino group of Orn is substituted by a DHB group. In heterobactin B, the α-amino group of Orn is free (R = H); in heterobactin A, R is probably a 2-hydroxybenzoxazolyl-carbonyl group.
Rhodobactin (31) was isolated from Rhodococcus rhodochrous (86). A sequence of four Orn units derivatized in different ways is linked together. The nitrogen atoms of the N-terminal Orn are substituted with DHB groups, the N-terminal Orn is followed by two Orn moieties, for which the N5-amino groups are transformed into urea units (NH2CONH-), and the C-terminus is cOHOrn. The stereochemistry of the Orn units was not determined. Rhodobactin forms a 1:1 Fe3+/Lig complex. Iron uptake was studied with 55Fe3+.
Thermobifida fusca, belonging to the Actinomycetales, produces three closely related siderophores, namely, the fuscachelins (92). Fuscachelin B starts with the sequence DHB-Arg-Gly-Gly-Ser, which is bound to the hydroxylated N5-amino group of Orn. Its N2-amino group (the carboxyl group is free) is bound to the C-terminus of the sequence Gly-Gly-Arg-DHB (32). Fuscachelin A is considered to be the genuine metabolite, with B and C degradation products.
In fuscachelin C the carboxyl group of Orn forms an amide, while in fuscachelin A an ester bond occurs between the carboxyl group of Orn and the hydroxy group of Ser.
2.8 Lipopeptidic Siderophores
From Burkholderia cepacia (formerly Pseudomonas cepacia) three siderophores named ornibactins (33) were isolated for which the structures were determined by degradation and NMR studies (335, 336) as containing 3-hydroxy fatty acid residues and putrescine that blocks the C-terminus, with acyl = R-CHOH-CH2-CO (R = CH3, C3H7, C5H11).
The three ornibactins are accompanied by minor components, which contain an additional oxygen atom. Their structure has not been investigated. Ornibactins are the main siderophores of a series of Burkholderia strains accompanied in part by pyochelin (Sect. 5) and cepabactin (Sect. 6) (235). A further B. cepacia siderophore is cepaciachelin (Sect. 3.2) (15). The iron acquisition by the various siderophores of B. cepacia has been discussed in detail (359).
From Nocardia strains several closely related compounds (nocobactins, formobactin, amamistatins) were isolated that contain three typically Fe3+ binding sites, two hydroxamate units, and a hydroxyphenyloxazole structure (cf. Sect. 3.2 below). The C-terminus is N-hydroxy-cyclo-Lys bound to a long chain 3-hydroxy fatty acid, whose hydroxy group is esterified by N 6-acyl-N 6-hydroxy-Lys, the α-amino group of which is bound to 2-o-hydroxyphenyl-5-methyl-oxazole-4-carboxylic acid (Table 4). For the amamistatins the configuration of the cyclic lysine was determined as l, the open one as d, and that of C-3 of the fatty acid as (S). The involvement in the iron metabolism was not investigated.
Structurally related with the nocobactin family are the mycobactins and carboxymycobactins (the latter were also referred to as exochelins, Sect. 2.5 (128)) from Mycobacterium spp. For reviews see (85, 331, 369). They have the same basic skeleton as the nocobactins, but the 4,5-double bond of the oxazole ring is saturated. A series of differently substituted representatives has been isolated (see Table 5). The major group comprises mixtures carrying saturated and unsaturated long-chain fatty acid residues as substituents of the hydroxamic acid unit formed by the N6-amino group of lysine. For some (“J”, M, and N), the fatty acid residues are located in the chain, as for the nocobactins. Representatives of the MAIS group (Mycobacterium avium, M. intracellulare, M. scrofulaceum) possess two long chain fatty acid residues. The stereochemistry of most chiral centers has been determined. For the Fe3+ complex of mycobactin P an X-ray analysis is available (157). For the carboxymycobactins the residues R3 in Table 5 are saturated or unsaturated alkyl groups with terminal carboxyl groups or their methyl esters.
Transvalencin Z (245a) from Nocardia transvalensis could be a precursor or side product of mycobactin biosynthesis, possibly acquired from a vagabonding gene. It comprises the left part of the serine/salicylic acid based molecules (Table 5 R4 = R5 = H) and ends with N 6-formyl-Lys (R3 = H, no N-hydroxy group). The stereochemistry of the two chiral centers was not determined. Transvalencin Z seems not to bind Fe3+.
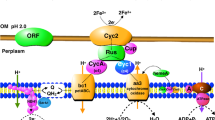
Iron uptake of Mycobacterium smegmatis involving mycobactin S was studied with 55Fe3+ (293). Mycobactin is not given off into the surrounding medium but is located instead in the lipid envelope of the cell and is active in the trans-membrane transport of Fe3+ (taxi mechanism). Iron is relased at the inside of the membrane by a reductive mechanism. There is some evidence that salicylic acid is the extracellular siderophore.
Corrugatin (34) (Fig. 9) is the siderophore of Pseudomonas corrugata (302). It was also found as secondary siderophore of several pyoverdin producing Pseudomonas strains as P. fluorescens, occasionally in slightly modified forms such as ornicorrugatin (35) where one Dab is replaced by Orn (218), or with OHHis instead of OHAsp as the C-terminus (S. Matthijs, unpublished).
A group of amphiphilic siderophores was isolated from marine bacteria (410), the marinobactins (Fig. 10, 36), from Marinobacter sp., aquachelins (Fig. 10, 37), from Halomonas aquamarina (215, 217), the amphibactins (Fig. 10, 38), from Vibrio sp. (216), and the loihichelins (Fig. 10, 39), from Halomonas sp. (150). They all comprise series of related molecules differing in the nature of the saturated or unsaturated fatty acid (for amphibactins and loihichelins also 3-hydroxy fatty acids) linked to the N-terminus (see also ochrobactins and synechobactins, Sect. 4.1). Structure elucidations were effected by spectroscopic methods and degradation studies. For the marinobactins a N-terminal nine-membered lactam ring was suggested to be formed by an amide bond between the carboxyl group of Asp and the C-4 amino group of Dab (Fig. 10, a). It may be suggested that rather a condensation with the amide carbonyl group had occurred (Fig. 10, b; cf. Chart 1). This would keep the α-hydroxycarboxyl grouping of OHAsp intact, which acts as a binding site for Fe3+ and is essential for photolytic degradation. The rather scarce structural data presented do not allow a decision to be made. The siderophores show a strong affinity to lipid membranes (389). The Fe3+ complexes of aquachelins and marinobactins suffer degradation under sunlight irradiation. For the Fe3+-aquachelin complexes the formation of Fe2+, of hydrophobic and of hydrophylic cleavage products was observed. For the latter a N-formyl-Ser terminus was suggested based on mass spectral data (Chart 2) (12). There is evidence that this type of photolytic degradation is common for siderophores containing α-hydroxycarboxyl ligands (13, 150, 401).


Light-induced degradation of Fe3+-aquachelins


Corrugatin (n = 2, l-Dab) (34) and ornicorrugatin (n = 3, D-Orn) (35)


Amphiphilic marine siderophores
2.9 Pseudomonas mendocina Siderophores
From Pseudomonas mendocina five siderophores were isolated by chromatography. They are reported to have identical molecular masses of 1,152 Da (the also reported (3a) value of 929 Da is an error; L. E. Hersman, private communication) and an identical amino acid composition, which has not been revealed (141a). Color reactions show the presence of a hydroxamate, but not of a catecholate grouping. A gene analysis suggests a partial sequence acyl-Asp-Dab-Ser-formylOHOrn-Ser-formylOHOrn where asparagine could be OHAsp and the C-terminal ornithine cOHOrn (9b). In which way the five isomeric siderophores with identical molecular masses differ from each other is not clear.
3 Siderophores Based on Diamino- and Triaminoalkane Skeletons
3.1 Rhizobactin
Rhizobactin (40) is the siderophore of Rhizobium meliloti (328). It contains one α-hydroxycarboxylic acid and two α-amino acid units as probable binding sites for Fe3+. Acid hydrolysis yields inter alia l-malic acid. The stereochemistry of the other two chiral centers is not known.
3.2 Catecholate Siderophores
For other catecholate siderophores, see the peptide-based siderophores above (Sect. 2.7) and the citric acid derivatives below (Sect. 4.3); for a review on syntheses, see (24), for a general review, (38).
The tricatecholate siderophore protochelin (41) (Fig. 11) was obtained from a methanol – bacterium (351). Subsequently it was also found to be produced by Azotobacter vinelandii (72, 360) together with its constituents 2,3-dihydroxybenzoic acid, azotochelin (bis-DHB lysine) (70) and aminochelin (mono-DHB cadaverine) (273). Cepaciachelin from Burkholderia cepacia (15) lacks the DHB residue from the aminochelin part of protochelin. The amino acid in all compounds is l-lysine. Azotobacter vinelandii shows an interesting rationale when confronted with a deficiency in iron supply. At concentrations >7 µM, 2,3-dihydroxybenzoic acid is secreted, between 3 and 7 µM, the di- and tricatecholate siderophores are produced, and at still lower concentrations, it is resorted to azotobactin D (see above Sect. 2.2) (72). Myxochelin A from Angiococcus disciformis (197) and Nonomuraea pusilla (239a) can be considered as a reduction product of azotochelin (lysinol instead of Lys). The absolute configuration of lysinol (S) was determined by synthesis. Both antipodes show about the same antitumor activity (239a).


Protochelin (41)
Pistillarin was first isolated from Clavariadelphus pistillaris and from several Ramaria spp. (Basidiomycetes) (334). Recently, it was found to be produced also by the marine fungus Penicillium bilaii (56). Like siderochrome II below it is a spermidine derivative substituted only at the terminal NH2-groups (N 1, N 10-di-(3,4-dihydroxy)benzoyl-spermidine). Its synthesis and that of siderochrome II was reported, their siderophore activity and their complexation with Fe3+ (1:1 complexes) was investigated (102, 299). A derivative of pistillarin substituted at all three amino functions has not been reported yet.
When DHB is bound to serine or threonine cyclization may occur resulting in an oxazoline ring (cf. above anachelin, Sect. 2.3, and mycobactins, Sect. 2.8). It has been discussed whether the oxazoline nitrogen atom may act as a ligand site (see below, (303)). This would explain why DHB is replaced by a salicylic acid residue in some cases.
To this group of siderophores belong photobactin (42a) from Photorhabdus luminescens (Fig. 12) (66), derived from 1,4-diaminobutane substituted by DHB and by cyclized DHB-Thr (1H-NMR data indicate that the substituents of the oxazoline ring are in trans positions; the absolute stereochemistry is not known), and its lower homolog serratiochelin (42b) from Serratia marcescens derived from 1,3-diaminopropane. Its structure including the absolute stereochemistry (L-Thr) was confirmed by synthesis (101).


Photobactin (42a), serratiochelin (42b)
Spermidine derivatives are agrobactin from Agrobacterium tumefaciens (Fig. 13, 43) (268), for which the structure was confirmed by X-ray analysis (109) and synthesis of the hydrolyzed form (DHB-Thr) agrobactin A (283), and parabactin (Fig. 13, 44) from Paracoccus denitrificans (284). Two syntheses are reported for parabactin (28, 28c, 255) (see Sect. 8.3). The open form (parabactin A) as well as the precursors 2,3-dihydroxybenzoic acid and a compound with a free central NH group (N 1, N 10-di-DHB-spermidine, siderochrome II) were also found (347). The 1:1 Ga3+/Lig complex shows Λ-cis configuration (28a). Parabactin also forms a 1:1 complex with Fe3+ (347) for which the structure was investigated by X-ray photoelectron and electron spin resonance spectroscopy. In particular, the question as to whether the oxazoline nitrogen acts as a binding site has been discussed. An experimental proof seemed not to be possible (303).


Agrobactin (43), parabactin (44), fluvibactin (45)
Iron transport was studied using the 55Fe3+- and 3H-complexes of parabactin (25). After a quick uptake of 10% of both labels there was a continuing steady uptake of 55Fe3+ while the amount of 3H remained constant. This could either mean that after binding to the cell surface 55Fe3+ only is transferred into the cell (“taxi mechanism”) or there is a fast re-export of the ligand. A decision in favor of the taxi-mechanism could be reached by offering the Ga3+ complex of [3H]-parabactin (Ga3+ cannot be released reductively in the cell and hence a re-export of the ligand is not possible). The uptake curve resembled that of ferri-[3H]-parabactin: a small amount of complex is bound to the cell surface, but there is no transport of the ligand in the cell. This is in agreement with temperature studies (30 and 4°C). While the uptake of 55Fe3+ decreases that of 3H is not influenced.
Fluvibactin (Fig. 13, 45) from Vibrio fluvialis (391) differs from agrobactin by replacement of spermidine by norspermidine. Also here the precursor with a free central NH group could be isolated. Vibriobactin from Vibrio cholerae (Fig. 14, 46) contains two cyclized DHB-Thr substituents (129). Syntheses of agrobactin, fluvibactin and vibriobactin are published (26, 30, 308). In vulnibactin from Vibrio vulnificus (Fig. 14, 47) (264) two DHB groups are replaced by salicylic acid units. The precursor with a free central NH group was also found.


Vibriobactin (46), vulnibactin (47)
3.3 Hydroxamic Acid Siderophores
Bisucaberin (48) from Alteromonas haloplanktis (181) is a cyclic dimer of succinyl-(N-hydroxycadaverin) (348); cf. the cyclic trimer proferrioxamine E (Table 6).
In putrebactin from Shewanella putrefaciens (201) cadaverine is replaced by putrescine (49, R = H). For the cyclic trimer, see proferrioxamine X2 in Table 6. The arctic S. gelidimarina living in a habitat with extremely low iron supply produces a cell-associated hydroxamic acid siderophore with the mass 977 Da for [M+H]+ of unknown structure (274).
Alcaligin from Alcaligenes denitrificans (260) and from Bordetella spp. (244) is a cyclic dimer of succinyl-N 1,3S-dihydroxyputrescine (49, R = H) confirmed by synthesis (402).
Alcaligin forms at pH 2.0 a 1:1 and at pH 6.0 a 2:3 Fe-to-ligand complex. The structure (Plate 3) of the (Fe3+)2Lig3 complex was studied by X-ray analysis (156). One ligand bridges two metal ions while the remaining two are coordinated with a single Fe3+ each. The metal centers show Λ-configuration.


X-ray structure of ferri-alcaligin (ferri-49)
Alcaligin E from Alcaligenes eutrophus is described from color tests as a phenolic siderophore (126a). According to a recent publication (90a) it is identical with staphyloferrin B, a citrate siderophore (Sect. 4.2). No further information is given to resolve these discrepancies.
A group of related siderophores comprises the desferri- or deferriferrioxamines (occasionally abbreviated as desferrioxamines) or proferrioxamines. Originally they were obtained from Actinomycetes, mainly Nocardia and Streptomyces spp. (187) and later found to be produced also by Erwinia spp. (several representatives) (e.g. (30a, 113, 115, 180)), Arthrobacter simplex (B), Chromobacterium violaceum (E) (246a), and by Pseudomonas stutzeri (several) (229a, 246, 398). They consist of three (or in rare cases four) mono-N-hydroxy-1,4-diaminobutane (putrescine), mono-N-hydroxy-1,5-diaminopentane (cadaverine) or (rarely) mono-N-hydroxy-1,3-diaminopropane units connected by succinic acid links. The hydroxylated terminus carries an acetyl or a succinyl (as in the structural formula heading Table 6) residue, and in the latter case the free carboxyl group and the free N-terminus may form a macrolactam. The terminal acid residue can also be missing (referred to as “truncated”) (115, 398). By feeding of suitable diamino precursors to the culture medium unnatural analogs can be obtained (111, 194, 227). At pH values above 6.5 (Fe3+)2Lig3 complexes prevail, in more acidic media Fe3+Lig is formed (194). The crystals of the Fe3+Lig complexes of ferrioxamine D1 and E are racemic mixtures of Λ-cis and Δ-cis coordination isomers (154, 366a). The outer membrane receptor protein of Erwinia amylovora was structurally determined (180). Siderophore activity was demonstrated for 55Fe-labeled ferrioxamine E (30a). For the mass spectrometric analysis, see (112a).
Originally the various natural representatives had been designated by capital letters, but later a nomenclature system was proposed (110). In short, the indices and modifications as listed in Table 6 (p = 0 means that the entire fourth diaminoalkane-succinyl unit is missing) are grouped around the acronym pFO. The system is essentially self-explanatory; for details and possible extensions see the original publication.
4 Citrate Siderophores
For a review, see (39). Some citrate siderophores are accompanied by cyclic imide structures formed by the loss of water from the central carboxyl group and a lateral amide NH (Chart 3). They are usually designated by an A following the name of the siderophore. Free citric acid can be a true siderophore, e.g. for Bradyrhizobium spp. (205), Pseudomonas aeruginosa (213), and Mycobacterium smegmatis (228a). The mode of the uptake differs. Bradyrhizobium and Pseudomonas incorporate ferric citrate but Pseudomonas shows also a citrate mediated Fe2+ uptake, while in the case of Mycobacterium no citrate enters the cell. Ferric citrate is a complex system depending on the pH of the solution and the relative concentration of the two constituents (333a, 333b). In an acidic milieu equimolar concentrations form [FeCit]-, at about pH 4 polymerization starts resulting at pH 8–9 in an insoluble complex with an iron hydroxide core and citrate ions bound to the surface. With a citrate excess species like [FeCit2]5- are discussed.


Cyclization of citrate siderophores to amidic structures
It should be mentioned that the central carbon atom of citric acid becomes chiral when the two peripheral carboxy groups are substituted differently (examples will be found below). For enzyme reactions it is a prochirality center. This has been shown for vibrioferrin (58) and staphyloferrin B (59).
4.1 Siderophores with Two Hydroxamic Acid Units
In siderophores of this series, 1,3-diaminopropane, 1,5-diaminopentane, or lysine (by its α-amino group) is connected to the outer two carboxyl groups of citric acid. These spacers, in turn, are acylated and derivatized by a N-hydroxy group thus forming hydroxamic acids. For a synthesis concept see (404).
Schizokinen (Fig. 15, 50) was first isolated from Bacillus megaterium (53), subsequently from Ralstonia solanacearum (43), Rhizobium leguminosarum (339), and several species of the cyanobacterium Anabena (e.g. (326)). It was named after its cell division promoting effect observed with Bacillus cultures (200). Its structure was elucidated by degradation and spectral data and confirmed by synthesis (43, 202, 237, 248). For a compilation of details on structural data the review (39) should be consulted. Both natural and synthetic schizokinen is accompanied by the cyclized schizokinen A (43, 202, 237, 248). Schizokinen forms a 1:1 complex with Fe3+, but at the central hydroxy group acetylated schizokinen yields (Fe3+)2Lig3. This proves that the central unit is one of the binding sites (285). Also N-deoxyschizokinen from Bacillus megaterium lacking one hydroxamic acid unit still binds Fe3+ (158). Whether it acts as a siderophore is not known.


