
Abstract
In chronic liver disease, excess hepatic deposition of extracellular matrix, the ensuing development of cirrhosis, the associated renal dysfunction, which ranges from pre-ascitic sodium retention to hepatorenal syndrome, are all dependent, to large extent, on altered function of the renin-angiotensin-aldosterone system (RAAS). The RAAS, once believed to be a hormonal system for blood pressure control and extracellular fluid volume regulation, is now considered a flexible and branching network of enzymes, peptides and receptors that regulates, in addition to arterial circulation, local and systemic inflammation, development of fibrosis in several organs, tumorigenesis, and even bodily reactions to common viruses. In patients with liver disease, besides production of angiotensin II by angiotensin converting enzyme in the vessel wall, there are adaptable synthesis and degradation of bioactive peptides within several tissues by means of enzymes that may be different from those located in arterial endothelium and smooth muscle cells. These ‘nonclassical’ RAAS metabolic pathways that lead to arterial vasodilatation, increased natriuresis and blunting of inflammation (e.g. through angiotensin 1-7 or 3-8) have been identified and can be manipulated pharmacologically, with foreseeable advantages in the treatment of circulatory and renal complications of liver cirrhosis and portal hypertension. Therefore, a comprehensive understanding of the classical and nonclassical RAAS pathways together with the enzymes and peptides involved, especially those that operate inside the liver and the kidney, will provide insights into disease pathogenesis and help to devise treatment strategies for the various disease processes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Renin angiotensin system
- Liver cirrhosis
- Sodium retention
- Liver fibrosis
- Aldosterone
- Chymase
- ACE
- ACE2
- Neprilysin
Introduction
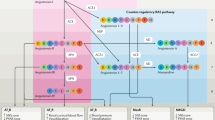
Ever since Tigerstedt and Bergman observed in 1898 that an extract of canine kidney, which they called renin, led to increased arterial blood pressure when injected into another animal, a challenge has been presented to scientists and renewed many times over [1]. The dilemma facing the investigators was not elucidated until 1940, when Prinzmetal showed the presence of renin in ischemic kidneys. Concepts were made clearer when Braun‐Menendez identified the octapeptide, angiotensin II (Ang II), as the actual vasoactive compound leading to increased arterial pressure through its binding to a single cell membrane receptor, later called angiotensin type 1 receptor (AT1R) [1]. Soon the ‘classical’ renin‐angiotensin‐aldosterone system (RAAS) was characterized (Fig. 11.1), only to become, over the following decades, less important as many other ‘nonclassical’ pathways of the RAAS were identified (Fig. 11.2) through the discovery of many more critical enzymes capable of producing a further series of angiotensin peptides. These peptides of different lengths, most of which endowed with specific functions, interact with at least four, and maybe five, different cell membrane receptors.

Schematic representation of ‘classical’ renin-angiotensin-aldosterone system, with peptide receptors. Ang: angiotensin; AT1Rs: angiotensin type1 receptors; AT4Rs: angiotensin type4 receptor; MasR: Mas receptor, ACE: angiotensin converting enzyme; Ang: angiotensin; AT1Rs: angiotensin type1 receptors; AT4Rs: angiotensin type4 receptor; JGA: juxtaglomerular apparatus

Schematic representation of the ‘non-classical’ renin-angiotensin system, with peptide receptors. ACE: angiotensin converting enzyme; AT1Rs: angiotensin type1 receptors; AT2Rs: angiotensin type2 receptor; MasR; Mas receptor; MrgD: Mas-related G protein-coupled receptor member D
The challenge in this review is to explain concisely the role of the new and expanded RAAS in relation to the development of chronic liver disease up until the end stage of liver insufficiency, along with the related progressive derangement of renal and circulatory function that ensues. Several aspects of this system are worthy of note: (a) This complicated endocrine, paracrine, and even intracrine (i.e. intracellular) system of bioactive peptides of RAAS is now known for being deeply involved not only in the control of arterial blood pressure, as once was exclusively believed. It is also involved in the development of local and systemic inflammation, extracellular matrix deposition (fibrogenesis) in several organs, tumorigenesis, and even bodily reactions to common, sometimes lethal, viruses such as hRSV, SARS-CoV and SARS-CoV-2. (b) Many enzymes we will discuss (Figs. 11.1 and 11.2) can adapt their synthetic function when manipulated pharmacologically. Examples of such variable functional capability are shown in several alternative metabolic pathways: when angiotensin converting enzyme type 2 (ACE2) is blocked by metallopeptidase inhibitors, neprilysin starts converting Angiotensin I (Ang I) into angiotensin 1-7 (Ang1-7), the usual peptide product of ACE2 itself [2, 3]; when angiotensin converting enzyme (ACE) is blocked by ACE inhibitors (ACEis), chymase and cathepsin G start producing Ang II, with the result of a paradoxical increase in aldosterone plasma levels (‘the aldosterone escape’ phenomenon) during prolonged ACEi administration [3]; when renin is blocked by specific non-peptide inhibitors like aliskiren, Ang I and II are cleaved by chymase from the newly described angiotensins 1-25 (Ang1-25) and 1-12 (Ang1-12), which are polypeptides generated through non-renin pathways [4]. (c) These various enzymatic reactions sometimes occur in the systemic circulation (i.e. in the arterial endothelium and smooth muscle cells), sometimes in the local circulation (e.g. in heart, liver and kidney), sometimes in single cells that both produce and react to a peptide, sometimes inside the cytoplasm of definite cells (e.g. in cardiomyocytes). (d) Finally, these complex systems of ‘classical’ and ‘nonclassical’ systems (all cleaved from liver-derived α2 globulin angiotensinogen) can interfere with other endocrine or paracrine systems such as those of endothelins, kinins, plasmins, and with the secretion and function of catecholamines themselves [1].
In this review, we shall try to summarize all of this in relation to the development of chronic liver disease, mostly liver cirrhosis, and the cirrhosis-associated derangement of kidney function. Introduction to the basic physiology of the many protagonists of RAAS is essential to understand their perturbation in the setting of liver cirrhosis.
Physiology Considerations
-
(a)
Prorenin and its receptor
The triggers to renin release are hypoperfusion of afferent glomerular arterioles, low chloride content in the macula densa segment of the nephron and stimulation of β1 adrenergic receptors [5]. Losing a 43-amino acid N-terminal segment, a variable amount of the precursor prorenin is cleaved into the protease renin in the juxtaglomerular (JG) cells of the kidney, which are the only cells secreting active renin into blood, but a remarkable share of integral prorenin is also secreted into blood by the same JG cells and by adrenal glands [6], so much so that, normally, circulating prorenin levels are ten times higher than plasma renin concentrations. This ratio increases further in patients with diabetes and arterial hypertension [7]. One quarter of the circulating prorenin is proteolytically converted into renin in plasma by cathepsin B and proconvertase I or undergoes nonproteolytic activation into mature renin by binding to cell surface prorenin/renin receptors [(P)RRs]. Finally, half of circulating prorenin binds to nonspecific clearance receptors [8, 9] (Fig. 11.3).

Proteolytic and non-proteolytic activation of plasma prorenin into active renin. AngI: angiotensin I; AngII: angiotensin II
(P)RR is a 350-aminoacid protein that shows higher affinity for prorenin rather than active renin (Fig. 11.4). It is expressed in vascular smooth muscle cells and in mesangial, distal convoluted tubule and collecting duct cells of the kidney. In the distal nephron, (P)RRs are functionally associated with H+-ATPases: these proton pumps transport protons across plasma membranes in the intercalated cells of collecting ducts and acidify urine in exchange with aldosterone-dependent Na+ reabsorption [9].

