
Abstract
Autophagy is a self-degrading process that helps determine the fate of cells in response to stress. Although the role of autophagy in promoting cell survival is well understood, the connection between autophagy and cell death is still not entirely clear. Ferroptosis is a type of cell death that is driven by iron-mediated lipid peroxidation and plasma membrane rupture. Recent studies suggest that the level of autophagy in a cell can impact its sensitivity to ferroptosis. The selective destruction of certain proteins or organelles through autophagy can trigger ferroptosis by promoting iron accumulation and lipid peroxidation. Some of the genes and proteins involved in regulating autophagy-dependent ferroptosis may differ from those involved in starvation-induced autophagy. To develop effective treatments for related diseases, a deeper understanding of the mechanisms and regulation of autophagy in ferroptosis is essential.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
4.1 Introduction
Autophagy plays a critical role in maintaining cellular homeostasis by breaking down and disposing of intracellular waste, including unused proteins and damaged organelles (Dikic and Elazar 2018). There are different types of autophagy, including microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy, which can be further classified into bulk and selective autophagy. By degrading these waste products, autophagy provides cells with the resources they need to perform their biological functions and survive in the face of stressors, such as starvation, hypoxia, and drug toxicity (Kroemer et al. 2010). However, excessive autophagy can lead to cell damage or death, known as autophagy-dependent cell death (Tang et al. 2019). This process is regulated by the autophagy machinery, including autophagy-associated genes and protein complexes (Galluzzi et al. 2018; Xie et al. 2015). Autophagy-dependent cell death has been linked to various diseases, including cancer (Kriel and Loos 2019; Li et al. 2021b). Despite recent advancements, the underlying signaling pathways and regulatory networks that govern different types of selective autophagy-mediated cell death remain to be fully understood.
Ferroptosis is a regulated form of cell death that is triggered by oxidative damage from iron accumulation, leading to lipid peroxidation (Tang and Kroemer 2020). Despite its historical classification as a type of autophagy-independent cell death due to the inability of the autophagy inhibitor chloroquine to block the activity of ferroptosis activators in Rat sarcoma (RAS)-mutated cancer cells (Dixon et al. 2012), recent genetic evidence suggests that autophagy plays a role in the initiation and execution of ferroptosis through the triggering of iron accumulation and lipid peroxidation (Zhou et al. 2020; Liu et al. 2020a; Zhang et al. 2022; Xie et al. 2020a). The depletion of key components of the autophagy machinery (such as ATG5, ATG7, or BECN1) decreases the sensitivity of cells to ferroptosis, while the activation of autophagy-related factors accelerates ferroptosis in both cancer and non-cancer cells (Liu et al. 2020a; Chen et al. 2021a).
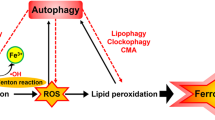
In this chapter, we present the fundamental mechanisms behind autophagy and ferroptosis. Our focus is on how dysregulated autophagy impacts ferroptosis (Fig. 4.1). This understanding may offer valuable insights into the possibility of targeting autophagy-dependent ferroptosis in diseases (Chen et al. 2023).

Role of autophagy in ferroptosis. Autophagy is a type of cellular degradation that is dependent on the lysosome and can occur in both selective and non-selective forms. Ferroptosis, on the other hand, is a type of cell death that is driven by oxidative stress and is dependent on iron. This type of cell death is characterized by lipid peroxidation, which leads to the production of toxic lipids. The role of selective autophagy in ferroptosis is complex, as it can both enhance and inhibit the process through the degradation of various organelles or proteins. Moreover, the BECN1 protein has been found to bind directly to SLC7A11, thereby inhibiting cystine uptake and maintaining low levels of the antioxidant glutathione
4.2 Mechanism of Autophagy
Autophagy is a complex, lysosome-dependent cellular degradation process that involves the formation of intracellular membrane structures, including phagophore, autophagosome, and autolysosome. On the other hand, other processes like CMA and microautophagy have different mechanisms. CMA specifically degrades proteins with the KFERQ motif, which is recognized by the heat shock protein 70 (HSP70) and regulated by lysosome-associated membrane protein 2 (LAMP2A) in lysosomes (Kaushik and Cuervo 2018). Microautophagy involves the direct phagocytosis of degraded cargo through lysosomes or late endosomes (Wang et al. 2022). This section provides a detailed description of the dynamic process and regulation of macroautophagy (hereinafter referred to as autophagy) in mammalian cells.
4.2.1 Phagophore
Autophagy is activated by various external (such as starvation, hypoxia, and pH changes) and internal factors (such as metabolic stress, aging, accumulation of misfolded proteins, and DNA damage). The activation of autophagy is triggered by the activation or impairment of multiple signaling pathways, such as the mitogen-activated protein kinase (MAPK) pathway, the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta (PI3K) pathway, the adenosine monophosphate (AMP)-activated protein kinase (AMPK) pathway, and the mammalian target of rapamycin (MTOR) pathway (Xie et al. 2015; Lahiri et al. 2021). These signals cause the formation of a cup-shaped phagophore structure, which is formed by membranes from various sources (such as the endoplasmic reticulum [ER], mitochondria, Golgi apparatus, and plasma membrane), and engulfs cytoplasmic molecules.
The formation of the phagophore structure is facilitated by two protein complexes, namely the unc-51-like autophagy-activating kinase (ULK) complex and the class III phosphatidylinositol 3-kinase (PtdIns3K) complex (Mizushima 2010). The ULK complex includes the core component ULK1 (a homolog of yeast Atg1), ATG13, and the scaffolding protein RB1-inducible coiled-coil 1 (RB1CC1, also known as FIP200, a homolog of yeast Atg17). The PtdIns3K complex consists of phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3, an ortholog of yeast Vps34), BECN1 (a mammalian homolog of yeast Vps30/Atg6), and phosphoinositide-3-kinase regulatory subunit 4 (PIK3R4, a mammalian homolog of yeast Vps15).
The ULK1 complex integrates signals from two key regulators of nutrient and energy levels, MTOR and AMPK, to initiate autophagy in response to changes in intracellular energy and nutrient levels. ULK1 kinase activity then activates the PtdIns3K complex, which is responsible for the formation of phagophore assembly sites, membrane nucleation, membrane elongation, and the formation of autophagosomes (Mizushima 2010). BECN1 is a key player in the regulation of autophagy, as it binds to many proteins and serves as a hub to control the induction of autophagy and phagophore formation (Kang et al. 2011).
4.2.2 Autophagosome
Autophagosomes, double-membrane structures derived from phagophore elongation, are formed by two ubiquitin-like conjugation systems. The first system involves the covalent conjugation of ATG12 and ATG5, which is catalyzed by ATG7 and ATG10. This conjugation forms a complex with ATG16L, which acts as an E3 enzyme to facilitate the conjugation of ubiquitin-like microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3)-family proteins (Scherz-Shouval et al. 2019; Nakatogawa 2013).
The second system involves the processing and lipidation of pro-MAP1LC3. The C-terminal polypeptide is cleaved by ATG4, a cysteine protease, to form MAP1LC3-I (Scherz-Shouval et al. 2007). This protein is then covalently associated with phosphatidylethanolamine (PE) in the presence of ATG7 and ATG3, resulting in the formation of MAP1LC3-II, the membrane-associated protein that is essential for autophagosome formation and degradation of cargo proteins. MAP1LC3-II serves not only as a marker for autophagosomes but also as a bridge between degraded substrates and autophagy receptors like sequestosome 1 (SQSTM1/p62). In addition to these processes, ATG9A-containing vesicles also contribute to starvation-induced autophagosome formation but not ferroptosis-associated autophagosome formation (Liu et al. 2022a). Therefore, different stimulatory signals and altered membrane sources are involved in the formation of autophagosomes.
