
Abstract
RNA virus infections have been a leading cause of pandemics. Aided by global warming and increased connectivity, their threat is likely to increase over time. The flaviviruses are one such RNA virus family, and its prototypes such as the Japanese encephalitis virus (JEV), Dengue virus, Zika virus, West Nile virus, etc., pose a significant health burden on several endemic countries. All viruses start off their life cycle with an infected cell, wherein a series of events are set in motion as the virus and host battle for autonomy. With their remarkable capacity to hijack cellular systems and, subvert/escape defence pathways, viruses are able to establish infection and disseminate in the body, causing disease. Using this strategy, JEV replicates and spreads through several cell types such as epithelial cells, fibroblasts, monocytes and macrophages, and ultimately breaches the blood-brain barrier to infect neurons and microglia. The neurotropic nature of JEV, its high burden on the paediatric population, and its lack of any specific antivirals/treatment strategies emphasise the need for biomedical research-driven solutions. Here, we highlight the latest research developments on Japanese encephalitis virus-infected cells and discuss how these can aid in the development of future therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Autophagy
- Blood-brain barrier
- Cell death
- ER stress
- Flavivirus
- Innate immunity
- Japanese encephalitis virus
- Neuroinflammation
- Neurotropic
- Unfolded protein response
Introduction
Japanese encephalitis virus (JEV) remains one of the leading global causes of viral encephalitis. It poses a major threat to more than 2 billion people living in endemic regions like southeast Asian countries (van den Hurk et al. 2009; Pan et al. 2011) and is still evolving to new ecological niches of Europe, northern Australia and Africa (Simon-Loriere et al. 2017; Gao et al. 2019). According to a 2019 WHO report, JEV causes almost 68,000 cases with 13,600–20,400 deaths annually. The virus is neurotropic, and its clinical manifestations range from febrile illness to central nervous system (CNS) disorders and death (Sips et al. 2012). A majority of JEV infections remain asymptomatic, and less than 1% of infections develop into the disease, which is either mild or neuroinvasive. Of the diseased cases, one-third recover completely, one-third develop severe lifelong neurological complications, and one-third ultimately succumb to the disease (Solomon 2004). Children aged 0–15 years are the most affected group and are likely to have more neurological complications than adults (Campbell et al. 2011). Currently, there is no antiviral therapy available and existing vaccines are struggling to control the JEV burden due to lack of long-term protection and cross-protection against newly emerging genotypes. Treatment is only supportive and limited clinical trials have been conducted for testing antiviral and drug therapies (Turtle and Solomon 2018).
JEV is an arthropod-borne flavivirus, which is transmitted in an enzootic life cycle between birds, pigs and other vertebrates by Culex mosquitoes. Humans are dead-end hosts due to low viremia. Following inoculation upon a blood-feeding mosquito bite, the virus first replicates in skin keratinocytes and then propagates to nearby lymph nodes and other tissues or organs like the liver and kidney, causing transient low viremia. If the virus is not restricted to the periphery, it can cross the blood-brain barrier (BBB) to gain entry into the CNS, which results in the neurological manifestations of the disease. The viral tropism and subsequent host responses govern disease pathogenesis and severity. JEV is known to infect diverse cell types such as epithelial cells, fibroblasts, monocytes, macrophages, dendritic cells (DCs), endothelial cells, brain resident microglial and neuronal cells, and activates an array of cellular responses. A detailed understanding of virus-host crosstalk is important for delineating crucial host responses and identifying cellular factors involved in disease pathogenesis and antiviral development. Herein we review the interactions of JEV with the mammalian host at the cellular and system level, and their role in disease biology.
Epidemiology
JE was first reported in Japan, with more than 6000 cases during the 1924 epidemic. The prototype Nakayama strain was isolated from the brain of a fatal case in 1935, and since then, the disease has been recognised across Asia (Miyake 1964; Solomon 2003). Genetic studies have proposed that JEV evolved from an African ancestral virus that spread to the Indonesia-Malaysia region many centuries ago, from where it further spread throughout Asia (Solomon et al. 2003). In the first half of the twentieth century, JEV was recognised in the temperate regions of Asia such as Japan, Korea, Taiwan and mainland China; and continued to spread to southeast Asia, India, Bangladesh, Sri Lanka and Nepal over the next decades. By the 1990s, JEV showed its presence even in the non-Asian regions, Saipan and Australia (Filgueira and Lannes 2019; Mulvey et al. 2021).
JEV has a total of five genotypes (I–V), arising from the ancestor virus of the Indonesia-Malaysia region. Genotype III was reported to be responsible for most human cases in Asia up to the 1990s, whereas genotype I is now likely to become the dominant strain and the major cause of JE disease in the region (Pan et al. 2011). In a 2022 outbreak in Australia, a JEV genotype IV was identified (Sikazwe et al. 2022). Epidemiological and genetic studies have reported geographical expansion of different genotypes and JEV emergence in non-epidemic regions (van den Hurk et al. 2009; Gao et al. 2019; Mulvey et al. 2021). The wide distribution of the virus over the years has been due to changes in climate, ecology, agricultural and animal practices. Migratory bird patterns and population shifts can potentially further contribute to virus expansion to non-endemic regions. Increased surveillance and reporting of JEV infections need to be undertaken to assess the true burden of JE.
Transmission Cycle
The Culex tritaeniorrhynchus mosquito which breeds in stagnant water (such as rice paddy fields), is the most important vector for human infections (Solomon et al. 2003). Other domestic (cows, dogs, chickens, goats and horses) and wild animals (flying foxes, frogs, snakes and ducks) can also be infected with the virus, but ones with high viral loads (birds and pigs) maintain the virus (Mansfield et al. 2017). Due to brief and low viremia, humans do not transmit the disease further (Turtle and Solomon 2018). However, a recent report suggested possible JEV transmission via blood transfusion in humans (Cheng et al. 2018). Birds maintain and amplify the virus in the environment, and migratory/seasonal birds are responsible for JEV spread/expansion to new geographical areas (Johnsen et al. 1974; Rodrigues et al. 1981; Yoshikawa et al. 2016; Bae et al. 2018; Preziuso et al. 2018; Turtle and Solomon 2018; Mulvey et al. 2021). Pigs are the natural hosts with prolonged and high viremia, and the virus can also transmit directly via the intra-nasal route in pigs (Ricklin et al. 2016; Garcia-Nicolas et al. 2018). The virus replicates and remains in the porcine tonsils for up to 25 days enabling its persistence in seasons when mosquitoes are inactive (Garcia-Nicolas et al. 2018). JEV can also persist in vaginal mucosa for several days and is shed in vaginal secretions in pigs, suggesting a potential for sexual transmission (Chapagain et al. 2022).
Clinical Features
The majority of the JEV infections in humans are either asymptomatic or cause febrile illness with mild flu-like symptoms such as fever, sore throat, headache, muscle pain, diarrhoea and vomiting, that lasts for 5–15 days. Neurologic manifestations depend upon the site of infection in the CNS. Patients who develop symptoms of encephalitis suffer significant morbidity and mortality. Encephalitis is characterised by neck stiffness, disorientation, seizures, paralysis, coma and in severe cases, leads to death (Misra and Kalita 2010; Salimi et al. 2016).
JEV Molecular Biology
JEV belongs to the Flaviviridae family that also contains several other pathogenic arboviruses such as West Nile virus (WNV), Zika virus (ZIKV), Dengue virus (DENV), Yellow fever virus (YFV), Murray valley Encephalitis (MVE), St. Louis encephalitis virus (SLEV) and Tick-borne encephalitis virus (TBEV). The enveloped virus contains a single-stranded positive-sense RNA genome of ̴11 kb (Vashist et al. 2011), which is a single open reading frame (ORF), flanked by 5′ and 3′ non-coding regions (NCR). The viral polyprotein ( ̴3400 aa) is cleaved into three structural proteins – Nucleocapsid (C), Membrane (M) and Envelope protein (E), and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5), by the action of viral and host proteases (Chambers et al. 1990). The viral nucleocapsid is enclosed in a membrane containing the envelope (E) glycoprotein. A near-atomic structure of JEV has revealed various structural determinants associated with virus stability and neurovirulence (Wang et al. 2017b, c). The non-structural viral proteins are an integral part of the replication complex and also interact with diverse host factors involved in multiple cellular pathways to create an infection-supportive environment.
Infection Route: A Cellular Overview
The JEV virus life cycle is an orchestration of six major steps: receptor binding, entry, polyprotein translation, genome replication, assembly and egress. Infection begins with the non-specific binding of the viral glycoprotein E to one or more cellular attachment factors, that enhance the avidity and facilitates specific interaction with the receptor. Studies in different cell types have identified attachment factors (Heparan sulphate proteoglycans, glycosaminoglycans) and several potential receptors: Heat shock protein 70, vimentin, laminin receptor, CD4, α5β3 integrin, Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN), Glucose-regulated protein 78 (GRP78), T cell immunoglobulin and mucin domain 1 (TIM-1), C type lectin member 5A (CLEC5A), plasmalemma vesicle-associated protein and gastrokine 3 (Chiou et al. 2005; Chien et al. 2008; Chen et al. 2012b; Nain et al. 2016; Nain et al. 2017; Mukherjee et al. 2018; Niu et al. 2018).
Virus-receptor interaction results in receptor-mediated endocytosis. JEV is seen to exploit different endocytic routes in a cell-type-dependent manner. Studies have now established that the virus utilises clathrin-mediated endocytosis (CME) to infect fibroblasts and epithelial cells, and under conditions of CME inhibition, it can employ clathrin-independent endocytosis (CIE) to infect neuronal cells (Zhu et al. 2012; Kalia et al. 2013; Yang et al. 2013; Xu et al. 2016; Liu et al. 2017; Khasa et al. 2019; Khasa et al. 2020). RNA interference-based screens have identified several membrane trafficking proteins such as the ARP2/3 complex, RhoA, Cdc42, Pak1, Rab5, Rab11, ezrin and valosin-containing protein (VCP) to be involved in JEV entry (Xu et al. 2016; Khasa et al. 2019; Khasa et al. 2020; Liu et al. 2020; Sehrawat et al. 2021; Zhou et al. 2021). Characterisation of JEV entry in neuronal cells, and its interaction with host factors, is an important research domain and can generate potential therapeutic targets to combat the virus at an early time of infection.
A low pH achieved in the endosome induces conformational changes in the viral E glycoprotein, which triggers the fusion of viral and host endosomal membranes and releases the viral RNA genome into the cytoplasm. The positive sense RNA is directly translated via the host translational machinery into two precursor polyproteins (with or without a ribosomal frameshifting at the beginning of NS2A-coding region), that are cleaved into three structural (C, prM and E) and seven non-structural (NS1 to NS5) proteins, along with NS1’.
The virus non-structural proteins: NS4A, NS4B, NS1, NS2A, NS2B, NS3 and NS5 are known to interact/associate with several host factors, majorly ER-associated proteins and lipids to form the ER-derived replication complexes (Arakawa and Morita 2019). Distinct structures referred to as convoluted membranes (CMs) and vesicle packets (VPs) can be seen in flavivirus-infected cells and are the sites for polyprotein translation/processing and viral RNA replication, which proceeds through the formation of the viral dsRNA intermediate. The signal peptidase complex subunit 1 (SPCS1) is a crucial host factor that interacts with NS2B and impacts the post-translational processing of JEV proteins and virus assembly (Ma et al. 2018). The ER-associated degradation pathway (ERAD) proteins such as microtubule-associated protein 1 light chain 3-I (MAP1LC3-I, hereafter LC3-I), EDEM1 and Sel1L are also found enriched in the JEV VPs and CMs (Sharma, Bhattacharyya et al. 2014; Sarkar et al. 2020). The replication complex has a pore opening into the cytosol for entry of nucleotides and exit of the positive-strand RNA for packaging. Interestingly, the production of the plus strands is ̴10–100 fold higher than minus strand RNA, showing an asymmetric and semi-conservative process of replication (Uchil and Satchidanandam 2003). The viral capsid protein, along with LC3-I (autophagy-independent form), is observed to be concentrated on lipid droplets (LDs), and this is likely to be the site of RNA packaging (Sarkar et al. 2020). Interestingly, the number of LDs decreases in JEV-infected cells suggesting a negative regulation of lipid metabolism, an observation which is also supported by proteome studies describing the down-modulation of lipid metabolic proteins in JEV-infected cells (Sarkar et al. 2020; Sharma et al. 2021a).
