
Abstract
The influenza A viruses are dangerous pathogens with the potential to provoke devastating disease. The challenge for the medical research community is to design preventive measures and therapeutic interventions that will limit the severe consequences of pandemic influenza A virus infections. Vaccines have long been available, but there is considerable scope for improvement as they target only the prevailing influenza A virus strains, do not give broad immunity, and work poorly in the elderly, the target group that is most at risk of fatal disease. Improved vaccines will only emerge if the development strategy is based on a firm understanding of the host immune response to the virus. Here, we summarize the research to date that details immune mechanisms participating in the control and elimination of influenza A viruses.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Major Histocompatibility Complex Class
- Avian Influenza
- Neutralize Antibody
- Natural Antibody
- Antigenic Drift
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
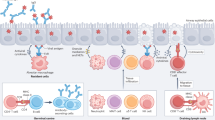
The influenza viruses are Orthomyxoviruses with an eight-segmented, negative-sense, single-stranded RNA genome. There are three types: influenza A, B, and C. The influenza A viruses that cause the most serious problems in humans are the subject of this review. These pathogens are classified according to their two major surface glycoproteins: hemagglutinin (HA or H) and neuraminidase (NA or N). Infecting both mammalian and avian species, the highly contagious influenza A viruses are responsible for widespread morbidity and mortality [1]. In mammals, infection is established in the upper and lower respiratory tracts, provoking an illness that is associated with fever, myalgia, congestion, pharyngitis, and, in severe cases, pneumonia. Early on, some of the very virulent influenza A viruses can induce a “cytokine shock” syndrome mediated via the innate immune response pathway. Fortunately, infection also elicits potent adaptive immunity and long-term memory, though the virus can mutate readily, allowing strains with variant HA molecules to cause successive pandemics. The current killed or subunit vaccines induce effective antibody responses in normal adults, though they do not promote a virus-specific CD8+ T-cell response and memory and they are poorly immunogenic in those who are even marginally immunologically compromised. The major task for immunologists interested in the problem that influenza virus poses is to develop better vaccines. Most of our detailed knowledge about immunity to the influenza A viruses is derived from the murine model that allows rigorous analysis due to the availability of an extensive panel of defined analytical reagents. Here, we provide a comprehensive summary of a large body of research examining the immune mechanisms that act to control influenza A virus infection (Fig. 1). This information should provide a useful basis for the informed design of novel, next generation influenza A virus vaccines.

Summary of the host immune response to influenza A virus
2 Detection of Influenza A Virus
Invading influenza A viruses are detected in the host environment by “pattern recognition receptors” (PRRs) [2]. Previously, the molecular target was considered to be double-stranded viral RNA (dsRNA) recognized by the PRR, toll-like receptor 3 (TLR3) [3, 4]. A role for TLR3 was questioned, however, given that the concentration of dsDNA is unlikely to be sufficient to signal TLR3 [5]. It is now considered that influenza A virus infection does not generate dsRNA at all [6]. Instead, the influenza A virus polymerase generates single-stranded RNA (ssRNA) with an uncapped 5′-phosphate that serves as the molecular signature identified by the immune system [6]. The cytoplasmic RNA helicase, RIG-1 [6, 7], but not MDA5 [6, 8], is responsible for influenza A virus recognition, which occurs independently of viral replication [7]. In addition to RIG-1, TLR7 is implicated in influenza A virus detection. Expressed in the endosomal compartments of plasmacytoid dendritic cells (DCs) and B cells, TLR7 detects influenza A virus ssRNA [9, 10]. The participation of multiple PRRs in the surveillance of influenza A virus may reflect cell type-specific roles [11]. Influenza A virus infection also activates NOD-like receptor-associated inflammasomes that are critical for the processing and release of IL-1β [12–14]. Once influenza A virus is recognized, PRRs initiate multiple signaling cascades that facilitate both innate and adaptive immunity to enable viral eradication.
3 Innate Immunity and the Influenza A Viruses
Innate immunity directed against influenza A virus provides an immediate and rapid response to the pathogen. The pulmonary infiltrate of innate immune cells is comprised mainly of natural killer (NK) cells, neutrophils, and macrophages. The NK cell represents the major innate response element and is detected in the infected lung as early as 48 h following influenza A virus infection [15, 16]. Protection is thought to be mediated by both cytokine production (IFN-γ and TNF-α) and direct cytotoxicity of virus-infected cells [17]. Influenza A virus-infected cells are recognized by NKp46 [18] and NKp44 [19] interaction with HA. The critical role for this pathway in influenza control is illustrated by the fatal infection that occurs in mice that lack NKp46 [20]. Together with NK cells, neutrophils also contribute to influenza A virus clearance through the secretion of an array of proinflammatory molecules that serve to limit viral replication [21–23]. Finally, alveolar macrophages (AMs) are also present in the innate pulmonary infiltrate, although they form only a small contribution early, they are recruited in large numbers later by the T-cell response. AMs represent the major phagocytic cell type resident in the lung [24], acting to scavenge influenza A virus-derived antigen [25]. In addition, AMs secrete proinflammatory cytokines including tumor necrosis factor (TNF)-α, interleukin (IL-1)-β, IL-6, and interferon (IFN)-α/β [26, 27] together with the chemokines macrophage inflammatory protein (MIP)-1α, monocyte chemotactic protein (MCP)-1, RANTES, and IFN-inducible protein (IP)-10 [21, 26, 28–30]. The magnitude and duration of the potent AM inflammatory response are negatively regulated via CD200R/CD200 [31]. The AM can also modulate adaptive T-cell immunity to influenza A viruses [32]. Present in the lung during active viral replication, AMs are fully susceptible to influenza A virus infection [26]. Unlike in epithelial cells, however, the infection is nonproductive with little, if any, virion release [26, 33], though it does lead to subsequent apoptosis [33]. Depletion of macrophages during influenza A virus infection results in elevated viral titers and increased morbidity and mortality [21]. In contrast, macrophages can elicit damage to the infected respiratory tissue [34]. Therefore, multiple immune cell types participate in the immediate innate response to influenza A viruses.
The pulmonary infiltrate releases a torrent of innate immune molecules that are considered to limit influenza A virus infection. A long list of cytokines and chemokines are potentially involved. A major player is type I IFN, representing the most potent cytokine attack against the virus [35]. So potent is the IFN response that the influenza A viruses encode a protein (NS2) to disable this pathway (described in Sect. 6). Nasal and pulmonary IFN-α and -β rise rapidly following influenza A virus infection [36] and act to directly limit viral replication and induce further cytokines and/or chemokine secretion that enhances recruitment and activation of multiple immune cell types. Type I IFN serves to enhance macrophage function, promote antigen presentation by antigen-presenting cells (APCs), and modulate adaptive immunity. The importance of this pathway is exemplified by the severe pulmonary disease that develops following influenza A virus infection of mice with disrupted type I IFN signaling [37, 38]. Plasmacytoid DCs are the major producers of type 1 IFN in response to many viruses, including influenza A virus [39–42]. Other cytokines implicated in influenza A virus immunity include TNF-α [43], IL-6 [44, 45], IL-1 [46], IL-18 [47], and IL-12 [48, 49]. In contrast, mice that lack functional IFN-γ can efficiently clear influenza A viruses, suggesting only a minor or redundant role for IFN-γ in the response [50–52]. Chemokines with defined roles in influenza A virus immunity include MIP-1α [53] and CCR5 [54], as illustrated by the elevated disease burden following infection of the chemokine-deficient mice. Finally, while cytokines and chemokines are important in the immune control of influenza A virus infections, their contribution can be detrimental as they elicit potentially fatal “cytokine shock” [55]. Recent studies dramatically illustrate the devastating impact of increased inflammatory infiltrates on viral-induced pathology. In animal models, infection with the reconstructed 1918 influenza A virus promotes massive inflammatory infiltrates with significantly higher levels of cytokines (IFN-γ, TNF-α, IL-1, IL-6, IL-12, IL-18, and granulocyte-colony-stimulating factor) and chemokines (MIP-2, MIP-1α/β, MCP-1) [21, 56–58]. Therefore, particularly early on, potent inflammatory antiviral activity may be dangerous, rather than protective, to the host due to the deleterious impact on lung pathology.
Collectins are collagen-like lectins that participate in innate immunity to viral pathogens [59]. Collectin family members, the surfactant proteins A (SP-A), and SP-D, are constitutively present in the fluids that line the respiratory tract [60]. Together with the mannan-binding lectin (MBL), SP-A and SP-D contribute to influenza A virus clearance via a number of mechanisms. Hemagglutination and viral infectivity are inhibited by SP-A [61, 62], SP-D [61, 63], and MBL [61, 64, 65]. In addition, complement-mediated lysis of influenza A virus-infected cells is enhanced by MBL [66], while SP-A and SP-D promote the binding and uptake of influenza A viruses by neutrophils [61, 67] and SP-A promotes opsonization and phagocytosis of influenza A virus by the AM population [68]. The sensitivity of different influenza A viral strains to collectin-mediated defense correlates with the degree of glycosylation of the HA glycoprotein [66, 69].