Citrate siderophores with two hydroxamic acid units
The schizokinen-mediated Fe3+ transport in Bacillus megaterium was studied by double labelling with 59Fe and 3H (8). At 37°C, uptake of 59Fe and of 3H are parallel during the first 30 sec, then that of 59Fe continues until it levels off after 2 min, while that of [3H]-schizokinen drops to a low constant level. At 0°C, uptake of both labels reaches this low level which is obviously due to the binding of the ferri-siderophore to the cell surface. At 37°C, transport into the cell, release of iron, and re-export of the ligand follow. Apparently a shuttle mechanism takes place, cf. the experimental results obtained with parabactin (Sect. 3.2) indicative of a taxi mechanism.
Arthrobactin (Fig. 15, 51) was obtained from Arthrobacter spp. and originally described as the growth factor of A. terregens, the “terregens factor” (51). Its structure was elucidated (207) and confirmed by synthesis (202). Also the structure of acinetoferrin from Acinetobacter haemolyticus was established (Fig. 16, 52) (265) and confirmed by synthesis (375). It shows strong interaction with lipid membranes like the marine liposiderophores above (211) (Sect. 2.8).


Vibrioferrin (cyclic form) (58)
Aerobactin (Fig. 15, 53) was first isolated from Aerobacter (Enterobacter) aerogenes (126), Enterobacter cloacae (368) and subsequently from various enterobacteria such as Escherichia (376), Salmonella (225), Shigella (277), Yersinia (340), but also from Erwinia carotovora (163), Pseudomonas sp. (52) and Vibrio spp. (141, 266). Aerobactin is an important virulence factor for enterobacteria (75). Aerobactin contains l-lysine. A synthesis is described (222). The bright orange Fe3+ complex was investigated in detail (predominant Λ configuration in solution, stability constant, redox potential) (138). Fe3+ transport was studied by double labelling (59Fe and 3H) (8). The results corresponded to those obtained with schizokinen. Aerobactin binds to the same receptor as the bacteriocin cloacin DF13 and thus alleviates the growth inhibiting effect of the latter (368).
Nannochelin C (Fig. 15, 54) from the myxobacterium Nannocystis exedens contains two l-Lys and two (E)-cinnamic acid units. The reported mono- and di-methyl esters (nannochelin B and A) may be artifacts from the work-up (198). A synthesis is described (29) (see Sect. 8.4). The ochrobactins (Fig. 15, 55) isolated from the sea-shore bacterium Ochrobactrum sp. (214) with the spacer l -lysine are membrane active due to the fatty acid residues (saturated C8 and (2E)-unsaturated C8 and C10); cf. lipopeptidic siderophores in Sect. 2.8.
Rhizobactin 1021 (Fig. 15, 56) (for rhizobactin, see diaminoalkane-based siderophores, Sect. 3.1) from Rhizobium meliloti (281), contains an acetyl and an (E)-decenoyl group. Its Fe3+ complex in aqueous solution is Λ-configured and forms an equilibrium between a monomeric and a dimeric form that can be separated by chromatography. A synthesis is described (404).
Synechobactins (Fig. 15, 57) from the cyanobacterium Synechococcus (165), contain an acetyl and C12-, C10-, and C8-saturated acid residues and thus belong to the amphiphilic marine siderophores (cf. Sect. 2.8). Both rhizobactin 1021 and the synechobactins are substituted unsymmetrically. Hence, for each, the central C-atom of citric acid is chiral, but its stereochemistry has not been determined.
Awaitins are synthetic homologs of siderophores (A: 53, n = 3; B: 50, n = 3; C: 53, n = 2) “awaited” to be found in nature, so far without success (405).
4.2 Siderophores with 2-Oxoglutaric Acid Units
N-Alkylated 2-oxoglutaric acid derivatives cyclize at neutral pH values to two epimeric 5-carboxy-5-hydroxy-2-oxopyrolidine structures (Chart 4). In this way, α-hydroxycarboxylic acid groupings are formed that can act as ligand sites for Fe3+.


Cyclization of 2-oxoglutaric acid substituents
Vibrioferrin (58, Fig. 16) was isolated from Vibrio parahaemolyticus. The stereochemistry of the central citric acid C-atom is R, that of the alanine part is S as shown by stereospecific synthesis (411). Iron uptake was studied with 55Fe3+ proving that vibrioferrin acts as a siderophore despite the fact that it has only five ligand sites, the two α-hydroxy acids and the free citric acid carboxyl group. Possibly a solvent molecule satisfies the eighth octahedral position (393, 411). Vibrioferrin is also formed by Marinobacter spp. It is a week Fe3+ chelator (complexing constant 1024). Its Fe3+ complex is very susceptible to photodegradation by oxidative decarboxylation of the cyclized 2-oxoglutaric acid unit yielding a succinimide ring. This species cannot bind Fe3+. The concomitantly formed Fe2+ (cf. Chart 2) is reoxidized to fairly soluble Fe3+ hydroxo complexes, which are readily taken up by the bacteria (410).
Staphyloferrin B (59, Fig. 17) is produced together with staphyloferrin A (see below Sect. 4.4) by Staphylococcus hyicus and other staphylococci (94, 131), by Ralstonia eutropha (250) (= Cupriavidus metallidurans (90a)). Comparison of its CD spectrum with those of model compounds suggests the (S)-configuration of the central citric acid C-atom. Mass spectral investigations show a 1:1 Fe3+-to-ligand ratio, and NMR studies of the Ga3+ complex confirm the participation of the two α-hydroxy- and of the α-amino acid functions in complex formation. Uptake studies with 55Fe3+ showed that staphyloferrin B acts as a siderophore, but it is less efficient than staphyloferrin A.


Staphyloferrin B (cyclic form) (59)
Achromobactin (60, Fig. 18) is produced by Erwinia chrysanthemi in addition to chrysobactin (see above under the catecholate siderophores, Sect. 2.7). It has two chiral centers, a l-Dab unit and the central citric acid C-atom (not determined) (249). Recently, achromobactin was also found to be produced by Pseudomonas syringae (30b), a very versatile bacterial species (see pyoverdin, Sect. 2.1, and yersiniabactin, Sect. 5).


Achromobactin (cyclic form) (60)
4.3 Siderophores with Two Catecholate Units
In petrobactin, spermidine residues are bound to citric acid substituted with 3,4-dihydroxybenzoyl (27), and not 2,3-dihydroxybenzoyl units (Fig. 19, 61), as assumed originally (14). One or both of the substituents can carry a sulfonic acid group in the 2-position of the aromatic ring (Fig. 19, 62 and 63) (142, 149); cf. also (40). Petrobactin was originally obtained from Marinobacter hydrocarbonoclasticus (14) and subsequently from Bacillus anthracis (195, 382), B. cereus and B. thuringiensis (195a), its sulfonated derivatives from Marinobacter spp. It is probably identical with the incompletely characterized anthrachelin (123).


Petrobactin (61), petrobactin monosulfonic acid (62), petrobactin disulfonic acid (63)
4.4 Siderophores with Two Citric Acid Units
(S,S)-(enantio)-Rhizoferrin (Fig. 20, 64) was obtained from Ralstonia pickettii (251). It is the optical antipode of the fungal (R,R)-rhizoferrin first isolated from Rhizopus microsporus (93) and subsequently found to be a common siderophore of Zygomycetes (358). It is accompanied by two dehydration products, which are due to the formation of one or two imide rings (cf. Chart 3). UV spectral studies revealed that rhizoferrin forms a 1:1 Fe3+-to-ligand complex despite the fact that it has only two α-hydroxy acid binding sites (95). NMR studies of the Ga3+ complex proved the twofold symmetry of the complex and showed that only the carboxyl groups, but not the hydroxy groups are deprotonated between pH 5.5 and 9.0. The Fe3+ complex is chiral and shows Λ-configuration (58). Uptake studies suggest a shuttle mechanism (61). While Ralstonia accepts both antipodes with equal rates Rhizopus shows a clear preference for its native (R,R)-enantiomer (251).


Rhizoferrin (64), staphyloferrin A (65)
Staphyloferrin A (Fig. 20, 65) is a second siderophore of Staphylococcus spp. (226). d-Ornithine connects the two citric acid parts. Due to the unsymmetrical link the central C-atoms of the citric acid units are chiral, but their stereochemistry has not been determined. Another consequence of the asymmetric structure is that two mono- and one di-dehydration products are observed. Staphyloferrin A forms a 1:1 Fe3+-to-ligand complex, which is preferentially Λ-configured. For steric considerations only cis-(SR′) or cis-(RS′) arrangements can be considered. Uptake experiments with 55Fe showed that it is a true siderophore (193).
4.5 Legiobactin
Legionella pneumophila produces a siderophore named legiobactin, which shows no catecholate or hydroxamate reactions (206). Enzymatic studies suggest a citrate structure in agreement with the data obtained by mass spectrometry (molecular mass ca. 350 Da) and NMR (three carbonyl and ten aliphatic C atoms). It is not clear yet as to whether legiobactin is essential for the iron acquisition in the aqueous habitat of the bacterium or during lung infection (2, 65).
5 Pyochelin and Related Structures
This group comprises condensation products of salicylic acid with cysteine giving a thiazoline ring. For a review, see (310). Some structurally related compounds will also be mentioned here. Salicylic acid isolated from Burkholderia (Pseudomonas) cepacia was named azurochelin (333). It was found to act as a siderophore, e.g. for Pseudomonas fluorescens (230) and P. syringae (178); see also Mycobacterium smegmatis (Sect. 2.8). For details on the siderophore activity of salicylic acid, see (359).
The structure of pyochelin (for a detailed bibliography, see (37)), a secondary siderophore of Pseudomonas aeruginosa and of Burkholderia cepacia was established (73) as 2-(2-o-hydroxyphenyl-2-thiazolin-4-yl)-3-methylthiazolidine-4-carboxylic acid. It consists of a mixture of two easily interconvertible stereoisomers (pyochelin I and II) differing in the configuration of C-2″. They can be separated by chromatography, but in methanolic solution (not in DMSO) the equilibrium (ca. 3:1) is restored quickly. For a discussion of the mechanism of isomerization, see (37, 317).
The relative and absolute stereochemistry (4′R,2″R,4″R) of pyochelin I (Fig. 21, 66) were established by an X-ray analysis of its Fe3+ complex (316). Fe3+ is associated with the phenolate and the carboxylate oxygen ions and with the two nitrogen atoms. Two of these units are bridged by an acetate ion and a water molecule satisfying the remaining two ligand loci of Fe3+ (Plate 4). However, by titration a (Fe3+)/pyochelin ratio of 1:2 has been determined at pH 2.5 (370). This may be due to a partial protonation of the complexing sites. From Burkholderia cepacia, a mixed complex was obtained comprising Fe3+/pyochelin/cepabactin 1:1:1 (see Sect. 6 below) (188). An X-ray analysis has been performed of ferri-pyochelin bound to its outer membrane receptor (67a). Pyochelin II has the configuration (4′R,2″S,4″R). It does not complex Fe3+ (140).


Pyochelin I (66), aeruginoic acid (67)


X-ray structure of ferri-pyochelin I (ferri-66)
Several syntheses resulting in mixtures of stereoisomers (C-4′ and C-2″) have been developed (6, 301, 397) (Sect. 8.5). Pseudomonas fluorescens CHA0 produces enantio-pyochelin (394). The two optical antipodes are not accepted reciprocally by the two Pseudomonas species.
Pyochelin is a non-ribosomal condensation product of salicylic acid with two molecules of cysteine (289). Intermediates with one cysteine unit are aeruginoic acid (Fig. 21, 67) first isolated from Pseudomonas aeruginosa (390), and (+)-(S)-4,5-dihydroaeruginoic acid, from Pseudomonas fluorescens (57). Detailed studies (274a) suggest that N-hydroxybenzoyl-l-cysteine bound to the synthetase racemizes, that bound dihydroaeruginoic acid is still a racemate, and that in the further steps only the 4′(R) isomer is used.
Micacocidin (Fig. 22, 68) from Pseudomonas sp. complexes Fe3+ and other metal ions (189, 190). Whether it acts as a siderophore has not been investigated. A stereospecific synthesis was elaborated (161, 161a), but the same isomerization problems at C-4′ and C-2″ were encountered as had been observed with pyochelin (see Note 14 in (161)).


Micacocidin (68)
Yersiniabactin (Fig. 23, 69) was obtained from Yersinia spp., and is produced also by Pseudomonas syringae (49) and Escherichia coli (178). Its structure was elucidated independently by two groups and given the names yersiniabactin (96) and yersiniophore (64). The configurations of the four chiral centers were not determined, but epimerization probably at C-10 (corresponding to C-2″ of pyochelin) was indicated. A recent X-ray analysis (Plate 5) of the Fe3+ complex (238) established the absolute stereochemistry [N-2 (R), C-9 (R), C-10 (R) as for pyochelin, C-12 (R), C-13 (S), C-19 (S)], with Δ-configuration.


Yersiniabactin (69)


X-ray structure of ferri-yersiniabactin (ferri-69)
Anguibactin (Fig. 24, 70) from Vibrio anguillarum (171) contains DHB condensed with Cys (stereochemistry not determined). It is accompanied by a biosynthetic by-product (311) without the histamine part as its methyl ester.


Anguibactin (70), pre-acinetobactin (78), pre-pseudomonine (79)
6 Miscellaneous Siderophores
Desferri-ferrithiocin from Streptomyces antibioticus (Fig. 25, 71) (4, 254) is structurally related to the pyochelin group. It is (S)-configured and forms a Fe3+Lig2 complex (131a).


Desferri-ferrithiocin (71), cepabactin (72)
Cepabactin (Fig. 25, 72) from Burkholderia cepacia (232) forms a (Fe3+)Lig3 complex (386) and a mixed Fe3+ complex with pyochelin (Sect. 5).
Pyridine-2,6-di(monothiocarboxylic acid) (Fig. 26, 73) [for a review, see (36), cf. also (37)] was obtained from Pseudomonas putida (262) and later from Pseudomonas stutzeri (203). It forms a brown Fe3+ complex and a blue Fe2+ complex (both FeLig2) (143), which may be accompanied by complexes carrying two additional cyanide ions (145). An X-ray analysis (Plate 6) of the Fe3+ complex of 73 shows a distorted octahedral symmetry (143). There is evidence that a sulfenic acid residue (-CO-SOH) is the biosynthetic link between -COOH and -COSH (144).


Pyridine-di(monothiocarboxylic acid) (73), thioquinaldic (74), quinaldic acid (75)


X-ray structure of the Fe3+ -complex of 73
From iron-deficient cultures of Pseudomonas fluorescens, 8-hydroxy-4-methoxy-monothioquinaldic acid (thioquinolobactin) together with the corresponding quinaldic acid (quinolobactin) (Fig. 26, 74 and 75), could be isolated (258). Quinolobactin can act as an alternative siderophore of Pseudomonas fluorescens (245), although it is the hydrolysis product of the thioacid (220). Its synthesis and complex formation as (Fe3+)Lig2 was described (98).
Pseudomonine (Fig. 27, 76) is produced by Pseudomonas fluorescens strains (7, 228) and by P. entomophila, where it can act as a secondary siderophore (209). The substituents on C-4 and C-5 of the isoxazolinone ring are in trans positions (311). The complex formation has not been studied. In vitro enzyme-catalyzed synthesis studies (311, 388) showed that initially the intermediate pre-pseudomonine (Fig. 24, 79) is formed, which non-enzymatically rearranges to pseudomonine.


Pseudomonine (76), acinetobactin (77)
An analogous set of studies demonstrated that acinetobactin from Acinetobacter baumannii (392) has actually the structure 77 shown in Fig. 27 and that the one originally proposed (Fig. 24, 78) is that of pre-acinetobactin. In contrast, the thiazoline ring of anguibactin (Fig. 24, 70) (see above Sect. 5) is stable. Acinetobactin forms a 1:1 complex with Fe3+.
Domoic acid (Fig. 28, 80) (263) is a neuro-phycotoxin responsible for the mortality of wildlife and for amnesic shellfish poisoning (ASP) of humans during algal bloom. Domoic acid was first isolated from the red alga Chondria armata (“domoi” in Japanese), and it is produced also by diatoms, such as Pseudo-nitzschia spp. For the latter, evidence has been presented that it is involved in iron acquisition (307).


Domoic acid (80)
The smallest hydroxamate siderophore is N-methyl-N-thioformylhydroxylamine, CH3-N(OH)-CHS, named thioformin (100) or fluopsin (325). The synthesis was described (CH3-N(OH)-CHO + P2S5 or CH3-N(OH)-H + HCSSK) (100, 166a). It forms a purple Fe3+Lig3 complex. Pseudomonas mildenbergii produces N-methyl-N-phenylacetylhydroxylamine (CH3-N(OH)-CO-CH2-C6H5) (159), which also forms a purple Fe3+ complex.
7 Fe2+ Binding Ligands
Pseudomonas roseus fluorescens (288), Pseudomonas GH (324) and Erwinia rhapontici (113) produce pro-ferrorosamine A (81), also named pyrimine, which forms a red (Fe2+)Lig3 complex. Under acidic conditions, an open form of pro-ferrorosamine A prevails, which cannot bind Fe2+ (Chart 5). Pro-ferrorosamine B is probably an artifact produced by condensation of pro-ferrorosamine A with CHO-COOH. Pro-ferrorosamine A is essential for iron uptake by Pseudomonas (367) and for the pathogenicity of Erwinia (114).


Proferrorosamin A (81)
Structurally closely related is the Nocardia metabolite, siderochelin, for which the structure and relative and absolute stereochemistry were all established by X-ray crystallography (208, 267). It is a mixture of two epimers A and B (Fig. 29, 82 and 83). Siderochelin C, with an ethyl residue (Fig. 29, 84), was obtained from a different actinomycete, tentatively identified as Streptoalloteichus sp. (239).


Siderochelin A (R = CH3) (82), B (C-3 epimer) (83) and C (R = C2H5) (84)
The green pigments produced by Streptomyces spp. chelating Fe2+ with o-nitrosophenolate residues are occasionally referred to as siderophores, but whether they are really involved in iron metabolism has not been investigated. Ferroverdin A (11) forms a (Fe2+)Lig3 complex (55), with the ligand being p-vinylphenyl-3-nitroso-4-hydroxybenzoate (Fig. 30, 85). In ferroverdin B and C, one of the three ligands is substituted at the vinyl group (Fig. 30, 86 and 87) (346, 361). From Streptomyces murayamaensis, a precursor of ferroverdin was obtained (Fig. 30, 88) (69).


Ferroverdins
For a further chelator of Fe2+, see pyridine-2,6-di(monothiocarboxylic acid) above (Sect. 6).
8 Selected Syntheses
In this section the syntheses of several typical siderophores will be presented in a summarized form pointing out interesting features.
8.1 Anachelin H (10)
The challenge lay in the stereochemically correct synthesis of the polyketide part of the molecule. Starting from l-serine (89) (Chart 6) by C2-elongation steps, reduction of the obtained keto functions including adequate protection and deprotection, and introduction of the salicylic acid residue the four stereoisomeric 3,5-diols (90) were obtained. Comparison of the 1H-NMR data with those of anachelin (10) showed that the isomer with (3R,5S,6S) configuration was the correct starting material.