4.2.3 Autolysosome
Autophagosomes fuse with lysosomes to form autolysosomes, although the exact molecular mechanism in mammalian cells remains unclear. Several lysosomal and membrane proteins have been implicated in this process, including lysosome-associated membrane protein 2 (LAMP2), the GTPase-activating protein RAB7A, the homotypic fusion and protein sorting (HOPS) complex, the soluble N-ethylmaleimide–sensitive fusion factor attachment protein receptor (SNARE) family, integration of autophagosomal syntaxin 17 (STX17), and lysosomal vesicle-associated membrane protein 8 (VAMP8) (Chi et al. 2019; Shen et al. 2021; Tian et al. 2020, 2021; Bernard and Klionsky 2015). The inner membrane of autophagosomes, including MAP1LC3-II, is also degraded by lysosomal enzymes. This degradation of MAP1LC3-II does not necessarily indicate inhibition of autophagy, and measuring autophagic flux in conjunction with late-stage autophagy inhibitors is a reliable method for assessing autophagic activity.
The cargo in lysosomes is typically degraded by lysosomal acid enzymes, such as cathepsin B (CTSB), cathepsin D (CTSD), cathepsin L (CTSL), acid phosphatases, and lipases. The selectivity of the cargo degradation remains unknown. The degraded cargo can be recycled to construct cellular components for self-renewal. However, excessive lysosomal degradation can be harmful to cellular function, so maintaining a healthy balance of degradation is essential for maintaining normal cellular homeostasis.
4.3 Selective Autophagy
Compared to bulk autophagy, selective autophagy is a targeted form of autophagy where specific cellular components, such as damaged organelles, misfolded proteins, or invading pathogens, are selectively degraded. Selective autophagy relies on specific autophagy receptors, which recognize and bind to these components to deliver them to autophagosomes for degradation (Vargas et al. 2022). The specificity of selective autophagy allows cells to maintain cellular homeostasis and regulate the quality control of cellular components. Autophagy receptors can be divided into two categories: ubiquitin-binding receptors, which recognize ubiquitinated cargoes, and cargo-localizing receptors, which bind directly to the degraded cargoes. Selective autophagy receptors contain a motif known as the Atg8-interacting motif or LC3-interacting region (AIM/LIR) to bind to the LC3/GABARAP/Atg8 family of lipidated proteins.
In yeast, the first identified form of selective autophagy was the cytoplasm-to-vacuole transport, which involved Atg11- and Atg19-dependent processing of aminopeptidase 1, aspartate aminopeptidase, and α-mannosidase (Baba et al. 1997). In mammalian cells, the receptors involved in selective autophagy are more diverse, and over 30 have been studied. Mitophagy, the selective removal of mitochondria, is the most studied form of autophagy receptor in mammals and involves the recognition of ubiquitinated mitochondrial proteins (Xie et al. 2020b, 2021). PTEN-induced putative kinase 1 (PINK1) and Parkin RBR E3 ubiquitin protein ligase (Parkin) proteins play a key role in this process, with PINK1 being a mitochondrial serine/threonine kinase and Parkin being an E3 ubiquitin ligase (Lazarou et al. 2015).
LC3 proteins or LC3 receptors can induce autophagy through a direct association with mitochondrial proteins in a ubiquitin-independent mechanism. Different receptors are involved in different types of selective autophagy and play a role in regulating cellular homeostasis. The binding of cargo-containing autophagy receptors to the Atg8/LC3 family of proteins ensures selectivity. In mammalian cells, there are two Atg8 protein families: MAP1LC3 and gamma-aminobutyric acid (GABA) type A receptor-associated protein (GABARAP), which are further divided into seven isoforms (Schaaf et al. 2016). Selective autophagy receptors bind LC3/GABARAP proteins through conserved LIR and GABARAP-interacting motifs, with the core motif being [W/F/Y]-XX-[L/V/I]. LIRs also contain residues that can be phosphorylated to increase interaction with the Atg8/LC3 family (Rogov et al. 2017).
4.4 Mechanism of Ferroptosis
Ferroptosis is a type of iron-dependent oxidative cell death that is largely regulated by the body’s antioxidant defenses (Kuang et al. 2020). However, other signals or molecules also play a role in determining a cell’s sensitivity to ferroptosis. In the following section, we will provide an overview of the key mechanisms and antioxidant systems that govern ferroptosis (Tang et al. 2021; Liu et al. 2021b).
4.4.1 Iron Toxicity
Iron is a vital trace element for all living organisms that exist in two redox states: ferrous (Fe2+) and ferric (Fe3+). The balance of iron in the body is crucial for proper functioning, including oxygen transport, electron transfer, and DNA synthesis. This balance is achieved through the processes of uptake, utilization, storage, and export of iron (Torti et al. 2018; Crielaard et al. 2017).
Cells acquire Fe3+ through transferrin receptor (TFRC)-mediated endocytosis, which is bound to transferrin (TF). Fe3+ is then reduced to Fe2+ by the STEAP3 metalloreductase within the endosome and released into the cytosol via the solute carrier family 11 member 2 (SLC11A2) pathway. Fe2+ can be stored in ferritin, including the ferritin heavy polypeptide 1 (FTH1) and ferritin light polypeptide 1 (FTL1) subunits.
Fe2+ also plays a role in oxygen transport and the creation of mitochondrial iron–sulfur clusters. Excess Fe2+ can be exported from the cell by solute carrier family 40 member 1 (SLC40A1), which converts Fe2+ to Fe3+. As such, any disruptions in iron metabolism may impact ferroptosis sensitivity (Chen et al. 2021c).
Iron contributes to ferroptosis not only by creating reactive oxygen species (ROS) through a non-enzymatic Fenton reaction but also by serving as a co-factor for metabolic enzymes, such as arachidonate lipoxygenase (ALOX) and cytochrome P450 oxidoreductase (POR), which can induce lipid peroxidation (Zou et al. 2020; Li et al. 2021a). This is why an increase in intracellular iron can trigger ferroptosis (Ajoolabady et al. 2021).
For instance, the degradation of ferritin through ferritinophagy, which is facilitated by nuclear receptor coactivator 4 (NCOA4), leads to iron release from ferritin and ferroptosis in pancreatic cancer cells (Hou et al. 2016). Conversely, glutamate oxaloacetate transaminase 1 (GOT1) can inhibit ferroptosis by inhibiting ferritinophagy (Kremer et al. 2021). Overall, the regulation of iron homeostasis in relation to ferroptosis is complex and delicate.
4.4.2 Lipid Toxicity
Ferroptosis is characterized by excessive lipid peroxidation and the formation of toxic lipid metabolites (Lin et al. 2021). The lipids most susceptible to peroxidation, arachidonic acid (AA) and adrenic acid (AdA), can lead to cell membrane rupture. The synthesis of polyunsaturated fatty acid phospholipids (PUFA-PLs) is governed by two key mediators, acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) (Yuan et al. 2016; Dixon et al. 2015). ACSL4 links PUFAs (AA or AdA) to CoA to form PUFA-CoAs, which are then esterified with PLs by LPCAT3. The carboxylation of acetyl CoA to malonyl-CoA by acetyl CoA carboxylase (ACC) is also needed for some PUFA synthesis. Inhibition of ACSL4 or LPCAT3 can, therefore, prevent or reduce sensitivity to ferroptosis. The lipid flippase solute carrier family 47 member 1 (SLC47A1) deficiency can also use the ACSL4-sterol O-acyltransferase 1 (SOAT1) pathway instead of the ACSL4-LPCAT3 pathway to produce PUFA-containing cholesterol esters and promote ferroptosis (Lin et al. 2022).
