
Abstract
DNA methylation is a hot topic in basic and biomedical research. Despite tremendous progress in understanding the structures and biochemical properties of the mammalian DNA methyltransferases (DNMTs), principles of their targeting and regulation in cells have only begun to be uncovered. In mammals, DNA methylation is introduced by the DNMT1, DNMT3A, and DNMT3B enzymes, which are all large multi-domain proteins containing a catalytic C-terminal domain and a complex N-terminal part with diverse targeting and regulatory functions. The sub-nuclear localization of DNMTs plays an important role in their biological function: DNMT1 is localized to replicating DNA and heterochromatin via interactions with PCNA and UHRF1 and direct binding to the heterochromatic histone modifications H3K9me3 and H4K20me3. DNMT3 enzymes bind to heterochromatin via protein multimerization and are targeted to chromatin by their ADD, PWWP, and UDR domains, binding to unmodified H3K4, H3K36me2/3, and H2AK119ub1, respectively. In recent years, a novel regulatory principle has been discovered in DNMTs, as structural and functional data demonstrated that the catalytic activities of DNMT enzymes are under a tight allosteric control by their different N-terminal domains with autoinhibitory functions. This mechanism provides numerous possibilities for the precise regulation of the methyltransferases via controlling the binding and release of the autoinhibitory domains by protein partners, chromatin interactions, non-coding RNAs, or posttranslational modifications of the DNMTs. In this chapter, we summarize key enzymatic properties of DNMTs, viz. their specificity and processivity, and afterwards focus on the regulation of their activity and targeting via allosteric processes, protein interactions, and posttranslational modifications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- DNA methyltransferase
- DNMT1
- DNMT3A
- DNMT3B
- Enzyme mechanism
- Enzyme regulation
- Enzyme specifcity
- DNA methylation
4.1 Introduction
The expression of genes in multicellular organisms is coordinated during development and cellular differentiation by epigenetic information comprising DNA methylation, histone tail posttranslational modifications (PTMs), and non-coding RNAs [for general reviews on Molecular Epigenetics cf. (Allis and Jenuwein 2016)]. In mammals, DNA methylation mainly occurs at the C5-position of the cytosine residues, primarily in CpG dinucleotide sequences [for general reviews on DNA methylation cf. (Ambrosi et al. 2017; Schubeler 2015; Jeltsch and Jurkowska 2014)]. However, only certain CpG sites are methylated, resulting in the establishment of a tissue- and cell type-specific pattern of DNA methylation consisting of modified and unmodified sites. In different cell types, approximately 60–80% of all CpGs in the human genome are modified (3–8% of all cytosines). Notably, the correct methylation pattern is essential for development and human health, and several diseases, including cancer, are associated with aberrant DNA methylation [for reviews cf. (Zhao et al. 2021; Weinberg et al. 2019; Bergman and Cedar 2013; Suva et al. 2013; Hamidi et al. 2015)].
In mammals, DNA methylation patterns are introduced during early development and maturation of germ cells by DNA methyltransferases (MTases) DNMT3A and DNMT3B, with the help of the stimulatory factor DNMT3L (Jurkowska et al. 2011a; Jeltsch and Jurkowska 2016). DNMT3A and DNMT3B have been traditionally designated as de novo DNA MTases, as they do not display any significant preference between hemimethylated and unmethylated DNA (Okano et al. 1998; Gowher and Jeltsch 2001). In agreement with this role, they are highly expressed in undifferentiated cells and germ cell precursors, and present at much lower levels in somatic cells. In the cell nucleus, they localize to pericentromeric heterochromatin (Chen et al. 2004; Ge et al. 2004; Barau et al. 2016), where they are tightly bound to nucleosomes containing methylated DNA (Jeong et al. 2009; Sharma et al. 2011). Mice and other rodents also contain an additional DNMT3B-related DNA methyltransferase called DNMT3C, which is specifically expressed in testis. It is required for methylation and silencing of retrotransposons during spermatogenesis and hence critical for male fertility in mice (Barau et al. 2016; Jain et al. 2017).
After their establishment, DNA methylation patterns are perpetuated through cell divisions, with small tissue-specific changes. The palindromic nature of the CpG sites provides an elegant mechanism for the inheritance of the DNA methylation mark because the methylation information is encoded in both DNA strands. During DNA replication the fully methylated CpG sites are converted into a hemimethylated state, with the parental strand carrying the original methylation marks and the daughter strand devoid of methylation. The methylation pattern is copied after each round of DNA replication by the maintenance methyltransferase DNMT1. This enzyme is present at the replication fork, where it quickly methylates hemimethylated CpG dinucleotides, thereby restoring the original DNA methylation pattern (Petryk et al. 2021). DNMT1 is ubiquitously and highly expressed in proliferating cells, representing the major DNA MTase activity in somatic tissues throughout mammalian development, but it is present only at low levels in non-dividing cells (Robertson et al. 1999).
However, recent data showed that this traditional division of tasks into de novo and maintenance methyltransferases is an oversimplification. DNA methylation is more correctly described as a dynamic process of ongoing methylation and demethylation, and DNMT1, DNMT3A, and DNMT3B all play roles in both de novo and maintenance methylation (Jeltsch and Jurkowska 2014). Hence, the dynamic regulation and targeting of DNMTs and Ten-eleven translocation (TET) DNA demethylating enzymes controls the methylation state of each CpG site, thereby governing all the biological processes associated with DNA methylation. Consequently, the complex role of DNA methylation in human biology cannot be decoded without a thorough mechanistic understanding of the properties of the DNMTs, including their regulation, targeting, and interaction with chromatin and other epigenetic factors.
4.2 General Features of Mammalian DNMTs
4.2.1 Structure and Domain Composition of Mammalian DNMTs
Structural and biochemical data provided compelling evidence that the arrangement of the specific domains in DNMTs plays a central role in the regulation of the biological functions of these enzymes. The general architecture of all mammalian DNMTs is similar. They all are multi-domain proteins, in which two functional parts can be distinguished, a large N-terminal regulatory part and a smaller C-terminal part, required for catalysis (Fig. 4.1) (Jeltsch 2002; Hermann et al. 2004a; Jurkowska et al. 2011a). The N-terminal parts of variable size are different between DNMT1 and DNMT3 proteins. They guide the nuclear localization of the enzymes and mediate their interaction with other proteins, regulatory nucleic acids (like non-coding RNAs), and chromatin. They are also subject to posttranslational modifications (PTMs) and are involved in the allosteric regulation of the enzymes’ activity and specificity.

Domain structure of the mammalian DNMT enzymes. Abbreviations used: DMAPD DNA methyltransferase-associated protein 1 interacting domain, PDB PCNA binding domain, RFTD replication foci targeting domain, CXXC CXXC domain, BAH1 and BAH2 Bromo-adjacent homology domains 1 and 2, GK glycine lysine repeats, UBR ubiquitin-dependent recruitment domain, PWWP PWWP domain, ADD ATRX-DNMT3-DNMT3L domain. DNMT3C is a rodent-specific enzyme
The C-terminal domains harboring the catalytic centers of the enzymes are required for binding of the S-adenosyl-L-methionine (AdoMet) cofactor and the DNA substrate. They contain ten conserved amino acid motifs characteristic of the common structure of all DNA-(cytosine-C5)-MTases, called the “AdoMet-dependent MTase fold”, which consists of a mixed seven-stranded β-sheet, formed by six parallel β-strands and a seventh strand inserted in an anti-parallel orientation into the sheet between strands 5 and 6. Six α-helices surround the central β-sheet on both sides (Cheng and Blumenthal 2008; Jeltsch 2002). The C-terminal domain is involved in the cofactor binding (motifs I and X), binding of the flipped substrate cytosine base, and the methyl group transfer (motifs IV, VI, and VIII). The non-conserved region between motifs VIII and IX, the so-called target recognition domain (TRD), is involved in substrate DNA recognition and specificity.
4.2.2 Catalytic Mechanism of C5-MTases
DNA-(cytosine C5)-methyltransferases catalyze the transfer of the methyl group from an AdoMet cofactor molecule to the C5-position of cytosine residues. In this reaction, 5-methylcytosine (5mC) is created and the AdoMet is converted into S-adenosyl-L-homocysteine (AdoHcy), which is then released from the enzyme. The transfer of the activated methyl group from AdoMet to the C5-position of the cytosine requires a close contact between the enzyme’s active site and the substrate base. Such proximity is not possible while the base is located in the DNA double helix; therefore, DNA methyltransferases flip their target base out of the DNA during catalysis and bury it into a hydrophobic pocket of their active center. This base flipping mechanism was first discovered in 1994 for the bacterial DNA C5-MTase M.HhaI (Klimasauskas et al. 1994). Later, it became clear that it is common to all DNA methyltransferases, including the mammalian enzymes (Cheng and Roberts 2001; Jeltsch 2002) and flipping of the cytosine base was observed in different crystal structures of DNMT1, DNMT3A, and DNMT3B with bound substrate DNA (Song et al. 2012; Adam et al. 2020; Zhang et al. 2018; Gao et al. 2020b; Lin et al. 2020).
The methylation of the C5-position of cytosine is not an easy chemical task, because the cytosine is an electron-poor aromatic system. Therefore, its C5-atom is not intrinsically reactive and it will not attack the activated methyl sulfonium group of the AdoMet spontaneously. Hence, a key step in the catalysis of DNA-(cytosine C5)-methyltransferases is the nucleophilic attack of the catalytic cysteine residue located in a PCQ motif (motif IV) on the C6 position of the cytosine ring, leading to the formation of a covalent bond between the enzyme and the substrate base. Thereby, the negative charge density at the C5-atom of the cytosine increases, so that it can attack the methyl group of the cofactor. It has been postulated that the nucleophilic attack of the cysteine might be facilitated by a transient protonation of the cytosine ring at the endocyclic nitrogen atom (N3) by an enzyme-derived acid; the conserved glutamate residue from an ENV motif (motif VI) has been proposed to carry out this reaction. In addition, it stabilises the flipped cytosine by forming an H-bond to the N4-amino group. The second arginine residue from an RXR motif (motif VIII) may be involved in the stabilization of both the glutamate and the cytosine base as well. The addition of the methyl group to the base is followed by a deprotonation of the C5-atom, catalyzed by a so far unknown proton acceptor, which resolves the covalent bond between the enzyme and the base in an elimination reaction and re-establishes aromaticity (Cheng and Roberts 2001; Jeltsch 2002). For DNMT1, kinetic isotope effects confirmed this two-step mechanism (Du et al. 2016). For DNMT3A, mutations of the key catalytic residues reduced the catalytic activity, confirming their critical role in catalysis (Reither et al. 2003; Gowher et al. 2006; Lukashevich et al. 2016).
Unexpectedly, DNA-(cytosine C5)-methyltransferases, including DNMT3A, also introduce low levels of methylation at the N3-atom of the cytosine ring, forming 3-methylcytosine (3mC) (Rosic et al. 2018), which is a toxic DNA alkylation lesion that interferes with RNA synthesis and DNA replication. 3mC is removed by members of the ALKB2 family of DNA alkylation repair enzymes that are evolutionarily strongly connected to DNA methyltransferases, reflecting their close functional link (Rosic et al. 2018). Mechanistically, the 3mC methylation is likely introduced after positioning the flipped cytosine base in an inverted conformation into the active site pocket of the DNMT (Dukatz et al. 2019b). Further mechanistic details of DNMTs, including their sequence specificity, processivity, oligomerization, and the mechanism of DNA and chromatin binding will be discussed below.
4.2.3 Regulation and Targeting of DNMTs
Despite tremendous progress in understanding the biochemical properties of the mammalian DNA methyltransferases, their genomic targeting combined with regulation of their activity is still insufficiently understood. Recent discoveries demonstrated the involvement of the N-terminal parts of the mammalian DNMTs in enzyme targeting and regulation. In this context, different domains of DNMT3A, DNMT3B, and DNMT1 were shown to directly bind modified histone H3 tails. Moreover, various domains (ADD domain in DNMT3A and CXXC and RFT domains in DNMT1) engage in autoinhibitory interactions with the catalytic domain, demonstrating that the activity of the enzymes is under precise allosteric control. Similarly, the interactions of the N-terminal domains of DNMTs with other proteins regulate the enzymes’ activities and genome targeting. Thus, allosteric control represents a unifying concept in the regulation of DNMTs, which sets the stage for additional regulatory cues. By influencing the allosteric conformational changes of DNMTs, interacting proteins or RNAs, chromatin modifications or PTMs can affect key enzymatic properties of DNMTs, including their activity and eventually specificity (Jeltsch and Jurkowska 2016).
Several interaction partners of DNMTs have been described so far and their effect on the MTases has been studied mechanistically. This includes PCNA (Chuang et al. 1997), DNMT3L (Bourc'his et al. 2001; Hata et al. 2002; Chedin et al. 2002; Gowher et al. 2005a), UHRF1 (Sharif et al. 2007; Bostick et al. 2007; Meilinger et al. 2009), MeCP2 (Fuks et al. 2003b; Kimura and Shiota 2003; Rajavelu et al. 2018), p53 (Wang et al. 2005; Sandoval and Reich 2019), or USP7 (Du et al. 2010; Felle et al. 2011). Other important interaction partners like HP1-beta (Fuks et al. 2003a), Mbd3 (Datta et al. 2005), MYC (Brenner et al. 2005), PU.1 and RP58 transcription factors (Suzuki et al. 2006; Fuks et al. 2001), zinc-finger proteins ZHX1 and Trim28 (Kim et al. 2007; Quenneville et al. 2011), protein lysine methyltransferases (PKMTs) G9a, SUV39H1 (Fuks et al. 2003a), EZH2 (Vire et al. 2006), and SETDB1 (Li et al. 2006), histone deacetylase (HDAC1) (Fuks et al. 2000; Fuks et al. 2001), and remodeling factors HELLS (Myant and Stancheva 2008; Zhu et al. 2006), SMARCA4 (Datta et al. 2005), and hSNF2 (Geiman et al. 2004) have been reported, but their interaction with DNMTs has not yet been mechanistically investigated in great details. Finally, various aspects of the biological function of DNMTs, including their targeting and activity in cells, are regulated by posttranslational modifications (PTMs). Until now, several PTMs, including phosphorylation, acetylation, ubiquitination, SUMOylation, and methylation, have been identified on mammalian DNMTs in proteomic studies (http://www.phosphosite.org). PTMs are ideally suited to mediate regulation of DNMTs’ function, either by direct effects on catalytic activity or by recruiting modification-specific readers that could influence the enzymes’ stability, activity, localization, or interaction with other proteins. Notably, the few modifications that have been functionally characterized revealed the important regulatory potential of the PTMs, opening the field for future research.
Finally, non-coding RNA (ncRNA) is an emerging player in chromatin regulation (Holoch and Moazed 2015; Rinn and Chang 2012) and RNA molecules have been shown to influence DNA methylation. In plants, a process of RNA-dependent DNA methylation exists, in which the RNA sequence directly guides DNA methylation (Matzke and Mosher 2014). Though this pathway is absent in mammals, binding of small and long non-coding RNAs (lncRNA) to mammalian DNMTs has been shown to guide and regulate their activity. In addition, the piRNA-mediated DNA methylation in the germline of many animals, including mammals (Iwasaki et al. 2015), recapitulates many features of an RNA-directed DNA methylation pathway. Recently, SPOCD1 has been identified to bind to the PIWI protein MIWI2 and DNMT3A/DNMT3L and to play an essential role in targeting DNA methylation to piRNA binding sites (Zoch et al. 2020), but many further details of piRNA-directed DNA methylation process are not yet well understood at the molecular level. The direct regulation of DNA methylation by genome-encoded non-coding RNAs adds another fascinating dimension to the complex interplay between the genetic information (encoded in the DNA sequence) and the epigenetic information (encoded in the chromatin modification pattern, including DNA methylation), urging more research in this direction.
4.3 Structure, Function, and Mechanism of DNMT1
4.3.1 Domain Composition of DNMT1
DNMT1 is a large enzyme, comprising 1620 amino acids in mice and 1616 amino acids in humans, but different isoforms of DNMT1, resulting from alternative splicing or use of an alternative promoter have been described (Hermann et al. 2004a; Jurkowska et al. 2011a). DNMT1 contains multiple functional domains located in the N-terminal part that is joined to the C-terminal part by a flexible linker composed of lysine-glycine (KG) repeats (Fig. 4.1). The N-terminal part serves as a platform for the assembly of various proteins involved in the control of chromatin structure and gene regulation.
The very N-terminus of DNMT1 contains the DNA methyltransferase-associated protein 1 (DMAP1) interaction domain that is involved in the interaction of DNMT1 with DMAP1, a transcriptional repressor, mediating the stability of DNMT1 in cells (Rountree et al. 2000). Next to it, the proliferating cell nuclear antigen (PCNA) binding domain (PBD) has been mapped (Chuang et al. 1997). The interaction with PCNA is involved in the targeting and tethering of DNMT1 to the replication fork during S-phase, which supports DNA methylation in the cell (Egger et al. 2006). The same region also contains an AT-hook-like DNA binding motif (Suetake et al. 2006). The replication foci targeting domain (RFTD) following next is involved in the targeting of DNMT1 to replication foci (Leonhardt et al. 1992) and centromeric chromatin (Easwaran et al. 2004). This domain interacts with UHRF1 (ubiquitin-like with PHD and ring finger domains 1), which harbors an SRA (SET and RING-associated) domain that specifically binds to hemimethylated DNA (see below). Moreover, the RFTD binds to ubiquitinated histone H3 tails, a modification introduced by the RING domain of UHRF1 (Nishiyama et al. 2013; Qin et al. 2015), in the context of H3K9me3 (Ren et al. 2020), a major heterochromatic histone PTM in the mammalian epigenome (Jeltsch et al. 2019). Next, the N-terminal part of DNMT1 contains a CXXC domain that binds unmethylated DNA and is implicated in DNMT1 regulation (Pradhan et al. 2008; Song et al. 2011; Bashtrykov et al. 2012a). The CXXC domain is followed by the BAH1 and BAH2 (Bromo-adjacent homology 1 and 2) domains. BAH1 binds H4K20me3 (Ren et al. 2021), another key heterochromatic histone PTM. Hence, through its N-terminal part, DNMT1 interacts with other proteins and specific histone marks, contributing to the crosstalk between DNA methylation and other epigenetic modifications.
The C-terminal domain of DNMT1 contains the catalytic center of the enzyme, but is not active in an isolated form, both in vitro and in vivo, despite the presence of all motifs required for catalysis (Fatemi et al. 2001; Margot et al. 2003). The structural arrangement of the particular domains in DNMT1 has been revealed by crystallographic studies (Song et al. 2011, 2012; Takeshita et al. 2011; Syeda et al. 2011) (Fig. 4.2). They demonstrated that the various domains in the N-terminal part of DNMT1 contact the C-terminal catalytic domain from different sides, explaining why the isolated terminal domain lacks catalytic activity.

