
Abstract
Fungi are key players in vital ecosystem services, spanning carbon cycling, decomposition, and varied plant symbioses. Due to their cryptic lifestyle, it was difficult to assess their diversity until the advent of methods of high-throughput sequencing. Based on the papers utilizing high-throughput sequencing approaches to study fungi in natural habitats using the nuclear ribosomal internal transcribed spacer 2 (ITS2) contained in the public open database GlobalFungi (https://globalfungi.com), the current estimate of global fungal diversity is 6.3 million species, considering 97% sequence similarity as a species-level threshold. Of the observed fungi, most belong to Ascomycota and Basidiomycota: 57% and 37% of taxa, respectively. Soil and litter represent the habitats with the highest alpha diversity of fungi followed by air, plant shoots, plant roots, and deadwood. Based on the high-throughput sequencing data, the highest proportion of unknown fungal species is associated with samples of lichen and plant tissues. Climate was identified as the key driver of fungal biogeography. In contrast to plants and most other taxa, fungal diversity in tropics appears to be lower than at high latitudes. Despite limitations, the use of high-throughput sequencing is an important tool for the assessment of diversity, biogeography, and ecology of fungi.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Fungi represent one of the most diverse groups of organisms in the world (Purvis and Hector 2000; Hawksworth 2001). However, the cryptic lifestyle of most fungal species makes estimates of global biodiversity challenging. Given their critical roles in many ecosystem processes (Peay et al. 2016), understanding the extent of global fungal biodiversity and the factors that determine it can provide crucial information about the stability and functioning of ecosystems. In the past, mycology has mostly relied on morphological characteristics, but early studies of amplified molecular markers derived from high-throughput sequencing (HTS) (Buéé et al. 2009; Amend et al. 2010; Baldrian et al. 2012) have suggested that traditional approaches greatly underestimate the diversity of fungi. The ability to recognize and classify fungi based on appropriate molecular markers obtained by DNA sequencing has opened up the opportunity to explore the composition and diversity of fungal communities (Koljalg et al. 2013), and HTS-based metabarcoding has demonstrated its scientific predictive power for studying global fungal diversity and biogeography (Tedersoo et al. 2014; Davison et al. 2015) as well as for elucidating regional drivers of fungal community diversity and composition (Talbot et al. 2014; Tedersoo et al. 2020b; Odriozola et al. 2021). Moreover, mycological efforts supported by HTS are able to address a wide range of questions regarding ecological and functional aspects of fungal communities (Baldrian et al. 2012; Peay et al. 2016; Nilsson et al. 2019a). Metabarcoding of fungal communities has become a well-established method readily available to the broader scientific community with a solid methodological backing (Lindahl et al. 2013; Větrovský and Baldrian 2013; Anslan et al. 2018; Nilsson et al. 2019a). As a result, several hundred papers have been published to date using high-throughput sequencing to analyse fungal community composition (Větrovský et al. 2020).
With the exception of a few large-scale attempts (Tedersoo et al. 2014; Davison et al. 2015), the power of HTS to determine the global diversity of fungi by assembling data from multiple studies is only now beginning to be exploited. This cumulative approach allows for more precise estimates of global fungal diversity (Větrovský et al. 2019; Větrovský et al. 2020; Baldrian et al. 2022).
2 Global Database of Fungal Metabarcoding Data
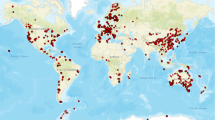
An appreciation of the number of studies that use metabarcoding to characterize fungal communities in terrestrial and aquatic communities has led to the creation of a community resource: a database of results from all available studies that use fungal internal transcribed spacer (ITS) as a metabarcoding marker—the GlobalFungi database (Větrovský et al. 2020). This curated database (https://globalfungi.com) is a public repository of metabarcoding data that continuously catalogues the fungal records generated by high-throughput sequencing studies (Fig. 10.1). To achieve the goal of making published data findable, accessible, interoperable, and reusable (FAIR), the web interface allows users to search the entire database or within a selected set of samples for individual sequences, fungal species hypotheses—groups of sequences that share similarity approximately corresponding to a species level created using the data of the UNITE database (Koljalg et al. 2013), species, or genera; obtain a visual representation of their distribution in the environment; and access and download sequence data and metadata (Fig. 10.1). In addition, the user interface allows authors to submit data from studies not yet included, thereby contributing to building a resource for the community of researchers in fungal systematics, biogeography, and ecology (Větrovský et al. 2020).