Synthesis scheme of anachelin H (10)
The chromophore part was prepared from Boc-protected N, N-dimethyl-l-DOPA (91), reduction to the diamine 92 and tellurium-mediated oxidative ring closure (93). The free amino group of 94 was coupled with protected l-Ser and l-Thr-d-Ser (95) and then the two constituent parts were connected and deprotected yielding 10 (121).
8.2 Alterobactin (26)
Several building blocks were prepared separately (Chart 7). Methyl trans-cinnamate gave by Sharpless enantiocontrolled dihydroxylation a diol from which by a series of stereo- and regioselective transformations (96) and Ru-catalyzed oxidation for transformation of the phenyl into a carboxyl group accompanied by adequate protection (97) and deprotection steps the protected OHAsp derivative 98 was obtained.


Synthesis scheme of alterobactin (26)
The protected (S)-4,8-diamino-3-oxooctanoic acid 99 was reduced with NaBH4, the resulting mixture of diastereomers was separated and the (3R,4S)-product was derivatized with benzylated DHB (100). Then derivatized d-Ser-Gly was added and the serine OH-group was esterified with the protected OHAsp (101). The Gly carboxyl group was finally set free.
The synthesis of the remaining part of the molecule started from a condensation of protected Gly with the OHAsp derivative 98, and subsequently with protected Arg (102). In the resulting protected tripeptide the Boc group from the Arg residue was removed. Connection of the two building blocks between Gly and Arg was followed by ring closure between Ser and Gly. Deprotection yielded finally alterobactin (26) (83).
8.3 Parabactin (44)
Here the critical step is the formation of the oxazoline ring. Both the stereochemistry of the two chiral centers and its acid lability had to be considered. Two approaches have been published. They can be modified for other members of this class.
The terminal NH2-groups of N 5-benzylspermidine (Chart 8) were acylated with 2,3-dimethoxybenzyol chloride and the benzyl group was removed by hydrogenolysis (28b). N 1, N 10-bis(2,3-dimethoxy)benzoylspermidine (103) was then reacted with protected l-threonine (104). The Boc group was removed with CF3COOH and the methoxy groups were cleaved with BF3 (105). Subsequent reaction with 2-hydroxybenzimidoethyl ether (106) gave parabactin (44) (28, 28c).


Synthesis I of parabactin (44)
In the second synthesis (Chart 9) of 44 the carboxyl group of benzoyl-protected salicylic acid was activated by transformation into the 1,2-thiazolidine-2-thione derivative 107 and reacted with d-threonine. The methyl ester was debenzoylated reductively (108). Treatment with SOCl2 resulted in cyclization accompanied by stereoinversion of Cβ of threonine. The resulting cis-oxazoline derivative 109 was epimerized at Cα with C2H5ONa. Subsequent hydrolysis of the ester function gave the trans-carboxylic acid 110 which was reacted with N 1, N 10-bis(benzyloxycarbonyl)spermidine by treatment with phenylbis-(2-thioxo-1,3-thiazolidine-3-yl)phosphinoxide (111). The remaining steps leading to 44 (removal of the N-protecting groups, reaction with 2,3-diacetoxybenzyl chloride and cleavage of the acetoxyl groups) were standard operations (255).


Synthesis II of parabactin (44)
In a recent modification of the second synthesis (308) effected for fluvibactin (45) an o-xylene protection group was proposed (reaction of 2,3-dihydroxybenzoic acid methyl ester with 1,2-di(bromomethyl)benzene) which could be removed later by hydrogenolysis. The formation of the oxazoline ring from protected DHB-l-threonine methyl ester was achieved with Mo(VI) catalysts (e.g. (NH4)2MoO4) without affecting the chiral centers. Derivatization of the primary amino groups of norspermidine with the protected DHB methyl ester was catalyzed by Sb(OC2H5)3.
8.4 Nannochelin A
For the condensation with the properly derivatized lysine part (112) 3′-tert-butyl-1,5-di-N-hydroxysuccinimidyl citrate (113) was used (Chart 10). It was prepared from 1,5-dimethyl citrate by reaction with tert-butyl acetate, alkaline hydrolysis of the methyl ester and coupling with N-hydroxysuccinimide by DCCI (237).


Synthesis of nannochelin A (54-dimethyl ester)