Structures of DNMT1 with different N-terminal domains. (a) DNMT1 in an active conformation with DNA (green) bound in the active site (Song et al. 2012) (pdb 4D4A). Removal of the autoinhibitory RFTD can be triggered by UHRF1 interaction (Berkyurek et al. 2014; Bashtrykov et al. 2014a). (b) DNMT1 with unmethylated DNA bound to the autoinhibitory CXXC domain (Song et al. 2011) (pdb 3PTA). (c) DNMT1 with the RFT domain blocking access to the active site (Takeshita et al. 2011) (pdb 3AV4). (d) Flanking sequence-dependent base flipping mechanism observed in different DNMT1 structures. In the CCG structure (Adam et al. 2020) (pdb 6W8W) only the target cytosine (light blue) is rotated out of the double helix and bound by the enzyme (symbolized by the orange circle). In the ACG structure (Adam et al. 2020) (pdb 6W8V), both the target cytosine and the orphaned G (dark blue) are rotated out of the double helix and in the GCG structure (Song et al. 2012) (pdb 4DA4), the orphaned G forms a non-canonical base pair with the G(−1) and the corresponding C(−1′) (pink) is flipped out in the opposite direction. Catalytic activity of the complexes was inversely correlated with the extent of the conformational changes of the DNA upon complex formation. DNMT1 is symbolized by an orange circle. Reprinted in modified version from Jeltsch et al. (2021) with permission from Elsevier
4.3.2 Structures of DNMT1 and Allosteric Regulation
In recent years, several structures of truncated DNMT1 proteins (lacking various parts of the N-terminus) have been solved (Song et al. 2011, 2012; Takeshita et al. 2011; Zhang et al. 2015b; Adam et al. 2020). They all confirmed that the catalytic domain of DNMT1 adopts the typical AdoMet-dependent MTase fold described above. These studies also revealed that the enzyme unexpectedly undergoes large domain rearrangements, which allosterically regulate its catalytic activity (Fig. 4.2).
A DNMT1 C-terminal fragment lacking the RFT and CXXC domains adopted an open conformation, in which the enzyme was able to bind the hemimethylated substrate DNA (with a GGCGGC sequence) and showed high catalytic activity (Song et al. 2012) (Fig. 4.2a). This complex represented a real breakthrough in the field, as it provided the first example of a mammalian DNMT structure solved with substrate DNA bound in the active site. As expected, it showed the target cytosine flipped out of the DNA helix and bound to DNMT1 in a manner reminiscent of other DNA MTases. Moreover, this structure also revealed additional unforeseen rearrangements in the DNMT1–DNA structure, including the formation of a non-Watson/Crick base pair of the orphan G residue with a G flanking the CpG site. The (then orphaned) C of the flanking G:C base pair was rotated out of the DNA helix in a direction roughly opposite to the target C flipping (Fig. 4.2d, GCG complex). Several contacts of the enzyme to the target CpG site observed in the structure were validated in kinetic studies as essential for the enzyme activity and the recognition of the CpG site (described in detail below in Sect. 4.3.3) (Bashtrykov et al. 2012b).
Additional recent structures of this DNMT1 fragment in complex with different DNA sequences strikingly revealed a DNA sequence-dependent base flipping mechanism (Adam et al. 2020) (Fig. 4.2d). The structure of DNMT1 bound to a hemimethylated TACGGA substrate showed flipping of the target C and its Watson/Crick partner G, but no formation of a non-canonical base pair. In turn, the structure of DNMT1 bound to a hemimethylated TCCGTA substrate only showed target base flipping (Adam et al. 2020). A kinetic analysis uncovered strong differences in the methylation rates of DNMT1 depending on the sequences flanking the target CG site. A comparison of the kinetic and structural data showed that the extent of the conformational rearrangements during base flipping was anti-correlated with the methylation rates of the corresponding substrates.
A structure of a larger C-terminal fragment of DNMT1 also containing the CXXC domain showed a CpG site-specific binding of an unmethylated DNA, but this interaction surprisingly occurred not at the C-terminal domain containing the active center, but at the CXXC domain (Song et al. 2011) (Fig. 4.2b). This observation led to the proposal that the CXXC domain has an autoinhibitory function and acts as a specificity filter in DNMT1 by preventing unmethylated DNA from accessing the active site. Kinetic experiments with this DNMT1 version indeed revealed an influence of the CXXC domain on the specificity of DNMT1 (Song et al. 2011). Surprisingly, similar experiments conducted with the full-length DNMT1 did not provide evidence for a role of the CXXC domain in the specificity of DNMT1 (Bashtrykov et al. 2012a), indicating that this point deserves further attention.
Finally, a crystal structure of an almost complete DNMT1 fragment, but without DNA provided additional seminal insight into the mechanism of DNMT1 by showing that the RFT domain inhibits the enzyme through binding to the active site cleft of the catalytic domain (Takeshita et al. 2011) (Fig. 4.2c). The autoinhibition was observed in biochemical studies as well (Takeshita et al. 2011; Syeda et al. 2011) and engineering of this interface altered the conformation of DNMT1, generating a methyltransferase that was hyperactive in vitro and in cells (Bashtrykov et al. 2014b).
Importantly, the arrangement of different domains in DNMT1 is controlled by long linker regions, which form tight interactions with surface clefts of the domains. Both the linkers and the clefts are subject to many reported PTMs in DNMT1, including phosphorylation, acetylation, and ubiquitination (http://www.phosphosite.org), which might directly control the positioning of these domains in DNMT1 and thereby enzymatic activity. Accumulating evidence indicates that the autoinhibitory mechanism of the RFT domain plays a central role as an allosteric trigger in DNMT1 (Fig. 4.2) that can be influenced by protein partners and chromatin binding. Indeed, the interaction of the RFTD with UHRF1 stimulates the activity of DNMT1 by relieving autoinhibition (Berkyurek et al. 2014; Bashtrykov et al. 2014a). Similarly, its interaction with ubiquitinated H3 and H3K9me3 also leads to DNMT1 activation (Nishiyama et al. 2013; Qin et al. 2015; Ren et al. 2020) and the binding of H4K20me3 to the BAH1 domain modulates the conformation of autoinhibitory linker regions connecting the different domains of DNMT1 (Ren et al. 2021).
Structural studies combined with molecular dynamics simulations showed that the helix following the catalytic loop in DNMT1 can adopt either a kinked or straight conformation. Mutational data suggested that the structural transition between these states is necessary for DNMT1 activity (Ye et al. 2018). Later, it was shown that these conformational changes are also dependent on the DNA sequence flanking the target sites and that the most active complex shows the least conformational changes (Adam et al. 2020). Hence, protein partners and chromatin interactions can regulate DNMT1 activity by influencing the allosteric conformation of the enzyme.
4.3.3 Specificity of DNMT1
DNMT1 shows a preference for hemimethylated DNA over unmethylated substrates, supporting its role as a maintenance MTase (Bashtrykov et al. 2012a; Bashtrykov et al. 2012b; Fatemi et al. 2001; Goyal et al. 2006; Song et al. 2012). Its intrinsic preference for hemimethylated DNA has been estimated to be about 30–40 fold (Jeltsch 2006), but it depends on the exact substrate sequence, its length, and the reaction conditions. This preference has been investigated for decades, as it is one of the mechanistic foundations of the role of DNA methylation in the transfer of epigenetic information. We know now that it is molecularly based on the sequence-specific interaction of hemimethylated CpG sites with the active center of the enzyme that is mediated by the interaction of the methyl group with a hydrophobic pocket formed by the enzyme (Song et al. 2012). More precisely, the methyl group of the 5mC is placed into a pocket formed by C1501, L1502, W1512, L1515, and M1535, which explains the preference of the enzyme for hemimethylated target sites. Further details of this process could be uncovered once a structure of DNMT1 with an unmethylated DNA bound to the active center becomes available. The recognition of the 5mC-G base pair is based on side-chain- and backbone-mediated H-bonds of M1535, K1537, Q1538, and R1237 to the edges of the CpG base pair in the major and minor groove (Song et al. 2012). These interactions explain why the 5mC and the corresponding G in the target DNA strand are very accurately recognized by DNMT1 and cannot be exchanged by other nucleotides (Bashtrykov et al. 2012b). The requirement for a close contact between the catalytic domain of DNMT1 and its substrate DNA also explains the finding that the activity of DNMT1 on nucleosomal DNA is restricted to the linker DNA regions (Mishima et al. 2017).
Two recent studies investigated DNA replication-coupled maintenance of DNA methylation by DNMT1, providing novel evidence for de novo methylation activity of DNMT1 post-replication (Wang et al. 2020b; Ming et al. 2020). Genetic studies showed that the de novo activity of DNMT1 is particularly strong at intracisternal A particles (IAP) retrotransposons, possibly contributing to their stable silencing (Haggerty et al. 2021). This activity was dependent on UHRF1 acting as a universal cofactor of DNMT1, as well as H3K9me3 and TRIM28, suggesting that it crosstalks with the KRAB/TRIM28/SETDB1 silencing complex, which delivers H3K9me3 at retrotransposons (Haggerty et al. 2021; Markouli et al. 2021). In this context, H3K9me3 interaction could be mediated by the tandem Tudor domain (TTD) of UHRF1 (Nady et al. 2011) or by its direct interaction with the RFT domain of DNMT1 (Ren et al. 2020) (see below).
Recent evidence suggests that besides specificity for hemimethylated sites, Dnmt1 also has a preference for certain sequence contexts flanking the target CpGs. Biochemical experiments investigating the methylation of CpG sites in a randomized sequence context uncovered about 100-fold differences in the methylation rates of hemimethylated CpG sites placed in a variable NNCGNN sequence context (Adam et al. 2020). A comparison of the DNMT1–DNA structures on preferred and disfavored substrates with the kinetic data revealed the mechanistic basis for some of the observed flanking sequence preferences (Adam et al. 2020; Jeltsch et al. 2021). The disfavor for a G in the target strand in the −1 flanking base pair can be explained, because it allows the formation of the non-canonical G-G base pair with the orphaned G, which is accompanied by base flipping of the C in the non-target strand seen in the low-activity GGCGGC complex. The observed disfavor for a G in the non-target strand at the −2 flanking base pair could be explained because it could stack to the non-canonical G:G base pair and further stabilize this low-activity conformation. In turn, the preference for a G in the non-target strand at the −1 site could be related to its ability to stack with the orphaned G, keeping it inside of the DNA helix and thereby stabilizing the highly active conformation seen in the TCCGTA complex. In addition to these direct effects, minor groove width at the +1 to +3 flank correlated with DNMT1 activity as well. Notably, the comparison with genomic methylation data from various sources showed that the flanking sequence preferences of DNMT1 highly correlate with the flanking site-dependent modulation of genomic DNA methylation levels in human and mouse cells, indicating that the preferences determined in vitro affect genomic DNA methylation patterns in cells (Adam et al. 2020).
4.3.4 Processivity of DNMT1
DNMT1 is a highly processive enzyme, able to methylate long stretches of hemimethylated DNA without dissociation from the substrate, a property that fits perfectly to its function as a molecular copy machine at the replication fork (Goyal et al. 2006; Hermann et al. 2004b; Vilkaitis et al. 2005). A recent study revealed that DNMT1 undergoes a conformational change after DNA binding from an open into a closed conformation capable of processive methylation. Once the enzyme has adopted the closed conformation, it has a 97% chance of staying on the DNA and continuing processive DNA methylation after each methylation event (Adam et al. 2020). Interestingly, processive methylation is possible only in one strand of the DNA, which indicates that DNMT1 does not exchange DNA strands while moving along its substrate (Hermann et al. 2004b). These biochemical findings are in perfect agreement with the structure of DNMT1 with bound substrate DNA (Song et al. 2012), showing that the enzyme enwraps the DNA, which enables it to slide along the substrate and catalyze several successive methylation reactions without dissociation from the DNA. Due to its high processivity, DNMT1 is a very effective enzyme, ideally suited to follow DNA replication and methylate the newly synthetized DNA strand before the chromatin is reassembled.
4.3.5 Allosteric Regulation and Targeting of DNMT1
The sub-nuclear localization of DNMT1 changes dynamically during the cell cycle (Hermann et al. 2004a; Jurkowska et al. 2011a). The enzyme is diffusely distributed in the nucleus during interphase (when cells are not replicating) but localizes to replication foci in the early and mid-S-phase (in cells actively synthesizing DNA). During progression of the S-phase, the sub-nuclear pattern of DNMT1 changes from small, punctuate, and abundant structures in early S-phase to fewer, large, toroidal structures in late S-phase, which co-localized with late replicating heterochromatic satellite DNA (Leonhardt et al. 1992; O'Keefe et al. 1992; Easwaran et al. 2004; Liu et al. 2013; Schneider et al. 2013). In addition, some DNMT1 remains associated with the centromeric heterochromatin in G2 phase even after heterochromatin replication. In murine ESC cells, DNMT1 shows a heterochromatic distribution (Ren et al. 2020, 2021). Three regions of DNMT1 have been implicated in the targeting of the enzyme to the replication foci during S-phase, namely the PCNA binding domain (PBD) (Chuang et al. 1997), the replication foci targeting domain (RFTD) (Leonhardt et al. 1992), and the BAH domains (Liu et al. 1998), which will be described in the following chapters in more detail (Figs. 4.1 and 4.3).

Regulatory networks controlling the activity and stability of DNMT1 and heterochromatic DNA methylation. (a) Schematic illustration of the complex interplay between DNMT1, UHRF1, replication forks, and chromatin. Enzymatic activities are indicated by solid lines with arrows. Binding (“reading”) interactions are symbolized by dotted lines. For details cf. the text. Abbreviations used: CD catalytic domain, PCNA Proliferating cell nuclear antigen, UBL Ubiquitin-like domain, TTD Tandem Tudor domain, PHD Plant homeodomain, SRA SET and RING-associated domain, RING Really interesting new gene, Ub ubiquitinated H3 tail. For DNMT1 domain abbreviations, refer to the legend of Fig. 4.1. (b) Schematic illustration of the four different chromatin modification sub-networks involved in the establishment and maintenance of the heterochromatic DNA methylation, H3K9me3 (light blue circle) and H4K20me3 (pink circle). Catalytic activities are shown as blue arrows, chromatin reading interactions as dark red lines and protein/protein interaction as gray lines. (c) Selection of known PTMs on human DNMT1. Phosphorylations, acetylations, methylations, and ubiquitinations are represented by red circles labeled with P, A, M, or U, respectively. RFTD is shown in green, CXXC as a red loop, BAH1 in orange, BAH2 in violet, and the catalytic domain in blue. Reprinted from Jeltsch and Jurkowska (2016) with permission from Oxford University Press
4.3.5.1 The DNMT1–PCNA Interaction
Deletion of RFTD or BAH domains did not affect the delivery of DNMT1 to the replication fork (Easwaran et al. 2004), suggesting that the PBD domain has a central role in this process. Through this domain, DNMT1 directly interacts with PCNA, the so-called processivity factor of the replication machinery that forms a ring around the DNA helix (Chuang et al. 1997). In addition, both proteins co-localize in vivo, indicating that PCNA might recruit DNMT1 to the replication fork and load it onto DNA. Indeed, the expression of a truncated DNMT1, which lacked parts of the PBD domain, led to a delay in the re-methylation of DNA after replication (Egger et al. 2006). However, it did not cause massive defects in DNA methylation, indicating that the interaction of PCNA with DNMT1 contributes to the efficiency of DNA re-methylation, but it is not essential for this process. In addition, in vitro experiments provided evidence that the interaction with PCNA increases the DNA binding and catalytic activity of DNMT1 (Iida et al. 2002).
The interaction of DNMT1 with heterochromatin occurs in a replication-independent manner (Easwaran et al. 2004) and is mediated in part by the PBD domain of DNMT1 and by UHRF1, as described in the next paragraph. Direct interactions with heterochromatic histone marks mediated by the RFT and BAH1 domains of DNMT1 further contribute to DNMT1 genomic targeting and methylation of heterochromatic regions (Ren et al. 2020, 2021).
4.3.5.2 The DNMT1–UHRF1 Interaction
Another key pathway of DNMT1 targeting was discovered with the finding that UHFR1 is essential for maintaining DNA methylation in mammals (Bostick et al. 2007; Sharif et al. 2007). UHRF1 specifically binds to hemimethylated DNA via its SET and RING-associated (SRA) domain (Bostick et al. 2007; Avvakumov et al. 2008; Hashimoto et al. 2008; Arita et al. 2008) and its localization to replicating heterochromatin is dependent on the presence of hemimethylated DNA (Sharif et al. 2007) and specific histone PTMs (Nady et al. 2011; Rothbart et al. 2012). UHFR1 co-localizes with DNMT1 and PCNA at replicating heterochromatic regions during mid to late S-phase and DNMT1 association with chromatin is lost in UHFR1 knock out (KO) cells (Sharif et al. 2007; Bostick et al. 2007). It interacts with DNMT1 through the RFTD domain, partially explaining the central role of this domain in the localization of DNMT1 to the replication foci (Leonhardt et al. 1992). Remarkably, the phenotype of the UHFR1 KO in mice mimics that of DNMT1 KO, as UHRF1-deficient embryos die shortly after gastrulation and show significantly reduced levels of DNA methylation (Sharif et al. 2007), indicating that UHRF1 has a central role in the maintenance of DNA methylation. These data led to a model that UHFR1 recruits DNMT1 to the replicated hemimethylated DNA to facilitate its efficient re-methylation (Jeltsch 2008) (Figs. 4.2 and 4.3).
Later, it was found that two domains of UHRF1 recognize histone marks: the tandem Tudor domain (TTD) of UHRF1 binds methylated lysine 9 and unmethylated lysine 4 on histone 3 tail (Nady et al. 2011; Rothbart et al. 2012) and the plant homeodomain (PHD) of UHRF1 binds to unmodified arginine 2 of the H3 tail (Hu et al. 2011; Rajakumara et al. 2011; Wang et al. 2011). The interaction with H3K9me3 is required for the proper localization of UHRF1 to heterochromatin and maintenance of DNA methylation, since a mutation in TTD, which prevents binding to H3K9me3, abolished both functions (Nady et al. 2011; Rothbart et al. 2012). Similarly, disruption of H3R2 binding in UHRF1 abolished DNA methylation by DNMT1 in cells (Qin et al. 2015). These data indicate that the coordinated recognition of two histone marks, H3K9me3 and H3R2, as well as the interaction with hemimethylated DNA by UHRF1, are all necessary for the guidance of DNMT1 and faithful maintenance of DNA methylation (Rothbart et al. 2013; Liu et al. 2013) (Fig. 4.3).
In addition to its role in the targeting of DNMT1, UHRF1 also directly stimulates the catalytic activity of DNMT1, by interacting with the RFT domain of DNMT1 and preventing the autoinhibitory conformation (Berkyurek et al. 2014; Bashtrykov et al. 2014a). Moreover, the RING domain of UHRF1 ubiquitinates H3 at K18 and K23 (Nishiyama et al. 2013; Qin et al. 2015). Ubiquitinated H3 is bound by DNMT1 as described in the next paragraph, increasing its methyltransferase activity. Furthermore, UHRF1 is involved in the ubiquitination of DNMT1, which reduces DNMT1’s stability (see below).
However, UHRF1 also forms a stable interaction with DNA Ligase 1 methylated at K126 (which is bound to the TTD instead of H3K9me3) (Ferry et al. 2017). This interaction occurs at replication forks, where the ligase is needed to seal the Okazaki fragments. Interestingly, the replication-coupled maintenance activity of DNMT1 is determined by the UHRF1–Ligase 1 and PCNA–DNMT1 interactions, while its replication-independent activity depends on nucleosome occupancy and the interaction between UHRF1 and methylated H3K9 (Ming et al. 2020). This finding is in agreement with the observation that the activity of DNMT1 is inhibited by nucleosome formation as mentioned above (Mishima et al. 2017). All these observations demonstrate that UHRF1 is a key multifaceted regulator of DNMT1 and the entire maintenance DNA methylation machinery (Fig. 4.3).
