
Abstract
The discovery of antibiotics in the early nineteenth century and their introduction in the 1940s into clinical practice has revolutionised the global healthcare system. Infectious diseases were controlled, various medical procedures became attainable and the pharmaceutical industry greatly benefited. However, the indiscriminate overuse of these antibiotics has contributed to an emerging bacterial resistance. Hence, it encouraged the search for new antimicrobial peptides (AMPs) and nano-drug delivery systems, which can leverage these bioactive therapeutics with a tuneable controlled delivery system for clinical use against microbial infections. This review presents a brief history on the treatment of infectious diseases during the pre-antibiotic era, the golden era of conventional antibiotics, the emergence of the superbugs, and the surge in the AMP research and pharmaceutical exploration to offer a new class of pharmacotherapy through a wide range of different nano-vehicles as alternative options for antibiotics to treat various infectious diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Antibiotics
- Antibiotic resistance
- Infectious diseases
- Antimicrobial peptides
- Nanobiotechnology
- Nano-drug delivery systems
1.1 Introduction
Humans, alongside animal species, have shared the planet since the very beginning. Thus, it is fair to assume that human ancestors have adopted similar survival and reproducibility strategies that animal species successfully employed in their behavioural defence mechanisms and self-medicating habits against pathogens and parasites (Hart 2011, Shurkin 2014). For thousands of years, flora and herbal remedies played a crucial role in traditional medicine in eradicating a long list of historical pandemics, which includes the Plague of Athens in 430 BC, the Plague of Cyprian between 250 AD and 270 AD, the Plague of Justinian in 540 AD, the Black Death of China and then Russia in the fourteenth century, the Great Plague of London in 1665, the Cholera in Paris in 1832 and the Malaria in 1897, amongst many others (Swenson 1988; Hassel et al. 2002; Aminov 2010; Holmes 2011; Wray 2012; Spinney 2019). Nevertheless, inquisitive scientists’ and medical historians’ revelations often argue the distinctive abilities and the active adaptation of human beings for survival throughout history. Case in point, numerous research projects have shown how moulds were commonly used by different civilisations, including the Greek (during the sixteenth-century BC) and the Chinese (3000 years ago) to treat wounded soldiers with infected skin wounds (Wainwright 1989a; Bentley 2005). Moreover, several archaeological studies showed traces of tetracycline in human bones that stretch back to the Nubia and Egyptian civilisations that inhabited Southeast Africa more than 1400 years ago (Bassett et al. 1980; Cook et al. 1989; Nelson et al. 2010). These findings argue that different civilisations had different views and experiences in survival and fighting of the unknown microbial diseases long before biomedical sciences took control of disease diagnoses and treatments during the early decades of the twentieth century.
In 1928, Professor Alexander Fleming, a physician-scientist, experimented with the mutual biological interaction between Staphylococcus aureus (an aerobic bacterium) and airborne mould spores. His attempt was based on previous observations of Staphylococcus aureus colony lysis in a contaminated petri dish with fungal infection (Fleming 1929). Subsequently, Professor Fleming has stated that “It was found that broth in which the mould had been grown at room temperature for 1 or 2 weeks had acquired marked inhibitory, bactericidal and bacteriolytic properties to many of the more common pathogenic bacteria” which, thereupon, have changed the modern medical practice beyond recognition.
Thus, the antibiotic era started almost 90 years ago based on the “magic bullet” concept that Paul Ehrlich introduced in 1910 and the discovery of penicillin by Alexander Fleming in 1928 (Strebhardt and Ullrich 2008; Aminov 2010). Paul Ehrlich has connected chemistry to biology by introducing the “magic bullet” concept, which became the cornerstone of the pharmaceutical industry, and many medicines have been produced, including antimicrobials based on this concept (Strebhardt and Ullrich 2008). The “magic bullet” concept refers to the specificity of drug activity through its affinity to a specifically targeted receptor exerting its effect on the target cell or pathogen without affecting the rest of the biological system (Elliott 2010).
The golden age of antimicrobial pharmacotherapy, which was initiated by the discovery of penicillin in 1928, started gaining momentum in the early 1930s of the twentieth century when the German scientist Gerhard Domagk discovered the sulpha drugs in 1932 (Oesper 1954). Soon after, this group of drugs was developed to sulphonamides, one of the most effective antibiotics against a wide range of infections, including pneumonia and gonorrhoea (Stanwell-Smith 2007). However, penicillin stayed in the lead, and during the Second World War, it continued to do wonders in treating various infections between skin injuries and systemic infections. Bacteriologists continued their research and have listed different penicillins, streptomycin, chloramphenicol and tetracyclines in their list of discoveries during the 1940s and the 1950s (Rebstock et al. 1949; Daniel 2005; Stanwell-Smith 2007; Liu and Myers 2016). Benjamin Duggar discovered aureomycin (chlortetracycline) in 1948, which was approved in 1950 for human use, followed with natural tetracycline in the early 1950s, such as terramycin, which Pfizer produced in 1950 (Finlay et al. 1950; Liu and Myers 2016). In 1952, Selman Abraham Waksman was awarded the Nobel Prize in medicine for his discovery of streptomycin (Daniel 2005). Streptomycin was tested in vivo against tuberculosis and proved successful in January 1945 at the Mayo Clinic after it was administered to Patricia T, who suffered from that pulmonary infectious disease. Moreover, in 1948, the Italian pharmacologist Giuseppe Brotzu efficaciously isolated Cephalosporium acremonium from sewage water in South Italy which resulted in the development of different classes of broad-spectrum cephalosporins (Bo 2000; Chizhik 2014).
However, the confidence in antibiotics and their effectiveness swiftly became sceptical. Since the mid-1970s and early 1980s, bacterial resistance to different antibiotics started breaking the surface leading to the regression of the nineteenth century in terms of infectious disease infestation, which became untrammelled (Stanwell-Smith 2007; Gould 2016). Over the second half of the previous millennium, a random, inconsiderable, unquestionable and unnecessary prescribing and administering of antibiotics to humans, animals, animal food and plants were practised for treatment and prophylactic purposes (Stanwell-Smith 2007; Landers et al. 2012; Stockwell and Duffy 2012). This irrational behaviour of healthcare specialists, pharmaceutical industry workforce and within agriculture has led to profound genetic mutations resulting in virulent strains to survive and thrive. Such risks require immediate and fast interventions to prevent imminent global health crisis.
Hence, in addition to the strict policies concerning antibiotic prescription and use that have been adopted by the World Health Organisation (WHO) and other health authorities around the world, such as controlled supply chains and drug cycling (Stallmann et al. 2006), scientists have started looking for an alternative to the conventional antibiotics. One way to do so is to go back to the early days of humanity and ask the question: what was the critical factor that helped humans and other prokaryotic life forms in their battle against nasty pathogens during their early primitive days? No doubt, it is our innate defence system. So AMPs are inherited natural antibiotics born with us and are available as part of our immune system to fight microbial infections with minimal chance of bacterial resistance (Boman 2003; Stallmann et al. 2006). Such atypical therapeutic molecules endure unique pharmacodynamic behaviours accompanied with fewer side effects (Leader et al. 2008). They are one of the leading categories of nonconventional molecules that exert a remarkable remedial potential against infectious diseases. They demonstrate their pharmacological effects via specific cellular pathways resembling natural molecular signalling.
Nevertheless, these peptides are immensely susceptible to physicochemical and biological degradations preventing them from reaching the microbial pathogen at the vital bioavailability. Poor luminal permeability, high cytosolic metabolism, gastric degradation and the first hepatic clearance of these molecules have executed most of therapeutic peptides including antimicrobial from being translated into medicines (Gupta et al. 2013; Muheem et al. 2016). Moreover, all of the currently used antimicrobial peptides and proteins are formulated in a parenteral form that requires high-cost cold chains for storage and transport.
This chapter aims to shed some light on the history and the process of developing antibiotics and the emerging bacterial resistance. However, our focus will be on antimicrobial peptides as the inherent and intrinsic self-defence mechanism behind our powerful immune system and the latest developments in nanobiotechnology as an adopted strategy to develop systemic and topical targeted delivery vehicles.
1.2 Antibiotics: Key Events from Prehistory to the Golden Age
The awareness of the invisible pathogens, their effect on the human body and how to eradicate them was evident since the early days of our ancient history. Substantial historical evidence has shown that ancient civilisations, including Ancient Egypt and Southeast Africa, Middle East, China, Greece and the Romans, used different antimicrobial remedies including honey, herbs, clay and mouldy bread to treat systemic and skin infections (Wainwright 1989a; Martin and Ernst 2003; Newman and Cragg 2007; Falkinham III et al. 2009; Eteraf-Oskouei and Najafi 2013; Gould 2016; Kirchhelle 2018; Qiao et al. 2018). Surprisingly, even tetracycline was detected in human bones exhumed from Sudanese and Egyptian cemeteries going back to the Nubia and Roman occupation era (Nelson et al. 2010; Gould 2016).
Archaeologists, anthropologists and biologists frequently and silently travel for their adventures and share their knowledge to uncover the discreet. In the early 1980s of the last century, different archaeological scientific projects, carried out in Southeast Africa, uncovered human bone remains that exhibit traces of a 1400-year-old tetracycline (Bassett et al. 1980; Cook et al. 1989; Nelson et al. 2010). Fluorescence microscopy tests showed an identical fluorescence imaging between the antibacterial agent of tetracycline and the contamination from the bone recovered from the X-group cemetery (Bassett et al. 1980). Historically, the X-group cemetery belongs to one of the Sudanese Nubia tribes who lived on the west bank of the Nile River across from Wadi Halfa in Sudan (Bassett et al. 1980; Nelson et al. 2010). The counteracting claim argued that the fluorescence microscopy results from the bone discoloration are merely due to fossilisation or the so-called taphonomic process and the decomposition of the dead bone tissue performed by fungi (e.g. Stachybotrys and Cladosporium). This fossilisation mechanism produces an intermediate substrate with a leaching effect (Piepenbrink 1986; Schaller et al. 2015) rather than calcium complex formation between the tetracycline, as a chelating agent, and the calcium that exists in the bone tissue (Menon 2014; Shasmitha 2014). However, Bassett et al. have dismissed this possibility since the fluorescence was patterned in the same manner as the modern tetracycline-labelled and not diffused as it appears in the post-mortem mould infestation.
Moreover, the bones were found undamaged, reducing the chances of mould infestation (Bassett et al. 1980). Furthermore, a histological ageing study performed by Cook et al. on femoral mid-shafts goes back to the Roman period in Dakhleh Oasis, Egypt, has shown a distinct fluorochrome labelling (Cook et al. 1989). These fluorochromes were also traced within the enamel matrix of some individuals of that population. Moreover, the fluorescent patterns obtained from these fluorochromes correspond to those obtained from patients treated regularly with tetracycline. These findings argue that the fluorochrome is most likely tetracycline that was administered in vivo over a prolonged time. In addition, tracing the fluorochrome in the enamel supports Bassett’s theory, which argues that the fluorochrome found in the remains of the Nubia tribes was an ingested tetracycline rather than a fungi contamination based on the fact that enamel is an avascular body compartment and, unlike the bone, is more resistant to contaminations introduced by taphonomic processes (Cook et al. 1989). Finally, according to Cook’s observations, including periosteal examinations, the effect of the tetracycline on these populations was for antibacterial and prophylactic treatment.
Based on the above, it is easy to agree that the usage of some antibiotics was available a long time before the discovery of penicillin. However, the argument is that the revolutionised treatment of infectious diseases, which started less than 100 years ago, was validated after introducing novel antibiotics into a scientifically regulated medical field (owing to Paul Ehrlich and Alexander Fleming). Statistical data from the USA has shown a significant annual decline of 8.2% in the infectious disease mortality rate between 1938 and 1952 (Armstrong et al. 1999). The early bright of this era started when Rudolf Emmerich has revealed the first antibacterial substance in modern history (Gould 2016). The pyocyanase, a proteolytic substance extracted from Bacillus pyocyaneus, exerts autolytic activity, against its source, and the lytic activity against Staphylococcus and Streptococcus species (Waksman 1948; Gould 2016).
Further advancements in the golden era of antibiotics are owed to the German chemist, Paul Ehrlich, and his team in 1909, after their famous discovery of the arsenic derivative, Salvarsan or dioxydiamidoarsenobenzol, for the treatment of syphilis (Scovil 1912). The yellowish powder produced in an injectable form has been selected from 606 other compounds despite its high level of toxicity, which pushed towards the new formulation (neosalvarsan) in 1912. The neosalvarsan was an optimised derivative of the original Salvarsan after improving its solubility. Also, the acidity of the new product was reduced, which eliminated the need to use soda as an alkaline base additive before the injection (McIntosh et al. 1912).
During the 1920s, Joseph Klarer, Fritz Mietzsch and Gerhard Domagk discovered the antibacterial sulpha agents or sulphonamides. This group consisted of three main categories of sulphonamide series of antibacterial drugs that have been given the generic name of Prontosil by the Bayer Products Ltd (Nature 1938). At that time, Dr. Doris Brown from Royal Maternity Hospital, Belfast, reported that Prontosil drugs were significantly effective against septicaemia caused by haemolytic Streptococci. Three years of comparison trials have found that Prontosil has reduced the death rate caused by general septicaemia from 87.5% to 28.57% and the mortality rate caused by Streptococcus spp. from 23.5% to 6.6%. There were further encouraging results reported on the remarkable effectiveness of Prontosil usage against urinary tract infections caused by E. coli (Nature 1937).
During the same period and precisely on the third day of September 1928, Alexander Fleming witnessed the birth of penicillin. He observed accidental mould contamination in a petri dish-contained bacteria for influenza research purposes, inhibiting the growth of the nearby Staphylococcus colonies. This mould was Penicillium notatum (initially identified as Penicillium rubrum) which produces a substance against bacterial growth, which was named penicillin (Marshall 1946; Wainwright 1989a; Wainwright 2002). Indeed, the discovery of penicillin by Alexander Fleming was one of the most remarkable scientific achievements in the modern history of the medical field. However, Professor Fleming himself underestimated this attainment at the start. His evaluation of the initial product was described as good as a topical antiseptic substance, and after few years from his initial observations, he started consulting chemists for purification techniques and chemical stability studies of the product (Aminov 2010; Lobanovska and Pilla 2017).
Nevertheless, Fleming’s publication in 1929 about the penicillin discovery has reached the hands of two gurus, Howard Florey and Ernst Chain, from Oxford’s Sir William Dunn School of pathology. They followed Fleming’s discoveries, including his 1922 work on antibacterial lysosome, trying to look for a product invention (Swann 1983). The Oxford team started the purification process of penicillin, and in 1940, they conducted their first trial on infected animals with haemolytic Streptococci where they found that the treated group survived as much as threefold of the untreated control (Swann 1983). Soon after, Florey and Chain discovered that penicillin is effective against different Staphylococcus spp., Streptococcus spp. and Gonococcus strains. These observations have advantaged penicillin over sulphonamides, which was limited to Staphylococci strains (Swann 1983). Consequently, in 1941, penicillin was commercialised and started saving the lives of millions of civilians and soldiers on the battlefields around the world during the Second World War (Aminov 2017).
Further advancement in the penicillin world occurred in 1959 when Batchelor, Doyle, Nayler and Rolinson from Beecham Research Laboratories, Ltd, reported the amine 6-amino-penicillanic acid (6-APA) in penicillin fermentation (Batchelor et al. 1959; Batchelor et al. 1961). Acetylation of 6-APA results in synthesising various semisynthetic penicillins, including penicillins, against Gram-negative bacteria such as Escherichia coli, Haemophilus, Listeria, Neisseria, Proteus mirabilis, Shigella and Salmonella. Other examples include ampicillin, amoxicillin, bacampicillin and penicillins against Enterobacteriaceae and Pseudomonas aeruginosa such as carbenicillin and ticarcillin penicillinase-resistant penicillins including methicillin, oxacillin and nafcillin (Wright 1999; Aminov 2017).
Moreover, in 1947, John Ehrlich and his team discovered chloramphenicol, the first member of the amphenicol group to be discovered (Ehrlich et al. 1948). They found that Streptomyces venezuelae, which was extracted from a soil sample near Caracas in Venezuela, is the source of chloramphenicol (Ehrlich et al. 1948). In 1954, Gottlieb et al. have described the natural production process of the chloramphenicol by S. venezuelae, which is encouraged by adding P-nitrophenylserinol to a synthetic glycerol lactate medium, and soon after, it became the first antibiotic to be chemically synthesised (Gottlieb et al. 1954; Gottlieb et al. 1956; Dinos et al. 2016). The physiochemical characteristics of chloramphenicol support its permeability through the blood-brain barrier, and it was found to have a bactericidal effect against meningitis-related bacterial species including Haemophilus influenzae, Streptococcus pneumoniae and Neisseria meningitidis (Dinos et al. 2016). However, neurotoxicity and haematological disorders such as bone marrow depression and aplastic anaemia, amongst others, limited its usage (Aminov 2017).
This notable scientific success in antibiotic discovery in the golden era and the tremendous impact of the clinical translation on the medical field has inspired entrepreneurial scientists and commercial pharmaceutical companies worldwide for more innovations and blooming in the antimicrobial arena. Waksman’s argument in the early 1940s that “a considerable proportion of all actinomycetes that can be isolated from soils or other natural substrates have the capacity of inhibiting the growth of, and even of destroying, bacteria and other microorganisms” has captivated the minds of many scientists as well as the attention of the pharmaceutical industry with the antibiotic rush (Waksman et al. 1946; Nelson and Levy 2011). In the early 1940s, a retired professor and a world specialist named Benjamin Minge Duggar has joint the Cyanamid as the head of the soil department looking for actinomycetes in a wide range of soil samples sent to him from all around the world with a vision to discover a novel antibiotic (Nelson and Levy 2011).
In 1945, Duggar and his team had witnessed an unusual antimicrobial activity of a yellow coloured sample that wiped off all the Gram-positive and Gram-negative bacteria in the tested group. Duggar named this broad-spectrum substance aureo, meaning gold (yellow) in Italian (Duggar 1948; Yan and Song 2014). Aureomycin, or chlortetracycline, was the first of the tetracycline class of antibiotics to be discovered and 3 years later was experimented successfully on a five-year-old Tobey Hockett who had a life-threatening post-operative wound infection at the John Hopkins Children Hospital in Washington, DC (Nelson and Levy 2011). Nowadays, tetracyclines belong to a large family of antimicrobials and are the second widely used antibiotic in humans, animals and agriculture (Zhang et al. 2011; Yan and Song 2014).
After discovering streptomycin, scientists have intensified the screening of actinomycetes resulting in further discoveries (Benedict 1953). In 1949, a new antibacterial agent was exposed in Iloilo City on the Philippine Island. Dr. Abelardo Aguilar collected a soil sample analysed at the Eli Lilly Research Laboratories, where they isolated the antibiotic erythromycin-A (Robertsen and Musiol-Kroll 2019). Erythromycin-A is a polyketide antibacterial substance produced by Saccharopolyspora erythraea, a Gram-positive soil bacterium (Jiang et al. 2013). Erythromycin-A has shown a great deal of similarity with penicillin. It is effective against a wide range of bacteria including Gram-positives and acid-fast rods, in addition to some of the following Gram-negative genera, Brucella, Haemophilus and Neisseria, thereby providing an alternative treatment for patients with penicillin allergy (Benedict 1953).
Furthermore, in 1947, cyclic peptide with a hydrophobic backbone chain was named colistin, or polymyxin-E, after its discovery in Japan (Storm et al. 1977; Aminov 2017). Colistin is a member of the polymyxins family of antibacterial peptides produced by Paenibacillus polymyxa, a Gram-positive bacterium found in soil, plants and marine precipitants (Ainsworth et al. 1947; Stansly and Schlosser 1947). Colistin was found to be a remarkable substance as an active antibacterial agent against Gram-negative bacteria (Stansly and Schlosser 1947). Colistin’s cationic cyclohepta peptide ring targets the anionic lipopolysaccharide (LPS) molecules in the outer cell membrane of the Gram-negative bacteria. Attacking the LPS by the cationic moiety of the colistin leading to a non-enzymatic disturbance of the magnesium and calcium ions’ positioning (two metals that stabilise the membrane phospholipids by bridging the negatively charged phosphor-sugar molecule) destabilises the negatively charged LPS molecules, leading to cell membrane damage. Moreover, destabilisation of the cytoplasmic membrane allows the antimicrobial peptides to interact with the cytoplasmic organelles (Zasloff 2002; Falagas and Kasiakou 2005; Zhang and Gallo 2016). This cationic intervention results in local destruction of the outer membrane integrity, promoting membrane permeability and leakage of the cell content and cell lysis of the targeted bacteria (Newton 1956; Laporte et al. 1977). Based on the above results, it was approved in 1949 for clinical use, and it was utilised against pathogens resistant to standard treatments such as ꞵ-lactams, aminoglycosides and fluoroquinolones (Evans et al. 1999; Falagas et al. 2008). Moreover, colistin was the last resort antibiotic against life-threatening infections caused by multidrug-resistant bacterial pathogens including Acinetobacter baumannii, Klebsiella pneumoniae and Pseudomonas aeruginosa (Endo et al. 1987; Falagas et al. 2008). However, due to systemic severe side effects such as nephrotoxicity, neuromuscular blockade and acute airway obstruction, colistin’s usage was banned except for patients with cystic fibrosis (Evans et al. 1999; Falagas and Kasiakou 2006).
