
Abstract
This chapter discusses microextraction using liquid membranes immobilized in a porous support membrane, including two- and three-phase hollow fibre liquid-phase microextraction (HF-LPME), 96-well LPME (or parallel artificial liquid membrane extraction (PALME)), and electromembrane extraction (EME). These techniques are essentially two- or three-phase liquid extraction systems, but downscaled to the level where the consumption of organic solvent per sample is less than 10 µL. Such microextraction systems are interesting for several reasons. First, they are ideal for green sample preparation, and therefore they are expected to be important in the near future in the context of sustainability. In addition, due to size and technical arrangement, they are easily implemented in microchip systems. Recently, several research papers have been investigating such microchip systems in combination with smartphone detection. This research has the potential to move analytical measurements out of today’s specialized laboratories. The fundamentals are discussed, to underline that microextraction with liquid membranes can be performed in partition-based systems, or in systems controlled by an external electrical field. In addition, this chapter discusses novel developments and new applications, based on examples from recent literature. The chapter is not comprehensive but is intended to give a flavour of the field.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The interest for miniaturized extraction systems was initiated by the invention of solid-phase microextraction (SPME) in 1990 [1]. In SPME, a thin fibre covered with a polymeric coating serves as extraction phase. The fibre can be immersed directly in the sample for extraction or can be inserted in the headspace for extraction of volatile compounds. Polydimethylsiloxane (PDMS) is the most popular coating, but a range of commercial alternatives are available. SPME is often combined with gas chromatography (GC), after thermal desorption of the extracted material. SPME can also be combined with liquid chromatography (LC), and in such cases the extracted material is desorbed from the fibre using organic solvent. SPME was commercialized a few years after the first scientific paper and has gained substantial interest. SPME is solvent-free, automated, and enables soft extraction.
In parallel with the development of SPME, different approaches to liquid-phase microextraction were introduced. In 1996, single-drop microextraction (SDME) was introduced [2, 3]. In SDME, target analytes are extracted from the sample, and into a small droplet of organic solvent located at the needle tip of a micro-syringe. The volume is only a few micro litres, and the droplet can be injected directly into GC after extraction. Equipment for SDME is very simple and includes a micro-syringe and a magnetic stirrer. SDME is simple and inexpensive, but the droplet is unprotected and may be lost in the sample during extraction. Therefore, hollow-fibre liquid-phase microextraction (HF-LPME) was introduced in 1999, where the droplet was located inside the lumen of a hollow fibre as illustrated in Fig. 1 [4]. The purpose of the hollow fibre was to mechanically protect the droplet. The droplet (termed acceptor) was in liquid contact with a thin layer of organic solvent immobilized in the porous wall of the hollow fibre, and this was again in liquid contact with the sample. Thus, analytes were extracted from the sample, through the liquid membrane located in the wall of the hollow fibre, and into the acceptor inside the lumen of the hollow fibre.

Principles of two- and three-phase hollow-fibre liquid-phase microextraction (HF-LPME)
HF-LPME has been conducted in 96-well systems [5], but unfortunately no such systems are commercially available. However, the idea of 96-well HF-LPME with commercial equipment was realized using commercial 96-well filter plates [6]. In this configuration, originally termed parallel artificial liquid membrane extraction (PALME), the samples are pipetted into separate wells in a 96-well sample plate (Fig. 2a). The membrane solvent is then pipetted onto the filters in a 96-well filter plate (Fig. 2b). The membrane solvent is immobilized by capillary forces in the pores of the filter, and by such the liquid membrane is established. Finally, the reservoir above each filter in the filter plate is filled with acceptor (Fig. 2c), the filter plate and the sample plate are clamped, and extraction is facilitated by agitation of the clamped plates (Fig. 2d). Although the 96-well plates used are not intended for 96-well LPME, they are excellent for this purpose [6].

96-well LPME equipment and procedure
Mass transfer in HF-LPME is based on passive diffusion and is driven by partition coefficients. For this reason, extraction is relatively slow, and extraction times may be up to 45 min to reach equilibrium. To improve kinetics, electromembrane extraction (EME) was introduced in 2006 [7]. The setup for EME is very similar to HF-LPME or 96-well LPME, but electrodes are placed in the sample and acceptor, and are connected to an external power supply (Fig. 3). In this way, an electrical field can be sustained across the liquid membrane, and positively and negatively charged compounds can be extracted across the liquid membrane.

Principle of electromembrane extraction (EME)
This chapter will focus on HF-LPME, 96-well LPME, and EME. Related approaches are found in literature, including membrane bag-assisted-liquid-phase microextraction [8], solvent bar microextraction [9], solvent stir-bar microextraction [10], in-line supported liquid membrane extraction in capillary electrophoresis [11], hollow fibre-supported dispersive liquid–liquid microextraction [12], and micro-fluidic liquid–liquid microextraction [13]. Literature also include important approaches not based on SLMs, such as dispersive liquid–liquid microextraction [14], but these techniques are not discussed in this chapter.
2 Fundamentals
In HF-LPME and 96-well LPME, mass transfer across the liquid membrane is due to passive diffusion, and transfer across the phase-boundaries in and out of the organic liquid membrane is controlled by partition coefficients. HF-LPME can be performed either in two-phase or three-phase mode (Fig. 1). In two-phase mode, the liquid membrane and the acceptor is an organic solvent. Thus, the analyte is extracted from the aqueous sample, through the immobilized organic solvent (liquid membrane) in the wall of the hollow fibre, and into the bulk liquid of the same solvent located in the lumen of the hollow fibre. The acceptor is organic, and the bulk part of this may be collected after extraction and injected into gas chromatography (GC). Thus, two-phase HF-LPME is very convenient in combination with GC. Two-phase HF-LPME can also be combined with liquid chromatography (LC). In those cases, the analyte is extracted into the liquid membrane, followed by desorption into a polar organic solvent and injection in LC. For LC analysis, however, three-phase HF-LPME is more convenient. Here, the analyte is extracted from aqueous sample, through the liquid membrane, and into aqueous acceptor located in the lumen of the hollow fibre. Three-phase LPME can be performed for basic and acidic analytes. For basic analytes, the sample is made alkaline, to suppress the ionization of the analyte, and by such increase the partition into the organic solvent. The acceptor is acidic, and the analyte is protonated in contact with the acceptor, and trapped there. Bases are thus extracted from high to low pH across the liquid membrane. For acidic analytes, the pH gradient is reversed, and they are extracted from low pH to high pH.
In EME, mass transport across the liquid membrane is mainly by electrokinetic migration. For this reason, EME is faster than LPME under optimized conditions [15]. Mass transfer across the phase-boundaries in and out of the organic liquid membrane is controlled by electro-partition. Basic analytes are extracted as cations, and pH in both sample and acceptor should be neutral or acidic, to ensure that the analytes are completely protonated during extraction. The negative electrode (cathode) is placed in the acceptor and the positive electrode (anode) is placed in the sample. Acidic analytes are extracted as anions; the direction of the electrical field is reversed, and pH is neutral or alkaline to ensure the analytes are deprotonated during extraction.
The liquid membrane is a crucial component in both LPME and EME systems. The liquid membrane separates the sample and acceptor, and mass transfer is to a large degree controlled by the chemical composition and properties of the liquid membrane both in LPME and EME. The membrane solvent should be immiscible with water, and solubility in water should not exceed 0.5 mg/mL. The reason for this is to avoid leakage of membrane solvent into the acceptor during extraction. In addition, the membrane solvent should be non-volatile to avoid evaporative losses during extraction. Typical liquid membranes in two-phase LPME are dihexyl ether, dodecyl acetate, and octanol, but a large range of other solvents have also been reported to be successful [16]. Mass transfer in LPME is affected by the chemical composition and polarity of the membrane solvent, but also viscosity plays a very important role. In EME, the selection of membrane solvent is more critical than in LPME, because EME involves extraction of charged species. Thus, while a large range of solvents can work in LPME, the number of efficient membrane solvents for EME is much more limited. For basic analytes, 2-nitrophenyl octyl ether (NPOE) has established as a first choice, and NPOE is efficient for bases in the log P range 2.0 to 6.0. For more polar bases, 2-undecanone can be used in the log P range 1.0 to 2.0, while bases with log P < 1.0 normally are extracted with NPOE mixed with di(2-ethylhexyl) phosphate (DEHP) [17]. The latter compound is added to the liquid membrane as ionic carrier. For acidic analytes, EME is normally performed with higher alcohols such as 1-octanol, 1-nonanol, and 1-decanol, and polar acids are in similar way extracted with higher alcohols mixed with ionic carriers [17].
The pH-value in the sample is an important experimental parameter. In LPME, target analytes should be in neutral form in order to transfer across the liquid membrane. Therefore, acidic analytes require low pH in the sample, while basic analytes requires high pH. For EME, the analyte should be in ionic form; therefore, samples are acidified or neutral for extraction of bases, and is neutral or alkaline for extraction of acids. Adjustment of pH in samples solutions has been accomplished using a broad range of buffer systems, or with strong acids or bases.
Acceptor pH is equally important, and in both LPME and EME, the analyte should be ionized within the acceptor. This prevents back-diffusion into the liquid membrane, and is an important action in order to maximize extraction recovery. Aqueous solutions of formic acid, acetic acid, or ammonia are recommended when the acceptor is to be analysed by LC–MS. In cases where the analytical instrument is less sensitive to acidic or alkaline solutions, phosphate buffers and dilute solutions of hydrochloric acid or sodium hydroxide can be used.