4.3.5.3 Binding of the DNMT1-RFTD to Ubiquitinated H3 Tails
The UHRF1-dependent ubiquitination of histone H3 has an essential role in DNMT1 function, as the catalytically inactive UHRF1 RING mutant failed to recruit DNMT1 to the replication sites (Nishiyama et al. 2013). The molecular mechanism of this finding has begun to be uncovered with the observation that DNMT1 preferentially associates with monoubiquitinated H3 through its RFT domain and that this interaction leads to the activation of the methyltransferase (Nishiyama et al. 2013; Qin et al. 2015). The binding to the monoubiquitinated H3 peptide increased the methylation activity of DNMT1 on a substrate with multiple hemimethylated CpG sites (Mishima et al. 2020), indicating that it may contribute to the efficiency of DNA methylation maintenance. This stimulatory effect was reduced by mutations in the RFTD of DNMT1 that are linked to human autosomal dominant cerebellar ataxia, deafness, and narcolepsy (ADCA-CN) (Mishima et al. 2020).
The ubiquitination of the H3 tail is introduced by the RING domain of UHRF1, which is an E3 ligase (Nishiyama et al. 2013; Qin et al. 2015). Monoubiquitination of H3 has been detected at K14, K18, and K23. Structural and biochemical studies showed that the dual monoubiquitinated H3 (at K18 and K23) peptide is bound preferentially by DNMT1 in a binding cleft located in the RFT domain (Ishiyama et al. 2017). The binding of H3-K18Ub/23Ub results in a conformational change of the RFTD, leading to an increase in DNMT1 activity. In addition, the RFTD also binds the ubiquitin-like (UBL) domain of UHRF1, further strengthening the DNMT1–UHRF1 interaction. Notably, both the ubiquitin ligase and the ubiquitin-like domain of UHRF1 are required for the heterochromatic localization of DNMT1 and DNA methylation of repeat elements (Li et al. 2018). Consistently, the USP7 deubiquitinase, which removes histone ubiquitination, has been shown to suppress DNMT1 recruitment and DNA methylation (Li et al. 2020). In addition, PCNA-associated factor 15 (PAF15) undergoes dual mono-ubiquitination by UHRF1 in a DNA replication-coupled manner and it thereby recruits DNMT1 to replicating chromatin (Nishiyama et al. 2020). Strikingly, during early S-phase, UHRF1 preferentially ubiquitinates PAF15, whereas H3Ub2 predominates during late S-phase, suggesting that the mechanism of DNMT1 recruitment changes between the early and late replicating DNA regions. Taken together, these data indicate an important additional connection between the chromatin interactions of DNMT1 and UHRF1, which is essential for efficient maintenance methylation to occur (Fig. 4.3).
4.3.5.4 Binding of DNMT1 to Heterochromatic Chromatin Marks
DNA methylation, H3K9me3 and H4K20me3 together constitute a characteristic modification state called constitutive heterochromatin (Jeltsch et al. 2019). Recent work has demonstrated that DNMT1 directly binds to both, H3K9me3 and H4K20me3, which explained the heterochromatic localization of DNMT1 and provided novel connections between these chromatin modifications and DNA methylation. A structural and biochemical study demonstrated that the RFT domain of DNMT1 preferentially interacts with ubiquitinated H3 peptides if they also contain H3K9me3, leading to the stimulation of DNMT1 activity (Ren et al. 2020). Structural analysis revealed the H3 tail bound in a surface cleft of the RFT domain with the interaction sites for ubiquitin on the surface of RFTD and H3K9me3 binding mediated by a non-conventional binding site formed by RFTD and one ubiquitin moiety. The mutation of a tryptophan residue critical for the H3K9me3 interaction led to a global reduction of DNA methylation in cells, underscoring the functional relevance of this interaction. Another recent study discovered that the first BAH domain of DNMT1 (BAH1) binds H4K20me3, contributing to heterochromatin targeting of DNMT1 and DNA methylation (Ren et al. 2021). Structural analysis revealed binding of the H4 tail to the BAH1 domain and recognition of H4K20me3 by an aromatic half-cage. The binding of the H4 tail led to the displacement of the autoinhibitory linker between the CXXC and BAH1 domains, causing an allosteric activation of DNMT1. Disruption of the H3K9me3 or H4K20me3 binding led to a loss of the heterochromatic localization of DNMT1 in murine ES cells (Ren et al. 2020, 2021).
DNA methylation, H3K9me3 and H4K20me3 form an interconnected network of chromatin modifications that defines the constitutive heterochromatin state (Fig. 4.3b). Previous work has already identified several molecular connections between readers (HP1ß and UHRF1 for H3K9me3 and MBD1 for DNA methylation) and writers of these modifications (SUV39H1/H2 and SETDB1 for H3K9 methylation, SET8 and SUV420H1/H2 for H4K20 methylation). For example, H3K9 methylation stimulates H4K20 methylation, because HP1 recruits SUV420H enzymes (Schotta et al. 2004) and SUV39H1 stimulates the activity of SET8 (Kudithipudi et al. 2017). Similarly, HP1 stimulates further spreading of H3K9 methylation by interaction with SUV39H enzymes (Raurell-Vila et al. 2017). UHRF1 functions as a critical cofactor of DNMT1 (Liu et al. 2013) and DNA methylation recruits SETDB1 via MDB1 binding (Markouli et al. 2021). The data showing that DNMT1 also directly binds to H3K9me3 and H4K20me3 connect this network even more, ensuring efficient methylation and silencing of heterochromatin and repetitive sequences. These complex interactions elegantly illustrate the cooperation between various layers of epigenetic modifications that all establish and reinforce specific epigenetic states and biological outcomes.
4.3.5.5 Regulation of Activity and Specificity of DNMT1 by Nucleic Acid Binding
DNMT1 possesses multiple DNA binding sites, which contribute to the allosteric regulation of its activity and specificity. Many groups reported that the enzyme shows reduced specificity in the presence of methylated DNA (Fatemi et al. 2001, 2002; Christman et al. 1995; Bacolla et al. 1999). This effect was due to an increase in the rate of de novo methylation of unmodified DNA, while the methylation of hemimethylated DNA was weakly inhibited (Fatemi et al. 2001; Goyal et al. 2006). The increase in the methylation efficiency of unmethylated DNA indicates that the binding of the methylated DNA to the N-terminal domain of the enzyme induces an allosteric activation for the methylation of unmethylated substrates. The molecular mechanism of the allosteric activation of DNMT1 is not well understood, the CXXC domain (Fatemi et al. 2001) and the residues 284–287 of the murine DNMT1 (Pradhan and Esteve 2003) have been implicated in this process. Therefore, it is likely that DNA binding to the CXXC domain is involved in these effects. In addition, an inhibitory effect of unmethylated DNA was demonstrated in several studies (Svedruzic and Reich 2005; Flynn et al. 2003; Bacolla et al. 1999), suggesting that binding of an unmethylated DNA to the N-terminal part of DNMT1 leads to a repression of the enzymatic activity on hemimethylated DNA. The binding site for this substrate inhibition effect was localized in the first 501 amino acids of DNMT1 (Bacolla et al. 2001). Additional evidence suggests that binding of the methylated DNA to the N-terminal inhibition site also caused de-repression of the enzyme (Bacolla et al. 2001). Whether the inhibition and stimulation effects observed in these various studies are due to binding to the same or different sites and to what extent different DNAs compete for the different sites is not clear.
Interestingly, all studies agree that binding to unmethylated DNA at a secondary site reduces the activity of DNMT1, while binding to methylated DNA increases its activity. This observation could be related to the fact that DNA methylation patterns in the human genome are highly bimodal (Eckhardt et al. 2006; Meissner et al. 2008; Zhang et al. 2009), meaning that the genomic regions tend to be either highly methylated or almost unmethylated. The occurrence of the bimodal methylation patterns could be explained by the allosteric binding of the substrate DNA to a secondary site because DNMT1 would be activated on methylated regions and inactivated on unmethylated DNA. Consequently, highly methylated regions will tend to gain methylation, whereas lowly methylated regions will tend to lose even their residual methylation.
In addition to DNA, DNMT1 binds various RNA molecules. Initial studies showed that DNMT1 purified from insect cells contains inhibitory RNA (Glickman et al. 1997a). Later, it was discovered that RNA binding regulates the activity of DNMT1 in a locus-specific manner. A long non-coding RNA (lncRNA) originating from the CEBPA locus was observed to bind and inhibit DNMT1 and prevent the methylation of this locus. Similar effects were observed for several other loci on a genomic scale (Di Ruscio et al. 2013). Based on these findings, the authors proposed a model, in which the ncRNAs transcribed at one locus function as a shield for this locus preventing its methylation. Thereby, the expression of the locus would be perpetuated. Later, it was also reported that DNMT1 binds to miRNAs like miR-155-5p (Zhang et al. 2015a). Other studies showed regulation of DNMT1 by DNMT1-associated lncRNAs, leading to global changes in DNA methylation and gene regulation in cancer cells (Merry et al. 2015; Somasundaram et al. 2018). A specific example of this mechanism is the DACOR1 lncRNA, which is a positive regulator of DNA methylation (Somasundaram et al. 2018). Similar to lncRNAs, miRNAs function as inhibitors of DNMT1 and the transfection of miRNAs to cells caused changes in cellular methylation (Zhang et al. 2015a). RNA binding was mapped to the catalytic domain of DNMT1 (Di Ruscio et al. 2013; Zhang et al. 2015a), and it was reported that miRNAs can act as DNA competitive inhibitors (Zhang et al. 2015a). These findings suggest that the inhibition of DNMT1 by miRNAs is based on a direct competition of the RNA and DNA for access to the catalytic center. However, it is well conceivable that the additional DNA binding sites described above bind regulatory RNAs as well. These important features of the interaction of DNMT1 with regulatory DNA and RNA are not well understood at a molecular level and deserve additional experimental work.
4.3.6 PTMs of DNMT1
4.3.6.1 Phosphorylation of DNMT1
DNMT1 is subject to several posttranslational modifications like phosphorylation, methylation, ubiquitination, acetylation, and SUMOylation, (Fig. 4.3c). Following the initial identification of S515 as a major phosphorylation site in DNMT1 purified from insect cells (Glickman et al. 1997b), several more phosphorylated serine and threonine residues have been identified in targeted and high-throughput proteomics approaches with DNMT1 purified from human or mouse cells. Currently, >60 phosphorylation sites have been mapped on human and mouse DNMT1 (http://www.phosphosite.org), but only a few of them have been functionally studied. The phosphorylated S515 is involved in the interaction between the N-terminal and catalytic domains of DNMT1 which is necessary for the activity of the enzyme (Goyal et al. 2007). Phosphorylation of S146 introduced by casein kinase 1 delta/epsilon decreases the DNA binding affinity of DNMT1 (Sugiyama et al. 2010), and phosphorylation of S127 and S143 regulates the interaction of DNMT1 with PCNA and UHRF1 (Hervouet et al. 2010). Moreover, phosphorylation of DNMT1 by PKC has been reported, but the target sites have not yet been identified (Lavoie et al. 2011). S143 of DNMT1 is phosphorylated by AKT1, which leads to the stabilization of the methyltransferase (Esteve et al. 2011). A specific 14–3-3 family reader protein for this modification has been identified (Esteve et al. 2016). It binds phosphorylated DNMT1, leading to the inhibition of DNMT1 activity, aberrant DNA methylation, and cell invasion (Esteve et al. 2016). The functional significance of many of the other phosphorylations in DNMT1 still awaits elucidation. In particular, the influence of the PTMs on the allosteric regulation of DNMT1 activity and specificity needs to be studied.
4.3.6.2 Acetylation and Ubiquitination of DNMT1
Multiple acetylation sites have been identified on DNMT1 up to date in proteomics analyses (Kim et al. 2006; Choudhary et al. 2009; Peng et al. 2011) (http://www.phosphosite.org); however, their functional significance has only begun to be revealed. Initial experiments with deacetylase inhibitors demonstrated the involvement of acetylation in the control of DNMT1 stability (Zhou et al. 2008; Peng et al. 2011). Based on this, an elegant mechanism regulating the abundance of DNMT1 during cell cycle was identified. It starts with the acetylation of DNMT1 in the KG linker by the acetyltransferase Tip60, followed by UHRF1-mediated ubiquitination, resulting in proteasomal degradation of DNMT1 at the end of DNA replication. In turn, histone deacetylase 1 (HDAC1) and deubiquitinase ubiquitin-specific peptidase 7 (USP7, also known as HAUSP) have an opposite effect and increase the stability of DNMT1 (Du et al. 2010; Qin et al. 2011). The crystal structure of DNMT1 in complex with USP7 revealed that this interaction is dependent on the KG linker of DNMT1, explaining why acetylation of this region impairs complex formation and promotes degradation of DNMT1 (Cheng et al. 2015). In addition, SIRT1 deacetylates DNMT1 at several sites and thereby regulates the activity and function of the methyltransferase (Peng et al. 2011).
4.3.6.3 Lysine Methylation of DNMT1
DNMT1 is methylated by SET7/9, both in vivo and in vitro. The monomethylation of human DNMT1 by SET7/9 occurs at K142 mainly during late S-phase and promotes proteasomal degradation of the enzyme in a cell cycle-dependent manner (Esteve et al. 2009). Recent work demonstrated that proteasomal targeting of DNMT1 is mediated by the L3MBTL3 methyl-binding protein that recruits CRL4(DCAF5) ubiquitin ligase (Leng et al. 2018). Methylation of DNMT1 is reversible and can be removed by LSD1 (Wang et al. 2009; Leng et al. 2018). In addition, it is antagonistic with phosphorylation of DNMT1 at S143 by AKT1 kinase described above (Esteve et al. 2011). The existence of these complex mechanisms to regulate DNMT1 stability underscores the biological requirement for tight regulation of cellular DNMT1 levels.
4.4 Structure, Function, and Mechanism of DNMT3 Enzymes
4.4.1 Domain Composition of DNMT3 Proteins
In most mammals, the DNMT3 family contains three members: DNMT3A, DNMT3B, and DNMT3L, which in humans comprise 912 aa, 853 aa, and 387 aa, respectively. In addition, a DNMT3B paralog called DNMT3C (739 aa) has been identified in rodents, where it has a specific role in transposon repression in the male germline (Barau et al. 2016; Jain et al. 2017) (Fig. 4.1). Several isoforms of DNMT3A and DNMT3B, resulting from alternative splicing or use of alternative start codons, have been identified both in mice and humans (Jurkowska et al. 2011a). In the case of DNMT3A, the DNMT3A2 isoform lacks the first 223 amino acid residues (Qiu et al. 2002). For DNMT3B, multiple isoforms have been found (Weisenberger et al. 2004); among them the inactive splicing isoform DNMT3B3, which lacks parts of the linker between the PWWP and ADD domains and a region of the catalytic domain containing the target recognition domain (Fig. 4.1). Besides the C-terminal domain required for catalysis, DNMT3A and DNMT3B possess an N-terminal part with domains involved in the targeting of the enzymes to chromatin and regulation of their function (Jurkowska et al. 2011a). In this part, three functional domains are present: a UDR (ubiquitin-dependent recruitment region) which is present specifically in DNMT3A1, a PWWP domain in DNMT3A and DNMT3B, and an ADD (ATRX-DNMT3-DNMT3L) domain, also known as PHD (Plant homeodomain) domain that is present in all four proteins.
The ADD domain is a cysteine-rich region that binds zinc ions and creates a platform for protein–protein interactions. This domain mediates the interaction of DNMT3 enzymes with histone H3 tails unmethylated at lysine K4 (Ooi et al. 2007; Otani et al. 2009; Zhang et al. 2010; Guo et al. 2015). In addition, it is involved in the interaction of DNMT3A with various components of the epigenetic machinery, like protein lysine methyltransferases SUV39H1 (Fuks et al. 2003a), SETDB1 (Li et al. 2006), EZH2 (Vire et al. 2006), and deacetylase HDAC1, reading domain proteins, including HP1ß (Fuks et al. 2003a), Mbd3 (Datta et al. 2005), and MeCP2 (Kimura and Shiota 2003; Fuks et al. 2003b; Rajavelu et al. 2018), as well as transcription factors PU.1 (Suzuki et al. 2006), MYC (Brenner et al. 2005), and RP58 (Fuks et al. 2001), and chromatin remodeling factors hSNF2 (Geiman et al. 2004) and SMARCA4 (Datta et al. 2005). The ADD domain has been implicated in the allosteric control of DNMT3A, as it interacts with the catalytic domain of the methyltransferase and inhibits its activity (see below), indicating that ADD-mediated interactions with other proteins and chromatin could have direct regulatory effects on the catalytic activity of DNMT3A and DNMT3B.
The PWWP domain of DNMT3A and DNMT3B is a region of 100–150 amino acids, containing a conserved proline–tryptophan motif (hence the name PWWP). PWWP domains belong to the Royal domain superfamily, members of which interact with histone tails in various modification states (Qin and Min 2014). The PWWP domains of DNMT3A and DNMT3B specifically recognize the H3K36 di- and trimethylation mark (H3K36me2/3) (Dhayalan et al. 2010). This domain is essential for the targeting of DNMT3 enzymes to pericentromeric chromatin (Chen et al. 2004; Ge et al. 2004). The structures of the PWWP domains from both DNMT3A and DNMT3B have been solved (Qiu et al. 2002; Rondelet et al. 2016; Dukatz et al. 2019a). A biochemical study revealed that the PWWP domain synergistically binds the H3K36me2/3-modified histone tail and DNA through its conserved aromatic cage for H3K9me2/3 binding and a positively charged surface for DNA binding. Both interfaces were found to be necessary for chromatin targeting of DNMT3A1 (Dukatz et al. 2019a). In addition, the ZHX1 (zinc-finger and homeobox protein 1) interacts with the PWWP domain of DNMT3B and enhances DNMT3B-mediated transcriptional repression (Kim et al. 2007). Interestingly, although DNMT3C arose from a duplication of the DNM3B gene, it lost the PWWP domain. This may prevent targeting of the enzyme to H3K36me2/3-rich regions, potentially contributing to the specific localization of DNMT3C to retrotransposon promoters (Barau et al. 2016).
The part of DNMT3A and DNMT3B N-terminal to the PWWP domain is the least conserved region between both enzymes. This domain binds DNA (Suetake et al. 2011) and it is important for anchoring the enzymes to nucleosomes (Jeong et al. 2009; Baubec et al. 2015). In DNMT3A1, a small, folded domain called ubiquitin-dependent recruitment (UDR) domain has been recently identified. It is responsible for the interaction of DNMT3A1 with H2AK119ub1 (Weinberg et al. 2021). DNMT3A2 and DNMT3B enzymes lack this domain.
The C-terminal domains of DNMT3A and DNMT3B, which enclose the catalytic centers of the enzymes, share approximately 85% sequence homology. In contrast to the catalytic domain of DNMT1 they are active in an isolated form (Gowher and Jeltsch 2002) and have been used as a model system to study the catalytic mechanism and specificity of the DNMT3 proteins. Interestingly, isolated catalytic domains of DNMT3A and DNMT3B show higher enzymatic activity than the full-length proteins, indicating that the N-terminal domains allosterically inhibit the activity of the enzymes (Li et al. 2011). The molecular mechanism underlying this observation was revealed by a structural study, which demonstrated that the ADD domain of DNMT3A, which directly interacts with the catalytic domain of the methyltransferase in two different binding modes (see below), is responsible for this inhibition in the absence of histones (Guo et al. 2015) (Fig. 4.4b). This model is further supported by kinetic experiments, showing that the binding of ADD domain of DNMT3A to H3 tail stimulates the activity of the enzyme (Li et al. 2011; Zhang et al. 2010).

