
Abstract
Acute adrenal emergencies are rare but potentially life-threatening conditions with associated high morbidity and mortality if unrecognized or untreated. Endocrine emergencies related to the adrenal gland present in the traumatic or nontraumatic setting. Hemorrhage may occur in the setting of trauma or may be due to severe illness or tumor rupture. Adrenal tumors such as an adrenal adenoma, adrenocortical carcinoma, pheochromocytoma, or ectopic tumors may result in a crisis state. High clinical suspicion and prompt initiation of treatment are critical in the diagnosis and management of adrenal emergencies. In general, if a high index of suspicion occurs in the setting of an adrenal emergency, treatment should not be delayed for diagnostic confirmation. This chapter outlines the medical and surgical interventions for these rare but life-threatening emergencies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Adrenal injury in trauma
- Adrenal hemorrhage
- Acute adrenal crisis
- Pheochromocytoma crisis
- Acute
- Severe hypercortisolism
2.1 Introduction
Adrenal emergencies occur in both the traumatic and nontraumatic settings. Adrenal gland hemorrhage may be a direct result of blunt or penetrating trauma, whereas nontraumatic hemorrhage may occur from spontaneous rupture of an adrenal gland tumor or in the setting of acute illness or coagulopathy. Additionally, patients with known tumor pathology such as a pheochromocytoma or Cushing’s syndrome are at risk for developing rare but life-threatening adrenal crises. The clinical manifestations and the degree of hemodynamic stability are key factors in determining the appropriate evaluation and intervention.
2.2 Adrenal Hemorrhage
Adrenal hemorrhage occurs in the traumatic and nontraumatic setting. Traumatic adrenal hemorrhage is usually discovered on computed tomography (CT) imaging during routine trauma workup and is very rarely an isolated finding. Traumatic adrenal hemorrhage is present in approximately 0.5% of blunt trauma cases and very rarely occurs in penetrating trauma. Recent studies have demonstrated that the presence of blunt adrenal gland injury is not a marker of severe injury or associated with increased mortality rate [1]. Blunt adrenal trauma is usually seen in high-impact mechanisms and is typically associated with other intra-abdominal injuries. Patient presentation may vary widely from hemodynamically stable to critically ill depending on other associated injuries.
Nontraumatic adrenal hemorrhage may present in the setting of ruptured adrenal tumor, illness, and coagulopathies including antiphospholipid antibody syndrome. Tumors associated with spontaneous hemorrhage include pheochromocytoma, myelolipoma, metastasis, carcinoma, and adenoma. Patients may present in hemorrhagic shock associated with flank pain and fever. Typically, unilateral hemorrhage is seen in tumors and blunt trauma, whereas bilateral hemorrhage may be seen in the setting of acute illness and coagulopathies. The evaluation and management of adrenal hemorrhage are based on the clinical presentation and stability of the patient.
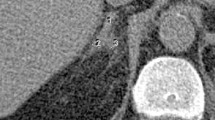
Adrenal hemorrhage often presents with nonspecific symptoms including abdominal, flank, or loin pain as well as nausea, vomiting, weakness, and lethargy, making the diagnosis challenging. In cases of severe hemorrhage, symptoms of adrenal insufficiency may be present. Adrenal hemorrhage on CT scan appears as a round-to-ovoid lesion [2]. In the setting of acute adrenal hemorrhage, peri-adrenal fat stranding and bleeding into the perinephric space may be present (Fig. 2.1a). Attenuation is dependent on the age of the hematoma with acute hematomas having high attenuation. Adrenal congestion (adrenal gland thickening, peri-adrenal fat stranding) on CT may indicate impending adrenal hemorrhage [3]. Laboratory evaluation ranges from patients with normal laboratory parameters to those with leukocytosis and anemia as well as evidence of adrenal insufficiency including hyponatremia and hyperkalemia. Derangements in levels of cortisol, ACTH, and catecholamine levels may be present and aid in identifying the extent of gland destruction/involvement.