One of the significant breakthroughs during that golden era was the discovery of vancomycin in 1953. Vancomycin was purified from a soil sample sent from Borneo, in Southeast Asia, to Dr. Edmund C. Kornfeld, a chemist at Eli Lilly (Levine 2006), which represented, at that point, a new class of antibiotics produced by Streptomyces orientalism. Vancomycin is constructed from peptides with a carbohydrate moiety attached to the amino acid residues forming glycopeptides. These peptides possess high selective toxicity against different bacterial pathogens including the Gram-positive penicillin-resistant Staphylococcus spp., and the anaerobic Clostridium spp., in addition to the Gram-negative bacterium Neisseria gonorrhoeae (Reynolds 1989; Levine 2006; Chen et al. 2011; Aminov 2017). Vancomycin, like other glycopeptides, is a large and rigid molecule that configurationally fits into the particular structure of the bacterial cell wall precursor, sequenced as L-Ala-D-Ala-D-Ala, forming a stable hydrogen bonding which results in blocking of the trans-glycosylation process of bacterial cell wall peptidoglycan biosynthesis (Allen and Nicas 2003). Therefore, in 1956, the FDA has approved vancomycin as an antibacterial drug; however, its usage was limited until the emergence of the methicillin-resistant Staphylococci aureus (MRSA) in the mid-1980s (Chen et al. 2011).
In 1959, a new class of antibiotics against one of the most indelible and debilitating pulmonary infectious diseases had been discovered. Sensi et al. from Lepetit laboratories in Milan, Italy, have managed to isolate an antibacterial substance which they named rifamycin based on its initial nickname (Rififi), via a process of broth fermentation performed by Amycolatopsis mediterranei – a member of Actinomycetaceae group (Lester 1972; Sensi 1983; Murray et al. 2015). Further studies have identified rifamycin as a constituent of five different substances, including rifampicins A, B, C, D and E (Lester 1972). Rifamycin-B portion composed less than 10% of the whole mixture, and it was the only substance that had been purified in a crystalline form where the rest were unstable to be isolated or characterised (Sensi 1975; Sensi 1983). Moreover, the purified rifampicin-B showed a low level of activity in its initial chemical form; nevertheless, it demonstrated an insignificant degree of toxicity and a notable therapeutic effect against pathogenic infections in animals which, in turn, inspired further pharmacotherapeutic-related chemical modifications of the molecule (Lester 1972; Sensi 1983). Hence, 1963 rifamycin SV was developed based on rifampicin-B’s spontaneous activation chemical process (Sensi 1975, Sensi 1983). Rifamycin-B has demonstrated a unique ability to undergo a spontaneous and reversible transformation into a rifamycin-O in an oxygenated aqueous medium. In turn, rifamycin-O goes through a hydrolysis process which forms the so-called rifamycin-S by losing a glycolic acid, thereafter a mild reduction of the S form results in rifamycin SV. Rifamycin SV exerts a significant bactericidal effect against Gram-positive pathogens, including the acid-fast Mycobacterium tuberculosis, in addition to moderate antibacterial effect against Gram-negative bacteria (Sensi 1983).
The great activity of rifamycin SV against M. tuberculosis has inspired scientists to develop an oral form of the medicine which has seen the light of day in 1966. Maggi et al. have innovated a modifiable, semisynthetic and orally active substance (N-methyl-piperazine derivative of rifamycin SV) at the Lepetit Research Laboratories, which was named rifampin or rifampicin. Rifampicin has demonstrated a defined pharmacokinetic profile, including significant levels of solubility and stability throughout the gastrointestinal tract system, as well as an elevated transepithelial absorption. Such acceptable stability and solubility are attributed to unique physicochemical properties such as high lipophilicity of the molecule (Maggi et al. 1968; Lester 1972; Di Stefano et al. 2011; Grobbelaar et al. 2019).
The mechanism of action of rifampicin culminates in inhibiting the bacterial RNA polymerase synthesis, which is a crucial enzyme in bacterial RNA transcription. The bacterial RNA polymerase catalytic core consists of five different subunits including 2α, ꞵ, ꞵ’, β and σ as an initiation unit. Rifampicin binds to the ꞵ-subunit (adjacent to DNA/RNA channel), leading to a physical blockade and, consequently, inhibition of the DNA-dependent RNA elongation process (Sensi 1983; Pang et al. 2013; Nusrath Unissa and Hanna 2017).
Nevertheless, another pivotal event occurred during the late stages of the golden era of antibiotics: the accidental discovery of metronidazole as a treatment for gingivitis. In 1962, the D. L. S. Shinn from King’s College Hospital Dental School, Denmark Hill, London, reported that a treatment with metronidazole 200 mg, three times daily, for trichomonal vaginitis cured, also, acute marginal gingivitis, which was diagnosed at the same time (Shinn 1962). Before that, in 1959, the Rhone-Poulenc Labs in France produced a substance called nitroimidazole as a secondary agent of the extracted antibiotic azomycin from Streptomyces species. Then, the nitroimidazole was synthetically derived from metronidazole (a b-hydroxyethyl-2-methyl-5-nitroimidazole) for chronic trichomonas infections (Petrin et al. 1998; Samuelson 1999).
In addition to the activity of metronidazole against anaerobic Gram-positive bacteria Clostridium spp. and anaerobic Gram-negative bacteria like Bacteroides spp. by disrupting the bacterial DNA synthesis, it is effective against parasitic infections (Samuelson 1999). Metronidazole has demonstrated significant efficacy against the anaerobic protozoan parasite Entamoeba histolytica, which causes dysentery and liver abscess (Ravdin 1995). Moreover, it is highly efficacious against the protozoan luminal parasite Giardia lamblia, which stands behind severe malabsorption and epigastric pain conditions in developing countries (Zaat et al. 1997) (Fig. 1.1).

Timeline of the key events in the history of the discovery of the natural antimicrobials and the introduction of the semi-synthetic and synthetic conventional antibiotics
1.3 Antibiotic Resistance: Health Crisis and Solutions
The habitation of Earth for more than 200,000 years by its inhabitants utilising its resources, coexisting amongst a remarkable diversity of a wide range of different species includes prokaryotes and eukaryotes and, concurrently, preserving the homoeostatic cycle of the life forms is owing to the symbiotic fashion of interaction between the various species. Nevertheless, predatism is the alternative way of practice when competing over limited essential resources for survival (Gauze 1934). Along with that, the victorious species are the elite which will continue to prevail. This instinctive principle, the essence of Darwin’s theory of evolution, has been empirically applied to quantify the interaction between pathogens and their hosts, including the epidemiological outcomes of the struggling course for existence (Gauze 1934).
Gauze et al. argued that the relationship between the prey and the predator (i.e. the host and the pathogen) results in a cyclical oscillation of their numbers. Moreover, the species will never be able to eliminate each other (Gauze 1934). Hence, the declaration that was made by the clinician William H. Stewart in the early 1960s, “it is time to close the book on infectious diseases and declare the war against pestilence won” (Spellberg et al. 2008), was an invalid scientific statement. Furthermore, needless to say that the unsupported overconfidence of the medical practitioners in prescribing and the uncontrollable usage of antibiotics in livestock as well as in the agricultural industry has contributed, overwhelmingly, to the emergence of mutated and stubborn-resistant predators (Stockwell and Duffy 2012; Venter et al. 2017). Such microbial resistance counteracts the so-called magic bullet impact, or the therapeutic effect of the antibiotic, and aborts the host’s combat during the downfalls of these oscillatory cycles (Sengupta et al. 2013).
Admittedly, acquiring resistance genes to antibiotics is a defensive biological mechanism adopted by single-cell microorganisms more than 30,000 years ago (Vanessa et al. 2011). Bacteria develop antibiotic substances to compete against other bacterial species over common living resources (Venter et al. 2017). Nevertheless, our argument concerns the augmented prevalence of bacterial resistance amongst communities, which correlates with the highest records of antibiotic consumption (Sun et al. 2012). Goossens et al. have found that antibiotic resistance incidences are significantly higher in southern and eastern European countries versus the northern ones where the prescribing rate in primary healthcare was 32.2 per 1000 residents compared to 10 per 1000 residents, respectively (Goossens et al. 2005).
Antibiotic-resistant infections have been declared by WHO and other global health organisations as a health “crisis” which demonstrates an imminent potential of a “catastrophic consequence” on the human race (WHO 2014; Sivalanka et al. 2016; Alam et al. 2019; Sprigg and Pietrangeli 2019). Hospital-acquired infections are one the most prominent infections that attack immunocompromised patients and are caused by different pathogens, including antibiotic-resistant bacteria such as the methicillin-resistant Staphylococcus aureus (MRSA), which is responsible for more than 11,000 deaths annually in the USA alone (Gross 2013; Venter et al. 2017; Podolsky 2018). MRSA was initially encountered in 1962 (Sprigg and Pietrangeli 2019). Nowadays, MRSA has spread worldwide, and it is one of the most frequent and severe infections arising out of antibiotic-resistant bacteria (Ventola 2015). However, there are reports acknowledged by some European health authorities from the United Kingdom and the Netherlands stating that hospital-acquired MRSA incidences were declined by almost 31% between 2005 and 2011 due to the adoption of strict hygienic measures, while the trend in the community-acquired MRSA is still increasing (Ventola 2015).
MRSA is sensitive to glycopeptide treatment such as vancomycin (Rai et al. 2005), which prevents the biosynthesis of the bacterial cell wall by blocking the peptidoglycan production throughout a complex formation with the D-Ala-D-Ala residues of the building blocks, which, in turn, inhibits the trans-glycosylation process (Allen and Nicas 2003). Such a mechanism of action was considered highly immunised against bacterial resistance. In 1979, Sengupta et al. showed the development of resistance to vancomycin in coagulase-negative Staphylococci (Sengupta et al. 2013). Currently, vancomycin-resistant Enterococci (VER) pose a significant concern, predominantly, because of the wide range of hospital-acquired infections caused by these pathogens, such as post-operative and urinary tract infections, besides the limited options of specific treatments against them (Handwerger and Skoble 1995; Ventola 2015). Furthermore, in 1997, Hiramatsu et al. demonstrated the reduction of vancomycin susceptibility in a methicillin-resistant S. aureus (VISA) strain isolated from an infected surgical wound (Hiramatsu et al. 1997). This reduction in susceptibility to vancomycin by MRSA has been, also, extended to different advanced cephalosporins (e.g. ceftaroline) and vancomycin analogues (e.g. telavancin, oritavancin and dalbavancin) that have been used as alternative treatments (Venter et al. 2017).
Cross-transfer of antibiotic-resistant coding genes may happen between different species throughout plasmids or transposons (Handwerger and Skoble 1995; Podolsky 2018). A new vancomycin-resistant S. aureus (VRSA) strain has emerged due to conjugative transfer of a plasmid transposon from a vancomycin-resistant Enterococcus faecalis strain to the MRSA bacterium. In turn, these transconjugants depict the contributor’s role regarding vancomycin resistance after their incorporation into the recipient’s chromosome, via changing the bacterial cell wall precursor from D-Ala-D-Ala to D-Ala-D-Lac (Handwerger and Skoble 1995; Venter et al. 2017).
To sum up, during the golden era of antibiotic history between the early 1940s and the early 1960s, medical practitioners and the pharmaceutical industry took initial control over pathogenic infections but ignored critical scientific clues and the extraordinary and evolutionarily skills of the biological cell. The euphoria of victory ended prematurely and was buried under the new imminent threat of the superbugs with potentially catastrophic consequences. Between the early 1960s and the 1980s, scientists have comprehended the magnitude of the antibiotic resistance crisis; they have witnessed the transmission of resistant genes horizontally, such as the emergence of the VRSA throughout the plasmid transformation between two different species they have cried out in desperation for help. Consequently, in 1985, Sweden was the first to ban the usage of antibiotics as promoters in animal farming (Wierup 2001), followed by Denmark in 1994 in an attempt to eliminate the resistance reservoir and to reduce the chances of transforming antibiotic-resistant genes into human pathogens (Hayes and Jensen 2003). Thereafter, in the 1990s, the awareness of drug resistance became a global concern, and in 2006, the EU imposed a total ban on the usage of antibiotics in animal farming. Moreover, in 2014, WHO has recognised the antibiotic resistance condition as a global crisis (Alam et al. 2019).
For the time being, hospital-acquired infectious diseases remain a serious risk. Increased antibiotic resistance reduces the treatment options and incurs painful financial costs worth billions of dollars of expenses on management. Moreover, the excessive use of antiseptics in hospitals and communities can lead to resistance increment against these sanitizers and increase the chances for cross-resistance to antibiotics. Hence, comprehensive management and control programmes are in desperate need of a comprehensive solution in the clinical and veterinary fields and for antimicrobials used in agriculture. Finally, reducing the frequent usage of conventional antibiotics requires a new alternative of less resistible pharmacotherapeutics such as antimicrobial peptides (AMPs).
1.4 Antimicrobial Peptides (AMPs): Conception and History
Humanity has branched out into different societies experiencing distinctive plagues along the way reshaping our history. Nevertheless, our long-term coexistence with microorganisms proved to be in a continual dynamic ecological equilibrium until a retaliation had taken place, as it happened when the microbial resistance to antibiotics emerged following the excessive use of antibiotics in the 1960s and 1970s (Anderson 2004). Hence, dealing with infectious diseases from the ecological and evolutionary point of view will be more advantageous than handling them from a solo and narrow clinical angle (Burnet 1941). As Dr. Joshua Lederberg has stated: “Perhaps one of the most important changes we can make is to supersede the twentieth-century metaphor of ware for describing the relationship between people and infectious agents. A more ecological informed metaphor, which includes the germ’s-eye view of infection, might be more fruitful… yet they are equally part of the superorganism genome with which we engaged the rest of the biosphere” (Lederberg 2000).
The relationships and interactions between microorganisms and microorganisms/host involve a complex of ecological aspects, including cellular signalling amongst others, as an integral feature of an instructive and controlling co-evolutionarily system that supports a peaceful adaptation (Braga et al. 2016). This part of the present review focuses on the AMPs and their roles in bacterial control’s intercellular/intracellular signalling mechanism.
AMPs are a diverse class of natural and multifunctional small molecules ranging between 10 and 150 amino acids that exert their biological function either constitutively or through an external factor inducement (Lei et al. 2019; Lazzaro et al. 2020). AMPs are evolutionarily preserved indigenous molecules that originated and are employed by all different life forms of prokaryotes and eukaryotes of the human and animal kingdom for different cellular functions, including antimicrobial protection (Epand 2016; Zhang and Gallo 2016; Wu et al. 2018). Hence, they serve as the first line of defence against different microbes, including viruses, bacteria, fungi and protozoa. Moreover, in higher life forms, AMPs are ubiquitously dispersed in epithelial cells, and some of them can be expressed as immunomodulatory agents (Boman 2003; Gordon et al. 2005; Lai and Gallo 2009; Lee et al. 2015; Scorciapino et al. 2017; Pfalzgraff et al. 2018; Xia et al. 2018) (Fig. 1.2).

Summary illustrating the general and various functions of the AMPs
The initial detection of AMPs was in 1921 when Alexander Fleming observed a lysis process in a bacterial culture obtained from nasal mucosa of a patient with the common acute cold (Fleming 1932; Tan and Tatsumura 2015). Dr. Fleming named the responsible effector for the bacterial lysis, lysosome, which is known, nowadays, as a generic name of various proteolytic organelles that possess more than 50 different intracellular digestive enzymes (Bainton 1981). Soon after this discovery, Alexander Fleming stated that this bacteriolytic element is dispersed throughout the human body, confirming Metchnikoff’s synopsis: “Nature, to protect the skin and mucous membranes does not use antiseptics. The fluids which bathe the surface of the mouth and other mucous membranes are not bactericidal or only very imperfectly so. Nature removes from the mucous membranes and the skin quantities of microbes, eliminating them by epithelial desquamations and expelling them with the secretions and liquid excretions. Nature has chosen this mechanical procedure just as the surgeons who replace the antisepsis of the mouth, the intestine and other organs by lavage with physiological saline” (Fleming 1932).
The first defined AMP was found in 1939 in a culture of an aerobic sporulating bacilli species. Dubos and his group isolated an alcohol-soluble substance named tyrothricin, which was bactericidal against a large number of Gram-negative and Gram-positive bacteria (Dubos 1939). After that, two distinct crystalline substances have been purified from the tyrothricin, including tyrocidine, which showed bactericidal activity against both Gram-positive and Gram-negative pathogens, and gramicidin which showed apparent effectiveness against Gram-positive species when it was applied topically (Dubos and Hotchkiss 1941; Mootz and Marahiel 1997).
The euphoria of the golden era of antibiotics between the 1940s and the 1960s has overshadowed the discovery of AMPs. However, the redemption of the AMPs to be considered a novel remedy for infectious diseases started gaining momentum in the 1960s through the exploration course for a new ecological resolution to counteract the emerging bacterial resistance for conventional antibiotics (Davies and Davies 2010; Aminov 2017).
A sequence of exclusive studies conducted by Hans G. Boman and his group in the 1970s and the 1980s has led to the discovery of insect-based antimicrobial peptides with an overwhelming stimulant effect on the immunity response in Drosophila (Faye and Lindberg 2016). Boman’s great deal of interest in the functionality of the human immune system has diverted his focus towards the preliminary defensive processes in the vertebrates’ immune system that take place before the maturity of the required antibodies during the early stages of infections. Thus, he employed insect species, which lack lymphocytes and immunoglobulins, as a fundamental model for his manipulative studies of the immune system via selected stimulus (Hultmark et al. 1982). In 1981, Boman and his group identified and characterised a new group of AMPs named cecropins that deliver a significant antibacterial effect against Gram-negative bacteria (Steiner et al. 1981).
Moreover, in 1987, a new family of AMPs which consists of two similar small peptides (23 amino acids) has been discovered by Zasloff et al., for which he named magainins (Zasloff 1987). They have isolated the broad-spectrum amphiphilic peptides with antimicrobial activity against a wide range of bacteria, fungi and protozoa, from the skin of the Xenopus laevis, an African frog. Furthermore, in 1994, scientists have isolated the first mammalian skin of AMPs, which belongs to a larger group of small cationic antimicrobials named cathelicidins, which was found in humans and other animals, birds and marine species (e.g. cattle, sheep, pigs, chicken and some fish) and exert a broad-spectrum antimicrobial activity against bacteria, viruses and fungi (Kościuczuk et al. 2012).
Undoubtfully, such fascinating research, alongside other studies that identified and characterised a wide range of AMPs for the last 60 years, has instigated a widespread campaign behind the comprehensive appreciation of the role of the AMPs in our innate immune system (Hultmark et al. 1982; Tracey et al. 1995; Lung et al. 2001; Faye and Lindberg 2016; Wu et al. 2018).
1.5 Diversification, Structural Characterisation and Mechanisms of Action of AMPs
AMPs are circulated and diversified in nature across and within the different species of vertebrates, bacteria, fungi and plants (Lei et al. 2019). The AMP database is updated regularly, and it includes natural AMPs with a defined sequence and biological activity (Wang et al. 2016). The latest version of the AMP database shows the sum of 2169 antibacterial peptides in addition to 277 antivirals, including anti-HIV, 959 antifungals, 80 antiparasitics and 185 bioactive peptides listed for different anticancer activities (Wang et al. 2016). In humans, the defensin family is the more significant ubiquitous type of AMPs followed by the cathelicidins, the first family to be identified (Dhople et al. 2006; Mojsoska and Jenssen 2015). Moreover, cathelicidins were discovered ubiquitously in monkeys, rats, mice, rabbits, pigs, cattle, goats, sheep and horses (Gudmundsson et al. 1995; Zanetti 2004; Lei et al. 2019).
The characterisation of AMPs is very complex (Zasloff 2002) due to their diversified origin and the crossover of functionalities against different microorganisms and target cells (e.g. cancer cells). However, there is a simple classification approach based on the geometrical (structural) features of the AMPs upon their contact with the microbial membrane (Zasloff 2002). This approach broadly categorises the AMPs into four main types based on their secondary structure, including the alpha-helix, the beta-sheet, the loop (mixed) and the extended conformation (Steinberg et al. 1997; Mojsoska and Jenssen 2015). The formation of these different structural characteristics of the AMPs depends on the blend of the different amino acids throughout the peptide, which supports the folding of the AMP that leads to the formation of its three-dimensional conformation, shaping the physicochemical characteristics, and consequently influences the biological activity based on the structure-activity relationship (SAR) concept (Zasloff 2002; Bahar and Ren 2013; Phoenix et al. 2013; Lei et al. 2019).