Structure and allosteric regulation of DNMT3A. (a) Structure of the DNMT3A/DNMT3L complex with bound DNA (pdb 5YX2) (Zhang et al. 2018). Subunits and interfaces are annotated. AdoHcy is shown in yellow as a ball and stick model. (b) Allosteric regulation of DNMT3A. The ADD domain of the dark blue DNMT3A subunit is shown in both the autoinhibitory (orange) and in the active conformation (red) (pdb 4U7P and 4U7T) (Guo et al. 2015). The ADD domain of the second DNMT3A subunit (gray) has been omitted for clarity. Binding of the H3 peptide (green) to the ADD domain occurs with the residues involved in the autoinhibitory-binding interface. Therefore, H3 peptide binding is only possible in the active conformation and this conformation is consequently stabilized in the presence of the H3 peptide (Guo et al. 2015; Li et al. 2011). (c) Cryo-EM structure of the DNMT3A/DNMT3B3 nucleosome complex (pdb 6PA7) (Xu et al. 2020). Nucleosomal DNA is shown in green and AdoHcy is shown in yellow as ball and stick model
DNMT3L, the third member of the DNMT3 family, lacks parts of the N-terminal region including the PWWP domain. Strikingly, it also carries amino acid exchanges and deletions within the conserved DNA-(cytosine C5)-MTase motifs, which contain the catalytic residues, indicating that while it still adopts the typical AdoMet-dependent MTase fold described above, it cannot have catalytic activity and is unable to bind AdoMet. The same is true for one splicing isoform of DNMT3B, DNMT3B3, which also contains a deletion in the C-terminal domain and lacks catalytic activity (Weisenberger et al. 2004; Zeng et al. 2020). While DNMT3L is mainly expressed in ES cells and the germline (Bourc'his and Bestor 2004; Bourc'his et al. 2001; Hata et al. 2002), DNMT3B3 shows expression in differentiated cells (Zeng et al. 2020). Despite being inactive, both DNMT3L and DNMT3B3 interact with the active members of the DNMT3 family and stimulate their catalytic activity.
4.4.2 Structures of DNMT3A and DNMT3B
The structure of the complex of the C-terminal domains of DNMT3A/DNMT3L was solved in 2007 and represented the first structure published for a mammalian DNMT. It showed that the complex forms a linear heterotetramer consisting of two DNMT3L subunits (at the edges of the tetramer) and two DNMT3A subunits (in the center) (Jia et al. 2007) (Fig. 4.4a). The heterotetrameric structure of the complex was confirmed in solution (Jurkowska et al. 2008). The structure also revealed that the C-terminal domain of DNMT3A contains two interfaces for protein–protein contacts: a hydrophobic one generated by the stacking interaction of two phenylalanine residues (called FF interface), which mediates the DNMT3A/DNMT3L interaction, and a polar interface generated by a hydrogen bonding network between arginine and aspartate residues from both subunits (called RD interface), which can only mediate DNMT3A/DNMT3A interactions since the corresponding region is absent in DNMT3L (Fig. 4.5). DNA binding studies showed that the central DNMT3A/DNMT3A interface in the tetramer creates the DNA binding site, while both interfaces are essential for AdoMet binding and catalytic activity (Jurkowska et al. 2008). The dimerization of DNMT3A/DNMT3L complexes via the RD interface increases the size of the DNA interface and compensates for the small TRD of DNMT3A.

Multimerization of DNMT3A and DNMT3A/DNMT3L complexes. (a) Structure of the DNMT3A/DNMT3L complex with bound DNA (pdb 5YX2) (Zhang et al. 2018). Subunits and interfaces are annotated. (b) Schematic models of DNMT3A multimerization on DNA, protein multimerization and binding to several DNA molecules, and the combination of both processes. (c) Hypothetical binding of a DNMT3A hexamer to the two linker DNAs emerging from one nucleosome
Later, the structure of the DNMT3A/DNMT3L C-terminal domain heterotetramer was solved in complex with a DNA molecule containing two CpG sites spaced in a distance of 12 base pairs (Zhang et al. 2018) (Fig. 4.4a). It provided the first mechanistic insights into the DNA interaction and specificity of DNMT3A. Zebularine was incorporated into the DNA instead of the target cytosines in the upper strand of the left CpG site and the lower strand of the right CpG site. This base analog leads to the formation of stable covalent complexes between the DNMT and the DNA because the nucleophilic attack of the active site cysteine residue is catalyzed, but its later elimination is blocked. The complex showed base flipping of both zebularine bases, indicating that the heterotetramer could potentially co-methylate CpG sites at this distance. Biochemical studies confirmed that the 12 bp distance is the preferred one for covalent DNA complex formation of DNMT3A/DNMT3L and DNMT3B/DNMT3L (Gao et al. 2020a), further supported by strong peaks of co-methylation at CpG sites placed in this distance in substrates containing two CpG sites (Emperle et al. 2021). The DNMT3A–DNA interaction involves a target recognition domain (TRD) loop, a catalytic loop following the catalytic PCN motif, and a helix of the RD tetramer interface (Zhang et al. 2018). The TRD loop (which is unfolded in the DNA free complex) contains the R836 residue, which recognizes the guanine of the CpG sites, ensuring the preference of DNMT3A towards CpG observed in previous studies (Gowher and Jeltsch 2001; Aoki et al. 2001; Ramsahoye et al. 2000). V716 from the catalytic loop approaches the DNA from the minor groove and fills the DNA cavity generated by the flipping of the zebularine base. The RD interface loop contains R882, which is often mutated in acute myeloid leukaemia (AML) (see below). It contacts the DNA backbone at several phosphate residues on the 3′ side of the CpG site. The central part of the DNA shows about 40° bending and kinetic experiments demonstrated that enrichment of T in the region of bending stimulates methylation (Emperle et al. 2021).
Recent structures of DNMT3B/DNMT3L C-terminal domain heterotetramers with DNA revealed a very similar overall structure as the DNMT3A/DNMT3L complex (Gao et al. 2020b; Lin et al. 2020). Strikingly, despite similarities, the DNA recognition of both enzymes differs in the target recognition loop. In DNMT3B, N779 interacts specifically with the guanine in CpG sites, while in DNMT3A this interaction is mediated by R836. Moreover, DNMT3B contains a lysine residue (K777) which specifically interacts with the base at the +1 side of the CpG, mediating a strong preference for a G at this place, in particular during non-CpG methylation. The amino acid sequences and structures of DNMT3A and DNMT3B diverge most at the RD interface loop, as illustrated, for example, by a different conformation of R823 in DNMT3B, which corresponds to DNMT3A R882. These differences lead to distinct contacts to the DNA regions flanking the target CpG site and provide a mechanistic basis of the enzyme-specific flanking sequence preferences (see below) (Gao et al. 2020b).
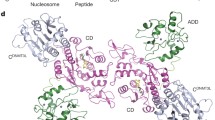
In a seminal publication, a cryo-EM structure of a DNMT3A2/DNMT3B3 heterotetramer bound to a mononucleosome was reported (Xu et al. 2020) (Fig. 4.4c). The complex formed a similar linear heterotetramer as the DNMT3A/DNMT3L complexes, but DNMT3B3 replaced DNMT3L at the outer complex positions. This study confirms previous biochemical data (Li et al. 2007) showing that the DNMT3 binding interfaces support the interaction of different DNMT3 members, offering the unique potential for regulating methyltransferase activity depending on the complex composition. Unexpectedly, two arginine residues in the C-terminal domain of one DNMT3B3 subunit formed a direct contact with the H2A/H2B acidic patch on the disc face of the histone octamer. Thereby, the DNMT3A2/DNMT3B3 tetramer was anchored on the nucleosome core particle positioning the DNA binding region and active sites of the central DNMT3A subunits right above the linker DNA strand near the dyad axis (Xu et al. 2020). The detailed functional consequences of this unexpected architecture are still unknown, but biochemical data showed that binding of DNMT3A and DNMT3A/DNMT3B3 complexes to the acidic patch of histones contributes to the methylation preferences of CpG sites within the linker DNA (Bröhm et al. 2022).
4.4.3 Allosteric Regulation of DNMT3A
Additional structures of a longer DNMT3A C-terminal fragment also including the ADD domain in complex with DNMT3L were solved, providing seminal insights into the mechanism of this enzyme. They showed that the ADD domain can bind to the catalytic domain at two distinct sites, creating two alternative conformations. ADD binding activates the enzyme in one conformation (allosteric binding), while it blocks access of the DNA to the active center and inhibits catalysis in the other (autoinhibitory binding) (Guo et al. 2015) (Fig. 4.4b). A modeling study suggested that a hinge-like property of the RD interface is important for the cooperative reorientations of the tetramer into the autoinhibitory or the active state (Liang et al. 2018).
These data indicate that the activity of DNMT3A, like DNMT1, is under precise allosteric control by domain rearrangements, illustrating a fascinating convergence of regulatory principles of these two enzymes. Similarly as in DNMT1, protein partners can influence the equilibrium of the active and inactive conformations, as it was shown that the stimulatory effect of H3 on DNMT3A depends on its binding to the ADD domain, leading to the stabilization of the ADD at the allosteric binding site (Li et al. 2011; Guo et al. 2015). Direct allosteric regulation of DNMT3A activity by the PWWP domain has not yet been shown. Moreover, so far it is not known if DNMT3B undergoes similar steps of allosteric regulation as DNMT3A.
4.4.4 Specificity of DNMT3 Enzymes
Consistent with their designation as de novo MTases, DNMT3A and DNMT3B do not display any significant preference between hemimethylated and unmethylated DNA (Okano et al. 1998; Gowher and Jeltsch 2001). However, in addition to their preference for the methylation of CpG sites, both DNMT3A and DNMT3B are very sensitive to the sequences flanking their target sites. This is illustrated by the finding that CpG sites in certain flanking sequences cannot be methylated by DNMT3A at all (Jurkowska et al. 2011c). It has been shown that purine bases are preferred at the 5′ side of the CpG sites, whereas pyrimidines are favored at their 3′ side (Lin et al. 2002; Handa and Jeltsch 2005; Jurkowska et al. 2011c). One further consequence of the strong flanking sequence preferences of DNMT3A and DNMT3B is that both DNA strands of a CpG site, which are embedded in an asymmetric flanking sequence context, usually differ strongly in their preference for DNMT3 methylation. This leads to the preferential methylation of one cytosine in each CpG site, meaning that DNMT3 enzymes tend to generate hemimethylated products. In vitro experiments showed that the products of DNMT3A methylation can be readily methylated by DNMT1 and that both enzymes can act synergistically in the efficient de novo methylation of unmethylated DNA (Fatemi et al. 2002). Mutational analysis of residues in the DNA binding site of DNMT3A demonstrated that exchanges of critical residues caused massive changes in flanking sequence preferences (Gowher et al. 2006). Interestingly, this includes the exchange at R882, a residue frequently mutated in acute myeloid leukemia (AML) cancer (Hamidi et al. 2015). An in-depth mechanistic understanding of the CpG recognition and flanking sequence preferences of DNMT3 enzymes was provided by the recent DNMT3 structures with bound substrate DNA (Zhang et al. 2018; Gao et al. 2020b; Lin et al. 2020), as described below.
Although DNMT3A and DNMT3B methylate cytosine residues predominantly in the context of CpG dinucleotides, they can also introduce methylation in a non-CpG context (CA > > CT > CC) (Gowher and Jeltsch 2001; Aoki et al. 2001; Ramsahoye et al. 2000). Consistently, methylated non-CpG sites (mainly CpA) were detected in embryonic stem (ES) cells and the brain, where DNMT3A and DNMT3B enzymes are highly expressed, but not in cells where DNMT3 enzymes are downregulated (Lister et al. 2009, 2013; Varley et al. 2013; Guo et al. 2014). However, another survey of the human body epigenomes identified low levels of non-CpG methylation in almost all human tissues (Schultz et al. 2015). Studies with DNMT KO cell lines confirmed that DNMT3 enzymes introduce the non-CpG methylation (Ziller et al. 2011; Arand et al. 2012). The exact mechanism for the propagation of DNA methylation outside of the CpG context is unknown, but it cannot be maintained by DNMT1, which is very specific for CpG sites (Fatemi et al. 2001). First insights into the biological function of non-CpG methylation were provided with the observation that it can repress expression of long genes in the brain by recruiting MeCP2 (Guo et al. 2014; Gabel et al. 2015; Chen et al. 2015), disruption of which is implicated in the Rett syndrome.
Detailed biochemical studies demonstrated that the flanking sequence preferences of DNMT3A and DNMT3B differ (Gao et al. 2020b; Mallona et al. 2021; Jeltsch et al. 2021). This is due to the DNMT3B specific readout of the +1 flanking site by K777 and the differences in the conformation of the RD interface loop (Gao et al. 2020b; Lin et al. 2020). DNMT3B displays strong and characteristic preferences for CpG sites located in a sequence context that resembles the SatII minor satellite repeats, which lose methylation in the immunodeficiency-centromeric instability-facial anomalies (ICF) syndrome (Xu et al. 1999). This finding explains previous observations showing that 1) insufficient DNMT3B activity causes the ICF syndrome and 2) DNMT3A apparently cannot take over the function of DNMT3B in minor satellite methylation. Currently, specific details of how DNMT3A and DNMT3B interact with the CpG sites in different flanking sequence contexts are unknown. A recent DNMT3B mutational study indicated that the interaction with different flanking sequences involves complex and sequence-dependent contact networks of enzyme residues with the DNA (Dukatz et al. 2020). These adaptive interaction modes could help to balance the interaction of DNMT3B with different flanking sites, allowing a more equal methylation of CpG sites in different contexts, which is required for the function of DNA methylation as a system for storage and processing of epigenetic information (Jeltsch et al. 2021).
The differences in the flanking sequence preferences of DNMT3A and DNMT3B were even more pronounced in the context of non-CpG methylation (Gao et al. 2020b; Dukatz et al. 2020; Jeltsch et al. 2021). Here, DNMT3B showed a strong preference for a G at the +1 flanking site, mainly generating methylated CAG, while DNMT3A preferred a C instead, yielding predominantly methylated CAC. These preferences are in agreement with the cellular distribution of non-CpG methylation obtained in triple DNMT1/DNMT3A/DNMT3B KO cells reconstituted with DNMT3A- or DNMT3B (Gao et al. 2020b) and other cellular methylation data (Lister et al. 2009, 2013; Laurent et al. 2010; Xie et al. 2012; Lee et al. 2017).
Mutations of R882 in DNMT3A, most prominently R882H, are observed at a high frequency in the AML tumors (Hamidi et al. 2015). As described above, R882 is located in the RD loop at the DNA binding interface of DNMT3A and it is involved in flanking sequence DNA contacts on the 3′ side of the CpG site (Zhang et al. 2018). This loop shows conformational differences between DNMT3A and DNMT3B, which are related to the distinct flanking sequence preferences of both enzymes (Gao et al. 2020b). The structure of the DNMT3A(R882H)/DNMT3L heterotetramer bound to DNA showed enhanced dynamics of the TRD loop (Anteneh et al. 2020), suggesting that this loop recognizes CpG dinucleotides in a + 1 flanking site-dependent manner. Accordingly, the R882H mutation leads to strong changes in the flanking sequence preferences of DNMT3A (Emperle et al. 2018, 2019). Other mutations of R882 were shown to cause similar effects, indicating that the loss of the R882 side chain is responsible for the effect, rather than the amino acid side chain introduced instead (histidine 882 in case of the R882H mutation) (Emperle et al. 2019). Interestingly, detailed analyses revealed that the DNMT3A R882H flanking sequence preferences differ from wildtype DNMT3A mainly on the 3′-side of the CpG site, where they change into a DNMT3B-like pattern (Emperle et al. 2019; Norvil et al. 2020). Hence, the changes in flanking sequence preferences are one potential reason for the pathogenic effect of this mutation.
4.4.5 Kinetic Mechanism of DNMT3 Enzymes
Initial studies with the C-terminal domains of DNMT3A and DNMT3B showed an interesting difference in the catalytic mechanism of both enzymes. Whereas DNMT3B was able to methylate multiple CpG sites in a processive manner, DNMT3A was distributive (Gowher and Jeltsch 2002). Later, Reich and colleagues reported that DNMT3A methylates DNA in a processive manner (Holz-Schietinger and Reich 2010). However, at the same time, DNMT3A was shown to bind cooperatively to DNA forming large multimeric protein/DNA fibers (Jia et al. 2007; Jurkowska et al. 2008; Rajavelu et al. 2012) (Fig. 4.5). These properties appear mutually exclusive because the concept of a processive turnover is based on isolated enzyme complexes moving along a DNA substrate, which is not compatible with protein complexes multimerizing on DNA. Other biochemical studies did not detect processive DNA methylation by DNMT3A (Emperle et al. 2014).
4.4.6 Oligomerization of DNMT3 Enzymes
The DNMT3 enzymes exhibit a complex oligomerization and multimerization potential including two independent orthogonal multimerization reactions [for a review cf. (Jeltsch and Jurkowska 2013)]. First, DNMT3A multimerizes on DNA and cooperatively binds to DNA, and second, it can form protein oligomers able to bind to more than one DNA molecule. These two processes will be further described in the next sub-chapters (Fig. 4.5).
4.4.6.1 Protein Multimerization of DNMT3 Enzymes
Besides forming heterotetrameric complexes with DNMT3L, DNMT3A alone also forms homotetrameric structures and higher aggregates. The reason for this is that the FF interface of the DNMT3A/DNMT3L tetramer is symmetric so it also supports the homotypic interaction of two DNMT3A molecules in addition to the heterotypic interaction of DNMT3A with DNMT3L. Hence, each DNMT3A subunit contains two interfaces for homotypic interactions, the RD interface and the FF interface, explaining why it can form tetramers in which two additional DNMT3A subunits replace DNMT3L. Biochemical studies indeed demonstrated that DNMT3A catalytic domain and DNMT3A2 homotetramers are formed in the absence of DNMT3L (Jurkowska et al. 2011b; Nguyen et al. 2019). At higher protein concentrations further multimerization can occur and generate protein fibers (Fig. 4.5b), which can lead to reversible aggregation of DNMT3A as observed in different studies (Jurkowska et al. 2011b; Kareta et al. 2006). Notably, the addition of DNMT3L directs the preferential formation of defined DNMT3A/DNMT3L heterotetramers that cannot extend anymore, because DNMT3L does not contain an RD interface, and therefore functions as a cap in protein multimerization. As described below, this process has been implicated in the release of DNMT3A from heterochromatic sites by the addition of DNMT3L (Jurkowska et al. 2011b).
Since each RD interface of a multimeric DNMT3A oligomer constitutes a potential DNA binding site, the protein oligomers can bind to more than one DNA molecule, provided that they are oriented roughly in parallel, as shown by biophysical experiments (Jurkowska et al. 2011b). Strikingly, modeling suggests that a DNMT3A hexamer could simultaneously bind to the two linker DNAs emerging from one nucleosome (Fig. 4.5c). The ability to form protein oligomers apparently plays a central role in the heterochromatin localization of DNMT3A, as non-oligomerizing DNMT3A mutants affected at these interfaces lost the ability to bind to heterochromatin, despite the presence of intact PWWP and ADD domains. Since heterochromatic DNA is densely packed, it can provide several DNA strands for DNMT3A interaction in matching geometry, and this might contribute to guiding DNMT3A to pericentromeric chromatin.
Recently, a biochemical and molecular dynamics study revealed that the R882H cancer mutation also has a direct effect on the multimerization of DNMT3A as it was found that R882H/R822H RD interfaces were more preferred than WT/WT RD interfaces (Mack et al. 2022). Interestingly, one consequence of this finding is that R882H/WT FF-heterodimers preferentially assemble WT-R882H-R882H-WT heterotetramers, in which all enzymatic activity comes from the two central R882H subunits. Hence, this mechanism provides an elegant explanation why in R882H/WT heterozygous cells the R882H mutant behaves dominantly.