(a) CT images of bilateral adrenal myelolipomas; the left has fat stranding and active extravasation of contrast suggestive of acute hemorrhage. (b) Angiography of the same adrenal tumor showing two areas of active hemorrhage
In a hemodynamically unstable patient with concerns for acute blood loss anemia and expanding hemorrhage, it is advisable for admission to an intensive care unit for close hemodynamic monitoring, blood product resuscitation, serial labs, and arterial embolization by interventional radiology. Angioembolization with interventional radiology is first-line therapy [4] (Fig. 2.1b). However, if angioembolization is not feasible or fails, proceeding to the operating room for an emergent adrenalectomy is the next best course of action.
For adrenal hemorrhage in the clinically stable patient (regardless of traumatic vs. nontraumatic etiology) identified on imaging, conservative management with admission for observation, IV fluid resuscitation, monitoring labs, and serial abdominal exams is advisable. Patients who have had a nontraumatic hemorrhage should ultimately be referred to an experienced adrenal surgeon for elective adrenalectomy, particularly in large tumors at risk for rebleeding (Fig. 2.2a, b).

(a) CT image of a giant left adrenal myelolipoma that had bled about 6 months prior to obtaining this CT. (b) Giant left adrenal myelolipoma surgical specimen
In the setting of trauma, when emergent exploratory laparotomy is indicated for other reasons and an adrenal injury is incidentally discovered during surgical exploration, the decision of whether to repair versus proceed with resection should be based on the clinical status of the patient, viability of the adrenal tissue, and presence of a contralateral gland [5].

2.3 Pheochromocytoma
Pheochromocytoma is a rare catecholamine-secreting tumor of the adrenal gland, and symptoms manifest due to excessive catecholamine release. Symptoms may occur at any age, although the sporadic form is most common in the fourth and fifth decades. However, hereditary pheochromocytoma may present in younger patients with persistently elevated blood pressure despite maximal medical therapy. This may be seen in the setting of associated genetic syndromes such as multiple endocrine neoplasia (MEN) type 2, neurofibromatosis type 1, tuberous sclerosis, and ataxia telangiectasia. The classic triad of symptoms associated with a pheochromocytoma consists of episodic headache, sweating, and tachycardia, whereas sustained or paroxysmal hypertension is the most common sign of a pheochromocytoma. Patients with pheochromocytomas are at risk for developing a pheochromocytoma crisis or pheochromocytoma multisystem crisis. Crises typically present as a hypertensive emergency and may be associated with multiorgan failure and cardiopulmonary collapse. It is important to recognize that while the classic presentation is a hypertensive emergency, patients may also present with severe hypotension. Other manifestations include metabolic derangements, encephalopathy, and hyperthermia. Pheochromocytoma crisis may be triggered by acute stress (mechanical or psychological) or may be drug induced (glucocorticoids, dopamine receptor antagonist, opioids, norepinephrine reuptake inhibitors, neuromuscular blocking agents) [6]. Moreover, patients with a known pheochromocytoma are at risk for tumor rupture resulting in potential life-threatening adrenal hemorrhage and subsequent transient hypocortisolism.
Pheochromocytoma crisis should be considered in patients who present in a hypertensive crisis with associated relatively vague complaints of abdominal pain, headache, and tachycardia, and suspicion should be raised in those with laboratory values demonstrating end-organ damage. While 24-h urine metanephrine and plasma metanephrines are diagnostic for a pheochromocytoma and should be collected in the emergent setting, treatment of a presumed pheochromocytoma crisis should not be delayed for diagnostic confirmation. Imaging evaluation with CT abdomen and pelvis may demonstrate a large heterogeneous mass on the adrenal gland, and the gland itself may have evidence of hemorrhagic changes (Fig. 2.3a). Pertinent relevant history including refractory blood pressure control despite optimal medical management in conjunction with imaging findings concerning for an adrenal mass raises suspicion for a pheochromocytoma crisis. In the inpatient setting, it is important to consider a pheochromocytoma multisystem crisis (hypertension, hypothermia, and encephalopathy) in critically ill patients with concomitant comorbidities, evidence of end-organ damage, and hypertension refractory to typical medical management [7].