User interface of the GlobalFungi database (https://globalfungi.com) provides a quick yet complex access to the data in the GlobalFungi Database. It is possible to search for individual fungal taxa—genera, species, or Species Hypotheses (Taxon search) or to search for sequences and taxa by sequence similarity (Sequence search). The list of studies can be accessed through the Studies button and regional mycoflora can be retrieved using Geosearch. There is also the option to submit new data using the Submit your study option
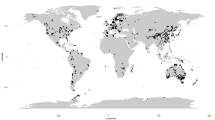
The GlobalFungi database includes high-throughput studies of fungal community composition published since 2009. All studies that fulfilled the following criteria are included: (1) the samples represent natural habitats that have not been experimentally treated in a laboratory or greenhouse, (2) the exact geographic location of each sample is available, (3) the primers used for amplicon sequencing were designed to target the entire fungal community (rather than subsets of it), (4) internal transcribed spacer regions (ITS1, ITS2, or both) were the subject of amplification, and (5) the sequencing data were publicly available. In its third release, the database covered up to 36,684 samples of fungal communities from 367 studies that contained over 1.1 billion sequence records from more than 200 million unique variants (Fig. 10.2).

Location of 36,684 samples covered in GlobalFungi database, release 3 (https://globalfungi.com)
3 Global Fungal Diversity Estimate
It was long in the past when scientists realized that fungi are extremely diverse. Since then, there have been repeated to estimate the total number of fungal species on Earth. The estimates were originally obtained by quantifying of host-specific fungi and multiplying them with putative number of fungal specialists per host (i.e. plant or insect) species (Hawksworth and Lücking 2017; Hyde et al. 2020). The high-throughput sequencing opened in the recent years another opportunity to assess the global fungal richness, and with increasing numbers of studies, the opportunities offered by this approach increase.
As GlobalFungi is currently the largest collection of fungal ITS marker data, it can serve as a data source for estimating global fungal diversity. Based on 97% sequence similarity clustering of full-length fungal ITS2 sequences, we recently identified more than a million fungal nonsingleton Operational Transcription Units (OTUs) that were already reported and are contained in the database. The total global diversity of fungi was estimated to be approximately 6.3 million OTUs based on the Chao1 estimate calculated from the entire dataset (Baldrian et al. 2022).
Although this estimate is significantly higher than some previously proposed estimates of global fungal diversity, e.g., 2.2 or 3.8 million species (Hawksworth and Lücking 2017; Hyde et al. 2020), there are several reasons why this might be a rather conservative value. First, the threshold for judging a sequence as fungal was set at a conservative e-value of e-50 after using BLAST. Second, although some of the global doubletons and OTUs with higher sequence counts are likely to be undetected chimaeras or otherwise compromised data, many of the global singletons that were excluded from the analysis as potential technical sequencing errors or chimeric sequences are likely to represent true fungal species. Third, although some fungal species harbour several different copies of ITS2 with less than 97% similarity (Stockinger et al. 2010; Lindner et al. 2013; Větrovský et al. 2016), and some OTUs may represent rRNA pseudogenes (Glass et al. 2013), identical or very similar ITS sequences shared between different species are known from many species-rich genera of Pezizomycotina, including Cladosporium, Penicillium, Fusarium, and Aspergillus (Schoch et al. 2012). It should also be noted that there are several geographical gaps in knowledge where our information about fungal communities is limited. The high-throughput sequencing work published to date shows considerable geographic variation with one sample per 4000 km2 reported from Europe and only one sample per 200,000 km2 in Africa. Habitat sampling has thus far been largely dominated by studies focusing on soils, particularly forest soils (Větrovský et al. 2020). It is very likely that more representative sampling covering a wider range of environmental conditions would lead to higher estimates of overall diversity. In addition, there is for sure additional fungal diversity associated with other habitats: marine environments, animal-associated microbiomes, or engineered systems. As has been shown for bacteria, these habitats contain a high proportion of specific microbial taxa (Thompson et al. 2017), and the same pattern should be true in fungi. It should be noted that metabarcoding studies on fungal communities in marine habitats are even fewer than studies on bacteria. Additionally, surveys of animal-host-associated mycobiomes are much less frequent than studies on host-associated bacteria (Peay et al. 2016; Nilsson et al. 2019a; Seibold et al. 2019). Finally, many fungal taxa, such as the phylum Glomeromycota, show limited recovery when general fungal primers are used (Kohout et al. 2014).