Despite significant progress in dissecting protein multimerization of the DNMT3 enzymes, many questions are still not resolved. For example, DNMT3A has been shown to form catalytically active heterodimers with DNMT3B that use the same interfaces as described above for DNMT3A (Li et al. 2007). In addition, a recent cryo-EM structure illustrated that DNMT3B3, a splice isoform of DNMT3B, can replace DNMT3L at the outer complex positions, forming linear DNMT3A2/DNMT3B3 heterotetramers (Xu et al. 2020). However, the relative affinities for the homotypic DNMT3A and DNMT3B, as compared to the heterotypic interaction of DNMT3A and DNMT3B at the two interfaces are currently unknown. Moreover, the relative preferences for binding DNMT3L at the FF interface are also unknown, although the formation of defined heterotetramers of DNMT3A and DNMT3L suggests that the DNMT3A/DNMT3L interaction is preferred over the DNMT3A/DNMT3A interaction. Finally, the direct proof for the existence of DNMT3 protein multimers in cells that are larger than the tetrameric structure observed in the DNMT3A/DNMT3L complex still needs to be provided. Nevertheless, the availale biochemical and structural data confirmed that the DNMT3 binding interfaces support the interaction of different DNMT3 members, offering a unique potential for regulating methyltransferase activity depending on the complex composition.
4.4.6.2 Multimerization of DNMT3A and DNMT3A/DNMT3L on DNA
As described above, DNMT3A forms a linear heterotetrameric complex with DNMT3L, in which two central DNMT3A subunits interact via the RD interface and generate the DNA binding pocket (Fig. 4.5a) (Jia et al. 2007; Jurkowska et al. 2008). DNA binding by DNMT3A is non-specific (Rajavelu et al. 2012) and DNMT3A (and DNMT3A/DNMT3L) complexes polymerize on DNA by binding next to each other and forming DNMT3A–DNA filaments (Jurkowska et al. 2008; Rajavelu et al. 2012) (Fig. 4.5). A productive interaction of DNMT3A complexes with neighboring CpG sites is possible if they are present approximately 10–12 bps apart, due to the spacing of the two active centers at the RD interface of the individual DNMT3 complex subunits. Indeed, in vitro methylation experiments demonstrated that there is a correlation of methylation between sites localized ~10 bps apart (Jia et al. 2007; Jurkowska et al. 2008). Interestingly, the enrichment of CpG sites in such distance is observed in the differentially methylated regions (DMRs) of 12 maternally imprinted mouse genes, which are the biological substrates of the DNMT3A/DNMT3L complex, suggesting that the favorable CpG spacing could make these sequences good substrates for the MTase complex (Jia et al. 2007). As mentioned above, co-methylation of CpG sites at a distance of 12 bps by DNMT3A and DNMT3A/DNMT3L complexes was experimentally shown. Kinetic experiments supported with atomic force microscopy provided evidence that dimers of DNMT3A homotetramers or DNMT3A/DNMT3L heterotetramers can also form and interact with the CpG sites in distances of 9 or 5–6 base pairs. These complexes induce stronger DNA bending and represent either a tetramer swap or a side-by-side binding structure (Emperle et al. 2021). These additional complex conformations may explain how DNMT3A can methylate natural DNA, which does not present CpG sites in regular 12 bp spacings.
Multimerization of DNMT3A or DNMT3A/DNMT3L tetramers on DNA leads to a cooperative DNA binding, as confirmed by different methods, including cooperative binding detected in gel retardation assays, sigmoidal binding curves of DNA substrates observed in solution DNA binding experiments, and direct imaging of DNMT3–DNA filaments by atomic force microscopy (Jia et al. 2007; Jurkowska et al. 2008; Rajavelu et al. 2012; Emperle et al. 2014). The interface of adjacent DNMT3A complexes bound to DNA has been mapped to a loop within the TRD of DNMT3A and mutation of residues within this interface disrupted multimerization (Rajavelu et al. 2012). Interestingly, it also led to the loss of heterochromatic enrichment of DNMT3A, suggesting that cooperative DNA binding and multimerization of DNMT3A complexes on DNA contribute to the heterochromatic localization of the enzyme in cells. Biochemical studies have further shown that the cooperative binding of DNMT3A to long DNA substrates increases the rate of DNA methylation (Emperle et al. 2014), indicating that it is important for DNA methylation by DNMT3A. However, the exact role of cooperative DNA binding of DNMT3A in cells needs further investigation because the sizes of DNMT3A–DNA filaments in living cells are currently unknown; one may speculate that binding of up to 5 complexes would be possible in the linker DNA regions between neighboring nucleosomes. This is in agreement with biochemical data showing preferential methylation of linker DNA by DNMT3 enzymes in vitro (Gowher et al. 2005b; Takeshima et al. 2008; Felle et al. 2011; Bröhm et al. 2022). In vivo studies confirmed this observation, showing that DNMT3B expressed in yeast preferentially methylates linker DNA (Morselli et al. 2015) and a similar pattern was also observed after reintroduction of the DNMT3 enzymes into KO cell lines (Baubec et al. 2015). Longer filaments may form if DNMT3 binding is coupled to nucleosome remodeling. Consistently, DNMT3s form complexes with various chromatin remodelers, including SMARCA4 (Datta et al. 2005), CHD4 (Cai et al. 2014), hSNF2 (Geiman et al. 2004), and HELLS (Zhu et al. 2006; Myant and Stancheva 2008) and the interaction with HELLS is essential for DNA methylation (Muegge 2005). In line with this model, the remodeling activity has been shown to promote the methylation of nucleosomal DNA (Felle et al. 2011). One important functional aspect of the cooperative DNA binding of DNMT3A may be that it increases the DNA binding affinity and reduces the rate of dissociation, which may help to anchor the MTase on the DNA, in agreement with its strong binding to methylated chromatin (Jeong et al. 2009; Sharma et al. 2011).
4.4.7 Direct Chromatin Interaction of DNMT3 Enzymes
4.4.7.1 Binding of the DNMT3 ADD Domain to H3 Tails
The ADD domains of DNMT3A, DNMT3B, and DNMT3L proteins interact specifically with histone H3 tails unmethylated at lysine 4 (Fig. 4.4), and the binding is disrupted by H3K4me2, H3K4me3, H3K4ac or the acetylation of the N-terminus of H3 (Ooi et al. 2007; Otani et al. 2009; Zhang et al. 2010; Noh et al. 2015). Interestingly, H3K4me1, which is observed at enhancers, does not hinder the binding of ADD much, but the phosphorylation of T6 does (Zhang et al. 2010; Noh et al. 2015). The structures of the ADD domains from DNMT3A and DNMT3L in complex with histone H3 tail peptides were solved (Ooi et al. 2007; Otani et al. 2009). Notably, binding to H3 tails stimulates the methylation of chromatin-bound DNA by DNMT3A in vitro (Zhang et al. 2010) and directly activates DNMT3A by an allosteric mechanism (Li et al. 2011). This regulatory mechanism has been confirmed in a structural analysis by Xu and colleagues, which showed that the ADD domain could bind to the catalytic domain of DNMT3A at two sites, an allosteric site and an autoinhibitory site. H3 peptide binding stabilizes the active conformation, leading to allosteric activation of DNMT3A (Guo et al. 2015) (Fig. 4.4b). These results indicate that the ADD domain of DNMT3A can guide DNA methylation in response to specific histone modifications and provided the first evidence that DNA methyltransferases could be targeted to chromatin carrying specific marks. Indeed, a strong correlation of DNA methylation with the absence of H3K4me3 was observed in several genome-wide studies (Hodges et al. 2009; Meissner et al. 2008; Weber et al. 2007; Zhang et al. 2009), suggesting that this mechanism plays an important role in the generation of the genomic DNA methylation pattern. This hypothesis was experimentally verified, when it was shown that 1) a DNMT3A enzyme with an engineered ADD domain able to tolerate K4 methylation or T6 phosphorylation generated abnormal DNA methylation patterns in cells (Noh et al. 2015), and 2) DNMT3B artificially introduced in yeast did not methylate genomic regions with high H3K4me3 content (Morselli et al. 2015). The stimulation of DNMT3A activity by histone H3 PTMs interaction is reminiscent of the DNMT1 interaction with double ubiquitinated H3 tails, H3K9me3 or H4K20me3, further illustrating the common principles of regulatory mechanisms of DNMT1 and DNMT3 enzymes.
4.4.7.2 Binding of DNMT3 PWWP Domain to H3 Methylated at K36
The PWWP domain is essential for the targeting of DNMT3A and DNMT3B to pericentromeric chromatin (Chen et al. 2004; Ge et al. 2004) and gene bodies via specific recognition of histone H3 tails di- or tri-methylated at lysine 36 (H3K36me2/me3) (Dhayalan et al. 2010). In addition, the interaction of DNMT3A with H3K36me3 increases the activity of DNMT3A on chromatin, which carries H3K36me2/3 (Dhayalan et al. 2010). These findings explain the genome-wide correlation of DNA methylation and H3K36me3 methylation in gene bodies. H3K36me3 accumulates in euchromatin in the body of active genes and its distribution is anti-correlated with H3K4me3 (Barski et al. 2007; Edmunds et al. 2008; Guenther et al. 2007; Larschan et al. 2007; Vakoc et al. 2006). DNA methylation of gene bodies mirrors that pattern, with gene bodies of active genes showing high and those of inactive genes low methylation (Ball et al. 2009; Hellman and Chess 2007). Additionally, a correlation between H3K36me3 and DNA methylation was observed at exon–intron boundaries, with exons showing increased levels of both H3K36me3 (Kolasinska-Zwierz et al. 2009) and DNA methylation (Hodges et al. 2009). Moreover, a subset of heterochromatin containing repetitive sequences with copy number variations is strongly enriched in H3K36me3 (Ernst et al. 2011), which may explain the role of the DNMT3A PWWP domain in the heterochromatic localization of the enzyme and the strong correlation of DNA methylation, absence of H3K4me3 and presence of H3K36me3 observed in genome-wide DNA methylation studies (Meissner et al. 2008; Hodges et al. 2009).
The central role of K36 methylation in targeting DNA methylation has been experimentally confirmed in yeast (Morselli et al. 2015) and in a study showing that the methylation of gene bodies by DNMT3B directly depends on H3K36me3 methylation and requires an intact DNMT3B PWWP domain (Baubec et al. 2015). In contrast, the PWWP domain of DNMT3A mediates binding and DNA methylation at H3K36me2-containing intergenic regions (Weinberg et al. 2019), in agreement with a preference of the DNMT3A PWWP domain for binding to H3K36me2 over H3K36me3 in vitro (Weinberg et al. 2019; Dukatz et al. 2019a).
Finally, mutations within the PWWP domain cause aberrant DNMT3A localization and genomic DNA methylation (Heyn et al. 2019; Dukatz et al. 2019a; Remacha et al. 2018; Weinberg et al. 2021), further emphasizing the critical role of H3K36me2/3 interaction for DNMT3A targeting and function. The PWWP domains of DNMT3A and DNMT3B were also shown to interact with DNA to a variable degree, with DNMT3B PWWP binding DNA more strongly (Qiu et al. 2002; Purdy et al. 2010). Moreover, a synergistic interaction of the DNMT3A PWWP domain with methylated H3K36 and DNA has been observed (Dukatz et al. 2019a). In this study, a basic surface region on the PWWP domain was identified which mediates DNA interaction and is essential for the cellular localization of DNMT3A. This finding is not unexpected, as the K36 side chain emerges from the nucleosome body next to the exit site of the linker DNA. H3K36me2/3 and DNA binding by PWWP domains are mediated by two adjacent interfaces, one featuring an aromatic cage for peptide binding and the other one a basic region for DNA interaction, similarly as observed in complexes of the LEDGF PWWP domain bound to a nucleosome (Wang et al. 2020a).
4.4.7.3 H2AK119ub Binding of DNMT3A1
Interaction of the DNMT3A PWWP domain with H3K36me2/3 is believed to limit DNA methylation in Polycomb-marked regions. Mutations in the PWWP domain that disrupt K36me2/3 or DNA binding were identified in microcephalic dwarfism (Heyn et al. 2019) and paraganglioma, a rare neuroendocrine neoplasm (Remacha et al. 2018). In these diseases, hypermethylation of the DNA was observed at Polycomb regulated regions (Heyn et al. 2019; Weinberg et al. 2021), similarly as observed in mouse cells with knock-in of a DNMT3A PWWP carrying a mutation that blocks H3K36me2/3 binding (Sendzikaite et al. 2019). The aberrant DNA methylation was accompanied by the mistargeting of DNMT3A1 to Polycomb-marked chromatin. Further experiments showed that this localization is dependent on PRC1-deposited H2AK119ub, but not directly on H3K27me3 (Weinberg et al. 2021). The UDR domain in the N-terminus of DNMT3A1 has been identified as responsible for ubiquitin interaction and DNMT1A1 targeting to H2AK119-marked regions (Weinberg et al. 2021). The UDR domain of DNMT3A1 is similar to a region in 53BP1 that mediates the interaction with H2AK15ub-modified nucleosomes. H2AK119ub1 is a repressive Polycomb histone modification that occurs together with H3K27me3, hence the DNMT3A–H2AK119ub1 interaction provides a direct molecular link between DNA methylation and Polycomb silencing, two very important repressed chromatin states, explaining the association of both signals in somatic cells and cancer (Jeltsch et al. 2019). Moreover, the data indicating that the disruption of the PWWP domain promotes DNA methylation in Polycomb regions can be explained by an increased role of UDR domain mediated DNMT3A1 targeting after the functional loss of the PWWP domain. The fact that the UDR domain is absent in the DNMT3A2 splicing isoform may explain an earlier observation that the DNMT3A1 isoform is preferentially localized to bivalent CpG island promoters (Manzo et al. 2017), which likely contain H2AK119ub. Hence, a misbalance between DNMT3A recruitment mediated by distinct reader domains may contribute to the abnormal methylation patterns observed in diseases. Overall, the multivalent interaction of the DNMT3 enzymes with chromatin through multimerization, UBD, ADD, and PWWP domains can explain the strong binding of these enzymes to nucleosomal heterochromatic DNA (Jeong et al. 2009; Sharma et al. 2011).
4.4.8 Interaction Partners of DNMT3s
Up to date, the interaction of DNMT3 enzymes with DNMT3L and MeCP2 has been studied in detail, revealing important roles in targeting, allosteric regulation and control of DNMT3 multimerization. Unfortunately, for most other DNMT3 interacting proteins, detailed information about their function is not yet available.
4.4.8.1 DNMT3A/DNMT3L Interaction
DNMT3L co-localizes with both DNMT3A and DNMT3B in mammalian cells (Hata et al. 2002). It directly interacts with its C-terminal domain with the catalytic domains of DNMT3A and DNMT3B and stimulates the activity of both enzymes in vivo (Chedin et al. 2002; Chen et al. 2005) and in vitro (Suetake et al. 2004; Gowher et al. 2005a; Kareta et al. 2006). DNMT3L is expressed during gametogenesis and embryonic stages (Bourc'his et al. 2001; Hata et al. 2002; Bourc'his and Bestor 2004), where it functions as a stimulatory factor of DNTM3A needed to establish DNA methylation patterns in the developing germline cells. The structure of the complex of the C-terminal domains of DNMT3A and DNMT3L (Fig. 4.4a) provided a mechanistic explanation for the observed stimulatory effect of DNMT3L. It revealed that the interaction of DNMT3A with DNMT3L through the FF interface influences the structure of DNMT3A via the α-helices C, D, and E. Residues from these helices directly interact with the key catalytic and AdoMet binding residues, which may explain the stimulatory effect DNMT3L exerts on AdoMet binding and the catalytic activity of DNMT3A (Jia et al. 2007). Systematic studies indicated that DNMT3L increased the activity of DNMT3A and DNMT3B without changing their flanking sequence preferences (Gao et al. 2020b; Mao et al. 2020), different from an earlier study that was based on the analysis of a much smaller number of CpG sites with different flanking context (Wienholz et al. 2010). Recently, it was shown that the DNMT3 splice isoform, DNMT3B3, can replace DNMT3L in heterotetramer formation and stimulate the activity of DNMT3A and DNMT3B as well (Zeng et al. 2020). Hence, different complex formation between DNMT3 family members and their splice variants provides an additional layer regulating the activity of DNMT3 enzymes.
As described above, binding of DNMT3L to DNMT3A leads to the disruption of DNMT3A protein oligomers and changes the sub-nuclear localization of DNMT3A in cells (Fig. 4.6). In vivo, DNMT3L was shown to release DNMT3A from heterochromatin, by disrupting large DNMT3A oligomers and converting them into defined tetramers, which are homogeneously distributed in the cell nucleus (Jurkowska et al. 2011b). The redistribution of DNMT3A may be important for the methylation of imprinted differentially methylated regions (DMRs) and other targets in gene promoters, which generally are euchromatic. This finding goes in line with the discovery that DNMT3L favors DNA methylation in gene bodies (Neri et al. 2013). Hence, DNMT3L, which was originally discovered as a stimulator of DNMT3A (Gowher et al. 2005a), also changes the sub-nuclear localization of this enzyme (Jurkowska et al. 2011b). It needs to be investigated whether DNMT3B3 has similar effects as well. Additional evidence indicates that the combined regulation of the activity and localization of DNMT3A also applies to other regulatory cues (see below for MeCP2 interaction and CK2-mediated phosphorylation of DNMT3A) and might be a general mechanism of regulation for this family of enzymes (Fig. 4.6).

Mechanisms regulating the activity and localization of DNMT3A. Different interactors and PTMs regulate the activity and localization in a concerted fashion. DNMT3L stimulates DNMT3A and promotes its euchromatic localization. Contrarily, MeCP2- and CK2-mediated phosphorylation downregulate the activity of DNMT3A and promote its heterochromatic localization, where the interaction with modified H3 tails allosterically stimulates the enzyme
4.4.8.2 Interaction of DNMT3A with MeCP2
The chromatin protein MeCP2, which binds methylated DNA with its methyl-binding domain (MBD), was identified as a direct and strong interactor of DNMT3A and DNMT3B (Rajavelu et al. 2018). The interaction was mapped to the transcriptional repression domain (TRD) of MeCP2 and the ADD domain of the DNMT3 enzymes. Binding of MeCP2 resulted in a strong reduction of the DNA methylation activity of DNMT3A in vitro, and overexpression of MeCP2 in human cells led to a global reduction of DNA methylation. Biochemical experiments revealed that the binding of MeCP2 allosterically stabilizes the autoinhibitory conformation of DNMT3A. Interestingly, this interaction and its resulting inhibition were relieved by the binding of histone H3 to DNMT3A. In addition, MeCP2 contributed to the heterochromatic targeting of DNMT3A. These findings led to a model of an allosteric control of the target site specificity of DNMT3A by the combined effects of its interacting partners, like MeCP2 and histone H3 tails (Fig. 4.6). In this model, MeCP2 binding inactivates DNMT3A, thereby preventing aberrant methylation of bulk DNA. At the same time, it helps to deliver DNMT3A to heterochromatin. After binding to chromatin, which presents H3 tails modified in a PTM pattern matching the specificity of DNMT3A, the MeCP2 is released from DNMT3A, the allosteric inhibition is relieved and the activated DNMT3A enzyme can methylate its target sites. By this mechanism, MeCP2 generates a self-enhancing feedback loop that contributes to the deposition of DNA methylation at heterochromatic sites (Fig. 4.3b).