Mechanisms of prorenin-induced tissue fibrogenesis. Agn II: angiotensin II; ERK 1 and 2: extracellular signal-regulated kinase 1 and 2; HRP: handle region of prorenin [(P)RR blocker]; MAPK: mitogen-activated protein kinase; PAI-1: plasminogen activator inhibitor-1; (P)RRs: tissue prorenin receptors; TGF-β1: Transforming growth factor β1
Prorenin binding to (P)RRs promotes generation of Ang II in tissues. Independently of local Ang II, (P)RRs stimulation directly causes activation of stress related kinases such as mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) 1 and 2, which upregulate transcription of pro-fibrogenic genes such as TGF-β1, plasminogen activator inhibitor-1 (PAI-1), fibrillar collagen and fibronectin [10,11,12] (Fig. 11.4). Interestingly, estrogens increase ERK 1 and 2 phosphorylation and function through the same MAPK-dependent mechanism [13].
The peptide known as the ‘handle region of prorenin’ (HRP) prevents prorenin/renin binding to (P)RR (Fig. 11.4). Therefore, in experimental murine diabetes, HRP infusion reduces the glomerulosclerosis index and the renal content of TGF-β1 and Ang II [14].
-
(b)
Role of calcium in renin secretion and renal sodium metabolism
Pressor responses to sodium chloride loading in salt-sensitive essential hypertensive patients are preceded by a decrease in serum total and ionized calcium [15], and hypocalcemia with secondary hyperparathyroidism promotes arterial hypertension in chronic renal insufficiency [16]. Conversely, activation of the vitamin D receptor by 1,24-(OH)2 vitamin D, which increases serum Ca++ concentrations through augmented intestinal absorption and decreased urine excretion of Ca++, reduces renin secretion and indirectly sodium tubular retention [17]. Once again, the MAPK/ERK 1/2 pathway, notably stimulated by estrogens, was demonstrated to upregulate vitamin D receptors and therefore blunt renin secretion [13]. This means, as mentioned above, that fibrotic and sodium-retentive mechanisms might be regulated in opposite directions through the same sex hormone-dependent mechanism.
Plasma hypercalcemia stimulates the plasma membrane-associated receptors for extracellular calcium (calcium-sensing receptors or CaSR) in kidney JG cells, thus decreasing prorenin gene transcription and renin release through inhibition of adenylate cyclase, stimulation of phospholipase C, and production of diacylglycerol and inositol 1,4,5-triphosphate [18, 19]. CaSR stimulation by hypercalcemia also suppresses gene transcription and expression of vasopressin-dependent water channels in the kidney collecting duct [20] and reduces the content of sodium–potassium-chloride co-transporters in the thick ascending limb of the Henle’s loop [21]. In other words, extracellular calcium acts as a natriuretic and diuretic agent.
-
(c)
Key endopeptidases and peptides of RAAS
The 13-amino acid N-terminal sequence of angiotensinogen in humans is essential to understand the role of all RAAS peptidases [22]: N-Asp1-Arg2-Val3-Tyr4-Ile5-His6-Pro7-Phe8-His9-Leu10-Val11-Ile12-His13…-C. Four different peptidases are the source of most bioactive peptides of the RAAS. The four peptidases are: ACE, a peptidyl-dicarboxypeptidase (EC 3.4.15.1 according to the EC system); ACE2, a peptidyl-monocarboxypeptidase (EC 3.4.17.23); chymase, a serine endopeptidase (EC 3.4.21.39); neprilysin, a Zn-metallo-endopeptidase (NEP, neutral endopeptidase, EC 3.4.24.11). ACE, ACE2 and NEP are classified as metallopeptidases. These four enzymes have membrane-anchoring domains that orient their active sites on the extracellular surface of the cell [23].
ACE. Once renin has cut the Leu10-Val11 peptide bond of angiotensinogen to generate Ang I (Ang 1-10), dicarboxypeptidase ACE cleaves the Phe8-His9 bond of the decapeptide to make Ang II (Ang 1-8). ACE also cleaves the newly described angiotensin 1-12 (Ang1-12) into Ang I, angiotensin 1-9 (Ang1-9) into angiotensin 1-7 (Ang1-7) and, finally, Ang1-7 into inactive angiotensin 1-5 (Ang1-5) [24, 25]. Outside RAS, ACE degrades enkephalins, substance P and luteinizing hormone releasing hormone [26]. Kidney ACE is found in most tubular cells, vascular endothelial cells and glomerular mesangial cells. Outside the kidney, ACE is located in endothelial cells, especially in the lung [24].
ACE2. This monocarboxypeptidase is the main source of the vasodilator and natriuretic peptide Ang1-7 since it cleaves the Pro7-Phe8 bond of Ang II almost ubiquitously. Inside non-classical pathways of RAAS, ACE2 cleaves Ang I into Ang1-9 [24, 25]. ACE2 metabolizes also other peptide substrates (apelin, kinins and endorphins) and regulates the level of tryptophan in the blood [27].
NEP. This enzyme is a membrane-bound Zn-metallo-endopeptidase. It is also called atriopeptidase because it leads to the proteolytic clearance of urodilatin, atrial, brain-derived and C-type natriuretic peptides in the kidney, lung, brain, and heart [28]. NEP degrades opioid-peptides [29], bradykinin [30], bombesin-like peptide [31], substance P [32] and adrenomedullin [33]. Inside the RAAS, NEP cleaves Ang1-9, Ang1-12 and Ang I into Ang1-7 and, finally, the latter vasodilator and natriuretic peptide into the inactive by-product angiotensin 1-4 (Ang1-4) [24, 25]. In other words, NEP generates Ang1-7, but continues to metabolize Ang1-7 at the Tyr4-Ile5 bond into Ang1-4 (Fig. 11.2), as shown in studies employing vasopeptidase inhibitors (i.e. combined ACE and NEP inhibitors) in patients or animal models with arterial hypertension [34, 35].
NEP also produces the vasoconstrictor, profibrogenic and anti-natriuretic polypeptide endothelin-1 (ET-1) from circulating precursors (big ET-1 and ET 1-31) [36, 37].
Chymase. Serine endopeptidase chymase, in heart, liver, renal tubules and mast cells, converts Ang I into Ang II; ACE catalyzes this same reaction in the arterial endothelium [38]. In tissues with chronic inflammation, chymase is over-expressed and converts big ET-1 into ET-1 [39] and activates TGF-β1 through potentiation of Ang II action [40]. 80% of Ang II synthesized in kidney, heart and blood vessels is produced by chymase [41], but chymase inhibitors do not lower blood pressure and do not increase active renin [42] because chymase is confined to mast cells of the vascular adventitia of arterial vessels. Moreover, systemic plasma contains serine endopeptidase inhibitors [42]. Chymase also cleaves Ang1-12 into the Ang II. Upstream in this atypical metabolic pathway, Ang1-12 is not cleaved from angiotensinogen by renin, but through a hitherto unidentified protease that cuts the Ile12-His13 bond of angiotensinogen in humans [43, 44]. Ang1-12 may be a precursor of Ang I through ACE [45, 46], of Ang1-7 and then Ang1-4 (in the tubular nephron) through NEP [46], and, above all, of Ang II through chymase in heart and kidney [43]. Recent studies underline that, alongside Ang1-12, another polypeptide derived from angiotensinogen through non-renin pathways (i.e. angiotensin 1-25) may be a suitable source of tissue Ang I and II by means of chymase action, at least in the heart [47, 48].
-
(d)
Angiotensin receptors so far described
The cell surface receptors identified so far as binding sites of this host of angiotensins are five: AT1-2-4Rs, Mas receptors (MasRs) and Mas-related G protein-coupled receptor member D (MrgD) (Figs. 11.1 and 11.2). The main endogenous ligand of AT1R and AT2R in vascular endothelium, kidney, adrenals, brain, heart, liver and testis is Ang II. Ang 1-9 is also a ligand of AT2R (7). The main endogenous ligands of AT4R and MasR are, respectively, angiotensin 3-8 (Ang3-8) and Ang1-7 [25]. Putative ligand of MrgD is newly described heptapeptide alamandine (Fig. 11.2).