The role of lipoxygenases (ALOXs), a non-heme iron-dependent enzyme that can directly oxidize PUFA-containing lipids in membranes, in ferroptosis is content-dependent. For example, ALOX12 and ALOX15 are not essential for ferroptosis in mice, suggesting that other regulators, such as cytochrome P450 oxidoreductase (POR), contribute to lipid peroxidation during ferroptosis (Zou et al. 2020). Monounsaturated fatty acids (MUFAs) produced by stearoyl-CoA desaturase (SCD) can completely inhibit PUFA-mediated ferroptosis (Magtanong et al. 2019). Additionally, 4-hydroxy-2-nonenal (4-HNE), a byproduct of lipid peroxidation, has recently been implicated as a direct mediator of ferroptosis (Chen et al. 2022b), highlighting that the effector of ferroptosis may not necessarily be a protein.
4.4.3 GPX4 Antioxidant System
Glutathione peroxidase 4 (GPX4) is the sole GPX member found in mammalian cells, and it converts phospholipid (PL) hydroperoxides into alcohols. Studies have revealed that cytosolic GPX4 plays a crucial role in inhibiting ferroptosis (Yang et al. 2014), as evidenced by the fact that re-expression of cytosolic GPX4 rescues Gpx4 deletion-induced ferroptosis in mouse embryonic fibroblasts (Yant et al. 2003). In certain cases, mitochondrial GPX4 also helps in blocking mitochondrial oxidative damage-induced ferroptosis in cancer cells (Mao et al. 2021b).
The anti-ferroptotic effects of GPX4 are dependent on reduced glutathione (GSH), a tripeptide derived from glycine, glutamate, and cysteine, with cysteine being the rate-limiting precursor. Most cells obtain cysteine through the system xc−-mediated uptake and subsequent transformation of cystine, which involves solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2). Deprivation of cysteine through dietary means or pharmacological inhibition of the SLC7A11-GPX4 pathway (using erastin or RSL3) can trigger ferroptosis in various cells. While GPX4 is considered a key suppressor of ferroptosis, the conditional inactivation of GPX4 does not always result in ferroptosis (Viswanathan et al. 2017; Kang et al. 2018a), pointing to the presence of a GPX4-independent pathway.
4.4.4 AIFM2 Antioxidant System
Screening with CRISPR-Cas9 has identified apoptosis-inducing factor mitochondria-associated 2 (AIFM2, also known as FSP1) as a defense protein against ferroptosis, which operates in parallel to the GPX4 system (Doll et al. 2019). The mechanism involves N-myristoylation, which is necessary for the translocation of AIFM2 from mitochondria to the plasma membrane, thereby reducing lipid peroxidation and ferroptosis. This is achieved by reducing ubiquinone to ubiquinol (CoQH2) through its NADH:ubiquinone oxidoreductase activity, which traps lipid peroxyl radicals and prevents lipid autoxidation or indirectly oxidized α-tocopheryl radicals (vitamin E, a lipid antioxidant) (Doll et al. 2019). As a result, AIFM2 functions as a non-mitochondrial electron transport chain.
AIFM2 also serves as a vitamin K reductase, converting vitamin K into hydroquinone (VKH2) and thereby protecting GPX4-depleted cells from harmful lipid peroxidation and ferroptosis (Mishima et al. 2022). In addition to its reported enzymatic functions, AIFM2 can activate endosomal sorting complexes required for transport (ESCRT)-III–dependent membrane repair mechanisms to inhibit ferroptosis (Dai et al. 2020c).
In conclusion, these findings underscore the presence of three different molecular and cellular mechanisms through which AIFM2 protects against ferroptosis.
4.4.5 DHODH Antioxidant System
Mitochondria play a crucial role in regulating cellular metabolism and death, and they generate a substantial amount of ROS during oxidative phosphorylation. An imbalance in the mitochondrial antioxidant system can lead to lipid peroxidation. Dihydroorotate dehydrogenase (DHODH) is an enzyme that plays a crucial role in the de novo synthesis of pyrimidine nucleotides, which are the building blocks of DNA and RNA (Zhou et al. 2021). DHODH catalyzes the oxidation of dihydroorotate to orotate, which is an essential step in the formation of uracil and cytosine, two of the four nucleotides that make up the DNA molecule. In ferroptosis, DHODH controls pyrimidine synthesis in the inner mitochondrial membrane and can transfer electrons to CoQ to reduce it to CoQH2 (Mao et al. 2021a). An increase in DHODH-mediated CoQH2 production compensates for lipid peroxidation caused by the inactivation of GPX4. This mechanism can only be rescued by mitochondrial GPX4 or DHODH, not cytosolic GPX4. This highlights the existence of a mitochondrial antioxidant defense mechanism that helps prevent ferroptosis.
4.4.6 NFE2L2 Antioxidant System
NFE2L2, also known as NRF2, is a crucial transcription factor in the cap’n’collar (CNC) family (Dai et al. 2020a). It plays a vital role in maintaining redox homeostasis and regulating antioxidant genes by binding to antioxidant response elements (AREs). Under normal circumstances, the activity of NFE2L2 is suppressed as it is bound by Kelch-like ECH-associated protein 1 (KEAP1) in the cytoplasm, leading to its degradation. However, under oxidative and electrophilic stress, NFE2L2 is dissociated from KEAP1 and travels to the nucleus, where it partners with small v-maf musculoaponeurotic fibrosarcoma oncogene homolog (sMaf) proteins to activate the expression of ARE-dependent target genes.
In addition, NFE2L2 also provides cellular protection against ferroptosis activators like erastin and sorafenib (Sun et al. 2016b). It increases the stability of the NFE2L2 protein by suppressing the formation of the NFE2L2-KEAP1 complex. This process is regulated by SQSTM1, an autophagy receptor, which activates NFE2L2 expression through the inactivation of KEAP1. The activation of the SQSTM1-KEAP1-NFE2L2 pathway, therefore, blocks ferroptosis induced by erastin and sorafenib in cancer cells by upregulating multiple target genes (Sun et al. 2016b). Along with the well-known NFE2L2-targeted genes, such as nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) quinone dehydrogenase 1 (NQO1), GPX4, SLC7A11, and ferritin heavy chain 1 (FTH1), the cysteine-rich metallothionein 1G (MT1G) protein has been identified as a novel NFE2L2-targeted gene that provides resistance to ferroptosis in vitro and in xenograft mouse models (Sun et al. 2016a; Houessinon et al. 2016).
4.4.7 Membrane Repair System
The plasma membrane’s integrity and function are essential for cell survival and communication, and the membrane repair system helps preserve them. This system includes regrouping lipids, forming lipid rafts, and involving proteins, such as phospholipases, lipases, and flippases. A lipid flippase, SLC47A1, can prevent ferroptosis in cancer cells by restructuring lipids and has been observed at elevated levels in cancers (Wittwer et al. 2013). Targeting SLC47A1 may overcome resistance to ferroptosis in cancer cells.
The ESCRT complexes play a crucial role in repairing the plasma membrane and preventing various forms of regulated necrosis, including ferroptosis (Liu et al. 2021a). Inhibiting charged multivesicular body protein 5 (CHMP5) or CHMP6, components of ESCRT-III, increases ferroptosis susceptibility in HCC cells (Dai et al. 2020b). Calcium signaling and endoplasmic reticulum stress regulate ferroptosis through the activation of ESCRT-III on cell membranes.
4.5 Autophagy in Ferroptosis
Autophagy can either inhibit or promote cell death depending on the context. This section discusses the mechanisms by which autophagy regulates ferroptosis in cancer cells (Xie et al. 2016).