Interestingly, by this mechanism MeCP2 acts as a perfect antagonist of DNMT3L, which increases the activity of DNMT3A and leads to its release from heterochromatin. Moreover, the regulation of DNMT3A by CK2 (which is described below) resembles the MeCP2 effect, since it reduces the activity of DNMT3A and contributes to the heterochromatic sequestering of the methyltransferase. This illustrates an unexpected mechanistic convergence in the regulation and targeting of DNMT3A by interactors and posttranslational modifications (Fig. 4.6).
4.4.8.3 Other DNMT3A Interacting Proteins
Biochemical studies revealed that DNMT3A interacts with the tumor suppressor protein p53 in vitro and in cells (Wang et al. 2005; Sandoval and Reich 2019). This interaction reduces the activity of DNMT3A by interfering with tetramer formation in a DNMT3L competitive manner (Sandoval and Reich 2019). In turn, DNMT3A binding suppresses p53-mediated transcriptional activation in cells (Wang et al. 2005). The recent discovery of the SPOCD1 protein as an interactor of DNMT3A/DNMT3L and a key mediator in piRNA-directed DNA methylation has started to shed light on this fascinating process (Zoch et al. 2020).
4.4.9 Phosphorylation of DNMT3A
The regulation of the DNMT3 enzymes by phosphorylation has not been studied much, even though >70 phosphorylation sites have been identified in DNMT3A and DNMT3B in global proteomics studies (http://www.phosphosite.org). A unique example has been provided for casein kinase 2 (CK2) (Deplus et al. 2014). CK2 is a so-called survival protein kinase, which suppresses cancer cell death and is often upregulated in cancers. It was shown that CK2 phosphorylates DNMT3A at two sites, S386 and S389, located next to the PWWP domain. CK2-mediated phosphorylation increased the heterochromatic targeting of DNMT3A and reduced its DNA methylation activity. This effect was reflected by changes in the cellular DNA methylation after CK2 knockout, which may explain global hypomethylation in cancer cells overexpressing CK2. These data further support the view that the combined regulation of enzymatic activity and localization is a general principle in the regulation of DNMT3A (as already described for DNMT3L and MeCP2 interaction above) (Fig. 4.6).
4.4.10 Binding of Regulatory DNA and RNA to DNMT3 Enzymes
Similar to DNMT1, additional nucleic acid binding sites have been identified in the N-terminal part of DNMT3 enzymes. As described above, the isolated PWWP domain of DNMT3B has a DNA binding activity (Qiu et al. 2002). In DNMT3A, an additional DNA binding site was detected and connected to the PWWP domain (Purdy et al. 2010; Dukatz et al. 2019a). Moreover, the very N-terminal part of DNMT3A had been shown to bind DNA (Suetake et al. 2011). Furthermore, it was observed that lncRNAs bind strongly to the catalytic domain of DNMT3A, causing inhibition of the enzyme (Holz-Schietinger and Reich 2012). The authors also detected binding of RNA to allosteric sites, which did not change the catalytic activity. In addition, it was shown that a ncRNA derived from the rDNA promoter binds to the promoter forming RNA/DNA triplex structures that are specifically recognized by DNMT3B, establishing a novel pathway of RNA-directed DNA methylation (Schmitz et al. 2010; Bierhoff et al. 2010). Conversely, the r-loop formation has been reported to protect promoters from DNMT3B engagement and DNA methylation in early development (Ginno et al. 2012). Future work will show whether the recruitment by regulatory DNA and RNA emerges as a new and general principle of DNMT targeting and regulation.
4.5 Outlook
After more than 40 years of intensive research in the DNA methylation field, we have learned many fascinating details regarding the biochemical, structural, and enzymatic properties of the mammalian DNA methyltransferases. With the availability of additional DNMT structures with bound substrate DNA, we begin to understand the molecular determinants of the enzymes’ specificities. Strong effects of flanking sequences on the activity of DNMTs have been partially explained and structural studies of DNMT1 discovered a striking flanking sequence-dependent complex conformation. A better understanding of these effects will require the generation of additional DNMT structures with different DNA substrates, potentially combined with molecular dynamic simulations.
Despite progress regarding the mechanism of DNMTs, their regulation in cells has only begun to be uncovered. Importantly, it has been lately realized that the precise control of DNMT activity is critically involved in the generation and maintenance of the dynamic DNA methylation patterns in living cells. Crystallographic studies with DNMT1 and DNMT3A revealed that both enzymes unexpectedly undergo large domain rearrangements, which allosterically regulate their catalytic activity. This unforeseen discovery leads to the important conclusion that by influencing domain rearrangements any posttranslational modification and interaction partner, be it a protein, allosteric DNA or non-coding RNA, could directly regulate the enzymatic activity, specificity, and localization of DNMTs via allosteric effects. This novel insight provides fascinating perspectives on the investigation of the effects of interactors and PTMs on these enzymes. More and more DNMTs interaction partners are discovered and investigated, emphasizing the key roles of other protein factors and chromatin modifications in targeting and regulation of DNMTs. To better understand the mechanisms and functions of these interactions, more cryo-EM structures of larger protein complexes, including DNMTs and nucleosomes will be needed.
Abbreviations
- 5mC:
-
5-methylcytosine
- ADD domain:
-
ATRX-Dnmt3-DNMT3L domain
- AdoHcy:
-
S-adenosyl-L-homocysteine
- AdoMet:
-
S-adenosyl-L-methionine
- AML:
-
acute myeloid leukaemia
- BAH domain:
-
Bromo-adjacent homology domain
- CpG:
-
cytosine-guanine dinucleotide sequence
- DMAP1:
-
DNA methyltransferase-associated protein 1
- DMR:
-
differentially methylated region
- DNMT:
-
(mammalian) DNA nucleotide methyltransferase
- ES cells:
-
embryonic stem cells
- HDAC:
-
histone deacetylase
- ICF:
-
Immunodeficiency-centromeric instability-facial anomalies syndrome
- lncRNA:
-
long non-coding RNA
- KG repeats:
-
lysine-glycine repeats
- KO:
-
knock out
- MBD:
-
methyl-binding domain
- miRNA:
-
micro-RNA
- MTase:
-
methyltransferase
- ncRNA:
-
non-coding RNA
- PCNA:
-
proliferating cell nuclear antigen
- PBD:
-
PCNA binding domain
- PHD:
-
plant homeodomain
- PTM:
-
posttranslational modification
- RING:
-
really interesting new gene
- RFTD:
-
replication foci targeting domain
- SIRT1:
-
sirtuin 1
- SRA domain:
-
SET and RING-associated domain
- TET:
-
Ten-eleven translocation
- TRD:
-
target recognition domain
- TTD:
-
tandem Tudor domain
- UBL:
-
ubiquitin-like domain
- UDR:
-
ubiquitin-dependent recruitment
- UHRF1:
-
ubiquitin-like with PHD and ring finger domains 1
- USP7:
-
ubiquitin-specific peptidase 7
References
Adam S, Anteneh H, Hornisch M, Wagner V, Lu J, Radde NE et al (2020) DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation. Nat Commun 11(1):3723. https://doi.org/10.1038/s41467-020-17531-8
Allis CD, Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat Rev Genet 17(8):487–500. https://doi.org/10.1038/nrg.2016.59
Ambrosi C, Manzo M, Baubec T (2017) Dynamics and context-dependent roles of DNA methylation. J Mol Biol 429(10):1459–1475. https://doi.org/10.1016/j.jmb.2017.02.008
Anteneh H, Fang J, Song J (2020) Structural basis for impairment of DNA methylation by the DNMT3A R882H mutation. Nat Commun 11(1):2294. https://doi.org/10.1038/s41467-020-16213-9
Aoki A, Suetake I, Miyagawa J, Fujio T, Chijiwa T, Sasaki H et al (2001) Enzymatic properties of de novo-type mouse DNA (cytosine-5) methyltransferases. Nucleic Acids Res 29(17):3506–3512
Arand J, Spieler D, Karius T, Branco MR, Meilinger D, Meissner A et al (2012) In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet 8(6):e1002750. https://doi.org/10.1371/journal.pgen.1002750
Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M (2008) Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature 455(7214):818–821. https://doi.org/10.1038/nature07249
Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C et al (2008) Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 455(7214):822–825. https://doi.org/10.1038/nature07273
Bacolla A, Pradhan S, Roberts RJ, Wells RD (1999) Recombinant human DNA (cytosine-5) methyltransferase. II. Steady-state kinetics reveal allosteric activation by methylated dna. J Biol Chem 274(46):33011–33019
Bacolla A, Pradhan S, Larson JE, Roberts RJ, Wells RD (2001) Recombinant human DNA (cytosine-5) methyltransferase. III. Allosteric control, reaction order, and influence of plasmid topology and triplet repeat length on methylation of the fragile X CGG.CCG sequence. J Biol Chem 276(21):18605–18613. https://doi.org/10.1074/jbc.M100404200
Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH et al (2009) Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 27(4):361–368. https://doi.org/10.1038/nbt.1533
Barau J, Teissandier A, Zamudio N, Roy S, Nalesso V, Herault Y et al (2016) The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 354(6314):909–912. https://doi.org/10.1126/science.aah5143
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z et al (2007) High-resolution profiling of histone methylations in the human genome. Cell 129(4):823–837. https://doi.org/10.1016/j.cell.2007.05.009
Bashtrykov P, Jankevicius G, Smarandache A, Jurkowska RZ, Ragozin S, Jeltsch A (2012a) Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem Biol 19(5):572–578. https://doi.org/10.1016/j.chembiol.2012.03.010
Bashtrykov P, Ragozin S, Jeltsch A (2012b) Mechanistic details of the DNA recognition by the Dnmt1 DNA methyltransferase. FEBS Lett 586(13):1821–1823. https://doi.org/10.1016/j.febslet.2012.05.026
Bashtrykov P, Jankevicius G, Jurkowska RZ, Ragozin S, Jeltsch A (2014a) The UHRF1 protein stimulates the activity and specificity of the maintenance DNA methyltransferase DNMT1 by an allosteric mechanism. J Biol Chem 289(7):4106–4115. https://doi.org/10.1074/jbc.M113.528893
Bashtrykov P, Rajavelu A, Hackner B, Ragozin S, Carell T, Jeltsch A (2014b) Targeted mutagenesis results in an activation of DNA methyltransferase 1 and confirms an autoinhibitory role of its RFTS domain. Chembiochem 15(5):743–748. https://doi.org/10.1002/cbic.201300740
Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR et al (2015) Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520(7546):243–247. https://doi.org/10.1038/nature14176
Bergman Y, Cedar H (2013) DNA methylation dynamics in health and disease. Nat Struct Mol Biol 20(3):274–281. https://doi.org/10.1038/nsmb.2518
Berkyurek AC, Suetake I, Arita K, Takeshita K, Nakagawa A, Shirakawa M et al (2014) The DNA methyltransferase Dnmt1 directly interacts with the SET and RING finger-associated (SRA) domain of the multifunctional protein Uhrf1 to facilitate accession of the catalytic center to hemi-methylated DNA. J Biol Chem 289(1):379–386. https://doi.org/10.1074/jbc.M113.523209
Bierhoff H, Schmitz K, Maass F, Ye J, Grummt I (2010) Noncoding transcripts in sense and antisense orientation regulate the epigenetic state of ribosomal RNA genes. Cold Spring Harb Symp Quant Biol 75:357–364. https://doi.org/10.1101/sqb.2010.75.060
Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317(5845):1760–1764. https://doi.org/10.1126/science.1147939
Bourc'his D, Bestor TH (2004) Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431(7004):96–99. https://doi.org/10.1038/nature02886
Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294(5551):2536–2539. https://doi.org/10.1126/science.1065848
Brenner C, Deplus R, Didelot C, Loriot A, Vire E, De Smet C et al (2005) Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J 24(2):336–346. https://doi.org/10.1038/sj.emboj.7600509
Bröhm A, Schoch T, Dukatz M, Graf N, Dorscht F, Mantai E et al (2022) Methylation of recombinant mononucleosomes by DNMT3A demonstrates efficient linker DNA methylation and a role of H3K36me3. Commun Biol 5(1):192
Cai Y, Geutjes EJ, de Lint K, Roepman P, Bruurs L, Yu LR et al (2014) The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene 33(17):2157–2168. https://doi.org/10.1038/onc.2013.178
Chedin F, Lieber MR, Hsieh CL (2002) The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci U S A 99(26):16916–16921. https://doi.org/10.1073/pnas.262443999
Chen TP, Tsujimoto N, Li E (2004) The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol Cell Biol 24(20):9048–9058. https://doi.org/10.1128/Mcb.24.20.9048-9058.2004
Chen ZX, Mann JR, Hsieh CL, Riggs AD, Chedin F (2005) Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J Cell Biochem 95(5):902–917. https://doi.org/10.1002/jcb.20447
Chen L, Chen K, Lavery LA, Baker SA, Shaw CA, Li W et al (2015) MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc Natl Acad Sci U S A 112(17):5509–5514. https://doi.org/10.1073/pnas.1505909112
Cheng X, Blumenthal RM (2008) Mammalian DNA methyltransferases: a structural perspective. Structure 16(3):341–350. https://doi.org/10.1016/j.str.2008.01.004
Cheng X, Roberts RJ (2001) AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res 29(18):3784–3795
Cheng J, Yang H, Fang J, Ma L, Gong R, Wang P et al (2015) Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat Commun 6:7023. https://doi.org/10.1038/ncomms8023
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC et al (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325(5942):834–840. https://doi.org/10.1126/science.1175371
Christman JK, Sheikhnejad G, Marasco CJ, Sufrin JR (1995) 5-Methyl-2′-deoxycytidine in single-stranded DNA can act in cis to signal de novo DNA methylation. Proc Natl Acad Sci U S A 92(16):7347–7351
Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF (1997) Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 277(5334):1996–2000
Datta J, Majumder S, Bai S, Ghoshal K, Kutay H, Smith DS et al (2005) Physical and functional interaction of DNA methyltransferase 3A with Mbd3 and Brg1 in mouse lymphosarcoma cells. Cancer Res 65(23):10891–10900. https://doi.org/10.1158/0008-5472.CAN-05-1455
Deplus R, Blanchon L, Rajavelu A, Boukaba A, Defrance M, Luciani J et al (2014) Regulation of DNA methylation patterns by CK2-mediated phosphorylation of Dnmt3a. Cell Rep 8(3):743–753. https://doi.org/10.1016/j.celrep.2014.06.048
Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S et al (2010) The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem 285(34):26114–26120. https://doi.org/10.1074/jbc.M109.089433
Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J et al (2013) DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 503(7476):371–376. https://doi.org/10.1038/nature12598
Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S et al (2010) DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci Signal 3(146):ra80. https://doi.org/10.1126/scisignal.2001462
Du Q, Wang Z, Schramm VL (2016) Human DNMT1 transition state structure. Proc Natl Acad Sci U S A 113:2916. https://doi.org/10.1073/pnas.1522491113
Dukatz M, Holzer K, Choudalakis M, Emperle M, Lungu C, Bashtrykov P et al (2019a) H3K36me2/3 binding and DNA binding of the DNA methyltransferase DNMT3A PWWP domain both contribute to its chromatin interaction. J Mol Biol 431(24):5063–5074. https://doi.org/10.1016/j.jmb.2019.09.006
Dukatz M, Requena CE, Emperle M, Hajkova P, Sarkies P, Jeltsch A (2019b) Mechanistic insights into cytosine-N3 methylation by DNA methyltransferase DNMT3A. J Mol Biol 431(17):3139–3145. https://doi.org/10.1016/j.jmb.2019.06.015
Dukatz M, Adam S, Biswal M, Song J, Bashtrykov P, Jeltsch A (2020) Complex DNA sequence readout mechanisms of the DNMT3B DNA methyltransferase. Nucleic Acids Res 48(20):11495–11509. https://doi.org/10.1093/nar/gkaa938
Easwaran HP, Schermelleh L, Leonhardt H, Cardoso MC (2004) Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep 5(12):1181–1186. https://doi.org/10.1038/sj.embor.7400295
Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M et al (2006) DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 38(12):1378–1385. https://doi.org/10.1038/ng1909
Edmunds JW, Mahadevan LC, Clayton AL (2008) Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J 27(2):406–420. https://doi.org/10.1038/sj.emboj.7601967
Egger G, Jeong S, Escobar SG, Cortez CC, Li TW, Saito Y et al (2006) Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc Natl Acad Sci U S A 103(38):14080–14085. https://doi.org/10.1073/pnas.0604602103
Emperle M, Rajavelu A, Reinhardt R, Jurkowska RZ, Jeltsch A (2014) Cooperative DNA binding and protein/DNA fiber formation increases the activity of the Dnmt3a DNA methyltransferase. J Biol Chem 289(43):29602–29613. https://doi.org/10.1074/jbc.M114.572032
Emperle M, Rajavelu A, Kunert S, Arimondo PB, Reinhardt R, Jurkowska RZ et al (2018) The DNMT3A R882H mutant displays altered flanking sequence preferences. Nucleic Acids Res 46(6):3130–3139. https://doi.org/10.1093/nar/gky168
Emperle M, Adam S, Kunert S, Dukatz M, Baude A, Plass C et al (2019) Mutations of R882 change flanking sequence preferences of the DNA methyltransferase DNMT3A and cellular methylation patterns. Nucleic Acids Res 47(21):11355–11367. https://doi.org/10.1093/nar/gkz911
Emperle M, Bangalore DM, Adam S, Kunert S, Heil HS, Heinze KG et al (2021) Structural and biochemical insight into the mechanism of dual CpG site binding and methylation by the DNMT3A DNA methyltransferase. Nucleic Acids Res 49(14):8294–8308. https://doi.org/10.1093/nar/gkab600
Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB et al (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473(7345):43–49. https://doi.org/10.1038/nature09906
Esteve PO, Chin HG, Benner J, Feehery GR, Samaranayake M, Horwitz GA et al (2009) Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc Natl Acad Sci U S A 106(13):5076–5081. https://doi.org/10.1073/pnas.0810362106
Esteve PO, Chang Y, Samaranayake M, Upadhyay AK, Horton JR, Feehery GR et al (2011) A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat Struct Mol Biol 18(1):42–48. https://doi.org/10.1038/nsmb.1939
Esteve PO, Zhang G, Ponnaluri VK, Deepti K, Chin HG, Dai N et al (2016) Binding of 14-3-3 reader proteins to phosphorylated DNMT1 facilitates aberrant DNA methylation and gene expression. Nucleic Acids Res 44(4):1642–1656. https://doi.org/10.1093/nar/gkv1162
Fatemi M, Hermann A, Pradhan S, Jeltsch A (2001) The activity of the murine DNA methyltransferase Dnmt1 is controlled by interaction of the catalytic domain with the N-terminal part of the enzyme leading to an allosteric activation of the enzyme after binding to methylated DNA. J Mol Biol 309(5):1189–1199. https://doi.org/10.1006/jmbi.2001.4709