For the different AMPs’ structures to exert their biological activities, they need to achieve the following:
-
(i)
Matchmaking between the length of the AMP residues and the thickness of bacterial membrane dimensions (Tossi et al. 2000). Given that the reaction is not based on receptor-ligand mediation, complete configurational matchmaking (lock-key theory) is not required; however, for the alpha-helical structure, it is optimal to have a complete penetration through the bacterial membrane (see discussion under AMPs formed in alpha-helix type));
-
(ii)
An ideal amphiphilic balance between the hydrophilicity and the lipophilicity ratios, as well as an appropriate cationic level of charge to govern the efficacy of the antibacterial activity (Lei et al. 2019).
As we have seen thus far in the present review, AMPs exert their antibacterial effect in various ways, including disruption of protein synthesis and cell wall formation, inhibition of genetic material expression and hindering different enzymatic activities. However, the primary mechanism of action is based on the disintegration of the bacterial membrane (Strömstedt et al. 2010). Moreover, the above particulars argue that the physicochemical characteristics of AMPs are the primary key behind their biological activity against the integrity of the bacterial membrane (Lei et al. 2019). The AMPs are predominantly short cationic (net charge between +2 and +9) peptides due to amino acids such as lysine and arginine with an amphiphilic composition (Bahar and Ren 2013; Pushpanathan et al. 2013; Zhang and Gallo 2016; Mirski et al. 2018). The amphiphilic nature governs the peptides’ pharmacokinetic profile of peptides through the balancing between the required optimal solubility in an aqueous physiological medium supported by the hydrophilic chemical groups versus the membrane permeability and the therapeutic bioavailability of the AMPs owing to the hydrophobic moiety. However, pharmacodynamically, this amphiphilic property of the AMPs, also, allows a direct interaction between the peptide and the microbial membrane rather than a receptor-mediated interaction which plays a major role in the bacterial resistance development (discussed below) (Tossi et al. 2000). On the other hand, the cationic property of the AMPs owing to lysin and arginine residues (Strömstedt et al. 2010) controls the pharmacodynamic effect of the AMP through its interaction with the negatively charged bacterial membrane (Zasloff 2002).
To exhaust our discussion within the scope of this review, we present a selected debate for each structural type with an example of a peptide and a related mechanism of action based on the expected physicochemical characteristics.
1.5.1 Alpha-Helical AMPs
Arguably, the effectiveness of the AMPs depends on the partitioning of the peptide molecule (distribution at physiological pH) into the phospholipid membrane of the microorganism. Thus, the higher the partitioning into the lipid phase, the more influential the peptide. Moreover, this partitioning is proportional to the molecule’s hydrophobicity, or it is inversely related to the availability of hydrogen bonding. This hypothesis has been tested by quantifying the free energy cost (using Gibbs free energy module) for partitioning the native bee venom peptide melittin, which is in alpha-helical form of 12 residues versus the unfolded form D4,L-melittin. The results showed a reduction of 0.4 Kcal mole−1 in the free energy cost per each alpha-helical residue distribution into the lipid phase (Ladokhin and White 1999). Hence, these results confirm that the secondary alpha-helical structure reduces the availability of hydrogen bonding and exposes the hydrophobic groups of the peptide to the lipophilic interfaces of the membrane’s phospholipids, helping in tunnelling through the membrane (Almeida et al. 2012). Nevertheless, the alpha-helical structural characteristic of the peptide plays only a partial role in its biological activity, which is determined by several other effectors, including the flexibility and self-assembly of the peptide, the amphiphilic balance of the peptide, the cationic charge of the peptide and the ionicity of the bacterial membrane (Juretić and Simunić 2019).
The alpha-helical segment results from the peptide sequence enrichment with specific amino acids such as arginine, alanine, phenylalanine, isoleucine, leucine and lysine (Dathe and Wieprecht 1999; Phoenix et al. 2013). These AMPs, generally, are more frequent and more prolonged peptides that can channel the bacterial membrane (Juretić and Simunić 2019). The disintegration of the bacterial membrane results from different ways of AMP activity, including the barrel-stave mechanism, the toroidal mechanism and the surfactant-induced lysis (Fig. 1.3) (Shai 2002; Strömstedt et al. 2010).

Summary illustrating the multistep of the mechanism of action of the AMPs
Usually, the alpha-helical type of AMPs with more significant residues (over 22 amino acids) adopts the barrel-stave mechanism, causing few transmembrane pores, dissociating the membrane integrity and depleting the transmembrane potential (Shai 2002). The peptide must present enough hydrophobic moiety which faces the hydrophobic bilayer of the bacterial membrane after the electrostatic attraction between the cationic sides of the peptide, and the anionic charge of the bacterial membrane occurs. Magainin, the clawed frog of Xenopus laevis, is one of the prototypes of alpha-helical AMPs that inhibits bacterial and fungal growth and mediates protozoa lysis (Zasloff 1987).
Another mechanism of action adopted by different structures of AMPs includes the alpha-helical type, called the carpet-like mechanism, which can progress to either the toroidal or the surfactant-induced mechanisms depending on the AMP. The carpet-like mechanism is an alternative module to the barrel-stave mechanism but with fewer requirements for activation in terms of peptide’s specifications such as transmembrane insertion and porosity forming or a minimal peptide length or the number of amino acids (Shai 2002). Therefore, it is available for a broader range of AMPs. In this module, the peptide accumulates parallelly on the surface of the pathogen’s membrane and causes a lysis-type reaction due to surfactant effect and micellisation (Pokorny and Almeida 2004; Strömstedt et al. 2010; Bertelsen et al. 2012). However, some other peptides augment their concentration until they reach a critical level on the membrane surface which results in an adsorbent reaction of the peptide on the polar surface of the membrane due to the development of chemical imbalance in the ionic surface of the membrane (Zasloff 2002). Following the peptide’s adsorption on the membrane surface, a translocation process of the peptide takes place across the membrane throughout the inner components, which leads to toroidal pore formation (Fig. 1.3) (Strömstedt et al. 2010).
Furthermore, the incorporation of the peptide into the outer layer of the membrane expands the surface of the membrane laterally, which, in turn, thins the membrane to the cleaving point (Mecke et al. 2005). The human cathelicidin (LL-37) is another example of the alpha-helical structure. However, it adopts the carpet-like mechanism of action against bacterial membrane (Pokorny and Almeida 2004). LL-37 is produced by phagocytes and skin keratinocytes and released into phagocytic vacuoles and skin wound fluids (Tossi et al. 2000).
1.5.2 Beta-Sheet AMPs
Beta-sheet family of AMPs adopts the anti-parallel beta-sheet structure upon their contact with aqueous medium due to disulphide bond formation between the cysteine residues positioned on the adjacent beta-strands (Kier et al. 2015; Moravej et al. 2018). Folding adjoining beta-sheets with no less than two disulphide covalent bonds creates a rigid platform that harbours the peptide’s essential functional moieties, including the cationic and the hydrophobic groups (Zasloff 2002). Generally, beta-sheet AMPs such as human defensins, bactenecin, cateslytin, protegrin and tachyplesin are less abundant than alpha-helix peptides. They are ubiquitous across the different levels of life forms (Phoenix et al. 2013; Moravej et al. 2018).
Although the beta-sheet family of antimicrobials employs the same barrel-stave and carpet-like mechanisms in their attack on bacterial membrane, different studies have presented various arguments concerning the structure-activity relationship. Generally, the highly rigid constructed beta-strands are occupied by polar and non-polar domains (cationic and hydrophobic groups). The cationic component initiates the electrostatic interaction between the peptide and the bacterial membrane, settling the hydrophobic residues on the membrane surface ready for partitioning and disrupting the membrane integrity via the formation of transmembrane channels (Yeaman and Yount 2003).
Amongst the most comprehensively studied peptides that fit as a model of SAR for this debate are the tachyplesins, which exist in three main types, including tachystatins A, B and C, consisting of 44, 42 and 41 amino acids, respectively (Osaki et al. 1999). Tachyplesins have been isolated from Japanese horseshoe crap T. tridentatus (Powers and Hancock 2003). The structural motif of tachystatin C is distinctively different from A and B. It can form an amphiphilic beta-sheet on terminal C which drives the partitioning of the peptide into the microbial membrane and enables the peptide to function as a haemolytic and cell lysis effector against Pichia pastoris. Moreover, tachystatin C shows an unusual antimicrobial activity against Gram-positive, Gram-negative and fungi throughout specific recognition and interaction with the lipoteichoic acids, the lipopolysaccharide and the chitin, respectively (Osaki et al. 1999).
1.5.3 Loop AMPS
Cyclic or loop peptides, in general, are linear peptides that adopt a loop-shaped segment due to either a single disulphide bridging or other types of bonds such as isopeptide, ester or amide, which lead to the so-called heterodetic cyclic peptide (Fehlbaum et al. 1996; Powers and Hancock 2003; Davies 2003). Restriction and freedom reduction of the amino acids’ residues by cyclisation leads to a higher degree of configurational rigidity. Subsequently, a higher affinity between the ligand and the target site results in a notable improvement in the biological activity of the peptide (Davies 2003).
Mika et al. have shown in a comparative study between the biological activities of a linear AMP named BPC194 and its analogue, a de novo designed cyclic decapeptide, against the plant pathogens Erwinia amylovora and Xanthomonas vesicatoria (Mika et al. 2011). The cyclic peptide demonstrated adoption of a beta-sheet structure which supported a higher affinity and more significant partitioning into the bacterial membrane whereas the linear analogue resided on the surface of the membrane (Mika et al. 2011).
Moreover, Hirakura et al. examined the relationship between the structural diversity of AMPs and specific antimicrobial activities (Hirakura et al. 2002). They tested the activity of the cyclic tachyplesin versus the activity of the alpha-helical magainin. The cyclic beta-stranded tachyplesin demonstrated higher affinity (by 280-folds) towards the lipopolysaccharide component of the cell membrane compared to its affinity to the acidic phospholipids, whereas the linear alpha-helical magainin acted equally towards both membrane components (Hirakura et al. 2002).
1.5.4 Extended AMPs
The extended type of AMPs lacks a steady as well as specific structural shape. However, they are rich in certain amino acids such as arginine, glycine, histidine, proline and tryptophan (Powers and Hancock 2003; Mishra et al. 2018). Moreover, their active configuration is an outcome of their electrostatic interaction, such as Van der Waals forces, with the microbial membrane rather than their intrinsic chemical/physical bonding amongst the amino acid residues (Powers and Hancock 2003). Thereupon, the extended conformational structure of certain AMPs has minimal impact on the microbial membrane integrity (Mika et al. 2011).
Indolicidin is an example of an extended cationic AMP isolated from cow neutrophils that exert its antimicrobial effect through its interaction with the bacterial membrane due to the amphiphilic cloud of the side chains around peptide (Mishra et al. 2018). It consisted of 5 tryptophan residues out of 13 and had demonstrated antibacterial activity against E. coli. Nevertheless, its affinity to lipopolysaccharides was notably low compared to the beta-sheet type of AMPs (Powers and Hancock 2003). Thus, the [AMPs] mode of action is derived from the amino acid composition rather than on a presumed secondary structure. Tryptophan is one of the most common amino acids that has been identified in the extended AMPs (Powers and Hancock 2003). Tryptophan residue is a critical interactive element in the interfacial vicinity of the lipid bilayers (Chan et al. 2006). It can form hydrogen bonding with the microbial membrane promptly. Likewise, arginine residues enhance the peptide interaction with the microbial cell membrane by providing the cationic characteristic and hydrogen bonding capabilities to attract the anionic bacterial membrane as an initiation for the peptide partitioning across the microbial membrane (Chan et al. 2006). On the other hand, proline disrupts the protein synthesis throughout its interaction with the 70S ribosomal subunit, which inhibits specific molecular signals and the production of certain constructive microbial proteins (Gennaro et al. 2002; Mishra et al. 2018).
Bellamy et al. demonstrated the chemical characteristic-activity relationship via an experimental measurement of lactoferrin B (47 amino acids) as an AMP against different bacteria, including Pseudomonas fluorescens and Enterococcus faecalis and Bifidobacterium bifidum (Bellamy et al. 1992). Lactoferrin B is one of the two active forms of lactoferrin (Bellamy et al. 1992), which is an extended broad-spectrum AMP found in most exocrine secretions in mammalians (Wakabayashi et al. 2014). Bellamy’s group reported two main findings (Bellamy et al. 1992): (i) the activity of the lactoferrin B was pH-dependent; hence, the peptide was more active at pH 7.5 compared to pH 5.5, and (ii) the antibacterial activity against the tested panel was reduced with the addition of Na+, K+, Mg++ or Ca++ ions. Bellamy et al. argued that the reduction in the peptide’s susceptibility was related to the changes in the ionicity of the membrane after the introduction of the cationic minerals. This argument was supported by their report about the higher effectiveness of the lactoferrin B activity in an alkaline environment compared to the acidic conditions (Bellamy et al. 1992), where the sensitivity of the peptide was reduced in the protonated medium presumably because of the changes in the ionicity of the bacterial membrane.
1.6 Immunomodulatory Signalling of AMPs
AMPs’ functional roles are widely versatile between the innate and the acquired immune systems of the complex life form. The ubiquitous presence of the indigenous AMPs in circulating and epithelial barrier cells across the different compartments of the living physiological system capacitates them as immunomodulators throughout various signalling pathways of the immunological and inflammatory processes (Hancock et al. 2016). That is to say, AMPs can serve as antibiotics and controllers of inflammatory mechanisms via immunomodulation and up−/downregulation of different cytokines (Phoenix et al. 2013).
In 1989, Mary C. Territo was the first to relate the AMPs and the immunomodulatory concept. Mary’s team argued that monocytes’ recruitment at inflammatory sites by neutrophils ought to be due to the AMP defensin mediation (Territo et al. 1989). Their argument was based on the observation that showed an unusual monocyte chemotactic activity that resulted after releasing HNP-1 (a human defensin) from neutrophil granules (Territo et al. 1989). Thereafter, further studies evidenced that alpha-defensins isolated from circulating human neutrophils stimulate T cells which, in turn, express CD4/CD45RA and CD8 antigens (Zasloff 2002). Nevertheless, the involvement of certain AMPs, such as the human cathelicidins in the signalling pathways of the immune system, can be even more convoluted. For instance, the human cathelicidin LL-37 interacts with more than 16 different proteins resulting in over 1000 successive interactive signalling molecules due to an expression of more than 900 different genes (Lau et al. 2005; Hancock et al. 2016).
Based on the mentioned above, AMPs play vital roles in mediating an array of cellular regulatory signalling during microbial infection and the subsequent inflammation (Scott et al. 2007). The roles of these cellular signals range over the preparation of the acquired immune response, such as the attraction of monocytes as well as the formation of antibodies against the invading pathogens, and the up-/downregulation of the pro-inflammatory cytokines such as interleukins (IL) (6, 8 and 18) and tumour necrosis factor-alpha (TNF-alpha), in addition to the anti-inflammatory cytokine interleukin-10 (Chaudhry et al. 2013; Li et al. 2017; Muñoz-Carrillo et al. 2018).
Inflammation is a complex process; nevertheless, it is a crucial segment of the immune system’s response to an infection. It is a vital signal for the immune system to prepare the body for protection and a successive healing process (Muñoz-Carrillo et al. 2018). However, inflammation can be too dangerous and more harmful, in some cases, than the actual bacterial infection. The role of bacterial infection in the pathophysiology of inflammation is related to the bacterial cell wall and membrane-associated LPS endotoxin (Ginsburg 2002). The release of the LPS into the bloodstream enhances the production and release of pro-inflammatory cytokines (e.g. TNF-alpha and interleukin-6 and interleukin-8) from monocytic and phagocytic cells (Sun and Shang 2015). Overreaction of the immune system to the titre level of LPS in the blood system can lead to overexpression of cytokines which, in turn, ought to lead to sepsis followed by multiple visceral organ injuries or what so-called flesh-eating syndrome (over 700,000 cases in the USA alone, with mortality rate up to 50%) (Ginsburg 2002).
Cathelicidins family of AMPs, particularly LL-37, were found to reduce inflammation by neutralising the endotoxin LPS via a direct interaction between the cationic moiety of the AMP and the anionic groups of the glycolipid (Nagaoka et al. 2001). Moreover, the antiseptic activity of the LL-37 is resulted from the expression of anti-inflammatory cytokines such as interleukin-8 (Scott et al. 2002). Scott et al. took their study further by performing gene expression experiments aiming to reveal the effect of LL-37 on the modulation of macrophages (Scott et al. 2002). The results revealed that LL-37 directly affects the downregulation of 20 genes; nevertheless, it causes upregulation of 29 genes, some of which coding for chemokines and their receptors, in addition to the anti-inflammatory interleukin-8 (Scott et al. 2002).
Besides the antimicrobial activity and the mediation/regulation of the inflammatory process of the AMPs, LL-37, the human beta-defensin (hBDs) group plays a significant role in the wound pathophysiology by controlling the healing process (Diamond et al. 2009). The skin produces and enhances the activation of LL-37 and hBDs to prevent and eliminate microbial infections and, also, to support the healing of cutaneous injuries (Lehrer and Ganz 1999). It promotes the production of cytokines/chemokines, attracting keratinocyte migration, angiogenesis and cell proliferation (Heilborn et al. 2003). Niyonsaba et al. have found that three out of four different hBD peptides that are produced in the skin mediate the production of IL-6, IL-10, interferon-gamma-inducible protein (IP-10), macrophage inflammatory protein-3-alpha (MIP-3alpha) chemokine, monocyte chemoattractant protein-1 and RANTES, by stimulating the epidermal keratinocyte cells which, in turn, increase their gene expression (Niyonsaba et al. 2007). Moreover, hBDs induce the phosphorylation process of the epidermal growth factor receptor (EGFR), which promotes skin wound healing via the induction of the epidermal and the rejuvenation of the dermal cells (Niyonsaba et al. 2007; Bodnar 2013).
Along with the significant role of the hBDs and the LL-37, which are expressed in leukocytes and epithelial cells (Koczulla et al. 2003), they are immensely filtrated into wound bed during the resorptive phase of the injury before it starts declining towards the end of the regeneration phase and the closure of the wound (Heilborn et al. 2003). Heilborn’s team argued that LL-37 has a great deal of influence in wound healing and closer mediation. They validated their hypothesis by showing the induction of LL-37 during the re-epithelialisation of the skin wound. Furthermore, this re-epithelialisation was halted by antagonising the LL-37 with specific antibodies (Heilborn et al. 2003).
The human cathelicidin LL-37 has been further presented as a critical intrinsic factor in cutaneous wound healing. Koczulla et al. demonstrated that LL-37 directly manipulates the angiogenesis and arteriogenesis processes via the activation of the formyl peptide receptor-like-1 on the endothelial cells (Koczulla et al. 2003). They showed that administration of LL-37 into the chorioallantoic membrane assay resulted in neovascularisation. Furthermore, they demonstrated a reduction in wound bed vascularisation in mice with cathelin-related antimicrobial peptide (CRAMP) (LL-37 murine homologue) deficiency (Koczulla et al. 2003).
To sum up, the AMPs’ portfolio of pleiotropic bioactivities involves a wide range of vital roles and mechanisms. They can exert direct action against microbes based on the incapacitating potential of the physicochemical characteristics of the AMP, adopting chemotactic mediation to assist the migration of different leukocytes to the site of the infection and stimulating the production of cytokines and chemokine to promote keratinocyte migration and prefoliation in wound beds. Concurrently, AMPs mediate a homoeostatic equilibrium in which all the biological processes are subject to perpendicular regulatory feedback.
1.7 Bacterial Resistance to AMPs
The focus on AMPs as a feasible solution for antibiotic resistance has been intensified in recent years. AMPs have been in the scientists’ spotlight since the early days of the emerging of bacterial resistance to conventional antibiotics. The initial assumptions stated that the probability of bacterial resistance development against AMPs is negligible. This hypothesis was based on the preliminary perception of AMPs, as they are simple and lack specificity in their mechanisms of action against microbes (Lazzaro et al. 2020). Furthermore, the early descriptive module of AMPs’ antimicrobial activity was presented as a platform involving various effectors that target different microbial biological aspects (Zasloff 2002). However, recent comprehensive research findings have revealed the inaccuracy of that early approach concerning the AMPs’ pharmacodynamic ligand binding modelling. The new studies argue that AMPs execute their biological activities based on high specificity and affinity to their target sites, which occur synergistically with other AMPs (Lazzaro et al. 2020). Pharmacodynamically, these findings provide more evidence that the development of microbial resistance against AMPs is possible; nevertheless, it can be counteracted by employing various types of peptides to achieve a broader spectrum and mode of activities for a specific target (Diamond et al. 2009).