AT1 receptors. AT1Rs greatly exceed the number of AT2Rs after birth [25], leading to vasoconstriction, aldosterone secretion from the glomerulosa cells of adrenal glands, tubular sodium retention and increased arterial blood pressure, when stimulated by Ang II (classical RAAS).
AT1R signaling is primarily mediated through G-proteins, leading to adenylyl cyclase activation and intracellular cAMP generation, activation of phospholipase C, production of inositol-1,4,5-triphosphate (IP3), Ca++ release from sarcoplasmic reticulum into the cytoplasm, and final Ca++/calmodulin-dependent vasoconstriction. Further AT1R signaling is mediated through small GTPase proteins, G-protein independent β-arrestin, reactive oxygen species (ROS) (through NADPH-oxidase [NOX] activation, leading to tissue fibrogenesis) [49], non-receptor type tyrosine kinases, transactivation of receptor tyrosine kinases [50]. AT1Rs also undergo homo and hetero oligomerization with other receptors, including AT2Rs, bradykinin B2 receptors, β2 adrenergic receptors, and dopamine D2 receptors [51]. Recently, it has been shown that AT2Rs directly bind to AT1Rs, inhibiting AT1R functions. Bradykinin B2 receptors potentiate AT1R signaling, enhancing the vasoconstrictive effects of Ang II. Evidence also exists of direct interaction between the β2-adrenergic receptors and AT1Rs. β-blockers have been shown to interfere with Ang II signaling in heart failure patients and have become a mainstay of therapy in patients with chronic heart failure [52].
AT2 receptors. These G protein-coupled receptors share only 34% amino acid sequence homology with AT1Rs [53]. AT2R signaling involves G protein, specific protein phosphatases (MKp-1, PP2A, etc.) and scaffolding proteins, nitric oxide/cGMP ion channel protein, and constitutive activity (i.e. ligand independent activity of AT2R) [50]. Stimulation of AT2Rs in interlobular arterioles and the tubular nephron of the kidney leads to vasodilatation and natriuresis. The latter effect is mediated by stimulation of nitric oxide/cGMP/Sp 1 pathways that inhibit the proximal tubule Na+/K+-ATPase [25, 54]. Moreover, it appears that specific stimulation of AT2R can down-regulate expression of AT1Rs, resulting in the finding that, based on the AT1R/AT2R balance, Ang II itself can be hyper- or hypotensive and natriuretic or anti-natriuretic [54, 55].
AT4 receptors. AT4Rs are mainly in brain, heart, kidney, adrenals and blood vessels. This receptor is the Angiotensin 3-8 (Ang3-8) binding site (Fig. 11.1). Ang3-8 binding protein was identified as insulin-regulated amino peptidase (IRAP, EC 3.4.11.3), which is a type 2 trans-membrane protein of the gluzincin amino peptidase family [56, 57].
Mas receptors. MasR is a G protein-coupled receptor and the binding site for Ang1-7. The action of Ang1-7 through MasR causes production of arachidonic acid and activation of nitric oxide synthase. MasRs exhibit the highest expression in brain and testis [50]. In common with AT4R, stimulation of vasodilator and natriuretic MasR leads to nitric oxide production via enhanced phosphorylation of protein kinase B and increased cell levels of cyclic GMP [24, 25].
Mas-related G protein-coupled receptor member D (MrgD). MrgD expression is detected in arterial smooth muscle cells, endothelial nitric oxide synthase (eNOS)-positive endothelial cells, and in atherosclerotic plaques [58]. MrgD stimulation is thought to elicit phospholipase C activation and increased expression of nitric oxide synthase (NOS) enzymes [58]. MrgD is the putative binding site for alamandine, an heptapeptide derived from Ang1-7 through decarboxylation of the N-terminal aspartate residue (Fig. 11.2).
-
(e)
Intracrine RAAS
Over the last decades, a large amount of literature has shown that not only do tissue renin-angiotensin systems exist, but so do intracellular (i.e. intracrine) renin-angiotensin systems. Various reports have identified intracellular location and actions of such RAAS components as Ang II, Ang1-7, prorenin receptor, angiotensinogen, several isoforms of renin, AT1Rs, AT2Rs, MasRs, ACE, ACE2 and chymase [59]. Perhaps intracellular RAAS alone would warrant a separate review. What matters here may be summarized as follows. Ang II treatment produces a significant increase in nitric oxide (NO) and superoxide/H2O2 production in isolated nuclei (Fig. 11.5). These effects are inhibited by losartan (an AT1R inhibitor) but not by an AT2R blocker [60]. The likely sources of these intracellular NO and reactive oxygen species are intranuclear NOS and NADPH oxidase 4 (NOX 4). At least in diabetic rats, the intracellular Ang II content in the heart is correlated with cardiomyocyte apoptosis, oxidative stress and extracellular matrix deposition [61] (Fig. 11.5). There is strong experimental evidence to support the view that intracrine Ang II activity may function independent of the circulating RAAS [47]. Whether these findings can be transferred to the model of liver fibrosis is a matter of debate.

Synthetic representation of intracrine RAAS inside cardiomyocytes. Ang II: angiotensin II; AT1R: angiotensin type 1 receptor; NOS: nitric oxide synthase; NOX 4: NADPH oxidase 4
-
(f)
Aminopeptidases and Ang II clearance
In the systemic circulation, degradation of Ang II may lead to Ang1-7 generation through ACE2, but, in wild-type mice and normal humans, low systemic levels of Ang1-7 and much higher levels of angiotensin 2-8 (Ang2-8 or Ang III) and angiotensin 3-8 (Ang3-8 or Ang IV) [62] emphasize that the actual clearance of Ang II is through the sequential actions of plasma aminopeptidases (Fig. 11.1). In plasma, aspartyl-aminopeptidase or aminopeptidase A (APA) cleaves the Asp1-Arg2 bond at the N-terminal end of Ang II to generate Ang2-8, which in turn is cleaved at the new N-terminal Arg-Val bond by arginyl-aminopeptidase or aminopeptidase N (APN), to form Ang3-8 [25]. The kidney synthesizes and secretes most APA and APN found in blood [62, 63].
Ang2-8 and Ang3-8, have important hormonal activities. Ang2-8 binds AT1R, AT2R and MasR, and Ang3-8 binds mostly AT4R. AT4R stimulation by Ang3-8 causes arterial vasodilatation and natriuretic responses [25].
In summary, all RAAS peptides that are generated downstream of Ang II are either vasodilating and natriuretic agents (Ang1-7, Ang3-8, alamandine and even Ang2-8 when stimulating AT2Rs or MasR) or inactive by-products (Ang1-5, Ang1-4) (Figs. 11.1 and 11.2).
Liver Cirrhosis. Prorenin and Renin Regulation by Extracellular Calcium
By comparing normal and CCl4 cirrhotic rats, it is found that (P)RR content in the liver is significantly lower, not higher, in the cirrhotic group (western blot analysis). Conversely, plasma concentrations of prorenin can be derived empirically from the ratio direct renin (DR)/plasma renin activity (PRA) [1], and DR/PRA ratios were 3.3 ± 0.8 and 7.9 ± 1.6 (P < 0.03) in healthy and cirrhotic rats respectively, showing more plasma prorenin in the latter group [64]. Significantly lower content of (P)RRs in the cirrhotic liver along with increased circulating prorenin may be the expression of physiological receptor downregulation after prolonged agonist stimulation. To summarize, it is clear that (P)RR is expressed also in the liver and, as such, its role as a pathogenic factor, among many others, of hepatic fibrogenesis cannot be excluded (Figs. 11.3 and 11.4).
Sansoè and Wong observed significant natriuretic and aquaretic responses to intravenous calcium loading in human compensated cirrhosis [65] and to intravenous administration of CaSR agonists (i.e. poly-L-arginine) in experimental pre-ascitic cirrhosis [66]. Of course, these calcium-driven diuretic responses were not accompanied by any increase in plasma renin activity, due to the already described downregulating effects of CaSR stimulation on renin gene transcription and secretion by JG cells [18, 19].