Virion assembly in the ER lumen follows as the viral genome and proteins assemble abundantly. The nucleocapsid is further enclosed by E and prM proteins decorated on ER membranes to form immature virus particles. These undergo maturation before budding out of the cell membrane, via furin protease activity that cleaves prM to M during exocytosis via the trans-golgi network (Stadler et al. 1997; Li et al. 2008; Yu et al. 2008). Mapping the virus-host interactome and developing inhibitors to block the virus-receptor/host dependency factor interaction are attractive targets for antiviral drug development.
Cellular Response to Infection
Viruses have evolved strategies to interfere with cellular signalling pathways and exploit organellar compartments, which perturbs cellular homeostasis and triggers the activation of stress responses in infected cells. Flaviviruses are shown to induce stress responses such as the formation of stress granules, oxidative stress, and ER stress leading to the activation of the unfolded protein response (UPR), autophagy and activation of innate immunity. Cross-communication between these pathways regulates the antiviral and cell survival response, in addition to other cellular functions such as translation, metabolism, cytoskeletal organisation and inflammation, and therefore influences the viral pathogenesis and disease outcome.
Alteration of Signalling Pathways
Activation of PI3-kinase/Akt signalling has been observed early during JEV infection and is thus likely to be a result of virus-receptor interaction (Das et al. 2010). JEV attachment can also specifically activate EGFR-PI3K signalling (Xu et al. 2016), resulting in phosphorylation of EGFR, and infection can be blocked by using EGFR inhibitors (Zhang et al. 2022). MAPK signalling including ERK, p38, MAPK and JNK plays an important role in JEV-induced caspase activation (Gupta et al. 2011) and neuroinflammation (Ye et al. 2016; He et al. 2017). JEV has also been shown to modulate several tyrosine phosphorylation-mediated signalling events in infected cells (Raung et al. 2005, 2007; Yang et al. 2012).
PKR Activation and Formation of Stress Granules
Recognition of viral dsRNA activates the interferon (IFN)-induced protein kinase R (PKR), which phosphorylates the eukaryotic translation initiation factor 2α subunit (eIF2α), and results in a block of protein translation. PKR activation also results in the sequestration of actively transcribing mRNA into cytoplasmic foci called stress granules (SG), and this process is circumvented by nearly all viruses to enable their propagation. The JEV NS2A protein can counteract PKR activation and eIF2α-phosphorylation (Tu et al. 2012). JEV capsid protein has been shown to inhibit the SG formation by binding to the RNA-binding protein, Caprin-1 which is an essential component of the stress granules (Katoh et al. 2013). JEV NS4B can also recruit the VCP-NPL4 complex and block stress granule formation (Arakawa et al. 2022).
Induction of Oxidative Stress
Oxidative stress is generated when there is an imbalance between the production and neutralisation of reactive oxygen species (ROS). In general, ROS is produced as a by-product of normal aerobic metabolism, by a variety of enzymes present in mitochondria, ER, and peroxisomes, which are simultaneously taken care of by the antioxidant system (Go and Jones 2008; Roy et al. 2017; Zhang et al. 2019). ROS acts as a double-edged sword in cell health, as its maintenance to a certain level is necessary for signalling activation and survival. However, stress conditions such as pathogen infection, accumulate intracellular ROS, resulting in cell injury or death.
JEV has been demonstrated to trigger oxidative stress in infected cells together with the production of toxic oxygen species in neutrophils (Srivastava et al. 1999), human astrocytoma and astroglioma cell lines (Mishra et al. 2008), the superoxide anion and nitric oxide species in rat cortical glial cells (Liao et al. 2002), and ROS intermediates in murine neuroblastoma cells (Raung et al. 2001). The elevated ROS level during JEV infection is implicated in virus-induced cell death (Ghoshal et al. 2007; Ghosh and Basu 2009; Kumar et al. 2009b; Yang et al. 2010). Alterations in mitochondrial health (Lin et al. 2004), and high levels of proinflammatory mediators secreted by activated microglia (Ghoshal et al. 2007), are major drivers of JEV-induced oxidative stress. Even UV-inactivated JEV (replication incompetent) damages actively growing neuronal cells through a ROS-mediated pathway (Lin et al. 2004). ROS is thus a major contributor of JEV pathogenesis and therapeutic modulation of JEV-induced oxidative stress could be beneficial for the host (Zhang et al. 2014). Oxidative stress has also been linked to UPR activation, autophagy, immunity, inflammation and cell death pathways (Olagnier et al. 2014; Chen et al. 2018a; Sharma et al. 2018; de Almeida et al. 2020).
Activation of the Unfolded Protein Response
The ER is a ubiquitous and versatile organelle involved in multiple cellular functions including protein production, folding, trafficking and turnover; lipid synthesis and distribution, calcium homeostasis, cell signalling and innate immunity. The ER is central to the JEV life cycle as it provides both a scaffold for viral protein translation and ER resident proteins and lipids for virus replication complex biogenesis and virion assembly. This poses a significant burden on the organelle, resulting in the induction of ER stress and activation of UPR (Yu et al. 2006; Blazquez et al. 2014). The three main pathways that modulate UPR are protein kinase-like ER resident kinase (PERK), the activating transcription factor 6 (ATF6), and the inositol-requiring enzyme 1 (IRE-1) (Liu and Kaufman 2003). The activation of the PERK pathway follows eIF2α-phosphorylation and causes global translational arrest. The insufficiency of translational arrest in reducing ER stress, leads to the activation of the IRE1 and ATF6 pathways, which upregulate the expression of ER chaperons and ERAD machinery components, to boost protein folding capacity and degrade terminally misfolded proteins. The UPR signalling attempts to restore ER homeostasis, however, prolonged ER stress and high-level signalling through PERK and eIF2α, results in ATF4 activation and expression of transcription factor GADD153 (CHOP), which altogether arrests the cell-cycle and induces apoptosis (Rozpedek et al. 2016). Activation of ER stress has also been linked to other crucial cellular processes such as lipid metabolism, autophagy, innate immunity and differentiation (McLean et al. 2011; Blazquez et al. 2014; Datan et al. 2016; Chan and Ou 2017; Sharma et al. 2017; Carletti et al. 2019), expanding its role in generating the integrative stress response against viral infections.
Studies have demonstrated that JEV infection generates ER stress and triggers the activation of all three sensors (PERK, ATF6 and IRE-1) of the UPR. PERK activation and CHOP expression have been linked to virus-induced apoptosis and disease pathogenesis (Su et al. 2002; Wang et al. 2019). The regulated IRE1-dependent decay (RIDD) pathway has been shown to benefit viral replication and enhance cell death (Bhattacharyya et al. 2014). In contrast, the XBP1 and ATF6-mediated UPR pathways exert a protective role against JEV-induced cell death via upregulating autophagy in neuronal cells (Yu et al. 2006; Sharma et al. 2017). The cellular ERAD pathway degrades the extra membrane-anchored JEV NS proteins in convoluted membranes and this process is essential for optimal virus replication (Tabata et al. 2021).
Upregulation of Autophagy
Autophagy is a highly conserved, multi-step degradative process which is greatly involved in maintaining cellular homeostasis by degrading misfolded/faulty proteins and damaged organelles through lysosomal compartments. Autophagy has basal housekeeping functions and is induced by stress conditions such as hypoxia, pathogen infection, ER stress, oxidative stress and accumulation of aggregated proteins and damaged organelles. In the context of viruses, autophagy can either restrict or enhance infection, depending on the virus and cell type (Ahmad et al. 2018). To restrict virus proliferation, autophagy either directly targets the viral components for degradation or indirectly modulates other host antiviral and survival pathways (Lennemann and Coyne 2015; Abdoli et al. 2018; Choi et al. 2018; Sharma et al. 2019).
During JEV infection, autophagy has been shown to be induced in both in vitro and in vivo model systems. JEV-induced autophagy has been shown to support virus replication by suppressing the cellular immune environment (Li et al. 2012a; Jin et al. 2013), however, some studies have suggested otherwise (Sharma et al. 2014; Xu et al. 2017). Significant enhancement of JEV replication and titres has been observed in autophagy-deficient (ATG5/ATG7 depleted) mouse fibroblasts and neuronal cells (Sharma et al. 2014). The E3 ubiquitin ligase Nedd4 protein also restricts JEV-induced autophagy and facilitates JEV replication in human neuroblastoma cells (Xu et al. 2017). Autophagy thus appears to play an antiviral role for JEV by restricting virus replication and virus-induced cell death (Sharma et al. 2014). JEV-infected cells also show enhanced mitophagy flux and a decrease in mitochondria number through the interaction of the viral NS4A protein with PTEN-induced kinase 1 (Agarwal et al. 2022). Interestingly, based on the anti-inflammatory and neuroprotective properties, autophagy activators have the potential to be repurposed as antivirals against JEV infection.
Innate Immune Activation
The cell recognises a virus infection through its pathogen recognition receptors (PRRs) that bind specific pathogen-associated molecular patterns (PAMPs). The PRR-PAMP association triggers downstream effectors and the production of type-I interferons (IFN-α and IFN-β) and inflammatory cyto/chemokines, which further initiates the JAK-STAT signalling in an autocrine and paracrine fashion. The IFN-driven JAK-STAT pathway ultimately induces the expression of a wide array of genes collectively referred to as interferon-stimulated genes (ISGs) that function to inhibit virus infection by directly acting on the virus itself or by enhancing the cellular antiviral state.
Various cell line and animal model studies have established the crucial role of numerous PRRs such as RIG-I, MDA-5, MyD88 (Kato et al. 2006), TLR3 (Han et al. 2014) and TLR7 (Nazmi et al. 2014; Awais et al. 2017), in sensing JEV components for innate immune activation. The quality and magnitude of the antiviral response of brain resident cells govern JE pathogenesis. Activation of RIG-I, MDA-5 and TLR-3 in JEV-infected neuronal and microglial cells is critical for virus inhibition, as their deletion compromised antiviral immunity and increased viral load (Nazmi et al. 2011; Jiang et al. 2014). Various reports have demonstrated the activation of a wide variety of ISGs including PKR, OAS, TRIM21, ISG15, IFITs, IFITMs, GBPs, MX1, etc., upon JEV infection (Clarke et al. 2014; Sharma et al. 2021a, b) (Fig. 10.1). Some of these such as IFNα, ISG15, MX2 and OAS-L have been shown to have an antiviral role against JEV (Hsiao et al. 2010; Liu et al. 2013; Zheng et al. 2016). Type-I IFN production by astrocytes restricts viral spread in the CNS and virus-induced cytopathic effects (Lindqvist et al. 2016).