In both LPME and EME, mass transfer in the sample solution is mainly by passive diffusion. Therefore, agitation is important. Extraction efficiency normally increases with increasing agitation rate up to 750 to 1000 rpm. Agitation rates above this level tend to be less efficient. Agitation is applied during the entire extraction process. The extraction time also plays a key role in both techniques, and recoveries increase with time until the systems are in equilibrium. The typical equilibrium time in EME is between 5 and 30 min, depending on the polarity and charge of the analyte. Extraction times are also affected by operational parameters including the volumes of sample, liquid membrane, and acceptor respectively, and by the extraction potential. Equilibrium times in LPME are longer, and typically in the range 30 to 60 min. The optimal extraction time (equilibrium time) is normally established experimentally during method development.
In EME, the electrical field serves as driving force for extraction. Therefore, the extraction efficiency increases with increasing extraction potential up to a certain limit (typically 50–100 V), from where the extraction potential is no longer the limiting factor. The optimal voltage is found by optimization experiments. EME is normally performed at optimal voltage to maximize extraction recovery. EME at lower voltages requires longer extraction time, but may increase the selectivity.
Under optimized conditions, recoveries in LPME an EME are often above 85%. Samples are normally exhausted for analyte, but small amounts of analyte may be trapped in the liquid membrane upon equilibrium. Therefore, extractions with recovery at 85% or higher are considered exhaustive. For polar analytes (log P < 0), recoveries are often lower than 85%, due to poor partition into the liquid membrane. When compared to SPME, extraction recoveries are normally higher with LPME and EME. Enrichment can be obtained if the volume of sample exceeds the volume of the acceptor. While most EME systems are operated with low enrichment (< five times), 27.000 times enrichment has been reported with LPME, where pharmaceuticals were extracted from 2 litres of sea water [18]. Due to the lipophilic nature of the liquid membrane, and due to the electrical field, selectivity is high in EME. Selectivity can easily be tuned by the direction and magnitude of the electrical field, by the chemical composition of the liquid membrane, and by pH in the sample and acceptor. Also LPME provides high selectivity, due to the discriminative nature of liquid membrane and the pH gradient. The consumption of organic solvent is less than 10 μL per sample, and LPME and EME are therefore future candidates for green and sustainable sample preparation.
3 Novel Developments
HF-LPME has since its introduction been demonstrated to have several advantages over traditional liquid–liquid extraction (LLE), namely the miniaturization of the extraction process which has enabled a substantially reduced consumption of organic solvents, as well as provide clean extracts and potentially great enrichment of analytes. HF-LPME however also has several disadvantages that have limited its adoption outside of academia; for example slower extraction kinetics compared to LLE. HF-LPME (Fig. 1) also requires a lot of manual handling in preparation of extraction, and therefore has limited applicability for automated and/or high-throughput sample preparation configurations. These limitations have however been addressed in various ways in recent years. HF-LPME is fundamentally a non-exhaustive technique, meaning that extraction recoveries rarely reach 100%. Strategies to improve the extraction efficiency and range of compounds that can be extracted with high efficiency have therefore also been explored. This includes the investigation of new membrane materials, and solvents to be used as SLM. A major trend in recent years has likewise been the development of environmentally friendly, or so-called “green”, solvents and materials. These novel developments are discussed in more detail in the following sections. For each subject, a few representative examples are given, with emphasis on the most relevant aspect for the specific subject. For more detailed examples of HF-LPME applications, the reader is referred to Sect. 4.
3.1 External Force-Assisted HF-LPME and EME
As previously discussed, one limitation of HF-LPME is the slow extraction kinetics. Over time, several approaches involving the application of external force to the extraction system have therefore been developed, one example being the application of an electric field across the SLM to stimulate mass transport (termed electromembrane extraction (EME). Another example is increased temperature. In HF-LPME and EME, the formation of boundary diffusion layers also constitute a mechanism for slow kinetics. Specifically, as analytes are extracted into the SLM a thin layer of sample solution surrounding the SLM becomes depleted, and must therefore be replenished by new analyte molecules to make the extraction progress. This process is diffusion-driven and therefore slow. Similarly, a boundary layer with high analyte concentration is formed on the acceptor-side of the SLM, creating a gradient that slows the transport out of the SLM. Good convection of the solutions is therefore important to ensure that these boundary layers are as thin as possible. In HF-based extractions, stirring the sample solution with a magnetic stirrer is a very common approach; however, other novel approaches have also been introduced.
Vortexing the sample instead of stirring has been suggested as a method for improving kinetics. Wang et al. for example compared vortex-assisted (VA)-HF-LPME to stirring-based HF-LPME, and found the VA-HF-LPME system to reach extraction equilibrium (88–94% recovery) after only two minutes [19]. The stirring-based system, on the other hand, had only achieved 6–10% recovery in the same time, but reached 86–95% after 40 min. If reviewing the academic literature, one however quickly finds that vortexing only occasionally is used for agitation in HF-LPME. There may be several good reasons for this, namely that it typically requires the operator to manually hold the sample as it is being vortexed, whereas a stirring-based system can be left while the extraction progresses. The strong shear forces of sample against SLM may also put greater requirements on the SLM stability; else, parts of the SLM solvent may be dissolved or emulsified in the sample.
Ultrasound has also been suggested to improve extraction kinetics, and was likewise compared to stirring by Wang et al. [19]. Compared to stirring, ultrasound-assisted HF-LPME had, like VA-HF-LPME, much faster kinetics with 76–82% recovery after two minutes. However, there was also a clear loss of SLM integrity that made the sample solution clouded. Compared to stirring and vortexing, ultrasound however has the advantage of also providing convection to the acceptor solution as well, which may help decrease the thickness of the boundary layer on the acceptor side of the SLM. Ultrasound has also recently been applied in EME, with the dual aim of decreasing the thickness of the diffusion boundary layer and the electrical double layer [20, 21]. The latter arises from ions that cannot enter the SLM due to poor partitioning, such as salts or buffer ions, and therefore accumulate in the boundary layers in either side of the SLM. Due to the electric field, these ions will have the same charge as the analyte(s), and therefore exert a charge-repulsive effect on approaching analyte ions, which limits their partitioning into the SLM. In the report be Seyfinejad et al. [20], the impact of ultrasound power was investigated. It was found that increasing recoveries were obtained until 20 W. Above 20 W, the SLM (2-nitrophenyl octyl ether) was dissolved into the sample and acceptor solutions. However, at 20 W both the extraction kinetics and steady-state recovery were higher than a conventional stirring-based method, which was attributed to the reduced thickness of diffusion and electric double layers. This was confirmed experimentally by Shang et al. [21].
Microwave-assisted extraction (MAE) is another emerging method that has particularly been successful for solid–liquid extraction, for example of plant material. However, to the best of the authors’ knowledge, no reports on microwave-assisted HF-LPME or EME have been published.
3.2 Novel Membrane Materials
In the vast majority of HF-LPME and EME reports published so far, polypropylene (PP) has been the preferred material for the support membrane (i.e. the hollow fiber) due to its excellent properties such as chemical inertness and being very inexpensive. However, alternative membranes and materials are currently also in development. Some of these are discussed in this section.
3.2.1 Polymer Inclusion Membranes (PIMs)
PIMs are a novel class of functionalized membranes that, among other applications, are emerging for microextraction purposes. Typically, PIMs are composed of a base polymer, in example cellulose triacetate (CTA) or polyvinyl chloride (PVC), a plasticizer, and a carrier. The base polymer serves as the backbone of the material, providing mechanical strength, while plasticizers make the membrane flexible and allow it to be formed into different shapes. Plasticizers act as the solvent, and may serve to enhance the transport analyte molecules, in tandem with carriers that enhance the analyte transport by complexation. In some cases, the carrier may also function as a plasticizer. The selection of all three components may affect the extraction properties of the membrane, though the carrier generally is considered the most important. Carriers may be acidic, basic, neutral, have chelating properties, and other characteristics, which enables quite different extraction selectivity. Practically, a PIM is prepared by dissolving the base polymer in an organic solvent and adding plasticizer and carrier, after which the mixture is cast into the desired shape. Flat sheet PIMs are most common, though hollow fiber PIMs also are made.
An example application of a PIM in EME was reported by Román-Hidalgo et al. in 2018 [22]. The PIM was synthesized by dissolving CTA in dichloromethane and adding Aliquat® 336, a technical mixture of alkyl-substituted quaternary ammonium chlorides, as carrier. The mixture was poured into a glass Petri dish and left for the dichloromethane to evaporate, after which the PIM could be peeled out and applied as a 25 µm thick, flat membrane. The final PIM composition was 29% CTA and 71% Aliquat® 336. Prior to extraction, the PIM was added 1-octanol as extraction solvent and applied in a homemade extraction device for EME of polar acidic substances. In this case, the positive charge on the ammonium group of Aliquat® 336 could form an ion-pair with the negatively charged acidic analytes, to stimulate the transfer across the PIM.
3.2.2 Alternative Membrane Materials
Various other materials have been applied as supporting membrane. Hydrophobic membranes, similar to PP, have included polytetrafluoroethylene (PTFE) and polyvinylidene fluoride (PVDF). The latter has been suggested to improve extraction kinetics [16, 23], but is still not widely used. Other, more polar, materials have also been used, for example nylon and electrospun acrylic-based nanofiber sheets. Biopolymers such as chitosan and agarose have also emerged as environmentally friendly materials.
For a more detailed overview of supporting membrane materials in HF-LPME and EME the reader is referred to Ocaña-González et al. [16].