Defensins are cationic peptides produced by both leukocytes and epithelial cells. Defensins can exert direct microbial activity or promote immunity by acting as chemotactic agents. Examples of defensin-mediated anti-influenza A virus activity include retrocyclin-2 (o-defensin) and human β defensin 3 inhibition of HA-mediated membrane fusion [70]. The human neutrophil peptide (HNP) 1 (α-defensin) directly inactivates influenza A virus [65, 71].
4 Humoral Immunity and the Influenza A Viruses
Humoral immunity provides host defense through B lymphocyte secretion of antibody. Protective antibodies target antigenic structures exposed on the pathogen surface. Antibody-mediated immunity contributes to defense against the influenza A viruses [72–75] but is not always essential for optimal viral clearance [76, 77]. In any case, the influenza A viruses elicit a diverse spectrum of antiviral antibody responses. Natural antibodies present the first line of antibody-mediated defense [78]. These are low-affinity antibodies that restrict early virus dissemination [78] and promote the recruitment of viral antigen to the secondary lymphoid organs [79]. Natural antibodies reduce the overall load of influenza A virus and, as such, are required for optimal specific IgG antibody responses [75, 80]. Secretion of natural antibodies requires the transcriptional repressor Blimp-1: mice with Blimp-1-deficient B cells are more susceptible to influenza A virus infection [81]. Although natural antibodies are involved in the primary response to influenza A viruses, they are not required for optimal protection from secondary challenge [82]. Furthermore, while natural antibodies clearly display antiviral properties, effective virus clearance requires the induction of neutralizing antibody. Such neutralizing antibodies can be rapidly induced and possess high affinity (or avidity) for viral antigen. Mostly, virus neutralization is thought to be optimally achieved via antibody-mediated interference with viral binding to the host receptors required for cell entry or egress. Consequently, the influenza virus HA is heavily targeted by neutralizing antibodies [83, 84]. Crystallographic examination of HA in complex with neutralizing antibodies shows that antibody binding can occur at the same site as host receptor binding [85] or in distal regions where receptor binding is obstructed by steric hindrance [86]. Anti-HA neutralizing antibodies can also interfere with HA-mediated membrane fusion [87]. Similar to HA, NA is also targeted by neutralizing antibodies [88]. Neutralizing antibodies represent the major target of current influenza A virus vaccine strategies. While most neutralizing antibody strategies target HA or NA [89], the matrix protein 2 (M2) represents an interesting potential vaccine candidate [90]. M2 is a transmembrane protein expressed at the infected cell surface [91], but in contrast to HA and NA, is highly conserved among influenza A virus strains. Unfortunately thus far, M2-targeted vaccine strategies have elicited only weak immunity that does not protect mice from lethal challenge [92].
CD4+ T-helper cells contribute to humoral immunity by promoting B-cell differentiation into immunoglobulin class-switched, antibody-secreting cells. In most studies, the production of anti-influenza A virus antibody is CD4+ T-cell dependent [74, 93–95], although exceptions are reported [73, 74]. Classically, CD4+ T-cell help involves (1) the recognition of viral antigen and (2) the delivery of an activation signal to the B cell via the TNFR family member, CD40. Mice deficient in CD40 generate significantly impaired influenza A virus-specific antibody responses [93, 96]. Of interest, CD4+ T cells can help B lymphocytes by noncognate interactions that do not require specific influenza A virus antigen recognition [93].
5 T-Cell Immunity and the Influenza A Viruses
5.1 Dendritic Cells
DCs enable pathogen-derived antigens to be presented in a context that facilitates successful T-cell immunity [97]. Specialized in antigen presentation, the DCs facilitate (1) the acquisition of antigen, (2) processing and presentation of antigenic peptides in the context of host major histocompatibility complex (MHC) molecules, and (3) the provision of costimulatory signals. Immunity to influenza A virus infection requires DCs for both primary [98] and secondary T-cell responses [99, 100]. Many DC subsets are involved including the CCR2-dependent “inflammatory” DCs [101, 102], while plasmacytoid DCs are dispensable for influenza A virus clearance [103]. DC can control the magnitude of influenza A virus-specific T-cell immunity via FasL-mediated apoptosis [104]. In the respiratory tract, an extensive network of DC populations is present both in the lung [105] and in the draining lymph node [106]. Furthermore, pulmonary infection recruits additional DC populations into the lung [107–109]. To acquire influenza A virus antigen, DC may simply be directly infected with the virus. Infection induces the maturational changes (upregulation of costimulatory molecules and MHC class II) that are necessary for DC stimulation of T cells [110–112]. Infection can result in the expression of influenza NA at the DC surface, with NA-mediated removal of sialic acids serving to both enhance and inhibit DC function depending on the multiplicity of infection [113, 114]. DCs can also acquire influenza A virus-derived antigen released following the apoptotic lysis of infected respiratory cells [115, 116]. Once antigen is acquired, lung DCs migrate to the lymph node that drains the respiratory tract [107, 117, 118]. Migration occurs early after infection (24–48 h), and then the DCs display a refractory state to further inflammatory stimuli [107]. The lymph node also contains a resident DC set that has no direct access to the airways. Despite this, these resident DCs can also present influenza A virus-derived antigen [117]. Therefore, antigen transfer between the resident and migratory lung DC subsets must occur [119, 120]. Most experiments indicate that MHC class I presentation of influenza A virus-derived antigen in the lung draining lymph node ceases beyond 12–14 days [121, 122], although recently it has been suggested that antigen presentation can occur for up to 2 months following infection [123]. MHC class II presentation is also reported to persist for as long as 4 weeks after infection [124]. This is surprising given that infectious virus is cleared by day 10 [125]. Therefore, it has been postulated that the respiratory lymph node DCs can serve as a reservoir for antigen, with a depot being maintained well beyond the clearance of pathogen from the infected respiratory tissue [123, 126]. This, however, remains a contentious issue as the presence of an influenza A virus antigen depot was not detected in a separate independent study [127].
5.2 Costimulation
The participation of DCs in adaptive immunity is critical due to the rich array of costimulatory molecules expressed at the cell surface. A growing list of costimulatory molecules has been identified, most of which belong to either CD28/B7 [128] or TNFR [129] families. Costimulation serves to enhance the antigen-specific signals that are delivered through the T-cell receptor (TCR). As such, costimulation is required for optimal T-cell immunity in many viral infections [130]. The major pathway of costimulation is via the CD28/B7 interaction that plays an important role in influenza A virus immunity. This signal contributes to the generation of influenza A virus-specific T-cell immunity at multiple levels. For CD8+ T cells, CD28/B7 contributes to expansion [131–133], cytotoxicity, and/or effector cytokine production [131, 134, 135], recruitment to the infected airways [134], and survival [135]. In contrast, the hierarchy of T-cell response magnitude to individual influenza A virus-derived epitopes (a phenomenon termed immunodominance [136, 137]) is not altered in the absence of CD28/B7 signaling [138]. Mice deficient in CD28/B7 also display impaired influenza-specific neutralizing antibody responses [133]. While CD28/B7 plays a prominent part early in response to influenza A virus infection, 41BB/41BBL is important for sustained CD8+ T-cell expansion and is critical for optimal recall responses [131, 133, 139]. Effective CD4+ T-cell immunity during influenza A virus infection also requires CD28/B7 [133], OX40/OX40L [140], and ICOS/ICOSL [141]-mediated costimulation. The accumulation of T cells in influenza A virus-infected lungs depends on CD27/CD70 signaling [132, 142]. This is due to its impact on T-cell survival and/or migration to the infected respiratory tract [132]. Together, multiple costimulatory signals are delivered via the DCs to promote optimal adaptive immunity and, in turn, influenza A virus elimination.