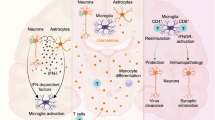
JEV infection-induced changes in fibroblasts. Upon binding, the virus is recognised by distinct pathogen recognition receptors (PRRs), initiating the downstream signalling by phosphorylation and nuclear translocation of IRF-3 and IRF-7 thereby resulting in the production of type 1-IFN. The newly synthesised IFN binds to IFNAR in an autocrine fashion and activates the downstream JAK-STAT signalling pathway. Nuclear translocation of STAT dimmers induces several ISGs that encode for different antiviral effector molecules thereby building a virus-resistant cellular state. Virus infection in the fibroblast results in remodelling of cellular proteome, causing activation of innate immune response (including the enhanced levels of PRR, IRFs, production of type-I interferon & ISGs and MHC presentation), cell death pathways and enhanced immunoproteasome and PARPs. Virus infection also induces the activation of DNA sensor cGAS which plays a crucial role in restricting virus replication. Different cellular pathways modulating lipid and sterol metabolism, focal adhesion, transporters, ECM organisation and lysosomal proteins get downregulated in the infected cell, thereby impacting the cellular metabolism
The virus also engages to counteract the IFN response and establish a replication niche. JEV NS5 is a potent antagonist of IFN-induced Jak-STAT signalling through abrogation of nuclear translocation and tyrosine phosphorylation of Tyk2 and STAT1 (Lin et al. 2006). JEV NS4A also functions as an IFN-antagonist through inhibition of STAT phosphorylation (Lin et al. 2008). JEV NS1’ has been shown to inhibit mitochondrial antiviral signalling protein (MAVS) mediated IFNβ induction by blocking dephosphorylation of CDK1 (Li et al. 2021b).
The transcription factor IRF8 modulates microglial activation during infection and enhances IFN-γ production resulting in reduced viral loads in the brain (Tripathi et al. 2021). Activated microglial cells and astrocytes also produce a strong wave of pro/anti-inflammatory cytokines including RANTES, TNF-α, IL1-α, IL-6, IL-12, IL-18, IL1-β, CCL2, CXCL9, CXCL10, CXCL11 and pro-inflammatory enzymes like cyclooxygenase-2 and iNOS during JEV infection (Chen et al. 2004; Bhowmick et al. 2007; Ghoshal et al. 2007; Das et al. 2008; Gupta et al. 2010a, b; Chen et al. 2011a, b; Kaushik et al. 2011; Fadnis et al. 2013; Lannes et al. 2017a; Yu et al. 2019), which is thought to be the major driver of JEV-induced neuroinflammation and bystander neuronal cell death (Ye et al. 2016; He et al. 2017; Singh et al. 2020; Ashraf et al. 2021). The suppression of anti-inflammatory cytokine IL-10 has also been observed during JEV infection (Swarup et al. 2007). A quantitative phosphoproteomic analysis identified JNK1 cascade activation upon JEV infection as a major contributor to virus-induced encephalitis and lethality (Ye et al. 2016).
Viral infections including JEV, also activate an epithelial-mesenchymal transition (EMT)-like process as an antiviral strategy through activation of the Snail transcription factor, via coregulation with type-I IFN (Vedagiri et al. 2021).
All flaviviruses, including JEV, produce a unique subgenomic flavivirus RNA (sfRNA) as a result of incomplete degradation of genomic RNA by the cellular exoribonuclease XRN1. This consists of the highly structured 3′UTR and plays an important role in viral pathogenesis through the regulation of cellular mRNA decay and IFN responses (Clarke et al. 2015). Activation of the RNA decay pathways are an important aspect of the innate immune response to virus infection. The monocyte chemoattractant protein 1-induced protein 1 (MCPIP1) ribonuclease binds JEV RNA and targets it for degradation (Lin et al. 2013). The zinc finger protein ZFP36L1 recognises the AUUUA motif in the 3′-UTR of the JEV genome and destabilises it by degrading the viral RNA through the 5′-3′ XRN1 and 3′-5′RNA-exosome RNA decay pathways (Chiu et al. 2022).
These studies highlight the importance of active innate immunity hubs in virus restriction and necessitate their detailed exploration for understanding disease severity. An optimal innate immune response is also crucial for limiting JEV at the periphery and blocking viral entry to CNS, a decisive checkpoint in JE pathogenesis.
Cell Death Pathways
An infected cell exposed to unrecoverable intracellular perturbations activates cell death pathways. Depending on the cell type and host response, viruses lead to three major regulated cell death modalities: apoptosis and inflammation programmed – necroptosis and pyroptosis (Dhuriya and Sharma 2018; Imre 2020). Cell death may repress virus replication and alter the local and systemic immune responses by releasing a variety of death-associated molecular patterns (DAMPS). Viruses have evolved strategies to inhibit or activate cell death pathways to escape the defence mechanism or to kill certain cell populations to dysregulate host immune responses.
Direct neuronal infection and prolonged microglial activation (gliosis) driven inflammatory environment trigger diverse markers of cell death pathways in JEV-infected neuronal cells (Chen et al. 2012a, b; Mukherjee et al. 2019). Several studies have supported the role of prolonged ER stress and virus-induced UPR pathways in neuronal cell death (Su et al. 2002; Mukherjee et al. 2017; Wang et al. 2019). JEV also activates mitochondrial stress leading to ROS production and cytochrome-c release in the cytoplasm and activation of Caspase-8, -9 and -3 (Tsao et al. 2008; Yang et al. 2009, 2010; Wongchitrat et al. 2019). Recent evidence points to the role of inflammation-driven necroptosis and pyroptosis in the pathogenesis of JEV infection. The levels of the MLKL protein, a marker of necroptosis, increase in neuronal cells upon JEV infection, and its deletion reduces JE progression and inflammatory cytokines in the mouse model (Bian et al. 2017). JEV-infected fibroblasts and mouse brain showed upregulated levels of Gasdermin D and MLKL, along with increased transcript levels of genes of the necroptosis pathways (Sharma et al. 2021a). JEV-infected-pyroptotic macrophages release IL1-α, which is shown to be responsible for viral neuroinvasion (Wang et al. 2020a, b). Transcriptomic analysis of macrophages has also shown the launch of diverse programmed cell death pathways upon JEV infection (Wang et al. 2020a, b). A crucial role of necroptosis has been shown in neuroinflammation and cell death in other neurological disorders, signifying that targeting these cell death pathways can potentially reduce disease severity.
Downregulation of Cell Adhesion Molecules
JEV infection downregulates several collagens, laminin and other cell adhesion proteins involved in proteoglycan binding and ECM organisation (Sharma et al. 2021a). This could be a host-driven immune activation strategy for the generation of potential ligands for T and NK cell activation (Fig. 10.1).
Metabolic Reprogramming
All virus infections reprogramme the cellular metabolome to meet the high energy and resource demands of virus replication. In parallel, the cell also regulates its metabolism as an innate immune defence programme. A proteome wide-study has shown that JEV infection of fibroblasts down-modulates several metabolic enzymes of sterol and lipid biosynthetic pathways and transporters (solute carrier and ATP-binding cassette transporter) involved in shuttling metabolites and ions across membranes (Sharma et al. 2021a). Downmodulation of cholesterol/lipid biosynthetic activities is likely to be intimately linked with the IFN and inflammatory response. Increased glycolysis and pentose phosphate pathway flux, impaired oxidative phosphorylation and catabolic patterns of lipid metabolism are hallmarks of JEV replication in neurons (Li et al. 2021a). The unique metabolic signature of JEV infection is still an underexplored area, but we are likely to see this field expanding rapidly in the coming years with antiviral treatment strategies based on targeted metabolic modulation.
Infection Route: A Host Overview
Infection Route in the Periphery
JEV enters through an infected-mosquito bite, where the dermis layer acts as a primary site of infection. The local dermal cells (fibroblasts, endothelial, tissue-resident dendritic cells and pericytes) surrounding the mosquito bite area become infected and spread the virus to local lymph nodes, resulting in primary asymptomatic viremia (Filgueira and Lannes 2019; Ashraf et al. 2021). The virus escapes through either a hematogenous route or efferent lymphatic system and might infect multiple organs (liver, spleen, heart, muscle, kidney), generating secondary symptomatic viremia. Very little is known about how JEV affects the cardiovascular, respiratory, digestive, reproductive and urinary systems in humans (Qi et al. 2020; Chapagain et al. 2022); however, mouse model studies have clearly indicated that JEV infects several visceral organs in addition to the brain (Li et al. 2017). In the periphery, JEV mainly replicates in the monocytes/macrophages and DCs (Aleyas et al. 2009; Terry et al. 2012; Wang et al. 2016; Garcia-Nicolas et al. 2019). The virus migrates through the body either as free virions or via migratory infected DCs. In most cases, virus infection is cleared by an effective peripheral immune response. However, the virus can utilise various immune evasion strategies to escape peripheral immune surveillance to cross the BBB (Aleyas et al. 2010; Aleyas et al. 2012; Adhya et al. 2013; Manocha et al. 2014; Sood et al. 2017; Wang et al. 2017a; Banerjee and Tripathi 2019).
Peripheral Immune Response to Infection
Antigen-presenting cells (APCs) such as DCs and macrophages are the first cells to generate a robust immune response with the production of various anti-/pro-inflammatory cytokines and lowering of blood viremia (Solomon 2004). The IFN-β response in macrophages and their migration to the CNS is regulated by the RNA-binding protein quaking (QKI), which functions as an immune suppressor (Liao et al. 2021; Deng et al. 2022). DCs initiate the adaptive immune response by stimulating T-cell activation (Aleyas et al. 2009; Li et al. 2011; Sooryanarain et al. 2012; Sharma et al. 2021b). The virus infection can also result in inflammatory demyelination in the peripheral nervous system (Wang et al. 2022).
JEV also replicates in monocytes and upregulates various antiviral and immune factors, resulting in their activation and differentiation into monocyte-derived dendritic cells (MoDCs) and monocyte-derived macrophages (MoDMs) (Cao et al. 2011; Sooryanarain et al. 2012; Gupta et al. 2014; Garcia-Nicolas et al. 2019). JEV induces functional impairment of DCs through MyD88-dependent and -independent pathways, which leads to poor CD4(+) and CD8(+) T cell responses, and boosts viral survival and dissemination in the body (Aleyas et al. 2009). The virus also reduces the expression of co-stimulatory cytokines in human and mouse DCs, leading to suppressed T cell activation and enhanced Treg (T regulatory cell) differentiation (Cao et al. 2011; Gupta et al. 2014) (Fig. 10.2). Transcriptional profiling of JEV-infected human MoDCs has demonstrated the activation of antiviral and inflammatory pathways, and expansion of Tregs in an allogenic response (Chauhan et al. 2021). Overall, the enhanced Tregs response can exert a neuroprotective effect by reducing excessive inflammatory response as seen in another virus-induced encephalitis (Lund et al. 2008; Anghelina et al. 2009; Lanteri et al. 2009; James et al. 2016). Numerous mouse studies have exhibited the contribution of DCs in protection against JE via T cell-dependent and -independent mechanisms. CD11chi DCs regulate the innate CD11b+Ly-6Chi monocyte differentiation to protect immune-privileged CNS during JEV infection (Kim et al. 2015). The ablation of CD11chi DC is seen to lead to a higher ratio of CD4+ Th17/Treg cells and CD11b+Ly-6Chi/Ly-6Clo monocytes in the lymphoid tissue and CNS leading to enhanced permeability of the BBB (Choi et al. 2017).