Synthesis of pyochelin stereoisomer mixture
For the synthesis of the lysine part (112) N 2-Boc-l-lysine methyl ester (114) was treated with benzoylperoxide/Na2CO3 (115) and subsequently with trans-cinnamoyl chloride yielding 116. The hydroxamate ester was deprotected with NH3/CH3OH at −23°C and the Boc group was removed with CF3COOH. Condensation with the citric acid 3-tert-butyl ester was effected with (C2H5)3N. After cleavage of the ester with CF3COOH nannochelin A (54-dimethyl ester) was obtained (29). The difficulties in the synthesis lay in the various functional and protecting groups, which had to be introduced and removed in a deliberate sequence.
8.5 Pyochelin
The problem encountered with all published syntheses (6, 301, 397) is the non-stereospecific formation of C-4′ and the facile conversion of C-2″. The common approach (Chart 11) consists in the reaction of 2-hydroxybenzonitrile (117) with l-cysteine (118) giving dihydroaeruginoic acid (119), reduction of the carboxyl group to the aldehyde 121 and condensation of the latter with l-N-methyl-cysteine. Details will be given for the procedure worked out by Zamri and Abdallah (397). The first condensation step was effected in a phosphate buffer (pH 6.4) to minimize epimerization at C-4′. Then the carboxyl group was reacted with N,O-dimethylhydroxylamine (120) using diethylcyanophosphonate as condensation agent. Reduction with LiAlH4 yielded the aldehyde 121, which then was treated with l- N-methyl-cysteine. A mixture of the four stereoisomers of (66), (4′R,2″S,4″R), (4′S,2″S,4″R), (4′R,2″R,4″R), (4′S,2″R,4″R) in a ratio of 2:1:2:5 was obtained.
9 Epilog
The history of siderophores actually began towards the end of the nineteenth century when laboratories engaged in bacteriological research observed that certain bacterial cultures showed a green fluorescence, and when in 1891 the first attempts were reported to isolate the fluorescent pigment (later namend pyoverdin, Sect. 2.1) produced by Bacterium fluorescens liquefaciens (Pseudomonas fluorescens), although it was not before 1978 that J.-M. Meyer demonstrated its being involved in the iron transport into the bacterial cell (37). Pyoverdins were among the centers of interest during the last decades, and other preferred topics were the fungal siderophores (Sect. 2.6), and more recently the marine lipopeptides (Sect. 2.8).
This review is mainly concerned with structural aspects of siderophores and their iron transport, intended to give a status report of what has been achieved up to late-2009. But the fields of interest in siderophores are much wider, spreading into
-
genetics (identification of the genes responsible for the synthesis of the siderophores and their receptors (e.g. (403, 409)),
-
medicine (siderophores as virulence factors (e.g. (75)), but serving also as carriers for antibiotics in a Trojan Horse strategy (e.g. (35a)),
-
agriculture (starving phytopathogenic bacteria by binding iron (e.g. (187b, 406)),
-
environmental problems (binding heavy metal ions (e.g. (90a)), degrading detrimental compounds (e.g. (205a)), mobilizing uranium and trans-uranium elements in contanimated soils (e.g. (242a)),
just to mention some areas. The diversity of scientific journals to be found in the References Section gives an idea of where information on siderophores can be hidden.
Structural work may take its time. Examples are Pseudomonas mendocina (Sect. 2.9) where the first structural data were reported in 2000 and the next pertinent publication appeared in 2008, or Legionella pneumophila (Sect. 4.5) whose legiobactin was first characterized in 2000, further details followed in 2007 and 2009, with loose ends in both cases. Only partially characterized siderophores are mentioned wherever data were available in order to stimulate further work. This would be worthwhile: siderophore research is a fascinating branch of natural products chemistry promising sometimes surprising results (e.g. (311, 388)).
References
Adapa S, Huber P, Keller-Schierlein W (1982) Stoffwechselprodukte von Mikroorganismen. 216. Mitteilung. Isolierung, Strukturaufklärung und Synthese von Ferrioxamin H. Helv Chim Acta 65: 1818
Adolphs M, Taraz K, Budzikiewicz H (1996) Catecholate Siderophores from Chryseomonas luteola. Z Naturforsch 51c: 281
Allard KA, Dao J, Sanjeevaiah P, McCoy-Simandle K, Chatfield CH, Crumrine DS, Castignetti D, Cianciotto NP (2009) Purification of Legiobactin and the Importance of this Siderophore in Lung Infection by Legionella pneumophila. Infect Immun 77: 2887
Amann C, Taraz K, Budzikiewicz H, Meyer JM (2000) The Siderophores of Pseudomonas fluorescens 18.1 and the Importance of Cyclopeptidic Substructures for the Recognition at the Cell Surface. Z Naturforsch 55c: 671
Ams DA, Maurice PA, Hersman LE, Forsythe JH (2002) Siderophore Production by an Aerobic Pseudomonas mendocina Bacterium in the Presence of Kaolinite. Chem Geol 188: 161
4. Anderegg G, Räber M (1990) Metal Complex Formation of a New Siderophore Desferrithiocin and of Three Related Ligands. J Chem Soc Chem Commun 1194
Anke H, Kinn J, Bergquist KE, Sterner O (1991) Production of Siderophores by Strains of the Genus Trichoderma. Isolation and Characterization of the New Lipophilic Coprogen Derivative, Palmitoylcoprogen. Biol Metals 4: 176
Ankenbauer RG, Toyokuni T, Staley A, Rinehard KL Jr, Cox CD (1988) Synthesis and Biological Activity of Pyochelin, a Siderophore of Pseudomonas aeruginosa. J Bacteriol 170: 5344
Anthoni U, Christophersen C, Nielsen PH, Gram L, Petersen BO (1995) Pseudomonine, an Isoxazolidone with Siderophoric Activity from Pseudomonas fluorescens AH2 Isolated from a Lake Victorian Nile Perch. J Nat Prod 58: 1786
Arceneau JEL, Davis WB, Downer DN, Haydon AH, Byers BR (1973) Fate of Labeled Hydroxamates during Iron Transport from Hydroxamate-Iron Chelates. J Bacteriol 115: 919
Arnow LE (1937) Colorimetric Determination of the Components of 3,4-Dihydroxyphenylalanine-Tyrosine Mixtures. J Biol Chem 118: 531
Atkin CL, Neilands JB (1968) Rhodotorulic Acid, a Diketopiperazine Dihydroxamic Acid with Growth-Factor Activity. I. Isolation and Characterization. Biochemistry 7: 3734
Atkin CL, Neilands JB, Phaff HJ (1970) Rhodotorulic Acid from Species of Leucosporidium, Rhodosporidium, Rhodotorula, Sporidiobolus, and Sporobolomyces, and a New Alanine-containing Ferrichrome from Cryptococcus melibiosum. J Bacteriol 103: 722
Awaya JD, DuBois JL (2008) Identification, Isolation, and Analysis of a Gene Cluster Involved in the Iron Acquisition by Pseudomonas mendocina ymp. Biometals 21: 353
Bachhawat AK, Ghosh S (1987) Iron Transport in Azospirillum brasiliense: Role of the Siderophore Spirilobactin. J Gen Microbiol 133: 1759
Ballio A, Bertholdt H, Carilli A, Chain EB FRS, Di Vittorio V, Tonolo A, Vero-Barcellona L (1963) Studies on Ferroverdin, a Green Iron-containing Pigment Produced by a Streptomyces Wak. Species. Proc Royal Soc London; Ser B; Biol Sci 158: 43
Barbeau K, Rue EL, Bruland KW, Butler A (2001) Photochemical Cycling of Iron in the Surface Ocean Mediated by Microbial Iron(III)-Binding Ligands. Nature 413: 409
Barbeau K, Rue EL, Trick CG, Bruland KW, Butler A (2003) Photochemical Reactivity of Siderophores Produced by Marine Heterotrophic Bacteria and Cyanobacteria Based on Characteristic Fe(III) Binding Groups. Limnol Oceanogr 48: 1069
Barbeau K, Zhang G, Live DH, Butler A (2002) Petrobactin, a Photoreactive Siderophore Produced by the Oil-Degrading Marine Bacterium Marinobacter hydrocarbonoclasticus. J Am Chem Soc 124: 378
Barelmann I, Meyer JM, Taraz K, Budzikiewicz H (1996) Cepaciachelin, a New Catecholate Siderophore from Burkholderia (Pseudomonas) cepacia. Z Naturforsch 51c: 627
Barelmann I, Taraz K, Budzikiewicz H, Geoffroy V, Meyer JM (2002) The Structures of the Pyoverdins from Two Pseudomonas fluorescens Strains Accepted Mutually by their Respective Producers. Z Naturforsch 57c: 9
Barelmann I, Uría Fernández D, Budzikiewicz H, Meyer JM (2003) The Pyoverdine from Pseudomonas chlororaphis D-TR133 Showing Mutual Acceptance with the Pyoverdine from Pseudomonas fluorescens CHA0. BioMetals 16: 263
Barklay R, Ewing DF, Ratledge C (1985) Isolation, Identification, and Structural Analysis of the Mycobactins of Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium scrofulaceum, and Mycobacterium paratuberculosis. J Bacteriol 164: 896
Barry SM, Challis GL (2009) Recent Advances in Siderophore Biosynthesis. Curr Opin Chem Biol 13: 205
Beiderbeck H (1997) Untersuchung der Pyoverdine aus Pseudomonas aeruginosa ATCC 15152, Pseudomonas fluorescens CFBP 2392 und Pseudomonas putida 2461. Diplomarbeit, Universität zu Köln, and unpublished results
Beiderbeck H, Risse D, Budzikiewicz H, Taraz K (1999) A New Pyoverdin from Pseudomonas aureofaciens. Z Naturforsch 54c: 1
Beiderbeck H, Taraz K, Budzikiewicz H, Walsby AE (2000) Anachelin, the Siderophore of the Cyanobacterium Anabena cylindrica CCAP 1403/2A. Z Naturforsch 55c: 681
Beiderbeck H, Taraz K, Meyer JM (1999) Revised Structures of the Pyoverdins from Pseudomonas putida CFBP 2461 and from Pseudomonas fluorescens CFBP 2392. BioMetals 12: 331-338
Bergeron RJ (1987) Synthesis and Properties of Polyamine Catecholamide Chelators. In: Winkelmann G, van der Helm D, Neilands JB (eds) Iron Transport in Microbes, Plants and Animals. VCH: Weinheim, p 285
Bergeron RJ, Dionis JB, Elliot GT, Kline SJ (1985) Mechanism and Stereospecificity of the Parabactin-mediated Iron-Transport System in Paracoccus denitrificans. J Biol Chem 260: 7936
Bergeron RJ, Garlich JR, McManis JS (1985) Total Synthesis of Vibriobactin. Tetrahedron 41: 507
Bergeron RJ, Huang G, Smith RE, Bharti N, McManis JS, Butler A (2003) Total Synthesis and Structure Revision of Petrobactin. Tetrahedron 59: 2007
Bergeron RJ, Kline SJ (1982) Short Synthesis of Parabactin. J Am Chem Soc 104: 4489
Bergeron RJ, Kline SJ (1984) 300-MHz 1H NMR Study of Parabactin and its Ga(III) Chelate. J Am Chem Soc 106: 3089
Bergeron RJ, Kline SJ, Stolowich NJ, McGovern KA, Burton PS (1981) Flexible Synthesis of Polyamine Catecholamides. J Org Chem 46: 4524
Bergeron RJ, McManis JS, Dionis JB, Garlich JR (1985) An Efficient Total Synthesis of Agrobactin and its Ga(III) Chelate. J Org Chem 50: 2780
Bergeron RJ, Phanstiel O iv (1992) The Total Synthesis of Nannochelin, a Novel Cinnamoyl Hydroxamate-containing Siderophore. J Org Chem 57: 7140
Bergeron RJ, Xin MG, Weimar WR, Smith RE, Wiegand J (2001) Significance of Asymmetric Sites in Choosing Siderophores as Deferration Agents. J Med Chem 44: 2469
Berner I, Konetschny-Rapp S, Jung G, Winkelmann G (1988) Characterization of Ferrioxamine E as the Principal Siderophore of Erwinia herbicola (Enterobacter agglomerans). Biol Metals 1: 51
Berti AD, Thomas MG (2009) Analysis of Achromobactin Biosynthesis by Pseudomonas syringae pv. syringae B728a. J Bacteriol 191: 4594
Bertrand S, Larcher G, Landreau A, Richomme P, Duval O, Bouchara JP (2009) Hydroxamate Siderophores of Scedosporium apiospermum. Biometals 22: 1019
Bickel H, Bosshardt R, Gäumann E, Reusser P, Vischer E, Voser W, Wettstein A, Zähner H (1960) Stoffwechselprodukte von Actinomyceten. 26. Mitteilung. Über die Isolierung und Charakterisierung der Ferrioxamine A–F, neuer Wuchsstoffe der Sideramingruppe. Helv Chim Acta 43: 2118
Bickel H, Hall GE, Keller-Schierlein W, Prelog V, Vischer E, Wettstein A (1960) Stoffwechselprodukte von Actinomyceten. 27. Mitteilung. Über die Konstitution von Ferrioxamin B. Helv Chim Acta 43: 2129
Bister B, Bischoff D, Nicholson GJ, Valdebenito M, Schneider K, Winkelmann G, Hantke K, Süssmuth RD (2004) The Structure of Salmochelins: C-Glucosylated Enterobactins of Salmonella enterica. BioMetals 17: 471
Boopathi E, Rao KS (1999) A Siderophore from Pseudomonas putida Type A1: Structural and Biological Characterization. Biochim Biophys Acta 1435: 30
Boukhalfa H, Reilly SD, Michalczyk R, Iyer S, Neu P (2006) Iron (III) Coordination Properties of a Pyoverdin Siderophore Produced by Pseudomonas putida ATCC 33015. Inorg Chem 45: 5607
Braun V, Pramanik A, Gwinner T, Köberle M, Bohn E (2009) Sideromycins: Tools and Antibiotics. Biometals 22: 3
35. Briskot G, Taraz K, Budzikiewicz H (1989) Pyoverdin-Type Siderophores from Pseudomonas aeruginosa. Liebigs Ann Chem 375
Budzikiewicz H (2001) Siderophore-Antibiotic Conjugates used as Trojan Horses against Pseudomonas aeruginosa. Curr Top Med Chem 1: 73
Budzikiewicz H (2003) Heteroaromatic Monothiocarboxylic Acids from Pseudomonas spp. Biodegradation 14: 65
Budzikiewicz H (2004) Siderophores of the Pseudomonadaceae sensu stricto (Fluorescent and Non-fluorescent Pseudomonas spp.). Progr Chem Org Nat Prod 87: 81
Budzikiewicz H (2004) Bacterial Catecholate Siderophores. Mini-Rev Org Chem 1: 163
Budzikiewicz H (2005) Bacterial Citrate Siderophores. Mini-Rev Org Chem 3: 119
Budzikiewicz H (2006) Bacterial Aromatic Sulfonates – a Bucherer Reaction in Nature? Mini-Rev Org Chem 3: 93
Budzikiewicz H, Bössenkamp A, Taraz K, Pandey A, Meyer JM (1997) Corynebactin, a Cyclic Catecholate Siderophore from Corynebacterium glutamicum ATCC 14067 (Brevibacterium sp. DSM 20411). Z Naturforsch 52c: 551
Budzikiewicz H, Kilz S, Taraz K, Meyer JM (1997) Identical Pyoverdines from Pseudomonas fluorescens 9AW and from Pseudomonas putida 9BW. Z Naturforsch 52c: 721
Budzikiewicz H, Münzinger M, Taraz K, Meyer JM (1997) Schizokinen, the Siderophore of the Plant Deleterious Bacterium Ralstonia (Pseudomonas) solanacearum ATCC 11969. Z Naturforsch 52c: 496
Budzikiewicz H, Schäfer M, Meyer JM (2007) Siderotyping of Fluorescent Pseudomonads – Problems in the Determination of Molecular Masses by Mass Spectrometry. Mini-Rev Org Chem 4: 246
Budzikiewicz H, Schäfer M, Uría Fernández D, Matthijs S, Cornelis P. (2007) Characterization of the Chromophores of Pyoverdins and Related Siderophores by Electrospray Tandem Mass Spectrometry. BioMetals 20: 135
Budzikiewicz H, Schäfer M, Uría Fernández D, Meyer JM (2006) Structure Proposal for a New Pyoverdin from Pseudomonas sp. PS 6.10. Z Naturforsch C 61c: 815
Budzikiewicz H, Schröder H, Taraz K (1992) Zur Biogenese der Pseudomonas-Siderophore: Der Nachweis analoger Strukturen eines Pyoverdin-Desferriferribactin-Paares. Z Naturforsch 47c: 26
Budzikiewicz H, Uría Fernández D, Fuchs R, Michalke R, Taraz K, Ruangviriyachai C (1999) Pyoverdins with a Lys ε-Amino Link in the Peptide Chain? Z Naturforsch 54c: 1021
Bultreys A, Gheysen I, de Hoffmann E (2006) Yersiniabactin Production by Pseudomonas syringae and Escherichia coli, and Description of a Second Yersiniabactin Locus Evolutionary Group. Appl Environ Microbiol 72: 3814
Bultreys A, Gheysen I, Wathelet B, Maraite H, de Hoffman E (2003) High-Performance Liquid Chromatography Analyses of Pyoverdin Siderophores Differentiate among Phytopathogenic Fluorescent Pseudomonas Species. Appl Environ Microbiol 69: 1143 and unpublished material
Bultreys A, Gheysen I, Wathelet B, Schäfer M, Budzikiewicz H (2004) The Pyoverdins of Pseudomonas syringae and Pseudomonas cichorii. Z Naturforsch 59c: 613
Burton MO, Sowden FJ, Lochhead AG (1954) The Isolation and Nature of the “Terregens Factor”. Can J Biochem Physiol 32: 400 (Chem Abstr 48, 10839d, 1954).
Buyer JS, de Lorenzo V, Neilands JB (1991) Production of the Siderophore Aerobactin by a Halophilic Pseudomonad. Appl Environ Microbiology 57: 2246
Byers BR, Powell MV, Lankford CE (1967) Iron-Chelating Hydroxamic Acid (Schizokinen) Active in Initiation of Cell Division in Bacillus megaterium. J Bacteriol 93: 286
Calugay RJ, Takeyama H, Mukoyama D, Fukuda Y, Suzuki T, Kanoh K, Matsunaga T (2006) Catechol Siderophore Excretion by Magnetotactic Bacterium Magnetospirillum magneticum AMB-1. J Biosci Bioeng 101: 445
Candeloro S, Gardenić D, Taylor N, Thompson B, Viswamitra M, Hodgkin DC (1969) Structure of Ferroverdin. Nature 224: 589
Capon RJ, Steward M, Ratnayake R, Lacey E, Gill JH (2007) Citromycetins and Bilains A-C: new Aromatic Polyketides and Diketopiperazines from Australian Marine-derived and Terrestrial Penicillium spp. J Nat Prod 70: 1746
Carmi R, Carmeli S, Levy E, Gough FJ (1994) (+)-(S)-Dihydroaeruginoic Acid, an Inhibitor of Septoria tritici and other Phytopathogenic Fungi and Bacteria, Produced by Pseudomonas fluorescens. J Nat Prod 57: 1200
Carrano CJ, Drechsel H, Kaiser D, Jung G, Matzanke B, Winkelmann G, Rochel N, Albrecht-Gary AM (1996) Coordination Chemistry of the Carboxylate Type Siderophore Rhizoferrin: the Iron(III) Complex and its Metal Analogs. Inorg Chem 35: 6429
Carrano CJ, Jordan M, Drechsl H, Schmid DG, Winkelmann G (2001) Heterobactins: a New Class of Siderophores from Rhodococcus erythropolis IGTS8 Containing both Hydroxamate and Catecholate Donor Groups. BioMetals 14: 119
Carrano CJ, Raymond KN (1978) Coordination Chemistry of Microbial Iron Transport Compounds. 10. Characterization of the Complexes of Rhodotorulic Acid, a Dihydroxamate Siderophore. J Am Chem Soc 100: 5371
Carrano CJ, Thieken A, Winkelmann G (1996) Specificity and Mechanism of Rhizoferrin-Mediated Metal Ion Uptake. BioMetals 9: 185
Carson KC, Glenn AR, Dilworth MJ (1994) Specificity of Siderophore-mediated Transport of Iron in Rhizobia. Arch Microbiol 161: 333
Chakraborty RN, Patel HN, Desai SB (1990) Isolation and Partial Characterization of Catechol-Type Siderophore from Pseudomonas stutzeri. Curr Microbiol 20: 283