5.3 CD8+ T Cells
Effector CD8+ T cells, also known as cytolytic T lymphocytes (CTLs), are important in the normal clearance of influenza A viruses [143]. Mice deficient in CD8+ T cells show delayed influenza A virus clearance, though they eventually control infection with all but the most virulent viruses [144]. The influenza A virus-specific CD8+ T-cell response has been extensively characterized utilizing murine models of infection, particularly with the HKx31 (H3N2) and PR/8 (H1N1) influenza A viruses. CD8+ T cells are primed, are activated, and expand in the lung draining lymph nodes during the first week or so after primary infection [121, 145]. Activated CD8+ T cells then traffic to the respiratory airways and the infected lung to mediate viral clearance [146]. The trafficking [147] and retention of CD8+ T cells in the lung [148] are dependent on LFA-1 expression. At the site of infection, CD8+ T cells target virus-infected cells that express peptide derived from influenza A virus protein associated with major histocompatibility complex class I (MHC I). An array of epitopes is recognized in the C57BL/6 (B6) mouse model, with the dominant epitopes (in terms of response magnitude) seen by CD8+ T cells being provided by the viral polymerase A (PA224-233) [149] and nucleoprotein (NP366-374) [150, 151]. Subdominant epitopes are derived from the basic polymerase subunit 1 (PB1703-711) [152], the mitochondrial protein PB1-F262-70 [152, 153], nonstructural protein 2 (NS2114-121) [151], and matrix protein 1 (M1128-135) [154]. In the absence of the dominant epitopes, subdominant epitope-specific CD8+ T cells account for a compensatory response, although a slight delay in viral clearance is observed [155, 156]. Depending on the experimental model, 30–90% of CD8+ T cells recovered from the respiratory tract are influenza A virus specific at the peak of the primary response, illustrating their enrichment in the pneumonic lung [137, 151, 152, 157]. Epitope-specific CD8+ T cells can be found widely dispersed throughout various body organs, including the lung, spleen, bone marrow, blood, liver, and nondraining lymph nodes [157, 158]. Once their target antigen is recognized, CD8+ T cells exert multiple effector functions. Cytokines such as IFNγ, TNF-α, and IL-2 are secreted by influenza A virus-specific CD8+ T cells [159]. In addition, CD8+ T cells mediate direct cytolysis of influenza A virus-infected target cells by the exocytosis of cytolytic granules that contain perforin and granzymes [160–163] and/or through the expression of Fas-ligand (FasL) [164–166]. CD8+ T cells also exert regulation of the inflammatory process via the production of IL-10 [167].
Following influenza A virus clearance, virus-specific CD8+ T cells decrease in number until a plateau is reached approximately 2 months following infection [122, 157]. After primary infection, the codominant DbNP366-374 and DbPA224-233-specific CD8+ T-cell populations contract at the same rate [157] to memory pools that are approximately equivalent in number and represent 10% of the population at the peak of the response [168]. Influenza A virus-specific CD8+ T cells persist as a stable population for the life of a laboratory mouse [157, 169, 170]. Retention of memory CD8+ T cells in nonlymphoid tissue, such as the lung, is mediated by T-cell expression of VLA-1 [171]. Secondary challenge recruits the memory CD8+ T cells that expand in the lymph nodes and promote viral clearance approximately 2 days earlier than after primary infection [157]. During secondary infection, the NP366-374 CD8+ T-cell population is clearly dominant representing up to 80% of the virus-specific CTL responses [122, 137, 151, 152]. This dominance is maintained in the memory populations that persist following the peak of the secondary response (day 8) [122]. The skewed immunodominance hierarchy observed in secondary versus primary influenza A virus infection was initially thought to be largely a consequence of differential antigen presentation [172], though it is now considered that T-cell precursor frequency and antigen dose are likely to be important determining variables [173].
5.4 CD4+ T Cells
Virus-specific CD4+ T cells are important participants in influenza immunity [174, 175]. Although, acting alone, these cells do not normally eliminate virus [176], they exert distinct roles in both humoral immunity (as discussed) and CD8+ T-cell responses. A vigorous, heterogenous CD4+ T-cell response is elicited following influenza A virus infection [175]. Again, the process of clonal expansion and differentiations is initiated in the lung draining lymph node, with the peak response in the respiratory airways occurring 6–7 days following infection [175]. This is dominated by producers of the Th1 cytokines, such as IL-2, IFN-γ, and TNF-α [177]. CD4+ T cells also secrete IL-10 contributing to the regulation of the inflammatory response [167]. Following influenza A virus clearance, CD4+ T cells demonstrate increased contraction in the respiratory tract compared with influenza A virus-specific CD8+ T cells [178, 179]. A major role for CD4+ T cells is the provision of “help” for optimal CD8+ T-cell immunity. Although CD4+ T cells are not required for primary influenza-specific CD8+ T-cell responses, presumably due to the direct activation of DC by viral infection [180–182], they are critical for the optimal establishment of CD8+ T-cell memory. The absence of CD4+ T cells during primary influenza A virus infections leads to a significant reduction in the size and magnitude of the secondary response and impaired viral clearance [77, 180]. Activation of CD4+ T cells requires antigen-specific signaling via TCR recognition of antigens presented in the context of MHC class II molecules. Until recently, the spectrum of influenza A virus CD4+ T-cell epitopes was much less well characterized than the panel known for the CD8+ subset. Recently however, 20–30 peptides were identified for the influenza-specific CD4+ T-cell response in C57BL/6 mice, with the majority being derived from the NP and HA proteins [183]. There is some evidence that influenza MHC class II epitopes are persisting for a substantial interval after the virus has been cleared from the host [124]. Overall, the adaptive immune response to the influenza A viruses involves complex interactions between a spectrum of functionally different cell types and their secretions.
6 Influenza A Virus Escape
The major influenza A virus escape mechanism rests in the inherent genetic variation of these RNA viruses, combined with the selective pressure exerted by HA-specific neutralizing antibody [184–186]. This process is known as “antigenic drift.” Lacking proof reading capacity, the influenza A virus RNA polymerase promotes the accumulation of nucleotide point mutations. Such mutations generate approximately 3.5 amino acid substitutions per year [187]. Circulating viral subtypes are then selected where substitutions have occurred and maintain viral fitness [188] but abrogate immune recognition. For example, virus escape mutants are poorly recognized by neutralizing antibody due to (1) introduced steric interference with antibody binding [85], (2) virus conformational changes that render antibody binding energetically unfavorable [86], or (3) the introduction of new oligosaccharide attachment sites to surface glycoproteins that obscure antibody binding [189, 190]. Retention of amino acid substitutions at the HA membrane distal surface, an area targeted by antibodies, is favored over those buried within the protein [83]. Virus-specific CTL immunity can also be targeted by antigenic drift [191]. Here, viruses are selected with mutations that interfere with epitope binding to MHC class I or with epitopes that are no longer recognized by the TCR. Both NP388-391 [192, 193] and NP418-426 [194, 195] CTL peptides have shown evidence of antigenic drift. Hypervariability within a CTL epitope correlates with the functional avidity of the TCR [196]. Such antigenic drift can function to limit cross-protective immunity against multiple influenza A virus strains and, as a consequence, contribute to seasonal epidemics.
While antigenic drift represents a subtle mode of immune escape, influenza A viruses can also undergo major antigenic variation to outmaneuver the immune system. This takes place by “antigenic shift,” where infection of the same cell with two distinct influenza A virus strains allows reassortment of the viral genomic segments, generating a new hybrid influenza A virus. Reassortment can occur following infection with different species-adapted viruses. For example, pigs can be infected with both human and avian influenza A viruses. Simultaneous infection may thereby generate a reassortment virus where the “human” pathogen acquires an “avian” virus HA or NA gene. In this case, for the HA and NA in particular, there would be no prevailing immunity in the human population, leading to the possibility of a human pandemic [197, 198]. Such antigenic shift involving avian and human strains has been implicated in two of the influenza A virus pandemics that have occurred in the twentieth century; the 1957 H2N2 [199, 200] and 1968 H3N2 [187, 200] infections. Of interest, the influenza A virus that provoked the 1918 pandemic did not arise through antigenic shift. Instead the 1918 H1N1 virus, which was responsible for millions of deaths worldwide, is believed to be an entirely avian viral strain that mutated in a way that allowed it to infect humans [201, 202].
The nonstructural protein 1 (NS1) encoded by influenza A virus provides a mode of immune escape that does not require manipulation of the genome. NS1 inhibits the host cell IFNα/β response [203, 204], a major pathway of immune defense against the virus (as discussed). Type 1 IFN induction is antagonized by NS1-mediated suppression of IFN-induced proteins dsRNA-activated protein kinase, 2′–5′-oligo (A) synthetase [205–207], the transcription factors NFκB [208], and the IFN regulatory factor-3 [209]. Containing an RNA-binding domain at its N-terminus [208], it was previously considered that NS1 sequestered influenza A virus dsRNA [210]. Instead, NS1 forms a complex with RIG-1, the cellular sensor of influenza A virus uncapped ssRNA [6]. Therefore, NS1 acts to disable the host mechanism for detection of viral-derived RNA and the induction of the IFN response. Influenza A viruses lacking the NS1 protein are good vaccine candidates as the absence of this immunomodulatory protein greatly enhances the immunogenicity of the virus [211].
7 Heterotypic Influenza A Virus Immunity
Heterotypic immunity in this system is defined by cross-reactive, protective responses between serologically different (HA-distinct) influenza A viruses. It would obviously be advantageous if, for example, prior infection with a human influenza A virus could generate immune memory that provides at least some resistance to a highly pathogenic avian virus that suddenly adapted to transmit between people [212, 213]. Clearly, promoting heterotypic immunity is a desirable strategy for influenza A virus vaccine development. Described many decades ago [214], heterotypic immunity has now been shown for many influenza A virus combinations [215–218]. At least in mice, heterotypic immunity can both be long lasting and provide protection against otherwise lethal virus challenge. The best understood component of such responses is CTL immunity directed at generally conserved, internal viral proteins [215, 217, 218]. However, there is also evidence for the retention of a measure of heterotypic immunity in mice lacking CD8+ T cells [216, 219]. In addition to the CD8+ T effectors, CD4+ T cells, nonneutralizing IgA antibody, NKT cells, and γδ T cells have all been considered as possible players [217]. Immunization with a low dose of a cold-adapted, attenuated influenza A virus provides one vaccination strategy that has the potential to induce at least some degree of long-term, heterotypic immunity [220]. The promotion of such responses is clearly a worthwhile focus for future vaccination strategies.