(a) CT scan showing right adrenal mass that was a pheochromocytoma. (b) Surgical specimen of the pheochromocytoma after it was removed
Pheochromocytoma crisis may result in severe cardiovascular collapse, pulmonary edema, and acute respiratory failure. Unfortunately, laboratory confirmation requires an extended period of time, and no rapid testing has yet to be established. In patients with a high index of suspicion for pheochromocytoma crisis, immediate and rapid treatment of blood pressure control should be initiated with a combination of alpha-blockade (phentolamine), calcium channel blockade (nicardipine), and direct vasodilators (nitroprusside). Beta-blockade may be initiated for residual/refractory tachycardia only once adequate alpha-blockade has been achieved as unopposed alpha-adrenergic activity can lead to cardiovascular collapse.
Despite maximal medical intervention, patients may continue to clinically deteriorate. In hemodynamically unstable patients who continue to deteriorate clinically, despite maximal medical treatment, consideration should be given to proceeding to the operating room for an emergent adrenalectomy [8] (Fig. 2.3b). Postoperatively, patients should be carefully monitored for transient hypocortisolism and subsequently treated with intravenous hydrocortisone as needed, and medical endocrinology consult is advised.


2.4 Hypercortisolism
Prolonged tissue exposure to grossly elevated concentrations of glucocorticoids, either from an endogenous or an exogenous source, may result in hypercortisolism and in severe cases constitutes an acute emergency. Exogenous sources include oral steroid medications, whereas endogenous sources may be caused by ACTH-secreting pituitary adenoma (Cushing’s disease), benign adrenal lesions (adrenal adenoma, micronodular hyperplasia), malignant adrenal lesions (adrenocortical carcinoma), or ectopic ACTH secretion tumors (small-cell carcinoma). Cushing’s disease is the most frequent cause of endogenous Cushing’s syndrome and is characterized by secretion of cortisol secondary to an underlying pituitary adenoma. However, acute, severe clinical presentations of hypercortisolism are more typical of ectopic ACTH-secreting lesions (secretion of ACTH by a non-pituitary tumor) and account for approximately 20% of dependent Cushing’s syndrome. The rapid control of severe cortisol excess is crucial and lifesaving. Patients with ectopic ACTH-secreting lesions in acute, hypercortisolism crises present with hypokalemia, hypertension, hyperglycemia, and muscle weakness.
The definition of acute, severe hypercortisolism is fluid and should be considered in the context of laboratory abnormalities, presenting symptoms, past history, and clinical exam. Severe hypercortisolism may present in conjunction with the onset of other acute morbidities including sepsis, heart failure, gastrointestinal hemorrhage, thromboembolism, myopathy, opportunistic infections, or ketoacidosis. Laboratory abnormalities of hyperglycemia, hypokalemia, and metabolic alkalosis with concomitant hypertension should raise suspicion for hypercortisolism, particularly in patients with established diagnoses known to elevate circulating glucocorticoids (i.e., Cushing’s syndrome, adrenocortical carcinoma, or ectopic paraneoplastic). However, patients may present with no prior history and relatively vague complaints. A broad differential and workup should be initiated including comprehensive laboratory evaluation with comprehensive metabolic panel, complete blood count, serum cortisol and ACTH, as well as multiphase CT imaging. Imaging findings must be taken into consideration, particularly those with no previously established diagnoses but with other clinical evidence concerning severe hypercortisolism. Ectopic ACTH-secreting tumors may be seen as lesions in the adrenal glands, lung, or pancreas.