3.3 Novel Green Solvents
In HF-LPME, dihexyl ether, 1-octanol, toluene, and dodecane are among the most commonly used extraction solvents. While these solvents have been used with great success in many applications, there are several drawbacks to these and other similar solvents. Firstly, some solvents have low boiling points, high vapor pressure, and undesirable toxicity profiles. Toluene, for example, may evaporate during preparation of extraction and expose the operator to toxic fumes. Secondly, the traditional solvents typically only have one or two different functional groups, which limits the diversity of chemical interactions that can happen between solvent and analyte(s). This consequently limits the span of analytes that can be extracted with one solvent. While this may be an advantage if selective extraction is important, it makes method development for new analytes more cumbersome since many solvents may have to be tested. The difficulty of extraction generally increases with increasing analyte polarity, since these exhibit low partitioning coefficients, resulting from a large offset in hydrophobicity between analyte and hydrophobic solvent. One way of overcoming this hydrophobic discrimination is to increase the strength and diversity of chemical interactions offered by the solvent. However, increasing strength of interactions generally also increases the melting point of a solvent, which may form a solid substance at room temperature. To mitigate this problem, different novel solvent systems have been introduced. The two most prominent are ionic liquids (ILs) and deep eutectic solvents (DESs), both of which often also are claimed to be green. However, it should be noted that one never should generalize the greenness of an entire solvent system, as it is entirely dependent on the composition of a specific solvent. Another novel solvent system often considered green are the nanostructured supramolecular solvents (SUPRASs).
While the formation of ILs, DESs, and SUPRASs relies on different principles, they share the potential to be used as task-specific “designer solvents”, with properties that can be tuned to be suited for specific analytes, extraction methods, or sample matrices. This prospect and some advantages/disadvantages of each solvent system is discussed in sections below.
3.3.1 Ionic Liquids (ILs)
An IL is a salt made from a bulky organic cation and a smaller organic or inorganic anion. Some examples are given in Fig. 4. In conventional salt (e.g. NaCl), strong electrostatic interactions between the cation and anion result in a very high melting point. However, because of the asymmetric size of ions in ILs the electrostatic interactions are decreased substantially, to a point where the salt may form a liquid below 100 °C or even room temperature. Because both the cation and anion can be varied the number of potential ILs are billions or more. By varying the ions used, ILs may offer electrostatic, hydrogen bond, π-π and other π-type, dipole, dispersion, and hydrophobic interactions. Physico-chemical properties can likewise be tuned; viscosity, conductivity, density, and water-miscibility are examples that are particularly important in HF-LPME and EME. ILs are generally non-volatile, which is one of the reasons why ILs are proclaimed to be green. However, it should be noted that some ILs also have been shown to be toxic and poorly biodegradable [24]. Considerations of greenness is therefore much dependent on the toxicity of the organic solvent the IL is meant to replace.

Examples of frequently used cations and anions in ILs. Reprinted from Hansen and Pedersen-Bjergaard [25] with permission from American Chemical Society
ILs have been applied in different configurations, both two-phase and three-phase HF-LPME, although the widespread use has been somewhat limited. In one representative example, Wang et al. [26] developed a three-phase HF-LPME system for determination of phthalate esters in tea beverages, using 1-nonanol as SLM and the IL 1-butyl-3-methyl-imidazolium hexafluorophosphate ([BMIm]PF6) as acceptor phase. The phthalates were subsequently quantified by HPLC–DAD. After 4 min of extraction, recoveries were near 100% and 200-fold enrichment of the analytes was achieved.
A major disadvantage of ILs in HF-LPME is their relatively high viscosity, which may hinder the penetration into the pores of the hollow fiber and slow the mass transfer kinetics. This may be one of the reasons why the use of ILs as extraction solvent has been somewhat limited in HF-LPME. ILs have also been applied as SLM in a few instances of three-phase EME [27, 28], but is generally disfavored due to high conductivity leading to current-induced instability. ILs may also pose a problem in chromatography, where they may give high background signals or cause ion suppression in electrospray ionization mass spectrometry-based analysis [29].
3.3.2 Deep Eutectic Solvents (DESs)
DESs are in many ways similar to ILs in terms of properties and potential as “designer solvents”. While ILs form a liquid due to weakened electrostatic interactions, the principle of DESs is based on hydrogen bonding. A eutectic mixture is composed of two or more solid components, one hydrogen bond donor (HBD) and one acceptor (HBA), that when mixed in specific molar ratios form intermolecular hydrogen bonds that lower the melting point below that of the individual components. This is also seen from the phase diagram in Fig. 5a. If the melting point depression is large and the mixture forms a liquid at room temperature, it may be termed a deep eutectic solvent (DES) [30]. In example, the first DES discovered, in 2003, was composed of choline chloride and urea in 1:2 molar ratio, and exhibited a melting point 290 °C lower than that of choline chloride [31]. Until 2015, only hydrophilic (water-miscible) DESs were reported. The first hydrophobic and water-immiscible DESs were based on alkylated quaternary ammonium salts and fatty acids, for example tetrabutylammonium chloride (HBA) and decanoic acid (HBD) [32]. Other examples include phosphonium-based cations (Fig. 5b). Recently, hydrophobic DESs based on naturally occurring monoterpenes have also been developed [33]. Some examples of components include camphor and coumarin as HBA components and thymol, menthol, and different fatty acids as HBD components [34].

Adapted from Hansen and Pedersen-Bjergaard [25] with permission from American Chemical Society
a Illustration of a phase diagram for a deep eutectic two-component mixture. b Example HBA and HBD components.
DESs hold several advantages over ILs. They are easier to synthesize, less expensive and often readily available, have less toxicity and better biodegradability, and are generally less viscous then ILs [35]. The latter is an important advantage for HF-LPME. The naturally occurring DES components are particularly interesting, especially from a greenness perspective, since they are fully biodegradable and readily available at a low cost. The non-ionic structure of the above mentioned components further yields even lower viscosity, because of the weaker type of interactions. DESs also share the prospect of being “designer solvents”, since many solvents of different properties and chemical interactions can be produced.
DESs have recently become popular solvents for HF-LPME and EME. Hydrophilic DESs are however limited to be used as acceptor phase for aqueous samples, because they yield unstable SLMs in contact with aqueous samples. Seidi et al. for example used a DES of choline chloride (HBA) and ethylene glycol (HBD) in 1:4 molar ratio as acceptor solution, with an SLM of 1-octanol with cetyltrimethylammonium bromide (CTAB) (carrier-mediated HF-LPME), to extract raloxifine and ethinylestradiol from wastewater prior to HPLC–UV analysis [36]. Zhang et al. [37] used a hydrophobic DES of tetrabutylammonium chloride (HBA) and hexanoic acid (HBD) in 1:3 molar ratio for HF-LPME of some cinnamic acid derivatives from traditional Chinese medicine and human plasma. Because this DES was hydrophobic, it could be used as the SLM solvent, while an 85% solution of the DES in methanol was used as acceptor solution. The DES was reported to be fairly viscous, which hindered the mass transfer of analytes. However, by increasing the temperature during extraction to 55 °C the viscosity was reduced and the mass transfer was improved.
Different hydrophobic DESs have also been applied in EME. Because an electric field is applied across the SLM to stimulate the extraction in EME, the SLM solvent can only be slightly conductive to avoid excessive current and electrolysis. Thus, only non-ionic DESs have been reported. Solvents with coumarin or 6-methylcoumarin as HBA and thymol as HBD have in particular been successful for extraction of both non-polar drugs [38], polar drugs and endogenous metabolites [39, 40], and peptides [41] in human plasma samples. For these examples, the SLM was based on flat membranes either in 96-well format or in prototype equipment based on conductive vials. The EME systems were three-phase with aqueous sample and acceptor solutions, and 4–10 µL DES in the SLM. The wide range of analyte properties that have been successfully extracted with this DES is likely due to its structure being highly aromatic, thereby providing different π-type interactions in addition to hydrogen bonding.
3.3.3 Nanostructured Supramolecular Solvents (SUPRASs)
SUPRASs represent a third category of green solvents under development. SUPRASs are composed of amphiphilic surfactants (ionic or non-ionic) that in solution form nanostructured micelles or vesicles when above the critical micelle concentration (CMC). The formation of a SUPRAS happens by a change in environmental conditions that induces further aggregation and self-assembly of the nanostructures, which leads into phase-separation of the supramolecular aggregates from the bulk solution (Fig. 6). The process is termed coacervation, and the new supramolecular phase is termed a SUPRAS. The environmental change is made by addition of a coacervation reagent, which may be acids/bases (pH), organic solvent, or salts (ionic strength). Alternatively, temperature may be changed to induce coacervation. After assembly of aggregates, the SUPRAS structure is held together by non-covalent bonding. For application in HF-LPME, the SUPRAS is typically formed in a separate container and collected by pipette, before application to the hollow fiber. However, for other extraction techniques such as dispersive liquid–liquid microextraction (DLLME), the coacervation process may be performed in situ (in the sample solution itself). Here, the matrix may itself act as the coacervation agent, for example the high salt levels found in urine samples.

Formation process of SUPRASs. The final SUPRAS may have higher or lower density than the bulk solvent, which means it may sink or float. Reprinted from Ballesteros-Gómez et al. [42] with permission from Elsevier
The microscopic structure of SUPRAS provide them with very interesting properties, from an extraction point-of-view. The assembled SUPRAS has microenvironments with widely different properties, for example hydrophilic areas associated with the head of surfactants and water incorporated in the structure. Hydrophobic areas are on the other hand associated with the tail of the surfactants. SUPRASs thus have great potential for multi-residue extraction of analytes across a wide range of hydrophilicity. Similarly to ILs and DESs, the chemical interactions provided by a SUPRAS can be designed based on the type of surfactants used to make the solvent. Examples of chemical functionality for the polar head of surfactants include carboxylic acids, sulfonates, alcohols, ammonium and pyridinium ions. Most SUPRAS can thus be considered environmentally friendly, also considering that the formation is spontaneous and requires no energy input, albeit that organic solvents used to induce the coacervation are disfavored in this aspect. Another desirable feature of SUPRASs is a large surface area, because the coacervate remains as droplets. The mass transfer of extraction is therefore fast. Parts of the surface area is located within the internal structures of the droplets, and SUPRASs may therefore also possess restricted access properties (RAM). Macromolecules such as proteins may therefore be discriminated from extraction. The size of cavities can be tuned by different coacervation parameters, for example the amount of organic solvent used.