Challis GL (2005) A Widely Distributed Bacterial Pathway for Siderophore Biosynthesis Independent of Nonribosomal Peptide Synthetases. ChemBioChem 6: 601
Chambers CE, McIntyre DD, Mouck M, Sokol PA (1996) Physical and Structural Characterization of Yersiniophore, a Siderophore Produced by Clinical Isolates of Yersinia enterocolitica. BioMetals 9: 157
Cianciotto NP (2007) Iron Acquisition by Legionella pneumophila. Biometals 20: 323
Ciche TA, Blackburn M, Carney JR, Ensign JB (2003) Photobactin: a Catechol Siderophore Produced by Photorhabdus luminescens, an Entomopathogen Mutually Associated with Heterorhabditis bacteriophora NC1 Nematodes. Appl Environ Microbiol 69: 4706
Cobessi D, Celia H, Folschweiller N, Schalk IJ, Abdallah MA, Pattus F (2005) The Crystal Structure of the Outer Pyoverdine Membrane Receptor FpvA from Pseudomonas aeruginosa at 3.6 Å Resolution. J Mol Biol 347: 121
Cobessi D, Celia H, Pattus F (2005) Crystal Structure at High Resolution of Ferric-Pyochelin and its Membrane Receptor FptA from Pseudomonas aeruginosa. J Mol Biol 352: 893
Cody YS, Gross DC (1987) Characterization of Pyoverdinpss, the Fluorescent Siderophore Produced by Pseudomonas syringae pv. syringae. Appl Environ Microbiol 53: 928
Cone MC, Melville CR, Carney JR, Gore MP, Gould, SJ (1995) 4-Hydroxy-3-nitrosobenzamide and its Ferrous Chelate from Streptomyces murayamaensis. Tetrahedron 51: 3095
Corbin JL, Bulen WA (1969) The Isolation and Identification of 2,3-Dihydroxybenzoic Acid and 2-N,6-N-di(2,3-dihydroxybenzoyl)-L-lysine Formed by Iron-deficient Azozobacter vinelandii. Biochemistry 8: 757
71. Corey EJ, Bhattacharyya S (1977) Total Synthesis of Enterobactin, a Macrocyclic Iron Transporting Agent of Bacteria. Tetrahedron Lett 3919
Cornish AS, Page WJ (1995) Production of the Triacetocholate (sic!) Siderophore Protochelin by Azotobacter vinelandii. BioMetals 8: 332
Cox CD, Rinehart KL Jr, Moore ML, Cook, JC Jr (1981) Pyochelin: Novel Structure of an Iron-chelating Growth Promotor for Pseudomonas aeruginosa. Proc Natl Acad Sci USA 78: 4256
De M, Basu M, Chakrabartty PK (2003) Increased Synthesis of Dihydroxybenzoic Acid in the Presence of Aluminum by Rhizobium MO1. Acta Microbiol Polon 52: 195
De Lorenzo V, Martinez JL (1988) Aerobactin Production as a Virulence Factor: a Reevaluation. Eur J Clin Microbiol Infect Dis 7: 621
Demange P, Bateman A, Dell A, Abdallah MA (1988) Structure of Azotobactin D, a Siderophore of Azotobacter vinelandii Strain D (CCM 289). Biochemistry 27: 2745
Demange P, Bateman A, MacLeod JK, Dell A, Abdallah MA (1990) Bacterial Siderophores: Unusual 3,4,5,6-Tetrahydropyrimidine-Based Amino Acids in Pyoverdins from Pseudomonas fluorescens. Tetrahedron Lett 31: 7611
Demange P, Bateman A, Mertz C, Dell A, Piémont Y, Abdallah MA (1990) Bacterial Siderophores: Structure of Pyoverdins Pt, Siderophores of Pseudomonas tolaasii NCPPB 2192, and Pyoverdins Pf, Siderophores of Pseudomonas fluorescens CCM 2798. Identification of an Unusual Natural Amino Acid. Biochemistry 29: 11041
Demange P, Wendenbaum S, Bateman A, Dell A, Meyer JM, Abdallah MA (1986) Bacterial Siderophores: Structure of Pyoverdins and Related Compounds. In: Swinburne TR (ed) Iron, Siderophores, and Plant Diseases. Plenum, New York, p 131
Demange P, Wendenbaum S, Linget C, Bateman A, MacLeod J, Dell A, Albrecht AM, Abdallah MA (1989) Pseudomonas Siderophores: Structure and Physicochemical Properties of Pyoverdins and Related Peptides. Second Forum on Peptides 174: 95
Demange P, Wendenbaum S, Linget C, Mertz C, Cung MT, Dell A, Abdallah MA (1990) Bacterial Siderophores: Structure and NMR Assignment of Pyoverdins Pa, Siderophores of Pseudomonas aeruginosa ATCC 15692. Biol Metals 3: 155
Deml G, Voges K, Jung G, Winkelmann G (1984) Tetraglycylferrichrome – the First Heptapeptide Ferrichrome. FEBS 173: 53
Deng J, Hamada Y, Shioiri T (1995) Total Synthesis of Alterobactin A, a Super Siderophore from an Open-Ocean Bacterium. J Am Chem Soc 117: 7824
Dertz EA, Xu J, Stintzi A, Raymond KN (2006) Bacillibactin-mediated Iron Transport in Bacillus subtilis. J Am Chem Soc 128: 22
de Voss JJ, Rutter K, Schroeder BG, Barry CE iii (1999) Iron Acquisition and Metabolism by Mycobacteria. J Bacteriol 181: 4443
Dhungana S, Michalczyk R, Boukhalfa H, Lack JG, Koppisch AT, Fairlee JM, Johnson MT, Ruggiero CE, John SG, Cox MM, Browder CC, Forsythe JH, Vanderberg LA, Neu MP, Hersman LE (2007) Purification and Characterization of Rhodobactin: a Mixed Ligand Siderophore from Rhodococcus rhodochrous Strain OFS. Biometals 20: 853
Dhungana S, Miller MJ, Dong L, Ratledge C, Crumbliss AL (2003) Iron Chelation Properties of an Extracellular Siderophore Exochelin MN. J Am Chem Soc 125: 7654
Diekmann H (1967) Stoffwechselprodukte von Mikroorganismen. 56. Mitteilung. Fusigen – ein neues Sideramin aus Pilzen. Arch Mikrobiol 58: 1
Diekmann H (1970) Stoffwechselprodukte von Mikroorganismen. 81. Mitteilung. Vorkommen und Strukturen von Coprogen B und Dimerumsäure. Arch Microbiol 73: 65
Diekmann H, Krezdorn E (1975) Stoffwechselprodukte von Mikroorganismen. 150. Mitteilung. Ferricrocin, Triacetylfusigen und andere Sideramine aus Pilzen der Gattung Aspergillus, Gruppe Fumigatus. Arch Microbiol 106: 191
Diels L, Van Roy S, Taghavi S, Van Houdt R (2009) From Industrial Sites to Environmental Applications with Cupriavidus metallidurans. Antonie van Leeuwenhoek 96: 247
Dilworth MJ, Carson KC, Giles RGF, Byrne LT, Glenn AR (1998) Rhizobium leguminosarum bv. viciae Produces a Novel Cyclic Trihydroxamate Siderophore, Vicibactin. Microbiology 144: 781
Dimise EJ, Widboom PF, Bruner SD (2008) Structure Elucidation and Biosynthesis of Fuscachelins, Peptide Siderophores from the Moderate Thermophile Thermobifida fusca. Proc Natl Acad Sci USA 105: 15311
Drechsel H, Metzger J, Freund S, Jung G, Boelaert JR, Winkelmann G (1991) Rhizoferrin – a Novel Siderophore from the Fungus Rhizopus microsporus var. rhizopodiformis. Biol Metals 4: 238
Drechsel H, Freund S, Nicholson G, Haag H, Jung O, Zähner H, Jung G. (1993) Purification and Chemical Characterization of Staphyloferrin B, a Hydrophilic Siderophore from Staphylococci. BioMetals 6: 185
Drechsel H, Jung G, Winkelmann G (1992) Stereochemical Characterization of Rhizoferrin and Identification of its Dehydration Products. BioMetals 5: 141
96. Drechsel H, Stephan H, Lotz R, Haag H, Zähner H, Hantke K, Jung G (1995) Structure Elucidation of Yersiniabactin, a Siderophore from Highly Virulent Yersinia Strains. Liebigs Ann Chem 1727
Drechsel H, Winkelmann G (1997) Iron Chelation and Siderophores. In: Winkelmann G, Carrano CJ (eds) Transition Metals in Microbial Metabolism. Harwood Academic Publishers, Amsterdam, p 1
du Moulinet d’Hardemare A, Serratrice G, Pierre JL (2004) Synthesis and Iron-binding Properties of Quinolobactin, a Siderophore from a Pyoverdine-deficient Pseudomonas fluorescens. BioMetals 17: 691
Ecker DJ, Lancaster JR Jr., Emery T (1982) Siderophore Iron Transport Followed by Electron Paramagnetic Resonance Spectroscopy. J Biol Chem 257: 8623
Egawa Y, Umino K, Ito Y, Okuda T. (1971) Antibiotic YC 73 of Pseudomonas Origin. II. Structure and Synthsis of Thioformin and its Cupric Complex (YC 73). J Antibiot 24: 124
Ehlert G, Taraz K, Budzikiewicz H (1994) Serratiochelin, a New Catecholate Siderophore from Serratia marcescens. Z Naturforsch 49c: 11
102. El Hage Chahine JM, Bauer AM, Baraldo K, Lion C, Ramiandrasoa F, Kunesch G. (2001) Kinetics and Thermodynamics of Complex Formation between FeIII and Two Synthetic Chelators of the Dicatecholspermidine Family. Eur J Inorg Chem 2287
Emery T (1971) Role of Ferrichrome as a Ferric Ionophore in Ustilago sphaerogena. Biochemistry 10: 1483
Emery T (1976) Fungal Ornthine Esterases: Relationship to Iron Transport. Biochemistry 15: 2723
Emery T. (1980) Malonichrome, a New Iron Chelate from Fusarium roseum. Biochim Biophys Acta 629: 382
Emery T (1987) Reductive Mechanisms of Iron Assimilation. In: Winkelmann G, van der Helm D, Neilands JB (eds) Iron Transport in Microbes, Plants and Animals. VCH, Weinheim, p 235
Emery T, Neilands JB (1961) Structure of the Ferrichrome Compounds. J Am Chem Soc 83: 1626
Eng-Wilmot DL, Kerley EL, Perryman DD, Brown C, Noah WH, McDyer D, Gore M, Mergo PJ, Cockburn BA (1990) Pyoverdin Type Siderophores from Various Strains of Pseudomonas aeruginosa. Reported at the International Symposium on Iron Transport and Metabolism II, 20.–22. June 1990, Austin TX, USA
Eng-Wilmot DL, Rahman A, Mendenhall JV, Grayson SL, van der Helm D (1984) Molecular Structure of Ferric Neurosporin, a Minor Siderophore-like Compound Containing N δ-hydroxy-d-ornithine. J Am Chem Soc 106: 1285
Eng-Wilmot DL, van der Helm D (1980) Molecular and Crystal Structure of the Linear Tricatechol Siderophore, Agrobactin. J Am Chem Soc 102: 7719
Feistner GJ (1995) Suggestion for a New, Semirational Nomenclature for the Free Chelators of Ferrioxamines. BioMetals 8: 193
Feistner GJ (1995) Proferrioxamine Synthesis in Erwinia amylovora in Response to Precursor or Hydroxylysine Feeding: Metabolic Profiling with Liquid Chromatography-Electrospray Mass Spectrometry. BioMetals 8: 318
Feistner G, Beaman BL (1987) Characterization of 2,3-Dihydroxybenzoic Acid from Nocardia asteroides GUH-2. J Bacteriology 169: 3982
Feistner GJ, Hsieh LL (1995) On the Collision-activated Fragmentation of Proferrioxamines: Evidence for a Succinimide-mediated Mechanism. J Am Soc Mass Spectrom 6: 836
Feistner GJ, Korth H, Ko H, Pulverer G, Budzikiewicz H (1983) Ferrorosamine A from Erwinia rhapontici. Curr Microbiol 8: 239
Feistner GJ, Mavridis A, Rudolph K (1997) Proferrorosamines and Phytopathogenicity in Erwinia spp. BioMetals 10: 1
Feistner GJ, Stahl DC, Gabrik AH (1993) Proferrioxamine Siderophores of Erwinia amylovora. A Capillary Liquid Chromatographic/Electrospray Tandem Mass Spectrometric Study. Org Mass Spectrom 28: 163
Fekete FA, Lanzi RA, Beaulieu JB, Longcope DC, Sulya AW, Hayes RN, Mabbott GA (1989) Isolation and Preliminary Characterization of Hydroxamic Acids Formed by Nitrogen-fixing Azotobacter chroococcum B-8. Appl Envir Microbiol 55: 298
Filsak G, Taraz K, Budzikiewicz H. (1994) Untersuchungen zur Struktur und Derivatisierung von Kondensationsprodukten der 2,4-Diaminobuttersäure mit anderen Aminosäuren. Z Naturforsch 49c: 18
Frederick CB, Bentley MD, Shive W (1981) Structure of Triornicin, a New Siderophore. Biochemistry 20: 2436
Frederick CB, Bentley MD, Shive W (1982) The Structure of the Fungal Siderophore, Isotriornicin. Biochem Biophys Res Commun 105: 133
Fuchs R (2000) Massenspektrometrische Untersuchung cyclischer Pyoverdine: Strukturaufklärung und Siderotyping. Dissertation; Universität zu Köln.
Fukasawa K, Goto M, Sasaki K, Hirata Y, Sato S (1972) Structure of the Yellow-Green Fluorescent Peptide Produced by Iron-deficient Azotobacter vinelandii Strain O. Tetrahedron 28: 5359
Gademann K, Bethuel Y, Locher HH, Hubschwerlen C (2007) Biomimetic Total Synthesis and Antimicrobial Evaluation of Anachelin H. J Org Chem 72: 8361
Gademann K, Budzikiewicz H (2004) The Peptide Alkaloid Anachelin: NMR Spectroscopic Evidence for a β-Turn Formation in Aqueous Solution. Chimia 58: 212
Garner BL, Arceneaux JEL, Byers BR (2004) Temperature Control of a 3,4-dihydroxybenzoate (Protocatechuate)-based Siderophore in Bacillus anthracis. Curr Microbiol 49: 89
Geisen K, Taraz K, Budzikiewicz H (1992) Neue Siderophore des Pyoverdin-Typs aus Pseudomonas fluorescens. Monatsh Chem 123: 151
Georgias H, Taraz K, Budzikiewicz H, Geoffroy V, Meyer JM (1999) The Structure of the Pyoverdin from Pseudomonas fluorescens 1.3. Structural and Biological Relationship of Pyoverdins from Different Strains. Z Naturforsch 54c: 301
Gibson F, Magrath DI (1969) The Isolation and Characterization of a Hydroxamic Acid (Aerobactin) Formed by Aerobacter aerogenes 62-I. Biochim Biophys Acta 192: 175
Gilis A, Khan MA, Cornelis P, Meyer JM, Mergeay M, van der Lelie D (1996) Siderophore-mediated Iron Uptake in Alcaligenes eutrophus CH34 and Identification of aleB Encoding the Ferric Iron-Alcaligin E Receptor. J Bacteriol 178: 5499
Gipp S, Hahn J, Taraz K, Budzikiewicz H (1991) Zwei Pyoverdine aus Pseudomonas aeruginosa R. Z Naturforsch 46c: 534
Gobin J, Moore CH, Reeve JR Jr, Wong DK, Gibson BW, Horwitz MA (1995) Iron Acquisition by Mycobacterium tuberculosis: Isolation and Characterization of a Family of Iron-binding Exochelins. Proc Natl Acad Sci USA 92: 5189
Greenwald J, Nader M, Celia H, Gruffaz C, Geoffroy V, Meyer JM, Schalk IJ, Pattus F (2009) FpvA Bound to Non-cognate Pyoverdines: Molecular Basis of Siderophore Recognition by an Iron Transporter. Mol Microbiol 72: 1246
Griffiths GL, Sigel SP, Payne SM, Neilands JB (1984) Vibriobactin, a Siderophore from Vibrio cholerae. J Biol Chem 10: 383
Gwose I, Taraz K (1992) Pyoverdine aus Pseudomonas putida. Z Naturforsch 47c: 487
Haag H, Fiedler HP, Meiwes J, Drechsel H, Jung G, Zähner H (1994). Isolation and Biological Characterization of Staphyloferrin B, a Compound with Siderophore Activity from Staphylococci. FEMS Microbiol Lett 115: 125
Hahn FE, McMurry TJ, Hugi A, Raymond KN (1990) Coordination Chemistry of Microbial Iron Transport. 42. Structural and Spectroscopic Characterization of Diastereomeric Cr(III) and Co(III) Complexes of Desferriferrithiocin. J Am Chem Soc 112: 1854
Hancock DK (1991) Isolation and Structure of a Unique Pyoverdine-type Siderophore Containing l-threo-β-Hydroxyhistidine. Ph.D Thesis, University of Maryland
133. Hancock DK, Coxon B, Wang SY, White VE, Reeder DJ, Bellama JM (1993) l-threo-β-Hydroxyhistidine, an Unprecedented Iron (III) Ion-Binding Amino Acid in a Pyoverdine-Type Siderophore from Pseudomonas fluorescens 244. J Chem Soc Chem Commun 468
Hancock DK, Reeder DJ (1993) Analysis and Configuration Assignment of the Amino Acids in a Pyoverdine-Type Siderophore by Reversed-Phase High-Performance Liquid Chromatography. J Chromatogr 646: 335
Hantke K, Nicholson G. Rabsch W, Winkelmann G (2003) Salmochelins, Siderophores of Salmonella enterica and Uropathogenic Escherichia coli Strains, are Recognized by the Outer Membrane Receptor Iron. Proc Natl Acad Sci USA 100: 3677
Harada K, Tomita K, Fujii K, Masuda K, Mikami Y, Yazawa K, Komaki H (2004) Isolation and Structural Characterization of Siderophores, Madurastatins, Produced by a Pathogenic Actinomadura madurae. J Antibiot 57: 125
Harrington, JM, Crumbliss AL (2009) The Redox Hypothesis in the Siderophore-mediated Iron Uptake. Biometals 22: 679
Harris WR, Carrano CJ, Raymond KN (1979) Coordination Chemistry of Microbial Iron Transport Compounds. 16. Isolation, Characterization, and Formation Constants of Ferric Aerobactin. J Am Chem Soc 101: 2722
Haselwandter K, Dobernigg B, Beck W, Jung G, Cansier A, Winkelmann G (1992) Isolation and Identification of Hydroxamate Siderophores of Ericoid Mycorrhizal Fungi. BioMetals 5: 51
Hayen H, Volmer DA (2006) Different Iron-chelating Properties of Pyochelin Diastereoisomers Revealed by LC/MS. Anal Bioanal Chem 385: 606
Haygood MG, Holt PD, Butler A (1993) Aerobactin Production by a Planctonic Marine Vibrio sp. Limnol Oceanogr 38: 1091
Hersman LE, Huang A, Maurice PA, Forsythe JH (2000) Siderophore Production and Iron Reduction by Pseudomonas mendocina in Response to Iron Deprivation. Geomicrobiology J 17: 261
Hickford SHJ, Küpper FC, Zhang G, Carrano CJ, Blunt JW, Butler A (2004) Petrobactine Sulfonate, a New Siderophore Produced by the Marine Bacterium Marinobacter hydrocarbonoclasticus. J Nat Prod 67: 1897
Hildebrand U, Lex J, Taraz K, Winkler S, Ockels W, Budzikiewicz H (1984) Untersuchungen zum Redox-System Bis(pyridin-2,6-dicarbothioato)-ferrat(II)/ferrat(III). Z Naturforsch 39b: 1607
Hildebrand U, Taraz K, Budzikiewicz H (1986) 6-(Hydroxythio)carbonylpyridin-2-carbonsäure und Pyridin-2-carbonsäure-6-monothiocarbonsäure als biosynthetische Zwischenstufen bei der Bildung von Pyridin-2,6-di(monothiocarbonsäure) aus Pyridin-2,6-dicarbonsäure. Z Naturforsch 41c: 691
Hildebrand U, Taraz K, Budzikiewicz H, Korth H, Pulverer G. (1985) Dicyano-bis(pyridin-2,6-dicarbothioato)-ferrat (II)/ferrat (III), ein weiteres eisenhaltiges Redoxsystem aus der Kulturlösung eines Pseudomonas-Stammes. Z Naturforsch 40c: 201
Hoegy F, Celia H, Mislin GL Vicent M, Gallay J, Schalk IJ (2005) Binding of Iron-free Siderophore, a Common Feature of Siderophore Outer Membrane Transporters of Escherichia coli and Pseudomonas aeruginosa. J Biol Chem 280: 20222
Hohlneicher U, Hartmann R, Taraz K, Budzikiewicz H (1995) Pyoverdin, Ferribactin, Azotobactin – a new Triad of Siderophores from Pseudomonas chlororaphis ATTC 9446 and its Relation to Pseudomonas fluorescens ATCC 13525. Z Naturforsch 50c: 337
Holinsworth B, Martin JD (2009) Siderophore Production by Marine-derived Fungi. Biometals 22: 625
Holt PD, Reid RR, Lewis BL, Luther GW iii, Butler A (2005) Iron(III) Coordination Chemistry of Alterobactin A: a Siderophore from the Marine Bacterium Alteromonas luteoviolacea. Inorg Chem 44: 7671
Homann VV, Edwards KJ, Webb EA, Butler A (2009) Siderophores of Marinobacter aquaeolei: Petrobactin and its Sulfonated Derivatives. Biometals 22: 565
Homann VV, Sandy M, Tincu JA, Templeton AS, Tebo BM, Butler A (2009) Loihichelins A-F, a Suite of Amphiphilic Siderophores Produced by the Marine Bacterium Halomonas LOB-5. J Nat Prod 72: 884
Hopkinson BM, Morel FMM (2009) The Role of Siderophores in Iron Acquisition by Photosynthetic Marine Microorganisms. Biometals 22: 659