4.5.1 Ferritinophagy
Ferritinophagy is a type of autophagy, which is a process where cells break down and recycle their own components (Ajoolabady et al. 2021). Ferritinophagy specifically degrades ferritin, a complex made up of two proteins, FTH1 and FTL1. A cytosolic autophagy receptor called NCOA4 is involved in mediating ferritin degradation and promoting ferroptosis. Ferroptosis is a type of programmed cell death that occurs when cells accumulate toxic levels of iron.
The expression of NCOA4 can be regulated by different signaling pathways. For example, O-GlcNAcylation of FTH1 can inhibit its binding to NCOA4 and thus decrease ferritinophagy. On the other hand, inhibiting O-GlcNAcylation can enhance ferritinophagy (Yu et al. 2022). Another example is poly(rC) binding protein 1 (PCBP1), which can suppress autophagy and thus inhibit ferritinophagy-dependent ferroptosis (Lee et al. 2022). In contrast, ELAV-like RNA binding protein 1 (ELAVL1/HuR) can regulate ferritinophagy-dependent ferroptosis (Zhang et al. 2018).
Different factors can also affect ferritinophagy and ferroptosis. For example, hypoxia downregulates NCOA4 and increases FTH/FTL protein levels, which can inhibit ferritinophagy. On the other hand, transmembrane protein 164 (TMEM164), but not ATG9A, can selectively promote autophagosome formation to degrade ferritin during ferroptosis (Liu et al. 2022a). In addition, inhibiting the lysosomal V-ATPase with bafilomycin A1 can decrease ferritin degradation and ferroptosis (Gao et al. 2018), whereas dihydroartemisinin can induce ferritinophagy-dependent ferroptosis (Lin et al. 2016).
Overall, ferritinophagy plays a major role in promoting ferroptosis by increasing the accumulation of toxic iron. However, since NCOA4 is widely expressed, targeting NCOA4-dependent ferroptosis in cancer therapy may require careful monitoring of toxicity.
4.5.2 Lipophagy
Fatty acids are converted into triglycerides and cholesterol esters inside cells and stored in a spherical organelle called lipid droplets (LDs) (Maan et al. 2018). During times of stress, such as starvation or RSL3, LDs can be engulfed by autophagosomes for destruction in the lysosome, releasing free fatty acids. This process, called lipophagy, promotes RSL3-induced ferroptosis in primary mouse hepatocytes and the human liver cancer cell line HepG2. The regulation of this process is controlled by the LD cargo receptor RAB7A (Bai et al. 2019; Schroeder et al. 2015).
RAB7A selectively recognizes LDs and triggers the ATG5-dependent formation of autophagosomes (Bai et al. 2019). If ATG5 or RAB7A is knocked down, RSL3-induced ferroptosis is prevented both in vitro and in vivo (Bai et al. 2019). Similarly, knocking down the tumor protein D52 (TPD52), which regulates LD formation, increases RSL3-induced ferroptosis. On the other hand, overexpressing TPD52 limits ferroptosis (Bai et al. 2019). These findings suggest that increasing LD storage can protect against lipid toxicity, whereas lipophagy makes cells more sensitive to ferroptosis by releasing more lipids for lipid peroxidation. Further research on the signals and mediators of lipophagy is crucial for developing ferroptosis-related therapies in cancers with high levels of LDs, such as liver cancer.
4.5.3 Clockophagy
The circadian rhythm is an internal mechanism that is controlled by circadian clock proteins, such as aryl hydrocarbon receptor nuclear translocator-like protein (ARNTL/BMAL1) and CLOCK. These proteins regulate various cellular processes including iron and lipid metabolism. It has been found that ARNTL degradation can occur through an autophagy-dependent pathway called clockophagy (Liu et al. 2019), which specifically degrades ARNTL1 and promotes ferroptosis in cancer cells when exposed to GPX4 inhibitors (e.g., RSL3 and FIN56) but not when exposed to SLC7A11 inhibitors (e.g., erastin, sulfasalazine, and sorafenib) (Yang et al. 2019).
Mass spectrometry analysis showed that SQSTM1 acts as an autophagy receptor that binds ARNTL to ATG5- and ATG7-dependent autophagosomes for degradation in the lysosome (Yang et al. 2019). This process does not require ATG9A. The degradation of ARNTL represses the expression of the target gene EGLN2, which controls lipid metabolism through hypoxia-inducible factor 1-alpha (HIF1A) (Yang et al. 2019). Additionally, the knockdown of the circadian protein PER1 impedes autophagy in the hippocampus, which may increase susceptibility to ferroptosis during cerebral ischemia (Rami et al. 2017). The ferroptosis inhibitor liproxstatin-1 prevents acute pancreatitis in pancreatic Arntl-specific knockout mice induced by L-arginine (Liu et al. 2020b). Furthermore, extracellular SQSTM1 induces ACSL4 expression to enhance autophagy-dependent ferroptosis and subsequent pancreatitis (Yang et al. 2022), providing alternative strategies to mediate ferroptosis-induced sterile inflammation.
These findings show a relationship between circadian rhythms, autophagy, and ferroptosis. Further research is needed to determine if regulators of ferroptosis are influenced by circadian rhythms.
4.5.4 Mitophagy
Mitophagy, a mechanism for controlling the quality of mitochondria, regulates the production of mitochondrial ROS and the sensitivity to cell death (Jiao et al. 2021). PINK1-Parkin-dependent mitophagy is the central way of managing mitochondrial health. However, the connection between mitochondria and ferroptosis is unclear and can even seem paradoxical. For instance, the mitochondrial respiratory chain inhibitor BAY 87–2243 triggers excessive mitophagy and leads to an increase in mitochondrial ROS production and subsequent ferroptosis in BRAFV600E melanoma cell lines, but overexpression of GPX4 or administration of ferrostatin-1 reverses the ferroptosis induced by BAY 87–2243 (Basit et al. 2017). On the other hand, a loss of function in fumarate hydratase, a component of the mitochondrial tricarboxylic acid cycle, confers resistance to ferroptosis induced by cysteine deprivation. The compound WJ460, which interacts with myoferlin protein, promotes mitophagy and mitochondrial fission and heightens pancreatic cancer cell sensitivity to ferroptosis (Rademaker et al. 2022). Additionally, zalcitabine-induced stress on mitochondrial DNA triggers autophagy-dependent ferroptosis through the stimulator of interferon response CGAMP interactor 1 (STING1, also known as STING or TMEM173) pathway (Li et al. 2021a). STING1 also collaborates with MFN1/2 to enhance mitochondrial fusion during ferroptosis (Li et al. 2021a). These studies shed light on the intricate relationship between mitophagy, mitochondrial dynamics, and ferroptosis, yet the role of specific autophagy receptors in mediating mitophagy and ferroptosis remains to be investigated further.
4.5.5 Reticulophagy
The ER is a cell organelle responsible for making proteins and lipids. When there is an accumulation of unfolded proteins during protein synthesis, the cell activates the “unfolded protein response” (UPR) to prevent damage. Excess UPR can lead to cell death (Oakes and Papa 2015). Reticulophagy is a type of autophagy that helps to remove unnecessary parts of the ER or proteins that the UPR cannot process. Sorafenib, a type of drug, has been shown to limit ferroptosis in liver cancer cells by inducing reticulophagy through the reticulophagy regulator family member 3 (FAM134B) receptor (Liu et al. 2022b). The exact mechanism is still unknown, but it may involve the activation of FAM134B by the protein poly(A) binding protein cytoplasmic 1 (PABPC1) during ferroptosis (Liu et al. 2022b). These findings suggest that different types of selective autophagy can play a role in controlling ferroptosis.