Fatemi M, Hermann A, Gowher H, Jeltsch A (2002) Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur J Biochem 269(20):4981–4984
Felle M, Hoffmeister H, Rothammer J, Fuchs A, Exler JH, Langst G (2011) Nucleosomes protect DNA from DNA methylation in vivo and in vitro. Nucleic Acids Res 39(16):6956–6969. https://doi.org/10.1093/nar/gkr263
Ferry L, Fournier A, Tsusaka T, Adelmant G, Shimazu T, Matano S et al (2017) Methylation of DNA ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA methylation. Mol Cell 67(4):550–65 e5. https://doi.org/10.1016/j.molcel.2017.07.012
Flynn J, Fang JY, Mikovits JA, Reich NO (2003) A potent cell-active allosteric inhibitor of murine DNA cytosine C5 methyltransferase. J Biol Chem 278(10):8238–8243. https://doi.org/10.1074/jbc.M209839200
Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 24(1):88–91. https://doi.org/10.1038/71750
Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T (2001) Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J 20(10):2536–2544. https://doi.org/10.1093/emboj/20.10.2536
Fuks F, Hurd PJ, Deplus R, Kouzarides T (2003a) The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res 31(9):2305–2312
Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T (2003b) The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 278(6):4035–4040. https://doi.org/10.1074/jbc.M210256200
Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR et al (2015) Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522(7554):89–93. https://doi.org/10.1038/nature14319
Gao L, Anteneh H, Song J (2020a) Dissect the DNMT3A- and DNMT3B-mediated DNA co-methylation through a covalent complex approach. J Mol Biol 432(2):569–575. https://doi.org/10.1016/j.jmb.2019.11.004
Gao L, Emperle M, Guo Y, Grimm SA, Ren W, Adam S et al (2020b) Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat Commun 11(1):3355. https://doi.org/10.1038/s41467-020-17109-4
Ge YZ, Pu MT, Gowher H, Wu HP, Ding JP, Jeltsch A et al (2004) Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J Biol Chem 279(24):25447–25454. https://doi.org/10.1074/jbc.M312296200
Geiman TM, Sankpal UT, Robertson AK, Zhao Y, Robertson KD (2004) DNMT3B interacts with hSNF2H chromatin remodeling enzyme, HDACs 1 and 2, and components of the histone methylation system. Biochem Biophys Res Commun 318(2):544–555. https://doi.org/10.1016/j.bbrc.2004.04.058
Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F (2012) R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45(6):814–825. https://doi.org/10.1016/j.molcel.2012.01.017
Glickman JF, Flynn J, Reich NO (1997a) Purification and characterization of recombinant baculovirus-expressed mouse DNA methyltransferase. Biochem Biophys Res Commun 230(2):280–284. https://doi.org/10.1006/bbrc.1996.5943
Glickman JF, Pavlovich JG, Reich NO (1997b) Peptide mapping of the murine DNA methyltransferase reveals a major phosphorylation site and the start of translation. J Biol Chem 272(28):17851–17857
Gowher H, Jeltsch A (2001) Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpA sites. J Mol Biol 309(5):1201–1208. https://doi.org/10.1006/jmbi.2001.4710
Gowher H, Jeltsch A (2002) Molecular enzymology of the catalytic domains of the Dnmt3a and Dnmt3b DNA methyltransferases. J Biol Chem 277(23):20409–20414. https://doi.org/10.1074/jbc.M202148200
Gowher H, Liebert K, Hermann A, Xu G, Jeltsch A (2005a) Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J Biol Chem 280(14):13341–13348. https://doi.org/10.1074/jbc.M413412200
Gowher H, Stockdale CJ, Goyal R, Ferreira H, Owen-Hughes T, Jeltsch A (2005b) De novo methylation of nucleosomal DNA by the mammalian Dnmt1 and Dnmt3A DNA methyltransferases. Biochemistry 44(29):9899–9904. https://doi.org/10.1021/bi047634t
Gowher H, Loutchanwoot P, Vorobjeva O, Handa V, Jurkowska RZ, Jurkowski TP et al (2006) Mutational analysis of the catalytic domain of the murine Dnmt3a DNA-(cytosine C5)-methyltransferase. J Mol Biol 357(3):928–941. https://doi.org/10.1016/j.jmb.2006.01.035
Goyal R, Reinhardt R, Jeltsch A (2006) Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res 34(4):1182–1188. https://doi.org/10.1093/nar/gkl002
Goyal R, Rathert P, Laser H, Gowher H, Jeltsch A (2007) Phosphorylation of serine-515 activates the mammalian maintenance methyltransferase Dnmt1. Epigenetics 2(3):155–160
Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA (2007) A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130(1):77–88. https://doi.org/10.1016/j.cell.2007.05.042
Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B et al (2014) Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci 17(2):215–222. https://doi.org/10.1038/nn.3607
Guo X, Wang L, Li J, Ding Z, Xiao J, Yin X et al (2015) Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 517(7536):640–644. https://doi.org/10.1038/nature13899
Haggerty C, Kretzmer H, Riemenschneider C, Kumar AS, Mattei AL, Bailly N et al (2021) Dnmt1 has de novo activity targeted to transposable elements. Nat Struct Mol Biol 28(7):594–603. https://doi.org/10.1038/s41594-021-00603-8
Hamidi T, Singh AK, Chen T (2015) Genetic alterations of DNA methylation machinery in human diseases. Epigenomics 7(2):247–265. https://doi.org/10.2217/epi.14.80
Handa V, Jeltsch A (2005) Profound flanking sequence preference of Dnmt3a and Dnmt3b mammalian DNA methyltransferases shape the human epigenome. J Mol Biol 348(5):1103–1112. https://doi.org/10.1016/j.jmb.2005.02.044
Hashimoto H, Horton JR, Zhang X, Bostick M, Jacobsen SE, Cheng X (2008) The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature 455(7214):826–829. https://doi.org/10.1038/nature07280
Hata K, Okano M, Lei H, Li E (2002) Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129(8):1983–1993
Hellman A, Chess A (2007) Gene body-specific methylation on the active X chromosome. Science 315(5815):1141–1143. https://doi.org/10.1126/science.1136352
Hermann A, Gowher H, Jeltsch A (2004a) Biochemistry and biology of mammalian DNA methyltransferases. Cell Mol Life Sci 61(19–20):2571–2587. https://doi.org/10.1007/s00018-004-4201-1
Hermann A, Goyal R, Jeltsch A (2004b) The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem 279(46):48350–48359. https://doi.org/10.1074/jbc.M403427200
Hervouet E, Lalier L, Debien E, Cheray M, Geairon A, Rogniaux H et al (2010) Disruption of Dnmt1/PCNA/UHRF1 interactions promotes tumorigenesis from human and mice glial cells. PLoS One 5(6):e11333. https://doi.org/10.1371/journal.pone.0011333
Heyn P, Logan CV, Fluteau A, Challis RC, Auchynnikava T, Martin CA et al (2019) Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat Genet 51(1):96–105. https://doi.org/10.1038/s41588-018-0274-x
Hodges E, Smith AD, Kendall J, Xuan Z, Ravi K, Rooks M et al (2009) High definition profiling of mammalian DNA methylation by array capture and single molecule bisulfite sequencing. Genome Res 19(9):1593–1605. https://doi.org/10.1101/gr.095190.109
Holoch D, Moazed D (2015) RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 16(2):71–84. https://doi.org/10.1038/nrg3863
Holz-Schietinger C, Reich NO (2010) The inherent processivity of the human de novo methyltransferase 3A (DNMT3A) is enhanced by DNMT3L. J Biol Chem 285(38):29091–29100. https://doi.org/10.1074/jbc.M110.142513
Holz-Schietinger C, Reich NO (2012) RNA modulation of the human DNA methyltransferase 3A. Nucleic Acids Res 40(17):8550–8557. https://doi.org/10.1093/nar/gks537
Hu L, Li Z, Wang P, Lin Y, Xu Y (2011) Crystal structure of PHD domain of UHRF1 and insights into recognition of unmodified histone H3 arginine residue 2. Cell Res 21(9):1374–1378. https://doi.org/10.1038/cr.2011.124
Iida T, Suetake I, Tajima S, Morioka H, Ohta S, Obuse C et al (2002) PCNA clamp facilitates action of DNA cytosine methyltransferase 1 on hemimethylated DNA. Genes Cells 7(10):997–1007
Ishiyama S, Nishiyama A, Saeki Y, Moritsugu K, Morimoto D, Yamaguchi L et al (2017) Structure of the Dnmt1 reader module complexed with a unique two-mono-ubiquitin mark on histone H3 reveals the basis for DNA methylation maintenance. Mol Cell 68(2):350–60 e7. https://doi.org/10.1016/j.molcel.2017.09.037
Iwasaki YW, Siomi MC, Siomi H (2015) PIWI-interacting RNA: its biogenesis and functions. Annu Rev Biochem 84:405–433. https://doi.org/10.1146/annurev-biochem-060614-034258
Jain D, Meydan C, Lange J, Claeys Bouuaert C, Lailler N, Mason CE et al (2017) Rahu is a mutant allele of Dnmt3c, encoding a DNA methyltransferase homolog required for meiosis and transposon repression in the mouse male germline. PLoS Genet 13(8):e1006964. https://doi.org/10.1371/journal.pgen.1006964
Jeltsch A (2002) Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem 3(4):275–293
Jeltsch A (2006) On the enzymatic properties of Dnmt1: specificity, processivity, mechanism of linear diffusion and allosteric regulation of the enzyme. Epigenetics 1(2):63–66
Jeltsch A (2008) Reading and writing DNA methylation. Nat Struct Mol Biol 15(10):1003–1004. https://doi.org/10.1038/nsmb1008-1003
Jeltsch A, Jurkowska RZ (2013) Multimerization of the dnmt3a DNA methyltransferase and its functional implications. Prog Mol Biol Transl Sci 117:445–464. https://doi.org/10.1016/B978-0-12-386931-9.00016-7
Jeltsch A, Jurkowska RZ (2014) New concepts in DNA methylation. Trends Biochem Sci 39(7):310–318. https://doi.org/10.1016/j.tibs.2014.05.002
Jeltsch A, Jurkowska RZ (2016) Allosteric control of mammalian DNA methyltransferases - a new regulatory paradigm. Nucleic Acids Res 44(18):8556–8575. https://doi.org/10.1093/nar/gkw723
Jeltsch A, Broche J, Bashtrykov P (2019) Molecular processes connecting DNA methylation patterns with DNA methyltransferases and histone modifications in mammalian genomes. Genes 10(5):388. https://doi.org/10.3390/genes10050388
Jeltsch A, Adam S, Dukatz M, Emperle M, Bashtrykov P (2021) Deep enzymology studies on DNA methyltransferases reveal novel connections between flanking sequences and enzyme activity. J Mol Biol 433(19):167186. https://doi.org/10.1016/j.jmb.2021.167186
Jeong S, Liang GN, Sharma S, Lin JC, Choi SH, Han H et al (2009) Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol Cell Biol 29(19):5366–5376. https://doi.org/10.1128/Mcb.00484-09
Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X (2007) Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449(7159):248–251. https://doi.org/10.1038/nature06146
Jurkowska RZ, Anspach N, Urbanke C, Jia D, Reinhardt R, Nellen W et al (2008) Formation of nucleoprotein filaments by mammalian DNA methyltransferase Dnmt3a in complex with regulator Dnmt3L. Nucleic Acids Res 36(21):6656–6663. https://doi.org/10.1093/nar/gkn747
Jurkowska RZ, Jurkowski TP, Jeltsch A (2011a) Structure and function of mammalian DNA methyltransferases. Chembiochem 12(2):206–222. https://doi.org/10.1002/cbic.201000195
Jurkowska RZ, Rajavelu A, Anspach N, Urbanke C, Jankevicius G, Ragozin S et al (2011b) Oligomerization and binding of the Dnmt3a DNA methyltransferase to parallel DNA molecules: heterochromatic localization and role of Dnmt3L. J Biol Chem 286(27):24200–24207. https://doi.org/10.1074/jbc.M111.254987
Jurkowska RZ, Siddique AN, Jurkowski TP, Jeltsch A (2011c) Approaches to enzyme and substrate design of the murine Dnmt3a DNA methyltransferase. Chembiochem 12(10):1589–1594. https://doi.org/10.1002/cbic.201000673
Kareta MS, Botello ZM, Ennis JJ, Chou C, Chedin F (2006) Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J Biol Chem 281(36):25893–25902. https://doi.org/10.1074/jbc.M603140200
Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J et al (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23(4):607–618. https://doi.org/10.1016/j.molcel.2006.06.026
Kim SH, Park J, Choi MC, Kim HP, Park JH, Jung Y et al (2007) Zinc-fingers and homeoboxes 1 (ZHX1) binds DNA methyltransferase (DNMT) 3B to enhance DNMT3B-mediated transcriptional repression. Biochem Biophys Res Commun 355(2):318–323. https://doi.org/10.1016/j.bbrc.2007.01.187
Kimura H, Shiota K (2003) Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem 278(7):4806–4812. https://doi.org/10.1074/jbc.M209923200
Klimasauskas S, Kumar S, Roberts RJ, Cheng X (1994) HhaI methyltransferase flips its target base out of the DNA helix. Cell 76(2):357–369
Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J (2009) Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet 41(3):376–381. https://doi.org/10.1038/ng.322
Kudithipudi S, Schuhmacher MK, Kebede AF, Jeltsch A (2017) The SUV39H1 protein lysine methyltransferase Methylates chromatin proteins involved in heterochromatin formation and VDJ recombination. ACS Chem Biol 12(4):958–968. https://doi.org/10.1021/acschembio.6b01076
Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P et al (2007) MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell 28(1):121–133. https://doi.org/10.1016/j.molcel.2007.08.011
Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT et al (2010) Dynamic changes in the human methylome during differentiation. Genome Res 20(3):320–331. https://doi.org/10.1101/gr.101907.109
Lavoie G, Esteve PO, Laulan NB, Pradhan S, St-Pierre Y (2011) PKC isoforms interact with and phosphorylate DNMT1. BMC Biol 9:31. https://doi.org/10.1186/1741-7007-9-31
Lee JH, Park SJ, Nakai K (2017) Differential landscape of non-CpG methylation in embryonic stem cells and neurons caused by DNMT3s. Sci Rep 7(1):11295. https://doi.org/10.1038/s41598-017-11800-1
Leng F, Yu J, Zhang C, Alejo S, Hoang N, Sun H et al (2018) Methylated DNMT1 and E2F1 are targeted for proteolysis by L3MBTL3 and CRL4(DCAF5) ubiquitin ligase. Nat Commun 9(1):1641. https://doi.org/10.1038/s41467-018-04019-9
Leonhardt H, Page AW, Weier HU, Bestor TH (1992) A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 71(5):865–873
Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP (2006) The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem 281(28):19489–19500. https://doi.org/10.1074/jbc.M513249200
Li JY, Pu MT, Hirasawa R, Li BZ, Huang YN, Zeng R et al (2007) Synergistic function of DNA methyltransferases Dnmt3a and Dnmt3b in the methylation of Oct4 and Nanog. Mol Cell Biol 27(24):8748–8759. https://doi.org/10.1128/MCB.01380-07
Li BZ, Huang Z, Cui QY, Song XH, Du L, Jeltsch A et al (2011) Histone tails regulate DNA methylation by allosterically activating de novo methyltransferase. Cell Res 21(8):1172–1181. https://doi.org/10.1038/cr.2011.92
Li T, Wang L, Du Y, Xie S, Yang X, Lian F et al (2018) Structural and mechanistic insights into UHRF1-mediated DNMT1 activation in the maintenance DNA methylation. Nucleic Acids Res 46(6):3218–3231. https://doi.org/10.1093/nar/gky104
Li J, Wang R, Jin J, Han M, Chen Z, Gao Y et al (2020) USP7 negatively controls global DNA methylation by attenuating ubiquitinated histone-dependent DNMT1 recruitment. Cell Discov 6(1):58. https://doi.org/10.1038/s41421-020-00188-4
Liang Z, Hu J, Yan W, Jiang H, Hu G, Luo C (2018) Deciphering the role of dimer interface in intrinsic dynamics and allosteric pathways underlying the functional transformation of DNMT3A. Biochim Biophys Acta Gen Subj 1862(7):1667–1679. https://doi.org/10.1016/j.bbagen.2018.04.015
Lin IG, Han L, Taghva A, O'Brien LE, Hsieh CL (2002) Murine de novo methyltransferase Dnmt3a demonstrates strand asymmetry and site preference in the methylation of DNA in vitro. Mol Cell Biol 22(3):704–723
Lin CC, Chen YP, Yang WZ, Shen JCK, Yuan HS (2020) Structural insights into CpG-specific DNA methylation by human DNA methyltransferase 3B. Nucleic Acids Res 48(7):3949–3961. https://doi.org/10.1093/nar/gkaa111
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J et al (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462(7271):315–322. https://doi.org/10.1038/nature08514
Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND et al (2013) Global epigenomic reconfiguration during mammalian brain development. Science 341(6146):1237905. https://doi.org/10.1126/science.1237905
Liu Y, Oakeley EJ, Sun L, Jost JP (1998) Multiple domains are involved in the targeting of the mouse DNA methyltransferase to the DNA replication foci. Nucleic Acids Res 26(4):1038–1045
Liu X, Gao Q, Li P, Zhao Q, Zhang J, Li J et al (2013) UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat Commun 4:1563. https://doi.org/10.1038/ncomms2562
Lukashevich OV, Cherepanova NA, Jurkovska RZ, Jeltsch A, Gromova ES (2016) Conserved motif VIII of murine DNA methyltransferase Dnmt3a is essential for methylation activity. BMC Biochem 17(1):7. https://doi.org/10.1186/s12858-016-0064-y
Mack A, Emperle M, Schnee P, Adam S, Pleiss J, Bashtrykov P et al (2022) Preferential self-interaction of DNA methyltransferase DNMT3A subunits containing the R882H cancer mutation leads to dominant changes of flanking sequence preferences. J Mol Biol 434(7):167482. https://doi.org/10.1016/j.jmb.2022.167482
Mallona I, Ilie IM, Karemaker ID, Butz S, Manzo M, Caflisch A et al (2021) Flanking sequence preference modulates de novo DNA methylation in the mouse genome. Nucleic Acids Res 49(1):145–157. https://doi.org/10.1093/nar/gkaa1168
Manzo M, Wirz J, Ambrosi C, Villasenor R, Roschitzki B, Baubec T (2017) Isoform-specific localization of DNMT3A regulates DNA methylation fidelity at bivalent CpG islands. EMBO J 36(23):3421–3434. https://doi.org/10.15252/embj.201797038
Mao SQ, Cuesta SM, Tannahill D, Balasubramanian S (2020) Genome-wide DNA methylation signatures are determined by DNMT3A/B sequence preferences. Biochemistry 59(27):2541–2550. https://doi.org/10.1021/acs.biochem.0c00339
Margot JB, Ehrenhofer-Murray AE, Leonhardt H (2003) Interactions within the mammalian DNA methyltransferase family. BMC Mol Biol 4:7. https://doi.org/10.1186/1471-2199-4-7
Markouli M, Strepkos D, Piperi C (2021) Structure, activity and function of the SETDB1 protein methyltransferase. Life (Basel) 11(8):817. https://doi.org/10.3390/life11080817
Matzke MA, Mosher RA (2014) RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet 15(6):394–408. https://doi.org/10.1038/nrg3683
Meilinger D, Fellinger K, Bultmann S, Rothbauer U, Bonapace IM, Klinkert WE et al (2009) Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep 10(11):1259–1264. https://doi.org/10.1038/embor.2009.201
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A et al (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454(7205):766–770. https://doi.org/10.1038/nature07107
Merry CR, Forrest ME, Sabers JN, Beard L, Gao XH, Hatzoglou M et al (2015) DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum Mol Genet 24(21):6240–6253. https://doi.org/10.1093/hmg/ddv343
Ming X, Zhang Z, Zou Z, Lv C, Dong Q, He Q et al (2020) Kinetics and mechanisms of mitotic inheritance of DNA methylation and their roles in aging-associated methylome deterioration. Cell Res 30(11):980–996. https://doi.org/10.1038/s41422-020-0359-9