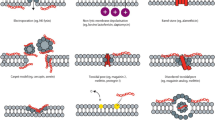
Different species of microorganisms adopt various techniques to evade the antagonism of antimicrobial peptides. In general, bacterial resistance to AMPs is categorised into two main types based on their broadness of spectrum. These types can be specific resistance against a particular type of AMP or broad resistance against multiple AMPs sharing the same motif (Nawrocki et al. 2014). For the scope of the present chapter, we reviewed the last 20 years’ research data related to, predominantly, bacterial mechanisms of resistance that can be adopted by Gram-positive and Gram-negative bacteria against AMPs to provide further insight into the tangled relationship between AMPs and microorganisms.
The AMPs’ efficacy, as well as the hindrance to them, is a complex of complicated mechanisms based on physiochemical characteristics of the peptides and the microbial wall/membrane, the vicinity of the environment including stressful parameters and the transport kinetics of the AMPs as well as the synergy between them (Groisman et al. 1992; Devine et al. 1999; Hancock 2001; Perron et al. 2006; Lazzaro et al. 2020). The AMP’s mode of action predominantly depends upon the molecule’s physicochemical characteristics and the microbe’s cell surface, where both, in turn, determine the magnitude of their mutual attraction to each other. Thus, the primary mechanisms of antagonism between the AMP biological activity and the microbial cell ought to be initiated through cell surface alteration or the so-called extracellular mechanisms of resistance which can be exerted via shielding of the binding site from the AMP and the enzymatic degradation of the peptide (Nawrocki et al. 2014).
1.7.1 Extracellular Mechanisms of Resistance
It is the predominant category of adopted bacterial mechanisms against AMPs’ bioactivities. Several studies have investigated the susceptibility of different bacterial species to the human cathelicidin LL-37. Schmidtchen et al. showed that human pathogens act on epithelial surfaces, including wound beds, such as Enterococcus faecalis, Proteus mirabilis, Pseudomonas aeruginosa and Streptococcus pyogenes, capable of producing proteinases against the cathelicidin LL-37 (Schmidtchen et al. 2002). A mass spectroscopy analysis demonstrated the cleavage of LL-37 at Arg-Ile and Asp-Phe, which took place in the presence of the elastase, an enzymatic product of P. aeruginosa in coetaneous wounds (Schmidtchen et al. 2001). Similarly, Sieprawska-Lupa et al. have examined the degradation of LL-37 by S. aureus-derived proteinases (Sieprawska-Lupa et al. 2005). Magdalena and her group have tested the susceptibility of LL-37 to metalloproteinase (aureolysin), which is generated by S. aureus. The results of the mass spectroscopy test demonstrated a cleaving enzymatic activity of aureolysin between Arg19-Ile20, Arg23-Ile24 and Leu31-Val32 peptide bonds of the LL-37, which inhibited the antibacterial activity of LL-37 in time- and concentration-dependent manner (Sieprawska-Lupa et al. 2005).
Exploring the extracellular mechanism of resistance against other AMPs, Schmidtchen and his group tested the susceptibility of alpha-defensin AMP against a mixed panel of bacterial species. They reported that pathogens that habituate connective tissues in wound beds, such as E. faecalis, P. aeruginosa and S. pyogenes, can evade the antimicrobial activity of the AMP alpha-defensin (Schmidtchen et al. 2001). Ultimately, these bacteria degrade the existing proteoglycans, such as decorin, biglycan and versican, in the host’s connective tissues via extracellular microbial proteases such as S. pyogenes cysteine proteinase, which, subsequently, leads to the generation of dermatan sulphate that binds and neutralizes the AMP alpha-defensin (Schmidtchen et al. 2001).
Another example of bacterial resistance to antimicrobial peptides concerning extracellular proteases is depicted in the products of Porphyromonas gingivalis and Prevotella species. It relates to oral anaerobic and highly proteolytic pathogens known to neutralize the antibacterial activity of some AMPs, including the wasp venom mastoparan and magainin II (Devine et al. 1999). The inhibition of the mastoparan and the magainin II was due to a specific structural cleavage at the Arg residues. Nevertheless, this type of inhibition was ceased after administering protease inhibitors, proving that the resistance’s nature is protease-based (Devine et al. 1999). It is worth noting that cecropin B’s activity was not affected by these proteases, which was most likely due to the higher rate of the cecropin B activity than the inhibition rate of the protease, which explains the superior efficacy of these AMPs (Devine et al. 1999).
Gelatinase is a notable representative of the extracellular proteases that contribute to bacterial resistance to AMPs in different ways. Gelatinase is produced by E. faecalis which is a prime nosocomial pathogen that causes various types of acquired infectious diseases such as urinary tract infections, post-surgical infections and endocarditis (Engelbert et al. 2004; Thurlow et al. 2010). Thurlow et al. showed that gelatinase is the primary factor behind the virulence of E. faecalis as the cause of endocarditis. Gelatinase was found to be an enhancer for bacterial resistance against the LL-37 peptide by cleaving it. Moreover, it was found to break down the anaphylatoxin complement C5a, reducing neutrophil migration and incrementing pathogen virulency. This action is in addition to its degradation bioactivity of the extracellular protein matrix in the connective tissue such as collagen and clotting factors, including fibrinogen and fibrin (Thurlow et al. 2010).
Furthermore, gelatinase is involved in biofilm formation as a valuable mechanism to evade the host’s defence mechanism (Hancock and Perego 2004). Hancock et al. showed that E. faecalis biofilms, which increase the resistance to the innate immune system and, hence, the bacterial virulence, are controlled through the production of gelatinase (Hancock and Perego 2004). The biofilm formation in E. faecalis was shown to follow the quorum sensing principle, a cell density controlling mechanism (Nakayama et al. 2001). Quorum sensing system regulates different characteristics of the E. faecalis, including biofilm development throughout the up-/downregulation of the extracellular gelatinase genes following the accumulation of the required threshold of a cyclic lactone peptide on the bacterial surface, which, typically, happens when the bacterial population is ready for an aggressive phenotype (Nakayama et al. 2001).
In addition to the proteases’ employment as a mechanism against AMPs’ hostile biological activity, isolation of the AMP can be another helpful mode of resistance. It has been found that some Gram-positive bacteria can produce surface-linked peptides that bind to the AMP and block its bioactivity against the bacterial membrane (Nawrocki et al. 2014). S. pyogenes, a Gram-positive bacterium that can lead to different infectious diseases in humans, is a typical example of human pathogens that adopt the sequestration methodology against mammal AMPs (Akesson et al. 1996). In addition, S. pyogenes was found to produce a streptococcal inhibitor of complement-mediated protein (SIC). This 31 KD extracellular protein lacks the typical structural characteristic (COOH-terminal) of the Gram-positive’s cell wall protein anchor, suggesting atypical biological functionality of the SIC (Akesson et al. 1996). After that, SIC was found to block different constituents of the innate immune system in the mucosal epithelial cells, including secretory leucocyte proteinase inhibitors (SLPI), cathelicidin LL-37, human alpha-defensin I and human beta-defensins I, II and III, throughout a complex formation with the target via ionic bonding, for example, interaction with the NH2-terminal of the SLPI group (Frick et al. 2003; Fernie-King et al. 2004; Pence et al. 2010).
Extracellular polysaccharides are polymeric carbohydrates produced by Gram-positive bacteria, which are attached to the microbial cell wall via covalent bonding forming capsular polysaccharides (CPS) (Fig. 1.4) (Nwodo et al. 2012), as an additional extracellular resistance mechanism, along with proteases and sequestration, to resist AMPs’ actions (Nawrocki et al. 2014). Campos et al. reported that Klebsiella pneumonia, a common nosocomial pathogen, upregulates the transcription of CPS when it is exposed to AMPs such as lactoferrin and polymyxin B aiming for limited interaction between the AMP and the bacterial cell surface (Campos et al. 2004). Hence, the prevention of the access of the AMPs into an orbital distance that allows an electrostatic interaction with the bacterial surface was hindered based on the amount of the produced CPS by the K. pneumonia rather than altering the surface chemical or serological characteristics of the LPS (Campos et al. 2004).

Illustration of the cell wall structures in Gram-positive and Gram-negative bacteria
1.7.2 Alterations of Cell Wall and Membrane Mechanism
Manipulations of the bacterial envelope are significant objectives in bacterial cells against AMPs (Zasloff 2002). These alterations include repulsion of the AMP through modifications of the anionic net charge of the cell membrane and alteration of the membrane fluidity (Hancock and Rozek 2002; Peschel 2002).
The anionic constituents in the cell membrane include the phosphatidylglycerol (PG), the phosphatidylserine (PS) and the di-phosphatidylglycerol or cardiolipins (CL). Moreover, the anionic constituents in the cell wall are the lipopolysaccharide (LPS) and lipoteichoic acid (LTA) (Rashid et al. 2016). On the other hand, most AMPs are amphiphilic molecules consisting of a cationic moiety with a net charge between +2 and +9 due to specific amino acids such as arginine and lysin (Pushpanathan et al. 2013). This cationic part develops an electrostatic attraction towards the negatively charged heads of the phospholipids in the bacterial membrane and the hydrophobic portion penetrating the inner hydrophobic section of the bacterial membrane (Zasloff 2002). Therefore, bacterial ionic charge alteration ought to reduce the interaction level between the AMP and the bacterial membrane and even resist the existence of the AMP in the vicinity of such membrane. The ionicity of the bacterial membrane can be disrupted or masked by adding positive amino acids to the surrounding environment via a multiple resistance factor protein (MprF) (Nawrocki et al. 2014). MprF is lysyl-phosphatidylglycerol synthetase located in the membrane which uses L-lysin to manipulate the anionic charge of the membrane by synthesising aminoacylated-phosphatidylglycerol and transforming it to the outer surface of the membrane, which, in turn, reduces the affinity of the membrane to AMPs, including the cathelicidin LL-37 and alpha-defensins (Ernst et al. 2009; Nawrocki et al. 2014). It is worth noting that the MprF mechanism of action is yet to be fully understood. However, it is known to have two required domains for full functionality, the C-terminal used for the lysinylation and the N-terminal employed for the exposure of the lysine on the surface (Ernst et al. 2009).
The DltABCD is another electrostatic repulsive pathway utilised by different pathogens against AMPs. The electrostatic forces of the bacterial envelope alongside other characteristics, for example, elasticity and porosity, are governed by the polyanionic network of the teichoic acid (TAs), which is made up of the composites of cell wall teichoic acid (WTA) and the lipoteichoic acid (LTA) (Neuhaus and Baddiley 2003). Scientists have reported that esterification of this matrix network by incorporating D-alanine through Dlt operon or (D-alanyl-LTA) pathway can modulate the properties of the envelope in many Gram-positive bacteria (Neuhaus and Baddiley 2003). Hence, the formation of the D-alanyl esters is found to be another enzymatic mediation that leads to ionicity masking the bacterial envelope and, consequently, results in AMPs’ resistance in Gram-positive pathogens like Bacillus, Enterococcus, Lactobacillus, Listeria, Staphylococcus and Streptococcus species (McBride and Sonenshein 2011; Nawrocki et al. 2014).
In an attempt to further elucidate the impact of D-alanylation of the bacterial cell wall on the susceptibility to the AMPs, Peschel et al. explored the insensitivity to AMPs in mutated Staphylococcus xylosus and Staphylococcus aureus species (Peschel et al. 1999). They reported that the bacteria lacking the D-alanine esters in the teichoic acids of the cell wall were the most sensitive to the AMPs’ panel, including the human defensin NHP1–3, tachplesins, magainin II, bacterial gallidermin, mammalian protegrins and nisins (Peschel et al. 1999).
Manipulation of the microbial cell wall and membrane constituents such as teichoic acids and phospholipids exerts a significant effect on the fluidity of the bacterial envelope, which, in turn, challenges the effectiveness of the AMPs (Peschel 2002). It has been speculated that the resistance of specific pathogens towards AMPs after D-alanylation of the envelope’s matrix is due to the changes in LTA’s configuration, which leads to an increment in the density and the rigidity of the bacterial cell wall, rather than the changes in electrostatic forces (Saar-Dover et al. 2012). By utilising atomic force microscopy, the researchers showed a drastic increase in the cell wall rigidity in wild-type group B Streptococci, which was escalated by 20-fold compared to the control group and, subsequently, reduced the wall’s flexibility and permeability to the peptide (Saar-Dover et al. 2012).
1.7.3 Efflux Mechanism
Although the bacterial interactions with the surrounding environment are directly aligned with the bacterial cell surface features and conditions, in some cases, they are mediated via cytoplasmic/periplasmic cell components. The ABC transporter, or the ATP-binding cassette, is a coupling system where ATP hydrolysis takes place as a driving force for an efflux mechanism that uptakes/reuptakes solutes from the cytoplasmic zone across the periplasmic space to the outer surface of the cell’s environment (Davidson and Chen 2004). ABC transporters are eukaryotic and prokaryotic features that consist of four distinct subunits, including two hydrophobic membrane-crossing subunits and two hydrophilic nucleotide-binding domains that generate the required energy for the active transport (Davidson and Chen 2004). ABC transport systems play a critical role in a wide range of cell functionalities by utilising specific binding proteins with high affinity for different substrates, including sugars, amino acids, vitamins, metals and ions (Davidson and Chen 2004; Davidson et al. 2008). Hence, ABC transporters can play a significant role in supporting some virulence determinants of bacteria. For example, ejecting substrates against AMPs, such as capsular polysaccharides, and presenting them on the bacterial surface can lead to electrostatic charge manipulation inhibiting the AMPs’ attraction to the bacterial envelope as discussed above (Liston et al. 2018).
Over the past 25 years, more studies were focused on mutagenesis of bacterial phenotypes associated with bacterial virulence, aiming to elucidate the significance of the membrane efflux pump concerning bacterial resistance to AMPs and virulence enhancement. Specifically, Jerse et al. tested the impact of the mutated multi-transferable resistance efflux system (mtrCDE) on the viability of N. gonorrhoea in the lower genital tract of female mice (Jerse et al. 2003). Jerse’s team confirmed the mutual relationship between the multidrug resistance efflux system and the survival rate of the bacterium. They reported that mtrCDE-deficient gonococci were more susceptible to different additives such as increased levels of progesterone which might exert an antibacterial effect (Jerse et al. 2003). Five years later, these results were confirmed through another study that investigated the effect of LL-37 on the manipulated gonococci. Shafer et al. reported that the loss of the mtrCDE increased the susceptibility of N. gonorrhoea to LL-37 (Shafer et al. 1998).
1.7.4 Biofilms
Biofilm formation is a different bacterial mechanism to combat AMPs’ action beyond the intrinsic mechanisms of the individual bacterial cell to inhibit the access or bioactivity of AMPs. These cellular actions include pumping AMPs out of the cell, breaking them down, diverting them to falsely produced surface targets and refusing their entry by changing the membrane. A biofilm is an organised group of bacterial cells in which cells become glued to each other due to a self-produced slim matrix composed of exopolysaccharides contributing to the adherence of the embedded cells to a live/lifeless surface, and this bacterial community can work collaboratively to form an organisational protection (Band and Weiss 2015) (Jolivet-Gougeon and Bonnaure-Mallet 2014). The construction of the biofilm yields a cell-to-cell communication process named quorum sensing, which depends on the density of the bacterial community. It is regulated on the molecular level involving the autoinducers and signalling molecules that control different features and functionalities such as biofilm formation, sporulation and bacterial virulence (Ashima et al. 2013).
Biofilms are of the most efficient features that contribute to bacterial resistance mechanisms against antimicrobial agents, including AMPs (Costerton et al. 1999), as they form a durable physical barrier around bacterial cells that prevents the infusibility of the AMPs towards the bacterial community zone (Costerton et al. 1999). Another possible reason for reducing biofilm susceptibility to antimicrobial agents can be directly related to their metabolic efficiency living in a cooperative protected community (Lazăr and Chifiriuc 2010). This structural way of living is manifested by a lower metabolic rate and, consequently, by a lower level of nutrient consumption, including uptake of existing AMPs below the minimum inhibitory concentration. Adding to that, some of the cells within the community can experience spatial deprivation in nutrient resources and other stress factors which might force them to develop a new mutated phenotype with less susceptibility to AMPs (Costerton et al. 1999; Jolivet-Gougeon and Bonnaure-Mallet 2014).
To sum up, bacteria differ in their susceptibility to AMPs which results in various mechanisms of action against bioactive molecules. Knowing the molecular signalling of the bacterial antagonism to AMPs with the appreciation of these molecules’ physicochemical and biological features may provide new ways to combat antimicrobial-resistant and highly virulent bacteria.
1.8 De Novo Designed AMPs as Potential Therapeutics
Studies focusing on high-throughput discovery and design of new AMPs, as alternative solutions for conventional antibiotics, were successful in the past decade. Scientists developed various screening and/or design techniques to generate innovative bioactive AMP analogues, whereon they have made remarkable progress in advancing novel technologies.
The outputs of protein screening assays are forked between cell-based and computational approaches, aiming to optimise the AMPs’ pharmacokinetics/dynamics, based on the current understanding of the mechanisms by which bacteria can circumvent the immune system. However, the persistent challenge of these different assays is preserving the therapeutic activity of the bioactive peptide with minimal adverse reactions. In the proceeding of this section, we present a couple of in silico approaches, including the Resonance Recognition Model (RRM) (Cosic 1994) and the Quantitative Structure-Activity Relationships (QSARs), (Taboureau et al. 2006) aiming to offer some insight into the recent advances in the field.
The RRM model is a physico-mathematical concept that employs digital signal processing approaches (Fourier and Wavelet transforms) in the translation of the peptide’s physicochemical features in the linear motif that determines the nature of the protein’s biological activity. It is based on the charge movement across the protein’s backbone that occurs at different energy levels depending on the sequence and the type of amino acids. The conductivity of the charge results in a resonated electromagnetic energy that transfers from the protein to the target molecule (Cosic 1994; Ćosić et al. 2006). The RRM methodology which is performed over two complementary stages, which include (i) conversion of the amino acid series into a numerical series and (ii) analysis of the resulting numerical sequences using a digital signal analysis approach. Translating the amino acid sequence into a numerical series is performed via the quantification of the electron-ion interaction potential (EIIP) value which represents the average energy level of the valence electrons for each amino acid. Subsequently, the following process is to apply the obtained numerical series to the digital signal analysis. Finally, the parent protein’s/peptide’s investigated bioactivity can be determined throughout the characteristic frequency of the digital signal analysis. The approach identifies the primary amino acid or the so-called hot spot that contributes to the bioactivity based on the prominent frequency shown on the characteristic frequency spectrum (Cosic 1994; Pirogova et al. 2011; Hu et al. 2013). For the last 10 years, the RRM approach has been utilised to design novel bioactive small molecular weight peptides, which proved to possess the intended bioactive therapeutic effects (Pirogova et al. 2009; Istivan et al. 2011; Almansour et al. 2012).
Another approach is the quantitative structure-activity relationship (QSAR) method, which is based on the correlation between the desired biological activity, the therapeutic agent’s toxicity and the physicochemical properties of an array of molecular atoms (Taboureau et al. 2006; Jenssen et al. 2008). It involves different physicochemical characteristics, named descriptors, presented by the AMP, such as amphiphilicity, hydrophobicity, helicity and surface area. These physicochemical properties govern the electrostatic attraction between the AMP and the envelope of the bacterial cells (Jenssen et al. 2005; Jenssen et al. 2007). Manipulating these characteristics through alteration of specific amino acids does not necessarily contribute further to the antibacterial effect, but it might reduce an undesired side effect of the peptide, for example, the impact on the erythrocytes (Taboureau et al. 2006). Hence, the QSAR approach is a regression module that attempts to establish a consistent correlation between the variations in the molecular characteristics’ values and the bioactivities for various compounds to rationalize the design of new chemical entities (Cherkasov and Jankovic 2004; Taboureau et al. 2006). Currently, QSAR analysis provides virtual screening for rapid design of in silico bioactive molecule libraries. Moreover, it can be utilised for data mining for novel AMPs (Cherkasov and Jankovic 2004).
1.9 Summary of the Current Knowledge on Antimicrobials Primes a Promising Upshot of Nanotechnology
The changeability and flexibility in the relationship between humankind and microbes have maintained the coexistence status quo since the early days of our history. Thus, up to a hundred years ago, this coexistence was balanced under the coevolution laws. Nevertheless, disruption evolved to this equilibrium after discovering antibiotics arguing in favour of the predominance of the modern human species as the winners over infectious diseases. Indeed, lack of knowledge concerns the microbial kingdom amongst different groups motivated by acquisitive instincts, such as pharmaceutical companies and health professionals, which has led to the emergence of stubborn resistance against the conventional antibiotics or the “magic bullet”, warning us of a challenging time to come.