Metabarcoding has previously been used to demonstrate the existence of so-far undescribed but widespread groups of fungi and provide their putative taxonomic placement (Nilsson et al. 2016; Tedersoo et al. 2020b). The present dataset also lends itself to such an endeavour. The majority of OTUs in our data belonged to Ascomycota (613,755 OTUs; 57%), followed by Basidiomycota (395,877; 37%), Glomeromycota (12,780; 1%), Mucoromycota (8263; 0.8%), Mortierellomycota (4621; 0.4%), Rozellomycota (3027; 0.3%), and fungal organisms with an unclear phylum-level classification (39,246 OTUs; 3.6%, Fig. 10.3). The most common families are listed in Table 10.1 (Baldrian et al. 2022).

The largest proportion of the terrestrial habitats was represented by soil samples—approximately 60% of all samples. Together with other samples from the rhizosphere or litter, these topsoil samples accounted for almost 70% of the total. Aboveground plant biomass (shoots) and plant roots were also frequently targeted by HTS (14% and 6%, respectively) as was deadwood (9% of samples). When examining the diversity across all samples, the Chao1 estimate increased with sampling depth across the dataset, but Chao1 estimates at fixed sampling depths (5000 sequences) were higher for soil than for deadwood, roots, and shoots (Baldrian et al. 2022) (Fig. 10.4).

Alpha diversity of fungal communities based on ITS2 sequencing. Correlation of the Chao1 diversity estimate and sequencing depth across soil (n = 9743), shoot (n = 2298), root (n = 1581), and deadwood (n = 1126) samples in the GlobalFungi database (Baldrian et al. 2022). The lines represent linear correlation fits
Accordingly, when comparing samples of different habitat types after random subsampling to 5000 sequences, the highest OTU richness was found in soil (mean ± SE: 366 ± 4), followed by litter, with deadwood showing the lowest value (82 ± 2). At this sampling depth, Chao1 estimates were highest for soil and lowest for shoots, roots, and deadwood (Table 10.2) (Baldrian et al. 2022). Although no significant correlation was found between the volume of material sampled and alpha diversity (Větrovský et al. 2019), we can assume that larger samples or samples that are composite and represent larger volume of material capture higher diversity of potential fungal habitats and thus more fungal species (Smith and Peay 2014).
The comparison of clustered sequences used in a study with the UNITE reference corpus (Baldrian et al. 2022) allowed us to estimate the relative proportion of taxa and sequences that belong to previously undescribed fungal species. Across all samples, 78% of sequences (representing 52% of OTUs) mapped to previously characterized taxa, showing a similarity of greater than 97% to any UNITE 8.2 species hypothesis. The relative proportion of sequences of known species ranged from 75% in soil samples to almost 97% in air samples. The lichen habitat appears to be the most promising for finding new fungal species, as 69% of recorded OTUs were not mapped to any characterized taxon; this share was significantly higher than in any other habitat. This observation is consistent with previous reports documenting a surprisingly high number of unrecognized species among lichenized fungi (Lücking et al. 2014). In the litter, the proportion of new taxa averaged a moderate 35%, perhaps reflecting the fact that this has been investigated by mycologists for several centuries. It should be noted that fungi associated with animal hosts were not included in the analysis due to limited data availability. These and other undersampled habitats may still harbour an unknown proportion of total fungal diversity (Wu et al. 2019) and definitely worth further exploration.
4 Global Patterns and Determinants of Fungal Diversity
In the context of ongoing global change, it is critical to determine how climate and other environmental factors affect the diversity and distribution of fungal communities (Hillebrand 2004). Fungal symbionts may benefit plants by mitigating abiotic stressors associated with climate change such as heat and drought (Kivlin et al. 2013), but the distribution of these symbionts may be controlled by mechanisms other than climate (Kreft and Jetz 2007; Bardgett and van der Putten 2014). Because the geographic distribution and environmental preferences of almost all fungi remain unknown (Tedersoo et al. 2014), it is difficult to assess their current status and future threats to their existence (Jetz et al. 2012; Joppa et al. 2016). Given the importance of plant–fungal interactions, the ability to predict shifts in fungal distribution could help to understand or predict ecosystem-level changes. Recently, fungal community composition has been found to be influenced by climatic (Wollan et al. 2008; Maestre et al. 2015; Newsham et al. 2016) and edaphic variables (Tedersoo et al. 2020b; Odriozola et al. 2021) as well as vegetation elements (Crowther et al. 2016).