4.5.6 GPX4 Degradation
Both CMA and autophagy play a role in the degradation of GPX4, leading to the enhancement of ferroptosis in cancer cells (Yang et al. 2020; Han et al. 2021). The autophagy inducer rapamycin and the ferroptosis inducer RSL3 can suppress MTOR and degrade GPX4, implying that ferroptosis might be regulated by GPX4 degradation through autophagy (Liu et al. 2021d). Exogenous copper leads to an increase in GPX4 ubiquitination, and the formation of GPX4 aggregates through direct binding to the GPX4 protein cysteines at positions 107 and 148. TAX1BP1, or Tax1 binding protein 1, functions as an autophagic receptor that mediates GPX4 degradation and initiates ferroptosis in response to copper stress (Xue et al. 2023). Heat shock protein 90 (HSP90) has been discovered as a molecular chaperone that interacts with the CMA cargo receptor LAMP2A, thereby mediating GPX4 degradation during ferroptosis (Wu et al. 2019). Conversely, inhibiting HSP90 stops CMA-induced ferroptosis in the mouse neuronal cell line HT-22. On the other hand, HSPA5, an ER-associated molecular chaperone, prevents erastin-induced ferroptosis in human pancreatic cancer cell lines by inhibiting GPX4 protein degradation (Zhu et al. 2017). These findings indicate that different HSP family proteins exert diverse molecular chaperone functions in either enhancing or inhibiting GPX4 degradation.
4.5.7 CDH2 Degradation
The role of specific autophagy receptors in ferroptosis is still an unresolved question in the field. However, recent research has shed light on this topic. A study that combined membrane protein screening with functional analyses showed that hippocalcin like 1 (HPCAL1), a calcin-like protein found in the hippocampus, serves as a specific autophagy receptor that promotes ferroptosis in cancer cells (Chen et al. 2022c). It does so by directly mediating the degradation of the cadherin 2 (CDH2) protein but not the degradation of ferritin, SLC40A1, or GPX4. HPCAL1 is not necessary for starvation-induced autophagy or cancer cell apoptosis, and its known role as a Ca2+-binding protein is not essential for its role in autophagy-dependent ferroptosis. The study also found that PRKCQ-mediated phosphorylation of Thr149 on HPCAL1 is necessary for its recognition and degradation of CDH2 (Chen et al. 2022c). This discovery provides new insights into the specific role of autophagy-dependent ferroptosis in cancer therapy.
4.5.8 SLC40A1 Degradation
SLC40A1 is found in cells that store and release iron, such as cells in the lining of the small intestine (enterocytes) and cells in the liver (hepatocytes). Mutations in the SLC40A1 gene have been linked to a type of iron overload disorder called hemochromatosis. This condition is characterized by the buildup of too much iron in the body, which can lead to damage to tissues and organs over time. Hemochromatosis has several forms, each with a different underlying cause, but mutations in the SLC40A1 gene are responsible for about 10% of cases. Autophagic degradation of SLC40A1 by SQSTM1 also increases iron accumulation and ferroptosis (Li et al. 2021c). It is necessary to further evaluate the relationship between autophagy, ferroptosis, and hemochromatosis in the future.
4.5.9 BECN1-Mediated System xc− Inhibition
The multifaceted protein BECN1 plays a crucial role in the regulation of ferroptosis (Kang et al. 2018b). Direct binding of BECN1 to SLC7A11, in response to inhibitors, such as erastin, sulfasalazine, and sorafenib but not GPX4 inhibitors, such as RSL3 and FIN56, results in cysteine deprivation-induced ferroptosis. Phosphorylation of BECN1 at S90 and S93 by AMPK promotes the formation of the BECN1-SLC7A11 complex, and mutations at these phosphorylation sites prevent ferroptosis. The BECN1 activator, Tat-beclin 1 protein peptide, boosts both autophagy and ferroptosis-induced tumor suppression in mice. However, the mechanism by which AMPK selectively activates BECN1 remains unclear.
The RNA binding protein ELAVL1/HuR can also mediate ferroptosis by promoting autophagy activation through binding to AU-rich elements within the 3′-untranslated region F3 of BECN1 mRNA in human hepatic stellate cells (Zhang et al. 2018). This process may be further enhanced by the exosome-mediated delivery of BECN1 secreted by human umbilical cord mesenchymal stem cells. The binding of BECN1 to ubiquitin-specific protease 11 (USP11) in spinal cord ischemia-reperfusion injury further demonstrates BECN1’s role in mediating autophagy-dependent ferroptosis (Rong et al. 2021). Overall, BECN1 can regulate ferroptosis by either limiting SLC7A11 activity directly or by activating an autophagy-dependent pathway.
4.5.10 Lysosomal Membrane Permeabilization
The accumulation of iron in the lysosomal lumen can trigger a Fenton response that increases lysosomal membrane permeabilization (LMP) and ultimately leads to cell death. In contrast, lysosomal ferritin can mitigate oxidative stress. LMP results in the release of lysosomal contents, such as CTSB, CTSD, and iron, which, in turn, increase ROS production and initiate ferroptosis. This process may also be regulated by V-ATPase-mediated selective autophagy (Chen et al. 2022a). Ferroptosis can also be induced by erastin-mediated lysosomal cell death. The signal transducer and activator of transcription 3 (STAT3) regulates this process by inducing CTSB expression, which is required for lysosomal cell death (Gao et al. 2018). However, inhibiting STAT3 pharmacologically (using the cathepsin inhibitor CA-074Me) or genetically limits ferroptosis in cancer cells. In 5-FU-resistant gastric cancer, STAT3 plays a negative role in ferroptosis by binding to the promoters of genes, such as GPX4, SLC7A11, and FTH1, that negatively regulate ferroptosis (Ouyang et al. 2022). These findings suggest that STAT3 has a context-dependent role in ferroptosis, and the receptors and mechanisms involved in mediating STAT3 degradation remain to be investigated.
4.6 Conclusions and Perspectives
Ferroptosis has seen significant advancements in research in recent years (Stockwell et al. 2017; Chen et al. 2021c; Chen et al. 2021b). Our perception of ferroptosis and how it is controlled has shifted as it has become known to have close ties to autophagy. Elevated autophagic activity can drive ferroptosis by selectively eliminating antioxidant proteins and cellular components, which raises questions about the molecular and metabolic checkpoints that influence cell survival or death. Ferroptosis and autophagy are complex processes that involve oxidative stress and membrane structural changes and are regulated by various mechanisms. However, more needs to be understood about the role of selective autophagy in regulating ferroptosis sensitivity.
Developing therapeutic strategies targeting cancer cells that involve autophagy or ferroptosis can be challenging as both processes can occur in normal cells and tissues. Further identification of biomarkers, such as damage-associated molecular patterns (DAMPs) (Zhang et al. 2013), high-mobility group protein 1 (HMGB1) (Wen et al. 2019), and decorin (DCN) (Liu et al. 2021c), is crucial for future clinical studies on ferroptosis. Moreover, gaining insight into the immune properties of ferroptotic cell death is crucial for the development of new immunotherapies in the fight against diseases (Chen et al. 2022c; Liu et al. 2021c, 2021d; Zhang et al. 2020; Yang et al. 2022).