Severe hypercortisolism is associated with a random serum cortisol higher than 40 μg/dL (normal range 5–25 μg/dL) or a 24-h urine free cortisol more than four times the upper limit of normal (normal range 10–100 mcg/24). This is often seen in the setting of severe hypokalemia (defined as a serum potassium of <3 mmol/L) (normal range 3.6–5.2 mmol/L). In the setting of acute, severe hypercortisolism, the classic features of Cushing’s syndrome are not evident; however, significant metabolic/electrolyte disturbances are apparent on laboratory analysis [9]. These laboratory abnormalities coupled with imaging findings and presenting symptoms should raise a high level of suspicion for severe hypercortisolism. Marked increase in serum cortisol and adrenocorticotropic hormone with other associated laboratory abnormalities in conjunction with concerning imaging findings should prompt rapid treatment. If the diagnosis of severe hypercortisolism is presumed, it is imperative to immediately initiate treatment and to not delay for diagnostic confirmation.
Treatment includes the rapid reduction of cortisol, correction of metabolic derangements, and management of any concomitant comorbidities. Rapid correction of cortisol should be initiated with oral ketoconazole and/or metyrapone. If enteral access is not available or if appropriate cortisol levels are not able to be achieved with oral medications, intravenous etomidate has been shown to be effective and safe in reducing elevated cortisol levels. Patients treated with etomidate should be carefully monitored in the intensive care unit with sedation scores, continuous hemodynamic monitoring, and serial serum cortisol to closely monitor levels (every 4–6 h) [10,11,12].
Intervention is directed at addressing the etiology of the severe hypercortisolism with optimal therapy being complete resection of the cortisol-secreting tumor or ACTH-secreting tumor. In the setting of severe hypercortisolism, admission to intensive care unit with close hemodynamic monitoring and rapid correction of elevated cortisol and electrolyte abnormalities with medical therapy should be initiated. Once the acute crisis is medically stabilized, then further treatment with either prolonged medical therapy or surgery may be initiated. Transsphenoidal surgery is the treatment of choice for ACTH-producing pituitary adenoma when a clearly circumscribed adenoma is identified. In patients with hypercortisolism secondary to ACTH secretion (pituitary adenoma or ectopic ACTH tumor) where the tumor is not resectable or non-localized, some patients may benefit from bilateral adrenalectomy, but careful patient selection and a multidisciplinary approach are required for this decision as medical management of lack of adrenal function is challenging.
In patients with primary adrenal disease, treatment is directed at removal of the adrenal gland or glands. Adrenal tumors should be removed with unilateral adrenalectomy, while bilateral adrenalectomy is indicated for bilateral micronodular and macronodular adrenal hyperplasia [13]. Unilateral tumors incidentally discovered on the adrenal gland should never be biopsied to determine etiology; rather, they should be removed with an oncologic reaction (en bloc with any nodes/invading structures) (Fig. 2.4a, b). In patients who continue to clinically deteriorate despite maximal medical therapy, emergent adrenalectomy may be considered as a lifesaving measure [14]. Finally, for tumors that are unable to be resected due to local invasion, palliative debulking may still be indicated to improve patient quality of life.

(a) CT scan of a right adrenal tumor in a patient with severe Cushing’s symptoms. (b) Surgical specimen of the same right adrenal tumor that turned out to be a low-grade adrenal cortical cancer
Either open or laparoscopic surgical approach is reasonable. While the laparoscopic approach is associated with less postoperative morbidity and mortality, ultimately surgical intervention should be at the discretion of the comfort level of the operative surgeon. Additionally, it is important to recognize that adrenal crisis is an emergent complication of bilateral adrenalectomy. Any patient who has bilateral adrenalectomy should wear a medic alert bracelet in case of trauma or other crises, as patients are unable to mount an acute stress response to trauma or severe illness. All patients receiving medical or surgical intervention should be monitored for adrenal insufficiency and treated accordingly with intravenous hydrocortisone with subsequent taper to oral steroids.