One limitation of SUPRASs is related to compatibility with HPLC and GC analysis. SUPRASs are very rich in non-volatile surfactants, and a back-extraction step into a volatile solvent may therefore be necessary prior to GC analysis. Alternatively, headspace GC may be utilized if the surfactants are thermally stable. SUPRAS are directly compatible with HPLC, as the nanostructures generally disassemble in the organic/aqueous mobile phase, and are retained and eluted as any compound normally would. However, the micelle or vesicle structure may be retained if the mobile phase is primarily aqueous, and may thus act as a pseudo-phase that disturbs the chromatography. Hyphenation of HPLC to MS is however more problematic, since the surfactants may foul the ion source and cause ion suppression. Chromatographic separation from the analytes is thus required, so the surfactants may be directed to the waste container.
There are several examples of SUPRASs applied in HF-LPME. However, because the SUPRAS is in an equilibrium with surrounding solutions, it may dissolve into the sample if the internal binding forces are too weak and the sample-to-SLM volume ratio is very large (as is typically the case in HF-LPME). HF-LPME has thus not been the primary microextraction technique SUPRASs have been applied for. However, a few applications have been developed in this way. One example of the successful application was presented by Rezaei et al. in 2013 [43]. In this work, a SUPRAS based on tetrabutylammonium and decanoic acid was used in a two-phase HF-LPME system for extraction of benzodiazepines from samples of fruit juice, human plasma, and urine. Quantitation was accomplished by HPLC–DAD analysis. The driving force of the extraction was proposed to be a mix of hydrogen bonding, cation-π, and hydrophobic interactions. Another example was reported by Li et al. in 2020 [44]. In this, the authors employed a magnetic solvent bar with a SUPRAS immobilized into the HF-wall to extract NSAID drugs from human serum, followed by LC–MS/MS determination. The solvent bar was prepared by fitting a stainless steel needle into the lumen of the fiber, after which the ends were closed with tweezers, and the fiber was immersed in the SUPRAS for 1 min to impregnate the pores. The SUPRAS was made from hexafluoroisopropanol and decanol. Extraction was performed by magnetically stirring the solvent bar in the sample for 33 min, after which the now analyte-enriched SUPRAS was eluted from the HF-walls using ethyl acetate and ultrasound. The eluate was subsequently evaporated to dryness, reconstituted, and injected into the LC–MS system.
Examples of SUPRASs applied for EME have so far not been reported. For more extensive overview of the SUPRAS concept and its applications, the reader is encouraged to see a recent review by Rubio [45].
3.4 Novel Sorbents
While substantial effort has gone into finding efficient and green solvents for HF-LPME, modification of HF-LPME systems by solid sorbents has also been investigated. The main idea of modifying the extraction system with solid sorbents is to improve extraction kinetics and/or selectivity by operating in mixed-mode. Practically, the addition of sorbent material can be done by dispersing into the acceptor solution, in which case an elution step must be performed after extraction. Alternatively, the sorbent may be added to the SLM itself, either by dispersing the sorbent in the SLM solvent or by decorating the surface and/or pores of the fiber with sorbent prior to immobilization. The latter may be done by submersing the hollow fiber into a solution of sorbent dispersed in a volatile solvent, and treating the solution with ultrasound to stimulate the movement of sorbent particles into the pores. Finally, the volatile solvent can be evaporated to leave a dry and decorated fiber. By keeping the sorbents inside the hollow fiber there is thus not any need for post-extraction elution steps.
Many different sorbent chemistries have over time been applied in HF-LPME. In the following sections, we discuss some of the most predominant materials.
3.4.1 Molecularly Imprinted Polymers (MIPs)
Much like ILs, DESs, and SUPRASs may be designed and applied with the aims of achieving specific analyte-solvent interactions, solid sorbents can be designed to offer desired chemical interactions, for example hydrogen bonding, π-π stacking, dispersion, dipole, etc. However, while these interactions may provide some selectivity they are fundamentally non-selective towards specific analytes, and the clean-up from matrix components may thus be unsatisfactory. Molecularly imprinted polymers (MIPs), on the other hand, have potential to offer much greater extraction selectivity, even between analytes with closely related structures/properties. MIPs are synthesized by polymerizing functional monomers (for analyte interaction) and cross-linking monomers (for cross-linking polymer) in the presence of a template molecule. The template is incorporated into the polymer by non-covalent interactions, and can subsequently be removed by appropriate washing steps, leaving cavities of the size, shape, and functionality of the analyte template. During extraction, the MIP functions as a restricted access material (RAM) to enable very selective extraction of target analytes. The template molecule may be the target analyte itself, but a structural analog that can be distinguished from the analyte in the analysis is often desirable to avoid contamination from leaching template molecules. Isotopes of the analyte may for example be distinguished by mass spectrometry. In addition to providing selective extraction, MIPs also have the benefit of being relatively inexpensive to prepare.
MIPs have been applied both for HF-LPME and EME, which has enabled greater extraction selectivity and efficiency. However, MIPs are more commonly applied in dispersive solid-phase extraction, where they are capable of extracting directly in the sample matrix. In this case, extraction recoveries are subject to decrease in the presence of a complex matrix due to blockage of the MIP binding sites. This may however be avoided by protecting the MIPs with an SLM that prevents for example proteins and larger suspended particles from getting contact with the MIP. Such a system can further offer very good clean-up because of dual-mechanism selectivity. An early example of the application of MIPs in HF-LPME was reported by Nemulenzi et al. [46], who extracted the hormone 17β-estradiol from wastewater (37 mL) using a two-phase flat membrane-based system with hexane:ethyl acetate in 3:2 ratio as SLM and acceptor solution [46]. 30 mg MIPs were suspended in the acceptor solution (2.5 mL). During 60 min of extraction, the analyte was extracted into the acceptor solution and partially bonded to the MIP particles. However, by subsequently transferring the entire acceptor solution to a vial and treating it with ultrasound for 15 min, the binding efficiency could be increased. Elution of analyte from the MIP was performed with acetonitrile, which was partially evaporated prior to injection on HPLC (to increase enrichment factor). The improved selectivity of the SLM-MIP-based method compared to simple SLM extraction was evident by cleaner chromatograms.
MIPs have also been dispersed in the SLM solvent itself in three-phase systems. In example, Yaripour et al. suspended MIPs in 1-octanol prior to immobilization in a hollow fiber, to perform EME of phenytoin and phenobarbital from biological fluids [47]. However, in the example, the extraction recovery was worse with MIPs added to the SLM, compared to the pure solvent. This may have been due to too strong bonding between the analytes and MIPs, which may have caused trapping inside the SLM.
3.4.2 Carbon-Based Nanoparticles
Another class of novel sorbent phases are carbon-based nanoparticles, such as carbon nanotubes (CNTs), graphite/graphene and related derivatives. These materials are attractive sorbents due to properties such as large surface-to-volume ratios and ability to provide different chemical interactions, for example hydrophobic interactions and π-π stacking. The materials can however also be functionalized to provide polar interactions, for example dipole–dipole and hydrogen bonding. For instance, graphene may by oxidized to graphene oxide (GO) to include epoxy, COOH, and OH groups on the surface. GO is sometimes chemically reduced to yield reduced graphene oxide (RGO), which contains fewer polar groups on its surface.
In 2020, Worawit et al. reported an application of HF-LPME for extraction of trihalomethanes (for example chloroform) in different water samples. Here, the effect of different carbon-based materials added to individual polypropylene hollow fibers was investigated [48]. This included graphite, graphene, and multi-walled carbon nanotubes (MWCNTs). The hollow fibers were prepared in advance by loading the sorbents into the pores. Preparation for extraction therefore only comprised the immobilization of 1-octanol as SLM solvent. All three materials provided equal or better extraction recoveries than a pure 1-octanol SLM, with the greatest improvement seen for graphite. This confirmed the potential benefit of operating such a mixed-mode extraction.
3.4.3 Metallic Nanoparticles
Metallic nanoparticles have also been used to decorate hollow fibers for HF-LPME and EME. Their benefits resemble that of carbon-based nanoparticles, including the potential for functionalization, and they are generally applied in a similar way. Various metals and metal oxides have so far been used to make the particles, including gold (Au), silver (Ag), copper (Cu), iron (Fe), and titanium dioxide (TiO2). The nanoparticles often enhance the extraction non-specifically (e.g. by enhancing surface area), but may also be selected to have more specific interactions with the analyte(s). In example, a hollow fiber decorated with copper nanoparticles (CuNPs) was applied for EME of thiouracil from urine [49]. CuNPs were in this case selected because they have high affinity for sulfhydryl compounds, such as thiols. Compared to AuNPs or AgNPs, CuNPs are further less expensive.
3.5 Automation and High Throughput
Automation and high throughput systems both represent one principle within the “Ten principles of green sample preparation” [50]. Automated systems are desirable because they free up personnel that otherwise would have performed the procedures manually, and any exposure of the operator to potentially toxic chemical is substantially reduced or eliminated. High throughput is desirable from a green chemistry point-of-view because the total analysis time, and thereby the operator’s exposure, as well as the energy consumption and cost typically is reduced for each sample. From a more practical point-of-view, high throughput also means that more samples can be processed in each working day. High throughput may be achieved by either very short analysis time of successive samples, or by analyzing multiple samples in parallel (for example in 96-well format).