Peripheral immune response to JEV infection. JEV enters the human body through the bite of an infected Culex mosquito. The virus first replicates in the skin and local lymph nodes and then infects the peripheral immune cells, primarily monocytes resulting in the production of TNF-α, which causes activation and differentiation of monocytes into dendritic cells and macrophages. These cells then result in T cell activation, secretion of inflammatory cytokines (IL-6 and TNF-α) and upregulation of co-stimulatory molecules. JEV also directly impairs DCs function leading to suppressed T cell activation and enhanced Tregs. After systemic infection, the virus crosses the blood-brain barrier and enters the brain either directly or through trans-endothelial migration of virus-infected monocytes
The exact function of T cells in JEV pathogenesis is still unclear as diverse mouse studies suggested different outcomes with partial or complete protection against JE. T cell activation leads to the production of various antiviral cytokines like IFN-γ, generation of T cell memory response, and humoral response (Sharma et al. 2021b) (Fig. 10.2). The involvement of human memory T cells in protection against human JE has been reported (Turtle et al. 2016). Interestingly, the responses of T cell subsets including CD4 (+) and CD8 (+) T cells are found to be associated with different clinical outcomes of JEV infection (Aleyas et al. 2009, 2010, 2012; Adhya et al. 2013; Turtle et al. 2016). The JEV-mediated cytolytic CD8 + T cell activation and associated IFN-γ response provide complete protection against JEV-induced morbidity (Larena et al. 2013; Jain et al. 2017). The CD4 (+) effector T cells are essential for B cell activation and in generating humoral response (Li et al. 2012b; Tarlinton 2019). In patients, IgM antibody response specific to JEV infection reaches a maximum within 7 days of infection and can be detected both in the serum and cerebrospinal fluid (Burke et al. 1985). JEV is also seen to modulate humoral response in mice by increasing myeloid-derived suppressor cell (MDSC) populations, which suppresses CD4+ T cell function and thus diminishes the splenic B cells (CD19+) and blood plasma cells (CD19 + CD138+) (Wang et al. 2017b, c). The generation of neutralising antibody response specific to JEV infection is shown to provide long-term immunity and protection (Lee et al. 1995; Lin et al. 1998; Konishi et al. 1999; Gupta et al. 2003; Plotkin 2010; Lee et al. 2016; Qiu et al. 2018). Animal studies have demonstrated that passive transfer of JEV-specific monoclonal antibodies could provide protection against JE (Kimura-Kuroda and Yasui 1988; Zhang et al. 1989; Beasley et al. 2004; Goncalvez et al. 2008; Van Gessel et al. 2011; Fernandez et al. 2018), and this has potential as an early treatment strategy.
Virus Entry into the CNS
The CNS is protected from peripheral contaminants by the BBB, which tightly regulates the selective transport of soluble factors and immune cells from blood to the brain. The BBB is comprised of tightly packed brain microvascular endothelial cells (BMECs) supported through interactions with microglial cells, astrocytes, pericytes and mast cells in the neurovascular unit that maintain the CNS microenvironment and neuronal function (Keaney and Campbell 2015; Villabona-Rueda et al. 2019).
In individuals who develop disease the virus crosses the BBB and replicates efficiently in the CNS. Mouse model studies suggest four possible mechanisms; (1) transcellular transport: virus infection in endothelial cells followed by passive transport of viral particles without affecting the cell viability; (2) diapedesis of virus-infected peripheral immune cells through endothelial cell junctions (“Trojan Horse” mechanism) or entry of infected immune cells through known physiological ways such as via the choroid plexus into the ventricular space; (3) virus transport via the peripheral nervous system (retrograde neuronal transport); and (4) virus entry upon BBB disruption due to virus-induced inflammatory mediators produced from cells present in both apical (blood) and basolateral (brain) sides of the BBB (Hsieh and St John 2020; Patabendige et al. 2018; Filgueira and Lannes 2019; Sharma et al. 2021b). A recent study using a BBB model of human brain endothelial cells and astrocytes suggested that JEV infection triggers the production of diverse host mediators, which regulate JEV production but disrupt BBB integrity, thus allowing virus to breach into the brain (Patabendige et al. 2018) (Fig. 10.3). Several inflammatory cytokines and metalloproteases produced from JEV-infected astrocytes and microglial cells trigger the proteasomal degradation of tight junction proteins (claudin-5 and ZO-1), leading to subsequent dysfunction of the endothelial barrier which promotes BBB leakage (Chen et al. 2014; Chang et al. 2015; Lannes et al. 2017b). Several studies also show that the virus gains CNS entry before BBB disruption (Li et al. 2015; Wang et al. 2018), and the breach is a fallout of a massive neuroinflammatory response in the brain.

CNS response to JEV infection. After systemic infection, JEV crosses the BBB and enters into CNS either directly or by Trojan horse mechanism (transmigration of monocytes containing virus). Virus infection then leads to the activation of pericytes, astrocytes and microglial cells (acting as virus reservoir), subsequently leading to the release of certain inflammatory cytokines and new virion particles. Virus infection of neurons causes neuronal cell death either directly or by causing excessive neuroinflammation resulting in neuronal damage. The increased levels of cytokines result in enhanced level of metalloproteases (MMP2/9) causing degradation of tight junction proteins including ZO-1 and Claudin-5 and disruption of BBB. This increases the permeability of BBB facilitating enhanced migration of JEV-infected leukocytes and JEV particles into the CNS, thereby increasing the neuroinflammation and causing excessive neuronal tissue damage
In the JEV-infected brain, the basal ganglia, thalamus and nuclei of the brainstem are the most affected regions (Kumar et al. 2009a). Virus-induced damage to the midbrain, brain stem, motor neurons in the spinal cord, periventricular tissue damage, etc., may result in different clinical pathologies (Misra and Kalita 2010; Suman et al. 2016).
CNS Response to Infection
Encephalitis is the hallmark of JEV pathogenesis. JEV infection of microglia (Thongtan et al. 2012; Gupta et al. 2017), astrocytes (Chen et al. 2011a, b) and neurons (Chen et al. 2018b; Yu et al. 2019), and subsequent upregulation of cell death and inflammatory responses contributes to virus-induced neuroinflammation. Genes associated with glutamate signalling are downregulated in JEV-infected mouse brains suggesting a potential negative impact on neurotransmission as well (Clarke et al. 2014). The virus can also modulate dopamine levels and can use dopamine-mediated neuronal communication to enhance infection of D2R neurons (Simanjuntak et al. 2017). Microglial activation in response to JEV PAMPS or DAMPs triggers various inflammatory factors and cytokines such as TNF-α, IL-1β, IL6, RANTES, MCP-1, etc., which upon overproduction leads to neuronal damage (Ghoshal et al. 2007; Chen et al. 2011a, b; Yang et al. 2012; Lannes et al. 2017a; Chen et al. 2018b) (Fig. 10.3). During infection, the initial microglial activation and subsequent production of cyto-/chemokines is necessary to eliminate the pathogen which can be executed either by directly targeting the virus or by recruiting immune cells (Chen et al. 2011a, b). In response to cytokines, the immune cells including inflammatory monocytes (Terry et al. 2012) and JEV-specific T cells, may also be recruited to the infected brain. However, prolonged microglial activation is detrimental as it leads to a magnified proinflammatory response and enhanced immune cell infiltration which causes bystander neuronal cell death (Ghoshal et al. 2007; Wang et al. 2019; Singh et al. 2020). In several cases, the virus is cleared from the brain with minimal collateral damage, but in rare cases, heightened inflammation and direct infection to neurons may lead to neuronal cell death and damage to key centres in the brain with long-term deficiencies or a fatal outcome (Sarkari et al. 2012; Shirai et al. 2015). Chronic JEV infection of microglial cells (Thongtan et al. 2010; Lannes et al. 2017a) and lymphocytes (Sharma et al. 1991) has been reported, which increases the possibility of virus reactivation.
Perspectives
Significant progress has been made in understanding the complex interplay of the JEV-host interaction at the cellular level. This has been augmented by high throughput omics studies that enable a holistic view of virus-driven changes in diverse cellular systems. The identification of crucial host dependency factors and pathways has also fuelled antiviral drug discovery. In addition, the cellular and animal model studies have given significant insights into the host immune response and disease pathogenesis. More epidemiological and molecular studies are required in the amplifying animal reservoirs (pigs and birds) and the transmitting insect vectors to better understand the virus propagation and spread. In the near future, we can expect to see advances driven by lipidomic, metabolomics, genomic and epigenomic studies that should enable biomarker development and enhance our understanding of their association with disease severity. These research-driven efforts will supplement disease management strategies and foster therapeutic development.
References
Abdoli A, Alirezaei M, Mehrbod P, Forouzanfar F (2018) Autophagy: the multi-purpose bridge in viral infections and host cells. Rev Med Virol 28(4):e1973
Adhya D, Dutta K, Kundu K, Basu A (2013) Histone deacetylase inhibition by Japanese encephalitis virus in monocyte/macrophages: a novel viral immune evasion strategy. Immunobiology 218(10):1235–1247
Agarwal A, Alam MF, Basu B, Pattanayak S, Asthana S, Syed GH, Kalia M, Vrati S (2022) Japanese encephalitis virus NS4A protein interacts with PTEN-induced kinase 1 (PINK1) and promotes Mitophagy in infected cells. Microbiol Spectr 10(3):e0083022
Ahmad L, Mostowy S, Sancho-Shimizu V (2018) Autophagy-virus interplay: from cell biology to human disease. Front Cell Dev Biol 6:155
Aleyas AG, George JA, Han YW, Rahman MM, Kim SJ, Han SB, Kim BS, Kim K, Eo SK (2009) Functional modulation of dendritic cells and macrophages by Japanese encephalitis virus through MyD88 adaptor molecule-dependent and -independent pathways. J Immunol 183(4):2462–2474
Aleyas AG, Han YW, George JA, Kim B, Kim K, Lee CK, Eo SK (2010) Multifront assault on antigen presentation by Japanese encephalitis virus subverts CD8+ T cell responses. J Immunol 185(3):1429–1441
Aleyas AG, Han YW, Patil AM, Kim SB, Kim K, Eo SK (2012) Impaired cross-presentation of CD8alpha+ CD11c+ dendritic cells by Japanese encephalitis virus in a TLR2/MyD88 signal pathway-dependent manner. Eur J Immunol 42(10):2655–2666
Anghelina D, Zhao J, Trandem K, Perlman S (2009) Role of regulatory T cells in coronavirus-induced acute encephalitis. Virology 385(2):358–367
Arakawa M, Morita E (2019) Flavivirus replication organelle biogenesis in the endoplasmic reticulum: comparison with other single-stranded positive-sense RNA viruses. Int J Mol Sci 20(9):2336
Arakawa M, Tabata K, Ishida K, Kobayashi M, Arai A, Ishikawa T, Suzuki R, Takeuchi H, Tripathi LP, Mizuguchi K, Morita E (2022) Flavivirus recruits the valosin-containing protein-NPL4 complex to induce stress granule disassembly for efficient viral genome replication. J Biol Chem 298(3):101597
Ashraf U, Ding Z, Deng S, Ye J, Cao S, Chen Z (2021) Pathogenicity and virulence of Japanese encephalitis virus: Neuroinflammation and neuronal cell damage. Virulence 12(1):968–980
Awais M, Wang K, Lin X, Qian W, Zhang N, Wang C, Wang K, Zhao L, Fu ZF, Cui M (2017) TLR7 deficiency leads to TLR8 compensative regulation of immune response against JEV in mice. Front Immunol 8:160
Bae W, Kim JH, Kim J, Lee J, Hwang ES (2018) Changes of epidemiological characteristics of Japanese encephalitis viral infection and birds as a potential viral transmitter in Korea. J Korean Med Sci 33(9):e70
Banerjee A, Tripathi A (2019). Recent advances in understanding Japanese encephalitis. F1000Res 8: F1000 Faculty Rev-1915.