2.5 Adrenal Crisis (Acute Adrenal Insufficiency)
Adrenal insufficiency results from a deficiency of adrenal cortisol production and is categorized as primary, secondary, or tertiary adrenal insufficiency. Primary adrenal insufficiency arises from a direct insult/failure of the adrenal gland. Secondary and tertiary adrenal insufficiency are due to disorders of the pituitary or hypothalamus. Adrenal crisis (acute adrenal insufficiency) most commonly occurs in patients with primary adrenal insufficiency; however, it may also be seen in those with secondary and tertiary adrenal insufficiency [15]. Adrenal crisis is a life-threatening emergency associated with significant morbidity and mortality and requires prompt treatment.
Primary adrenal insufficiency may occur with bilateral adrenal hemorrhage, bilateral adrenalectomy, congenital hyperplasia, drug-induced adrenal enzyme inhibition (i.e., mitotane, ketoconazole, and metyrapone), etomidate when continuously infused or frequently dosed, as well as other pharmacologic agents such as carbamazepine, phenytoin, and phenobarbitone [16]. Adrenal hemorrhage may occur in the setting of trauma, or spontaneously ruptured adrenal tumor. Although less common, acute adrenal insufficiency may occur in secondary and tertiary adrenal insufficiency and is typically precipitated by acute stress, pituitary infarct, or traumatic brain injury or following surgical cure of Cushing’s syndrome. Additionally, patients on exogenous steroids (prednisone 5 mg/day or equivalent for 4 weeks or longer across all routes of administration—oral, optical, inhaled, or intranasal) are at increased risk for adrenal insufficiency, and abrupt withdrawal from exogenous glucocorticoids may provoke adrenal crisis.
The clinical features of adrenal crisis include hypotension and hypovolemia with laboratory abnormalities of hyperkalemia and hyponatremia. However, patients often present with vague, nonspecific symptoms such as nausea, vomiting, fever, abdominal pain, fatigue, and altered consciousness.
In patients with a suspected adrenal crisis, prompt initiation of treatment is crucial. Serum chemistry, blood serum cortisol, ACTH, renin, and aldosterone should be collected as part of the initial evaluation; however, treatment should not be delayed for diagnostic confirmatory tests. Rapid correction of hypocortisolism can be done with bolus injection of 100 mg IV hydrocortisone (intramuscular may be substituted pending intravenous access) followed by 200 mg hydrocortisone per 24 h (continuous IV infusion or 50 mg of hydrocortisone intravenous injection every 6 h). Concurrently, rehydration should be started with isotonic saline or 5% dextrose in isotonic saline via continuous IV and titrated based on volume status/urine output. Patients should be admitted to an intensive care unit for close monitoring of vitals and serial serum chemistry laboratory tests to carefully monitor electrolyte abnormalities. Intravenous hydrocortisone replacement should continue at 50 mg every 6 h until the patient is clinically stable [16]. Following clinical stability (normalization of vitals, ability to tolerate oral intake/medication), parenteral glucocorticoid therapy may be tapered over 24–72 h and transitioned to oral stress or maintenance dose. Once the patient is clinically stable, tapering of intravenous hydrocortisone to replacement doses can be initiated within 24–72 h. Patients with primary adrenal insufficiency should additionally receive mineralocorticoid replacement when the total daily hydrocortisone dose is lower than 50 mg/24 h. Additionally, once a patient has stabilized, precipitating causes of the adrenal crisis should be investigated and appropriately treated.
It is important to note that all patients with adrenal insufficiency should take emergency precautions by wearing a medical alert bracelet/necklace as well as carry an emergency card with the diagnosis, daily medication, and doses listed. Patients and family/support members should be educated on the use of injectable glucocorticoids in the setting of an emergency and be instructed to inject the medication if symptoms of acute adrenal insufficiency occur, nausea/vomiting and inability to tolerate oral intake/medications, or if the patient is found unresponsive. Following medication administration, the family/support person(s) should seek medical help immediately [16].