8 Influenza A Virus Immunity and Vaccination
Ultimately, studies of the immune response to influenza A virus aim to provide the foundation for strategies that will combat influenza-mediated disease. Vaccination is the major weapon to enable reduced morbidity, mortality, and economic damage associated with widespread influenza A virus infection. The 2009 HINI pandemic highlights the urgency of developing safe and effective vaccines to emerging influenza A virus strains. H5N1 avian influenza A virus is another immediate concern. H5N1 is a highly pathogenic virus that possesses the capacity to provoke a debilitating pandemic of greater severity than that of H1NI. As such, much effort has been employed to design a suitable H5N1 vaccine. Eliciting high titer neutralizing antibody is a major priority of any vaccination strategy, although cell-mediated immunity is also considered important. Cell-mediated immunity is powerful in that it has the potential to provide universal protection against divergent viral strains [221, 222]. Many vaccine formulations have been tested to date, but the most widely utilized platform is the inactivated, attenuated H5N1 virus (whole virion, subvirion, or surface antigen). Studies indicate that two doses of this vaccine, together with an adjuvant such as MF59, elicit cross-protective immunogenic responses in healthy subjects [223–225]. Mechanisms underlying protection include the expansion of antigen-specific CD4+ T cells, which serves as a reliable correlate of vaccine protection [226]. H5N1 vaccination studies provide valuable lessons that are currently being harnessed for a swift and rapid response to the 2009 HINI pandemic.
9 Conclusion
The influenza A viruses pose intriguing challenges for vaccine design [227]. Moving beyond the currently available products will depend on exploiting our understanding of immune defense mechanisms against this important and potentially very dangerous group of human pathogens. Here, we have briefly summarized a current view of how these viruses are controlled by elements of both innate and adaptive host response, together with the escape strategies that influenza A viruses exploit to survive in nature and to maintain transmission at the species level. An ideal vaccine could be thought to induce high levels of neutralizing antibody and CTL memory. This might optimally be achieved by promoting more effective DC vaccination, perhaps via the pathway of driving the innate response in ways that enhance T-cell immunity. An important caveat is, though, that much of our understanding of (particularly) the innate and T-cell responses to the influenza A viruses is based on mouse experiments. As we go forward to develop vaccine candidates, it is important that the analysis of influenza virus cell-mediated immunity, in particular, should be greatly extended in human subjects.
References
Lewis DB (2006) Avian flu to human influenza. Annu Rev Med 57:139–154
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216
Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M (2006) Detrimental contribution of the toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2:e53
Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M (2005) Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem 280:5571–5580
Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB (2004) Does toll-like receptor 3 play a biological role in virus infections? Virology 322:231–238
Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001
Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G (2006) 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997
Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19–28
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–1531
Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA (2004) Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Natl Acad Sci USA 101:5598–5603
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105
Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A (2009) Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 206:79–87
Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30:556–565
Thomas PG, Dash P, Aldridge JR Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD (2009) The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30:566–575
Leung KN, Ada GL (1981) Induction of natural killer cells during murine influenza virus infection. Immunobiology 160:352–366
Stein-Streilein J, Bennett M, Mann D, Kumar V (1983) Natural killer cells in mouse lung: surface phenotype, target preference, and response to local influenza virus infection. J Immunol 131:2699–2704
Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP (1999) Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol 17:189–220
Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, Davis DM, Strominger JL, Yewdell JW, Porgador A (2001) Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409:1055–1060
Arnon TI, Lev M, Katz G, Chernobrov Y, Porgador A, Mandelboim O (2001) Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol 31:2680–2689
Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, Qimron U, Landau G, Greenbaum E, Zakay-Rones Z, Porgador A, Mandelboim O (2006) Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol 7:517–523
Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, Van Rooijen N, Katz JM, Basler CF (2005) Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79:14933–14944
Fujisawa H (2001) Inhibitory role of neutrophils on influenza virus multiplication in the lungs of mice. Microbiol Immunol 45:679–688
Ratcliffe DR, Nolin SL, Cramer EB (1988) Neutrophil interaction with influenza-infected epithelial cells. Blood 72:142–149
Sibille Y, Reynolds HY (1990) Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am Rev Respir Dis 141:471–501
Fujimoto I, Pan J, Takizawa T, Nakanishi Y (2000) Virus clearance through apoptosis-dependent phagocytosis of influenza A virus-infected cells by macrophages. J Virol 74:3399–3403
Hofmann P, Sprenger H, Kaufmann A, Bender A, Hasse C, Nain M, Gemsa D (1997) Susceptibility of mononuclear phagocytes to influenza A virus infection and possible role in the antiviral response. J Leukoc Biol 61:408–414
Gong JH, Sprenger H, Hinder F, Bender A, Schmidt A, Horch S, Nain M, Gemsa D (1991) Influenza A virus infection of macrophages. Enhanced tumor necrosis factor-alpha (TNF-alpha) gene expression and lipopolysaccharide-triggered TNF-alpha release. J Immunol 147:3507–3513
Kaufmann A, Salentin R, Meyer RG, Bussfeld D, Pauligk C, Fesq H, Hofmann P, Nain M, Gemsa D, Sprenger H (2001) Defense against influenza A virus infection: essential role of the chemokine system. Immunobiology 204:603–613
Sprenger H, Meyer RG, Kaufmann A, Bussfeld D, Rischkowsky E, Gemsa D (1996) Selective induction of monocyte and not neutrophil-attracting chemokines after influenza A virus infection. J Exp Med 184:1191–1196
Bussfeld D, Kaufmann A, Meyer RG, Gemsa D, Sprenger H (1998) Differential mononuclear leukocyte attracting chemokine production after stimulation with active and inactivated influenza A virus. Cell Immunol 186:1–7
Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, Gwyer E, Sedgwick JD, Barclay AN, Hussell T (2008) A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol 9:1074–1083
Wijburg OL, DiNatale S, Vadolas J, van Rooijen N, Strugnell RA (1997) Alveolar macrophages regulate the induction of primary cytotoxic T-lymphocyte responses during influenza virus infection. J Virol 71:9450–9457
Fesq H, Bacher M, Nain M, Gemsa D (1994) Programmed cell death (apoptosis) in human monocytes infected by influenza A virus. Immunobiology 190:175–182
Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, Mack M, Kuziel WA, Corazza N, Brunner T, Seeger W, Lohmeyer J (2008) Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med 205:3065–3077
Theofilopoulos AN, Baccala R, Beutler B, Kono DH (2005) Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 23:307–336
Wyde PR, Wilson MR, Cate TR (1982) Interferon production by leukocytes infiltrating the lungs of mice during primary influenza virus infection. Infect Immun 38:1249–1255
Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM, Vukmanovic S, Garcia-Sastre A, Levy DE (2000) Type I IFN modulates innate and specific antiviral immunity. J Immunol 164:4220–4228
Garcia-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, Durbin JE (1998) The role of interferon in influenza virus tissue tropism. J Virol 72:8550–8558
Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M (1999) Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 5:919–923