The search for new classes of atypical therapeutic molecules associated with specific pharmacodynamics and fewer side effects has been upheaved (Leader et al. 2008). Peptides are one of the leading categories of nonconventional molecules that demonstrate an outstanding remedial potential across various medical conditions, including metabolic disorders, degenerative neuronal diseases, cancer and infectious diseases. Over 200 proteins and 100 peptides have been introduced to the pharmaceutical market across numerous indications (Muheem et al. 2016) during the last three decades. They exert their pharmacological effects via specific cellular pathways resembling natural molecular signalling. However, they are utterly sensitive molecules to physicochemical and biological degradations, which prevent them from reaching their target sites at the relevant bioavailability. Poor luminal permeability, high cytosolic metabolism, gastric degradation and the first hepatic clearance of such molecules have resulted in the dismissal of most of these biotherapeutics from being translated into medicines (Gupta et al. 2013; Muheem et al. 2016). Moreover, most of the currently used peptides and proteins are formulated in a parenteral form that requires high-cost cold chains for storage and transport.
Pharmaceutical scientists and biophysicists have contributed remarkably to the area of conventional drug delivery systems (DDSs) in the last 70 years. They have developed controlled drug delivery systems that proved to be successful in the medical and pharmaceutical industries. However, nonconventional and larger molecules such as peptide-based molecules have presented very challenging physicochemical properties and yet have a comprehensive and sophisticated design of a controlled drug delivery system to carry AMPs and deliver them orally for acute microbial infections and vaccinate the human body for prophylaxis.
Smart hybrid nanogels alongside other nano-delivery vehicles possess unique physicochemical properties that manifest in various dynamic changes such as transformation into different sizes and shapes in response to environmental stimuli. These changes make such materials promising co-therapeutic candidates for different medical conditions such as cancer, metabolic disorders, rheumatoid arthritis, neurodegenerative diseases and infectious diseases. We can manipulate such characteristics for ideal application and crossover of usage in different medical and pharmaceutical applications. The options for researchers in terms of various designs to diversify existing drugs and medical conditions are unlimited. However, accomplishing such a complex task requires multidisciplinary collaborative work between biophysicists, chemists, material scientists, biologists, drug discovery scientists and clinicians.
1.10 Drug Delivery System Strategies for AMPs
Researchers have been focusing on customised drug delivery systems by which the AMP is delivered directly and actively to a specific target site. This strategy ought to minimise the unnecessary exposures of healthy cells to the AMP and increase its bioavailability in the vicinity of the target site. However, actualising these ultimate grails was faced with the size of the delivery system as a significant challenge. The size of the targeted drug delivery system must correlate with the cellular and the molecular size of the target site while it is enduring the physicochemical and biological impediments along the way in the complex biological system, which is discussed in the following section.
1.11 Nano-drug Delivery Systems for AMPs
Convoluted physicochemical properties of the different colloidal nanomaterials (˂1000 nm) or the so-called nanoparticles, made of various substances such as inorganic metals, hydrogels, lipids, inorganic polymers or self-assembled peptides/amino acids, empower them to become highly sophisticated chrono-spatially controlled drug delivery systems. They provide the desired pharmacokinetics and pharmacodynamics to many AMPs by bypassing several physicochemical and biological impediments such as degradable enzymes, proteases and membranous barriers between the different physiological compartments. Moreover, manipulation of the physicochemical properties of these colloidal vehicles can be reflected in their structural functionality. For example, controlling the surface lipophilicity and charge furthers the device’s chrono-spatial controlled drug release system and lowers the required dose and the frequency of administration (Makowski et al. 2019; Pinilla et al. 2021). Consequently, it increases the bioavailability of the AMPs and improves the anticipated therapeutic window by lowering their toxicity profile (Bozzuto and Molinari 2015; Sánchez-López et al. 2020; Nwabuife et al. 2021). Nevertheless, alterations to the size and shape that lads to surface deformities of the colloidal particle can impact their interactions with the surrounding environment and other biomolecules (Jeevanandam et al. 2018).
Based on the aforesaid, stability, homogeneity and consistency of distribution of the nanoparticles at the target site are the critical attributes for assessing their quality, safety and efficacy. Nevertheless, these elements can be impacted by the physicochemical characteristics of the nanoparticle device and the surrounding medium. Thus, these properties must be inspected and characterised meticulously during the validation process of every potential drug delivery system. Hence, alongside the conventional and routine analytical and characterisation methodologies such as scanning and transmission electron microscopy and different spectroscopic analyses that are typically employed in material sciences, there are a couple of critical techniques, including dynamic light scattering (DLS) technique and electrokinetic test that must be employed (Makowski et al. 2019).
DLS, also known as photon correlation spectroscopy (PCS), is a well-established and commonly used powerful tool to study the diffusion behaviour of the colloidal system by quantification of the diffusion coefficient and the hydrodynamic radii that are based on the size and the shape of the particles (Stetefeld et al. 2016). It allows for the measurement of the particle size in a limited size range lower than 1 nm, and also it permits the estimation of the distribution of the colloidal system in a submicron region based on measurement of the Brownian motion of the tested particles (Caputo et al. 2019). Hence, DSL methodology provides an estimated degree of homogeneity between the sizes of the colloidal particles, which is crucial for the dosing uniformity.
The surface charge is a different pivotal characteristic that affects the stability of the colloidal system. Each dispersed particle is an aqueous solution surrounded by a shell-like electrical double layer named EDL (Fig. 1.5). The surface charge of the particle forms the bordering layer of the particle. This charged layer attracts the oppositely charged liquid ions that exist in the continuous aqueous phase forming the outside layer of the shell. The potential difference between the external surface of the EDL and the aqueous solution is termed electrokinetic potential or the so-called zeta potential (Salopek et al. 1992). Generally, tiny particles are inclined to aggregate and form a cluster of particles due to Van der Waals attractive forces in the vicinity of zero zeta potential (approximately between −30 and 30 mV) (Makowski et al. 2019). Hence, the estimation of the zeta potential is a critical anchor in colloidal chemistry by which scientists can anticipate the potential stability of the colloidal system, that is, sustainable suspension versus flocculation.

Schematic illustration of the electrical double layer (EDL) formed around the positively charged surface of a nanoparticle
Just as importantly, the drug release from the nanosystem is a crucial element in the pharmacodynamic process. Hence, modelling the AMPs’ diffusion kinetics from the nano-vehicle is an imperative practice required to determine the therapeutic window of the used bioactive ingredient. Finding a general model to describe the diffusion behaviour of a therapeutic agent from the various nanosystems, so far, is unachievable due to the complexity of these systems and the number of physicochemical factors involved in the process. Having said that, the two primary general theoretical rate expressions include Noyes-Whitney dissolution law and Fick’s law of diffusion, in addition to over ten well-known models (Barzegar-Jalali 2008; Aulton et al. 2013) that can be utilised to study and model the different diffusion kinetics of various nanosystems based on their unique composites and characteristics.
With a view of shining some light on the current practice in the nanobiotechnology field concerning the delivery vehicles of AMPs, the following discussion depicts some examples of different nanoparticles representing the most common categories of nano-delivery systems that have been developed in recent years. These nano-delivery systems include inorganic metal, an inorganic polymer, hydrogel, lipid and self-assembled peptides/amino acid-based devices.
1.11.1 Inorganic Nanomaterial: Metal Nanoparticles (MNPs)
Deficient pharmacokinetics of AMPs could conceivably result from a high rate of degradation by different proteases or sub-absorption and transporting to the target site (Saghazadeh et al. 2018). The amalgamation of AMPs with MNPs’ carriers ought to enhance their absorption into the bloodstream, prolong their half-life, reduce the required dosage and frequency of administration and lessen their toxicity (Chenthamara et al. 2019).
MNPs can be fabricated in various shapes such as cubes, rods, cones, stars and spherical forms, which are the most common shapes (Jeevanandam et al. 2018). However, notwithstanding the different forms of MNPs, the principal concept in the MNP shape is the surface-to-volume ratio (Makowski et al. 2019). The nanosized scale characteristic provides such particles with a conspicuously sizeable surface-to-volume ratio that can adsorb a significant number of molecules on a small number of employed particles that enhance the bioavailability of the active ingredient and, subsequently, the therapeutic efficacy at the target site while maintaining lower toxicity levels of excipients (Patra et al. 2018). Moreover, due to the nanosized property of such particles, they can be the ideal sensitive agents to detect diminutive target sites on the cellular and molecular levels (Sun et al. 2008). Furthermore, unlike most nanomaterials that are utilised as DDSs, metal nanoparticles offer, exclusively, few unique and vital physical properties such as electrical conductivity, specific optical behaviour and high thermal stability in addition to their chemical durability (Khan et al. 2019; Coetzee et al. 2020).
Formulation of the nanostructure of metal particles can be obtained through various chemical methods. Wet chemical techniques have been used widely in the synthesis of MNPs by using excess reducing agents such as sodium borohydride (NaBH4) or trisodium citrate (Na3C6H5O7) in an aqueous solution (Panigrahi et al. 2004). However, there are an array of various chemical and biological techniques used for the synthesis of nanosized materials. To demonstrate one point, after the screening of different microorganisms, including bacteria, fungi and yeasts, scientists have reported the ability of some of these microorganisms to reduce gold ions onto nanoparticles of different sizes and shapes through unknown cellular mechanisms (Gericke and Pinches 2006).
Metal nanoparticles can be tailored to certain functionalities and interactions with the surrounding environment. For example, it is attainable to change the surface charge of the metal nanoparticle by attaching different functional chemical groups to promote the interaction with specific markers such as the negatively charged phospholipids on the cell membrane of the target cells (Fig. 1.6) (Abbasi et al. 2021). At the same time, conjugation of nanoparticles with different functional groups permits the MNP-AMP complex to cross biological membranes. Moreover, manipulating the surface structure of MNPs can be utilised to promote the so-called active targeting mechanisms (Attia et al. 2019). For instance, attaching specific biomolecules or ligands to the surface of the metal nanoparticle allows it to become uniquely interactive with a specific cell or microorganism. Furthermore, attaching biocompatible polymers such as polyethylene glycol (PEG) masks the exposure of the nanoparticles to the reticuloendothelial system leading to an increment in the circulation time and the stability of the MNPs (Fratoddi 2017).

Schematic illustration of a metal nanoparticle conjugated with various functional molecules
The stability of MNPs in different culture media is questionable due to their chemical interactions with several salts, whereby changes in structure (size) and surface charge are feasible (Pavlin and Bregar 2012). Consequently, the aggregation of multiple particles is becoming inevitable, which results in inhomogeneous biodistribution. Concurrently, in the biological system, the instability and clearance of the MNPs depend on the particle size. MNPs exceeding 200 nm are recognised earlier than smaller ones by the immune system and cleared out through phagocytosis. Nevertheless, the circulation lifetime for MNPs below 200 nm is longer, and they are eliminated by the spleen, followed by particles that are scaled down below 5 nm that cleared out by the renal system.
For the scope of this study, we present a couple of MNPs which include gold (Au) and silver (Ag) as representative models, which have been explored widely in recent years as DDSs for AMPs. Cationic MNPs such as Au can form an electrostatic bonding with the negatively charged phospholipids in prokaryotes’ cell membrane, leading to its disintegration and crumbling (Makowski et al. 2019). Moreover, MNPs can penetrate the cell and exert their anti-cellular activity by targeting intracellular molecules and different physiological processes by releasing their ions in the vicinity of the cytoplasm (Makowski et al. 2019). These ions can interfere and disrupt the electron transport chain known as the respiratory chain process leading to inhibition in ATP production and initiate the accumulation of reactive oxygen species (ROS) and consequently stimulate the oxidative stress mechanism resulting in bacterial death (Rai et al. 2009; Niikura et al. 2013). Victorious nanocarriers with optimised size such as MNPs can penetrate the bacterial cell membrane after crossing the epithelial cells and reaching the cell’s apical surface, waiting to be transported to the basal side. The dissemination of the MNPs into the cell occurs through the so-called transcytosis mechanism by which they are transported through the interior of the cell via an uptake mechanism, namely, endocytosis with the likelihood of withdrawal via exocytosis process, degradation by lysosomes or deposition into a specific cellular organelle, after releasing the load inside the cytoplasm (Reinholz et al. 2018). Several studies have shown that manipulation of the size or the surface of the MNP impacts exocytosis kinetics and efficacy. For example, PEGylated gold NPs were exocytosed significantly, faster than positively charged (cationic) AuNPs (gold nanoparticles) (Oh and Park 2014). In a separate study whereby Chithrani and Chan tested the exocytosis rate of transferrin-coated gold NPs with a size range from 14 to 100 nm, the smaller NPs were exocytosed faster and at higher volume/time compared to the bigger sizes (Chithrani and Chan 2007).
In light of the above discussion, MNPs can be tailor-made to eradicate bacterial infection by enhancing their intrinsic antimicrobial properties (Sánchez-López et al. 2020). Thus, the anticipated outcomes from the amalgamated MNP-AMP complex were set so far out.
The antibacterial activity of the conjugated complex of MNP-AMP was found to be significantly augmented (Makowski et al. 2019). Cationic AMPs possess a high affinity to the negatively charged bacterial cell membrane (Hancock 2001; Scorciapino et al. 2017; Pal et al. 2019). Pal and the group have presented even more hostile activities adopted by AMPs against the bacterial membrane through molecular dynamic simulation technique. They have shown that the bacterial lysis via the barrel-stave mechanism formation in the bacterial membrane bilayer was mediated through a hydrophobic collapse mechanism that was initiated by the physical attraction between the hydrophobic moieties of the cationic peptide named Odorranian-A-OA1, which was conjugated with AgNPs (silver nanoparticles) from one side and the Gram-negative E. coli cell membrane from the other side (Pal et al. 2019). Conjugation of AMPs with nanoparticles at the very least leads to a higher concentration of the AMP at the target site. However, it has been reported that binding peptides to a metal surface such as silver enhances the growth and stability of the AgNPs (Zeng et al. 2007; Pal et al. 2016). The high affinity of specific amino acids such as Arg, Cys and Met to silver stabilises the nanoparticle structure (Poblete et al. 2016); subsequently delivering the AMPs to the target cells will be prolonged.
Mohanty et al. have demonstrated the effect of conjugating the AMPs NK-2 and LLKK-18 with biogenic AgNPs produced by Alstonia macrophylla and Trichoderma sp. against Mycobacterium smegmatis and M. marinum as a combination in comparison to the effect of the MNP alone. They have found that combined AgNPs with the NK-2 or LLKK-18 have a significantly higher effect on the mycobacteria than the solo MNP without causing a notable cytotoxic or genotoxic effect on mammalian cells (Mohanty et al. 2013). Furthermore, Lee et al. have tested the effect of the AMP HPA3PHis loaded onto a gold nanoparticle-DNA aptamer against Vibrio vulnificus infection in mice (Lee et al. 2017). Lee and the group reported a full recovery of the infected mice after administering the complex AuNP-Apt-HPA3PHis intravenously. The AuNP-Apt-HPA3PHis has led to a total inhibition of the V. vulnificus in different organs, compared to the control group that died within 40 hours of being infected (Lee et al. 2017).
Based on the above discussion, MNPs present great potential as DDSs that can deliver AMPs for various applications and types of pathogens. However, such potential requires more collaborative work between scientists and clinicians to improve therapeutic outcomes and reduce the toxic effects that have been reported for some of MNPs, such as silver, on mammalian cells and the environment (Kulkarni and Muddapur 2014).
1.11.2 Hydrogel-Based Nanoparticles: Nanogels
Presently, the knowledge of hydrogels and their potential usage as responsive, intelligent controlled-release drug delivery systems is significantly rich. Hydrogel is a cross-linked hydrophilic polymer with a three-dimensional network that is capable of swelling and retaining significant amounts of water throughout its fabric, hence providing a degree of flexibility like natural tissues (Ahmed et al. 2013; Ahmed 2015; Caló and Khutoryanskiy 2015; Akhtar et al. 2016; Nassar et al. 2021). Simple physical or chemical reactions produce these cross-linked hydrocolloid systems. They are defined as diluted systems that might be categorised as weak or strong based on their rheological behaviour in their steady-state phase (Lauren et al. 1980). Hydrogels can be produced in different physical forms, including slabs, microparticles, nanoparticles, coatings and films. As a result, they can be commonly used in clinical practice and experimental medicine for a variety of applications, including biosensors, tissue engineering and regenerative medicine, separation of biomolecules or cells and hydrogel-based drug delivery devices that have become a significant area of research interest. Biopolymers, including hydrogels, can be manipulated in size, architecture and composites, leading to different physicochemical characteristics. Subsequently, they differ in responsiveness and functionality that determine the type of drug delivery system they control (Li and Mooney 2016).
These developments are due to an extensive effort worldwide by biophysicists and pharmaceutical scientists over the past few decades. They have delivered a fundamental understanding of polymeric science by establishing various critical concepts concerning the rheological behaviour and pharmacokinetic modules of such biomaterials (Korsmeyer et al. 1983; Peppas and Korsmeyer 1986; Peppas and Sahlin 1989; Siepmann et al. 1999; Kasapis 2006; Kasapis 2008). Advances in peptide screening and optimised in silico design of small molecular weight protein/peptide analogues in addition to developments in hydrogel system design for controlled drug delivery are likely to drive the commercialisation of AMPs as alternative antimicrobial medicines to improve human well-being.
1.11.2.1 Nanogels
Nanogels are one of the major three subdivisions of the hydrogels family, including macroscopic hydrogels, microgels and nanogels. Nanogels are designed at nanoscale sizes ranged between 10 and 100 nm depending on the requirements of the route of administration and the targeted delivery site of the active pharmaceutical ingredient (API) (Li and Mooney 2016). Nanogels, like hydrogels, have attracted the attention of drug delivery scientists by far and away for the sake of their versatile properties. They gained the reputation by being fastidious in their responsiveness to environmental stimuli such as pH, temperature and organic metabolites leading to the diffusion of the entrapped cargo at different and specific target sites (Soni and Yadav 2016). Fundamentally, they are three-dimensional hydrogels with the capacity of holding large amounts of water while sustaining their structural integrity (Ahmed et al. 2013; Zhang et al. 2016).
Furthermore, they are spherical with tunable physicochemical characteristics such as size, softness, porosity, electrostatic charges and chemically manipulative surface through alteration of its lipophilic properties (Soni and Yadav 2016; Soni et al. 2016). This structural flexibility and tunability allow for customised nanogels as exclusive controlled drug delivery systems in various formulations (e.g. oral and subcutaneous) for specific target sites at which they are subject to specific time-dependent stimuli (Li and Mooney 2016). Case in point, nanogels can be designed in an injectable form to deliver compatible APIs, including AMPs systemically due to their ability to infiltrate out of the small blood vessels via fenestrations across the endothelium towards different tissues (Li and Mooney 2016). The customised delivery via nanogel system is designed based on specific manipulation of the nanogel surface. They can be designed pharmacodynamically to interact with the surface of the targeted cell (Zhao et al. 2014). For example, few studies have successfully managed to deliver cytotoxic agents by utilising micellar nanogels directly to the tumour cells whereby a higher concentration of the APIs was achieved in the vicinity of the cancer cells and simultaneously lower drug availabilities were recorded nearby the normal cells (Qiu et al. 2014; Zhao et al. 2014).
Furthermore, nanogels can be utilised to optimise the stability, solubility and absorption of various AMPs. It is conceivable to enhance the solubility of hydrophobic drugs in an aqueous solution by encapsulating them within amphiphilic nanogels (Zhao et al. 2014). Moreover, such encapsulation can protect sensitive substances such as bioactive peptides and nucleic acids from chemical and biological enzymatic degradation throughout the delivery process via the bloodstream and other biological compartments (Soni et al. 2016).
Nanogels are uniquely defined by their morphology which consists of two main parts: (1) the inner micellar compartment, which holds and protect the AMP until its release, and (2) the corona shell that carries the ligands for specific targeting or self-protection (Fig. 1.7) (Alexis et al. 2008; Moritz and Geszke-Moritz 2015).

Schematic representation of the nanogel morphology conjugated with different functional molecules
Hence, the protection of the nanogel and subsequently the entrapped AMP within the physiological system (bloodstream and other body compartments) and the enhancement of the functionality of the outer shell of the nanogel are achievable via a specific design of the corona. To illustrate, grafting the hydrophilic polyethylene glycol (PEG) onto the surface of the nanogel provides a receptor-mediated drug delivery through PEG-conjugated agonists. This type of conjugation minimises the non-specific interactions with other proteins and reduces the adsorption to biodegradable enzymes leading to a reduction in the clearance rate of the nanogel (Otsuka et al. 2003; Romberg et al. 2007).
Nanogels are synthesised through chemical crosslinking or physical interactions between two low molecular weights of monomers (Soni et al. 2016). Chemical crosslinking between the different polymers/monomers is carried out via covalent bonding between the interactives, whereas the attractive physical forces between these polymers/monomers can be established due to relatively week forces such as hydrophobic attractions or ionic interactions (Soni et al. 2016). Nevertheless, conventional chemical synthesis through radical polymerisation provides nanogels with the required architectural structure, including the core-shell and the nanogel hollow (Chiang et al. 2012).