2.6 Conclusion
Adrenal emergencies are rare but potentially life-threatening diagnoses that present a challenge in the acute care setting. Due to their often vague presentation, a high index of suspicion for diagnoses is required. History, presentation, imaging, and laboratory values are helpful in aiding the clinician to ascertain the correct diagnosis. However, in the setting of a potentially life-threatening adrenal emergency, if the index of suspicion is high, initiation of treatment should not be delayed for diagnostic confirmation. Rapid intervention and treatment may be lifesaving in adrenal emergencies.
References
Di Gaiacomo JC, et al. Acute traumatic injuries of the adrenal gland: Pennsylvania trauma outcomes study registry. Trauma Surg Acute Care Open. 2020;15(5):e000487.
Bharucha T, Broderick C, Easom N, et al. Bilateral adrenal haemorrhage presenting as epigastric and back pain. JRSM Short Rep. 2012;3(3):15.
Godfrey RL, Clark J, Field B. Bilateral adrenal hemorrhagic infarction in patient with antiphospholipid syndrome. BMJ Case Rep. 2014;2014:bcr2014207050.
Madhusudhanan J, Robert JA, Sekar TV. Successful angioembolization for blunt adrenal gland trauma. Indian J Urol. 2020;36(1):59–61.
Gomez R, McAninch J, Carroll P. Adrenal gland trauma: diagnosis and management. J Trauma Injury Infect Crit Care. 1993;35(6):870–4.
Bartikoski S, Reschke D. Pheochromocytoma crisis in the emergency department. Cureus. 2021;13(3):e13683.
Katsura K, et al. Pheochromocytoma multisystem crisis treated with emergency surgery: a case report and literature review. BMC Res Notes. 2015;8:758.
Salinas C, Beltran O, Sanchez-Hidalgo J, et al. Emergency adrenalectomy due to acute heart failure secondary to complicated pheochromocytoma: a case report. World J Surg Oncol. 2011;9:49.
Lutgers HL, et al. Severe hypercortisolism: medical emergency requiring urgent intervention. Crit Care Med. 2010;38(7):1598–601.
Castinetti F, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab. 2014;99(5):1623–30.
Carroll TB, et al. Continuous etomidate infusion for the management of Severe Cushing Syndrome: validation of a standard protocol. J Endocr Soc. 2018;3(1):1–12.
Preda et at. Etomidate in the management of hypercortisolemia in Cushing’s syndrome: a review. Eur J Endocrinol. 2012;167(2):137–43.
Vieira Oberger Marques J, Boguszewski CL. Medical therapy in severe hypercortisolism. Best Pract Res Clin Endocrinol Metab. 2021;35(2):101487.
Davenport E, Lennard T. Acute hypercortisolism: what can the surgeon offer? Clin Endocrinol. 2014;81(4):498–502.
Huecker MR, Bhutta BS, Dominique E. Adrenal insufficiency. Treasure Island (FL): StatPearls; 2021.
Dineen R, Thompson C, Sherlock M. Adrenal crisis: prevention and management in adult patients. Ther Adv Endocrinol Metab. 2019;10:1–12.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Oberdoerster, M., Shahan, P., Elfenbein, D. (2023). Adrenal Emergencies in the Acute Care Setting. In: Tarasconi, A., Bui, S., Chirica, M., Roth, G., Nahmias, J. (eds) Oncologic Surgical Emergencies. Hot Topics in Acute Care Surgery and Trauma. Springer, Cham. https://doi.org/10.1007/978-3-031-36860-8_2
Download citation
DOI: https://doi.org/10.1007/978-3-031-36860-8_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-36859-2
Online ISBN: 978-3-031-36860-8
eBook Packages: MedicineMedicine (R0)