Beasley DW, Li L, Suderman MT, Guirakhoo F, Trent DW, Monath TP, Shope RE, Barrett AD (2004) Protection against Japanese encephalitis virus strains representing four genotypes by passive transfer of sera raised against ChimeriVax-JE experimental vaccine. Vaccine 22(27–28):3722–3726
Bhattacharyya S, Sen U, Vrati S (2014) Regulated IRE1-dependent decay pathway is activated during Japanese encephalitis virus-induced unfolded protein response and benefits viral replication. J Gen Virol 95(Pt 1):71–79
Bhowmick S, Duseja R, Das S, Appaiahgiri MB, Vrati S, Basu A (2007) Induction of IP-10 (CXCL10) in astrocytes following Japanese encephalitis. Neurosci Lett 414(1):45–50
Bian P, Zheng X, Wei L, Ye C, Fan H, Cai Y, Zhang Y, Zhang F, Jia Z, Lei Y (2017) MLKL mediated necroptosis accelerates JEV-induced Neuroinflammation in mice. Front Microbiol 8:303
Blazquez AB, Escribano-Romero E, Merino-Ramos T, Saiz JC, Martin-Acebes MA (2014) Stress responses in flavivirus-infected cells: activation of unfolded protein response and autophagy. Front Microbiol 5:266
Burke DS, Nisalak A, Ussery MA, Laorakpongse T, Chantavibul S (1985) Kinetics of IgM and IgG responses to Japanese encephalitis virus in human serum and cerebrospinal fluid. J Infect Dis 151(6):1093–1099
Campbell GL, Hills SL, Fischer M, Jacobson JA, Hoke CH, Hombach JM, Marfin AA, Solomon T, Tsai TF, Tsu VD, Ginsburg AS (2011) Estimated global incidence of Japanese encephalitis: a systematic review. Bull World Health Organ 89(10):766–774. 774A-774E
Cao S, Li Y, Ye J, Yang X, Chen L, Liu X, Chen H (2011) Japanese encephalitis virus wild strain infection suppresses dendritic cells maturation and function, and causes the expansion of regulatory T cells. Virol J 8:39
Carletti T, Zakaria MK, Faoro V, Reale L, Kazungu Y, Licastro D, Marcello A (2019) Viral priming of cell intrinsic innate antiviral signaling by the unfolded protein response. Nat Commun 10(1):3889
Chambers TJ, Hahn CS, Galler R, Rice CM (1990) Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44:649–688
Chan ST, Ou JJ (2017) Hepatitis C virus-induced autophagy and host innate immune response. Viruses 9(8):224
Chang CY, Li JR, Chen WY, Ou YC, Lai CY, Hu YH, Wu CC, Chang CJ, Chen CJ (2015) Disruption of in vitro endothelial barrier integrity by Japanese encephalitis virus-infected astrocytes. Glia 63(11):1915–1932
Chapagain S, Pal Singh P, Le K, Safronetz D, Wood H, Karniychuk U (2022) Japanese encephalitis virus persists in the human reproductive epithelium and porcine reproductive tissues. PLoS Negl Trop Dis 16(7):e0010656
Chauhan S, Rathore DK, Sachan S, Lacroix-Desmazes S, Gupta N, Awasthi A, Vrati S, Kalia M (2021) Japanese encephalitis virus infected human monocyte-derived dendritic cells activate a transcriptional network leading to an antiviral inflammatory response. Front Immunol 12:638694
Chen CJ, Chen JH, Chen SY, Liao SL, Raung SL (2004) Upregulation of RANTES gene expression in neuroglia by Japanese encephalitis virus infection. J Virol 78(22):12107–12119
Chen CJ, Ou YC, Chang CY, Pan HC, Liao SL, Raung SL, Chen SY (2011a) TNF-alpha and IL-1beta mediate Japanese encephalitis virus-induced RANTES gene expression in astrocytes. Neurochem Int 58(2):234–242
Chen CJ, Ou YC, Chang CY, Pan HC, Lin SY, Liao SL, Raung SL, Chen SY, Chang CJ (2011b) Src signaling involvement in Japanese encephalitis virus-induced cytokine production in microglia. Neurochem Int 58(8):924–933
Chen CJ, Ou YC, Chang CY, Pan HC, Liao SL, Chen SY, Raung SL, Lai CY (2012a) Glutamate released by Japanese encephalitis virus-infected microglia involves TNF-alpha signaling and contributes to neuronal death. Glia 60(3):487–501
Chen ST, Liu RS, Wu MF, Lin YL, Chen SY, Tan DT, Chou TY, Tsai IS, Li L, Hsieh SL (2012b) CLEC5A regulates Japanese encephalitis virus-induced neuroinflammation and lethality. PLoS Pathog 8(4):e1002655
Chen CJ, Ou YC, Li JR, Chang CY, Pan HC, Lai CY, Liao SL, Raung SL, Chang CJ (2014) Infection of pericytes in vitro by Japanese encephalitis virus disrupts the integrity of the endothelial barrier. J Virol 88(2):1150–1161
Chen Y, Zhou Z, Min W (2018a) Mitochondria, oxidative stress and innate immunity. Front Physiol 9:1487
Chen Z, Wang X, Ashraf U, Zheng B, Ye J, Zhou D, Zhang H, Song Y, Chen H, Zhao S, Cao S (2018b) Activation of neuronal N-methyl-D-aspartate receptor plays a pivotal role in Japanese encephalitis virus-induced neuronal cell damage. J Neuroinflammation 15(1):238
Cheng VCC, Sridhar S, Wong SC, Wong SCY, Chan JFW, Yip CCY, Chau CH, Au TWK, Hwang YY, Yau CSW, Lo JYC, Lee CK, Yuen KY (2018) Japanese encephalitis virus transmitted via blood transfusion, Hong Kong, China. Emerg Infect Dis 24(1):49
Chien YJ, Chen WJ, Hsu WL, Chiou SS (2008) Bovine lactoferrin inhibits Japanese encephalitis virus by binding to heparan sulfate and receptor for low density lipoprotein. Virology 379(1):143–151
Chiou SS, Liu H, Chuang CK, Lin CC, Chen WJ (2005) Fitness of Japanese encephalitis virus to neuro-2a cells is determined by interactions of the viral envelope protein with highly sulfated glycosaminoglycans on the cell surface. J Med Virol 76(4):583–592
Chiu H, Chiu HP, Yu HP, Lin LH, Chen ZP, Lin YL, Lin RJ (2022) Zinc finger protein ZFP36L1 inhibits Flavivirus infection by both 5′-3' XRN1 and 3′-5' RNA-exosome RNA decay pathways. J Virol 96(1):e0166521
Choi JY, Kim JH, Patil AM, Kim SB, Uyangaa E, Hossain FMA, Eo SK (2017) Exacerbation of Japanese encephalitis by CD11c(hi) dendritic cell ablation is associated with an imbalance in regulatory Foxp3(+) and IL-17(+)CD4(+) Th17 cells and in Ly-6C(hi) and Ly-6C(lo) monocytes. Immune Netw 17(3):192–200
Choi Y, Bowman JW, Jung JU (2018) Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol 16(6):341–354
Clarke P, Leser JS, Bowen RA, Tyler KL (2014) Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor a, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. MBio 5(2):e00902–e00914
Clarke BD, Roby JA, Slonchak A, Khromykh AA (2015) Functional non-coding RNAs derived from the flavivirus 3′ untranslated region. Virus Res 206:53–61
Das S, Mishra MK, Ghosh J, Basu A (2008) Japanese encephalitis virus infection induces IL-18 and IL-1beta in microglia and astrocytes: correlation with in vitro cytokine responsiveness of glial cells and subsequent neuronal death. J Neuroimmunol 195(1–2):60–72
Das S, Chakraborty S, Basu A (2010) Critical role of lipid rafts in virus entry and activation of phosphoinositide 3′ kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J Neurochem 115(2):537–549
Datan E, Roy SG, Germain G, Zali N, McLean JE, Golshan G, Harbajan S, Lockshin RA, Zakeri Z (2016) Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis 7:e2127
de Almeida A, de Almeida Rezende MS, Dantas SH, de Lima Silva S, de Oliveira J, de Lourdes Assuncao Araujo de Azevedo F, Alves R, de Menezes GMS, Santos PFD, Goncalves TAF, Schini-Kerth VB, de Medeiros IA (2020) Unveiling the role of inflammation and oxidative stress on age-related cardiovascular diseases. Oxidative Med Cell Longev 2020:1954398
Deng L, Wang W, Bian P, Wu M, Wang L, Lei Y, Lu Z, Zhai D (2022) QKI deficiency in macrophages protects mice against JEV infection by regulating cell migration and antiviral response. Mol Immunol 148:34–44
Dhuriya YK, Sharma D (2018) Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation 15(1):199
Fadnis PR, Ravi V, Desai A, Turtle L, Solomon T (2013) Innate immune mechanisms in Japanese encephalitis virus infection: effect on transcription of pattern recognition receptors in mouse neuronal cells and brain tissue. Viral Immunol 26(6):366–377
Fernandez E, Kose N, Edeling MA, Adhikari J, Sapparapu G, Lazarte SM, Nelson CA, Govero J, Gross ML, Fremont DH, Crowe JE Jr, Diamond MS (2018) Mouse and human monoclonal antibodies protect against infection by multiple genotypes of Japanese encephalitis virus. MBio 9(1):e00008-18
Filgueira L, Lannes N (2019) Review of emerging Japanese encephalitis virus: new aspects and concepts about entry into the brain and inter-cellular spreading. Pathogens 8(3):111
Gao X, Liu H, Li X, Fu S, Cao L, Shao N, Zhang W, Wang Q, Lu Z, Lei W, He Y, Cao Y, Wang H, Liang G (2019) Changing geographic distribution of Japanese encephalitis virus genotypes, 1935-2017. Vector Borne Zoonotic Dis 19(1):35–44
Garcia-Nicolas O, Braun RO, Milona P, Lewandowska M, Dijkman R, Alves MP, Summerfield A (2018) Targeting of the nasal mucosa by Japanese encephalitis virus for non-vector-borne transmission. J Virol 92(24):e01091-18
Garcia-Nicolas O, Lewandowska M, Ricklin ME, Summerfield A (2019) Monocyte-derived dendritic cells as model to evaluate species tropism of mosquito-borne Flaviviruses. Front Cell Infect Microbiol 9:5
Ghosh D, Basu A (2009) Japanese encephalitis-a pathological and clinical perspective. PLoS Negl Trop Dis 3(9):e437
Ghoshal A, Das S, Ghosh S, Mishra MK, Sharma V, Koli P, Sen E, Basu A (2007) Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 55(5):483–496
Go YM, Jones DP (2008) Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta 1780(11):1273–1290
Goncalvez AP, Chien CH, Tubthong K, Gorshkova I, Roll C, Donau O, Schuck P, Yoksan S, Wang SD, Purcell RH, Lai CJ (2008) Humanized monoclonal antibodies derived from chimpanzee Fabs protect against Japanese encephalitis virus in vitro and in vivo. J Virol 82(14):7009–7021
Gupta AK, Lad VJ, Koshy AA (2003) Protection of mice against experimental Japanese encephalitis virus infections by neutralizing anti-glycoprotein E monoclonal antibodies. Acta Virol 47(3):141–145
Gupta N, Lomash V, Rao PV (2010a) Expression profile of Japanese encephalitis virus induced neuroinflammation and its implication in disease severity. J Clin Virol 49(1):4–10
Gupta N, Santhosh SR, Babu JP, Parida MM, Rao PV (2010b) Chemokine profiling of Japanese encephalitis virus-infected mouse neuroblastoma cells by microarray and real-time RT-PCR: implication in neuropathogenesis. Virus Res 147(1):107–112
Gupta N, Bhaskar AS, Lakshmana Rao PV (2011) Transcriptional regulation and activation of the mitogen-activated protein kinase pathway after Japanese encephalitis virus infection in neuroblastoma cells. FEMS Immunol Med Microbiol 62(1):110–121