It is less clear whether global patterns of fungal biodiversity correspond to the higher diversity at low latitudes previously demonstrated for terrestrial macroorganisms and bacteria (Amundson et al. 2015; Thompson et al. 2017). The distribution of plant and animal species also exhibits strong biogeographic partitioning (Kreft and Jetz 2010). Although some biogeographic patterns have been observed for bacteria, other data indicate that dominant microbes are widely distributed and thus can occur in all regions of suitable environments (Hanson et al. 2012; Delgado-Baquerizo et al. 2018).
In a previous study (Větrovský et al. 2019), data from 3084 soil samples in 36 independent studies covering a wide range of climatic conditions were evaluated in terms of the diversity of these communities. Both species richness and the Chao1 index across the different sets consistently revealed relatively low fungal diversity in the tropics. In contrast, areas of higher latitude showed considerable variation in fungal diversity with the most diverse areas concentrated near the high latitudes (Fig. 10.5). Based on these results, there is no convincing support for high fungal diversity in the tropics, which contrasts dramatically with well-known patterns for plants, arthropods, vertebrates (Amundson et al. 2015), and some bacteria (Thompson et al. 2017). Moreover, fungal diversity showed a moderate inverse relationship with temperature, confirming higher diversity at higher latitudes with colder and more seasonal climates (Větrovský et al. 2019). Although these results will need to be further refined and re-evaluated as more data become available, particularly from currently understudied areas, the present comparisons strongly suggest surprisingly low fungal diversity in the tropics.

Inferred patterns of fungal species diversity (OTU richness) predicted by the best-subset GLM (Větrovský et al. 2019)
Fungi can be categorized into ecological guilds reflecting their nutrition or other features (Põlme et al. 2020). When comparing the distribution of fungal species-level taxa belonging to different ecological guilds, ectomycorrhizal fungi have a lower upper temperature limit than that of fungi from other guilds. The temperature valence, defined as a range of temperatures where 90% of all observations of a particular taxon were observed, averaged 4.2 °C in ectomycorrhizal fungi compared with those in other fungal guilds (6.7–9.6 °C). The precipitation valence (330 mm) of this group was also narrower than those of all other guilds (430 to 860 mm), except for endophytes and wood-associated saprotrophs, which was due to the relatively low abundance of ectomycorrhizal fungi in samples with low precipitation. The narrower climatic niche of ectomycorrhizal fungi suggests that these mutualistic plant symbionts may need to respond to climate change-related problems by altering their phenology, range of distribution, or physiology. If they fail to do so, they are likely to become extinct under future climatic conditions. Plant pathogens, in contrast, appear to have much broader climatic niches (Fig. 10.6) (Větrovský et al. 2019).

Climatic determinants of ecological guilds of fungi. (a) The first and ninth deciles of sample mean annual temperature and (b) annual precipitation for SHs belonging to selected ecological guilds with occurrence in >10 samples (Větrovský et al. 2019)
5 Limitations of Molecular Barcoding of Fungi
Although it is tempting to consider HTS as the best solution to explore fungal diversity and biogeography, molecular barcoding and metabarcoding approaches have several limitations that prevent their use as the sole tool for diversity studies. The first important problem is the definition of species. Species are biological entities whose existence is subject to assessment by experimental tools, although these tools may be difficult to apply to all species. Metabarcoding relies on the mathematical construction of taxa. These constructions are an approximation of reality and typically build on an average barcoding gap (sequence divergence) that delineates DNA sequences into molecular taxa or OTUs. The barcoding gap varies significantly among fungi, such that some biological species appear to be multiple OTUs, whereas others may be clustered into a single OTU (Schoch et al. 2012; Liu et al. 2015; Větrovský et al. 2016). Another important issue is the scale of observations. The distribution of fungi can be extremely heterogeneous in space, and samples collected over a small area may share only a few dominant taxa (Štursova et al. 2016). Metabarcoding is also prone to errors from technical sample processing as well as platform-specific sequencing issues, which adds more sources of bias (Nilsson et al. 2019a). One such potential bias is nucleic acid extraction, and another is the formation of chimeric sequences during marker amplification. The limited ability of bioinformatics tools to recognize chimaeras results in an unknown number of OTUs representing artificial taxa, whereas an unknown number of authentic OTUs are incorrectly removed as potential chimaeras (Schloss et al. 2011). For alpha diversity studies, cross-contamination of samples is a major and largely undescribed problem. There are also many important aspects related to the choice of molecular marker (Schoch et al. 2012). The ITS2 region has the advantage of high taxonomic resolution and limited length variability; moreover, a battery of well-tested PCR primers with varying degrees of specificity is available (Ihrmark et al. 2012; Schoch et al. 2012; Tedersoo et al. 2015). Despite the reasonable coverage of some ITS2 primers, all fungus-specific primer pairs used thus far in HTS studies, which were designed to be generally applicable to all fungi, miss at least a few of the 467 most abundant fungal taxa in GlobalFungi (Větrovský et al. 2019). It should be noted that existing ITS primers have been designed based on databases of available sequences, and the extent to which they work well for previously undiscovered and possibly genetically divergent fungal groups remains largely unknown.