Liver Cirrhosis. Endopeptidases and Peptides of RAAS Are Protagonists in Chronic Liver Disease and Its Renal Complications
Within the liver, low levels of ACE activity are detected in sham/control animals, while significantly increased levels are shown in areas of active fibrogenesis in bile duct ligated or CCl4-treated rats [67,68,69]. Inhibition of ACE reduces increased arterial blood pressure, and ACE inhibitors (ACEis) or AT1R antagonists (ARBs) can attenuate experimental liver fibrosis [68, 70], but these two classes of drugs have severe hypotensive effects in patients with established cirrhosis [71, 72]. Concentrations of ACE2, Ang1-7 and MasR (Ang1-7 specific receptor) (Fig. 11.2) are increased in splanchnic vessels from cirrhotic patients and rats compared to healthy controls [73]. Therefore, MasR blockade reduces portal pressure, indicating that activation of this receptor in splanchnic vasculature promotes mesenteric hyperdynamic circulation and increases portal inflow that contributes to portal hypertension [73]. However, non-peptidic MasR agonist AVE0991 reduces portal pressure without any change in arterial blood pressure [74]. At first sight, these data seem contradictory: it was apparently shown that both MasR blockers and MasR agonists reduce portal pressure. It is conceivable that MasR blockade reduces portal venous inflow, as stated, while MasR agonists reduce intrahepatic resistance to portal flow since ACE2 is upregulated in areas of active liver fibrogenesis [75]. As a matter of fact, recombinant ACE2 has anti-fibrogenic effects in bile duct ligated (BDL) and CCl4-treated rats, both acutely and long-term [76, 77], and diminazene aceturate, commonly used to treat human trypanosomiasis, enhances hepatic ACE2 activity and inhibits tumor necrosis factor-α (TNF-α) synthesis and gene expression of NADPH oxidase (NOX), a key source of fibrogenic reactive oxygen species (ROS) [78]. So doing, this drug exerts strong hepatic anti-fibrotic properties. ACE2, indeed, is thought to be a negative regulator of the RAAS and, in the liver, this enzyme functions to limit fibrosis [79].
In the kidney of cirrhotic rats with ascites, there is a mean 170% increase in NEP protein content, and NEP localizes mainly in proximal convoluted tubule and macula densa [80]; the NEP inhibitor candoxatrilat promptly increases urinary volume, and urinary excretion of sodium, atrial natriuretic peptide (ANP) and cyclic GMP (ANP second messenger), without significant changes in plasma renin activity or mean arterial pressure [80]. These overall results depend on the key contribution of NEP to ANP, Ang1-7, bradykinin clearance and to tissue ET-1 generation [24, 25, 28]. Notably, in patients with cirrhosis and ascites, renal plasma flow (RPF) and glomerular filtration rate (GFR) inversely correlate with plasma levels of ET-1 [81], and intravenous infusion of ET-1 results in prompt anti-natriuretic responses [82]. In the cytosol fraction of the cirrhotic rat liver, there is a even greater increase in NEP content, 280% to be exact. This enzyme is in the desmin-positive myofibroblast-like cells of the fibrotic septa. NEP inhibitor candoxatrilat, administered to rats with CCl4-dependent cirrhosis, acutely decreases portal pressure and increases liver plasma flow (evaluated through indocyanine green clearance) [83]. In the kidney of CCl4 cirrhotic animals, chymase protein content and activity are significantly increased in cortical arterioles and the tubular nephron. In cirrhotic rats and hamsters, chronic dosing of SF2809E, a specific chymase inhibitor, decreases renal Ang II content and increases natriuresis and aquaresis [84, 85]. In the liver of CCl4 cirrhotic rats, chymase is largely expressed in α-smooth muscle-positive myofibroblasts, while, in human cirrhosis, chymase is mainly found in hepatocytes of regenerative nodules. Moreover, chymase mRNA transcription is promptly upregulated by TGF-β1 in human HepG2 cells and activated hepatic stellate cells in vitro. Finally, SF2809E, specific chymase inhibitor, reduces liver Ang II content, hepatic fibrogenesis and portal pressure in CCl4-treated animals [84, 85].
To sum up, in the diseased liver, areas of active fibrogenesis express increased contents of ACE [67], chymase [85], and NEP [83], but also of ACE2 [75]. This leads to increased tissue levels of Ang II [85] and some five-fold increase in the Ang II/Ang1-7 ratio in the diseased liver [64]. Particularly critical is the role of desmin- and α-smooth muscle-positive liver myofibroblasts (HSC/MFs) of liver fibrotic septa: these cells host over-expressed NEP [83], the enzyme that degrades the vasodilating and anti-fibrogenic Ang1-7 [24, 25], and are active sources of Ang II and ET-1 through cellular over-expression of ACE and chymase [69, 85].
Moreover, in patients with liver cirrhosis, renal RAAS is aberrantly activated: angiotensinogen is secreted into proximal tubular fluid [25], active renin is massively produced in advanced cirrhosis with ascites, and ACE, NEP and chymase are upregulated and hyperactive in the tubular nephron even before clinical decompensation [24, 25, 83, 85]. This leads to Ang II concentrations in the kidney interstitial and tubular fluids being much higher than normal well before ascites development and before secondary hyper-reninism (i.e. irrespective of systemic levels of Ang II) [25, 85], producing a net effect of sodium retention along all segments of the tubular nephron (see later for the specific mechanisms of Ang II-dependent renal sodium retention).
Liver Cirrhosis. Receptors of Angiotensins and Post-receptor Mechanisms of Disease
Ang II is a key contributor (through binding to AT1Rs) to progression of liver fibrogenesis, cirrhosis development, and worsening of hepatic function in chronic liver disease. Liver fibrosis progression depends on interactions among injured hepatocytes, activated inflammatory cells, and hepatic myofibroblast (MFs)-like cells that originate mainly from activation of hepatic stellate cells (HSCs) or portal fibroblasts. Activated HSCs produce Ang II [67], Ang II binds to AT1R expressed by most myofibroblasts, and transcription of genes encoding for extracellular matrix components, pro-fibrogenic cytokines (e.g. TGF-β1) and collagenolysis inhibitors occurs [86,87,88].
The role of AT1R signaling in HSC activation and collagen deposition in chronically diseased liver is predominant (Fig. 11.6).

Mechanisms of liver disease mediated through AT1R stimulation by Ang II. Ang: angiotensin; AT1R: angiotensin type 1 receptor; ARHGEF1: Rho guanine nucleotide exchange factor 1; IκB: inhibitor of NF-κB; JAK2: intracellular Janus kinase-2; NF-κB: Nuclear factor κB; NO: nitric oxide; RhoA: Ras homolog gene family member A; Rock-1: Rho-associated coiled-coil-containing kinase protein-1; ROS: reactive oxygen species
-
RhoA/Rock-1 pathway. Among members of the Rho small GTPase superfamily (AT1R signaling mediators), Ras homolog gene family member A (RhoA) constitutes the RhoA/Rock-1 (Rho-associated coiled-coil-containing kinase protein-1) signaling pathway, with resultant activation of the small G protein Rac and reactive oxygen species (ROS) production, which plays a central role in the development of liver fibrosis [89]. Notably, one of the most important effects of ROS is the reduction of nitric oxide (NO) bioavailability: superoxide radical anion (O2−) reacts with NO, destroying it via its conversion to peroxynitrites [90] (Fig. 11.6). In BDL rats, liver collagen deposition can be blunted and portal pressure decreased through inhibition of the RhoA/Rock 1 signaling pathway, which is instead activated by Ang II through AT1R, [91].