Nanogels become highly sophisticated delivery systems after a unique tuning of their size, morphology and specific chemical manipulations leading to unique functionalities. Specific responsive morphological features concern different environmental stimuli such as pH, temperature, electromagnetic field, light, glucose, electrolytes and other metabolites and result in the so-called intelligent nanogels; however, it can be a convoluted chemical process. This complexity includes using different initiators that activate specific chemical reactions, allowing different functional groups to incorporate the structure (Sanson and Rieger 2010). Recently, the accumulated scientific data concerning stimuli-responsive polymers has been remarkably increased due to the advancement in polymerisation techniques. These new technologies, such as the atom transfer radical polymerisation (ATRP) and the reverse addition-fragmentation chain transfer polymerisation (RAFT), provide an excellent tolerance towards the addition of new chemical motifs (Jochum and Theato 2013). Generally, the stimuli-responsive behaviour is a downstream cascade of events initiated by either an indigenous environmental parameter in the vicinity of the nanogel such as pH or an exogenous stimulus including light or electromagnetic field (Soni et al. 2016). These stimuli initiate conformational changes in the nanogel structure based on the alteration in the balance between the surface hydrophobicity and the lipophilicity (Li and Mooney 2016; Soni et al. 2016), resulting in changes in the swelling ratio of the hydrogel and the expulsion of the trapped material (Eichenbaum et al. 1998).
Temperature-responsive nanogels were popular in drug delivery applications (Jochum and Theato 2013). Their type of response was studied intensively by introducing the lower critical solution temperature (LCST) concept, a unique chemical characteristic. Below, the LCST nanogels show complete miscibility in solution; however, above the LCST, clear phase separation occurs (Jochum and Theato 2013). For example, poly(N-isopropyl acrylamide) (PNIPAM) shows lower solubility in aqueous solution within the range of temperature between 30 and 35 °C due to macromolecular transition from hydrophilic to hydrophobic moiety around its LCST (Schild 1992). Hence, grafted nanogel with PNIPAM loaded with an AMP can be triggered externally to initiate the macromolecular transition back to hydrophobic nature, leading to the release of the drug. Hyaluronic acid (HA) is another example of a stimuli-responsive nanogel. HA is a common signal molecule in the human body, and it is ubiquitous in the extracellular matrix. Moreover, HA is targeted by hyaluronidase (HAdase), a highly expressed enzyme in metastatic cancer and lymph nodes. Based on that, nanogel made of HA was utilised to encapsulate indocyanine green (ICG) to be released post enzymatic stimuli-responsive mechanism in cancer diagnostic imaging (Mok et al. 2012).
Although the nanoscale size of nanogels enhances their diffusion through biological membranes and across the blood vessel endothelial linings with an average fenestra between 50 and 100 nm across most physiological compartments (Alexis et al. 2008); however, they are considered as foreign bodies to our immune system. Hence, manipulating the outer shell of the nanogel can reduce the attraction to digestive enzymes and natural immune peptides, leading to a long-circulating time in the different body compartments. On the other side, the clearance of nanoparticles from the biological system is performed via spleen and renal filtration or through macrophagic phagocytosis, determined by the particles’ size. Hence, particle size below 20 nm will be diverted to renal filtration, whereas larger particles up to 200 nm can be squeezed through the spleen (Moghimi et al. 1991; Zhang et al. 2012; Soni et al. 2016). Thus, phagocytosis, which targets larger sizes (0.5–10 μm), can be markedly halted as a leading clearance mechanism against nanogels by reducing the size below 0.5 μm (Alexis et al. 2008).
1.11.3 Lipid-Based Nanomaterials: Nanoliposomes and Nano-micelles
The name liposome is a combination of two Greek words lipos, meaning fat, and soma, meaning body. Liposomes were discovered and identified by Alec Bangham et al. in 1965 (Liu et al. 2016). Nanoliposomes are nanoscaled spherical liposomes. Generally, they are unilamellar chambers that consist of single amphiphilic bilayers made of cholesterol or nontoxic phospholipids (Fig. 1.8) (Akbarzadeh et al. 2013; Pinilla et al. 2021). Larger vesicles that can be tumid up to the micrometre range are, typically, made of multiple lipid bilayers (multilamellar vesicles). The spherical architecture of nanoliposomes offers an aqueous core as a suitable vacancy to lodge, protect and deliver sensitive cargos such as AMPs from the surrounding environment (Pinilla et al. 2021), which are diversified, based on their physicochemical properties, between hydrophobic, hydrophilic, anionic and cationic AMPs (Scorciapino et al. 2017; Magana et al. 2020).

Schematic illustration of the structural and functionalised liposome
Moreover, the phospholipid-based membrane of the liposome exhibits a unique platform for specific organic molecules that can protect the vesicle from different physical, chemical and biological injuries and provide customised guidance systems targeting specific delivery terminals (Manikkath et al. 2020). Furthermore, utilisation of liposomes for drug delivery systems has been proven to be utterly compatible and biodegradable in the biological system and significantly low in their cytotoxicity profiles (Akbarzadeh et al. 2013).
In light of the aforesaid information, these state-of-the-art properties depict liposomes as intelligent drug delivery systems that were significantly utilised commercially for various therapeutic agents, including AMPs (Liu et al. 2016). Nevertheless, apprehension and portrayal of the diversified physicochemical characteristics of liposomes that have been increased remarkably in recent years (Patil and Jadhav 2014) are imperative in demarcating their suitability and fitness as DDSs for the wide range of distinctive AMPs.
Liposomes are categorised concerning their function/response, such as ligand-triggered, controlled-release, exogenous/endogenous stimuli release and stealth. Moreover, the architecture of different liposomes is defined by the number and the size of the vesicles’ lamella (core). The completion of the different architectures occurs during the designing process based on the delivery material and the target site of the liposome. Furthermore, the degree of the fluidity of liposomes is another crucial feature that must be considered during the engineering process as the balancing point between the stability of the liposome and the diffusion of its load at the target site (Monteiro et al. 2014). Liposomes’ different groups and characteristics are controlled via their constituents and the employed technologies for their formulation. For this chapter’s scope, we will briefly discuss the conventional and the atypical formulation techniques; moreover, we will expound on some of the different customised functionalities only summarily, and further details may be found in the cited literature.
The formation of liposomes is a process that toes the line of the fundamental concept in pharmaceutical sciences, at which point oil and water can form a homogeneous mixture or the so-called oil-in-water emulsion by lowering the surface tension of the aqueous phase (Aulton et al. 2013). Based on that, the conventional technologies that are used in the formulation of liposomes can be achieved either by (i) forming a bulk of a yield via transferring a mass of organic phase of phospholipids into an aqueous (continuous) phase or (ii) depositing the organic phase on the surface of the aqueous solution forming a film leading to the deposition of liposome vesicles into the bulk of the continuous phase after reaching the critical micelle concentration point (CMC). This process is purported as a self-assembled mechanism at which a circular biological membrane is formed due to the phospholipids’ crowding of immiscible hydrocarbon chains due to their mutual hydrophobic attractions in aqueous solution (Shohda et al. 2015; Liu et al. 2016). Hindering of the hydrophobic moiety of the phospholipids within the membranous structure while revealing the hydrophilic polar heads of these phospholipids to the aqueous solution ought to increase the solubility and stability of the formed liposomes. Dynamically, the stability of the emulsion system is achieved by lowering the high energy status associated with the edges of the hydrophobic chains exposed in aqueous solution to a preferred conformational energy associate with the self-assembled hydrocarbons shielded from the water phase (Patil and Jadhav 2014). In completing the formulation process of the vesicle, the final size and the lamellarity of the formed liposome can be determined later through a wide range of typical and atypical techniques.
In general, inputting a more considerable amount of the used phospholipid into the system can lead to the size expansion of the vesicle. Nevertheless, fragmentation of that size is achieved through different dynamics. Moreover, allowing the bilayer edges to merge faster can lead to multilamellar vesicles and vice versa. For example, applying electric fields during the formulation process can reduce the hydrodynamic flow, causing a decline in the bilayer synthesis, and subsequently, unilamellar vesicles merge as the final product (Patil and Jadhav 2014; Rideau et al. 2018).
The formulation technique’s nomination depends on the size and the lamellarity of the vesicle that is required. For example, preparing giant unilamellar vesicles (GUVs) (>1 μm) can be achieved via hydration of a dried phospholipid film by employing different conventional methods. We can utilise the “gentle hydration of phospholipid film” method to produce a single GUVs made of a closed lipid bilayer membrane of diameter greater than 1 μm (Shohda et al. 2015). This method is known as the “natural swelling” method, whereby the formation of the liposome occurs spontaneously after hydration (Rodriguez et al. 2005), and it is the preferred technique for obtaining GUVs of charged phospholipids (Patil and Jadhav 2014). Furthermore, utilising this technique to prepare liposomes allows the formation of the GUV at ionic strength close to the physiological level after further improvement by Akashi et al. (Akashi et al. 1996).
Another technique used to formulate the GUVs is called the “electro-formation” method. It is a very similar technique to the gentle formation methodology. However, applying an electric current to the aqueous medium enhances the liposome formation from the dried phospholipid film (Rodriguez et al. 2005). Hence, an electromagnetic field is generated in the vicinity of hydrating solution after depositing the phospholipids on the surface of the electrodes, leading to the formation of a few to tens of micrometre liposomes with an improved rate of the unilamellar formation (Rodriguez et al. 2005).
Coalescence of small vesicles is another type of procedure to obtain GUVs. For instance, spontaneous clumping of small vesicles after prolonged incubation in suspension can lead to fusion between them and the formation of GUVs (Oku and Macdonald 1983). The fusion of small liposomes may be induced by various routes, including subjecting them to freeze-thaw cycles in highly concentrated solutions with electrolytes (Chavanpatil et al. 2006). In 2015, Motta et al. had developed a generic methodology by which GUVs were based on the free detergent coalescence of organic material in aqueous solution (Motta et al. 2015). The idea of this new method is based on the fragmentation of proteo-liposomes triggering the formation of proteo-GUVs. Hydration of dried small liposomes with purified water supposedly induces an osmotic shock causing the liposomes to burst before triggering the resealing process into GUV (Motta et al. 2015).
The giant unilamellar vesicles can be used as precursors to create large unilamellar vesicles (LUVs) and small unilamellar vesicles (SUCs) that fall within the nanoscale range. Nevertheless, these nanoliposomes can be, also, produced via specific conventional methods. The “reverse-phase evaporation” is a traditional preparation method that involves the hydration of phospholipids after their dissolution in an organic phase such as chloroform or ethanol. Inputting water into the organic solution throughout vigorous mixing results in the water-in-oil emulsion, transformed to an aqueous suspension containing LUVs after the evaporation of the organic phase (Deamer and Bangham 1976). Another technique used to produce LUVs is the detergent dialysis methodology (Patil and Jadhav 2014). In this methodology, the solubilisation of phospholipids in an aqueous solution occurs by utilising the amphiphilic property of detergents. Then stabilisation of the phospholipid-detergent nano-micellar system’s formed complex is enhanced by reducing the surface tension between the phospholipid micellar membrane and the aqueous solution. Elimination of the detergent from the micellar system increases the surface tension to a higher level, resulting in crowding of smaller phospholipid micelles leading to LUVs (Helenius et al. 1977).
SUVs’ formation can be achieved by injecting organic solvent with dissolved phospholipids into an aqueous phase (Nwabuife et al. 2021). The idea behind this technique is to force a self-assembly mechanism of phospholipids in an aqueous solution after dissolving them in an organic vehicle (Lasic 1995). When the organic solvent such as ether or ethanol is injected into a more significant amount of aqueous solution, the concentration of the organic phase is dissolved and diluted in the aqueous solution leading to a drastic reduction in their concentration below the critical dissolving forces that required to maintain the solubility of the phospholipids. Subsequently, the phospholipids are released into the aqueous solution and forced to self-assemble, forming SUVs (Lasic 1995).
Generally, liposomes provide great potential for a targeted and controlled drug delivery system, primarily attributed to the flexibility in size, lamellarity and charge alterations (Nwabuife et al. 2021). As has been mentioned earlier, the reduction in the size and lamellarity of the MUVs (>1 μm) is one of the methods that is employed to formulate SUVs (20–100 nm) and LUVs (>100 nm) (Yu et al. 2018). Sonication, homogenisation and membrane extrusion are common techniques used to downsize the MUVs to SUVs and LUVs (Patil and Jadhav 2014; Nwabuife et al. 2021).
Fabrication of multilamellar vesicles (MLVs) is achievable by utilising different conventional methods such as hydration of phospholipid film of stacked bilayers under strong hydrodynamic flow for few hours (Carugo et al. 2016). Moreover, the reverse-phase evaporation method, which is utilised to produce LUVs, can also fabricate MLVs. Providing higher concentrations of phospholipid content in the aqueous phase yields a higher fraction of MLVs than LUVs (Pidgeon et al. 1987).
Hydration of pro-liposomes is another conventional method that can be employed to formulate MLVs with the encapsulated bioactive agent. Pro-liposomes are dry and stable liposomes that consist of phospholipids with the encapsulated bioactive ingredient. They can be formed by drying an organic solution containing the phospholipids and the cargo, such as AMPs (Patil and Jadhav 2014), transformed to MLVs upon their dispersion in an aqueous solution (Nekkanti et al. 2015).
Since the commercial production of liposomes has been levelled up strikingly in the pharmaceutical industry and beyond, the range of liposome preparation techniques has been advanced significantly. Amongst these methodologies are the heating method, spray-drying, freeze-drying, supercritical reverse-phase evaporation and several modified ethanol injection techniques that are highly qualitative and engaging (Koynova and Tenchov 2015). Aside from that, in the early 1980s, an interesting multidisciplinary methodology called “microfluidic technology” has been introduced for different research purposes in various fields such as biochemistry, physics, biotechnology, engineering and nanotechnology. Microfluidic technology has been utilised for multiple drug screening in different body organs to the cell level. Nowadays, it is an atypical leading technique used to produce liposome-based DDSs for various conventional drugs, gene therapy and bioactive compounds, including AMPs (Carugo et al. 2016; Damiati et al. 2018; Dong et al. 2019).
The microfluidic formulation methodology is a nonconventional technique that provides unique advantages in the synthesis of drug delivery vesicles in terms of efficiency, homogeneous geometry and reproducibility over the conventional bulk methods that have been discussed above. It is a cost-effective technology that enables the fabrication of highly stable and uniformed vesicles at a fast rate with remarkable efficiency of encapsulation (Damiati et al. 2018). The microfluidic technology can be performed through different methods based on a hydrodynamic device that is consisted of microchannels and chambers to control the flow behaviour of a small volume of fluids and produce liposomes within the range of 50–75 nm (Dong et al. 2019), while, precisely, controlling the lipid hydration process as it has been reviewed extensively by Yu and the co-authors in Yu et al. (2009).
Structurally, liposomes can carry hydrophilic and lipophilic molecules; hence, they are suited for amphiphilic compounds such as AMPs. However, the uppermost role of the liposome in such an engaging process with the AMP is to permit and maintain its encapsulation within a stable and intact architecture. Moreover, maintaining the coexistence between the encapsulated AMP and the liposome is primarily controlled by the membrane’s fluidity. Generally, the fluidity of the biological membrane is regulated by the relative proportions of cholesterol to the constituents of the phospholipids (Charalampous 1979). Higher ratio levels of the phospholipids tend to increase the fluidity of the biological membrane leading to stability reduction, which can be counteracted by increasing cholesterol (Cooper 1978; Chabanel et al. 1983).
Material scientists could utilise different lipid-based materials with an amphiphilic property to formulate liposomal lamellar through self-assembly processes. Nevertheless, the uppermost aim is complete control over the fluidity (stability) of the membrane and the diffusivity of the incorporated AMPs. Huang and co-authors have reviewed a wide range of studies that dealt with the so-called amphiphilic mesogens, which can form lyotropic liquid crystals (LLCs) (Huang and Gui 2018). These LLCs are amphiphilic molecules formed through the self-assembly process in aqueous solutions producing unique physicochemical and internal structural properties (Boge et al. 2016). They have usually formed throughout various phases: the lamellar phase, the reversed bi-continuous cubic phase and the reversed hexagonal phase. The different yielded structures of these variable phases have presented different diffusion coefficients to a wide range of loaded cargos that were also managed to be released in a sustained manner. Moreover, they have shown varied responsiveness to different levels of pH and temperature, in addition to their abilities to load various sizes of cargos (Huang and Gui 2018).
Boge and co-researchers have examined the ability of a couple of lyotropic liquid crystalline (LC) structures incorporating (i) cubic glycerol monooleate in water and (ii) hexagonal glycerol and oleic acid in water, to carry different AMPs separately and maintain their antimicrobial effect while the LC structure is preserved (Boge et al. 2016). The results showed the antibacterial effectiveness of two of the tested AMPs, including AP114 and DPK-060, preserved in the cubic LC structure, whereas they noticed a reduction in the antibacterial effect of the LL-37. Moreover, the hydrophobic peptide (AP114) has induced an increment to the negative curvature of the cubic LC system. Conversely, the polar peptide (DPK-060) has reduced the negative curvature, while the LL-37 has not changed the LC phase. Interestingly, none of the peptides has affected the hexagonal LC phase; nevertheless, their antibacterial effect was significantly reduced (Boge et al. 2016).
The selection of the right combination of the liposomal constituents that are physiochemically compatible with the loaded AMP is another crucial aspect of liposomal formulation (Thapa et al. 2021). Nevertheless, scientists went beyond that by advancing liposomes to collaborate with AMPs and act synergistically, mounting the antimicrobial effect. Malheiros et al. used liposomes made of phosphatidylcholine (FC) and the cationic lipid named 1,2-dioleoyloxy-3-trimethylammonium propane (DOTAP) to encapsulate bacteriocin (Malheiros et al. 2016). The results showed that the encapsulation of the bacteriocin in the FC/DOTAP had advantaged the antibacterial activity of the bacteriocin in goat milk compared to the activity of the free bacteriocin. This synergistic effect between the liposome and the AMP might be attributed to the cationic effect of the DOTAP against the bacterial membrane (Malheiros et al. 2016).
Enriching the DDS with different selective physicochemical features as we know now can be advantageous in fighting microbial infections. Thus far, the hydrophobic attraction has been proven to be one of the effective mechanisms against bacterial cell integrity (Tian et al. 2015) and can be utilised to stabilise the AMPs and reduce their toxicity effects by confining them away from interacting with mammal cells (Chen et al. 2019). Hence, the inclusion of an augmented hydrophobic feature to a lipid-based vesicle might be an appealing choice for some AMPs for superior efficiency and stability. Based on this, utilising the concept of the micellar-based vesicle, a closed lipid monolayer with a polar core and hydrocarbon chain presenting the hydrophobic potential on the surface or a hydrophobic core with a polar surface (Fig. 1.9), has become one commonly used DDS as an alternative to the bilayer liposome.

Schematic illustration of revers versus normal micelle
Groo and co-researchers tested the effect of micellar nanosystem on the stability of an AMP called AP138 (Groo et al. 2018). They have developed reverse micelles incorporating lipid solution forming lipid nano-capsules (LNCs) by using the phase inversion process. The encapsulation rate of the AP138 into the complex of AP138-RM-LNCs was 97.8% which allowed them to conduct a comparison study, whereby they have tested the stability and the antibacterial effectiveness of the encapsulated peptide in comparison to the free AP138. They have shown that the encapsulated peptide (similar to the schematic illustration in Fig. 1.10) has demonstrated a significant resistance against trypsin compared to the free AP138; moreover, the antibacterial activity of the encapsulated peptide was preserved against Staphylococcus aureus, including MRSA (Groo et al. 2018). In short, micellar formulations loaded with antimicrobial peptides in Fig. 1.10 have already been applied into some dental applications due to their ability to bind to certain minerals integrated into teeth surfaces and to deliver encapsulated AMPs over a prolonged time against the formation of Streptococcus mutans biofilms (Carmona-Ribeiro and de Melo Carrasco 2014).

Schematic illustration depicting the formulation of a micelle loaded with an AMP
1.11.4 Inorganic Polymer-Based Nanomaterial: Nanofibres
Nanofibres are polymeric structures with the size of around 100 nm made of fibre that can be fabricated from inorganic materials such as ceramics, metal and metal oxides. Mixed metals such as Ba, Cu, Fe, Mn, Mo, Ni, Sb, Si, Sn and Ti and their oxides like CeO2, CuO, Fe2O3, MnO2, NiCo2O4, SnO2 and TiO2 culminate in various considerable types of synthesised nanofibres with captivating properties (Barhoum et al. 2019). Nanofibres, like most nanomaterials, exhibit physical properties such as high surface area, high porosity with small mesh size and high pore density over conventional fibres (Sousa et al. 2020). Nevertheless, the morphological properties and homogeneity of the system are hard to control during fabrication (Barhoum et al. 2019). Generally, nanofibres can be employed in different areas in the medical and pharmaceutical fields, such as wound dressings, tissue engineering scaffolding and controlled-release drug release systems (Sousa et al. 2021; Topcu et al. 2021).
In the proceeding chapter, we endeavour to unravel the latest status of drug delivery research which concerns the formulation of various AMPs via nanofibres as one of the widely explored strategies in recent years. However, it is imperative to briefly consider the different methodologies employed in the synthesis of nanofibres and their general compatibility with AMPs.