Due to the fact that the number of copies of rDNA (and thus ITS) per genome varies among fungi (Lofgren et al. 2019), single-copy markers were considered as an alternative metabarcoding marker. While some of them were proven successful for analysis of closely taxonomically related taxa and appeared to possibly better estimate species-level diversity, they typically miss a large proportion of taxa across the fungal tree of life and show highly variable barcoding gap (Větrovský et al. 2016).
6 Conclusions
Using a large number of metabarcoding datasets from the GlobalFungi database, the conservative estimate of global fungal species richness was 6.3 million species (Baldrian et al. 2022). The global distribution of fungal diversity is shaped by multiple environmental factors, but climate appears to be the major factor affecting the most common fungal species. This finding underscores how profound the impacts of ongoing climate change can be on ecosystem functioning and food security. For example, beneficial ectomycorrhizal fungi exhibit narrow climate niches compared with those of plant pathogens. Thus, the impact of climate on fungal distribution could seriously impair plant productivity. These results open the way for further such research and identify climate as a major determinant of biogeographic patterns in fungi, which appears to differ dramatically from those known for other eukaryotes and bacteria. It is clear that the estimation of diversity from HTS data needs to be viewed in light of the many potential sources of bias mentioned above. However, attempts to estimate extant fungal diversity without considering the HTS perspective appear unrealistic. Mycology as a whole stands to gain from the successful implementation of HTS, and classical mycology is entering the age of big data; there is considerable benefit for the future of fungal ecology and biogeography arising from this paradigm change.
References
Amend AS, Seifert KA, Samson R, Bruns TD (2010) Indoor fungal composition is geographically patterned and more diverse in temperate zones than in the tropics. Proc Natl Acad Sci U S A 107:13748–13753
Amundson R, Berhe AA, Hopmans JW, Olson C, Sztein AE, Sparks DL (2015) Soil and human security in the 21st century. Science 348:1261071
Anslan S, Nilsson RH, Wurzbacher C, Baldrian P, Tedersoo L, Bahram M (2018) Great differences in performance and outcome of high-throughput sequencing data analysis platforms for fungal metabarcoding. MycoKeys 39:29–40
Baldrian P, Kolařík M, Štursová M et al (2012) Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J 6:248–258
Baldrian P, Větrovský T, Lepinay C, Kohout P (2022) High-throughput sequencing view on the magnitude of global fungal diversity. Fungal Divers 114:539–547
Bardgett RD, van der Putten WH (2014) Belowground biodiversity and ecosystem functioning. Nature 515:505–511
Buéé M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, Martin F (2009) 454 pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol 184:449–456
Crowther TW, Todd-Brown KEO, Rowe CW et al (2016) Quantifying global soil carbon losses in response to warming. Nature 540:104–108
Davison J, Moora M, Opik M et al (2015) Global assessment of arbuscular mycorrhizal fungus diversity reveals very low endemism. Science 349:970–973
Delgado-Baquerizo M, Oliverio AM, Brewer TE, Benavent-González A, Eldridge DJ, Bardgett RD, Maestre FT, Singh BK, Fierer N (2018) A global atlas of the dominant bacteria found in soil. Science 359:320–325
Glass DJ, Takebayashi N, Olson LE, Taylor DL (2013) Evaluation of the authenticity of a highly novel environmental sequence from boreal forest soil using ribosomal RNA secondary structure modeling. Mol Phylogenet Evol 67:234–245
Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH (2012) Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol 10:497–506