-
JAK2 pathway. Through mechanisms that are not fully understood but probably involve Ca++ and PYK2 or Src kinase, stimulation of AT1Rs activates intracellular Janus kinase-2 (JAK2). JAK2 then phosphorylates Rho guanine nucleotide exchange factor 1 (ARHGEF1), which stimulates the RhoA-Rho kinase–myosin phosphatase target subunit cascade, and this inhibits myosin light chain phosphatase, leading to prolonged phosphorylation of myosin light chain and final contraction of vascular smooth muscle cells, the physiological effect of Ang II [92] (Fig. 11.6). Interestingly, JAK2 antagonists significantly attenuate HSC activation and collagen accumulation in experimental liver fibrosis models [93]. Notably, JAK2 phosphorylates and activates signal transducer and activator of transcription 3 (STAT3), and JAK2/STAT3 pathway is aberrantly expressed in tissues infected by SARS-CoV-2 during COVID-19 [2, 94], as well as in most malignancies: e.g., breast, pancreatic, bladder, colorectal, gastric cancers, lung adenocarcinoma, and natural killer/T-cell lymphoma [95].
-
NF-κB pathway. Activated liver HSCs express constitutive nuclear factor-κB (NF-κB), which promotes HSC survival by stimulating the expression of anti-apoptotic proteins. Specific inhibition of NF-κB is sufficient to provoke apoptosis of mature human HSCs and blunting of liver collagen deposition. Human HSC activation is accompanied by a sustained transcriptional repression of IκBα, the natural inhibitor of NF-κB. Moreover, upon stimulation of AT1Rs in activated HSCs, serine residues on IκBα are phosphorylated by the IκB kinase. This results in progressive degradation of IκBα, which releases NF-κB for nuclear transport and interaction with profibrogenic target genes, leading to their transcription [96] (Fig. 11.6).
In liver cirrhosis, intrarenal RAAS is activated earlier than its systemic counterpart, as confirmed in humans with pre-ascitic disease. In fact, lower-body negative pressure, which reduces central blood volume, enhances renal renin and Ang II secretion rates [97]. Moreover, despite baseline suppression of systemic RAAS, sodium overload induced by high sodium diet is reversed by the AT1R antagonist losartan administered at a dose not perturbing systemic hemodynamics, stressing exclusive intrarenal activation of renin-angiotensin system [98, 99].
In the kidney, Ang II constricts the efferent glomerular arteriole more than the afferent one, resulting in a tendency to preservation of GFR and filtration pressure. This occurs at the expense of reduction in renal plasma flow, increase in filtration fraction, and decrease in peritubular capillary hydrostatic pressure. The latter leads to retention of sodium and water in the tubular nephron [100].
In addition, Ang II causes direct sodium reabsorption in the proximal convoluted tubule through stimulation of tubular AT1R and activation of renal cortical Na+/H+ exchanger 3, a process involving an increase in intracellular Ca++ and activation of JAK2 and calmodulin [100, 101]. Enhanced release of Ang II and increase in oxidative stress (through activation of NOX and RhoA/Rock 1 kinase pathways) are also the key to further renal sodium retention via increased activity of thiazide-sensitive sodium chloride cotransporter in the later segments of the distal convoluted tubule [90]. Finally, increased systemic levels of Ang II and secondary aldosteronism lead to aldosterone-dependent Na+/K+-ATPase and epithelial sodium channels (ENaCs) upregulation in the collecting duct and stimulation of arginine vasopressin (AVP) secretion. In turn, increased plasma AVP and increased reactive oxygen species (ROS) (due to stimulation of kidney AT1Rs) enhance the activity of Na+-K+-2Cl− cotransporters in the thick ascending limb of the Henle’s loop [90]. In other words, increased renal content of Ang II, firstly, and increased systemic levels of Ang II with secondary aldosteronism, secondly, completely control sodium retention along all segments of the tubular nephron in cirrhosis, both in pre-ascitic and in ascitic patients.
Unfortunately, in patients with cirrhosis and ascites or end stage liver disease, oral ACEis or ARBs, due to their arterial vasodilatory activity, do not improve natriuresis and may aggravate arterial hypotension and hyper-reninism, leading to final fall in both RPF and GFR [102,103,104,105]. This is due to the systemic activation of RAAS, which nonetheless tries to compensate for the peripheral arterial vasodilatation of advanced cirrhosis [102]. Perhaps compensated patients with early cirrhosis and no systemic RAAS activation might take advantage of ARBs administration, at least to reduce liver fibrogenesis [67, 103]. In any event, recent systematic reviews of available trials show that ARBs, in patients with ascitic cirrhosis, do not reduce portal pressure significantly and increase the risk of symptomatic hypotension and renal failure [1, 104]. Moreover, it has long been known that ACEis in liver cirrhosis do not reduce portal pressure in Child–Pugh A cirrhotic patients [105,106,107] and, of course, are detrimental in decompensated cirrhotic patients [108].
Finally, in clinical settings characterized by enhanced systemic production of Ang II (e.g. in decompensated cirrhosis), AT1Rs-enriched exosomes transfer such receptors to peripheral target cells, in order to offset the physiological receptor downregulation after prolonged agonist stimulation. Exosomes are extracellular nanovescicles of 30–100 nm in size that are released into the extracellular space by cardiomyocytes through reverse budding of multivesicular intracellular bodies [109].
The discovery of non-peptidic AT2R agonists offers hope for new therapeutic approaches to modify the AT1R/AT2R balance [110]. Among these AT2R agonists, the most promising one, in relation to the management of cirrhosis complications, is Compound 21 (C21), which, in animal models of arterial hypertension, produces dose-dependent natriuretic and aquaretic effects but does not reduce blood pressure unless the AT1Rs are also blocked [111].
Moreover, agonists of AT2R do blunt fibrogenesis in chronic liver disease [55].
The putative MrgD ligand alamandine (Fig. 11.2) can attenuate arterial hypertension, alleviate cardiac hypertrophy in spontaneously hypertensive rat [58], and appears to attenuate hepatic fibrosis by regulating autophagy induced by NOX 4-derived reactive oxygen species [112]. Unfortunately, no human studies are available regarding this specific topic.
Liver Cirrhosis. Secondary Aldosteronism, Renal Sodium Retention and Progression of Liver Fibrosis
Patients with advanced liver cirrhosis and ascites display splanchnic and systemic hyperdynamic circulation, contraction of effective arterial blood volume, hyper-reninism and secondary aldosteronism [113]. Beyond the expected worsening of sodium retention because of secondary hyperaldosteronism itself, aldosterone, whose secretion by glomerulosa cells of adrenal glands is under Ang II control through stimulation of AT1Rs, has a definite role also in the initial development of cirrhotic ascites. In rats with CCl4-induced cirrhosis, pre-ascitic renal sodium retention is temporally related with increasing renal aldosterone excretion and is prevented by the aldosterone antagonist spironolactone [114]. In upright pre-ascitic cirrhotic patients, renal sodium retention is associated with a borderline elevation in plasma aldosterone and increased tubular sodium reabsorption by the distal nephron [115].
This traditional view of aldosterone as a trigger of clinical decompensation of liver cirrhosis has been recently enriched after the observation that patients with arterial hypertension chronically treated with ACEis show paradoxically high levels of circulating aldosterone because of the so called ‘aldosterone escape’: when ACE is blocked by ACE inhibitors, chymase and cathepsin G start producing Ang II, with the result of increased aldosterone plasma levels during prolonged ACEi administration. This ‘aldosterone escape phenomenon’ is thought to be the cause of ACEi treatment failure in the prevention of progressive renal fibrosis that occurs in subgroups of patients with arterial hypertension. Indeed, sustained increased levels of plasma aldosterone, as occur also in patients with advanced liver cirrhosis, induce ubiquitous plasminogen activator inhibitor-1 (PAI-1) expression, and treatment with mineralocorticoid receptor antagonists reverses this phenomenon. PAI-1 is a member of the serine protease inhibitor (serpine) gene family and the main inhibitor of tissue-type and urokinase-type plasminogen activators (tPA and uPA), and therefore of fibrinolysis. Unfortunately, the same tissue PAI-1, as such induced by increased plasma levels of aldosterone, is also a strong inhibitor of plasmin-dependent matrix metalloproteinases (MMPs) activation in the liver, where MMPs should provide the reabsorption of excess extracellular matrix deposition during chronic liver diseases [3, 116,117,118]. In brief, plasma aldosterone, through increased PAI-1 gene expression, is considered a relevant agent of progressive liver fibrosis in chronic liver disease [117, 118].