Considering classical HF-LPME, a typical procedure will, for each sample, involve manual preparation and cleaning of the hollow fiber, immobilization of the SLM, pipetting sample and acceptor solutions, immersing HF in sample and waiting during extraction, followed by removal of the analyte-enriched acceptor solution. It is clear that this is neither easily automated nor applicable for high throughput. However, research to transfer the principle of HF-LPME into suitable technical formats is currently ongoing, and in this section, we shall discuss some examples.
3.5.1 Approaches for Automation
In general, a system can be said to exhibit some degree of automation if one or more discrete steps in a procedure are performed without requiring any human intervention. In literature, terms such as “fully” or “semi” automated are frequently used. In accordance with established correct IUPAC terminology, a fully automated system involves the complete processing of consecutive samples without any human intervention [51]. Semi-automated systems, on the other hand, can perform one or more discrete steps automatically, but require some form of human intervention in the process. This may for example be manual transfer or replenishment of solutions between samples, or preparation of a new HF-SLM. The typical analytical workflow is illustrated in Fig. 7.

The typical five steps in an analytical workflow
In this workflow, a method’s degree of automation is normally considered from the raw, untreated sample (after sampling), to the detection (e.g. HPLC) has been performed. Data processing and interpretation is also sometimes included in the automated workflow. Considering extractions, this step can be performed off-line (i.e. not automated), at-line, on-line, or in-line. At-line refers to procedures performed in close proximity to the analytical instrument, with an automated transfer of the prepared sample, for example by robotics. In on-line systems, the sample preparation step is typically connected to the analytical instrument by means of tubing and a modulator (for example a switch-valve with a loop). Finally, in-line systems couple a continuous flow-system with an extraction device to a detector, without any flow-interruptions. Fully automated in-line HF-LPME is however rarely used when dealing with multiple samples, since it is mainly suitable for continuous process monitoring.
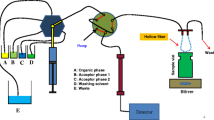
There are several practical challenges for automating HF-LPME and EME. Firstly, to avoid memory effects (i.e. carry-over and changing performance over time) it is normal to use a new HF-SLM for each extraction. Immobilization of the SLM solvent and placement of the fiber into the sample is however difficult to automate. Most literature reports have therefore comprised semi-automated systems, with the HF preparation steps performed manually. Secondly, handling of acceptor volumes of 5–20 µL inside the HF lumen is likewise difficult for robotic instrumentation. One example of this was reported by Cabal et al. in 2019 [52]. In the example, the authors constructed a semi-automated HF-LPME-HPLC-FLD (fluorescence detection) system based on a programmable syringe for loading and removal of acceptor solution from the lumen of the hollow fiber, and further transfer to the HPLC analysis. To prepare for operation, 8 mL urine sample was manually filled into a 10 mL vial with a stirrer, and a 10 cm long hollow fiber was introduced. One end was fitted manually to the syringe, via a three-way solenoid valve, which was also connected to a 6-port switch valve equipped with a 2 µL loop and coupled to the analytical column of the HPLC. The extraction steps are illustrated in Fig. 8.

Schematic illustration of the semi-automated extraction and HPLC analysis. Reprinted from Cabal et al. [52] with permission from Springer Nature
As the first step, 1-octanol (acceptor solution) was drawn into the syringe and used to fill the pores and lumen of the hollow fiber (two-phase HF-LPME). Extraction of some amphetamines from the urine was then performed for 14 min (not to steady-state), after which the analyte-enriched acceptor solution automatically was withdrawn from the HF-lumen, and flushed to the loop by the syringe. The 6-port valve was then switch to flush the loop with mobile phase towards the analytical column, and data acquisition was triggered. While the chromatographic separation was performed, the syringe underwent some washing steps and a new urine sample and hollow fiber was prepared for the next extraction. The present report is an example of a semi-automated extraction and chromatographic system, requiring for each sample manual preparation of the hollow fiber and manual fitting of the syringe to the fiber. For full automation, HF-LPME and EME systems are therefore mostly on-line. One such EME system was reported by Fuchs et al. in 2016 [53]. In this, a commercial autosampler equipped with a syringe holder, vial-wrack, and a wash station, was used to couple a custom EME probe to an LC–MS system. The EME probe was constructed by modifying a female luer lock adapter that was fitted to a 1 mL glass syringe (Fig. 9a). A hollow fiber with fluidic connections was then fixed inside the lumen of the luer lock adapter; specifically, a fused silica capillary was inserted into each end of a short hollow fiber segment that subsequently was fixed to the capillaries by heat-shrink tubing and heating (which also collapsed the HF pores), leaving a 3 mm segment with intact pores (Fig. 9b).

Adapted from Fuchs et al. [53] with permission from American Chemical Society
a Side-view illustration of EME probe. b Top view illustration with micrograph of the hollow fiber fixed to fused silica capillaries. c Schematic representation of the on-line hyphenation of EME to LC–MS.
The capillaries were connected to a 10-port switch valve that was set up with a 20 µL loop, flow of acceptor solution, and connection to an LC–MS instrument (Fig. 9c). A metal wire connected to a programmable voltage-sequencer was also inserted into the luer lock to provide an electric field across the SLM, with the 10-port valve serving as counter electrode (grounded). Before each working day, the SLM was prepared by immobilizing 2-nitrophenyl octyl ether (NPOE) in the pores of the 3 mm free HF segment, and the lumen was flushed with acidified aqueous acceptor solution (thus three-phase system). The extraction sequence for each sample was as follows: the syringe needle, with the modified adapter, was immersed into the sample solution and 100 µL was drawn up and down twice, thus passing the SLM inside the adapter lumen. A potential of 200 V was simultaneously applied to stimulate the extraction of protonated basic analytes through the SLM. The acceptor solution was kept stagnant during this step. Thereafter, the 10-port valve was switched to flush the analyte-enriched acceptor solution into the loop. Once loaded onto the loop, the valve was switched again to flush mobile phase towards the column and mass spectrometer. Meanwhile, the syringe needle moved to a wash port and washing was performed by repeated drawing and injecting of an aqueous acidic wash solution, while a potential of –200 V was applied to back-extract analytes trapped inside the SLM. Once the LC–MS separation was complete, the system would automatically progress to the next sample. The autosampler, 10-port valve, and voltage-sequencer were all controlled from the LC–MS by an inbuilt relay function. The total analysis time, including EME and LC–MS was only 5.5 min, and could therefore be considered as fairly high throughput. Considering that the same SLM was used for an entire sample sequence, potential memory effects were evaluated. Regarding stability, the system was reported to give stable signals for an entire day of work (>50 extractions), which may be attributed to a very low water solubility of NPOE. Carry-over was determined to be 0.9% when the method was applied for determination of methadone and its metabolite EDDP during an in vitro metabolism study. An interesting feature of the system was that only 4% recovery (termed soft extraction) was obtained during the extraction. The system can thus be useful when there may be a wish for analyzing the sample multiple times, when it is undesirable to disturb the chemical equilibrium of a biological system. For example during drug metabolism studies where soft extraction may be used to probe the drug and metabolite concentration over time. Soft extraction may however be disadvantageous if highly sensitive analysis is required.
3.5.2 Approaches for High Throughput
While high throughput can be achieved by analyzing samples in sequence with very short analysis time per sample, as exemplified in the above section, a more common approach is to analyze samples in parallel, due to typical duration of HF-LPME or EME extractions. The latter is commonly performed in 96-well format. Some examples of hollow fiber-based 96-well formats have been reported [54, 55], but these have required highly skilled operators performing considerable manual handling, and are therefore not very user-friendly. Alternatively, 96-well formats of HF-LPME and EME based on flat membranes have been developed [56,57,58]. These were discussed in Sect. 1. An example application of 96-well LPME is also given in Sect. 4.
Another strategy towards high throughput analysis based on HF-LPME was reported by Miková et al. in 2020, for at-line elution and extraction of analytes from dried blood spots (DBS) ahead of capillary electrophoresis—diode array detection (CE-DAD) quantitation [59]. DBSs are currently emerging as a new method for blood sampling that is inexpensive, as well as more convenient and less invasive to the donor than conventional blood sampling. Typically, a few drops of blood from a finger are place on a DBS card (paper) and dried, and may thereafter simply be mailed to the analytical laboratory. For analysis of DBSs, a typical workflow will comprise punching an area of the DBS and performing an elution of the dried blood by shaking with an elution solvent. This is followed by sample clean-up procedures and quantitative determination by for example HPLC. To improve the throughput of the procedure, the authors developed a simple and inexpensive device with a 9 mm long hollow fiber closed on one end and fixed to a pipette tip in the other end. While the fabrication was performed manually, devices were made in larger batches and stored until use. Prior to extraction, the SLM solvent was immobilized in the HF pores for 10 s, and 5 µL acceptor solution was pipetted into the lumen. The workflow is illustrated in Fig. 10.

The workflow of at-line DBS analysis by HF-LPME-CE-DAD. Reprinted from Miková et al. [59] with permission from American Chemical Society
The punched DBS was added to a CE-vial with 550 µL aqueous elution/donor solution, and the HF-device was placed into the CE-vial. The vial was then shaken for 10 min for simultaneous DBS elution and HF-LPME of some model basic drugs. Shaking was performed for up to 50 parallel sample vials that subsequently were transferred manually to the CE-instrument’s autosampler and analyzed. The extraction device was designed to allow immersion of the inlet capillary into the lumen of the HF, which enabled direct injection of the acceptor solution (Fig. 10f–g). The total CE-analysis time was 8 min, including regeneration steps between samples. For parallel DBS analysis, the total analysis time was thus approximately 10 min per sample. Despite the efficient combination of multiple sample preparation steps, the use of non-commercial equipment does represent a limitation. However, there is potential to make this system fully automated.