Nakano H, Yanagita M, Gunn MD (2001) CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med 194:1171–1178
Bruno L, Seidl T, Lanzavecchia A (2001) Mouse pre-immunocytes as non-proliferating multipotent precursors of macrophages, interferon-producing cells, CD8alpha(+) and CD8alpha(−) dendritic cells. Eur J Immunol 31:3403–3412
O’Keeffe M, Hochrein H, Vremec D, Caminschi I, Miller JL, Anders EM, Wu L, Lahoud MH, Henri S, Scott B, Hertzog P, Tatarczuch L, Shortman K (2002) Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J Exp Med 196:1307–1319
Seo SH, Webster RG (2002) Tumor necrosis factor alpha exerts powerful anti-influenza virus effects in lung epithelial cells. J Virol 76:1071–1076
Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J (2003) Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19:225–234
Lee SW, Youn JW, Seong BL, Sung YC (1999) IL-6 induces long-term protective immunity against a lethal challenge of influenza virus. Vaccine 17:490–496
Schmitz N, Kurrer M, Bachmann MF, Kopf M (2005) Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol 79:6441–6448
Denton AE, Doherty PC, Turner SJ, La Gruta NL (2007) IL-18, but not IL-12, is required for optimal cytokine production by influenza virus-specific CD8(+) T cells. Eur J Immunol 37(2):368–375
Bhardwaj N, Seder RA, Reddy A, Feldman MV (1996) IL-12 in conjunction with dendritic cells enhances antiviral CD8+ CTL responses in vitro. J Clin Invest 98:715–722
Monteiro JM, Harvey C, Trinchieri G (1998) Role of interleukin-12 in primary influenza virus infection. J Virol 72:4825–4831
Nguyen HH, van Ginkel FW, Vu HL, Novak MJ, McGhee JR, Mestecky J (2000) Gamma interferon is not required for mucosal cytotoxic T-lymphocyte responses or heterosubtypic immunity to influenza A virus infection in mice. J Virol 74:5495–5501
Bot A, Bot S, Bona CA (1998) Protective role of gamma interferon during the recall response to influenza virus. J Virol 72:6637–6645
Baumgarth N, Kelso A (1996) In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J Virol 70:4411–4418
Cook DN, Beck MA, Coffman TM, Kirby SL, Sheridan JF, Pragnell IB, Smithies O (1995) Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science 269:1583–1585
Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N (2000) Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol 156:1951–1959
La Gruta NL, Kedzierska K, Stambas J, Doherty PC (2007) A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85(2):85–92
Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, Feldmann F, Alimonti JB, Fernando L, Li Y, Katze MG, Feldmann H, Kawaoka Y (2007) Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445:319–323
Kobasa D, Takada A, Shinya K, Hatta M, Halfmann P, Theriault S, Suzuki H, Nishimura H, Mitamura K, Sugaya N, Usui T, Murata T, Maeda Y, Watanabe S, Suresh M, Suzuki T, Suzuki Y, Feldmann H, Kawaoka Y (2004) Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 431:703–707
Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, Swayne DE, Katze MG (2006) Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443:578–581
Holmskov U, Thiel S, Jensenius JC (2003) Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol 21:547–578
Crouch E, Hartshorn K, Ofek I (2000) Collectins and pulmonary innate immunity. Immunol Rev 173:52–65
Hartshorn KL, White MR, Shepherd V, Reid K, Jensenius JC, Crouch EC (1997) Mechanisms of anti-influenza activity of surfactant proteins A and D: comparison with serum collectins. Am J Physiol 273:L1156–L1166
Benne CA, Kraaijeveld CA, van Strijp JA, Brouwer E, Harmsen M, Verhoef J, van Golde LM, van Iwaarden JF (1995) Interactions of surfactant protein A with influenza A viruses: binding and neutralization. J Infect Dis 171:335–341
Hartshorn K, Chang D, Rust K, White M, Heuser J, Crouch E (1996) Interactions of recombinant human pulmonary surfactant protein D and SP-D multimers with influenza A. Am J Physiol 271:L753–L762
Hartshorn KL, Sastry K, White MR, Anders EM, Super M, Ezekowitz RA, Tauber AI (1993) Human mannose-binding protein functions as an opsonin for influenza A viruses. J Clin Invest 91:1414–1420
Daher KA, Selsted ME, Lehrer RI (1986) Direct inactivation of viruses by human granulocyte defensins. J Virol 60:1068–1074
Reading PC, Hartley CA, Ezekowitz RA, Anders EM (1995) A serum mannose-binding lectin mediates complement-dependent lysis of influenza virus-infected cells. Biochem Biophys Res Commun 217:1128–1136
Hartshorn KL, Reid KB, White MR, Jensenius JC, Morris SM, Tauber AI, Crouch E (1996) Neutrophil deactivation by influenza A viruses: mechanisms of protection after viral opsonization with collectins and hemagglutination-inhibiting antibodies. Blood 87:3450–3461
Benne CA, Benaissa-Trouw B, van Strijp JA, Kraaijeveld CA, van Iwaarden JF (1997) Surfactant protein A, but not surfactant protein D, is an opsonin for influenza A virus phagocytosis by rat alveolar macrophages. Eur J Immunol 27:886–890
Hartley CA, Reading PC, Ward AC, Anders EM (1997) Changes in the hemagglutinin molecule of influenza type A (H3N2) virus associated with increased virulence for mice. Arch Virol 142:75–88
Leikina E, Delanoe-Ayari H, Melikov K, Cho MS, Chen A, Waring AJ, Wang W, Xie Y, Loo JA, Lehrer RI, Chernomordik LV (2005) Carbohydrate-binding molecules inhibit viral fusion and entry by crosslinking membrane glycoproteins. Nat Immunol 6:995–1001
Doss M, White MR, Tecle T, Gantz D, Crouch EC, Jung G, Ruchala P, Waring AJ, Lehrer RI, Hartshorn KL (2009) Interactions of alpha-, beta-, and theta-defensins with influenza A virus and surfactant protein D. J Immunol 182:7878–7887
Graham MB, Braciale TJ (1997) Resistance to and recovery from lethal influenza virus infection in B lymphocyte-deficient mice. J Exp Med 186:2063–2068
Lee BO, Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Makris M, Sprague F, Lund FE, Randall TD (2005) CD4 T cell-independent antibody response promotes resolution of primary influenza infection and helps to prevent reinfection. J Immunol 175:5827–5838
Mozdzanowska K, Furchner M, Zharikova D, Feng J, Gerhard W (2005) Roles of CD4+ T-cell-independent and -dependent antibody responses in the control of influenza virus infection: evidence for noncognate CD4+ T-cell activities that enhance the therapeutic activity of antiviral antibodies. J Virol 79:5943–5951
Kopf M, Brombacher F, Bachmann MF (2002) Role of IgM antibodies versus B cells in influenza virus-specific immunity. Eur J Immunol 32:2229–2236
Topham DJ, Tripp RA, Hamilton-Easton AM, Sarawar SR, Doherty PC (1996) Quantitative analysis of the influenza virus-specific CD4+ T cell memory in the absence of B cells and Ig. J Immunol 157:2947–2952
Riberdy JM, Christensen JP, Branum K, Doherty PC (2000) Diminished primary and secondary influenza virus-specific CD8(+) T-cell responses in CD4-depleted Ig(-/-) mice. J Virol 74:9762–9765
Ochsenbein AF, Zinkernagel RM (2000) Natural antibodies and complement link innate and acquired immunity. Immunol Today 21:624–630
Ochsenbein AF, Pinschewer DD, Odermatt B, Ciurea A, Hengartner H, Zinkernagel RM (2000) Correlation of T cell independence of antibody responses with antigen dose reaching secondary lymphoid organs: implications for splenectomized patients and vaccine design. J Immunol 164:6296–6302
Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J (2000) B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med 192:271–280
Savitsky D, Calame K (2006) B-1 B lymphocytes require Blimp-1 for immunoglobulin secretion. J Exp Med 203:2305–2314
Harada Y, Muramatsu M, Shibata T, Honjo T, Kuroda K (2003) Unmutated immunoglobulin M can protect mice from death by influenza virus infection. J Exp Med 197:1779–1785
Skehel JJ, Wiley DC (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569
Kwong PD, Wilson IA (2009) HIV-1 and influenza antibodies: seeing antigens in new ways. Nat Immunol 10:573–578
Bizebard T, Gigant B, Rigolet P, Rasmussen B, Diat O, Bosecke P, Wharton SA, Skehel JJ, Knossow M (1995) Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 376:92–94
Fleury D, Barrere B, Bizebard T, Daniels RS, Skehel JJ, Knossow M (1999) A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nat Struct Biol 6:530–534
Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA (2009) Antibody recognition of a highly conserved influenza virus epitope. Science 324:246–251
Murphy BR, Kasel JA, Chanock RM (1972) Association of serum anti-neuraminidase antibody with resistance to influenza in man. N Engl J Med 286:1329–1332
Belshe RB, Gruber WC, Mendelman PM, Cho I, Reisinger K, Block SL, Wittes J, Iacuzio D, Piedra P, Treanor J, King J, Kotloff K, Bernstein DI, Hayden FG, Zangwill K, Yan L, Wolff M (2000) Efficacy of vaccination with live attenuated, cold-adapted, trivalent, intranasal influenza virus vaccine against a variant (A/Sydney) not contained in the vaccine. J Pediatr 136:168–175