Gupta N, Hegde P, Lecerf M, Nain M, Kaur M, Kalia M, Vrati S, Bayry J, Lacroix-Desmazes S, Kaveri SV (2014) Japanese encephalitis virus expands regulatory T cells by increasing the expression of PD-L1 on dendritic cells. Eur J Immunol 44(5):1363–1374
Gupta MK, Behera SK, Dehury B, Mahapatra N (2017) Identification and characterization of differentially expressed genes from human microglial cell samples infected with Japanese encephalitis virus. J Vector Borne Dis 54(2):131–138
Han YW, Choi JY, Uyangaa E, Kim SB, Kim JH, Kim BS, Kim K, Eo SK (2014) Distinct dictation of Japanese encephalitis virus-induced neuroinflammation and lethality via triggering TLR3 and TLR4 signal pathways. PLoS Pathog 10(9):e1004319
He W, Zhao Z, Anees A, Li Y, Ashraf U, Chen Z, Song Y, Chen H, Cao S, Ye J (2017) p21-activated kinase 4 signaling promotes Japanese encephalitis virus-mediated inflammation in astrocytes. Front Cell Infect Microbiol 7:271
Hsiao NW, Chen JW, Yang TC, Orloff GM, Wu YY, Lai CH, Lan YC, Lin CW (2010) ISG15 over-expression inhibits replication of the Japanese encephalitis virus in human medulloblastoma cells. Antivir Res 85(3):504–511
Hsieh JT, St John AL (2020) Japanese encephalitis virus and its mechanisms of neuroinvasion. PLoS Pathog 16(4):e1008260
Imre G (2020) Cell death signalling in virus infection. Cell Signal 76:109772
Jain N, Oswal N, Chawla AS, Agrawal T, Biswas M, Vrati S, Rath S, George A, Bal V, Medigeshi GR (2017) CD8 T cells protect adult naive mice from JEV-induced morbidity via lytic function. PLoS Negl Trop Dis 11(2):e0005329
James EA, Gates TJ, LaFond RE, Yamamoto S, Ni C, Mai D, Gersuk VH, O'Brien K, Nguyen QA, Zeitner B, Lanteri MC, Norris PJ, Chaussabel D, Malhotra U, Kwok WW (2016) Neuroinvasive West Nile infection elicits elevated and atypically polarized T cell responses that promote a pathogenic outcome. PLoS Pathog 12(1):e1005375
Jiang R, Ye J, Zhu B, Song Y, Chen H, Cao S (2014) Roles of TLR3 and RIG-I in mediating the inflammatory response in mouse microglia following Japanese encephalitis virus infection. J Immunol Res 2014:787023
Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y, Wang S, Wang W, Xiao G (2013) Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS One 8(1):e52909
Johnsen DO, Edelman R, Grossman RA, Muangman D, Pomsdhit J, Gould DJ (1974) Study of Japanese encephalitis virus in Chiangmia Valley, Thailand. V. Animal infections. Am J Epidemiol 100(1):57–68
Kalia M, Khasa R, Sharma M, Nain M, Vrati S (2013) Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J Virol 87(1):148–162
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis C, Sousa YM, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441(7089):101–105
Katoh H, Okamoto T, Fukuhara T, Kambara H, Morita E, Mori Y, Kamitani W, Matsuura Y (2013) Japanese encephalitis virus core protein inhibits stress granule formation through an interaction with Caprin-1 and facilitates viral propagation. J Virol 87(1):489–502
Kaushik DK, Gupta M, Basu A (2011) Microglial response to viral challenges: every silver lining comes with a cloud. Front Biosci (Landmark Ed) 16:2187–2205
Keaney J, Campbell M (2015) The dynamic blood-brain barrier. FEBS J 282(21):4067–4079
Khasa R, Vaidya A, Vrati S, Kalia M (2019) Membrane trafficking RNA interference screen identifies a crucial role of the clathrin endocytic pathway and ARP2/3 complex for Japanese encephalitis virus infection in HeLa cells. J Gen Virol 100(2):176–186
Khasa R, Sharma P, Vaidya A, Vrati S, Kalia M (2020) Proteins involved in actin filament organization are key host factors for Japanese encephalitis virus life-cycle in human neuronal cells. Microb Pathog 149:104565
Kim JH, Choi JY, Kim SB, Uyangaa E, Patil AM, Han YW, Park SY, Lee JH, Kim K, Eo SK (2015) CD11c(hi) dendritic cells regulate Ly-6C(hi) monocyte differentiation to preserve immune-privileged CNS in lethal Neuroinflammation. Sci Rep 5:17548
Kimura-Kuroda J, Yasui K (1988) Protection of mice against Japanese encephalitis virus by passive administration with monoclonal antibodies. J Immunol 141(10):3606–3610
Konishi E, Yamaoka M, Khin Sane W, Kurane I, Takada K, Mason PW (1999) The anamnestic neutralizing antibody response is critical for protection of mice from challenge following vaccination with a plasmid encoding the Japanese encephalitis virus premembrane and envelope genes. J Virol 73(7):5527–5534
Kumar S, Kalita J, Saxena V, Khan MY, Khanna VK, Sharma S, Dhole TN, Misra UK (2009a) Some observations on the tropism of Japanese encephalitis virus in rat brain. Brain Res 1268:135–141
Kumar S, Misra UK, Kalita J, Khanna VK, Khan MY (2009b) Imbalance in oxidant/antioxidant system in different brain regions of rat after the infection of Japanese encephalitis virus. Neurochem Int 55(7):648–654
Lannes N, Neuhaus V, Scolari B, Kharoubi-Hess S, Walch M, Summerfield A, Filgueira L (2017a) Interactions of human microglia cells with Japanese encephalitis virus. Virol J 14(1):8
Lannes N, Summerfield A, Filgueira L (2017b) Regulation of inflammation in Japanese encephalitis. J Neuroinflammation 14(1):158
Lanteri MC, O'Brien KM, Purtha WE, Cameron MJ, Lund JM, Owen RE, Heitman JW, Custer B, Hirschkorn DF, Tobler LH, Kiely N, Prince HE, Ndhlovu LC, Nixon DF, Kamel HT, Kelvin DJ, Busch MP, Rudensky AY, Diamond MS, Norris PJ (2009) Tregs control the development of symptomatic West Nile virus infection in humans and mice. J Clin Invest 119(11):3266–3277
Larena M, Regner M, Lobigs M (2013) Cytolytic effector pathways and IFN-gamma help protect against Japanese encephalitis. Eur J Immunol 43(7):1789–1798
Lee T, Komiya T, Watanabe K, Aizawa C, Hashimoto H (1995) Immune response in mice infected with the attenuated Japanese encephalitis vaccine strain SA14-14-2. Acta Virol 39(3):161–164
Lee EJ, Cha GW, Ju YR, Han MG, Lee WJ, Jeong YE (2016) Prevalence of neutralizing antibodies to Japanese encephalitis virus among high-risk age groups in South Korea, 2010. PLoS One 11(1):e0147841
Lennemann NJ, Coyne CB (2015) Catch me if you can: the link between autophagy and viruses. PLoS Pathog 11(3):e1004685
Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, Rossmann MG (2008) The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319(5871):1830–1834
Li Y, Ye J, Yang X, Xu M, Chen L, Mei L, Zhu J, Liu X, Chen H, Cao S (2011) Infection of mouse bone marrow-derived dendritic cells by live attenuated Japanese encephalitis virus induces cells maturation and triggers T cells activation. Vaccine 29(4):855–862
Li JK, Liang JJ, Liao CL, Lin YL (2012a) Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect 14(2):159–168
Li K, Li NL, Wei D, Pfeffer SR, Fan M, Pfeffer LM (2012b) Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 55(3):666–675
Li F, Wang Y, Yu L, Cao S, Wang K, Yuan J, Wang C, Wang K, Cui M, Fu ZF (2015) Viral infection of the central nervous system and Neuroinflammation precede blood-brain barrier disruption during Japanese encephalitis virus infection. J Virol 89(10):5602–5614
Li XF, Li XD, Deng CL, Dong HL, Zhang QY, Ye Q, Ye HQ, Huang XY, Deng YQ, Zhang B, Qin CF (2017) Visualization of a neurotropic flavivirus infection in mouse reveals unique viscerotropism controlled by host type I interferon signaling. Theranostics 7(4):912–925
Li M, Yang J, Ye C, Bian P, Yang X, Zhang H, Luo C, Xue Z, Lei Y, Lian J (2021a) Integrated metabolomics and transcriptomics analyses reveal metabolic landscape in neuronal cells during JEV infection. Virol Sin 36(6):1554–1565
Li Q, Zhou D, Jia F, Zhang L, Ashraf U, Li Y, Duan H, Song Y, Chen H, Cao S, Ye J (2021b) Japanese encephalitis virus NS1' protein interacts with host CDK1 protein to regulate antiviral response. Microbiol Spectr 9(3):e0166121
Liao SL, Raung SL, Chen CJ (2002) Japanese encephalitis virus stimulates superoxide dismutase activity in rat glial cultures. Neurosci Lett 324(2):133–136
Liao KC, Chuo V, Fagg WS, Modahl CM, Widen S, Garcia-Blanco MA (2021) The RNA binding protein quaking represses splicing of the fibronectin EDA exon and downregulates the interferon response. Nucleic Acids Res 49(17):10034–10045
Lin YL, Chen LK, Liao CL, Yeh CT, Ma SH, Chen JL, Huang YL, Chen SS, Chiang HY (1998) DNA immunization with Japanese encephalitis virus nonstructural protein NS1 elicits protective immunity in mice. J Virol 72(1):191–200
Lin RJ, Liao CL, Lin YL (2004) Replication-incompetent virions of Japanese encephalitis virus trigger neuronal cell death by oxidative stress in a culture system. J Gen Virol 85(Pt 2):521–533
Lin RJ, Chang BL, Yu HP, Liao CL, Lin YL (2006) Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J Virol 80(12):5908–5918
Lin CW, Cheng CW, Yang TC, Li SW, Cheng MH, Wan L, Lin YJ, Lai CH, Lin WY, Kao MC (2008) Interferon antagonist function of Japanese encephalitis virus NS4A and its interaction with DEAD-box RNA helicase DDX42. Virus Res 137(1):49–55
Lin RJ, Chien HL, Lin SY, Chang BL, Yu HP, Tang WC, Lin YL (2013) MCPIP1 ribonuclease exhibits broad-spectrum antiviral effects through viral RNA binding and degradation. Nucleic Acids Res 41(5):3314–3326
Lindqvist R, Mundt F, Gilthorpe JD, Wolfel S, Gekara NO, Kroger A, Overby AK (2016) Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflammation 13(1):277
Liu CY, Kaufman RJ (2003) The unfolded protein response. J Cell Sci 116(Pt 10):1861–1862
Liu K, Liao X, Zhou B, Yao H, Fan S, Chen P, Miao D (2013) Porcine alpha interferon inhibit Japanese encephalitis virus replication by different ISGs in vitro. Res Vet Sci 95(3):950–956
Liu CC, Zhang YN, Li ZY, Hou JX, Zhou J, Kan L, Zhou B, Chen PY (2017) Rab5 and Rab11 are required for Clathrin-dependent endocytosis of Japanese encephalitis virus in BHK-21 cells. J Virol 91(19):e01113-17
Liu K, Xiao C, Xi S, Hameed M, Wahaab A, Shao D, Li Z, Li B, Wei J, Qiu Y, Miao D, Zhu H, Ma Z (2020) Mosquito defensins enhance Japanese encephalitis virus infection by facilitating virus adsorption and entry within the mosquito. J Virol 94(21):e01164-20
Lund JM, Hsing L, Pham TT, Rudensky AY (2008) Coordination of early protective immunity to viral infection by regulatory T cells. Science 320(5880):1220–1224
Ma L, Li F, Zhang JW, Li W, Zhao DM, Wang H, Hua RH, Bu ZG (2018) Host factor SPCS1 regulates the replication of Japanese encephalitis virus through interactions with transmembrane domains of NS2B. J Virol 92(12):e00197-18