Mishima Y, Brueckner L, Takahashi S, Kawakami T, Arita K, Oka S et al (2017) RFTS-dependent negative regulation of Dnmt1 by nucleosome structure and histone tails. FEBS J 284(20):3455–3469. https://doi.org/10.1111/febs.14205
Mishima Y, Brueckner L, Takahashi S, Kawakami T, Otani J, Shinohara A et al (2020) Enhanced processivity of Dnmt1 by monoubiquitinated histone H3. Genes Cells 25(1):22–32. https://doi.org/10.1111/gtc.12732
Morselli M, Pastor WA, Montanini B, Nee K, Ferrari R, Fu K et al (2015) In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. elife 4:e06205. https://doi.org/10.7554/eLife.06205
Muegge K (2005) Lsh, a guardian of heterochromatin at repeat elements. Biochem Cell Biol 83(4):548–554. https://doi.org/10.1139/o05-119
Myant K, Stancheva I (2008) LSH cooperates with DNA methyltransferases to repress transcription. Mol Cell Biol 28(1):215–226. https://doi.org/10.1128/MCB.01073-07
Nady N, Lemak A, Walker JR, Avvakumov GV, Kareta MS, Achour M et al (2011) Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J Biol Chem 286(27):24300–24311. https://doi.org/10.1074/jbc.M111.234104
Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F et al (2013) Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 155(1):121–134. https://doi.org/10.1016/j.cell.2013.08.056
Nguyen TV, Yao S, Wang Y, Rolfe A, Selvaraj A, Darman R et al (2019) The R882H DNMT3A hot spot mutation stabilizes the formation of large DNMT3A oligomers with low DNA methyltransferase activity. J Biol Chem 294(45):16966–16977. https://doi.org/10.1074/jbc.RA119.010126
Nishiyama A, Yamaguchi L, Sharif J, Johmura Y, Kawamura T, Nakanishi K et al (2013) Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 502(7470):249–253. https://doi.org/10.1038/nature12488
Nishiyama A, Mulholland CB, Bultmann S, Kori S, Endo A, Saeki Y et al (2020) Two distinct modes of DNMT1 recruitment ensure stable maintenance DNA methylation. Nat Commun 11(1):1222. https://doi.org/10.1038/s41467-020-15006-4
Noh KM, Wang H, Kim HR, Wenderski W, Fang F, Li CH et al (2015) Engineering of a histone-recognition domain in Dnmt3a alters the epigenetic landscape and phenotypic features of mouse ESCs. Mol Cell 59(1):89–103. https://doi.org/10.1016/j.molcel.2015.05.017
Norvil AB, AlAbdi L, Liu B, Tu YH, Forstoffer NE, Michie AR et al (2020) The acute myeloid leukemia variant DNMT3A Arg882His is a DNMT3B-like enzyme. Nucleic Acids Res 48(7):3761–3775. https://doi.org/10.1093/nar/gkaa139
Okano M, Xie SP, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19(3):219–220
O'Keefe RT, Henderson SC, Spector DL (1992) Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol 116(5):1095–1110
Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z et al (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448(7154):714–717. https://doi.org/10.1038/nature05987
Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, Shirakawa M (2009) Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep 10(11):1235–1241. https://doi.org/10.1038/embor.2009.218
Peng L, Yuan Z, Ling H, Fukasawa K, Robertson K, Olashaw N et al (2011) SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol Cell Biol 31(23):4720–4734. https://doi.org/10.1128/MCB.06147-11
Petryk N, Bultmann S, Bartke T, Defossez PA (2021) Staying true to yourself: mechanisms of DNA methylation maintenance in mammals. Nucleic Acids Res 49(6):3020–3032. https://doi.org/10.1093/nar/gkaa1154
Pradhan S, Esteve PO (2003) Allosteric activator domain of maintenance human DNA (cytosine-5) methyltransferase and its role in methylation spreading. Biochemistry 42(18):5321–5332. https://doi.org/10.1021/bi034160+
Pradhan M, Esteve PO, Chin HG, Samaranayke M, Kim GD, Pradhan S (2008) CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry 47(38):10000–10009. https://doi.org/10.1021/bi8011725
Purdy MM, Holz-Schietinger C, Reich NO (2010) Identification of a second DNA binding site in human DNA methyltransferase 3A by substrate inhibition and domain deletion. Arch Biochem Biophys 498(1):13–22. https://doi.org/10.1016/j.abb.2010.03.007
Qin S, Min J (2014) Structure and function of the nucleosome-binding PWWP domain. Trends Biochem Sci 39(11):536–547. https://doi.org/10.1016/j.tibs.2014.09.001
Qin W, Leonhardt H, Spada F (2011) Usp7 and Uhrf1 control ubiquitination and stability of the maintenance DNA methyltransferase Dnmt1. J Cell Biochem 112(2):439–444. https://doi.org/10.1002/jcb.22998
Qin W, Wolf P, Liu N, Link S, Smets M, La Mastra F et al (2015) DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res 25(8):911–929. https://doi.org/10.1038/cr.2015.72
Qiu C, Sawada K, Zhang X, Cheng X (2002) The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat Struct Biol 9(3):217–224. https://doi.org/10.1038/nsb759
Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S et al (2011) In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell 44(3):361–372. https://doi.org/10.1016/j.molcel.2011.08.032
Rajakumara E, Wang Z, Ma H, Hu L, Chen H, Lin Y et al (2011) PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol Cell 43(2):275–284. https://doi.org/10.1016/j.molcel.2011.07.006
Rajavelu A, Jurkowska RZ, Fritz J, Jeltsch A (2012) Function and disruption of DNA methyltransferase 3a cooperative DNA binding and nucleoprotein filament formation. Nucleic Acids Res 40(2):569–580. https://doi.org/10.1093/nar/gkr753
Rajavelu A, Lungu C, Emperle M, Dukatz M, Brohm A, Broche J et al (2018) Chromatin-dependent allosteric regulation of DNMT3A activity by MeCP2. Nucleic Acids Res 46(17):9044–9056. https://doi.org/10.1093/nar/gky715
Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R (2000) Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A 97(10):5237–5242
Raurell-Vila H, Bosch-Presegue L, Gonzalez J, Kane-Goldsmith N, Casal C, Brown JP et al (2017) An HP1 isoform-specific feedback mechanism regulates Suv39h1 activity under stress conditions. Epigenetics 12(2):166–175. https://doi.org/10.1080/15592294.2016.1278096
Reither S, Li F, Gowher H, Jeltsch A (2003) Catalytic mechanism of DNA-(cytosine-C5)-methyltransferases revisited: covalent intermediate formation is not essential for methyl group transfer by the murine Dnmt3a enzyme. J Mol Biol 329(4):675–684
Remacha L, Curras-Freixes M, Torres-Ruiz R, Schiavi F, Torres-Perez R, Calsina B et al (2018) Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genetics in medicine : official journal of the American College of Medical Genetics 20:1644. https://doi.org/10.1038/s41436-018-0003-y
Ren W, Fan H, Grimm SA, Guo Y, Kim JJ, Yin J et al (2020) Direct readout of heterochromatic H3K9me3 regulates DNMT1-mediated maintenance DNA methylation. Proc Natl Acad Sci U S A 117(31):18439–18447. https://doi.org/10.1073/pnas.2009316117
Ren W, Fan H, Grimm SA, Kim JJ, Li L, Guo Y et al (2021) DNMT1 reads heterochromatic H4K20me3 to reinforce LINE-1 DNA methylation. Nat Commun 12(1):2490. https://doi.org/10.1038/s41467-021-22665-4
Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166. https://doi.org/10.1146/annurev-biochem-051410-092902
Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA et al (1999) The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 27(11):2291–2298
Rondelet G, Dal Maso T, Willems L, Wouters J (2016) Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J Struct Biol 194(3):357–367. https://doi.org/10.1016/j.jsb.2016.03.013
Rosic S, Amouroux R, Requena CE, Gomes A, Emperle M, Beltran T et al (2018) Evolutionary analysis indicates that DNA alkylation damage is a byproduct of cytosine DNA methyltransferase activity. Nat Genet 50(3):452–459. https://doi.org/10.1038/s41588-018-0061-8
Rothbart SB, Krajewski K, Nady N, Tempel W, Xue S, Badeaux AI et al (2012) Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat Struct Mol Biol 19(11):1155–1160. https://doi.org/10.1038/nsmb.2391
Rothbart SB, Dickson BM, Ong MS, Krajewski K, Houliston S, Kireev DB et al (2013) Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev 27(11):1288–1298. https://doi.org/10.1101/gad.220467.113
Rountree MR, Bachman KE, Baylin SB (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet 25(3):269–277. https://doi.org/10.1038/77023
Sandoval JE, Reich NO (2019) The R882H substitution in the human de novo DNA methyltransferase DNMT3A disrupts allosteric regulation by the tumor supressor p53. J Biol Chem 294(48):18207–18219. https://doi.org/10.1074/jbc.RA119.010827
Schmitz KM, Mayer C, Postepska A, Grummt I (2010) Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 24(20):2264–2269. https://doi.org/10.1101/gad.590910
Schneider K, Fuchs C, Dobay A, Rottach A, Qin W, Wolf P et al (2013) Dissection of cell cycle-dependent dynamics of Dnmt1 by FRAP and diffusion-coupled modeling. Nucleic Acids Res 41(9):4860–4876. https://doi.org/10.1093/nar/gkt191
Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G et al (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 18(11):1251–1262. https://doi.org/10.1101/gad.300704
Schubeler D (2015) Function and information content of DNA methylation. Nature 517(7534):321–326. https://doi.org/10.1038/nature14192
Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D et al (2015) Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523(7559):212–216. https://doi.org/10.1038/nature14465
Sendzikaite G, Hanna CW, Stewart-Morgan KR, Ivanova E, Kelsey G (2019) A DNMT3A PWWP mutation leads to methylation of bivalent chromatin and growth retardation in mice. Nat Commun 10(1):1884. https://doi.org/10.1038/s41467-019-09713-w
Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA et al (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450(7171):908–912. https://doi.org/10.1038/nature06397
Sharma S, De Carvalho DD, Jeong S, Jones PA, Liang G (2011) Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PLoS Genet 7(2):e1001286. https://doi.org/10.1371/journal.pgen.1001286
Somasundaram S, Forrest ME, Moinova H, Cohen A, Varadan V, LaFramboise T et al (2018) The DNMT1-associated lincRNA DACOR1 reprograms genome-wide DNA methylation in colon cancer. Clin Epigenetics 10(1):127. https://doi.org/10.1186/s13148-018-0555-3
Song J, Rechkoblit O, Bestor TH, Patel DJ (2011) Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331(6020):1036–1040. https://doi.org/10.1126/science.1195380
Song J, Teplova M, Ishibe-Murakami S, Patel DJ (2012) Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 335(6069):709–712. https://doi.org/10.1126/science.1214453
Suetake I, Shinozaki F, Miyagawa J, Takeshima H, Tajima S (2004) DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem 279(26):27816–27823. https://doi.org/10.1074/jbc.M400181200
Suetake I, Hayata D, Tajima S (2006) The amino-terminus of mouse DNA methyltransferase 1 forms an independent domain and binds to DNA with the sequence involving PCNA binding motif. J Biochem 140(6):763–776. https://doi.org/10.1093/jb/mvj210
Suetake I, Mishima Y, Kimura H, Lee YH, Goto Y, Takeshima H et al (2011) Characterization of DNA-binding activity in the N-terminal domain of the DNA methyltransferase Dnmt3a. Biochem J 437(1):141–148. https://doi.org/10.1042/BJ20110241
Sugiyama Y, Hatano N, Sueyoshi N, Suetake I, Tajima S, Kinoshita E et al (2010) The DNA-binding activity of mouse DNA methyltransferase 1 is regulated by phosphorylation with casein kinase 1delta/epsilon. Biochem J 427(3):489–497. https://doi.org/10.1042/BJ20091856
Suva ML, Riggi N, Bernstein BE (2013) Epigenetic reprogramming in cancer. Science 339(6127):1567–1570. https://doi.org/10.1126/science.1230184
Suzuki M, Yamada T, Kihara-Negishi F, Sakurai T, Hara E, Tenen DG et al (2006) Site-specific DNA methylation by a complex of PU.1 and Dnmt3a/b. Oncogene 25(17):2477–2488. https://doi.org/10.1038/sj.onc.1209272
Svedruzic ZM, Reich NO (2005) DNA cytosine C5 methyltransferase Dnmt1: catalysis-dependent release of allosteric inhibition. Biochemistry 44(27):9472–9485. https://doi.org/10.1021/bi050295z
Syeda F, Fagan RL, Wean M, Avvakumov GV, Walker JR, Xue S et al (2011) The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J Biol Chem 286(17):15344–15351. https://doi.org/10.1074/jbc.M110.209882
Takeshima H, Suetake I, Tajima S (2008) Mouse Dnmt3a preferentially methylates linker DNA and is inhibited by histone H1. J Mol Biol 383(4):810–821. https://doi.org/10.1016/j.jmb.2008.03.001
Takeshita K, Suetake I, Yamashita E, Suga M, Narita H, Nakagawa A et al (2011) Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc Natl Acad Sci U S A 108(22):9055–9059. https://doi.org/10.1073/pnas.1019629108
Vakoc CR, Sachdeva MM, Wang H, Blobel GA (2006) Profile of histone lysine methylation across transcribed mammalian chromatin. Mol Cell Biol 26(24):9185–9195. https://doi.org/10.1128/MCB.01529-06
Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F et al (2013) Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 23(3):555–567. https://doi.org/10.1101/gr.147942.112
Vilkaitis G, Suetake I, Klimasauskas S, Tajima S (2005) Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J Biol Chem 280(1):64–72. https://doi.org/10.1074/jbc.M411126200
Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C et al (2006) The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439(7078):871–874. https://doi.org/10.1038/nature04431
Wang YA, Kamarova Y, Shen KC, Jiang Z, Hahn MJ, Wang Y et al (2005) DNA methyltransferase-3a interacts with p53 and represses p53-mediated gene expression. Cancer Biol Ther 4(10):1138–1143. https://doi.org/10.4161/cbt.4.10.2073
Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J et al (2009) The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 41(1):125–129. https://doi.org/10.1038/ng.268
Wang C, Shen J, Yang Z, Chen P, Zhao B, Hu W et al (2011) Structural basis for site-specific reading of unmodified R2 of histone H3 tail by UHRF1 PHD finger. Cell Res 21(9):1379–1382. https://doi.org/10.1038/cr.2011.123
Wang H, Farnung L, Dienemann C, Cramer P (2020a) Structure of H3K36-methylated nucleosome-PWWP complex reveals multivalent cross-gyre binding. Nat Struct Mol Biol 27(1):8–13. https://doi.org/10.1038/s41594-019-0345-4
Wang Q, Yu G, Ming X, Xia W, Xu X, Zhang Y et al (2020b) Imprecise DNMT1 activity coupled with neighbor-guided correction enables robust yet flexible epigenetic inheritance. Nat Genet 52(8):828–839. https://doi.org/10.1038/s41588-020-0661-y
Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M et al (2007) Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39(4):457–466. https://doi.org/10.1038/ng1990
Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN et al (2019) The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573(7773):281–286. https://doi.org/10.1038/s41586-019-1534-3
Weinberg DN, Rosenbaum P, Chen X, Barrows D, Horth C, Marunde MR et al (2021) Two competing mechanisms of DNMT3A recruitment regulate the dynamics of de novo DNA methylation at PRC1-targeted CpG islands. Nat Genet 53(6):794–800. https://doi.org/10.1038/s41588-021-00856-5
Weisenberger DJ, Velicescu M, Cheng JC, Gonzales FA, Liang G, Jones PA (2004) Role of the DNA methyltransferase variant DNMT3b3 in DNA methylation. Mol Cancer Res 2(1):62–72
Wienholz BL, Kareta MS, Moarefi AH, Gordon CA, Ginno PA, Chedin F (2010) DNMT3L modulates significant and distinct flanking sequence preference for DNA methylation by DNMT3A and DNMT3B in vivo. PLoS Genet 6(9):e1001106. https://doi.org/10.1371/journal.pgen.1001106
Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J et al (2012) Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148(4):816–831. https://doi.org/10.1016/j.cell.2011.12.035
Xu GL, Bestor TH, Bourc'his D, Hsieh CL, Tommerup N, Bugge M et al (1999) Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402(6758):187–191. https://doi.org/10.1038/46052
Xu TH, Liu M, Zhou XE, Liang G, Zhao G, Xu HE et al (2020) Structure of nucleosome-bound DNA methyltransferases DNMT3A and DNMT3B. Nature 586(7827):151–155. https://doi.org/10.1038/s41586-020-2747-1
Ye F, Kong X, Zhang H, Liu Y, Shao Z, Jin J et al (2018) Biochemical studies and molecular dynamic simulations reveal the molecular basis of conformational changes in DNA Methyltransferase-1. ACS Chem Biol 13(3):772–781. https://doi.org/10.1021/acschembio.7b00890
Zeng Y, Ren R, Kaur G, Hardikar S, Ying Z, Babcock L et al (2020) The inactive Dnmt3b3 isoform preferentially enhances Dnmt3b-mediated DNA methylation. Genes Dev 34:1546. https://doi.org/10.1101/gad.341925.120
Zhang Y, Rohde C, Tierling S, Jurkowski TP, Bock C, Santacruz D et al (2009) DNA methylation analysis of chromosome 21 gene promoters at single base pair and single allele resolution. PLoS Genet 5(3):e1000438. https://doi.org/10.1371/journal.pgen.1000438
Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I et al (2010) Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res 38(13):4246–4253. https://doi.org/10.1093/nar/gkq147
Zhang G, Esteve PO, Chin HG, Terragni J, Dai N, Correa IR Jr et al (2015a) Small RNA-mediated DNA (cytosine-5) methyltransferase 1 inhibition leads to aberrant DNA methylation. Nucleic Acids Res 43(12):6112–6124. https://doi.org/10.1093/nar/gkv518
Zhang ZM, Liu S, Lin K, Luo Y, Perry JJ, Wang Y et al (2015b) Crystal structure of human DNA methyltransferase 1. J Mol Biol 427(15):2520–2531. https://doi.org/10.1016/j.jmb.2015.06.001
Zhang ZM, Lu R, Wang P, Yu Y, Chen D, Gao L et al (2018) Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 554(7692):387–391. https://doi.org/10.1038/nature25477
Zhao S, Allis CD, Wang GG (2021) The language of chromatin modification in human cancers. Nat Rev Cancer 21(7):413–430. https://doi.org/10.1038/s41568-021-00357-x
Zhou Q, Agoston AT, Atadja P, Nelson WG, Davidson NE (2008) Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res 6(5):873–883. https://doi.org/10.1158/1541-7786.MCR-07-0330
Zhu H, Geiman TM, Xi S, Jiang Q, Schmidtmann A, Chen T et al (2006) Lsh is involved in de novo methylation of DNA. EMBO J 25(2):335–345. https://doi.org/10.1038/sj.emboj.7600925
Ziller MJ, Muller F, Liao J, Zhang Y, Gu H, Bock C et al (2011) Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet 7(12):e1002389. https://doi.org/10.1371/journal.pgen.1002389
Zoch A, Auchynnikava T, Berrens RV, Kabayama Y, Schopp T, Heep M et al (2020) SPOCD1 is an essential executor of piRNA-directed de novo DNA methylation. Nature 584(7822):635–639. https://doi.org/10.1038/s41586-020-2557-5
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Jurkowska, R.Z., Jeltsch, A. (2022). Enzymology of Mammalian DNA Methyltransferases. In: Jeltsch, A., Jurkowska, R.Z. (eds) DNA Methyltransferases - Role and Function. Advances in Experimental Medicine and Biology, vol 1389. Springer, Cham. https://doi.org/10.1007/978-3-031-11454-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-031-11454-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-11453-3
Online ISBN: 978-3-031-11454-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)