Neirynck S, Deroo T, Saelens X, Vanlandschoot P, Jou WM, Fiers W (1999) A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat Med 5:1157–1163
Lamb RA, Zebedee SL, Richardson CD (1985) Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell 40:627–633
Jegerlehner A, Schmitz N, Storni T, Bachmann MF (2004) Influenza A vaccine based on the extracellular domain of M2: weak protection mediated via antibody-dependent NK cell activity. J Immunol 172:5598–5605
Sangster MY, Riberdy JM, Gonzalez M, Topham DJ, Baumgarth N, Doherty PC (2003) An early CD4+ T cell-dependent immunoglobulin A response to influenza infection in the absence of key cognate T-B interactions. J Exp Med 198:1011–1021
Scherle PA, Palladino G, Gerhard W (1992) Mice can recover from pulmonary influenza virus infection in the absence of class I-restricted cytotoxic T cells. J Immunol 148:212–217
Topham DJ, Tripp RA, Sarawar SR, Sangster MY, Doherty PC (1996) Immune CD4+ T cells promote the clearance of influenza virus from major histocompatibility complex class II -/- respiratory epithelium. J Virol 70:1288–1291
Lee BO, Moyron-Quiroz J, Rangel-Moreno J, Kusser KL, Hartson L, Sprague F, Lund FE, Randall TD (2003) CD40, but not CD154, expression on B cells is necessary for optimal primary B cell responses. J Immunol 171:5707–5717
Shortman K, Liu YJ (2002) Mouse and human dendritic cell subtypes. Nat Rev Immunol 2:151–161
Sigal LJ, Rock KL (2000) Bone marrow-derived antigen-presenting cells are required for the generation of cytotoxic T lymphocyte responses to viruses and use transporter associated with antigen presentation (TAP)-dependent and -independent pathways of antigen presentation. J Exp Med 192:1143–1150
Belz GT, Wilson NS, Smith CM, Mount AM, Carbone FR, Heath WR (2006) Bone marrow-derived cells expand memory CD8+ T cells in response to viral infections of the lung and skin. Eur J Immunol 36:327–335
Zammit DJ, Cauley LS, Pham QM, Lefrancois L (2005) Dendritic cells maximize the memory CD8 T cell response to infection. Immunity 22:561–570
Aldridge JR Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, Brown SA, Doherty PC, Webster RG, Thomas PG (2009) TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci USA 106:5306–5311
Nakano H, Lin KL, Yanagita M, Charbonneau C, Cook DN, Kakiuchi T, Gunn MD (2009) Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol 10:394–402
GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, Thielemans K, Bennett C, Clausen BE, Hoogsteden HC, Osterhaus AD, Rimmelzwaan GF, Lambrecht BN (2008) Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J Exp Med 205:1621–1634
Legge KL, Braciale TJ (2005) Lymph node dendritic cells control CD8+ T cell responses through regulated FasL expression. Immunity 23:649–659
Sung SS, Fu SM, Rose CE Jr, Gaskin F, Ju ST, Beaty SR (2006) A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing langerin and tight junction proteins. J Immunol 176:2161–2172
Henri S, Vremec D, Kamath A, Waithman J, Williams S, Benoist C, Burnham K, Saeland S, Handman E, Shortman K (2001) The dendritic cell populations of mouse lymph nodes. J Immunol 167:741–748
Legge KL, Braciale TJ (2003) Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity 18:265–277
McWilliam AS, Napoli S, Marsh AM, Pemper FL, Nelson DJ, Pimm CL, Stumbles PA, Wells TN, Holt PG (1996) Dendritic cells are recruited into the airway epithelium during the inflammatory response to a broad spectrum of stimuli. J Exp Med 184:2429–2432
Yamamoto N, Suzuki S, Shirai A, Suzuki M, Nakazawa M, Nagashima Y, Okubo T (2000) Dendritic cells are associated with augmentation of antigen sensitization by influenza A virus infection in mice. Eur J Immunol 30:316–326
Nonacs R, Humborg C, Tam JP, Steinman RM (1992) Mechanisms of mouse spleen dendritic cell function in the generation of influenza-specific, cytolytic T lymphocytes. J Exp Med 176:519–529
Macatonia SE, Taylor PM, Knight SC, Askonas BA (1989) Primary stimulation by dendritic cells induces antiviral proliferative and cytotoxic T cell responses in vitro. J Exp Med 169:1255–1264
Bhardwaj N, Bender A, Gonzalez N, Bui LK, Garrett MC, Steinman RM (1994) Influenza virus-infected dendritic cells stimulate strong proliferative and cytolytic responses from human CD8+ T cells. J Clin Invest 94:797–807
Oh S, Eichelberger MC (1999) Influenza virus neuraminidase alters allogeneic T cell proliferation. Virology 264:427–435
Oh S, McCaffery JM, Eichelberger MC (2000) Dose-dependent changes in influenza virus-infected dendritic cells result in increased allogeneic T-cell proliferation at low, but not high, doses of virus. J Virol 74:5460–5469
Albert ML, Sauter B, Bhardwaj N (1998) Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 392:86–89
Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, Young L, Forehan SP, Mount A, Steptoe RJ, Shortman KD, de Koning-Ward TF, Belz GT, Carbone FR, Crabb BS, Heath WR, Villadangos JA (2006) Systemic activation of dendritic cells by toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol 7:165–172
Belz GT, Smith CM, Kleinert L, Reading P, Brooks A, Shortman K, Carbone FR, Heath WR (2004) Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc Natl Acad Sci USA 101:8670–8675
Vermaelen KY, Carro-Muino I, Lambrecht BN, Pauwels RA (2001) Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J Exp Med 193:51–60
Carbone FR, Belz GT, Heath WR (2004) Transfer of antigen between migrating and lymph node-resident DCs in peripheral T-cell tolerance and immunity. Trends Immunol 25:655–658
Randolph GJ (2006) Migratory dendritic cells: sometimes simply ferries? Immunity 25:15–18
Lawrence CW, Braciale TJ (2004) Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J Immunol 173:1209–1218
Flynn KJ, Riberdy JM, Christensen JP, Altman JD, Doherty PC (1999) In vivo proliferation of naive and memory influenza-specific CD8(+) T cells. Proc Natl Acad Sci USA 96:8597–8602
Zammit DJ, Turner DL, Klonowski KD, Lefrancois L, Cauley LS (2006) Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity 24:439–449
Jelley-Gibbs DM, Brown DM, Dibble JP, Haynes L, Eaton SM, Swain SL (2005) Unexpected prolonged presentation of influenza antigens promotes CD4 T cell memory generation. J Exp Med 202:697–706
Doherty PC, Christensen JP (2000) Accessing complexity: the dynamics of virus-specific T cell responses. Annu Rev Immunol 18:561–592
Julia V, Hessel EM, Malherbe L, Glaichenhaus N, O’Garra A, Coffman RL (2002) A restricted subset of dendritic cells captures airborne antigens and remains able to activate specific T cells long after antigen exposure. Immunity 16:271–283
Mintern JD, Bedoui S, Davey GM, Moffat JM, Doherty PC, Turner SJ (2009) Transience of MHC Class I-restricted antigen presentation after influenza A virus infection. Proc Natl Acad Sci USA 106:6724–6729
Sharpe AH, Freeman GJ (2002) The B7-CD28 superfamily. Nat Rev Immunol 2:116–126
Croft M (2003) Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol 3:609–620
Bertram EM, Dawicki W, Watts TH (2004) Role of T cell costimulation in anti-viral immunity. Semin Immunol 16:185–196
Halstead ES, Mueller YM, Altman JD, Katsikis PD (2002) In vivo stimulation of CD137 broadens primary antiviral CD8+ T cell responses. Nat Immunol 3:536–541
Hendriks J, Xiao Y, Borst J (2003) CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med 198:1369–1380
Bertram EM, Lau P, Watts TH (2002) Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol 168:3777–3785
Lumsden JM, Roberts JM, Harris NL, Peach RJ, Ronchese F (2000) Differential requirement for CD80 and CD80/CD86-dependent costimulation in the lung immune response to an influenza virus infection. J Immunol 164:79–85
Liu Y, Wenger RH, Zhao M, Nielsen PJ (1997) Distinct costimulatory molecules are required for the induction of effector and memory cytotoxic T lymphocytes. J Exp Med 185:251–262
Yewdell JW, Del Val M (2004) Immunodominance in TCD8+ responses to viruses: cell biology, cellular immunology, and mathematical models. Immunity 21:149–153
Belz GT, Stevenson PG, Doherty PC (2000) Contemporary analysis of MHC-related immunodominance hierarchies in the CD8+ T cell response to influenza A viruses. J Immunol 165:2404–2409
Chen W, Bennink JR, Morton PA, Yewdell JW (2002) Mice deficient in perforin, CD4+ T cells, or CD28-mediated signaling maintain the typical immunodominance hierarchies of CD8+ T-cell responses to influenza virus. J Virol 76:10332–10337