Hawksworth DL (2001) The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 105:1422–1432
Hawksworth DL, Lücking R (2017) Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol Spectr 5:4
Hillebrand H (2004) On the generality of the latitudinal diversity gradient. Am Nat 163:192–211
Hyde KD, Jeewon R, Chen Y-J et al (2020) The numbers of fungi: is the descriptive curve flattening? Fungal Divers 103:219–271
Ihrmark K, Bodeker ITM, Cruz-Martinez K et al (2012) New primers to amplify the fungal ITS2 region - evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol 82:666–677
Jetz W, McPherson JM, Guralnick RP (2012) Integrating biodiversity distribution knowledge: toward a global map of life. Trends Ecol Evol 27:151–159
Joppa LN, O'Connor B, Visconti P et al (2016) Filling in biodiversity threat gaps. Science 352:416–418
Kivlin SN, Emery SM, Rudgers JA (2013) Fungal symbionts alter plant responses to global change. Am J Bot 100:1445–1457
Kohout P, Sudova R, Janouskova M, Ctvrtlikova M, Hejda M, Pankova H, Slavikova R, Stajerova K, Vosatka M, Sykorova Z (2014) Comparison of commonly used primer sets for evaluating arbuscular mycorrhizal fungal communities: is there a universal solution? Soil Biol Biochem 68:482–493
Koljalg U, Nilsson RH, Abarenkov K et al (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22:5271–5277
Kreft H, Jetz W (2007) Global patterns and determinants of vascular plant diversity. Proc Natl Acad Sci U S A 104:5925–5930
Kreft H, Jetz W (2010) A framework for delineating biogeographical regions based on species distributions. J Biogeogr 37:2029–2053
Lindahl BD, Nilsson RH, Tedersoo L et al (2013) Fungal community analysis by high-throughput sequencing of amplified markers - a user's guide. New Phytol 199:288–299
Lindner DL, Carlsen T, Nilsson RH, Davey M, Schumacher T, Kauserud H (2013) Employing 454 amplicon pyrosequencing to reveal intragenomic divergence in the internal transcribed spacer rDNA region in fungi. Ecol Evol 3:1751–1764
Liu XZ, Wang QM, Goker M et al (2015) Towards an integrated phylogenetic classification of the Tremellomycetes. Stud Mycol 81:85–147
Lofgren LA, Uehling JK, Branco S, Bruns TD, Martin F, Kennedy PG (2019) Genome-based estimates of fungal rDNA copy number variation across phylogenetic scales and ecological lifestyles. Mol Ecol 28:721–730
Lücking R, Dal-Forno M, Sikaroodi M, Gillevet PM, Bungartz F, Moncada B, Yynez-Ayabaca A, Chaves JL, Coca LF, Lawrey JD (2014) A single macrolichen constitutes hundreds of unrecognized species. Proc Natl Acad Sci U S A 111:11091–11096
Maestre FT, Delgado-Baquerizo M, Jeffries TC et al (2015) Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc Natl Acad Sci U S A 112:15684–15689
Newsham KK, Hopkins DW, Carvalhais LC, Fretwell PT, Rushton SP, O'Donnell AG, Dennis PG (2016) Relationship between soil fungal diversity and temperature in the maritime Antarctic. Nat Clim Change 6:182–186
Nilsson RH, Wurzbacher C, Bahram M et al (2016) Top 50 most wanted fungi. MycoKeys 12:29–40
Nilsson RH, Anslan S, Bahram M, Wurzbacher C, Baldrian P, Tedersoo L (2019a) Mycobiome diversity: high-throughput sequencing and identification of fungi. Nat Rev Microbiol 17:95–109
Nilsson RH, Larsson KH, Taylor AFS et al (2019b) The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res 47:D259–D264
Odriozola I, Navrátilová D, Tláskalová P, Klinerová T, Červenková Z, Kohout P, Větrovský T, Čížková P, Starý M, Baldrian P (2021) Predictors of soil fungal biomass and community composition in temperate mountainous forests in Central Europe. Soil Biol Biochem 161:108366
Peay KG, Kennedy PG, Talbot JM (2016) Dimensions of biodiversity in the earth mycobiome. Nat Rev Microbiol 14:434–447
Põlme S, Abarenkov K, Henrik Nilsson R et al (2020) Fungal traits: a user-friendly traits database of fungi and fungus-like stramenopiles. Fungal Divers 105:1–16