Liver Cirrhosis. Aminopeptidases and Chronic Liver Disease
Recently, it has been shown that plasma aminopeptidase A is significantly reduced in patients with liver cirrhosis [119]. In this clinical context, this means that lack of aminopeptidase A provides less Ang2-8 to aminopeptidase N, which in turn generates lesser amounts of the natriuretic Ang3-8 (Fig. 11.1). Furthermore, slowed degradation of Ang II itself means prolonged half-life of this key anti-natriuretic peptide, which perpetuates the vasoconstrictive and sodium retaining effects of Ang II.
Conclusions
With a more comprehensive understanding of the systemic and tissue RAAS, it is perhaps time to advance an updated theory of liver insufficiency and associated functional renal failure in cirrhosis. What was once thought of as secondary to mere hemodynamic abnormalities (i.e. the hyperdynamic circulation of liver cirrhosis with ensuing contraction of effective arterial blood volume) is now complementary to our understanding of the changes that occur both inside the diseased liver and inside the kidney: chymase, ACE, NEP are overexpressed and functioning in both organs, leading to a net imbalance towards too much Ang II and too little Ang1-7. The consequences in the organs are different: inflammation and progressive fibrosis inside the liver, vasoconstriction, tubular sodium retention and final GFR loss inside the kidney.
References
Sansoè G, Aragno M, Wong F (2020) Pathways of hepatic and renal damage through non-classical activation of the renin-angiotensin system in chronic liver disease. Liver Int 40:18–31
Sansoè G, Aragno M, Wong F (2021) COVID-19 and liver cirrhosis: focus on the nonclassical renin-angiotensin system and implications for therapy. Hepatology 74:1074–1080
Huang W, Xu C, Kahng KW et al (2008) Aldosterone and TGF-β1 synergistically increase PAI-1 and decrease matrix degradation in rat mesangial and fibroblast cells. Am J Physiol Renal Physiol 294:F1287–F1295
Moniwa N, Varagic J, Ahmad S et al (2013) Hemodynamic and hormonal changes to dual renin-angiotensin system inhibition in experimental hypertension. Hypertension 61:417–424
Lorenz JN, Weihprecht H, Schnermann J et al (1991) Renin release from isolated juxtaglomerular apparatus depends on macula densa chloride transport. Am J Physiol Renal Physiol 260:F486–F493
Krop M, Danser AHJ (2008) Circulating versus tissue renin-angiotensin system: on the origin of (pro)renin. Curr Hypertens Rep 10:112–118
Danser AHJ, Derkx F, Schalekamp M et al (1998) Determinants of interindividual variation of renin and prorenin concentration: evidence for a sexual dimorphism of (pro)renin levels in humans. J Hypertens 16:853–862
Batenburg WW, Krop M, Garrelds IM et al (2007) Prorenin is the endogenous agonist of the (pro)renin receptor. Binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor. J Hypertens 25:2441–2453
Nguyen G (2006) Renin/prorenin receptors. Kidney Int 69:1503–1506
Huang Y, Wongamorntham S, Kasting J et al (2006) Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int 69:105–113
Huang Y, Noble NA, Zhang J et al (2007) Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int 72:45–52
Zhang J, Wu J, Gu C et al (2012) Receptor-mediated nonproteolytic activation of prorenin and induction of TGF-β1 and PAI-1 expression in renal mesangial cells. Am J Physiol Renal Physiol 303:F11–F20
Gilad LA, Bresler T, Gnainsky J et al (2005) Regulation of vitamin D receptor expression via estrogen-induced activation of the ERK 1/2 signaling pathway in colon and breast cancer cells. J Endocrinol 185:577–592
Ichihara A, Hayashi M, Kaneshiro Y et al (2004) Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest 114:1128–1135
Shingu T, Matsuura H, Kusaka M et al (1991) Significance of intracellular free calcium and magnesium and calcium-regulating hormones with sodium chloride loading in patients with essential hypertension. J Hypertens 9:1021–1028
Gal-Moscovici A, Sprague SM (2010) Use of vitamin D in chronic kidney disease patients. Kidney Int 78:146–151
Sigmund CD (2002) Regulation of renin expression and blood pressure by vitamin D(3). J Clin Invest 110:155–156
Atchinson DK, Ortiz-Capisano MC, Beierwaltes WH (2010) Acute activation of the calcium-sensing receptor inhibits plasma renin activity in vivo. Am J Physiol Renal Physiol 299:R1020–R1026
Ortiz-Capisano MC, Reddy M, Mendez M et al (2013) Juxtaglomerular cell CaSR stimulation decreases renin secretion via activation of the PLC/IP3 pathway and the ryanodine receptor. Am J Physiol Renal Physiol 304:F248–F256
Procino G, Carmosino M, Tamma G et al (2004) Extracellular calcium antagonizes forskolin-induced aquaporin 2 trafficking in collecting duct cells. Kidney Int 66:2245–2255
Wang WH, Lu M, Hebert SC (1996) Cytochrome P-450 metabolites mediate extracellular Ca++-induced inhibition of apical Na+-K+-Cl− channels in the thick ascending limb of Henle. Am J Physiol Cell Physiol 271:C103–C111
Tewksbury DA, Dart RA, Travis J (1981) The amino terminal aminoacid sequence of human angiotensinogen. Biochem Biophys Res Commun 99:1311–2115
Wilson BA, Marshall AC, Alzayadneh EM et al (2014) The ins and outs of angiotensin processing within the kidney. Am J Physiol Regul Integr Comp Physiol 307:R487–R489
Chappell MC (2012) The non-classical renin-angiotensin system and renal function. Compr Physiol 2:2733–2752
Berard E, Niel O, Rubio A (2014) Is the renin-angiotensin system actually hypertensive? Pediatr Nephrol 29:951–960
Skidgel RA, Erdös EG (1987) The broad substrate specificity of human angiotensin I converting enzyme. Clin Exp Hypertens A 9:243–259
Alenina N, Bader M (2019) ACE2 in brain physiology and pathophysiology: evidence from transgenic animal models. Neurochem Res 44:1323–1329
Wilkins RM, Redondo I, Brown LA (1997) The natriuretic peptide family. Lancet 349:1307–1310
Malfroy B, Swerts JP, Guyon A et al (1978) High-affinity enkephalin-degrading peptidase in brain is increased after morphine. Nature 276:523–526
Deddish PA, Marcic BM, Tan F et al (2002) Neprilysin inhibitors potentiate effects of bradykinin on b2 receptor. Hypertension 39:619–623
Cohen AJ, King TEJr, Gilman LB et al (1998) High expression of neutral endopeptidase in idiopathic diffuse hyperplasia of pulmonary neuroendocrine cells. Am J Respir Crit Care Med 158:1593–1599
Erdos EG, Skidgel RA (1989) Neutral endopeptidase 24.11 (enkephalinase) and related regulators of peptide hormones. FASEB J 3:145–151
Lisy O, Jougasaki M, Schirger JA et al (1998) Neutral endopeptidase inhibition potentiates the natriuretic actions of adrenomedullin. Am J Physiol 275:F410–F414
Chappell MC, Allred AJ, Ferrario CM (2001) Pathways of angiotensin-(1-7) metabolism in the kidney. Nephrol Dial Transplant 16(Suppl. 1):22–26
Ferrario CM, Averill DB, Brosnihan KB et al (2002) Vasopeptidase inhibition and Ang-(1-7) in the spontaneously hypertensive rat. Kidney Int 62:1349–1357
Lebel N, D’Orleans-Juste P, Fournier A et al (1996) Role of the neutral endopeptidase 24.11 in the conversion of big endothelins in guinea-pig lung parenchyma. Br J Pharmacol 117:184–188