Hossein MB, Eng-Wilmot DL, Loghry RA, van der Helm D (1980) Circular Dichroism, Crystal Sructure, and Absolute Configuration of the Siderophore Ferric N,N′,N″-Triacetylfusarinine, FeC39H57N6O15. J Am Chem Soc 102: 5766
Hossein MB, Jalal MAF, Benson BA, Barnes CL, van der Helm D (1987) Structure and Conformation of Two Coprogen-type Siderophores: Neocoprogen I and Neocoprogen II. J Am Chem Soc 109: 4948
Hossein MB, Jalal MAF, van der Helm D (1986) The Structure of Ferrioxamine D1- Ethanol-Water (1/2/1) Acta Cryst C 42: 1305
Hossain MB, Jalal MAF, van der Helm D (1997) 6-l-Alanineferrirubin, a Ferrichrome-type from the Fungus Aspergillus ochraceous. Acta Cryst C 53: 716
Hossein MB, van der Helm D, Poling M (1983) The Structure of Deferriferrioxamine E (Norcardamin), a Cyclic Trihydroamate. Acta Cryst B 39: 258
Hou Z, Sunderland CJ, Nishio T, Raymond KN (1996) Preorganization of Ferric Alcaligin, Fe2L3. The First Structure of a Ferric Dihydroxamate Siderophore. J Am Chem Soc 118: 5148
Hough E, Rodgers D (1974) The Crystal Structure of Ferrimycobactin P, a Growth Factor for the Mycobacteria. Biochem Biophys Res Commun 57: 73
Howard DH (1999) Acquisition, Transport, and Storage of Iron by Pathogenic Fungi. Clin Microbiol Rev 12: 394
Howard DH, Rafie R, Tiwari A, Faull KF (2000) Hydroxamate Siderophores of Histoplasma capsulatum. Infect Immun 68: 2338
Hu X, Boyer GL (1995) Isolation and Characterization of the Siderophore N-Deoxyschizokinen from Bacillus megaterium ATCC 19213. BioMetals 8: 357
Hulcher FH (1982) Isolation and Characterization of a New Hydroxamic Acid from Pseudomonas mildenbergii. Biochemistry 21: 4491
Huschka HG, Jalal MAF, van der Helm D, Winkelmann G (1986) Molecular Recognition of Siderophores in Fungi: Role of Iron-surrounding N-Acyl Residues and the Peptide Backbone During Membrane Transport in Neurospora crassa. J Bacteriol 167: 1020
Huschka H, Naegeli HU, Leuenberger-Ryf H, Keller-Schierlein W, Winkelmann G (1985) Evidence for a Common Siderophore Transport System but Different Siderophore Receptors in Neurospora crassa. J Bacteriol 162: 715
Ino A, Hasegawa Y, Murabayashi A (1998) Total Synthesis of the Antimycoplasma Antibiotic Micacocidin. Tetrahedron Lett 39: 3509
Ino A, Hasegwa Y, Murabayashi A (1999) Synthetic Studies of Thiazoline- and Thiazolidine-Containing Natural Products. 2. Total Synthesis of Antimycoplasma Antibiotic Micacocidin. Tetrahedron 55: 10283
Inoue H, Takimura O, Kawaguchi K, Nitoda T, Fuse H, Murakami K, Yamaoka Y (2003) Tin-Carbon Cleavage of Organotin Compounds by Pyoverdine from Pseudomonas chlororaphis. Appl Environ Microbiol 69: 878
Ishimaru CA, Loper JE (1992) High-Affinity Iron Uptake Systems Present in Erwinia carotovora subsp. carotovora Include the Hydroxamate Siderophore Aerobactin. J Bacteriol 174: 2993
Ito T, Neilands JB (1958) Products of "Low-iron Fermentation" with Bacillus subtilis: Isolation, Characterization and Synthesis of 2,3-Dihydroxybenzoylglycine. J Am Chem Soc 80: 4645
Ito Y, Butler A (2005) Structure of Synechobactins, New Siderophores of the Marine Cyanobacterium Synechococcus sp. PCC 7002. Limnol Oceanogr 50: 1918
Ito Y, Ishida, K, Okada S, Murakami M (2004) The Absolute Stereochemistry of Anachelins, Siderophores from the Cyanobacterium Anabena cylindrica. Tetrahedron 60: 9075
Ito Y, Umino K, Sekiguchi T, Miyagishima T, Egawa Y (1971) Antibiotic YC 73 of Pseudomonas Origin. III. Synthesis of Thioformin Analogues. J Antibiot 24: 131
Itou Y, Okada S, Murakami M (2001) Two Structural Isomeric Siderophores from the Freshwater Cyanobacterium Anabena cylindrica (NIES-19). Tetrahedron 57: 9093
Jacques P, Ongena M, Gwose I, Seinsche D, Schröder H, Delphosse P, Thonart P, Taraz K, Budzikiewicz H (1995). Structure and Characterization of Isopyoverdin from Pseudomonas putida BTP1 and its Relation to the Biogenetic Pathway Leading to Pyoverdins. Z Naturforsch 50c: 622
Jalal MAF, Galles JL, van der Helm D (1985) Structure of Des(diserylglycyl)ferrirhodin, DDF, a Novel Siderophore from Aspergillus ochraceous. J Org Chem 50: 5642
Jalal MAF, Hossain MB, van der Helm D, Barnes CL (1988) Structure of Ferrichrome-type Siderophores with Dissimilar N δ-acyl Groups: Asperchrome B1, B2, B3, D1, D2 and D3. Biol Metals 1: 77
Jalal MAF, Hossain MB, van der Helm D, Sanders-Loehr J, Actis LA, Crosa JH (1989) Structure of Anguibactin, a Unique Plasmid-related Siderophore from Fish-pathogen Vibrio anguillarum. J Am Chem Soc 111: 292
Jalal MAF, Love SK, van der Helm D (1986) Siderophore Mediated Iron(III) Uptake in Gliocladium virens. 1. Properties of cis-Fusarinine, trans-Fusarinine, Dimerum Acid, and their Ferric Complexes. J Inorg Biochem 28: 417
Jalal MAF, Love SK, van der Helm D (1988) N α -Dimethylcoprogens. Three Novel Trihydroxamate Siderophores from Pathogenic Fungi. Biol Metals 1: 4
Jalal MAF, Mocharla R, Barnes CL, Hossain MB, Powell DR, Eng-Wilmot DL, Grayson SL, Benson BA, van der Helm D (1984) Extracellular Siderophores from Aspergillus ochraceous. J Bacteriol 158: 683
Jalal MAF, Mocharla R, van der Helm D (1984) Separation of Ferrichromes and Other Hydroxamate Siderophores of Fungal Origin by Reversed-phase Chromatography. J Chromatogr 301: 247
Jalal MAF, van der Helm D (1989) Siderophores of Highly Phytopathogenic Alternaria longipes. Structures of Hydroxycoprogens. Biol Metals 2: 11
Jalal MAF, van der Helm D (1991) Isolation and Spectroscopic Identification of Fungal Siderophores. In: Winkelmann G (ed) Handbook of Microbial Iron Chelates. CRC, Boca Raton, FL, p 235
Jones AM, Lindow SE, Wildermuth MC (2007) Salicylic Acid, Yersiniabactin, and Pyoverdin Production by the Model Phytopathogen Pseudomonas syringae pv. tomato DC3000: Synthesis, Regulation, and Impact on Tomato and Arabidopsis Host Plants. J Bacteriol 189: 6773
Jülich M, Taraz K, Budzikiewicz H, Geoffroy V, Meyer JM, Gardan L (2001) The Structure of the Pyoverdin Isolated from Pseudomonas syringae Pathovars. Z Naturforsch 56c: 687
Kachadourian R, Dellagi A, Laurent J, Bricard L, Kunesch G, Expert D (1996) Desferrioxamine-dependent Iron Transport in Erwinia amylovora CFBP1430: Cloning of the Gene Encoding the Ferrioxamine Receptor FoxR. BioMetals 9: 143
Kameyama T, Takahashi A, Kurasawa S, Ishizuka M, Okami Y, Takeuchi T, Umezawa H (1987) Bisucaberin, a New Siderophore, Sensitizing Tumor Cells to Macrophage-mediated Cytolysis. I. Taxonomy of the Producing Organism, Isolation and Biological Properties. J Antibiotics 40: 1664
Kamnev AA (1998) Reductive Solubilization of Fe(III) by Certain Products of Plant and Microbial Metabolism as a Possible Alternative to Siderophore Secretion. Dokl Biophys 358 - 360, 48 (translation from Dokl Akad Nauk 359: 691).
Kanoh K, Kamino K, Leleo G, Adachi K, Shizuri Y (2003) Pseudoalterobactin A and B, New Sidrophores Excreted by Marine Bacterium Pseudoalteromonas sp. KP20-4. J Antibiotics 56: 871
Keller-Schierlein W, Deér A (1963) Stoffwechselprodukte von Mikroorganismen. 44. Mitteilung. Zur Konstitution von Ferrichrysin und Ferricrocin. Helv Chim Acta 46: 1907
Keller-Schierlein W, Diekmann H (1970) Stoffwechselprodukte von Mikroorganismen. 85. Mitteilung. Zur Konstitution des Coprogens. Helv Chim Acta 53: 2035
Keller-Schierlein W, Hagmann L, Zähner H, Huhn W (1988) Stoffwechselprodukte von Mikroorganismen. 250. Mitteilung. Maduraferrin, ein neuartiger Siderophor aus Actinomadura madurae. Helv Chim Acta 71: 1528
Keller-Schierlein W, Mertens P, Prelog V, Walser A (1965) Stoffwechselprodukte von Mikroorganismen. 49. Mitteilung. Die Ferrioxamine A1, A2 und D2. Helv Chim Acta 48: 710
Keller-Schierlein W, Prelog V (1961) Stoffwechselprodukte von Actinomyceten. 29. Mitteilung. Die Konstitution des Ferrioxamins D1. Helv Chim Acta 44: 709
Keller-Schierlein W, Prelog V (1961) Stoffwechselprodukte von Actinomyceten. 30. Mitteilung. Über das Ferrioxamin E; ein Beitrag zur Konstitution des Nocardamins. Helv Chim Acta 44: 1981
Keller-Schierlein W, Prelog V (1961) Stoffwechselprodukte von Actinomyceten. 34. Mitteilung. Ferrioxamin G. Helv Chim Acta 45: 590
Keller-Schierlein W, Prelog V, Zähner H (1964) Siderochrome (Natürliche Eisen(III)-trihydroxamat-Komplexe). Progr Chem Org Nat Prod 22: 279
Kilz S, Lenz C, Fuchs R, Budzikiewicz H (1999) A Fast Screening Method for the Identification of Siderophores from Fluorescent Pseudomonas spp. by Liquid Chromatography/Electrospray Mass Specrtrometry. J Mass Spectrom 34: 281
Kloepper JW, Leong J, Teintze M, Schroth MN (1980) Pseudomonas Siderophores: A Mechanism Explaining Disease-Suppressive Soils. Curr Microbiol 4: 317
Klumpp C, Burger A, Mislin GL, Abdallah MA (2005) From a Total Synthesis of Cepabactin and its 3:1 Ferric Complex to the Isolation of a 1:1:1 Mixed Complex between Iron (III), Cepabactin and Pyochelin. Bioorg Med Chem Lett 15: 1721
Kobayashi S, Hidaka S, Kawamura Y, Ozaki M, Hayase Y (1998) Micacocidin A, B and C, Novel Antimycoplasma Agents from Pseudomonas sp. I. Taxonomy, Fermentation, Isolation, Physico-chemical Properties and Biological Activities. J Antibiotics 51: 323
Kobayashi S, Nakai H, Ikenishi Y, Sun WY, Ozaki M, Hayase Y, Takeda R (1998) Micacocidin A, B and C, Novel Antimycoplasma Agents from Pseudomonas sp. II. Structure Elucidation. J Antibiotics 51: 328
Kokubo S, Suenaga K, Shinohara C, Tsuji T, Uemura D (2000) Structures of Amamistatins A and B, Novel Growth Inhibitors of Human Tumor Cell Lines from Nocardia asteroides. Tetrahedron 56: 6435
Konetschny-Rapp S, Huschka HG, Winkelmann G, Jung G (1988) High-performance Liquid Chromatography of Siderophores from Fungi. Biol Metals 1: 9
Konetschny-Rapp S, Jung G, Meiwes J, Zähner H (1900) Staphyloferrin A: a Structurally New Siderophore from Staphylococci. Eur J Biochem 191: 65
Konetschny-Rapp S, Jung G, Raymond KN, Meiwes J, Zähner H (1992) Solution Thermodynamics of the Ferric Complexes of New Desferrioxamine Siderophores by Directed Fermentation. J Am Chem Soc 114: 2224
Koppisch AT, Browder CC, Moe AL, Shelley JT, Kinkel BA, Hersman LE, Iyer S, Ruggiero CE (2005) Petrobactin is the Primary Siderophore Synthesized by Bacillus anthracis str. Sterne under Conditions of Iron Starvation. BioMetals 18: 577
Koppisch AT, Dhungana S, Hill KK, Boukhalfa H, Heine HS, Colip LA, Romero RB, Shou Y, Ticknor LO, Marrone BL, Hersman LE, Iyer S, Riggiero CE (2008) Petrobactin is Produced by Both Pathogenic and Non-pathogenic Isolates of the Bacillus cereus Group of Bacteria. Biometals 21: 581
Korth H (1970) Über das Vorkommen von 2,3-Dihydroxybenzoesäure und ihrer Aminosäurederivate in Kulturmedien von Klebsiella oxytoca. Arch Microbiol 70: 297
Kunze B, Bedorf N, Kohl W, Höfle G, Reichenbach H (1989) Myxochelin A, a New Iron-Chelating Compound from Angiococcus disciformis (Myxobacterales). Production, Isolation, Physicochemical and Biological Properties. J Antibiotics 42: 14
Kunze B, Trowitzsch-Kienast W, Höfle G, Reichenbach H (1992) Nannochelins A, B and C, New Iron Chelating Compounds from Nannocystis exedens (Myxobacteria). Production, Isolation, Physico-chemical and Biological Properties. J Antibiotics 45: 147
Lane SJ, Marshall PS, Upton RJ, Ratledge C, Ewing M (1995) Novel Extracellular Mycobactins, the Carboxymycobactins from Mycobacterium avium. Tetrahedron Lett 36: 4129
Lankford CE, Walker JR, Reeves JB, Nabbut NH, Byers BR, Jones RJ (1966) Inoculum-Dependent Division Lag of Bacillus Cultures and its Relation to an Endogenous Factor(s) ("Schizokinen"). J Bacteriol 91: 1070
Ledyard KM, Butler A (1997) Structure of Putrebactin, a New Dihydroxamate Siderophore Produced by Shewanella putrefaciens. J Biol Inorg Chem 2: 93
Lee BH, Miller MJ (1983) Natural Ferric Ionophores: Total Synthesis of Schizokinen, Schizokinen A, and Arthrobactin. J Org Chem 48: 24
Lee CH, Lewis TA, Paszczynski A, Crawford RL (1999) Identification of an Extracellular Catalyst of Carbon Tetrachloride Dehalogenation from Pseudomonas stutzeri Strain KC as Pyridine-2,6-bis(thiocarboxylate). Biochem Biophys Res Commun 261: 562
Leong SA, Winkelmann G (1998) Molecular Biology of Iron Transport in Fungi. In: Sigel A, Sigel H (eds) Metal Ions in Biological Systems. Marcel Dekker, New York, 147
Lesueur D, Diem HG, Meyer JM (1993) Iron Requirement and Siderophore Production in Bradyrhizobium Strains Isolated from Acacia mangium. J Appl Bacteriol 74: 675
Lewis TA, Crawford RL (1995) Transformation of Carbon Tetrachloride via Sulfur and Oxygen Substitution by Pseudomonas sp. Strain KC. J Bacteriol 177: 2204
Liles MR, Scheel TA, Cianciotto NP (2000) Discovery of a Nonclassical Siderophore, Legiobactin, Produced by Strains of Legionella pneumophila. J Bacteriol 182: 749
Linke WD, Crueger A, Diekmann H (1972) Stoffwechselprodukte von Mitroorganismen. 106. Mitteilung. Zur Konstitution des Terregens-Faktors. Arch Mikrobiol 85: 44
Liu WC, Fisher SM, Wells JS Jr, Ricca CS, Principe PA, Trejo WH, Bonner DP, Gougoutos JZ, Toeplitz BK, Sykes RB (1981) Siderochelin, a New Ferrous-ion Chelating Agent Produced by Nocardia. J Antibiot 34: 791
Llinás L, Neilands JB (1976) The Structure of Two Alanine Containing Ferrichromes: Sequence Determination by Proton Magnetic Resonance. Biophys Struct Mechanism 2: 105
Loomis LD, Raymond KN (1991) Solution Equilibria of Enterobactin and Metal-enterobactin Complexes. Inorg Chem 30: 906
Luo M, Fadeev EA, Groves JT (2005) Membrane Dynamics of the Amphiphilic Siderophore, Acinetoferrin. J Am Chem Soc 127: 1726
MacDonald JC, Bishop GG (1984) Spectral Properties of a Mixture of Fluorescent Pigments Produced by Pseudomonas aeruginosa. Biochim Biophys Acta 800: 11
Maksimova NP, Blazhevich OV, Lysak VV, Fomichev YuK (1994) Characteristics of fluorescent pigment pyoverdin Pm produced by Pseudomonas putida bacteria. Microbiology 63: 587 (Russian original:Mikrobiologia 63: 1038)
Marshall B, Stintzi A, Gilmour C, Meyer JM, Poole K (2009) Citrate-mediated Iron-uptake in Pseudomonas aeruginosa: Involvement of the Citrate-inducible FecA Receptor and the FeoB Ferrous Iron Transporter. Microbiology 155: 305
Martin JD, Ito Y, Homann VV, Haygood MG, Butler A (2006) Structure and Membrane Affinity of New Amphiphilic Siderophores Produced by Ochrobactrum sp. SP18. J Biol Inorg Chem 11: 633
Martinez JS, Butler A (2007) Marine Amphiphilic Siderophores: Marinobactin Structure, Uptake, and Microbial Partitioning. J Inorg Biochem 101: 1692
Martinez JS, Carter-Franklin JN, Mann EL, Martin JD, Haygood MG, Butler A (2003) Structure and Membrane Affinity of a Suite of Amphiphilic Siderophores Produced by a Marine Bacterium. Proc Natl Acad Sci USA 100: 3754
Martinez JS, Zhang GP, Holt PD, Jung HT, Carrano CJ, Haygood MG, Butler A (2000) Self-assembling Amphiphilic Siderophores from Marine Bacteria. Science 287: 1245
Matthijs S, Budzikiewicz H, Schäfer M, Wathelet B, Cornelis P (2008) Ornicorrugatin, a New Siderophore from Pseudomonas fluorescens AF76. Z Naturforsch 63c: 8
Matthijs S, Laus G, Meyer JM, Abbaspour-Tehrani K, Schäfer M, Budzikiewicz H, Cornelis P (2009) Siderophore-mediated Iron Acquisition in the Entomopathogenic Bacterium Pseudomonas entomophila L48 and its Close Relative Pseudomonas putida KT2440. Biometals 22: 951
Matthijs S, Tehrani KA, Laus G, Jackson RW, Cooper RM, Cornelis P (2007) Thioquinolobactin, a Pseudomonas Siderophore with Antifungal and anti-Pythium Activity. Environ Microbiol 9: 425
Maurer B, Müller A, Keller-Schierlein W, Zähner H (1968) Stoffwechselprodukte von Mikroorganismen. 61. Ferribactin, ein Siderochrom aus Pseudomonas fluorescens Migula. Arch Microbiol 60: 326
Maurer PJ, Miller MJ (1982) Microbial Iron Chelators: Total Synthesis of Aerobactin and its Constituent Amino Acid, N 6-Acetyl-N 6-hydroxylysine. J Am Chem Soc 104: 3096
May JJ, Wendrich TM, Mahariel MA (2001) The dhb Operon of Bacillus subtilis Encodes the Biosynthetic Template for the Catecholic Siderophore 2,3-Dihydroxybenzoate-glycine-threonine Trimeric Ester Bacillibactin. J Biol Chem 276: 7209
McCullough WG, Merkal RS (1982) Structure of Mycobactin J. Curr Microbiol 7: 337
McDougall S, Neilands JB (1984) Plasmid- and Chromosome-coded Aerobactin Synthesis in Enteric Bacteria: Insertion Sequences Flank Operon in Plasmid-mediated Systems. J Bacteriol 159: 300
Meiwes J, Fiedler HP, Haag H, Zähner H, Konetschny-Rapp S, Jung G (1990) Isolation and Characterization of Staphyloferrin A, a Compound with Siderophore Activity from Staphylococcus hyicus DSM 20459. FEMS Microbiol Lett 67: 201
Meiwes J, Fiedler HP, Zähner H, Konetschny-Rapp S, Jung G (1990) Production of Desferrioxamine E and New Analogues by Directed Fermentation and Feeding Fermentation. Appl Microbiol Biotechnol 32: 505
Mercado-Blanco J, van der Drift KMGM, Olsson PE, Thomas-Oates JE, van Loon LC, Bakker PAHM (2001) Analysis of the pmsCEAB Gene Cluster Involved in Biosynthesis of Salicylic Acid and the Siderophore Pseudomonine in the Biocontrol Strain Pseudomonas fluorescens WCS374. J Bacteriol 183: 1909
Messenger AJM, Ratledge C (1982) Iron Transport in Mycobacterium smegmatis: Uptake of Iron from Ferric Citrate. J Bacteriol 149: 131
Meyer JM (2000) Pyoverdines: Pigments, Siderophores and Potential Taxonomic Markers of Fluorescent Pseudomonas Species. Arch Microbiol 174: 135
Meyer JM, Abdallah MA (1980) The Siderochromes of Non-fluorescent Pseudomonads: Production of Norcardamine by Pseudomonas stutzeri. J Gen Microbiol 118: 125
Meyer JM, Azelvandre P, Georges C (1992). Iron Metabolism in Pseudomonas: Salicylic Acid, a Siderophore of Pseudomonas fluorescens CHAO. BioFactors 4: 23