Polymeric fibres with a diameter range between nanometres and micrometres are complex biomaterials that can be fabricated through various methods such as electrospinning, dry spinning, laser spinning and gel spinning (Frenot and Chronakis 2003; Sousa et al. 2021). Electrospinning is a nonconventional methodology that can produce an inorganic polymer fibre in the nanometre diameter. Moreover, it is one of the current advanced primary methods to deliver AMPs (Sousa et al. 2021) and, hence, is of interest in this review.
Electrospinning or the electrostatic wiring technique, in essence, is based on a combined electric and hydrodynamics, whereon an electric force is applied to a polymer solution resulting in a formation of a continuous hydrofibre upon its release into the air. Precisely, the electrospinning process is involving the application of high voltage to a needle that is attached to a filled syringe with a polymeric solution (Fig. 1.11). The polymeric solution is set for rotation supported by an electrode after its ejection from the needle by a high electric field developed between the needle and the collector (Frenot and Chronakis 2003). That is to say, an electric charge develops on the surface of the first droplet coming out of the tip of the needle leading to higher surface tension. When the applied electrical field between the needle and the collector overcomes the surface tension of that first droplet, the ejection of the solution occurs, leading to instability during its traveling through the air to the collector, which results in a whipping-type flying (Sousa et al. 2021). During this traveling time and distance, the solvent evaporates, and the solidification of the polymer takes place, leading to a longer and thinner nanofibre (Shahriar et al. 2019).

Schematic representation of a traditional electrospinning system producing nanofibre loaded with AMP
Various exemplary physical and morphological characteristics of the constructed functional electrospun nanofibres (nanotubes/nanowires) such as high surface-to-volume ratio, high density of porosity and hollow diameter are subject to various elements, including the molecular weight and the concentration of the polymer; viscosity, conductivity and surface tension of the solution; the applied electric potential; the gauge and the angle of the fixed needle; the distance of the needle from the collector; and the feeding and releasing rate of the polymer (Frenot and Chronakis 2003; Peng et al. 2015; Ren et al. 2015; Shahriar et al. 2019; Sousa et al. 2020).
The incorporation of the AMPs into the electrospun nanofibre can be performed in different ways. In situ technique is a commonly used method to load AMPs into the drug delivery system by introducing them into the solution before forming the nanostructure (Truskewycz et al. 2021). Differently, AMPs can be adsorbed to the surface of the generated nanofibres, whereby they are actively attached to the polymeric system (Dart et al. 2019). The attachment of AMPs to the nanofibre’s surface can be obtained through (i) immersion of the nanofibre into a solution of the dissolved peptide until reaching a saturation point. This type of attachment occurs without generation of covalent bonding between the AMP and the surface (Sousa et al. 2021), (ii) grafting the AMP onto the surface via “graft-to” methodology by creating a covalent bonding that results in a stable and long-lasting attachment via a direct grafting of the AMP onto the surface of the nanofibre (Costa et al. 2011) or through “surface-initiated” strategy (Sousa et al. 2021). The “graft-to” process involves activation of the surface of the fibre through UV radiation and oxidation, which activates different functional groups such as carboxylic acid, amines, aldehydes or thiols leading to covalent bonding with the AMP (Felgueiras and Amorim 2017). In comparison, the “surface-initiated” strategy involves the attachment of the AMPs mediated by initiators covalently immobilised at the surface (Green et al. 2011).
In doing so, it is possible to create a compatible loaded vehicle with the biological system that can release the loaded AMP at the target site according to the chronospatial plan. However, to achieve the desired kinetics and the pharmacological effect of the AMP, it is essential to choose the appropriate combination of the solvent and the rest of the excipients based on the physicochemical characteristics of the employed AMP. Eriksen et al. have investigated the effect of the physicochemical properties of different molecules loaded into nanofibre material on their diffusion kinetics (Eriksen et al. 2013). They have tested the diffusion properties of a synthetic AMP named fluorescein-labelled inverse-Crabrolin (iCR-fluor) in comparison to the diffusion kinetics of tetracycline hydrochloride, where both were incorporated into the electrospun poly(ɛ-caprolactone) (PCL) nanofibres. The results showed that the release of iCR-fluor was only 40% compared to 85% of the tetracycline diffusion rate. Furthermore, the distribution density of the iCR-flour was less uniformed within the fibre compared to the distribution density of the tetracycline in the same nanofibre (Eriksen et al. 2013). The offered clarification by Eriksen and the group was based on the argument that the larger molecules of the iCR-flour were insoluble enough in the polymeric mix, which led to an integration of the AMP in the nanofibre structure followed by a significantly low rate of discharge from the fibre (Eriksen et al. 2013).
Just as significantly, unlike most of the nano-drug delivery systems, nanofibres can be utilised as a platform of more than one bioactive agent to be released either simultaneously or sequentially as each one of the bioactive agents is required along with the chronological development of the targeted medical conditions such as skin wound infections (Homaeigohar and Boccaccini 2020). Ahire and Dicks (2015) have successfully incorporated the antibiotic nisin and the 2,3-dihydroxybenzoic acid (DHBA) into nanofibre made of poly(D, L-lactide) and poly(ethylene oxide) that was diffused onto a biofilm formed by MRSA. They have demonstrated an apparent synergistic effect between the AMP and the DHBA against bacterial biofilm formation (Ahire and Dicks 2015). Furthermore, the different types of AMPs or bioactive agents can be loaded in different DDSs such as liposomes before incorporating them into the nanofibre for further protection (Xu et al. 2005). Alternatively, two or more syringes can coaxially produce multiple layers that result in a protective type of core-shell nanofibre (Calamak et al. 2017). Han et al. (2017) investigated the effect of the hygroscopic outer layer on the lifespan of the antimicrobial activity of the AMP nisin implanted in multiple syringes (Han et al. 2017). They found that nisin in a triaxial fibre membrane provides a more durable and sustainable antimicrobial activity that lasted over 7 days with over 99.99% of kills (4 log kills) compared to the coaxial fibre, which showed the exact duration of activity but with just 2 log cell death. The single nanofibre showed weaker antibacterial activity than the co- and obviously the triaxial, which lasted only for 1 day (Han et al. 2017).
1.11.5 Organic Polymer-Based Nanomaterials: Self-Assembled Peptides
Self-assembly is the process in which a spontaneous association of individual molecules or particles into a complex or functional structure through soft (non-covalent) chemical and physical interactions (Yadav et al. 2020). Peptides and proteins are amongst the first biological molecules, including carbohydrates, lipids and nucleic acids, to demonstrate the self-assembly phenomenon behind the cellular structure’s integrity (Misra et al. 2021). Hence, inventive material scientists were easily swayed to utilise this phenomenon in designing nano-blocks made of short peptides or amino acids for advanced DDSs. Researchers were invigorated by the diversified physicochemical properties of peptides and their inherited biocompatibility and biodegradability. Dipeptides and tripeptides have been identified to carry motifs with all the required information to compound sophisticated self-assembled nanostructures with unique functional characteristics such as mechanical rigidity, semi-conductivity, piezoelectricity and visible luminescence (Gazit 2015). Moreover, manipulating the self-assembled characteristics is attainable easily through a different assortment of the attached amino acids or by attaching different functional groups to the chain (Misra et al. 2021; Caruso et al. 2014). Nevertheless, peptides that are crafted from canonical amino acids are challenged by various issues concerning their stability and intramolecular interactions (Caruso et al. 2014).
Peptides, in general, are made of alpha-amino acids, and as such, they were called alpha-peptides which have a brief lifespan due to their high susceptibility to enzymatic degradation (Boto et al. 2018). Moreover, alpha-peptides are short chains, making it hard for them to form a stable secondary structure (Hecht and Huc 2007). Therefore, molecular engineering of durable DDSs for AMPs made of self-assembled peptides is still a convoluted process that requires a great deal of appreciation of the physicochemical characteristics of these small proteins. Notwithstanding, stable secondary structures made of alpha-helix, beta-sheet, beta-turn and beta-hairpin are determined by the sequence and the nature (canonical vs. non-canonical) of the amino acids and, eventually, play a pivotal role in the self-assembly process (Gopalan et al. 2015). Moreover, a prime arrangement for these secondary structures through soft chemical and physical interactions such as Van der Waals forces, hydrogen bonding or hydrophobic attraction can generate supramolecular design such as nanotubes, wormlike micelles, vesicles, fibrils and spherical micelles (Fig. 1.12) (Misra et al. 2021).

Schematic illustration of a self-assembled antimicrobial peptide forming various functional structures
This section will summarily discuss some of the different strategies adopted to counteract the significant elements upholding the poor quality of self-assembled peptides as a nano-DDS. Also, we will briefly shed the light on recent advances of this nanotechnology concerning the delivery of AMPs.
The instability of the natural alpha-peptides is an inherent property of the homochirality of the L-(alpha)amino acid enantiomer. It has been found that incorporation of the non-coded enantiomer (D-(alpha)amino), which possesses the same chemical and physical properties of the L structure but is rarely found in nature (in some venoms and antibiotics), enhances the peptide’s stability against proteolysis and prolongs its shelf-life significantly (Misra et al. 2021; Lu et al. 2020). Lu and co-workers have synthesised derivatives of a cationic AMP named Pep05 via the substitution of L-amino acid residues with the D structures, for example, D-arginine and D-lysine. They have revealed that the stability of the peptide made of the D structures of amino acids was enhanced significantly against trypsin and plasma proteases compared to the natural Pep05 (Lu et al. 2020). Moreover, formulation of a stable and self-assembled supramolecular structure or the so-called foldamer can be achieved through peptides comprising beta-amino acids (alpha-amino acids with an additional carbon atom) as an alternative to the natural alpha-amino acids (Gopalan et al. 2015). These beta-amino acids are regarded as “unnatural”, identified in mammals, living organisms and plants (Griffith 1986; Yasumoto and Satake 1998). Daura and co-researchers have found that beta-peptides are markedly chemically diversified and exhibit a more extraordinary secondary structural motif, resulting in higher resistance to enzymatic degradation (Daura et al. 1997).
Formulation of a unique functional architecture such as a vesicle, or a vesicular nanofibre, through the peptide’s self-assembly mechanism depends on the peptide’s amphiphilic nature and the specific secondary structure of that peptide (Rughani and Schneider 2008). For example, beta-hairpin and beta-sheet secondary structures lead to the formation of the fibrillar bilayers and higher-order laminates (Dong et al. 2007; Rughani and Schneider 2008). These secondary structures can be facilitated by combining D-proline with L-proline or glycine in the constructed peptide (Misra et al. 2021; Mahalakshmi and Balaram 2006).
In closing the formulation part, the advancement of the supermolecular assembly of peptides has experienced enormous input in recent years through the introduction of different alterations such as the incorporation of 3,4-dihydroxyl-L-phenylalanine, dehydro amino acids and 2-amino isobutyric acid to the peptide molecules, in addition to the introduction of gamma and δ-peptides and their hybrid analogues which have been reviewed in great details by Misra et al. (2021).
Another topic to highlight here is the toxicity which is the major hurdle in using AMPs as they may exert their cytotoxic activity against mammal cells upon interaction with the cell membrane (Hollmann et al. 2018). The cytotoxic effect of AMPs on mammal cells is preliminary based on configurational measures of the peptide that contribute to selective interactive forces with the eukaryotic cells (Ebenhan et al. 2014). Integration of such AMPs into self-assembly structures forming supramolecular nanofibres ought to alter their configurations, hindering their toxic effects by preventing them from interacting with eukaryotic cells (Chen et al. 2019). Chen and co-researchers have utilised the AMP melittin as a self-assembled natural model. They reported a dramatic change in its conformation when presented on the nanofibre surface, and consequently, the permeability of the bacterial and the mammalian cell membrane was modified (Chen et al. 2019). Precisely, positioning the melittin on the nanofibre’s surface has impacted the degree of freedom of the hydrophobic residues, which, in turn, has reduced its hydrophobic attractions to the lipids in the mammalian cells. Improving the selectivity of AMPs and enhancing the targeted cell responsiveness are examples illustrating the significance of the role of peptide self-assembly on the functionality of AMPs (Tian et al. 2015).
Finally, confining AMPs within a rigid supramolecular polymeric scaffold could introduce great features to them in terms of responsiveness, controlled release, enhancement of stability and lifespan and reduction of toxicity. Moreover, the peptide self-assembly engineering technique could be one of the promising methodologies in nanotechnology to promote AMPs for different therapeutic applications.
1.12 Conclusions
The relationship between humankind and microbes went through many hurdles since the early days of coexistence; however, it was balanced up to a hundred years ago under the coevolution laws. This equilibrium was disrupted after discovering antibiotics arguing for the predominance of the human species that won the war over an infectious disease. However, the lack of fundamental knowledge of the microbial world and the main aim of pursuing financial benefits led to the emergence of stubborn resistance against the indiscriminate and massive assault of the “magic bullet,” the antibiotics, teaching us a valuable lesson about other life forms on this planet. Over time, prokaryotes and eukaryotes have gone through countless evolutionary processes that support their survival and mutual relationships. AMPs are one of the yields of these protective mechanisms that have been developed, and we can take advantage of their availability to communicate precisely and gently with microbial infestations. They can be utilised as an alternative treatment for conventional antibiotics; nevertheless, they are too sensitive to handle complex biological systems abundant in physical and biological impediments. Advances in biomaterial sciences over the last 50 years allow the design of sophisticated biocompatible, biodegradable and chronospatial, nano-controlled drug delivery systems to carry AMPs and deliver them bioactively to the molecular level of the targeted microorganism cell at different physiological compartments of the eukaryotic biological system for effective remediation and self-healing.
References
Abbasi Z, Saeed W, Shah SM, Shahzad SA, Bilal M, Khan AF, Shaikh AJ (2021) Binding efficiency of functional groups towards noble metal surfaces using graphene oxide – metal nanoparticle hybrids. Colloids Surf A Physicochem Eng Asp 611:125858. https://doi.org/10.1016/j.colsurfa.2020.125858
Ahire JJ, Dicks LM (2015) Nisin incorporated with 2, 3-dihydroxybenzoic acid in nanofibers inhibits biofilm formation by a methicillin-resistant strain of Staphylococcus aureus. Probiotics Antimicrob Proteins 7(1):52–59. https://doi.org/10.1007/s12602-014-9171-5
Ahmed EM (2015) Hydrogel: preparation, characterization, and applications: a review. J Adv Res 6(2):105–121. https://doi.org/10.1016/j.jare.2013.07.006
Ahmed EM, Aggor FS, Awad AM, El-Aref AT (2013) An innovative method for preparation of nanometal hydroxide superabsorbent hydrogel. Carbohydr Polym 91(2):693–698. https://doi.org/10.1016/j.carbpol.2012.08.056
Ainsworth GC, Annie MB, Brownlee G (1947) ‘Aerosporin’, an antibiotic produced by Bacillus aerosporus Greer. Nature 160(4060):263. https://doi.org/10.1038/160263a0
Akashi K, Miyata H, Itoh H, Kinosita K (1996) Preparation of giant liposomes in physiological conditions and their characterization under an optical microscope. Biophys J 71(6):3242–3250. https://doi.org/10.1016/S0006-3495(96)79517-6
Akbarzadeh A, Rezaei-Sadabady R, Davaran S, Joo SW, Zarghami N, Hanifehpour Y, Samiei M, Kouhi M, Nejati-Koshki K (2013) Liposome: classification, preparation, and applications. Nanoscale Res Lett 8(1):102. https://doi.org/10.1186/1556-276X-8-102
Akesson P, Sjöholm AG, Björck L (1996) Protein SIC, a novel extracellular protein of streptococcus pyogenes interfering with complement function. J Biol Chem 271(2):1081–1088. https://doi.org/10.1074/jbc.271.2.1081
Akhtar MF, Hanif M, Ranjha NM (2016) Methods of synthesis of hydrogels … a review. Saudi Pharm J 24(5):554–559. https://doi.org/10.1016/j.jsps.2015.03.022
Alam M, Islam M, Wahab A, Billah M (2019) Antimicrobial resistance crisis and combating approaches. J Med 20(1):38–45. https://doi.org/10.3329/jom.v20i1.38842
Alexis F, Pridgen E, Molnar LK, Farokhzad OC (2008) Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm 5(4):505–515. https://doi.org/10.1021/mp800051m
Allen NE, Nicas TI (2003) Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol Rev 26(5):511–532. https://doi.org/10.1111/j.1574-6976.2003.tb00628.x
Almansour NM, Pirogova E, Coloe PJ, Cosic I, Istivan TS (2012) A bioactive peptide analogue for myxoma virus protein with a targeted cytotoxicity for human skin cancer in vitro. J Biomed Sci 19(1). https://doi.org/10.1186/1423-0127-19-65
Almeida PF, Ladokhin AS, White SH (2012) Hydrogen-bond energetics drive helix formation in membrane interfaces. BBA-Biomembranes 1818(2):178–182. https://doi.org/10.1016/j.bbamem.2011.07.019
Aminov R (2017) History of antimicrobial drug discovery: major classes and health impact. Biochem Pharmacol 133(C):4–19. https://doi.org/10.1016/j.bcp.2016.10.001
Aminov RI (2010) A brief history of the antibiotic era: lessons learned and challenges for the future. Front Microbiol 1:134–134. https://doi.org/10.3389/fmicb.2010.00134
Anderson W (2004) Natural histories of infectious disease: ecological vision in twentieth-century biomedical science. Osiris 19:39–61. https://doi.org/10.1086/649393
Armstrong GL, Conn LA, Pinner RW (1999) Trends in infectious disease mortality in the United States during the 20th century. JAMA 281(1):61–66. https://doi.org/10.1001/jama.281.1.61
Ashima KB, Kittappa V, Neha R (2013) Bacterial quorum sensing inhibitors: attractive alternatives for control of infectious pathogens showing multiple drug resistance. Recent Pat Antiinfect Drug Discov 8(1):68–83. https://doi.org/10.2174/1574891X11308010012
Attia MF, Anton N, Wallyn J, Omran Z, Vandamme TF (2019) An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J Pharm Pharmacol 71(8):1185–1198. https://doi.org/10.1111/jphp.13098