References
Ajoolabady A, Aslkhodapasandhokmabad H, Libby P, Tuomilehto J, Lip GYH, Penninger JM, Richardson DR, Tang D, Zhou H, Wang S, Klionsky DJ, Kroemer G, Ren J (2021) Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol Metab 32(7):444–462. https://doi.org/10.1016/j.tem.2021.04.010
Baba M, Osumi M, Scott SV, Klionsky DJ, Ohsumi Y (1997) Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J Cell Biol 139(7):1687–1695. https://doi.org/10.1083/jcb.139.7.1687
Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, Kang R, Wang X, Tang D, Dai E (2019) Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun 508(4):997–1003. https://doi.org/10.1016/j.bbrc.2018.12.039
Basit F, van Oppen LM, Schöckel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, Grefte S, Kopitz C, Heroult M, Hgm Willems P, Koopman WJ (2017) Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis 8(3):e2716. https://doi.org/10.1038/cddis.2017.133
Bernard A, Klionsky DJ (2015) Toward an understanding of autophagosome-lysosome fusion: the unsuspected role of ATG14. Autophagy 11(4):583–584. https://doi.org/10.1080/15548627.2015.1029220
Chen X, Kang R, Kroemer G, Tang D (2021a) Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol 18(5):280–296. https://doi.org/10.1038/s41571-020-00462-0
Chen X, Kang R, Kroemer G, Tang DL (2021b) Ferroptosis in infection, inflammation, and immunity. J Exp Med 218(6):e20210518. https://doi.org/10.1084/jem.20210518
Chen X, Li J, Kang R, Klionsky DJ, Tang D (2021c) Ferroptosis: machinery and regulation. Autophagy 17(9):2054–2081. https://doi.org/10.1080/15548627.2020.1810918
Chen F, Kang R, Liu J, Tang D (2022a) The V-ATPases in cancer and cell death. Cancer Gene Ther 29:1529–1541. https://doi.org/10.1038/s41417-022-00477-y
Chen X, Huang J, Yu C, Liu J, Gao W, Li J, Song X, Zhou Z, Li C, Xie Y, Kroemer G, Liu J, Tang D, Kang R (2022b) A noncanonical function of EIF4E limits ALDH1B1 activity and increases susceptibility to ferroptosis. Nat Commun 13(1):6318. https://doi.org/10.1038/s41467-022-34096-w
Chen X, Song X, Li J, Zhang R, Yu C, Zhou Z, Liu J, Liao S, Klionsky DJ, Kroemer G, Liu J, Tang D, Kang R (2022c) Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy 19:54. https://doi.org/10.1080/15548627.2022.2059170
Chen F, Cai X, Kang R, Liu J, Tang D (2023) Autophagy-dependent Ferroptosis in cancer. Antioxid Redox Signal. https://doi.org/10.1089/ars.2022.0202
Chi C, Leonard A, Knight WE, Beussman KM, Zhao Y, Cao Y, Londono P, Aune E, Trembley MA, Small EM, Jeong MY, Walker LA, Xu H, Sniadecki NJ, Taylor MR, Buttrick PM, Song K (2019) LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion. Proc Natl Acad Sci U S A 116(2):556–565. https://doi.org/10.1073/pnas.1808618116
Crielaard BJ, Lammers T, Rivella S (2017) Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discov 16(6):400–423. https://doi.org/10.1038/nrd.2016.248
Dai C, Chen X, Li J, Comish P, Kang R, Tang D (2020a) Transcription factors in Ferroptotic cell death. Cancer Gene Therapy 27:645. https://doi.org/10.1021/es026208x
Dai E, Meng L, Kang R, Wang X, Tang D (2020b) ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun 522(2):415–421. https://doi.org/10.1016/j.bbrc.2019.11.110
Dai E, Zhang W, Cong D, Kang R, Wang J, Tang D (2020c) AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun 523(4):966–971. https://doi.org/10.1016/j.bbrc.2020.01.066
Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19(6):349–364. https://doi.org/10.1038/s41580-018-0003-4
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, Stockwell BR (2015) Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol 10(7):1604–1609. https://doi.org/10.1021/acschembio.5b00245
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, Mourão A, Buday K, Sato M, Wanninger J, Vignane T, Mohana V, Rehberg M, Flatley A, Schepers A, Kurz A, White D, Sauer M, Sattler M, Tate EW, Schmitz W, Schulze A, O'Donnell V, Proneth B, Popowicz GM, Pratt DA, Angeli JPF, Conrad M (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575(7784):693–698. https://doi.org/10.1038/s41586-019-1707-0
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D'Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, Garcia-Saez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jaattela M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Munoz-Pinedo C, Nagata S, Nunez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G (2018) Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 25(3):486–541. https://doi.org/10.1038/s41418-017-0012-4
Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, Dai E (2018) Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun 503(3):1550–1556. https://doi.org/10.1016/j.bbrc.2018.07.078
Han L, Bai L, Fang X, Liu J, Kang R, Zhou D, Tang D, Dai E (2021) SMG9 drives ferroptosis by directly inhibiting GPX4 degradation. Biochem Biophys Res Commun 567:92–98. https://doi.org/10.1016/j.bbrc.2021.06.038
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, Tang D (2016) Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12(8):1425–1428. https://doi.org/10.1080/15548627.2016.1187366
Houessinon A, Francois C, Sauzay C, Louandre C, Mongelard G, Godin C, Bodeau S, Takahashi S, Saidak Z, Gutierrez L, Regimbeau JM, Barget N, Barbare JC, Ganne N, Chauffert B, Coriat R, Galmiche A (2016) Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol Cancer 15(1):38. https://doi.org/10.1186/s12943-016-0526-2
Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y, Li X, Sho T, Wang X, Li Y, Wu YT, Wei YH, Hu X, Yu L (2021) Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell 184(11):2896–2910.e2813. https://doi.org/10.1016/j.cell.2021.04.027
Kang R, Zeh HJ, Lotze MT, Tang D (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18(4):571–580. https://doi.org/10.1038/cdd.2010.191
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, Cao L, Xie M, Ran Q, Kroemer G, Wang H, Billiar TR, Jiang J, Tang D (2018a) Lipid peroxidation drives Gasdermin D-mediated Pyroptosis in lethal Polymicrobial sepsis. Cell Host Microbe 24(1):97–108e104. https://doi.org/10.1016/j.chom.2018.05.009
Kang R, Zhu S, Zeh HJ, Klionsky DJ, Tang D (2018b) BECN1 is a new driver of ferroptosis. Autophagy 14(12):2173–2175. https://doi.org/10.1080/15548627.2018.1513758
Kaushik S, Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19(6):365–381. https://doi.org/10.1038/s41580-018-0001-6
Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, Sajjakulnukit P, Myers A, Thurston G, Hou SW, Carpenter ES, Andren AC, Nwosu ZC, Cusmano N, Wisner S, Mbah NE, Shan M, Das NK, Magnuson B, Little AC, Savani MR, Ramos J, Gao T, Sastra SA, Palermo CF, Badgley MA, Zhang L, Asara JM, McBrayer SK, di Magliano MP, Crawford HC, Shah YM, Olive KP, Lyssiotis CA (2021) GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun 12(1):4860. https://doi.org/10.1038/s41467-021-24859-2
Kriel J, Loos B (2019) The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ 26:640. https://doi.org/10.1038/s41418-018-0267-4
Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40(2):280–293. https://doi.org/10.1016/j.molcel.2010.09.023
Kuang F, Liu J, Kang R, Tang D (2020) Oxidative damage and antioxidant defense in Ferroptosis. Front Cell Dev Biol 8:586578. https://doi.org/10.3389/fcell.2020.586578