Meyer JM, Gruffaz C, Raharinosy V, Bezverbnaya I, Schäfer M, Budzikiewicz, H (2008) Siderotyping of Fluorescent Pseudomonas: Molecular Mass Determination by Mass Spectrometry as a Powerful Pyoverdine Siderotyping Method. Biometals 21: 259
Meyer JF, Hohnadel D, Hallé F (1989) Cepabactin from Pseudomonas cepacia, a New Type of Siderophore. J Gen Microbiol 135: 1479
Meyer JM, Stintzi A, Coulanges V, Shivaji S, Voss JA, Taraz K, Budzikiewicz H (1998) Siderotyping of Fluorescent Pseudomonads: Characterization of Pyoverdines of Pseudomonas fluorescens and Pseudomonas putida Strains from Antarctica. Microbiology 144: 3119
Meyer JM, Stintzi A, De Vos D, Cornelis P, Tappe R, Taraz K, Budzikiewicz H (1997) Use of Siderophores to Type Pseudomonads: the Three Pseudomonas aeruginosa Pyoverdine Systems. Microbiology 143: 35
Meyer JM, Van VT, Stintzi A, Berge O, Winkelmann G (1995) Ornibactin Production and Transport Properties in Strains of Burkholderia vietnamensis and Burkholderia cepacia (formerly Pseudomonas cepacia). BioMetals 8: 309
Michalke R, Taraz K, and Budzikiewicz H (1996) Azoverdin – an Isopyoverdin. Z Naturforsch 51c: 772
Milewska MJ, Chimiak A, Glowacki Z (1987) Synthesis of Shizokinen, Homoschizokinen, its Imide and the Detection of Imide with 13C-N.M.R.-Spectroscopy. J Prakt Chem 329: 447
Miller MC, Parkin S, Fetherston JD, Perry RD, DeMoll E (2006) Crystal Structure of Ferric-yersiniabactin, a Virulence Factor of Yersinia pestis. J Inorg Biochem 100: 1495
Mitscher LA, Högberg T, Drake SD, Burgstahler AW, Jackson M, Lee B, Sheldon RI, Gracey HE, Kohl W, Theriault RJ (1984) Isolation and Structural Determination of Siderochelin C, a Fermentation Product of an Unusual Actinomycetes sp. J Antibiotics 37:1260
Miyanaga S, Obata T, Onaka H, Fujita T, Saito N, Sakurai H, Saiki I, Furumai T, Igarashi Y (2006) Absolute Configuration and Antitumor Activity of Myxochelin A Produced by Nonomuraea pusilla TP-A0861. J Antibiotics 59: 698
Modi M, Shah KS, Modi VV (1985) Isolation and Characterization of Catechol-like Siderophore from Cowpea Rhizobium RA-1. Arch Microbiol 141: 156
Mohn G, Koehl P, Budzikiewicz H, Lefèvre JF (1994) Solution Structure of Pyoverdin GM-II. Biochemistry 33: 2843
Mohn G, Taraz K, Budzikiewicz H (1990) New Pyoverdin-Type Siderophores from Pseudomonas fluorescens. Z Naturforsch 45b: 1437
Moll H, Glorius M, Bernhard G, Johnsson A, Pedersen K, Schäfer M, Budzikiewicz H (2008) Characterization of Pyoverdins Secreted by a Subsurface Strain of Pseudomonas fluorescens and their Interactions with Uranium(VI). Geomicrobiol J 25: 157
Moore RE, Emery T (1976) N α-Acetylfusarinines: Isolation, Characterization, and Properties. Biochemistry 15: 2719
Moore CH, Foster LA, Gerbig DG Jr, Dyer DW, Gibson BW (1995) Identification of Alcaligin as the Siderophore Produced by Bordetella pertussis and B. bronchiseptica. J Bacteriol 177: 1116
Mossialos D, Meyer JM, Budzikiewicz H, Wolf U, Koedam N, Baysse C, Anjaiah V, Cornelis P (2000) Quinolobactin, a New Siderophore of Pseudomonas fluorescens ATCC 17400, the Production of which is Repressed by the Cognate Pyoverdine. Appl Environ Microbiol 66: 487
Mukai A, Fukai T, Matsumoto Y, Ishikawa J, Hoshino Y, Yazawa K, Harada K, Mikami Y (2006) Transvalencin Z, a New Antimicrobial Compound with Salicylic Acid Residue from Nocardia transvalensis IFM 10065. J Antibiotics 59: 366
Mulet M, Gomila M, Gruffaz C, Meyer JM, Palleroni NJ, Lalucat J, García-Valdéz E (2008) Phylogenetic Analysis and Siderotyping as Useful Tools in the Taxonomy of Pseudomonas stutzeri: Description of a Novel Genovar. Int J System Evol Microbiol 58: 2309
Müller A, Zähner H (1968) Stoffwechselprodukte von Mikroorganismen. 65. Mitteilung. Ferrioxamine aus Eubacteriales. Arch Microbiol 62: 257
Müller SI, Valdebenito M, Hantke K (2009) Salmochelin, the Long-overlooked Catecholate Siderophore of Salmonella. Biometals 22: 691
Mullis KB, Pollack JR, Neilands JB (1971) Structure of Schizokinen, an Iron Transport Compound from Bacillus megaterium. Biochemistry 10: 4894
Münzinger M, Budzikiewicz H, Expert D, Enard C, Meyer JM (2000) Achromobactin, a New Citrate Siderophore of Erwinia chrysanthemi. Z Naturforsch 55c: 328
Münzinger M, Taraz K, Budzikiewicz H (1999) Staphyloferrin B, a Citrate Siderophore from Ralstonia eutropha. Z Naturforsch 54c: 867
Münzinger M, Taraz K, Budzikiewicz H, Drechsel H, Heymann P, Winkelmann G, Meyer JM (1999). S,S-Rhizoferrin (enantio-rhizoferrin) a Siderophore of Ralstonia (Pseudomonas) pickettii DSM 6297 – the Optical Antipode of R,R-Rhizoferrin Isolated from Fungi. BioMetals 12: 189
Murakami Y, Kato S, Nakajima M, Matsuoka M, Kawai H, Shin-Ya K, Seto H (1996) Formobactin, a Novel Free Radical Scavenging and Neuron Cell Protecting Substance from Nocardia sp. J Antibiotics 49: 839
Naegli HU, Keller-Schierlein W (1978) Stoffwechselprodukte von Mikroorganismen. 174. Mitteilung. Eine neue Synthese des Ferrichroms; enantio-Ferrichrom. Helv Chim Acta 61: 2088
Naegeli HU, Zähner H (1980) Stoffwechselprodukte von Mikroorganismen. 193. Mitteilung. Ferrithiocin. Helv Chim Acta 63: 1400
255. Nagao Y, Miyasaka T, Hagiwara Y, Fujita E 1984 Total Synthesis of Parabactin, a Spermidine Siderophore. J Chem Soc Perkin Trans I 183
Neilands JB (1981) Microbial Iron Compounds. Ann Rev Biochem 50: 715
Neilands JB, Ericson TJ, Rastetter WH (1981) Stereospecifity of the Ferric Enterobactin Receptor of Escherichia coli K-12. J Biol Chem 256: 3831
Nemoto A, Hoshino Y, Yazawa K, Ando A, Mikami Y, Komaki H, Tanaka Y, Gräfe U (2002) Asterobactin, a New Siderophore Group Antibiotic from Nocardia asteroides. J Antibiotics 55: 593
Neuenhaus W, Budzikiewicz H, Korth H, Pulverer G (1980) 8-Hydroxy-4-methoxymonothiochinaldinsäure – eine weitere Thiosäure aus Pseudomonas. Z Naturforsch 35b: 1569
Newkirk JD, Hulcher FH (1969) Isolation and Properties of a Fluorescent Pigment from Pseudomonas mildenbergii. Arch Biochem Biophys 134: 395
Nishio T, Tanaka N, Hiratake J, Katsube Y, Ishida Y, Oda J (1988) Isolation and Structure of the Novel Dihydroxamate Siderophore Alcaligin. J Am Chem Soc 110: 8733
O’Brian IG, Gibson F (1970) The Structure of Enterochelin and Related 2,3-Dihydroxy-N-benzoylserine Conjugates from Escherichia coli. Biochem Biophys Acta 215: 393
262. Ockels W, Römer A, Budzikiewicz H, Korth H, Pulverer G (1978) An Fe(II) Complex of Pyridine-2,6-di(monothiocarboxylic acid) – a Novel Bacterial Metabolic Product. Tetrahedron Lett 3341
Ohfune Y, Tomita M (1982) Total Synthesis of (−)-Domoic acid. A Revision of the Original Structure. J Am Chem Soc 104: 3511
Okujo N, Saito M, Yamamoto S, Yoshida T, Miyoshi S, Shinoda S (1994) Structure of Vulnibactin, a New Polyamine-containing Siderophore from Vibrio vulnificus. BioMetals 7: 109
Okujo N, Sakakibara Y, Yoshida T, Yamamoto S. (1994) Structure of Acinetoferrin, a New Citrate-based Dihydroxamate Siderophore from Acinetobacter haemolyticus. BioMetals 7: 170
Okujo N, Yamamoto S (1994) Identification of the Siderophores from Vibrio hollisae and Vibrio mimicus as Aerobactin. FEMS Microbial Lett 118: 187
Okuyama D, Nakamura H, Naganawa H, Takita T, Umezawa H, Iitaka, Y (1982) Isolation, Racemization and Absolute Configuration of Siderochelin A. J Antibiotics 35: 1240
Ong SA, Peterson T, Neilands JB (1979) Agrobactin, a Siderophore from Agrobacterium tumefaciens. J Biol Chem 25: 1860
Ongena M, Jacques P, Delfosse P, Thonart P (2002) Unusual Traits of the Pyoverdin-Mediated Iron Acquisition System in Pseudomonas putida Strain BTP1. BioMetals 15: 1
Ongena M, Jacques P, Thonart P, Gwose I, Uría Fernández D, Schäfer M, Budzikiewicz H (2001) The Pyoverdin of Pseudomonas fluorescens BTP2, a Novel Structural Type. Tetrahedron Lett 42: 5849
Ongena M, Jacques P, van Vyncht G, Charlier P, de Pauw E, Thonart P, Budzikiewicz H (1998) Structural Analysis of Two Pyoverdins by Electrospray and FAB Mass Spectrometry. J Mass Spectrom Soc Jpn 46: 53
Page WJ, Collinson SK, Demange P, Dell A, Abdallah MA (1991) Azotobacter vinelandii Strains of Disparate Origin Produce Azotobactin Siderophores with Identical Structures. Biol Metals 4: 217
Page WJ, von Tigerstrom M (1988) Aminochelin, a Catecholamine Siderophore Produced by Azotobacter vinelandii. J Gen Microbiol 134: 453
Pakchung AAH, Soe CZ, Codd R (2008) Studies of Iron-uptake Mechanisms in Two Bacterial Species of the Shewanella Genus Adapted to Middle-range (Shewanella putrefaciens) or Antarctic (Shewanella gelidimarina) Temperatures. Chem Biodivers 5: 2113
Patel HM, Tao J, Walsh CT (2003) Epimerization of an l-Cysteinyl to a d-Cysteinyl Residue during Thiazoline Ring Formation in Siderophore Chain Elongation by Pyochelin Synthetase from Pseudomonas aeruginosa. Biochem 42: 10514
Patel HN, Chakraborty RN, Desai SB (1988) Isolation and Partial Characterization of Phenolate Siderophore from Rhizobium leguminosarum IARI 102. FEMS Microbiol Lett 56: 131.
Pattus F, Abdallah MA (2000) Siderophores and Iron-transport in Microorganisms. J Chin Chem Soc 47: 1
Payne SM, Niesel DW, Peixotto SS, Lawlor KM (1983) Expression of Hydroxamate and Phenolate Siderophores by Shigella flexneri. J Bacteriol 155: 949
Persmark M, Expert D, Neilands JB (1989) Isolation, Characterization, and Synthesis of Chrysobactin, a Compound with Siderophore Activity from Erwinia chrysanthemi. J Biol Chem 264: 3187
Persmark M, Frejd T, Mattiasson B (1990) Purification, Characterization, and Structure of Pseudobactin 589A, a Siderophore from a Plant Growth Promoting Pseudomonas. Biochemistry 29: 7348
Persmark M, Neilands JB (1992) Iron(III) Complexes of Chrysobactin, the Siderophore of Erwinia chrysanthemi. BioMetals 5: 29
Persmark M, Pittman P, Buyer JS, Schwyn B, Gill PR Jr, Neilands JB (1993) Isolation and Structure of Rhizobactin 1021, a Siderophore from the Alfalfa Symbiont Rhizobium meliloti 1021. J Am Chem Soc 115: 3950
Peters WJ, Warren RAJ (1968) Itoic Acid Synthesis in Bacillus subtilis. J Bacteriol 95: 360
Peterson T, Falk KE, Leong SA, Klein MP, Neilands JB (1980). Structure and Behavior of Spermidine Siderophores. J Am Chem Soc 102: 7715
284. Peterson T, Neilands JB (1979) Revised Structure of a Catecholamide Spermidine Siderophore from Paracoccus denitrificans. Tetrahedron Lett 4805
Plowman JE, Loehr TM, Goldman SJ, Sanders-Loehr J (1984) Structure and Siderophore Activity of Ferric Schizokinen. J Inorg Biochem 20: 183
Pollak JR, Neilands JB (1970) Enterobactin, an Iron Transport Compound from Salmonella typhimurium. Biochem Biophys Res Commun 38: 989
Poppe K, Taraz K, Budzikiewicz H (1987) Pyoverdine Type Siderophores from Pseudomonas fluorescens. Tetrahedron 43: 2261
Pouteau-Thouvenot M, Gaudemer A, Barbier M (1968). Structure chimique de la pro-ferrorosamine. Bull Soc Chim Biol 50: 222
Quadri LEN, Keating TA, Patel HM, Walsh CT (1999) Assembly of Pseudomonas aeruginosa Nonribosomal Peptide Siderophore Pyochelin: in vitro Reconstitution of Aryl-4, 2-bisthiazoline Synthetase Activity from PchD, PchE, and PChF. Biochemistry 38: 14941
Rabsch W, Paul P, Reissbrodt R (1986) DHBA (2,3-Dihydroxybenzoesäure)-Ausscheidung durch einen enterobactinnegativen multiresistenten Salmonella typhimurium-Wildstamm. J Basic Microbiol 26: 113
Rabsch W, Paul P, Reissbrodt R (1987) A New Hydroxamate Siderophore for Iron Supply of Salmonella. Acta Microbiol Hungar 34: 85
Ratledge C (1964) Relationship between the Products of Aromatic Biosynthesis in Mycobacterium smegmatis and Aerobacter aerogenes. Nature 203: 428
Ratledge C, Ewing M (1996) The Occurrence of Carboxymycobactin, the Siderophore of Pathogenic Mycobacteria, as a Second Extracellular Siderophore in Mycobacterium smegmatis. Microbiology 142: 2207
Ratledge C, Marshall BJ (1972) Iron Transport in Mycobacterium smegmatis: the Role of Mycobactin. Biochim Biophys Acta 279: 58
Ratledge C, Patel PV (1976) The Isolation, Properties and Taxonomic Relevance of Lipid-soluble, Iron-binding Compounds (the Nocobactins) from Nocardia. J Gen Microbiology 93: 141
Ratledge C, Snow GA (1974) Isolation and Structure of Nocobactin NA, a Lipid-soluble Iron-binding Compound from Nocardia asteroides. Biochem J 139: 407
Raymond KN, Carrano CJ (1979) Coordination Chemistry and Microbial Iron Transport. Acc Chem Res 12: 183
Regenhardt D, Heuer H, Heim S, Fernández DU, Strömpl C, Moore ERB, Timmis KN (2002) Pedigree and Taxonomic Credentials of Pseudomonas putida Strain KT2440. Environ Microbiol 4: 912
Reid RT, Live DH, Faulkner DJ, Butler A (1993) A Siderophore from a Marine Bacterium with an Exceptional Ferric Ion Affinity Constant. Nature 366: 455
Reissbrodt R, Ramiandrasoa F, Bricard L, Kunesch G (1997) Siderophore Activity of Chemically Synthesized Dihydroxybenzoyl Derivatives of Spermidines and Cystamide. BioMetals 10: 95
Renshaw JC, Robson GD, Trinci APJ, Wiebe MG, Livens FR, Collison D, Taylor RJ (2002) Fungal Siderophores: Structures, Functions and Application. Mycol Res 106: 1123
Rinehart KL, Staley AL, Wilson SR, Ankenbauer RG, Cox CD (1995) Stereochemical Assignment of the Pyochelins. J Org Chem 60: 2786
Risse D, Beiderbeck H, Taraz K, Budzikiewicz H, Gustin D (1998) Corrugatin, a Lipopeptide Siderophore from Pseudomonas corrugata. Z Naturforsch 53c: 295
Robinson JP, Wawrousek EF, McArdle JV, Coyle G, Adler I (1984) X-ray Photoelectron and Electron Spin Resonance Spectra of Iron(III) Parabactin. Inorg Chim Acta 92: L19
Ruangviryachai C, Barelmann I, Fuchs R, Budzikiewicz H (2000) An Exceptionally Large Pyoverdin from a Pseudomonas Strain Collected in Thailand. Z Naturforsch 55c: 323
Ruangviriyachai C, Uría Fernández D, Fuchs R, Meyer JM, Budzikiewicz H (2001) A New Pyoverdin from Pseudomonas aeruginosa R’. Z Naturforsch 56c: 933
Ruangviriyachai C, Uría Fernández D, Schäfer M, Budzikiewicz H (2004) Structure Proposal for a New Pyoverdin from a Thai Pseudomonas putida Strain. Spectroscopy 18: 453
Rue E, Bruland K (2001) Domoic Acid Binds Iron and Copper: a Possible Role for the Toxin Produced by the Marine Diatom Pseudo-nitzschia. Mar Chem 76: 127
308. Sakakura A, Umemura S, Ishihara K (2008) Convergent Total Syntheses of Fluvibactin and Vibriobactin Using Molybdenum(vi) Oxide-catalyzed Dehydrative Cyclization as a Key Step. Chem Commun 3561
Salah-el-Din ALM, Kyslík P, Stephan D, Abdallah MA (1997) Bacterial Iron Transport: Structure Elucidation by FAB-MS and by 2D NMR (1H, 13C, 15N) of Pyoverdin G4R, a Peptidic Siderophore Produced by a Nitrogen-Fixing Strain of Pseudomonas putida. Tetrahedron 53: 12539
Sansinenea E, Ortiz A (2009) Bacterial Siderophores Containing a Thiazoline Ring. Mini-Rev Org Chem 6: 120
Sattely ES, Walsh CT (2008) A Latent Oxazoline Electrophile for N-O-C Bond Formation in Pseudomonine Biosynthesis. J Am Chem Soc 130: 12282
Saxena B, Modi M, Modi VV (1986) Isolation and Characterization of Siderophores from Azospirillum lipoferum D-2. J Gen Microbiol 132: 2219
Sayer JM, Emery TF (1968) Structures of the Naturally Occurring Hydroxamic Acids, Fusarinines A and B. Biochemistry 7: 184
Schaffner EM, Hartmann R, Taraz K, Budzikiewicz H (1996) Structure Elucidation of Azotobactin 87, Isolated from Azotobacter vinelandii ATCC 12837. Z Naturforsch 51c: 139
Schalk IJ, Hennard C, Dugave C, Poole K, Abdallah MA, Pattus F (2001) Iron-free Pyoverdin Binds to its Outer Membrane Receptor FpvA in Pseudomonas aeruginosa: a New Mechanism for Membrane Iron Transport. Mol Microbiol 39: 351
Schalk IJ, Kyslik P, Prome D, van Dorsselaer A, Poole K, Abdallah MA, Pattus F (1999) Copurification of the FpvA Ferric Pyoverdin Receptor of Pseudomonas aeruginosa with its Iron-Free Ligand: Implications for Siderophore-Mediated Iron Transport. Biochemistry 38: 9357
Schlegel K, Fuchs R, Schäfer M, Taraz K, Budzikiewicz H, Geoffroy V, Meyer JM (2001) The Pyoverdins of Pseudomonas sp. 96-312 and 96-318. Z Naturforsch 56c: 680
Schlegel K, Lex J, Taraz K, Budzikiewicz H (2006) The X-ray Structure of the Pyochelin Fe3+ Complex. Z Naturforsch 61c: 263
Schlegel K, Taraz K, Budzikiewicz H (2004) The Stereoisomers of Pyochelin, a Siderophore of Pseudomonas aeruginosa. BioMetals 17: 409
Schröder H, Adam J, Taraz K, Budzikiewicz H (1995) Dihydropyoverdinsulfonsäuren – Zwischenstufen bei der Biogenese? Z Naturforsch 50c: 616
Seinsche D, Taraz K, Budzikiewicz H, Gondol D (1993) Neue Pyoverdin-Siderophore aus Pseudomonas putida C. J Prakt Chem 335: 157
Shah S, Rao KK, Desai A (1993) Production of Catecholate Type of Siderophores by Azospirillum lipoferum M. Indian J Exp Biol 31: 41
321. Shanzer A, Libman J (1983) Total Synthesis of Enterobactin via an Organotin Template. J Chem Soc Chem Commun 846
Sharman GJ, Williams DH, Ewing DF, Ratledge C (1995) Isolation, Purification and Structures of Exochelin MS, the Extracellular Siderophore from Mycobacterium smegmatis. Biochem J 305: 187
Sharman GJ, Williams DH, Ewing DF, Ratledge C (1995) Determination of the Structure of Exochelin MN, the Extracellular Siderophore from Mycobacterium neoaurum. Chem Biol 2: 553
Shiman R, Neilands JB (1965) Isolation, Characterization and Synthesis of Pyrimine, an Iron(II) Binding Agent from Pseudomonas GH. Biochemistry 4: 2233
Shirahata K, Deguchi T, Hayashi T, Matsubara I, Suzuki T (1970). The Structures of Fluopsins C and F. J Antibiotics 23: 546
Simpson FB, Neilands JB (1976) Siderochromes in Cyanophyceae: Isolation and Characterization of Schizokinen from Anabena sp. J Phycol 12: 44