DeBenedette MA, Wen T, Bachmann MF, Ohashi PS, Barber BH, Stocking KL, Peschon JJ, Watts TH (1999) Analysis of 4-1BB ligand (4-1BBL)-deficient mice and of mice lacking both 4-1BBL and CD28 reveals a role for 4-1BBL in skin allograft rejection and in the cytotoxic T cell response to influenza virus. J Immunol 163:4833–4841
Kopf M, Ruedl C, Schmitz N, Gallimore A, Lefrang K, Ecabert B, Odermatt B, Bachmann MF (1999) OX40-deficient mice are defective in Th cell proliferation but are competent in generating B cell and CTL Responses after virus infection. Immunity 11:699–708
Bertram EM, Tafuri A, Shahinian A, Chan VS, Hunziker L, Recher M, Ohashi PS, Mak TW, Watts TH (2002) Role of ICOS versus CD28 in antiviral immunity. Eur J Immunol 32:3376–3385
Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J (2000) CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol 1:433–440
Doherty PC, Topham DJ, Tripp RA, Cardin RD, Brooks JW, Stevenson PG (1997) Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol Rev 159:105–117
Bender BS, Croghan T, Zhang L, Small PA Jr (1992) Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med 175:1143–1145
Tripp RA, Sarawar SR, Doherty PC (1995) Characteristics of the influenza virus-specific CD8+ T cell response in mice homozygous for disruption of the H-2lAb gene. J Immunol 155:2955–2959
Cerwenka A, Morgan TM, Dutton RW (1999) Naive, effector, and memory CD8 T cells in protection against pulmonary influenza virus infection: homing properties rather than initial frequencies are crucial. J Immunol 163:5535–5543
Galkina E, Thatte J, Dabak V, Williams MB, Ley K, Braciale TJ (2005) Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. J Clin Invest 115:3473–3483
Thatte J, Dabak V, Williams MB, Braciale TJ, Ley K (2003) LFA-1 is required for retention of effector CD8 T cells in mouse lungs. Blood 101:4916–4922
Belz GT, Xie W, Altman JD, Doherty PC (2000) A previously unrecognized H-2D(b)-restricted peptide prominent in the primary influenza A virus-specific CD8(+) T-cell response is much less apparent following secondary challenge. J Virol 74:3486–3493
Townsend AR, Rothbard J, Gotch FM, Bahadur G, Wraith D, McMichael AJ (1986) The epitopes of influenza nucleoprotein recognized by cytotoxic T lymphocytes can be defined with short synthetic peptides. Cell 44:959–968
Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC (1998) Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity 8:683–691
Belz GT, Xie W, Doherty PC (2001) Diversity of epitope and cytokine profiles for primary and secondary influenza a virus-specific CD8+ T cell responses. J Immunol 166:4627–4633
Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O’Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW (2001) A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 7:1306–1312
Vitiello A, Yuan L, Chesnut RW, Sidney J, Southwood S, Farness P, Jackson MR, Peterson PA, Sette A (1996) Immunodominance analysis of CTL responses to influenza PR8 virus reveals two new dominant and subdominant Kb-restricted epitopes. J Immunol 157:5555–5562
Andreansky SS, Stambas J, Thomas PG, Xie W, Webby RJ, Doherty PC (2005) Consequences of immunodominant epitope deletion for minor influenza virus-specific CD8+-T-cell responses. J Virol 79:4329–4339
Webby RJ, Andreansky S, Stambas J, Rehg JE, Webster RG, Doherty PC, Turner SJ (2003) Protection and compensation in the influenza virus-specific CD8+ T cell response. Proc Natl Acad Sci USA 100:7235–7240
Marshall DR, Turner SJ, Belz GT, Wingo S, Andreansky S, Sangster MY, Riberdy JM, Liu T, Tan M, Doherty PC (2001) Measuring the diaspora for virus-specific CD8+ T cells. Proc Natl Acad Sci USA 98:6313–6318
Turner SJ, Diaz G, Cross R, Doherty PC (2003) Analysis of clonotype distribution and persistence for an influenza virus-specific CD8+ T cell response. Immunity 18:549–559
La Gruta NL, Turner SJ, Doherty PC (2004) Hierarchies in cytokine expression profiles for acute and resolving influenza virus-specific CD8+ T cell responses: correlation of cytokine profile and TCR avidity. J Immunol 172:5553–5560
Johnson BJ, Costelloe EO, Fitzpatrick DR, Haanen JB, Schumacher TN, Brown LE, Kelso A (2003) Single-cell perforin and granzyme expression reveals the anatomical localization of effector CD8+ T cells in influenza virus-infected mice. Proc Natl Acad Sci USA 100:2657–2662
Liu B, Mori I, Hossain MJ, Dong L, Chen Z, Kimura Y (2003) Local immune responses to influenza virus infection in mice with a targeted disruption of perforin gene. Microb Pathog 34:161–167
Mintern JD, Guillonneau C, Carbone FR, Doherty PC, Turner SJ (2007) Cutting edge: tissue-resident memory CTL down-regulate cytolytic molecule expression following virus clearance. J Immunol 179:7220–7224
Moffat JM, Gebhardt T, Doherty PC, Turner SJ, Mintern JD (2009) Granzyme A expression reveals distinct cytolytic CTL subsets following influenza A virus infection. Eur J Immunol 39:1203–1210
Topham DJ, Tripp RA, Doherty PC (1997) CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J Immunol 159:5197–5200
Price GE, Huang L, Ou R, Zhang M, Moskophidis D (2005) Perforin and Fas cytolytic pathways coordinately shape the selection and diversity of CD8+-T-cell escape variants of influenza virus. J Virol 79:8545–8559
Fujimoto I, Takizawa T, Ohba Y, Nakanishi Y (1998) Co-expression of Fas and Fas-ligand on the surface of influenza virus-infected cells. Cell Death Differ 5:426–431
Sun J, Madan R, Karp CL, Braciale TJ (2009) Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med 15:277–284
Kedzierska K, La Gruta NL, Turner SJ, Doherty PC (2006) Establishment and recall of CD8+ T-cell memory in a model of localized transient infection. Immunol Rev 211:133–145
Hogan RJ, Usherwood EJ, Zhong W, Roberts AA, Dutton RW, Harmsen AG, Woodland DL (2001) Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J Immunol 166:1813–1822
Wiley JA, Hogan RJ, Woodland DL, Harmsen AG (2001) Antigen-specific CD8(+) T cells persist in the upper respiratory tract following influenza virus infection. J Immunol 167:3293–3299
Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, Doherty PC, de Fougerolles AR, Topham DJ (2004) The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity 20:167–179
Crowe SR, Turner SJ, Miller SC, Roberts AD, Rappolo RA, Doherty PC, Ely KH, Woodland DL (2003) Differential antigen presentation regulates the changing patterns of CD8+ T cell immunodominance in primary and secondary influenza virus infections. J Exp Med 198:399–410
La Gruta NL, Kedzierska K, Pang K, Webby R, Davenport M, Chen W, Turner SJ, Doherty PC (2006) A virus-specific CD8+ T cell immunodominance hierarchy determined by antigen dose and precursor frequencies. Proc Natl Acad Sci USA 103:994–999
Brown DM, Roman E, Swain SL (2004) CD4 T cell responses to influenza infection. Semin Immunol 16:171–177
Roman E, Miller E, Harmsen A, Wiley J, Von Andrian UH, Huston G, Swain SL (2002) CD4 effector T cell subsets in the response to influenza: heterogeneity, migration, and function. J Exp Med 196:957–968
Mozdzanowska K, Furchner M, Maiese K, Gerhard W (1997) CD4+ T cells are ineffective in clearing a pulmonary infection with influenza type A virus in the absence of B cells. Virology 239:217–225
Graham MB, Braciale VL, Braciale TJ (1994) Influenza virus-specific CD4+ T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. J Exp Med 180:1273–1282
Powell TJ, Brown DM, Hollenbaugh JA, Charbonneau T, Kemp RA, Swain SL, Dutton RW (2004) CD8+ T cells responding to influenza infection reach and persist at higher numbers than CD4+ T cells independently of precursor frequency. Clin Immunol 113:89–100
Homann D, Teyton L, Oldstone MB (2001) Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med 7:913–919
Belz GT, Wodarz D, Diaz G, Nowak MA, Doherty PC (2002) Compromised influenza virus-specific CD8(+)-T-cell memory in CD4(+)-T-cell-deficient mice. J Virol 76:12388–12393
Bender A, Bui LK, Feldman MA, Larsson M, Bhardwaj N (1995) Inactivated influenza virus, when presented on dendritic cells, elicits human CD8+ cytolytic T cell responses. J Exp Med 182:1663–1671
Larsson M, Messmer D, Somersan S, Fonteneau JF, Donahoe SM, Lee M, Dunbar PR, Cerundolo V, Julkunen I, Nixon DF, Bhardwaj N (2000) Requirement of mature dendritic cells for efficient activation of influenza A-specific memory CD8+ T cells. J Immunol 165:1182–1190
Crowe SR, Miller SC, Brown DM, Adams PS, Dutton RW, Harmsen AG, Lund FE, Randall TD, Swain SL, Woodland DL (2006) Uneven distribution of MHC class II epitopes within the influenza virus. Vaccine 24:457–467
Palese P, Young JF (1982) Variation of influenza A, B, and C viruses. Science 215:1468–1474