Manocha GD, Mishra R, Sharma N, Kumawat KL, Basu A, Singh SK (2014) Regulatory role of TRIM21 in the type-I interferon pathway in Japanese encephalitis virus-infected human microglial cells. J Neuroinflammation 11:24
Mansfield KL, Hernandez-Triana LM, Banyard AC, Fooks AR, Johnson N (2017) Japanese encephalitis virus infection, diagnosis and control in domestic animals. Vet Microbiol 201:85–92
McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z (2011) Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J Biol Chem 286(25):22147–22159
Mishra MK, Kumawat KL, Basu A (2008) Japanese encephalitis virus differentially modulates the induction of multiple pro-inflammatory mediators in human astrocytoma and astroglioma cell-lines. Cell Biol Int 32(12):1506–1513
Misra UK, Kalita J (2010) Overview: Japanese encephalitis. Prog Neurobiol 91(2):108–120
Miyake M (1964) The pathology of Japanese encephalitis. A review. Bull World Health Organ 30:153–160
Mukherjee S, Singh N, Sengupta N, Fatima M, Seth P, Mahadevan A, Shankar SK, Bhattacharyya A, Basu A (2017) Japanese encephalitis virus induces human neural stem/progenitor cell death by elevating GRP78, PHB and hnRNPC through ER stress. Cell Death Dis 8(1):e2556
Mukherjee S, Sengupta N, Chaudhuri A, Akbar I, Singh N, Chakraborty S, Suryawanshi AR, Bhattacharyya A, Basu A (2018) PLVAP and GKN3 are two critical host cell receptors which facilitate Japanese encephalitis virus entry into neurons. Sci Rep 8(1):11784
Mukherjee S, Akbar I, Kumari B, Vrati S, Basu A, Banerjee A (2019) Japanese encephalitis virus-induced let-7a/b interacted with the NOTCH-TLR7 pathway in microglia and facilitated neuronal death via caspase activation. J Neurochem 149(4):518–534
Mulvey P, Duong V, Boyer S, Burgess G, Williams DT, Dussart P, Horwood PF (2021) The ecology and evolution of Japanese encephalitis virus. Pathogens 10(12):1534
Nain M, Abdin MZ, Kalia M, Vrati S (2016) Japanese encephalitis virus invasion of cell: allies and alleys. Rev Med Virol 26(2):129–141
Nain M, Mukherjee S, Karmakar SP, Paton AW, Paton JC, Abdin MZ, Basu A, Kalia M, Vrati S (2017) GRP78 is an important host factor for Japanese encephalitis virus entry and replication in mammalian cells. J Virol 91(6):e02274-16
Nazmi A, Dutta K, Basu A (2011) RIG-I mediates innate immune response in mouse neurons following Japanese encephalitis virus infection. PLoS One 6(6):e21761
Nazmi A, Mukherjee S, Kundu K, Dutta K, Mahadevan A, Shankar SK, Basu A (2014) TLR7 is a key regulator of innate immunity against Japanese encephalitis virus infection. Neurobiol Dis 69:235–247
Niu J, Jiang Y, Xu H, Zhao C, Zhou G, Chen P, Cao R (2018) TIM-1 promotes Japanese encephalitis virus entry and infection. Viruses 10(11):630
Olagnier D, Peri S, Steel C, van Montfoort N, Chiang C, Beljanski V, Slifker M, He Z, Nichols CN, Lin R, Balachandran S, Hiscott J (2014) Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS Pathog 10(12):e1004566
Pan XL, Liu H, Wang HY, Fu SH, Liu HZ, Zhang HL, Li MH, Gao XY, Wang JL, Sun XH, Lu XJ, Zhai YG, Meng WS, He Y, Wang HQ, Han N, Wei B, Wu YG, Feng Y, Yang DJ, Wang LH, Tang Q, Xia G, Kurane I, Rayner S, Liang GD (2011) Emergence of genotype I of Japanese encephalitis virus as the dominant genotype in Asia. J Virol 85(19):9847–9853
Patabendige A, Michael BD, Craig AG, Solomon T (2018) Brain microvascular endothelial-astrocyte cell responses following Japanese encephalitis virus infection in an in vitro human blood-brain barrier model. Mol Cell Neurosci 89:60–70
Plotkin SA (2010) Correlates of protection induced by vaccination. Clin Vaccine Immunol 17(7):1055–1065
Preziuso S, Mari S, Mariotti F, Rossi G (2018) Detection of Japanese encephalitis virus in bone marrow of healthy young wild birds collected in 1997-2000 in Central Italy. Zoonoses Public Health 65(7):798–804
Qi ZL, Sun LY, Bai J, Zhuang HZ, Duan ML (2020) Japanese encephalitis following liver transplantation: a rare case report. World J Clin Cases 8(2):337–342
Qiu X, Lei Y, Yang P, Gao Q, Wang N, Cao L, Yuan S, Huang X, Deng Y, Ma W, Ding T, Zhang F, Wu X, Hu J, Liu SL, Qin C, Wang X, Xu Z, Rao Z (2018) Structural basis for neutralization of Japanese encephalitis virus by two potent therapeutic antibodies. Nat Microbiol 3(3):287–294
Raung SL, Kuo MD, Wang YM, Chen CJ (2001) Role of reactive oxygen intermediates in Japanese encephalitis virus infection in murine neuroblastoma cells. Neurosci Lett 315(1–2):9–12
Raung SL, Chen SY, Liao SL, Chen JH, Chen CJ (2005) Tyrosine kinase inhibitors attenuate Japanese encephalitis virus-induced neurotoxicity. Biochem Biophys Res Commun 327(2):399–406
Raung SL, Chen SY, Liao SL, Chen JH, Chen CJ (2007) Japanese encephalitis virus infection stimulates Src tyrosine kinase in neuron/glia. Neurosci Lett 419(3):263–268
Ricklin ME, Garcia-Nicolas O, Brechbuhl D, Python S, Zumkehr B, Nougairede A, Charrel RN, Posthaus H, Oevermann A, Summerfield A (2016) Vector-free transmission and persistence of Japanese encephalitis virus in pigs. Nat Commun 7:10832
Rodrigues FM, Guttikar SN, Pinto BD (1981) Prevalence of antibodies to Japanese encephalitis and West Nile viruses among wild birds in the Krishna-Godavari Delta, Andhra Pradesh, India. Trans R Soc Trop Med Hyg 75(2):258–262
Roy J, Galano JM, Durand T, Le Guennec JY, Lee JC (2017) Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J 31(9):3729–3745
Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I (2016) The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med 16(6):533–544
Salimi H, Cain MD, Klein RS (2016) Encephalitic arboviruses: emergence, clinical presentation, and Neuropathogenesis. Neurotherapeutics 13(3):514–534
Sarkar R, Sharma KB, Kumari A, Asthana S, Kalia M (2020) Japanese encephalitis virus capsid protein interacts with non-lipidated MAP1LC3 on replication membranes and lipid droplets. J Gen Virol 102(1):1–14
Sarkari NB, Thacker AK, Barthwal SP, Mishra VK, Prapann S, Srivastava D, Sarkari M (2012) Japanese encephalitis (JE) part II: 14 years’ follow-up of survivors. J Neurol 259(1):58–69
Sehrawat S, Khasa R, Deb A, Prajapat SK, Mallick S, Basu A, Surjit M, Kalia M, Vrati S (2021) Valosin-containing protein/p97 plays critical roles in the Japanese encephalitis virus life cycle. J Virol 95(11):e02336-20
Sharma S, Mathur A, Prakash V, Kulshreshtha R, Kumar R, Chaturvedi UC (1991) Japanese encephalitis virus latency in peripheral blood lymphocytes and recurrence of infection in children. Clin Exp Immunol 85(1):85–89
Sharma M, Bhattacharyya S, Nain M, Kaur M, Sood V, Gupta V, Khasa R, Abdin MZ, Vrati S, Kalia M (2014) Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 10(9):1637–1651
Sharma M, Bhattacharyya S, Sharma KB, Chauhan S, Asthana S, Abdin MZ, Vrati S, Kalia M (2017) Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J Gen Virol 98(5):1027–1039
Sharma M, Sharma KB, Chauhan S, Bhattacharyya S, Vrati S, Kalia M (2018) Diphenyleneiodonium enhances oxidative stress and inhibits Japanese encephalitis virus induced autophagy and ER stress pathways. Biochem Biophys Res Commun 502(2):232–237
Sharma KB, Sharma M, Aggarwal S, Yadav AK, Bhatnagar S, Vrati S, Kalia M (2019) Quantitative proteome analysis of Atg5-deficient mouse embryonic fibroblasts reveals the range of the autophagy-modulated basal cellular proteome. mSystems 4(6):e00481-19
Sharma KB, Chhabra S, Aggarwal S, Tripathi A, Banerjee A, Yadav AK, Vrati S, Kalia M (2021a) Proteomic landscape of Japanese encephalitis virus-infected fibroblasts. J Gen Virol 102(9):001657
Sharma KB, Vrati S, Kalia M (2021b) Pathobiology of Japanese encephalitis virus infection. Mol Asp Med 81:100994
Shirai K, Hayasaka D, Kitaura K, Takasaki T, Morita K, Suzuki R, Kurane I (2015) Qualitative differences in brain-infiltrating T cells are associated with a fatal outcome in mice infected with Japanese encephalitis virus. Arch Virol 160(3):765–775
Sikazwe C, Neave MJ, Michie A, Mileto P, Wang J, Cooper N, Levy A, Imrie A, Baird RW, Currie BJ, Speers D, Mackenzie JS, Smith DW, Williams DT (2022) Molecular detection and characterisation of the first Japanese encephalitis virus belonging to genotype IV acquired in Australia. PLoS Negl Trop Dis 16(11):e0010754
Simanjuntak Y, Liang JJ, Lee YL, Lin YL (2017) Japanese encephalitis virus exploits dopamine D2 receptor-phospholipase C to target dopaminergic human neuronal cells. Front Microbiol 8:651
Simon-Loriere E, Faye O, Prot M, Casademont I, Fall G, Fernandez-Garcia MD, Diagne MM, Kipela JM, Fall IS, Holmes EC, Sakuntabhai A, Sall AA (2017) Autochthonous Japanese encephalitis with yellow fever coinfection in Africa. N Engl J Med 376(15):1483–1485
Singh S, Singh G, Tiwari S, Kumar A (2020) CCR2 inhibition reduces neurotoxic microglia activation phenotype after Japanese encephalitis viral infection. Front Cell Neurosci 14:230
Sips GJ, Wilschut J, Smit JM (2012) Neuroinvasive flavivirus infections. Rev Med Virol 22(2):69–87
Solomon T (2003) Recent advances in Japanese encephalitis. J Neurovirol 9(2):274–283
Solomon T (2004) Flavivirus encephalitis. N Engl J Med 351(4):370–378
Solomon T, Ni H, Beasley DW, Ekkelenkamp M, Cardosa MJ, Barrett AD (2003) Origin and evolution of Japanese encephalitis virus in Southeast Asia. J Virol 77(5):3091–3098
Sood V, Sharma KB, Gupta V, Saha D, Dhapola P, Sharma M, Sen U, Kitajima S, Chowdhury S, Kalia M, Vrati S (2017) ATF3 negatively regulates cellular antiviral signaling and autophagy in the absence of type I interferons. Sci Rep 7(1):8789
Sooryanarain H, Ayachit V, Gore M (2012) Activated CD56(+) lymphocytes (NK+NKT) mediate immunomodulatory and anti-viral effects during Japanese encephalitis virus infection of dendritic cells in-vitro. Virology 432(2):250–260
Srivastava S, Khanna N, Saxena SK, Singh A, Mathur A, Dhole TN (1999) Degradation of Japanese encephalitis virus by neutrophils. Int J Exp Pathol 80(1):17–24
Stadler K, Allison SL, Schalich J, Heinz FX (1997) Proteolytic activation of tick-borne encephalitis virus by furin. J Virol 71(11):8475–8481
Su HL, Liao CL, Lin YL (2002) Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J Virol 76(9):4162–4171
Suman V, Roy U, Panwar A, Raizada A (2016) Japanese encephalitis complicated with obstructive hydrocephalus. J Clin Diagn Res 10(2):OD18-20