Plante M, Honorè JC, Neugebauer W et al (2002) Endothelin-1 (1-31) induces a thiorphan-sensitive release of eicosanoids via ETB receptors in the guinea pig perfused lung. Clin Sci 103(Suppl. 48):128S-131S
Dostal D, Baker KM (1999) The cardiac renin-angiotensin system: conceptual, or a regulator of cardiac function? Circ Res 85:643–650
Simard E, Jin D, Takai S et al (2009) Chymase-dependent conversion of big endothelin-1 in the mouse in vivo. J Pharmacol Exp Ther 328:540–548
Takai S, Jin D, Miyazaki M (2012) Multiple mechanisms for the action of chymase inhibitors. J Pharmacol Sci 118:311–316
Takai S, Sakaguchi M, Jin D et al (2001) Different angiotensin II-forming pathways in human and rat vascular tissues. Clin Chim Acta 305:191–195
Miyazaki M, Takai S (2001) Local angiotensin II-generating system in vascular tissues: the roles of chymase. Hypertens Res 24:189–193
Dell’Italia LJ, Ferrario CM (2013) The never-ending story of angiotensin peptides beyond angiotensin I and II. Circ Res 112:1086–1087
Ferrario CM, Ahmad S, Nagata S et al (2014) An evolving story of angiotensin II forming pathways in rodents and humans. Clin Sci 126:461–469
Nagata K, Kato J, Sasaki K et al (2006) Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun 350:1026–1031
Westwood BM, Chappell MC (2012) Divergent pathways for the angiotensin-(1-12) metabolism in the rat circulation and kidney. Peptides 35:190–195
Ferrario CM, Ahmad S, Varagic J et al (2016) Intracrine angiotensin II functions originate from noncanonical pathways in the human heart. Am J Physiol Heart Circ Physiol 311:H404–H414
Ferrario CM, Groban L, Wang H et al (2021) The angiotensin-(1-12)/chymase axis as an alternate component of the tissue renin angiotensin system. Mol Cell Endocrinol 529:111119. https://doi.org/10.1016/j.mce.2020.111119
Paik YH, Kim J, Aoyama T et al (2014) Role of NADPH oxidases in liver fibrosis. Antidox Redox Signal 20:2854–2872
Singh KD, Karnik SS (2016) Angiotensin receptors: structure, function signaling and clinical application. J Cell Signal 1. https://doi.org/10.4172/jcs.1000111
Metha PK, Griendling KK (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292:C82–C97
Barki-Harrington L, Luttrell LM, Rockman HA (2003) Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation 108:1611–1618
Karnik SS, Unal H, Kemp JR et al (2015) International Union of basic and clinical pharmacology. XCIX. Angiotensin receptors: interpreters of pathophysiological angiotensinergic stimuli. Pharmacol Rev 67:754–819
Shibata S, Ishizawa K, Uchida S (2017) Mineralocorticoid receptor as a therapeutic target in chronic kidney disease and hypertension. Hypertens Res 40:221–225
Nabeshima Y, Tazuma S, Kanno K et al (2006) Anti-fibrogenic function of angiotensin II type 2 receptor in CCl4-induced liver fibrosis. Biochem Biophys Res Commun 346:658–664
Albiston AL, McDowall SG, Matsacos D et al (2001) Evidence that the angiotensin IV (AT(4)) receptor is the enzyme insulin-regulated aminopeptidase. J Biol Chem 276:48623–48626
Nomura S, Ito T, Yamamoto E et al (2005) Gene regulation and physiological function of placental leucine aminopeptidase/oxytocinase during pregnancy. Biochim Biophys Acta 1751:19–25
Schleifenbaum J (2019) Alamandine and its receptor MrgD pair up to join the protective arm of the renin-angiotensin system. Front Med (Lausanne) 6:107. https://doi.org/10.3389/fmed.2019.00107
Re RN (2018) Role of intracellular angiotensin II. Am J Physiol Heart Circ Physiol 314:H766–H771
Villar-Cheda B, Costa-Besada MA, Valenzuela R et al (2017) The intracellular angiotensin system buffers deleterious effects of the extracellular paracrine system. Cell Death Dis 8:e3044. https://doi.org/10.1038/cddis.2017.439
Singh VP, Le B, Khode R et al (2008) Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 57:3297–3306
Wysocki J, Ye M, Batlle D (2015) Plasma and kidney angiotensin peptides: importance of the aminopeptidase A/angiotensin III axis. Am J Hypertens 28:1418–1426
Prieto I, Villarejo AB, Segarra AB et al (2015) Tissue distribution of CysAP activity and its relationship to blood pressure and water balance. Life Sci 134:73–78
Sansoè G, Aragno M, Mastrocola R et al (2018) Tissue renin-angiotensin system in the kidney of ascitic cirrhosis: an innocent bystander or a protagonist? Gastroenterology 154(6):S1178
Sansoè G, Wong F (2007) Natriuretic and aquaretic effects of i.v. infused calcium in pre-ascitic human cirrhosis: physiopathological and clinical implications. Gut 56:1117–1123
Sansoè G, Aragno M, Tomasinelli E et al (2010) Calcium-dependent diuretic system in preascitic liver cirrhosis. J Hepatol 53:856–862
Bataller R, Sancho-Bru P, Gines P et al (2003) Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 125:117–125
Jonsson JR, Clouston AD, Ando Y et al (2001) Angiotensin-converting enzyme inhibition attenuates the progression of rat hepatic fibrosis. Gastroenterology 121:148–155
Paizis G, Cooper ME, Schembri JM et al (2002) Up-regulation of components of the renin-angiotensin system in the bile duct-ligated rat liver. Gastroenterology 123:1667–1676
Hirose A, Ono M, Saibara T et al (2007) Angiotensin II type 1 receptor blocker inhibits fibrosis in rat nonalcoholic steatohepatitis. Hepatology 45:1375–1381
Gentilini P, Romanelli RG, La Villa G et al (1993) Effects of low-dose captopril on renal hemodynamics and function in patients with cirrhosis of the liver. Gastroenterology 104:588–594
Schepke M, Werner E, Biecker E et al (2001) Hemodynamics effects of the angiotensin II receptor antagonist irbesartan in patients with cirrhosis and portal hypertension. Gastroenterology 121:389–395
Grace JA, Klein S, Herath CB et al (2013) Activation of the MAS receptor by angiotensin-(1-7) in the renin-angiotensin system mediates mesenteric vasodilatation in cirrhosis. Gastroenterology 145:874–884
Klein S, Herath CB, Schierwagen R et al (2015) Hemodynamic effects of the non peptidic angiotensin-(1-7) agonist AVE0991 in liver cirrhosis. PLoS ONE 10:e0138732
Paizis G, Tikellis C, Cooper ME et al (2005) Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut 54:1790–1796
Osterreicher CH, Taura K, De Minicis S et al (2009) Angiotensin-converting-enzyme 2 inhibits liver fibrosis in mice. Hepatology 50:929–938
Mak KY, Chin R, Cunningham SC et al (2015) ACE2 therapy using adeno-associated viral vector inhibits liver fibrosis in mice. Mol Ther 23:1434–1443
Rajapaksa IG, Mak KY, Huang P et al (2018) The small molecule drug diminazene aceturate inhibits liver injury and biliary fibrosis in mice. Sci Rep 8:10175–10188
Wu HT, Chuang YW, Huang CP et al (2018) Loss of angiotensin converting enzyme II (ACE2) accelerates the development of liver injury induced by thioacetamide. Exp Anim 67:41–49
Sansoè G, Aragno M, Mastrocola R et al (2006) Overexpression of kidney neutral endopeptidase (EC 3.4.24.11) and renal function in experimental cirrhosis. Am J Physiol Renal Physiol 290:F1337–F1343
Bernardi M, Gulberg V, Colantoni A et al (1996) Plasma endothelin-1 and -3 in cirrhosis: relationship with systemic hemodynamics, renal function and neurohumoral systems. J Hepatol 24:161–168