Skorupska A, Choma A, Derylo M, Lorkiewicz Z (1988) Siderophore Containing 2,3-Dihydroxybenzoic Acid and Threonine Formed by Rhizobium trifolli (sic!). Acta Biochim Polon 35: 119
Smith MJ, Shoolery JN, Schwyn B, Holden I, Neilands JB (1985) Rhizobactin, a Structurally Novel Siderophore from Rhizobium meliloti. J Am Chem Soc 107: 1739
Snow GA (1965) The Structure of Mycobactin P, a Growth Factor for Mycobacterium johnei, and the Significance of its Iron Complex. Biochem J 94: 160
Snow GA (1965) Isolation and Structure of Mycobactin T, a Growth Factor from Mycobacterium tuberculosis. Biochem J 97: 166
Snow GA (1970) Mycobactins: Iron-chelating Growth Factors from Mycobacteria. Bacteriol Rev 34: 99
Snow GA, White AJ (1969) Chemical and Biological Properties of Mycobactins Isolated from Various Mycobacteria. Biochem J 115: 1031
Sokol PA, Lewis CJ, Dennis JJ (1992) Isolation of a Novel Siderophore from Pseudomonas cepacia. J Med Microbiol 36: 184
Spiro TG, Bates G, Saltman P (1989) The Hydrolytic Polymerization of Ferric Citrate. II. The Influence of Excess Citrate. J Am Chem Soc 89: 5559
Spiro TG, Pape L, Saltman P (1989) The Hydrolytic Polymerization of Ferric Citrate. I. The Chemistry of the Polymer. J Am Chem Soc 89: 5555
Steglich W, Steffan B, Stroech K, Wolf M (1984) Pistillarin, ein charakteristischer Inhaltsstoff der Herkuleskeule (Clavariadelphus pistillaris) und einiger Ramaria-Arten (Basidiomycetes). Z Naturforsch. 39c: 10
Stephan H, Freund S, Beck W, Jung G, Meyer JM, Winkelmann G (1993) Ornibactins – a New Family of Siderophores from Pseudomonas. BioMetals 6: 93
336. Stephan H, Freund S, Meyer JM, Winkelmann G, Jung G (1993) Structure Elucidation of the Gallium-Ornibactin Complex by 2D-NMR Spectroscopy. Liebigs Ann Chem 43
Stintzi A, Barnes C, Xu J, Raymond KN (2000) Microbial Iron Transport via a Siderophore Shuttle: a Membrane Ion Transport Paradigm. Proc Natl Acad Sci USA 97: 10691
Stoll A, Renz J, Brack A (1951) Beiträge zur Konstitutionsaufklärung des Nocardamins. 10. Mitteilung über antibakterielle Stoffe. Helv Chim Acta 34: 862
Storey EP, Boghozian R, Little JL, Lowman DW, Charkraborty R (2006) Characterization of ‘Schizokinen’; a Dihydroxamate-type Siderophore Produced by Rhizobium leguminosarum IARI 917. BioMetals 19: 637
Stuart SJ, Prpic JK, Robins-Browne RM (1986) Production of Aerobactin by some Species of the Genus Yersinia. J Bacteriol 166: 1131
Suenaga K, Kokubo S, Shinohara C, Tsuji T, Uemura D (1999) Structures of Amamistatins A and B, Novel Growth Inhibitors of Human Tumor Cell Lines from an Actinomycete. Tetrahedron Lett 40: 1945
Sultana R, Fuchs R, Schmickler H, Schlegel K, Budzikiewicz H, Siddiqui BS, Geoffroy V, Meyer JM (2000) A Pyoverdin from Pseudomonas sp. CFML 95-275. Z Naturforsch 55c: 857
Sultana R, Siddiqui BS, Taraz K, Budzikiewicz H, Meyer JM (2000) A Pyoverdine from Pseudomonas putida CFML 90-51 with a Lys ε-Amino Link in the Peptide Chain. BioMetals 13: 147
Sultana R, Siddiqui BS, Taraz K, Budzikiewicz H, Meyer JM (2001) An Isopyoverdin from Pseudomonas putida CFML 90-44. Z Naturforsch 56c: 303
Sultana R, Siddiqui BS, Taraz K, Budzikiewicz H, Meyer JM (2001) An Isopyoverdin from Pseudomonas putida CFML 90-33. Tetrahedron 57: 1019
Tabata N, Tomoda H, Ōmura S (1999) Ferroverdins, Inhibitors of Cholesteryl Ester Transfer Protein Produced by Streptomyces sp. WK-5344. II. Structure Elucidation. J Antibiotics 52: 1108
Tait GH (1975) The Identification and Biosynthesis of Siderochromes Formed by Micrococcus denitrificans. Biochem J 146: 191
Takahashi A, Nakamura H, Kameyama T, Kurasawa S, Naganawa H, Okami Y, Takeuchi T, Umezawa H, Iitaka Y (1987) Bisucaberin, a New Siderophore, Sensitizing Tumor Cells to Macrophage-mediated Cytolysis. II. Physico-chemical Properties and Structure Determination. J Antibiotics 40: 1671
Tappe R (1991) Ferribactin 3b, ein Siderophor von Pseudomonas aptata 3b. Diplomarbeit, Universität zu Köln
Tappe R, Taraz K, Budzikiewicz H, Meyer JM, Lefèvre JF (1993) Structure Elucidation of a Pyoverdin Produced by Pseudomonas aeruginosa ATCC 27853. J Prakt Chem 335: 83
Taraz K, Ehlert G, Geisen K, Budzikiewicz H, Korth H, Pulverer G (1990) Protochelin, ein Catecholat-Siderophor aus einem Bakterium (DMS Nr. 5746). Z Naturforsch 45b: 1327
Taraz K, Seinsche D, Budzikiewicz H (1991) Pseudobactin- und Pseudobactin A-Varianten: Neue Peptidsiderophore vom Pyoverdin-Typ aus Pseudomonas fluorescens “E2”. Z Naturforsch 46c: 522
Teintze M, Hossain MB, Barnes CL, Leong J, van der Helm D (1981) Structure of Ferric Pseudobactin, a Siderophore from a Plant Growth Promoting Pseudomonas. Biochemistry 20: 6446
Teintze M, Leong J (1981) Structure of Pseudobactin A, a Second Siderophore from Plant Growth Promoting Pseudomonas B10. Biochemistry 20: 6457
Telford JR, Leary JA, Tunstad LMG, Byers BR, Raymond KN (1994) Amonabactin: Characterization of a Series of Siderophores from Aeromonas hydrophila. J Am Chem Soc 116: 4499
Telford JR, Raymond KN (1997) Amonabactin: a Family of Novel Siderophores from a Pathogenic Bacterium. J Biol Inorg Chem 2: 750
Telford JR, Raymond KN (1998) Coordination Chemistry of the Amonabactins, Bis(catecholate) Siderophores from Aeromonas hydrophila. Inorg Chem 37: 4578
Temirov YuV, Esikova TZ, Kashporov IA, Balashova TA, Vinokurov LM, Alakhov YuB (2003) A Catecholic Siderophore Produced by the Thermoresistant Bacillus licheniformis VK21 Strain. Russian J Bioorg Chem 29: 542 (translated from Bioorg Khim 29: 597)
Thieken A, Winkelmann G (1992) Rhizoferrin: a Complexon Type Siderophore of the Mucorales and Entomophthorales (Zygomycetes). FEMS Microbiol Lett 94: 37
Thomas MS (2007) Iron Acquisition Mechanisms of the Burkholderia cepacia Complex. Biometals 20: 431
Tindale AE, Mehrotra M, Ottem D, Page WJ (2000) Dual Regulation of Catecholate Siderophore Biosynthesis in Azotobacter vinelandii by Iron and Oxidative Stress. Microbiology 146: 1617
Tomada H, Tabata N, Shinose M, Takahashi Y, Woodruff HB, Ōmura S (1999) Ferroverdins, Inhibitors of Cholesteryl Ester Transfer Protein Produced by Streptomyces sp. WK-5344. I. Production, Isolation and Biological Properties. J Antibiotics 52: 1101
Torres L, Pérez-Ortín JE, Tordera V, Beltrán JP (1986) Isolation and Characterization of an Fe(III)-Chelating Compound Produced by Pseudomonas syringae. Appl Environ Microbiol 2: 157
Uría Fernández D, Fuchs R, Schäfer M, Budzikiewicz H, Meyer JM (2003) The Pyoverdin of Pseudomonas fluorescens G173, a Novel Structural Type Accompanied by Unexpected Natural Derivatives of the Corresponding Ferribactin. Z Naturforsch 58c: 1
Uría Fernández D, Fuchs R, Taraz K, Budzikiewicz H, Munsch P, Meyer JM (2001) The Structure of a Pyoverdine Produced by a Pseudomonas tolaasii-like Isolate. BioMetals 14: 81
Uría Fernández D, Geoffroy V; Schäfer M, Meyer JM, Budzikiewicz H (2003) Structure Revision of Several Pyoverdines Produced by Plant-Growth Promoting and Plant-Deleterious Pseudomonas Species. Monatsh Chem 134: 1421
van der Helm D, Jalal MAF, Hossain MB (1987) The Crystal Structures, Conformations, and Configurations of Siderophores. In: Winkelmann G, van der Helm D, Neilands JB (eds) Iron Transport in Microbes, Plants and Animals. VCH, Weinheim, p 135
van der Helm D, Poling M (1976) The Crystal Structure of Ferrioxamine E. J Am Chem Soc 98: 82
van de Woestyne M, Bruyneel B, Mergeay M, Verstraete W (1991) The Fe2+ Chelator Proferrorosamine A is Essential for the Siderophore-mediated Uptake of Iron by Pseudomonas roseus fluorescens. Appl Environ Microbiol 57: 949
Van Tiel-Menkveld GJ, Mentjox-Vervuurt JM, Oudega B, de Graaf FK (1982) Siderophore Production by Enterobacter cloacae and a Common Receptor Protein for the Uptake of Aerobactin and Cloacin DF13. J Bacteriol 150: 490
Vergne AF, Walz AJ, Miller MJ (2000) Iron Chelators from Mycobacteria (1954-1999) and Potential Therapeutic Applications. Nat Prod Rep 17: 99
Visca P, Colotti G, Serino L, Verzili D, Orsi N, Chiancone E (1992) Metal Regulation of Siderophore Synthesis in Pseudomonas aeruginosa and Functional Effects of Siderophore-Metal Complexes. Appl Environ Microbiol 58: 2886
Voss J, Taraz K, Budzikiewicz H (1999) A Pyoverdin from the Antarctica Strain 51W of Pseudomonas fluorescens. Z Naturforsch 54c: 156
Voßen W, Fuchs R, Taraz K, Budzikiewicz H (2000) Can the Peptide Chain of a Pyoverdin be Bound by an Ester Bond to the Chromophore? – The Old Problem of Pseudobactin 7SR1. Z Naturforsch 55c: 153
Voßen W, Taraz K (1999) Structure of the Pyoverdin PVD 2908 – a New Pyoverdin from Pseudomonas sp. 2908. BioMetals 12: 323
Wallner A, Blatzer M, Schrettl M, Sarg B, Lindner H, Haas H (2009) Ferricrocin – a Siderophore Involved in Intra- and Transcellular Iron Distribution in Aspergillus fumigatus. Appl Envir Microbiol 75: 4194
Wang W, Chi Z, Liu G., Buzdar MA, Chi Z, Gu Q (2009) Chemical and Biological Characterization of Siderophores Produced by the Marine-derived Aureobasidium pullulans HN6.2 and its Antibacterial Activity. Biometals 22: 965
Wang QX, Phanstiel O iv (1998) Total Synthesis of Acinetoferrin. J Org Chem 63: 1491
Warner PJ, Williams PH, Bindereif A, Neilands JB (1981) CoIV Plasmid-Specific Aerobactin Synthesis by Invasive Strains of Escherichia coli. Infect Immunol 33: 540
Wasielewski E, Adkinson RA, Abdallah MA, Kieffer B (2002). The Three-Dimensional Structure of the Gallium Complex of Azoverdin, a Siderophore of Azomonas macrozytogenes ATCC 12334, Determined by NMR Using Residual Dipolar Coupling Constants. Biochemistry 41: 12488
Wawszkiewicz EJ, Schneider HA (1975) Control of Salmonellosis Pacifarin Biosynthesis by Iron. Infect Immunol 11: 69
Weber M, Taraz K, Budzikiewicz H, Geoffroy V, Meyer JM (2000) The Structure of a Pyoverdine from Pseudomonas sp. CFML 96.188 and its Relation to other Pyoverdines with a Cyclic C-terminus. BioMetals 13: 301
Westervelt P, Bloom ML, Mabbott GA, Fekete FA (1985) The Isolation and Identification of 3,4-Dihydroxybenzoic Acid Formed by Nitrogen-fixing Azomonas macrocytogenes. FEMS Microbiol Lett 30: 331
White AJ, Snow GA (1969) Isolation of Mycobactins from Various Mycobacteria. The Properties of Mycobactins S and H. Biochem J 111: 785
Wilson MK, Abergel RJ, Raymon J, Arceneaux JE, Byers BR (2006) Siderophores from Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis. Biochem Biophys Res Commun 348: 320
Winkelmann G (1990) Structural and Stereochemical Aspects of Iron Transport in Fungi. Biotech Adv 8: 207
Winkelmann G (1991) Specificity of Iron Transport in Bacteria and Fungi. In: Winkelmann G (ed) CRC Handbook of Microbial Iron Chelates. CRC, Boca Raton, FL, p 65
Winkelmann G, Huschka HG (1989) Molecular Recognition and Transport of Siderophores in Fungi. In: Winkelmann G, van der Helm D, Neilands JB (eds) Iron Transport in Microbes, Plants and Animals. VCH, Weinheim, p 317
Winkler S, Ockels W, Budzikiewicz H, Korth H, Pulverer G (1986) 2-Hydroxy-4-methoxy-5-methylpyridin-N-oxid, ein Al3+-bindender Metabolit von Pseudomonas cepacia. Z Naturforsch 41c: 807
Wong-Lun-Sang S, Bernardini JJ, Hennard C, Kyslík P, Dell A, Abdallah MA (1996) Bacterial Siderophores: Structure Elucidation, 2D 1H and 13C NMR Assignments of Pyoverdins Produced by Pseudomonas fluorescens CHA0. Tetrahedron Lett 37: 3329
Wuest WM, Sattely ES, Walsh CT (2009) Three Siderophores from one Bacterial Enzymatic Assembly Line. J Am Chem Soc 131: 5056
Xu G, Martinez JS, Groves JT, Butler A (2002) Membrane Affinity of the Amphiphilic Marinobactin Siderophores. J Am Chem Soc 124:13408
Yamada Y, Seki N, Kitahara T, Takahashi M, Matsui M (1970) Structure and Synthesis of Aeruginoic Acid (2-o-hydroxyphenylthiazole-4-carboxylic acid). Agric Biol Chem 34: 780
Yamamoto S, Okujo N, Fujita Y, Saito M, Yoshida T, Shinoda S (1993) Structures of Two Polyamine-containing Catecholate Siderophores from Vibrio fluvialis. J Biochem 113: 538
Yamamoto S, Okujo N, Sakakibara Y (1994) Isolation and Structure Elucidation of Acinetobactin, a Novel Siderophore from Acinetobacter baumannii. Arch Mikrobiol 162: 249
Yamamoto S, Okujo N, Yoshida T, Matsuura S, Shinoda S (1994) Structure and Iron Transport Activity of Vibrioferrin, a New Siderophore of Vibrio parahaemolyticus. J Biochem 115: 868
Youard ZA, Mislin GLA, Majcherczyk PA, Schalk IJ, Reimmann C (2007) Pseudomonas fluorescens CHA0 Produces enantio-Pyochelin, the Optical Antipode of the Pseudomonas aeruginosa Siderophore Pyochelin. J Biol Chem 282: 35546
Zähner H, Keller-Schierlein W, Hütter R, Hess-Leisinger K, Deér A (1963) Stoffwechselprodukte von Mikroorganismen. 40. Mitteilung. Sideramine aus Aspergillaceen. Arch Mikrobiol 45: 119
Zalkin A, Forrester JD, Templeton DH (1966) Ferrichrome A Tetrahydrate. Determination of Crystal and Molecular Structure. J Am Chem Soc 88: 1810
Zamri A, Abdallah MA (2000) An Improved Stereocontrolled Synthesis of Pyochelin, Siderophore of Pseudomonas aeruginosa and Burkholderia cepacia. Tetrahedron 56: 249
Zawadzka AM, Vandecasteele FPJ, Crawford RL, Paszczynski AJ (2006) Identification of Siderophores from Pseudomonas stutzeri. Can J Microbiol 52: 1164
Abergel RJ, Zawadzka AM, Hoette TM, Raymond KM (2009) Enzymatic Hydrolysis of Trilactone Siderophores: Where Chiral Recognition Occurs in Enterobactin and Bacillibactin Iron Transport. J Am Chem Soc 131: 12682
Amin SA, Green DH, Küpper FC, Carrano CJ (2009) Vibrioferrin, an Unusual Marine Siderophore: Iron Binding, Photochemistry and Biological Implications. Inorg Chem 48: 11451
Barbeau K (2006) Photochemistry of Organic Iron (III) Complexing Ligands in Oceanic Systems. Photochem Photobiol 82: 1505
Bergeron RJ, McManis JS, Perumal PT, Algee SE (1991) The Total Synthesis of Alcaligin. J Org Chem 56: 5560
Crosa JH, Walsh CT (2002) Genetics and Assembly Line Enzymology of Siderophore Biosynthesis in Bacteria. Microbiol Mol Biol Rev 66: 223
Fadeev E, Luo M, Groves JT (2005) Synthesis and Structural Modeling of the Amphiphilic Siderophore Rhizobactin-1021 and its Analogs. Bioorg Med Chem Lett 15: 3771
Ghosh A, Miller MJ (1993) Synthesis of Novel Citrate-Based Siderophores and Siderophore-β-lactam Conjugates. Iron Transport-mediated Drug Delivery Systems. J Org Chem 58: 7652
Lemenceau P, Expert D, Gaymard F, Bakker PAHM, Briat JF (2009) Role of Iron in Plant-Microbe Interaction. Adv Bot Res 51: 491
Mukai A, Komaki H, Takagi M, Shin-ya K (2009) Novel Siderophore, JBIR-16, Isolated from Nocardia tenerifensis 101015. J Antibiot 62: 601
Peuckert F, Miethke M, Albrecht AG, Essen LO, Marahiel MA (2009) Structural Basis and Stereochemistry of Tricatecholate Siderophore Binding by FeuA. Angew Chemie Int Ed 48: 7924
Ravel J, Cornelis P (2003) Genomics of Pyoverdin-mediated Iron Uptake in Pseudomonads. Trends Microbiol 11: 195
Sandy M, Butler A (2009) Microbial Iron Acquisition: Marine and Terrestrial Siderophores. Chem Rev 109: 4580
Takeuchi Y, Nagao Y, Toma K, Yoshikawa Y, Akiyama T, Nishioka H, Abe H, Harayama T, Yamamoto S (1999) Synthesis and Siderophore Activity of Vibrioferrin and One of its Diastereomeric Isomers. Chem Pharm Bull 47: 1284
Lautru S, Deeth RJ, Bailey LM, Challis GL (2005) Discovery of a New Peptide Natural Product by Streptomyces coelicolor Genome Mining. Nature Chem Biol 1: 65
Robbel L, Knappe TA, Linne U, Xie X, Marahiel MA (2009) Erythrochelin - a Hydroxamate-type Siderophore Predicted from the Genome of Saccharopolyspora erythraea. FEBS J published online DOI 10.1111/j.1742-4658.2009.07512.x
Acknowledgement
Many thanks are due to Dr. J. Neudörfl for preparing the Plates with siderophore X-ray structures. Data bases for the X-ray structures: Plate 1: FEPSBC 10; 3: TEQKQV; 4: YELJOP; 5: VENPAC; 6: CUHGUH.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendices
Appendix
Notes Added in Proof
2.1 Section 2.5
Coelichelin from Streptomyces coelicolor comprises D-N 5-formyl-N 5-hydroxy-Orn-D-aThr bound to N 5 of L-N 5-hydroxy-Orn whose N2 is acylated by D-N 5-formyl-N 5-hydroxy-Orn (412).
2.2 Section 2.6
Erythrochelin from Saccharopolyspora erythraea is a coprogen-type siderophore (Table 2) with Ac1 = i and Ac2 = D-Ser-D-N 2, N 5-diacetyl-N 5-hydroxy-Orn (413).
2.3 Section 2.7
The transport system of Bacillus subtilis accommodates the Fe3+ complexes of enterobactin (Δ-configured), enantio-D-enterobactin and of corynebactin (bacillibactin) (both Λ). Since only Λ complexes can be bound to the receptor a configurational change from Δ to Λ is induced. Only the natural ferri-L-siderophores can be degraded enzymatically (399, 408).
From Nocardia tenerifensis the heterobactin JBIR-16 was obtained (30, R = DHB). The stereochemistry of the two Orn residues was not established. By mass spectrometry a 1:1 Fe3+/Lig ratio was determined for the red complex (407).
Rights and permissions
Copyright information
© 2010 Springer-Verlag/Wien
About this chapter
Cite this chapter
Budzikiewicz, H. (2010). Microbial Siderophores. In: Kinghorn, A., Falk, H., Kobayashi, J. (eds) Fortschritte der Chemie organischer Naturstoffe / Progress in the Chemistry of Organic Natural Products, Vol. 92. Fortschritte der Chemie organischer Naturstoffe / Progress in the Chemistry of Organic Natural Products, vol 92. Springer, Vienna. https://doi.org/10.1007/978-3-211-99661-4_1
Download citation
DOI: https://doi.org/10.1007/978-3-211-99661-4_1
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-211-99660-7
Online ISBN: 978-3-211-99661-4
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)