4 Main Application
A large number of applications have been reported with two- and three-phase LPME, and with EME. LPME is well suited for extraction of analytes of low polarity from aqueous samples; neutral analytes are extracted with two-phase LPME, while acids and bases are extracted either with two- or three-phase LPME. With EME, both non-polar and polar acids and bases can be extracted, extractions are faster. Extraction of pharmaceuticals and drugs of abuse from biological fluids, organic micro-pollutants from environmental waters, and contaminants from food and beverages are typical applications for LPME and EME. It is outside the scope of this chapter to review all these, and readers with interest in a particular application should find more information in review articles. In the following, a few very recent applications are discussed. The idea of this is to give a flavour of current development and directions for LPME and EME, and to exemplify experimental conditions and performance.
96-Well technology is of high interest because it enables a large number of samples to be processed simultaneously. In one recent article, the hormones estrone, 17 β-estradiol, estriol, and 17 α-ethinylestradiol were extracted from human urine samples using two-phase HF-LPME conducted in a laboratory-built 96-well system [5]. The analytes were extracted from 1.5 mL urine samples, after addition of addition of 15% w/v sodium chloride. Sodium chloride was added to the samples to increase extraction recoveries, based on the salting out effect. Prior to extraction, the hollow fibres were dipped into 1-octanol, and the organic solvent quickly immobilized in the pores of the hollow fibre and formed a liquid membrane. In this case, no solvent was filled into the lumen of the hollow fibre. The hollow fibres were then inserted in the samples for 45 min, and the target analytes were extracted into the liquid membrane. After extraction, the hollow fibres were dipped into methanol for 10 min to desorb the analytes. Finally, the desorbed analytes were analysed by LC with fluorescence detection. The limits of detection were in the range 0.03 to 15 µg/L, intra-day precision was between 1 to 13% RSD, inter-day precision ranged between 7 and 18% RSD. Since hollow fibres are low-cost materials, they were used only for a single extraction. This eliminates carry over, which may be an issue in microextraction if the extraction phase is used for multiple extractions.
In another recent paper, 96-well LPME was reported in a three-phase PALME system for extraction of organophosphorus nerve agents from environmental water samples. The nerve agents included Soman acid, VX acid, Sarin acid, cyclohexyl-sarin acid, and Russian VX acid. These are acidic compounds with log P in the range from –0.5 to 0.8. Due to their polarity, partition into organic solvents is limited. For this reason, samples (350 μL) were pH adjusted to 1.0, and 30% sodium chloride was added to the samples. The pH adjustment served to keep the analytes 100% in their neutral form, while salting out increased their partition into the liquid membrane. The latter comprised 4.0 μL of 1-octanol. The acceptor was 50 μL of sodium hydroxide solution with pH 14, and strong alkaline conditions served to minimize back-diffusion into the liquid membrane. Extractions were conducted for 60 min under agitation at 1000 rpm. After EME, the acceptors were analysed by LC–MS/MS. LOQs were in the range 0.009–1.141 ng/mL, linearity was obtained for all analytes with r2 > 0.99, and precision was within 11% RSD. In spite of the fact that all the analytes were polar, the majority of the nerve agents were extracted exhaustively. Due to the high throughput capability of 96-well LPME, 192 samples were processed in 120 min (37.5 s per sample). Unlike previous application, 96-well LPME was in this case conducted with commercially available plates.
In addition to bioanalytical and environmental applications as discussed above, HF-LPME has been used also for food and beverages applications. In one example, HF-LPME was used to study migration of fluorescent whitening agents from plastic food contact materials [60]. This study involved detection of 4,4-bis(5-methylbenzoxazol-2-yl)stilbene, 1,4-bis(2-benzoxazolyl) naphthalene, 2,5-bis(5´-tert-butyl-2-benzoxazolyl) thiophene, 4,4´-bis(2-benzoxazolyl)stilbene, and 1,2-bis(5-methyl-2-benzoxazole)-ethylene. Extractions were conducted from different food simulants, including a 4% acetic acid aqueous solution, 20% aqueous ethanol solution, and isooctane. Hollow fibres of polypropylene were modified with sepiolite nanoparticles to enhance mass transfer into the liquid membrane. The liquid membrane was 15 μL of trichloromethane mixed with n-hexane in the ratio 1:1. Extraction was performed for 30 min under strong agitation. After extraction, the hollow fibres were transferred to centrifuge tubes containing acetonitrile, and the analytes were desorbed under ultrasonic treatment. Finally, the analytes were determined by UPLC-MS/MS. Due to the low volume of liquid membrane, high enrichment (71–205) was obtained. Correspondingly, limits of detection were in the range 0.3 to 0.9 ng/kg, and calibration curves were linear (r2 > 0.99). The intra-day and inter-day recoveries ranged between 83 and 112%, and precision was within 12%.
Unlike LPME, dedicated commercial equipment exists for EME [61]. The commercial equipment is based on the use of vials in conducting polymer as illustrated in Fig. 1. In this way the vials are used as electrodes for coupling the electrical field. In a recent paper, the commercial device was tested in a routine laboratory for clinical pharmacology, and performance was compared with a reference method for the determination of psychoactive drugs [62]. The sample preparation was based on EME, while the final analysis was done by UHPLC-MS/MS. The psychoactive drugs (analytes) included alimemazine, amitriptyline, atomoxetine, clomipramine, doxepin, duloxetine, fluvoxamine, levomepromazine, nortriptyline, and trimipramine, and the metabolites desmethyl clomipramine and dimethyl doxepin. The analytes are all bases, and are relatively hydrophobic. Extractions were conducted from serum samples, which were pipetted into sample vials. Prior to extraction, the samples (100 µL) were diluted 1:3 with 0.1% formic acid. Acidification of the samples ensured that the analytes were fully protonated in the sample. This is important in order to ensure their migration in the electrical field. The liquid membrane was NPOE, as this is the recommended liquid membrane for mono- and dibasic analytes in the log P range from 2.0 to 6.0. The volume of NPOE was 9 µL, and this was pipetted directly onto the solid support membrane. The acceptor was 300 µL of 0.1 M formic acid, and was filled into acceptor vials. Due to the acidic conditions, the analytes remained protonated in the acceptor, and therefore they were prevented from back-diffusion into the liquid membrane. Extractions were performed at 50 V for 15 min under agitation at 875 rpm. The positive electrode was coupled to the sample vials, while the negative electrode was coupled to the acceptor vials. With the commercial device, up to ten samples were extracted in parallel. The extraction potential of 50 V was the optimal voltage as determined experimentally, and further increase above 50 V did not improve the mass transfer. The extraction time was also established based on optimization experiments, and at 15 min extractions were more or less complete. After EME, the acceptors were analysed directly with UHPLC-MS/MS, without any evaporation and reconstitution, which is common practice after solid-phase extraction.
EME was highly efficient is this system, and recoveries for the analytes ranged between 75 and 117%. Extractions were thus either exhaustive or near-exhaustive. The EME method showed excellent precision and accuracy. Bias was within ±6%, CVs for intra- and inter-day ranged from 0.9–6% and 2–6% respectively. The EME method was applied on 30 different patient samples, and quantitative data were comparable with those obtained for the same samples using a routine method. While the routine method used close to 1 mL organic solvent per sample, only 9 µL was used with EME. The routine method used phospholipid removal plates after protein precipitation to eliminate ion-suppression during UHPLC-MS/MS, and such plates are expensive. With EME, the phospholipids did not migrate across the liquid membrane, and the acceptors were therefore free of phospholipids. Phospholipid removal was thus an integrated part of the EME process.
EME has been down-scaled and implemented in microchip systems, and combined with smartphone detection several times in recent years. In one example dyes were extracted from water by EME [63]. After EME, the extracted dyes were passed into a very small bed of ion exchanger sorbent, where they were retained and pre-concentrated based on micro solid phase extraction (μ-SPE). The colours were measured using a RGB colorimetric application on a smartphone (Fig. 11). Both EME and μ-SPE were integrated in the microchip. Erythrosine and crystal violet were used as acidic and basic model analytes, respectively. Erythrosine was extracted using 1-octanol as liquid membrane, while crystal violet was extracted using NPOE mixed with di(2-ethylhexyl) phosphate (DEHP) as liquid membrane. DEHP served as ion-pair reagent in the liquid membrane, and facilitated the mass transfer of crystal violet. Replicate extractions and smartphone measurements were within 7.8% RSD, and the dyes were measureable down to the 10–15 µg/L level. Although this work is preliminary in nature, it illustrates a potential of EME in a totally new direction, and further development in this direction is expected.

Modified from Zarghampour et al. [63] with permission from Elsevier
Illustration of EME chip combined with µ-SPE and smartphone-based RGB measurements of different concentrations of Erythrosine and crystal violet.
5 Conclusions and Future Trends
Up to date, LPME has been performed with laboratory-built equipment, or in commercial 96-well plates intended for filtration. The latter are neither produced nor intended for LPME. However, recently, commercial equipment was released for EME, and this can be used both for EME and LPME. This equipment is currently limited to extraction of 10 samples in parallel, but commercial 96-well equipment for EME is in progress. Thus, in very short time, analytical chemists can buy instrumentation and consumables for EME and LPME of industrial standard. This will be an important step forward for both techniques.