Swarup V, Ghosh J, Duseja R, Ghosh S, Basu A (2007) Japanese encephalitis virus infection decrease endogenous IL-10 production: correlation with microglial activation and neuronal death. Neurosci Lett 420(2):144–149
Tabata K, Arakawa M, Ishida K, Kobayashi M, Nara A, Sugimoto T, Okada T, Mori K, Morita E (2021) Endoplasmic reticulum-associated degradation controls virus protein homeostasis, which is required for Flavivirus propagation. J Virol 95(15):e0223420
Tarlinton D (2019) B cells still front and Centre in immunology. Nat Rev Immunol 19(2):85–86
Terry RL, Getts DR, Deffrasnes C, van Vreden C, Campbell IL, King NJ (2012) Inflammatory monocytes and the pathogenesis of viral encephalitis. J Neuroinflammation 9:270
Thongtan T, Cheepsunthorn P, Chaiworakul V, Rattanarungsan C, Wikan N, Smith DR (2010) Highly permissive infection of microglial cells by Japanese encephalitis virus: a possible role as a viral reservoir. Microbes Infect 12(1):37–45
Thongtan T, Thepparit C, Smith DR (2012) The involvement of microglial cells in Japanese encephalitis infections. Clin Dev Immunol 2012:890586
Tripathi A, Singh Rawat B, Addya S, Surjit M, Tailor P, Vrati S, Banerjee A (2021) Lack of interferon (IFN) regulatory factor 8 associated with restricted IFN-gamma response augmented Japanese encephalitis virus replication in the mouse brain. J Virol 95(21):e0040621
Tsao CH, Su HL, Lin YL, Yu HP, Kuo SM, Shen CI, Chen CW, Liao CL (2008) Japanese encephalitis virus infection activates caspase-8 and -9 in a FADD-independent and mitochondrion-dependent manner. J Gen Virol 89(Pt 8):1930–1941
Tu YC, Yu CY, Liang JJ, Lin E, Liao CL, Lin YL (2012) Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J Virol 86(19):10347–10358
Turtle L, Solomon T (2018) Japanese encephalitis - the prospects for new treatments. Nat Rev Neurol 14(5):298–313
Turtle L, Bali T, Buxton G, Chib S, Chan S, Soni M, Hussain M, Isenman H, Fadnis P, Venkataswamy MM, Satishkumar V, Lewthwaite P, Kurioka A, Krishna S, Shankar MV, Ahmed R, Begum A, Ravi V, Desai A, Yoksan S, Fernandez S, Willberg CB, Kloverpris HN, Conlon C, Klenerman P, Satchidanandam V, Solomon T (2016) Human T cell responses to Japanese encephalitis virus in health and disease. J Exp Med 213(7):1331–1352
Uchil PD, Satchidanandam V (2003) Characterization of RNA synthesis, replication mechanism, and in vitro RNA-dependent RNA polymerase activity of Japanese encephalitis virus. Virology 307(2):358–371
van den Hurk AF, Ritchie SA, Mackenzie JS (2009) Ecology and geographical expansion of Japanese encephalitis virus. Annu Rev Entomol 54:17–35
Van Gessel Y, Klade CS, Putnak R, Formica A, Krasaesub S, Spruth M, Cena B, Tungtaeng A, Gettayacamin M, Dewasthaly S (2011) Correlation of protection against Japanese encephalitis virus and JE vaccine (IXIARO((R))) induced neutralizing antibody titers. Vaccine 29(35):5925–5931
Vashist S, Bhullar D, Vrati S (2011) La protein can simultaneously bind to both 3′- and 5′-noncoding regions of Japanese encephalitis virus genome. DNA Cell Biol 30(6):339–346
Vedagiri D, Gupta D, Mishra A, Krishna G, Bhaskar M, Sah V, Basu A, Nayak D, Kalia M, Valiya Veettil M, Harshan KH (2021) Retinoic acid-inducible gene I-like receptors activate snail to limit RNA viral infections. J Virol 95(21):e0121621
Villabona-Rueda A, Erice C, Pardo CA, Stins MF (2019) The evolving concept of the blood brain barrier (BBB): from a single static barrier to a heterogeneous and dynamic relay center. Front Cell Neurosci 13:405
Wang P, Hu K, Luo S, Zhang M, Deng X, Li C, Jin W, Hu B, He S, Li M, Du T, Xiao G, Zhang B, Liu Y, Hu Q (2016) DC-SIGN as an attachment factor mediates Japanese encephalitis virus infection of human dendritic cells via interaction with a single high-mannose residue of viral E glycoprotein. Virology 488:108–119
Wang C, Zhang N, Qi L, Yuan J, Wang K, Wang K, Ma S, Wang H, Lou W, Hu P, Awais M, Cao S, Fu ZF, Cui M (2017a) Myeloid-derived suppressor cells inhibit T follicular helper cell immune response in Japanese encephalitis virus infection. J Immunol 199(9):3094–3105
Wang P, Li M, Lu W, Zhang D, Hu Q, Liu Y (2017b) DC-SIGN promotes Japanese encephalitis virus transmission from dendritic cells to T cells via virological synapses. Virol Sin 32(6):495–502
Wang X, Li SH, Zhu L, Nian QG, Yuan S, Gao Q, Hu Z, Ye Q, Li XF, Xie DY, Shaw N, Wang J, Walter TS, Huiskonen JT, Fry EE, Qin CF, Stuart DI, Rao Z (2017c) Near-atomic structure of Japanese encephalitis virus reveals critical determinants of virulence and stability. Nat Commun 8(1):14
Wang K, Wang H, Lou W, Ma L, Li Y, Zhang N, Wang C, Li F, Awais M, Cao S, She R, Fu ZF, Cui M (2018) IP-10 promotes blood-brain barrier damage by inducing tumor necrosis factor alpha production in Japanese encephalitis. Front Immunol 9:1148
Wang Q, Xin X, Wang T, Wan J, Ou Y, Yang Z, Yu Q, Zhu L, Guo Y, Wu Y, Ding Z, Zhang Y, Pan Z, Tang Y, Li S, Kong L (2019) Japanese encephalitis virus induces apoptosis and encephalitis by activating the PERK pathway. J Virol 93(17):e00887-19
Wang ZY, Zhen ZD, Fan DY, Qin CF, Han DS, Zhou HN, Wang PG, An J (2020a) Axl deficiency promotes the Neuroinvasion of Japanese encephalitis virus by enhancing IL-1alpha production from Pyroptotic macrophages. J Virol 94(17):e00602–e0e620
Wang ZY, Zhen ZD, Fan DY, Wang PG, An J (2020b) Transcriptomic analysis suggests the M1 polarization and launch of diverse programmed cell death pathways in Japanese encephalitis virus-infected macrophages. Viruses 12(3):356
Wang X, Wang G, Yang H, Fu S, He Y, Li F, Wang H, Wang Z (2022) A mouse model of peripheral nerve injury induced by Japanese encephalitis virus. PLoS Negl Trop Dis 16(11):e0010961
Wongchitrat P, Samutpong A, Lerdsamran H, Prasertsopon J, Yasawong M, Govitrapong P, Puthavathana P, Kitidee K (2019) Elevation of cleaved p18 Bax levels associated with the kinetics of neuronal cell death during Japanese encephalitis virus infection. Int J Mol Sci 20(20):5016
Xu Q, Cao M, Song H, Chen S, Qian X, Zhao P, Ren H, Tang H, Wang Y, Wei Y, Zhu Y, Qi Z (2016) Caveolin-1-mediated Japanese encephalitis virus entry requires a two-step regulation of actin reorganization. Future Microbiol 11:1227–1248
Xu Q, Zhu N, Chen S, Zhao P, Ren H, Zhu S, Tang H, Zhu Y, Qi Z (2017) E3 ubiquitin ligase Nedd4 promotes Japanese encephalitis virus replication by suppressing autophagy in human neuroblastoma cells. Sci Rep 7:45375
Yang TC, Shiu SL, Chuang PH, Lin YJ, Wan L, Lan YC, Lin CW (2009) Japanese encephalitis virus NS2B-NS3 protease induces caspase 3 activation and mitochondria-mediated apoptosis in human medulloblastoma cells. Virus Res 143(1):77–85
Yang TC, Lai CC, Shiu SL, Chuang PH, Tzou BC, Lin YY, Tsai FJ, Lin CW (2010) Japanese encephalitis virus down-regulates thioredoxin and induces ROS-mediated ASK1-ERK/p38 MAPK activation in human promonocyte cells. Microbes Infect 12(8–9):643–651
Yang CM, Lin CC, Lee IT, Lin YH, Yang CM, Chen WJ, Jou MJ, Hsiao LD (2012) Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K/Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J Neuroinflammation 9:12
Yang S, He M, Liu X, Li X, Fan B, Zhao S (2013) Japanese encephalitis virus infects porcine kidney epithelial PK15 cells via clathrin- and cholesterol-dependent endocytosis. Virol J 10:258
Ye J, Zhang H, He W, Zhu B, Zhou D, Chen Z, Ashraf U, Wei Y, Liu Z, Fu ZF, Chen H, Cao S (2016) Quantitative phosphoproteomic analysis identifies the critical role of JNK1 in neuroinflammation induced by Japanese encephalitis virus. Sci Signal 9(448):ra98
Yoshikawa A, Nabeshima T, Inoue S, Agoh M, Morita K (2016) Molecular and serological epidemiology of Japanese encephalitis virus (JEV) in a remote Island of western Japan: an implication of JEV migration over the East China Sea. Trop Med Health 44:8
Yu CY, Hsu YW, Liao CL, Lin YL (2006) Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J Virol 80(23):11868–11880
Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J (2008) Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319(5871):1834–1837
Yu SP, Ong KC, Perera D, Wong KT (2019) Neuronal transcriptomic responses to Japanese encephalitis virus infection with a special focus on chemokine CXCL11 and pattern recognition receptors RIG-1 and MDA5. Virology 527:107–115
Zhang MJ, Wang MJ, Jiang SZ, Ma WY (1989) Passive protection of mice, goats, and monkeys against Japanese encephalitis with monoclonal antibodies. J Med Virol 29(2):133–138
Zhang Y, Wang Z, Chen H, Chen Z, Tian Y (2014) Antioxidants: potential antiviral agents for Japanese encephalitis virus infection. Int J Infect Dis 24:30–36
Zhang Z, Rong L, Li YP (2019) Flaviviridae viruses and oxidative stress: implications for viral pathogenesis. Oxidative Med Cell Longev 2019:1409582
Zhang YG, Chen HW, Zhang HX, Wang K, Su J, Chen YR, Wang XR, Fu ZF, Cui M (2022) EGFR activation impairs antiviral activity of interferon signaling in brain microvascular endothelial cells during Japanese encephalitis virus infection. Front Microbiol 13:894356
Zheng S, Zhu D, Lian X, Liu W, Cao R, Chen P (2016) Porcine 2′, 5′-oligoadenylate synthetases inhibit Japanese encephalitis virus replication in vitro. J Med Virol 88(5):760–768
Zhou M, Wang S, Guo J, Liu Y, Cao J, Lan X, Jia X, Zhang B, Xiao G, Wang W (2021) RNA interference screening reveals requirement for platelet-derived growth factor receptor Beta in Japanese encephalitis virus infection. Antimicrob Agents Chemother 65(6):e00113-21
Zhu YZ, Xu QQ, Wu DG, Ren H, Zhao P, Lao WG, Wang Y, Tao QY, Qian XJ, Wei YH, Cao MM, Qi ZT (2012) Japanese encephalitis virus enters rat neuroblastoma cells via a pH-dependent, dynamin and caveola-mediated endocytosis pathway. J Virol 86(24):13407–13422
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sharma, K.B., Chhabra, S., Kalia, M. (2023). Japanese Encephalitis Virus-Infected Cells. In: Vijayakrishnan, S., Jiu, Y., Harris, J.R. (eds) Virus Infected Cells. Subcellular Biochemistry, vol 106. Springer, Cham. https://doi.org/10.1007/978-3-031-40086-5_10
Download citation
DOI: https://doi.org/10.1007/978-3-031-40086-5_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-40085-8
Online ISBN: 978-3-031-40086-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)