Kohan DE (1997) Endothelins in the normal and diseased kidney. Am J Kidney Dis 29:2–26
Sansoè G, Aragno M, Mastrocola R et al (2005) Neutral endopeptidase (EC 3.4.24.11) in cirrhotic liver: a new target to treat portal hypertension? J Hepatol 43:791–798
Komeda K, Takai S, Jin D et al (2010) Chymase inhibition attenuates tetrachloride-induced liver cirrhosis in hamsters. Hepatol Res 40:832–840
Sansoè G, Aragno M, Mastrocola R et al (2016) Role of chymase in the development of liver cirrhosis and its complications: experimental and human data. PLoS ONE 11:e0162644. https://doi.org/10.1371/journal.pone.0162644
Pinzani M, Failli P, Ruocco C et al (1992) Fat-storing cells as liver specific pericytes. Spatial dynamics of agonist-stimulated intracellular calcium transients. J Clin Invest 90:642–646
Bataller R, Schwabe RF, Choi YH et al (2003) NAPDH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest 112:1383–1394
Kamada Y, Tamura S, Kiso S et al (2003) Angiotensin II stimulates the nuclear translocation of Smad2 and induces PAI-1 mRNA in rat hepatic stellate cells. Hepatol Res 25:296–305
Tian L, Li W, Yang L et al (2017) Cannabinoid receptor 1participates in liver inflammation by promoting M1 macrophage polarization via RhoA/NF-kappaB p65 and ERK1/2 pathways, respectively, in mouse liver fibrogenesis. Front Immunol 8:1214. https://doi.org/10.3389/fimmu.2017.01214
Calò LA, Ravarotto V, Simioni F et al (2017) Pathophysiology of post transplant hypertension in kidney transplant: focus on calcineurin inhibitors induced oxidative stress and renal sodium retention and implications with RhoA/RhoKinase pathway. Kidney Blood Press Res 42:676–685
Chen Y, Tian Y (2019) Influence of miR-26b on hepatic cirrhosis and portal pressure in rats with cirrhotic portal hypertension by targeting hENT1 depending on RhoA/ROCK-1 pathway. Eur Rev Med Pharmacol Sci 23:1668–1673
Bernstein KE, Fuchs S (2010) Angiotensin II and JAK2 put on the pressure. Nat Med 16:165–166
Akcora BO, Dathathri E, Ortiz-Perez A et al (2019) TG101348, a selective JAK2 antagonist, ameliorates hepatic fibrogenesis in vivo. FASEB J 33:9466–9475
Matsuyama T, Kubli SP, Yoshinaga SK et al (2020) An aberrant STAT pathway is central to COVID-19. Cell Death Differ 27:3209–3225
Bai L, Zhou H, Xu R et al (2019) A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 36:498–511
Oakley F, Teoh V, Ching-A-Sue G et al (2009) Angiotensin II activates IκB kinase phosphorylation of Rela at Ser536to promote myofibroblast survival and liver fibrosis. Gastroenterology 136:2334–2344
Wong F, Sniderman K, Blendis L (1998) The renal sympathetic and renin-angiotensin response to lower body negative pressure in well-compensated cirrhosis. Gastroenterology 115:397–405
Girgrah N, Liu P, Collier J et al (2000) Hemodynamic, renal sodium handling, and neurohormonal effects of acute administration of low dose losartan, an angiotensin II receptor antagonist, in preascitic cirrhosis. Gut 46:114–120
Wong F, Liu P, Blendis L (2002) The mechanism of improved sodium homeostasis of low-dose losartan in preascitic cirrhosis. Hepatology 35:1449–1458
Ichikawa I, Pfeffer JM, Pfeffer MA et al (1984) Role of angiotensin II in the altered renal function in congestive heart failure. Circ Res 55:669–675
Banday AA, Lokhandwala MF (2011) Oxidative stress causes renal angiotensin type 1 receptor upregulation, Na+/H+ exchanger 3 overstimulation, and hypertension. Hypertension 57:452–459
Vlachogiannakos J, Tang AKW, Patch D et al (2001) Angiotensin converting enzyme inhibitors and angiotensin II antagonists as therapy in chronic liver disease. Gut 49:303–308
Schneider AW, Kalk JF, Klein CP (1999) Effect of losartan, an angiotensin II receptor antagonist, on the portal pressure in cirrhosis. Hepatology 29:334–339
Yao H, Zhang C (2018) angiotensin II receptor blockers for the treatment of portal hypertension in patients with liver cirrhosis: a systematic review and meta-analysis of randomized controlled trials. Ir J Med Sci 187:925–934
Pariente EA, Bataille C, Bercoff E et al (1985) Acute effect of captopril on systemic and renal hemodynamics and on renal function in cirrhotic patients with ascites. Gastroenterology 88:1255–1259
Ibarra FR, Afione C, Garzon D et al (1992) Portal pressure, renal function, and hormonal profile after acute and chronic captopril treatment in cirrhosis. Eur J Clin Pharmacol 43:477–482
Eriksson LS, Kagedal B, Wahren J (1984) Effect of captopril on hepatic venous pressure and blood flow in patients with liver cirrhosis. Am J Med 76:66–70
Tandon P, Abraldes JG, Berzigotti A et al (2010) Renin-angiotensin-aldosterone inhibitors in the reduction of portal pressure: a systematic review and meta-analysis. J Hepatol 53:273–282
Pironti G, Strachan RT, Abraham D et al (2015) Circulating exosomes induced by cardiac pressure overload contain functional angiotensin II type 1 receptors. Circulation 131:2120–2130
Bosnyak S, Jones ES, Christopoulos A et al (2011) Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin Sci 121:297–303
Tamargo M, Tamargo J (2017) Future drug discovery in renin-angiotensin-aldosterone system intervention. Expert Opin Drug Discov 12:827–848
Huang Y, Yang Li Y, Lou A et al (2020) Alamandine attenuates hepatic fibrosis by regulating autophagy induced by NOX 4-dependent ROS. Clin Sci 134:853–869
Alukal JJ, Savio J, Thuluvath PJ et al (2020) Hyponatremia in cirrhosis: an update. Am J Gastroenterol 115:1775–1785
Jiménez W, Martinez-Pardo A, Arroyo V et al (1985) Temporal relationship between hyperaldosteronism, sodium retention and ascites formation in rats with experimental cirrhosis. Hepatology 5:245–250
Sansoè G, Ferrari A, Baraldi E et al (1999) Renal distal tubular handling of sodium in central fluid volume homoeostasis in preascitic cirrhosis. Gut 45:750–755
Sawathiparnich P, Murphey LJ, Kumar S et al (2003) Effect of combined AT1 receptor and aldosterone receptor antagonism on plasminogen activator inhibitor-1. J Clin Endorinol Metab 88:3867–3873
Wang H, Zhang Y, Heuckeroth RO (2007) PAI-1 deficiency reduces liver fibrosis after bile duct ligation in mice through activation of tPA. FEBS Lett 581:3098–3104
Ghosh AK, Vaughan DE (2012) PAI-1 in tissue fibrosis. J Cell Physiol 227:493–507
Megias MJ, Alba-Araguez F, Luna JD et al (2015) Serum pyroglutamyl aminopeptidase activity: a promising novel biomarker candidate for liver cirrhosis. Endocr Regul 49:20–24
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
None to declare.
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sansoè, G., Wong, F. (2023). The Classical and Nonclassical Renin-Angiotensin-Aldosterone System in Liver Cirrhosis. In: Bhullar, S.K., Tappia, P.S., Dhalla, N.S. (eds) The Renin Angiotensin System in Cancer, Lung, Liver and Infectious Diseases. Advances in Biochemistry in Health and Disease, vol 25. Springer, Cham. https://doi.org/10.1007/978-3-031-23621-1_11
Download citation
DOI: https://doi.org/10.1007/978-3-031-23621-1_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-23620-4
Online ISBN: 978-3-031-23621-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)