Lahiri V, Metur SP, Hu Z, Song X, Mari M, Hawkins WD, Bhattarai J, Delorme-Axford E, Reggiori F, Tang D, Dengjel J, Klionsky DJ (2021) Post-transcriptional regulation of ATG1 is a critical node that modulates autophagy during distinct nutrient stresses. Autophagy 18:1694. https://doi.org/10.1080/15548627.2021.1997305
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524(7565):309–314. https://doi.org/10.1038/nature14893
Lee J, You JH, Roh JL (2022) Poly(rC)-binding protein 1 represses ferritinophagy-mediated ferroptosis in head and neck cancer. Redox Biol 51:102276. https://doi.org/10.1016/j.redox.2022.102276
Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D (2021a) Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy 17(4):948–960. https://doi.org/10.1080/15548627.2020.1739447
Li J, Chen X, Kang R, Zeh H, Klionsky DJ, Tang D (2021b) Regulation and function of autophagy in pancreatic cancer. Autophagy 17(11):3275–3296. https://doi.org/10.1080/15548627.2020.1847462
Li J, Liu J, Xu Y, Wu R, Chen X, Song X, Zeh H, Kang R, Klionsky DJ, Wang X, Tang D (2021c) Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 17(11):3361–3374. https://doi.org/10.1080/15548627.2021.1872241
Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, Wang L, Liang G (2016) Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett 381(1):165–175. https://doi.org/10.1016/j.canlet.2016.07.033
Lin Z, Liu J, Kang R, Yang M, Tang D (2021) Lipid metabolism in ferroptosis. Adv Biol (Weinh) 5(8):e2100396. https://doi.org/10.1002/adbi.202100396
Lin Z, Liu J, Long F, Kang R, Kroemer G, Tang D, Yang M (2022) The lipid flippase SLC47A1 blocks metabolic vulnerability to ferroptosis. Nat Commun 13(1):7965. https://doi.org/10.1038/s41467-022-35707-2
Liu J, Yang M, Kang R, Klionsky DJ, Tang D (2019) Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy 15(11):2033–2035. https://doi.org/10.1080/15548627.2019.1659623
Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D (2020a) Autophagy-dependent Ferroptosis: machinery and regulation. Cell Chem Biol 27(4):420–435. https://doi.org/10.1016/j.chembiol.2020.02.005
Liu Y, Wang Y, Liu J, Kang R, Tang D (2020b) The circadian clock protects against ferroptosis-induced sterile inflammation. Biochem Biophys Res Commun 525(3):620–625. https://doi.org/10.1016/j.bbrc.2020.02.142
Liu J, Kang R, Tang D (2021a) ESCRT-III-mediated membrane repair in cell death and tumor resistance. Cancer Gene Ther 28(1–2):1–4. https://doi.org/10.1038/s41417-020-0200-0
Liu J, Kang R, Tang D (2021b) Signaling pathways and defense mechanisms of ferroptosis. FEBS J 289:7038. https://doi.org/10.1111/febs.16059
Liu J, Zhu S, Zeng L, Li J, Klionsky DJ, Kroemer G, Jiang J, Tang D, Kang R (2021c) DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy 18:2036. https://doi.org/10.1080/15548627.2021.2008692
Liu Y, Wang Y, Liu J, Kang R, Tang D (2021d) Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther 28(1–2):55–63. https://doi.org/10.1038/s41417-020-0182-y
Liu J, Liu Y, Wang Y, Li C, Xie Y, Klionsky DJ, Kang R, Tang D (2022a) TMEM164 is a new determinant of autophagy-dependent ferroptosis. Autophagy 19:945. https://doi.org/10.1080/15548627.2022.2111635
Liu Z, Ma C, Wang Q, Yang H, Lu Z, Bi T, Xu Z, Li T, Zhang L, Zhang Y, Liu J, Wei X, Li J (2022b) Targeting FAM134B-mediated reticulophagy activates sorafenib-induced ferroptosis in hepatocellular carcinoma. Biochem Biophys Res Commun 589:247–253. https://doi.org/10.1016/j.bbrc.2021.12.019
Maan M, Peters JM, Dutta M, Patterson AD (2018) Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun 504(3):582–589. https://doi.org/10.1016/j.bbrc.2018.02.097
Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, Olzmann JA, Dixon SJ (2019) Exogenous monounsaturated fatty acids promote a Ferroptosis-resistant cell state. Cell Chem Biol 26(3):420–432e429. https://doi.org/10.1016/j.chembiol.2018.11.016
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, Poyurovsky MV, Olszewski K, Gan B (2021a) DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593(7860):586–590. https://doi.org/10.1038/s41586-021-03539-7
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, Poyurovsky MV, Olszewski K, Gan B (2021b) DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593:586. https://doi.org/10.1038/s41586-021-03539-7
Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, Tonnus W, Nepachalovich P, Eggenhofer E, Aldrovandi M, Henkelmann B, Yamada KI, Wanninger J, Zilka O, Sato E, Feederle R, Hass D, Maida A, Mourao ASD, Linkermann A, Geissler EK, Nakagawa K, Abe T, Fedorova M, Proneth B, Pratt DA, Conrad M (2022) A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608(7924):778–783. https://doi.org/10.1038/s41586-022-05022-3
Mizushima N (2010) The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22(2):132–139. https://doi.org/10.1016/j.ceb.2009.12.004
Nakatogawa H (2013) Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem 55:39–50. https://doi.org/10.1042/bse0550039
Oakes SA, Papa FR (2015) The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 10:173–194. https://doi.org/10.1146/annurev-pathol-012513-104649
Ouyang S, Li H, Lou L, Huang Q, Zhang Z, Mo J, Li M, Lu J, Zhu K, Chu Y, Ding W, Zhu J, Lin Z, Zhong L, Wang J, Yue P, Turkson J, Liu P, Wang Y, Zhang X (2022) Inhibition of STAT3-ferroptosis negative regulatory axis suppresses tumor growth and alleviates chemoresistance in gastric cancer. Redox Biol 52:102317. https://doi.org/10.1016/j.redox.2022.102317
Rademaker G, Boumahd Y, Peiffer R, Anania S, Wissocq T, Liegeois M, Luis G, Sounni NE, Agirman F, Maloujahmoum N, De Tullio P, Thiry M, Bellahcene A, Castronovo V, Peulen O (2022) Myoferlin targeting triggers mitophagy and primes ferroptosis in pancreatic cancer cells. Redox Biol 53:102324. https://doi.org/10.1016/j.redox.2022.102324
Rami A, Fekadu J, Rawashdeh O (2017) The hippocampal Autophagic machinery is depressed in the absence of the circadian clock protein PER1 that may Lead to vulnerability during cerebral ischemia. Curr Neurovasc Res 14(3):207–214. https://doi.org/10.2174/1567202614666170619083239
Rogov VV, Stolz A, Ravichandran AC, Rios-Szwed DO, Suzuki H, Kniss A, Löhr F, Wakatsuki S, Dötsch V, Dikic I, Dobson RC, McEwan DG (2017) Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep 18(8):1382–1396. https://doi.org/10.15252/embr.201643587
Rong Y, Fan J, Ji C, Wang Z, Ge X, Wang J, Ye W, Yin G, Cai W, Liu W (2021) USP11 regulates autophagy-dependent ferroptosis after spinal cord ischemia-reperfusion injury by deubiquitinating Beclin 1. Cell Death Differ 29:1164. https://doi.org/10.1038/s41418-021-00907-8
Schaaf MB, Keulers TG, Vooijs MA, Rouschop KM (2016) LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J 30(12):3961–3978. https://doi.org/10.1096/fj.201600698R
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26(7):1749–1760. https://doi.org/10.1038/sj.emboj.7601623
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2019) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 38(10). https://doi.org/10.15252/embj.2019101812
Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA (2015) The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 61(6):1896–1907. https://doi.org/10.1002/hep.27667
Shen Q, Shi Y, Liu J, Su H, Huang J, Zhang Y, Peng C, Zhou T, Sun Q, Wan W, Liu W (2021) Acetylation of STX17 (syntaxin 17) controls autophagosome maturation. Autophagy 17(5):1157–1169. https://doi.org/10.1080/15548627.2020.1752471