Research is in progress to define a set of generic methods for EME and LPME. Based on simple molecular descriptors, including charge and polarity, optimal extraction conditions can be derived for EME and LPME of a given analyte. This, in combination with commercial equipment, will make EME and LPME much more available for analytical scientists. Given the extremely low consumption of chemicals and organic solvents, green chemistry and sustainability will be one important justification for implementation of EME and LPME. Another justification will be implementation in microchip systems. Unlike the traditional extraction and sample preparation techniques, EME and LPME are very well suited for implementation in microchip systems. For both reasons, it is the opinion of the authors that EME and LPME will be used extensively in the future, and that research in this area will increase in the years to come.
Abbreviations
- EME:
-
Electromembrane extraction
- HF-LPME:
-
Hollow-fibre liquid-phase microextraction
- LPME:
-
Liquid-phase microextraction
- SDME:
-
Single drop microextraction
- SPME:
-
Solid-phase microextraction
References
Arthur CL, Pawliszyn J (1990) Solid phase microextraction with thermal desorption using fused silica optical fibers. Anal Chem 62(19):2145–2148. https://doi.org/10.1021/ac00218a019
Liu H, Dasgupta PK (1996) Analytical chemistry in a drop. Solvent extraction in a microdrop. Anal Chem 68(11):1817–1821. https://doi.org/10.1021/ac960145h
Jeannot MA, Cantwell FF (1996) Solvent microextraction into a single drop. Anal Chem 68(13):2236–2240. https://doi.org/10.1021/ac960042z
Pedersen-Bjergaard S, Rasmussen KE (1999) Liquid−Liquid−Liquid microextraction for sample preparation of biological fluids prior to capillary electrophoresis. Anal Chem 71(14):2650–2656. https://doi.org/10.1021/ac990055n
Lopes D, Morés L, da Silva M, Schneider M, Merib J, Carasek E (2022) Determination of hormones in urine by hollow fiber microporous membrane liquid–liquid extraction associated with 96-well plate system and HPLC-FLD detection. J Chromatogr B 1207. https://doi.org/10.1016/j.jchromb.2022.123406
Gjelstad A, Rasmussen KE, Parmer MP, Pedersen-Bjergaard S (2013) Parallel artificial liquid membrane extraction: micro-scale liquid-liquid-liquid extraction in the 96-well format. Bioanalysis 5(11):1377–1385. https://doi.org/10.4155/bio.13.59
Pedersen-Bjergaard S, Rasmussen KE (2006) Electrokinetic migration across artificial liquid membranes: new concept for rapid sample preparation of biological fluids. J Chromatogr A 1109(2):183–190. https://doi.org/10.1016/j.chroma.2006.01.025
Goh SXL, Goh HA, Lee HK (2018) Automation of ionic liquid enhanced membrane bag-assisted-liquid-phase microextraction with liquid chromatography-tandem mass spectrometry for determination of glucocorticoids in water. Anal Chim Acta 1035:77–86. https://doi.org/10.1016/j.aca.2018.07.031
Jiang X, Lee HK (2004) Solvent bar microextraction. Anal Chem 76(18):5591–5596. https://doi.org/10.1021/ac040069f
Fashi A, Salarian AA, Zamani A (2018) Solvent-stir bar microextraction system using pure tris-(2-ethylhexyl) phosphate as supported liquid membrane: a new and efficient design for the extraction of malondialdehyde from biological fluids. Talanta 182:299–305. https://doi.org/10.1016/j.talanta.2018.02.002
Pantůčková P, Kubáň P (2017) In-line coupling of supported liquid membrane extraction to capillary electrophoresis for simultaneous analysis of basic and acidic drugs in urine. J Chromatogr A 1519:137–144. https://doi.org/10.1016/j.chroma.2017.08.084
Lopes D, Dias AN, Simão V, Carasek E (2017) Determination of emerging contaminants in aqueous matrices with hollow fiber-supported dispersive liquid-liquid microextraction (HF-DLLME) and separation/detection by liquid chromatography: diode array detection. Microchem J 130:371–376. https://doi.org/10.1016/j.microc.2016.10.011
Asl YA, Yamini Y, Seidi S (2016) Development of a microfluidic-chip system for liquid–phase microextraction based on two immiscible organic solvents for the extraction and preconcentration of some hormonal drugs. Talanta 160:592–599. https://doi.org/10.1016/j.talanta.2016.07.063
Berijani S, Assadi Y, Anbia M, Milani Hosseini MR, Aghaee E (2006) Dispersive liquid-liquid microextraction combined with gas chromatography-flame photometric detection. Very simple, rapid and sensitive method for the determination of organophosphorus pesticides in water. J Chromatogr A 1123(1):1–9. https://doi.org/10.1016/j.chroma.2006.05.010
Gjelstad A, Andersen TM, Rasmussen KE, Pedersen-Bjergaard S (2007) Microextraction across supported liquid membranes forced by pH gradients and electrical fields. J Chromatogr A 1157(1):38–45. https://doi.org/10.1016/j.chroma.2007.05.007
Ocaña-González JA, Aranda-Merino N, Pérez-Bernal JL, Ramos-Payán M (2023) Solid supports and supported liquid membranes for different liquid phase microextraction and electromembrane extraction configurations: a review. J Chromatogr A 1691:463825. https://doi.org/10.1016/j.chroma.2023.463825
Huang CX, Gjelstad A, Pedersen-Bjergaard S (2016) Organic solvents in electromembrane extraction: recent insights. Rev Anal Chem 35(4):169–183. https://doi.org/10.1515/revac-2016-0008
Ho TS, Vasskog T, Anderssen T, Jensen E, Rasmussen KE, Pedersen-Bjergaard S (2007) 25,000-fold pre-concentration in a single step with liquid-phase microextraction. Anal Chim Acta 592(1):1–8. https://doi.org/10.1016/j.aca.2007.04.014
Wang P, Xiao Y, Liu W, Wang J, Yang Y (2015) Vortex-assisted hollow fibre liquid-phase microextraction technique combined with high performance liquid chromatography-diode array detection for the determination of oestrogens in milk samples. Food Chem 172:385–390. https://doi.org/10.1016/j.foodchem.2014.09.092
Seyfinejad B, Ozkan SA, Jouyban A (2021) Ultrasound-assisted electromembrane extraction of clonazepam from plasma and determination using capillary electrophoresis. J Chromatogr B 1181:122928. https://doi.org/10.1016/j.jchromb.2021.122928
Shang Q, Mei H, Feng X, Huang C, Pedersen-Bjergaard S, Shen X (2021) Ultrasound-assisted electromembrane extraction with supported semi-liquid membrane. Anal Chim Acta 1184:339038. https://doi.org/10.1016/j.aca.2021.339038
Román-Hidalgo C, Martín-Valero MJ, Fernández-Torres R, Bello-López MÁ (2018) Use of polymer inclusion membranes (PIMs) as support for electromembrane extraction of non-steroidal anti-inflammatory drugs and highly polar acidic drugs. Talanta 179:601–607. https://doi.org/10.1016/j.talanta.2017.11.066
Alsharif AMA, Tan G-H, Choo Y-M, Lawal A (2017) Efficiency of hollow fiber liquid-phase microextraction chromatography methods in the separation of organic compounds: a review. J Chromatogr Sci 55(3):378–391. https://doi.org/10.1093/chromsci/bmw188
Romero A, Santos A, Tojo J, Rodríguez A (2008) Toxicity and biodegradability of imidazolium ionic liquids. J Hazard Mater 151(1):268–273. https://doi.org/10.1016/j.jhazmat.2007.10.079
Hansen FA, Pedersen-Bjergaard S (2020) Emerging extraction strategies in analytical chemistry. Anal Chem 92(1):2–15. https://doi.org/10.1021/acs.analchem.9b04677
Wang J, Huang S, Wang P, Yang Y (2016) Method development for the analysis of phthalate esters in tea beverages by ionic liquid hollow fibre liquid-phase microextraction and liquid chromatographic detection. Food Control 67:278–284. https://doi.org/10.1016/j.foodcont.2016.03.015
Sun JN, Chen J, Shi YP (2014) Ionic liquid-based electromembrane extraction and its comparison with traditional organic solvent based electromembrane extraction for the determination of strychnine and brucine in human urine. J Chromatogr A 1352:1–7. https://doi.org/10.1016/j.chroma.2014.05.037
Sun JN, Shi YP, Chen J (2015) Development of ionic liquid based electromembrane extraction and its application to the enrichment of acidic compounds in pig kidney tissues. RSC Adv 5(47):37682–37690. https://doi.org/10.1039/c5ra01029b
De Boeck M, Dubrulle L, Dehaen W, Tytgat J, Cuypers E (2018) Fast and easy extraction of antidepressants from whole blood using ionic liquids as extraction solvent. Talanta 180:292–299. https://doi.org/10.1016/j.talanta.2017.12.044
Andruch V, Makoś-Chełstowska P, Płotka-Wasylka J (2022) Remarks on use of the term “deep eutectic solvent” in analytical chemistry. Microchem J 179:107498. https://doi.org/10.1016/j.microc.2022.107498
Abbott AP, Capper G, Davies DL, Rasheed RK, Tambyrajah V (2003) Novel solvent properties of choline chloride/urea mixtures. Chem Commun 1:70–71. https://doi.org/10.1039/B210714G
van Osch DJGP, Zubeir LF, van den Bruinhorst A, Rocha MAA, Kroon MC (2015) Hydrophobic deep eutectic solvents as water-immiscible extractants. Green Chem 17(9):4518–4521. https://doi.org/10.1039/C5GC01451D