Purvis A, Hector A (2000) Getting the measure of biodiversity. Nature 405:212–219
Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing Artifacts on 16S rRNA-based studies. PLoS One 6:14
Schoch CL, Seifert KA, Huhndorf S et al (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc Natl Acad Sci U S A 109:6241–6246
Seibold S, Müller J, Baldrian P, Cadotte MW, Štursová M, Biedermann PHW, Krah F-S, Bässler C (2019) Fungi associated with beetles dispersing from dead wood – Let's take the beetle bus! Fungal Ecol 39:100–108
Smith DP, Peay KG (2014) Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS One 9:e90234
Stockinger H, Kruger M, Schussler A (2010) DNA barcoding of arbuscular mycorrhizal fungi. New Phytol 187:461–474
Štursova M, Bárta J, Šantručková H, Baldrian P (2016) Small-scale spatial heterogeneity of ecosystem properties, microbial community composition and microbial activities in a temperate mountain forest soil. FEMS Microbiol Ecol 92:fiw185
Talbot JM, Bruns TD, Taylor JW et al (2014) Endemism and functional convergence across the north American soil mycobiome. Proc Natl Acad Sci 111:6341–6346
Tedersoo L, Bahram M, Polme S et al (2014) Global diversity and geography of soil fungi. Science 346:1256688
Tedersoo L, Anslan S, Bahram M et al (2015) Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 10:1–43
Tedersoo L, Sánchez-Ramírez S, Kõljalg U, Bahram M, Döring M, Schigel D, May T, Ryberg M, Abarenkov K (2018) High-level classification of the fungi and a tool for evolutionary ecological analyses. Fungal Divers 90:135–159
Tedersoo L, Anslan S, Bahram M, Kõljalg U, Abarenkov K (2020a) Identifying the ‘unidentified’ fungi: a global-scale long-read third-generation sequencing approach. Fungal Divers 103:273–293
Tedersoo L, Anslan S, Bahram M et al (2020b) Regional-scale in-depth analysis of soil fungal diversity reveals strong pH and plant species effects in Northern Europe. Front Microbiol 11:1953
Thompson LR, Sanders JG, McDonald D et al (2017) A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551:457–463
Větrovský T, Baldrian P (2013) Analysis of soil fungal communities by amplicon pyrosequencing: current approaches to data analysis and the introduction of the pipeline SEED. Biol Fertil Soils 49:1027–1037
Větrovský T, Kolařík M, Žifčáková L, Zelenka T, Baldrian P (2016) The rpb2 gene represents a viable alternative molecular marker for the analysis of environmental fungal communities. Mol Ecol Resour 16:388–401
Větrovský T, Kohout P, Kopecký M et al (2019) A meta-analysis of global fungal distribution reveals climate-driven patterns. Nat Commun 10:5142
Větrovský T, Morais D, Kohout P et al (2020) GlobalFungi, a global database of fungal occurrences from high-throughput-sequencing metabarcoding studies. Sci Data 7:228
Wollan AK, Bakkestuen V, Kauserud H, Gulden G, Halvorsen R (2008) Modelling and predicting fungal distribution patterns using herbarium data. J Biogeogr 35:2298–2310
Wu B, Hussain M, Zhang W, Stadler M, Liu X, Xiang M (2019) Current insights into fungal species diversity and perspective on naming the environmental DNA sequences of fungi. Mycology 10:127–140
Acknowledgements
This work was supported by the Czech Science Foundation (21-17749S).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Baldrian, P., Kohout, P., Větrovský, T. (2023). Global Fungal Diversity Estimated from High-Throughput Sequencing. In: Pöggeler, S., James, T. (eds) Evolution of Fungi and Fungal-Like Organisms. The Mycota, vol 14. Springer, Cham. https://doi.org/10.1007/978-3-031-29199-9_10
Download citation
DOI: https://doi.org/10.1007/978-3-031-29199-9_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-29198-2
Online ISBN: 978-3-031-29199-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)