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, Zhang DD (2017) Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171(2):273–285. https://doi.org/10.1016/j.cell.2017.09.021
Sun X, Niu X, Chen R, He W, Chen D, Kang R, Tang D (2016a) Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 64(2):488–500. https://doi.org/10.1002/hep.28574
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D (2016b) Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63(1):173–184. https://doi.org/10.1002/hep.28251
Tang D, Kroemer G (2020) Ferroptosis. Curr Biol 30:R1–R6. https://doi.org/10.3760/cma.j.issn.1001-9391.2019.10.002
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G (2019) The molecular machinery of regulated cell death. Cell Res 29(5):347–364. https://doi.org/10.1038/s41422-019-0164-5
Tang D, Chen X, Kang R, Kroemer G (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31(2):107–125. https://doi.org/10.1038/s41422-020-00441-1
Tian X, Zheng P, Zhou C, Wang X, Ma H, Ma W, Zhou X, Teng J, Chen J (2020) DIPK2A promotes STX17- and VAMP7-mediated autophagosome-lysosome fusion by binding to VAMP7B. Autophagy 16(5):797–810. https://doi.org/10.1080/15548627.2019.1637199
Tian X, Teng J, Chen J (2021) New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 17(10):2680–2688. https://doi.org/10.1080/15548627.2020.1823124
Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM (2018) Iron and cancer. Annu Rev Nutr 38:97–125. https://doi.org/10.1146/annurev-nutr-082117-051732
Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T (2022) The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol 24:167. https://doi.org/10.1038/s41580-022-00542-2
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, Viswanathan SR, Chattopadhyay S, Tamayo P, Yang WS, Rees MG, Chen S, Boskovic ZV, Javaid S, Huang C, Wu X, Tseng YY, Roider EM, Gao D, Cleary JM, Wolpin BM, Mesirov JP, Haber DA, Engelman JA, Boehm JS, Kotz JD, Hon CS, Chen Y, Hahn WC, Levesque MP, Doench JG, Berens ME, Shamji AF, Clemons PA, Stockwell BR, Schreiber SL (2017) Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547(7664):453–457. https://doi.org/10.1038/nature23007
Wang L, Klionsky DJ, Shen HM (2022) The emerging mechanisms and functions of microautophagy. Nat Rev Mol Cell Biol 24:186. https://doi.org/10.1038/s41580-022-00529-z
Wen Q, Liu J, Kang R, Zhou B, Tang D (2019) The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun 510(2):278–283. https://doi.org/10.1016/j.bbrc.2019.01.090
Wittwer MB, Zur AA, Khuri N, Kido Y, Kosaka A, Zhang X, Morrissey KM, Sali A, Huang Y, Giacomini KM (2013) Discovery of potent, selective multidrug and toxin extrusion transporter 1 (MATE1, SLC47A1) inhibitors through prescription drug profiling and computational modeling. J Med Chem 56(3):781–795. https://doi.org/10.1021/jm301302s
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, Shan B, Pan H, Yuan J (2019) Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A 116(8):2996–3005. https://doi.org/10.1073/pnas.1819728116
Xie Y, Kang R, Sun X, Zhong M, Huang J, Klionsky DJ, Tang D (2015) Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 11(1):28–45. https://doi.org/10.4161/15548627.2014.984267
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D (2016) Ferroptosis: process and function. Cell Death Differ 23(3):369–379. https://doi.org/10.1038/cdd.2015.158
Xie Y, Li J, Kang R, Tang D (2020a) Interplay between lipid metabolism and autophagy. Front Cell Dev Biol 8:431. https://doi.org/10.3389/fcell.2020.00431
Xie Y, Liu J, Kang R, Tang D (2020b) Mitophagy receptors in tumor biology. Front Cell Dev Biol 8:594203. https://doi.org/10.3389/fcell.2020.594203
Xie Y, Liu J, Kang R, Tang D (2021) Mitophagy in pancreatic cancer. Front. Oncologia 11:616079. https://doi.org/10.3389/fonc.2021.616079
Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ, Kroemer G, Chen X, Tang D, Liu J (2023) Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 19:1982. https://doi.org/10.1080/15548627.2023.2165323
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156(1–2):317–331. https://doi.org/10.1016/j.cell.2013.12.010
Yang M, Chen P, Liu J, Zhu S, Kroemer G, Klionsky DJ, Lotze MT, Zeh HJ, Kang R, Tang D (2019) Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv 5(7):eaaw2238. https://doi.org/10.1126/sciadv.aaw2238
Yang L, Chen X, Yang Q, Chen J, Huang Q, Yao L, Yan D, Wu J, Zhang P, Tang D, Zhong N, Liu J (2020) Broad Spectrum Deubiquitinase inhibition induces both apoptosis and Ferroptosis in cancer cells. Front Oncol 10:949. https://doi.org/10.3389/fonc.2020.00949
Yang L, Ye F, Liu J, Klionsky DJ, Tang D, Kang R (2022) Extracellular SQSTM1 exacerbates acute pancreatitis by activating autophagy-dependent ferroptosis. Autophagy 19:1733. https://doi.org/10.1080/15548627.2022.2152209
Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA (2003) The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med 34(4):496–502. https://doi.org/10.1016/s0891-5849(02)01360-6
Yu F, Zhang Q, Liu H, Liu J, Yang S, Luo X, Liu W, Zheng H, Liu Q, Cui Y, Chen G, Li Y, Huang X, Yan X, Zhou J, Chen Q (2022) Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discovery 8(1):40. https://doi.org/10.1038/s41421-022-00390-6
Yuan H, Li X, Zhang X, Kang R, Tang D (2016) Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478(3):1338–1343. https://doi.org/10.1016/j.bbrc.2016.08.124
Zhang Q, Kang R, Zeh HJ 3rd, Lotze MT, Tang D (2013) DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy 9(4):451–458. https://doi.org/10.4161/auto.23691
Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A, Zhang F, Zheng S (2018) Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 14(12):2083–2103. https://doi.org/10.1080/15548627.2018.1503146
Zhang Z, Guo M, Li Y, Shen M, Kong D, Shao J, Ding H, Tan S, Chen A, Zhang F, Zheng S (2020) RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 16(8):1482–1505. https://doi.org/10.1080/15548627.2019.1687985
Zhang R, Kang R, Klionsky DJ, Tang D (2022) Ion channels and transporters in autophagy. Autophagy 18(1):4–23. https://doi.org/10.1080/15548627.2021.1885147
Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D (2020) Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol 66:89–100. https://doi.org/10.1016/j.semcancer.2019.03.002
Zhou Y, Tao L, Zhou X, Zuo Z, Gong J, Liu X, Zhou Y, Liu C, Sang N, Liu H, Zou J, Gou K, Yang X, Zhao Y (2021) DHODH and cancer: promising prospects to be explored. Cancer Metab 9(1):22. https://doi.org/10.1186/s40170-021-00250-z
Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, Tang D (2017) HSPA5 regulates Ferroptotic cell death in cancer cells. Cancer Res 77(8):2064–2077. https://doi.org/10.1158/0008-5472.can-16-1979
Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, Schreiber SL (2020) Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 16(3):302–309. https://doi.org/10.1038/s41589-020-0472-6
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
The authors declare no competing interests.
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kang, R., Tang, D. (2023). The Role of Autophagy in Ferroptosis. In: Tang, D. (eds) Ferroptosis in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-031-39171-2_4
Download citation
DOI: https://doi.org/10.1007/978-3-031-39171-2_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-39170-5
Online ISBN: 978-3-031-39171-2
eBook Packages: MedicineMedicine (R0)