Yewdell JW, Webster RG, Gerhard WU (1979) Antigenic variation in three distinct determinants of an influenza type A haemagglutinin molecule. Nature 279:246–248
Webster RG, Laver WG, Air GM, Schild GC (1982) Molecular mechanisms of variation in influenza viruses. Nature 296:115–121
Bean WJ, Schell M, Katz J, Kawaoka Y, Naeve C, Gorman O, Webster RG (1992) Evolution of the H3 influenza virus hemagglutinin from human and nonhuman hosts. J Virol 66:1129–1138
Berkhoff EG, de Wit E, Geelhoed-Mieras MM, Boon AC, Symons J, Fouchier RA, Osterhaus AD, Rimmelzwaan GF (2005) Functional constraints of influenza A virus epitopes limit escape from cytotoxic T lymphocytes. J Virol 79:11239–11246
Wiley DC, Wilson IA, Skehel JJ (1981) Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289:373–378
Caton AJ, Brownlee GG, Yewdell JW, Gerhard W (1982) The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427
Price GE, Ou R, Jiang H, Huang L, Moskophidis D (2000) Viral escape by selection of cytotoxic T cell-resistant variants in influenza A virus pneumonia. J Exp Med 191:1853–1867
Rimmelzwaan GF, Boon AC, Voeten JT, Berkhoff EG, Fouchier RA, Osterhaus AD (2004) Sequence variation in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. Virus Res 103:97–100
Voeten JT, Bestebroer TM, Nieuwkoop NJ, Fouchier RA, Osterhaus AD, Rimmelzwaan GF (2000) Antigenic drift in the influenza A virus (H3N2) nucleoprotein and escape from recognition by cytotoxic T lymphocytes. J Virol 74:6800–6807
Boon AC, de Mutsert G, Graus YM, Fouchier RA, Sintnicolaas K, Osterhaus AD, Rimmelzwaan GF (2002) Sequence variation in a newly identified HLA-B35-restricted epitope in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. J Virol 76:2567–2572
Boon AC, de Mutsert G, van Baarle D, Smith DJ, Lapedes AS, Fouchier RA, Sintnicolaas K, Osterhaus AD, Rimmelzwaan GF (2004) Recognition of homo- and heterosubtypic variants of influenza A viruses by human CD8+ T lymphocytes. J Immunol 172:2453–2460
Boon AC, de Mutsert G, Fouchier RA, Osterhaus AD, Rimmelzwaan GF (2006) The hypervariable immunodominant NP418-426 epitope from the influenza A virus nucleoprotein is recognized by cytotoxic T lymphocytes with high functional avidity. J Virol 80:6024–6032
Webby RJ, Webster RG (2003) Are we ready for pandemic influenza? Science 302:1519–1522
Cox NJ, Subbarao K (2000) Global epidemiology of influenza: past and present. Annu Rev Med 51:407–421
Schafer JR, Kawaoka Y, Bean WJ, Suss J, Senne D, Webster RG (1993) Origin of the pandemic 1957 H2 influenza A virus and the persistence of its possible progenitors in the avian reservoir. Virology 194:781–788
Kawaoka Y, Krauss S, Webster RG (1989) Avian-to-human transmission of the PB1 gene of influenza A viruses in the 1957 and 1968 pandemics. J Virol 63:4603–4608
Reid AH, Taubenberger JK, Fanning TG (2004) Evidence of an absence: the genetic origins of the 1918 pandemic influenza virus. Nat Rev Microbiol 2:909–914
Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG (2005) Characterization of the 1918 influenza virus polymerase genes. Nature 437:889–893
Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T (1998) Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324–330
Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C (2003) Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 424:324–328
Bergmann M, Garcia-Sastre A, Carnero E, Pehamberger H, Wolff K, Palese P, Muster T (2000) Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol 74:6203–6206
Li S, Min JY, Krug RM, Sen GC (2006) Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349:13–21
Min JY, Krug RM (2006) The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′–5′ oligo (A) synthetase/RNase L pathway. Proc Natl Acad Sci USA 103:7100–7105
Wang X, Li M, Zheng H, Muster T, Palese P, Beg AA, Garcia-Sastre A (2000) Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol 74:11566–11573
Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, Garcia-Sastre A (2000) Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 74:7989–7996
Garcia-Sastre A (2001) Inhibition of interferon-mediated antiviral responses by influenza A viruses and other negative-strand RNA viruses. Virology 279:375–384
Ferko B, Stasakova J, Romanova J, Kittel C, Sereinig S, Katinger H, Egorov A (2004) Immunogenicity and protection efficacy of replication-deficient influenza A viruses with altered NS1 genes. J Virol 78:13037–13045
Jameson J, Cruz J, Terajima M, Ennis FA (1999) Human CD8+ and CD4+ T lymphocyte memory to influenza A viruses of swine and avian species. J Immunol 162:7578–7583
Jameson J, Cruz J, Ennis FA (1998) Human cytotoxic T-lymphocyte repertoire to influenza A viruses. J Virol 72:8682–8689
Schulman JL, Kilbourne ED (1965) Induction of partial specific heterotypic immunity in mice by A single infection with influenza A virus. J Bacteriol 89:170–174
Kreijtz JH, Bodewes R, van Amerongen G, Kuiken T, Fouchier RA, Osterhaus AD, Rimmelzwaan GF (2007) Primary influenza A virus infection induces cross-protective immunity against a lethal infection with a heterosubtypic virus strain in mice. Vaccine 25:612–620
Epstein SL, Lo CY, Misplon JA, Lawson CM, Hendrickson BA, Max EE, Subbarao K (1997) Mechanisms of heterosubtypic immunity to lethal influenza A virus infection in fully immunocompetent, T cell-depleted, beta2-microglobulin-deficient, and J chain-deficient mice. J Immunol 158:1222–1230
Benton KA, Misplon JA, Lo CY, Brutkiewicz RR, Prasad SA, Epstein SL (2001) Heterosubtypic immunity to influenza A virus in mice lacking IgA, all Ig, NKT cells, or gamma delta T cells. J Immunol 166:7437–7445
Nguyen HH, Moldoveanu Z, Novak MJ, van Ginkel FW, Ban E, Kiyono H, McGhee JR, Mestecky J (1999) Heterosubtypic immunity to lethal influenza A virus infection is associated with virus-specific CD8(+) cytotoxic T lymphocyte responses induced in mucosa-associated tissues. Virology 254:50–60
Nguyen HH, van Ginkel FW, Vu HL, McGhee JR, Mestecky J (2001) Heterosubtypic immunity to influenza A virus infection requires B cells but not CD8+ cytotoxic T lymphocytes. J Infect Dis 183:368–376
Powell TJ, Strutt T, Reome J, Hollenbaugh JA, Roberts AD, Woodland DL, Swain SL, Dutton RW (2007) Priming with cold-adapted influenza a does not prevent infection but elicits long-lived protection against supralethal challenge with heterosubtypic virus. J Immunol 178:1030–1038
Brown LE, Kelso A (2009) Prospects for an influenza vaccine that induces cross-protective cytotoxic T lymphocytes. Immunol Cell Biol 87:300–308
Thomas PG, Keating R, Hulse-Post DJ, Doherty PC (2006) Cell-mediated protection in influenza infection. Emerg Infect Dis 12:48–54
Lin J, Zhang J, Dong X, Fang H, Chen J, Su N, Gao Q, Zhang Z, Liu Y, Wang Z, Yang M, Sun R, Li C, Lin S, Ji M, Liu Y, Wang X, Wood J, Feng Z, Wang Y, Yin W (2006) Safety and immunogenicity of an inactivated adjuvanted whole-virion influenza A (H5N1) vaccine: a phase I randomised controlled trial. Lancet 368:991–997
Leroux-Roels I, Borkowski A, Vanwolleghem T, Drame M, Clement F, Hons E, Devaster JM, Leroux-Roels G (2007) Antigen sparing and cross-reactive immunity with an adjuvanted rH5N1 prototype pandemic influenza vaccine: a randomised controlled trial. Lancet 370:580–589
Galli G, Hancock K, Hoschler K, DeVos J, Praus M, Bardelli M, Malzone C, Castellino F, Gentile C, McNally T, Del Giudice G, Banzhoff A, Brauer V, Montomoli E, Zambon M, Katz J, Nicholson K, Stephenson I (2009) Fast rise of broadly cross-reactive antibodies after boosting long-lived human memory B cells primed by an MF59 adjuvanted prepandemic vaccine. Proc Natl Acad Sci USA 106:7962–7967
Galli G, Medini D, Borgogni E, Zedda L, Bardelli M, Malzone C, Nuti S, Tavarini S, Sammicheli C, Hilbert AK, Brauer V, Banzhoff A, Rappuoli R, Del Giudice G, Castellino F (2009) Adjuvanted H5N1 vaccine induces early CD4+ T cell response that predicts long-term persistence of protective antibody levels. Proc Natl Acad Sci USA 106:3877–3882
Doherty PC, Turner SJ, Webby RG, Thomas PG (2006) Influenza and the challenge for immunology. Nat Immunol 7:449–455
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Birkhäuser Basel
About this chapter
Cite this chapter
Mintern, J.D., Guillonneau, C., Turner, S.J., Doherty, P.C. (2011). The Immune Response to Influenza A Viruses. In: Rappuoli, R., Del Giudice, G. (eds) Influenza Vaccines for the Future. Birkhäuser Advances in Infectious Diseases. Springer, Basel. https://doi.org/10.1007/978-3-0346-0279-2_8
Download citation
DOI: https://doi.org/10.1007/978-3-0346-0279-2_8
Published:
Publisher Name: Springer, Basel
Print ISBN: 978-3-0346-0278-5
Online ISBN: 978-3-0346-0279-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)