van Osch DJGP, Dietz CHJT, van Spronsen J, Kroon MC, Gallucci F, van Sint AM, Tuinier R (2019) A search for natural hydrophobic deep eutectic solvents based on natural components. ACS Sustain Chem Eng 7(3):2933–2942. https://doi.org/10.1021/acssuschemeng.8b03520
Makoś P, Przyjazny A, Boczkaj G (2018) Hydrophobic deep eutectic solvents as “green” extraction media for polycyclic aromatic hydrocarbons in aqueous samples. J Chromatogr A 1570:28–37. https://doi.org/10.1016/j.chroma.2018.07.070
Makoś P, Słupek E, Gębicki J (2020) Hydrophobic deep eutectic solvents in microextraction techniques: a review. Microchem J 152:104384. https://doi.org/10.1016/j.microc.2019.104384
Seidi S, Alavi L, Jabbari A, Shanehsaz M (2019) Three-phase carrier-mediated hollow fiber microextraction based on deep eutectic solvent followed by HPLC–UV for determination of raloxifene and ethinylestradiol in pharmaceutical wastewater treatment plants. J Iran Chem Soc 16(5):1007–1018. https://doi.org/10.1007/s13738-018-01572-4
Zhang SM, Zhang XX, Chen X, Hu S, Bai XH (2020) Deep eutectic solvent-based hollow fiber liquid-phase microextraction for quantification of Q-markers of cinnamic acid derivatives in traditional Chinese medicines and research of their plasma protein binding rates. Microchem J 155:104696. https://doi.org/10.1016/j.microc.2020.104696
Hansen FA, Santigosa-Murillo E, Ramos-Payán M, Muñoz M, Leere Øiestad E, Pedersen-Bjergaard S (2021) Electromembrane extraction using deep eutectic solvents as the liquid membrane. Anal Chim Acta 1143:109–116. https://doi.org/10.1016/j.aca.2020.11.044
Hansen FA, Tirandaz S, Pedersen-Bjergaard S (2021) Selectivity and efficiency of electromembrane extraction of polar bases with different liquid membranes—link to analyte properties. J Sep Sci 44(13):2631–2641. https://doi.org/10.1002/jssc.202100167
Hay AO, Trones R, Herfindal L, Skrede S, Hansen FA (2022) Determination of methotrexate and its metabolites in human plasma by electromembrane extraction in conductive vials followed by LC-MS/MS. Adv Sample Prep 2:100011. https://doi.org/10.1016/j.sampre.2022.100011
Rye TK, Martinovic G, Eie LV, Hansen FA, Halvorsen TG, Pedersen-Bjergaard S (2021) Electromembrane extraction of peptides using deep eutectic solvents as liquid membrane. Anal Chim Acta 1175:338717. https://doi.org/10.1016/j.aca.2021.338717
Ballesteros-Gómez A, Sicilia MD, Rubio S (2010) Supramolecular solvents in the extraction of organic compounds: a review. Anal Chim Acta 677(2):108–130. https://doi.org/10.1016/j.aca.2010.07.027
Rezaei F, Yamini Y, Moradi M, Daraei B (2013) Supramolecular solvent-based hollow fiber liquid phase microextraction of benzodiazepines. Anal Chim Acta 804:135–142. https://doi.org/10.1016/j.aca.2013.10.026
Li X, Huang A, Liao X, Chen J, Xiao Y (2020) Restricted access supramolecular solvent based magnetic solvent bar liquid-phase microextraction for determination of non-steroidal anti-inflammatory drugs in human serum coupled with high performance liquid chromatography-tandem mass spectrometry. J Chromatogr A 1634:461700. https://doi.org/10.1016/j.chroma.2020.461700
Rubio S (2020) Twenty years of supramolecular solvents in sample preparation for chromatography: achievements and challenges ahead. Anal Bioanal Chem 412(24):6037–6058. https://doi.org/10.1007/s00216-020-02559-y
Nemulenzi O, Mhaka B, Cukrowska E, Ramström O, Tutu H, Chimuka L (2009) Potential of combining of liquid membranes and molecularly imprinted polymers in extraction of 17β-estradiol from aqueous samples. J Sep Sci 32(11):1941–1948. https://doi.org/10.1002/jssc.200800659
Yaripour S, Nojavan S, Khoshayand MR, Mohammadi A (2020) Electromembrane extraction of phenytoin from biological fluids: a survey on the effects of molecularly imprinted polymer and carbon nanotubes on extraction efficiency. Microchem J 156:104800. https://doi.org/10.1016/j.microc.2020.104800
Worawit C, Alahmad W, Miró M, Varanusupakul P (2020) Combining graphite with hollow-fiber liquid-phase microextraction for improving the extraction efficiency of relatively polar organic compounds. Talanta 215:120902. https://doi.org/10.1016/j.talanta.2020.120902
Tahmasebi Z, Davarani SSH, Asgharinezhad AA (2018) Highly efficient electrochemical determination of propylthiouracil in urine samples after selective electromembrane extraction by copper nanoparticles-decorated hollow fibers. Biosens Bioelectron 114:66–71. https://doi.org/10.1016/j.bios.2018.05.014
López-Lorente ÁI, Pena-Pereira F, Pedersen-Bjergaard S, Zuin VG, Ozkan SA, Psillakis E (2022) The ten principles of green sample preparation. Trends Anal Chem 148:116530. https://doi.org/10.1016/j.trac.2022.116530
Poole C, Mester Z, Miró M, Pedersen-Bjergaard S, Pawliszyn J (2016) Glossary of terms used in extraction (IUPAC Recommendations 2016). Pure Appl Chem 88(5):517–558. https://doi.org/10.1515/pac-2015-0903
Cabal LFR, Medina DAV, Costa JL, Lanças FM, Santos-Neto ÁJ (2019) Determination of ring-substituted amphetamines through automated online hollow fiber liquid-phase microextraction-liquid chromatography. Anal Bioanal Chem 411(29):7889–7897. https://doi.org/10.1007/s00216-019-02196-0
Fuchs D, Pedersen-Bjergaard S, Jensen H, Rand KD, Honore Hansen S, Petersen NJ (2016) Fully automated electro membrane extraction autosampler for LC-MS systems allowing soft extractions for high-throughput applications. Anal Chem 88(13):6797–6804. https://doi.org/10.1021/acs.analchem.6b01243
Lopes D, Dias AN, Merib J, Carasek E (2018) Hollow-fiber renewal liquid membrane extraction coupled with 96-well plate system as innovative high-throughput configuration for the determination of endocrine disrupting compounds by high-performance liquid chromatography-fluorescence and diode array detection. Anal Chim Acta 1040:33–40. https://doi.org/10.1016/j.aca.2018.07.032
Luo G, Li Y, Bao JJ (2016) Development and application of a high-throughput sample cleanup process based on 96-well plate for simultaneous determination of 16 steroids in biological matrices using liquid chromatography–triple quadrupole mass spectrometry. Anal Bioanal Chem 408(4):1137–1149. https://doi.org/10.1007/s00216-015-9213-1
Gjelstad A, Rasmussen KE, Parmer MP, Pedersen-Bjergaard S (2013) Parallel artificial liquid membrane extraction: micro-scale liquid–liquid–liquid extraction in the 96-well format. Bioanalysis 5(11):1377–1385. https://doi.org/10.4155/bio.13.59
Drouin N, Mandscheff JF, Rudaz S, Schappler J (2017) Development of a new extraction device based on parallel-electromembrane extraction. Anal Chem 89(12):6346–6350. https://doi.org/10.1021/acs.analchem.7b01284
Restan MS, Jensen H, Shen X, Huang C, Martinsen OG, Kuban P, Gjelstad A, Pedersen-Bjergaard S (2017) Comprehensive study of buffer systems and local pH effects in electromembrane extraction. Anal Chim Acta 984:116–123. https://doi.org/10.1016/j.aca.2017.06.049
Miková B, Dvořák M, Ryšavá L, Kubáň P (2020) Hollow fiber liquid-phase microextraction at-line coupled to capillary electrophoresis for direct analysis of human body fluids. Anal Chem 92(10):7171–7178. https://doi.org/10.1021/acs.analchem.0c00697
Zhou WL, Ding L, Cheng YH, Xu Z, Chen ML, Fu XS (2022) Application of an improved hollow fiber liquid phase microextraction technique coupled to LC-MS/MS to studying migration of fluorescent whitening agents from plastic food contact materials. Food Addit Contam Part A 39(7):1337–1347. https://doi.org/10.1080/19440049.2022.2066192
Drobnjak M, Hansen F, Øiestad EL, Løvli T, Trones R, Martinsen ØG, Pedersen-Bjergaard S (2020) Electromembrane extraction with vials of conducting polymer. LCGC North Am 38(8):435–439
Skaalvik TG, Øiestad EL, Trones R, Pedersen-Bjergaard S, Hegstad S (2021) Determination of psychoactive drugs in serum using conductive vial electromembrane extraction combined with UHPLC-MS/MS. J Chromatogr B 1183:122926. https://doi.org/10.1016/j.jchromb.2021.122926
Zarghampour F, Yamini Y, Alipanahpour Dil E, Shokrollahi A, Javadian G (2023) A new microfluidic-chip device followed by sensitive image analysis of smart phone for simultaneous determination of dyes with different acidic–basic properties. Talanta 254:124168. https://doi.org/10.1016/j.talanta.2022.124168
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2024 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hansen, F.A., Pedersen-Bjergaard, S. (2024). Hollow-Fibre Liquid-Phase Microextraction. In: Rodríguez-Delgado, M.Á., Socas-Rodríguez, B., Herrera-Herrera, A.V. (eds) Microextraction Techniques. Integrated Analytical Systems. Springer, Cham. https://doi.org/10.1007/978-3-031-50527-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-031-50527-0_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-50526-3
Online ISBN: 978-3-031-50527-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)