Aulton ME, Aulton ME, Taylor K (2013) Aulton's pharmaceutics : the design and manufacture of medicines, London, Edinburgh. https://doi.org/10.3109/9780203014356-17. New York, Elsevier Health Sciences UK
Bahar A, Ren D (2013) Antimicrobial Peptides. Pharmaceuticals 6(12):1543–1575. https://doi.org/10.3390/ph6121543
Bainton DF (1981) The discovery of lysosomes. J Cell Biol 91(3):66s–76s. https://doi.org/10.1083/jcb.91.3.66s
Band VI, Weiss DS (2015) Mechanisms of antimicrobial peptide resistance in gram-negative bacteria. Antibiotics (Basel, Switzerland) 4(1):18–41. https://doi.org/10.3390/antibiotics4010018
Barhoum A, Bechelany M, Makhlouf ASH (2019) Handbook of nanofibers. Springer
Barzegar-Jalali M (2008) Kinetic analysis of drug release from nanoparticles. J Pharm Pharm Sci 11(1):167–177. https://doi.org/10.18433/J3D59T
Bassett EJ, Keith MS, Armelagos GJ, Martin DL, Villanueva AR (1980) Tetracycline-labeled human bone from ancient Sudanese Nubia (A.D. 350). Science 209(4464):1532–1534
Batchelor FR, Chain EB, Hardy TL, Mansford KRL, Rolinson GN (1961) 6-Aminopenicillanic acid III. Isolation and purification. Royal Soc 154(957):498–508. https://doi.org/10.1098/rspb.1961.0047
Batchelor FR, Doyle FP, Nayler JHC, Rolinson GN (1959) Synthesis of penicillin: 6-Aminopenicillanic acid in penicillin fermentations. Nature 183(4656):257–258. https://doi.org/10.1038/183257b0
Bellamy W, Takase M, Wakabayashi H, Kawase K, Tomita M (1992) Antibacterial spectrum of lactoferricin-b, a potent bactericidal peptide derived from the n-terminal region of bovine lactoferrin. J Appl Bacteriol 73(6):472–479. https://doi.org/10.1111/j.1365-2672.1992.tb05007.x
Benedict R (1953) Antibiotics produced by actinomycetes. Bot Rev 19(5):229–320. https://www.jstor.org/stable/4353501
Bentley R (2005) The development of penicillin: genesis of a famous antibiotic. Perspect Biol Med 48:444–452. https://doi.org/10.1353/pbm.2005.0068
Bertelsen K, Dorosz J, Hansen SK, Nielsen NC, Vosegaard T (2012) Mechanisms of peptide-induced pore formation in lipid bilayers investigated by oriented 31P solid-state NMR spectroscopy. PLoS One 7(10):e47745. https://doi.org/10.1371/journal.pone.0047745
Bo G (2000) Giuseppe Brotzu and the discovery of cephalosporins. Clin Microbiol Infect 6(S3):6–8. https://doi.org/10.1111/j.1469-0691.2000.tb02032.x
Bodnar RJ (2013) Epidermal growth factor and epidermal growth factor receptor: the Yin and Yang in the treatment of cutaneous wounds and cancer. Adv Wound Care 2(1):24–29. https://doi.org/10.1089/wound.2011.0326
Boge L, Bysell H, Ringstad L, Wennman D, Umerska A, Cassisa V, Eriksson J, Joly-Guillou M-L, Edwards K, Andersson M (2016) Lipid-based liquid crystals as carriers for antimicrobial peptides: phase behavior and antimicrobial effect. Langmuir 32(17):4217–4228. https://doi.org/10.1021/acs.langmuir.6b00338
Boman HG (2003) Antibacterial peptides: basic facts and emerging concepts. J Intern Med 254(3):197–215. https://doi.org/10.1046/j.1365-2796.2003.01228.x
Boto A, Pérez de la Lastra JM, González CC (2018) The road from host-defense peptides to a new generation of antimicrobial drugs. Molecules 23(2):311. https://doi.org/10.3390/molecules23020311
Bozzuto G, Molinari A (2015) Liposomes as nanomedical devices. Int J Nanomedicine 10:975–999. https://doi.org/10.2147/IJN.S68861
Braga RM, Dourado MN, Araújo WL (2016) Microbial interactions: ecology in a molecular perspective. Br J Microbiol 47 Suppl 1(Suppl 1):86–98. https://doi.org/10.1016/j.bjm.2016.10.005
Burnet FM (1941) Biological aspects of infectious disease. Nature 147(3716):74–74. https://doi.org/10.1038/147074a0
Calamak S, Shahbazi R, Eroglu I, Gultekinoglu M, Ulubayram K (2017) An overview of nanofiber-based antibacterial drug design. Expert Opin Drug Discovery 12(4):391–406. https://doi.org/10.1080/17460441.2017.1290603
Caló E, Khutoryanskiy VV (2015) Biomedical applications of hydrogels: a review of patents and commercial products. Eur Polym J 65:252–267. https://doi.org/10.1016/j.eurpolymj.2014.11.024
Campos MA, Vargas MA, Regueiro V, Llompart CM, Albertí S, Bengoechea JA (2004) Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect Immun 72(12):7107–7114. https://doi.org/10.1128/IAI.72.12.7107-7114.2004
Caputo F, Clogston J, Calzolai L, Rösslein M, Prina-Mello A (2019) Measuring particle size distribution of nanoparticle enabled medicinal products, the joint view of EUNCL and NCI-NCL. A step by step approach combining orthogonal measurements with increasing complexity. J Control Release 299:31–43. https://doi.org/10.1016/j.jconrel.2019.02.030
Carmona-Ribeiro AM, de Melo Carrasco LD (2014) Novel formulations for antimicrobial peptides. Int J Mol Sci 15(10):18040–18083. https://doi.org/10.3390/ijms151018040
Carugo D, Bottaro E, Owen J, Stride E, Nastruzzi C (2016) Liposome production by microfluidics: potential and limiting factors. Sci Rep 6:25876–25876. https://doi.org/10.1038/srep25876
Caruso M, Gatto E, Placidi E, Ballano G, Formaggio F, Toniolo C, Zanuy D, Alemán C, Venanzi M (2014) A single-residue substitution inhibits fibrillization of Ala-based pentapeptides. A spectroscopic and molecular dynamics investigation. Soft Matter 10:2508–2519. https://doi.org/10.1039/C3SM52831F
Chabanel A, Flamm M, Sung KL, Lee MM, Schachter D, Chien S (1983) Influence of cholesterol content on red cell membrane viscoelasticity and fluidity. Biophys J 44(2):171–176. https://doi.org/10.1016/S0006-3495(83)84288-X
Chan DI, Prenner EJ, Vogel HJ (2006) Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. BBA-Biomembranes 1758(9):1184–1202. https://doi.org/10.1016/j.bbamem.2006.04.006
Charalampous FC (1979) Levels and distributions of phospholipids and cholesterol in the plasma membrane of neuroblastoma cells. Biochim Biophys Acta 556(1):38–51. https://doi.org/10.1016/0005-2736(79)90417-6
Chaudhry H, Zhou J, Zhong Y, Ali MM, McGuire F, Nagarkatti PS, Nagarkatti M (2013) Role of cytokines as a double-edged sword in sepsis. In vivo (Athens, Greece) 27(6):669–684
Chavanpatil MD, Jain P, Chaudhari S, Shear R, Vavia PR (2006) Novel sustained release, swellable and bioadhesive gastroretentive drug delivery system for ofloxacin. Int J Pharm 316(1–2):86–92. https://doi.org/10.1016/j.ijpharm.2006.02.038
Chen W, Yang S, Li S, Lang JC, Mao C, Kroll P, Tang L, Dong H (2019) Self-assembled peptide nanofibers display natural antimicrobial peptides to selectively kill bacteria without compromising cytocompatibility. ACS Appl Mater Interfaces 11(32):28681–28689. https://doi.org/10.1021/acsami.9b09583
Chen Y, Yang X, Zeckel M, Killian C, Hornbuckle K, Regev A, Voss S (2011) Risk of hepatic events in patients treated with vancomycin in clinical studies. Drug Saf 34(1):73–82. https://doi.org/10.2165/11539560-000000000-00000
Chenthamara D, Subramaniam S, Ramakrishnan SG, Krishnaswamy S, Essa MM, Lin F-H, Qoronfleh MW (2019) Therapeutic efficacy of nanoparticles and routes of administration. Biomater Res 23(1):20. https://doi.org/10.2165/11539560-000000000-00000
Cherkasov A, Jankovic B (2004) Application of ‘inductive’ QSAR descriptors for quantification of antibacterial activity of cationic polypeptides. Molecules (Basel, Switzerland) 9:1034–1052. https://doi.org/10.3390/91201034
Chiang W-H, Ho VT, Huang W-C, Huang Y-F, Chern C-S, Chiu H-C (2012) Dual stimuli-responsive polymeric hollow nanogels designed as carriers for intracellular triggered drug release. Langmuir 28(42):15056–15064. https://doi.org/10.1021/la302903v
Chithrani BD, Chan WC (2007) Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett 7(6):1542–1550. https://doi.org/10.1021/nl070363y
Chizhik U (2014) Cephalosporins. Pediatr Parents 30(7/8):22. https://doi.org/10.1351/goldbook.c00939
Coetzee D, Venkataraman M, Militky J, Petru M (2020) Influence of nanoparticles on thermal and electrical conductivity of composites. Polymers 12(4):742. https://doi.org/10.3390/polym12040742
Cook M, Molto E, Anderson C (1989) Fluorochrome labelling in roman period skeletons from dakhleh oasis, Egypt. Am J Phys Anthropol 80(2):137–143. https://doi.org/10.1002/ajpa.1330800202
Cooper RA (1978) Influence of increased membrane cholesterol on membrane fluidity and cell function in human red blood cells. J Supramol Struct 8(4):413–430. https://doi.org/10.1002/jss.400080404
Cosic I (1994) Macromolecular bioactivity: is it resonant interaction between macromolecules?—theory and applications. IEEE Trans Biomed Eng 41(12):1101–1114. https://doi.org/10.1109/10.335859
Ćosić I, Pirogova E, Vojisavljević V, Fang QJFT (2006) Electromagnetic properties of biomolecules. FME Trans 34(2):71–80. https://doi.org/10.1149/ma2006-02/24/1204
Costa F, Carvalho I, Montelaro R, Gomes P, Martins M (2011) Covalent immobilization of antimicrobial peptides (AMPs) onto biomaterial surfaces. Acta Biomater 7(4):1431–1440. https://doi.org/10.1016/j.actbio.2010.11.005
Costerton J, Stewart P, Greenberg E (1999) Bacterial biofilms: a common cause of persistent infections. Science (Washington) 284(5418):1318–1322. https://doi.org/10.1126/science.284.5418.1318
Damiati S, Kompella UB, Damiati SA, Kodzius R (2018) Microfluidic devices for drug delivery systems and drug screening. Genes 9(2):103. https://doi.org/10.3390/genes9020103
Daniel TM (2005) Selman Abraham Waksman and the discovery of streptomycin Founders of Our Knowledge. Int J Tuberc Lung Dis 9(2):120–122. https://doi.org/10.1111/j.1651-2227.2007.00182.x
Dart A, Bhave M, Kingshott P (2019) Antimicrobial peptide-based electrospun fibers for wound healing applications. Macromol Biosci 19(9):1800488. https://doi.org/10.1002/mabi.201800488
Dathe M, Wieprecht T (1999) Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim Biophys Acta Biomembr 1462(1):71–87. https://doi.org/10.1016/S0005-2736(99)00201-1
Daura X, van Gunsteren WF, Rigo D, Jaun B, Seebach D (1997) Studying the stability of a helical β-heptapeptide by molecular dynamics simulations. Chem Eur J 3(9):1410–1417. https://doi.org/10.1002/chem.19970030907
Davidson AL, Chen J (2004) ATP-binding cassette transporters in bacteria. Annu Rev Biochem 73:241. https://doi.org/10.1146/annurev.biochem.73.011303.073626
Davidson AL, Dassa E, Orelle C, Chen J (2008) Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev 72(2):317. https://doi.org/10.1128/MMBR.00031-07
Davies J, Davies D (2010) Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 74(3):417–433. https://doi.org/10.1128/MMBR.00016-10
Davies JS (2003) The cyclization of peptides and depsipeptides. J Peptide Sci 9:471–501. https://doi.org/10.1002/psc.491
Deamer D, Bangham AD (1976) Large volume liposomes by an ether vaporization method. Biochim Biophys Acta 443(3):629–634. https://doi.org/10.1016/0005-2787(76)90527-X
Devine D, Marsh P, Percival R, Rangarajan M, Curtis M (1999) Modulation of antibacterial peptide activity by products of Porphyromonas gingivalis and Prevotella spp. Microbiology 145(Pt 4):965–971. https://doi.org/10.1099/13500872-145-4-965
Dhople V, Krukemeyer A, Ramamoorthy A (2006) The human beta-defensin-3, an antibacterial peptide with multiple biological functions. BBA-Biomembranes 1758(9):1499–1512. https://doi.org/10.1016/j.bbamem.2006.07.007
Di Stefano AFD, Rusca A, Loprete L, Droge MJ, Moro L, Assandri A (2011) Systemic absorption of rifamycin SV MMX administered as modified-release tablets in healthy volunteers. Antimicrob Agents Chemother 55(5):2122. https://doi.org/10.1128/AAC.01504-10
Diamond G, Beckloff N, Weinberg A, Kisich KO (2009) The roles of antimicrobial peptides in innate host defense. Curr Pharm Des 15(21):2377–2392. https://doi.org/10.2174/138161209788682325
Dinos GP, Athanassopoulos CM, Missiri D, Giannopoulou PC, Vlachogiannis I, Papadopoulos G, Papaioannou D, Kalpaxis DL (2016) Chloramphenicol derivatives as antibacterial and anticancer agents: historic problems and current solutions. Antibiotics (Basel) 5(2). https://doi.org/10.3390/antibiotics5020020
Dong H, Paramonov SE, Aulisa L, Bakota EL, Hartgerink JD (2007) Self-assembly of multidomain peptides: balancing molecular frustration controls conformation and nanostructure. J Am Chem Soc 129(41):12468–12472. https://doi.org/10.1021/ja072536r
Dong YD, Tchung E, Nowell C, Kaga S, Leong N, Mehta D, Kaminskas LM, Boyd BJ (2019) Microfluidic preparation of drug-loaded PEGylated liposomes, and the impact of liposome size on tumour retention and penetration. J Liposome Res 29(1):1–9. https://doi.org/10.1080/08982104.2017.1391285
Dubos RJ (1939) Studies on a bactericidal agent extracted from a soil bacillus: II. Protective effect of the bactericidal agent against experimental pieuococcus infections in mice. J Exp Med 70(1):11–18. https://doi.org/10.1084/jem.70.1.11
Dubos RJ, Hotchkiss RD (1941) the production of bactericidal substances by Aerobic Sporulating Bacilli. J Exp Med 73(5):629–640. https://doi.org/10.1084/jem.73.5.629
Duggar BM (1948) Aureomycin: a product of the continuing search for new antibiotics. Ann N Y Acad Sci 51(2):177–181. https://doi.org/10.1111/j.1749-6632.1948.tb27262.x
Ebenhan T, Gheysens O, Kruger HG, Zeevaart JR, Sathekge MM (2014) Antimicrobial peptides: their role as infection-selective tracers for molecular imaging. Biomed Res Int 2014:867381–867381. https://doi.org/10.1155/2014/867381
Ehrlich J, Gottlieb D, Burkholder PR, Anderson LE, Pridham TG (1948) Streptomyces venezuelae, n. sp., the source of chloromycetin. J Bacteriol 56(4):467–477. https://doi.org/10.1128/jb.56.4.467-477.1948
Eichenbaum GM, Kiser PF, Simon SA, Needham D (1998) pH and ion-triggered volume response of anionic hydrogel microspheres. Macromolecules 31(15):5084–5093. https://doi.org/10.1021/ma970897t
Elliott R (2010) Combination cancer immunotherapy “expanding Paul Ehrlich's magic bullet concept”. Surg Oncol 21:53–55. https://doi.org/10.1016/j.suronc.2010.02.002
Endo Y, Tani T, Kodama M (1987) Antimicrobial activity of tertiary amine covalently bonded to a polystyrene fiber. Appl Environ Microbiol 53(9):2050. https://doi.org/10.1128/aem.53.9.2050-2055.1987
Engelbert M, Mylonakis E, Ausubel FM, Calderwood SB, Gilmore MS (2004) Contribution of gelatinase, serine protease, and fsr to the pathogenesis of enterococcus faecalis endophthalmitis. Infect Immun 72(6):3628. https://doi.org/10.1128/IAI.72.6.3628-3633.2004
Epand RM (2016) Host defense peptides and their potential as therapeutic agents. Springer, Cham. https://doi.org/10.1007/978-3-319-32949-9
Eriksen THB, Skovsen E, Fojan P (2013) Release of antimicrobial peptides from electrospun nanofibres as a drug delivery system. J Biomed Nanotechnol 9(3):492–498. https://doi.org/10.1166/jbn.2013.1553
Ernst C, Staubitz P, Mishra N, Yang S-J, Hornig G, Kalbacher H, Bayer A, Kraus D, Peschel A (2009) The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog 5:e1000660. https://doi.org/10.1371/journal.ppat.1000660
Eteraf-Oskouei T, Najafi M (2013) Traditional and modern uses of natural honey in human diseases: a review. Iran J Basic Med Sci 16(6):731–742. https://doi.org/10.34172/apb.2022.026
Evans ME, Feola DJ, Rapp RP (1999) Polymyxin B sulfate and colistin: old antibiotics for emerging multiresistant gram-negative bacteria. Ann Pharmacother 33:960–967. 10.1345%2Faph.18426
Falagas ME, Grammatikos AP, Michalopoulos A (2008) Potential of old-generation antibiotics to address current need for new antibiotics. Expert Rev Anti-Infect Ther 6(5):593–600. https://doi.org/10.1586/14787210.6.5.593
Falagas ME, Kasiakou SK (2005) Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin Infect Dis 40(9):1333–1341. https://doi.org/10.1086/429323
Falagas ME, Kasiakou SK (2006) Toxicity of polymyxins: a systematic review of the evidence from old and recent studies. Crit Care 10(1):R27–R27. https://doi.org/10.1186/cc3995
Falkinham JO III, Wall TE, Tanner JR, Tawaha K, Alali FQ, Li C, Oberlies NH (2009) Proliferation of antibiotic-producing bacteria and concomitant antibiotic production as the basis for the antibiotic activity of Jordan's red soils. Appl Environ Microbiol 75(9):2735. https://doi.org/10.1128/AEM.00104-09
Faye I, Lindberg BG (2016) Towards a paradigm shift in innate immunity-seminal work by Hans G. Boman and co-workers. Philos Trans Royal Soc Lond Ser B Biol Sci 371(1695):20150303. https://doi.org/10.1098/rstb.2015.0303
Fehlbaum P, Bulet P, Chernysh S, Briand JP, Roussel JP, Letellier L, Hetru C, Hoffmann JA (1996) Structure-activity analysis of thanatin, a 21-residue inducible insect defense peptide with sequence homology to frog skin antimicrobial peptides. Proc Natl Acad Sci U S A 93(3):1221. https://doi.org/10.1073/pnas.93.3.1221
Felgueiras HP, Amorim MTP (2017) Functionalization of electrospun polymeric wound dressings with antimicrobial peptides. Colloids Surf B: Biointerfaces 156:133–148. https://doi.org/10.1016/j.colsurfb.2017.05.001
Fernie-King BA, Seilly DJ, Lachmann PJ (2004) The interaction of streptococcal inhibitor of complement (SIC) and its proteolytic fragments with the human beta defensins. Immunology 111(4):444–452. https://doi.org/10.1111/j.0019-2805.2004.01837.x
Finlay AC, Hobby GL, P'An SY, Regna PP, Routien JB, Seeley DB, Shull GM, Sobin BA, Solomons IA, Vinson JW, Kane JH (1950) Terramycin, a new antibiotic. Science 111(2874):85. https://doi.org/10.1126/science.111.2874.85
Fleming A (1929) On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzæ. Br J Exp Pathol 10(3):226–236
Fleming A (1932) Lysozyme. Proc R Soc Med 26(2):71–84. https://doi.org/10.1177/003591573202600201
Fratoddi I (2017) Hydrophobic and hydrophilic Au and Ag nanoparticles. Breakthroughs and perspectives. Nanomaterials (Basel) 8(1). https://doi.org/10.3390/nano8010011
Frenot A, Chronakis IS (2003) Polymer nanofibers assembled by electrospinning. Curr Opin Colloid Interface Sci 8(1):64–75. https://doi.org/10.1016/S1359-0294(03)00004-9
Frick I-M, Akesson P, Rasmussen M, Schmidtchen A, Björck L (2003) SIC, a secreted protein of streptococcus pyogenes that inactivates antibacterial peptides. J Biol Chem 278(19):16561–16566. https://doi.org/10.1074/jbc.M301995200
Gauze GF (1934) The struggle for existence. The Williams & Wilkins Company. https://doi.org/10.5962/bhl.title.4489
Gazit E (2015) Searching sequence space. Nat Chem 7(1):14–15. https://doi.org/10.1038/nchem.2140
Gennaro R, Zanetti M, Benincasa M, Podda E, Miani M (2002) Pro-rich antimicrobial peptides from animals: structure, biological functions and mechanism of action. Curr Pharm Des 8(9):763–778. https://doi.org/10.2174/1381612023395394
Gericke M, Pinches A (2006) Biological synthesis of metal nanoparticles. Hydrometallurgy 83(1–4):132–140. https://doi.org/10.1016/j.hydromet.2006.03.019
Ginsburg I (2002) The role of bacteriolysis in the pathophysiology of inflammation, infection and post-infectious sequelae. APMIS 110(11):753–770. https://doi.org/10.1034/j.1600-0463.2002.1101101.x
Goossens H, Ferech M, Vander Stichele R, Elseviers M (2005) Outpatient antibiotic use in Europe and association with resistance: a cross-national database study. Lancet 365(9459):579–587. https://doi.org/10.1016/S0140-6736(05)17907-0
Gopalan RD, Del Borgo MP, Mechler AI, Perlmutter P, Aguilar M-I (2015) Geometrically precise building blocks: the self-assembly of β-peptides. Chem Biol 22(11):1417–1423. https://doi.org/10.1016/j.chembiol.2015.10.005
Gordon YJ, Romanowski EG, McDermott AM (2005) Mini review: a review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr Eye Res 30(7):505–515. https://doi.org/10.1080/2F02713680590968637
Gottlieb D, Carter HE, Legator M, Gallicchio V (1954) The biosynthesis of chloramphenicol I.: precursors stimulating the synthesis. J Bacteriol 68(2):243. https://doi.org/10.1128/jb.68.2.243-251.1954
Gottlieb D, Robbins PW, Carter HE (1956) The biosynthesis of chloramphenicol: II. Acetylation of p-nitrophenylserinol. J Bacteriol 72(2):153. https://doi.org/10.1128/jb.72.2.153-156.1956
Gould K (2016) Antibiotics: from prehistory to the present day. J Antimicrob Chemother 71(3):572–575. https://doi.org/10.1093/jac/dkv484
Green J-B, Fulghum T, Nordhaus M (2011) Immobilized antimicrobial agents: a critical perspective. 109:84–98. https://doi.org/10.1116/1.3645195
Griffith OW (1986) Β-amino acids: mammalian metabolism and utility as α-amino acid analogues. Ann Rev Biochem 55(1):855–878. https://doi.org/10.1146/annurev.bi.55.070186.004231
Grobbelaar M, Louw GE, Sampson SL, van Helden PD, Donald PR, Warren RM (2019) Evolution of rifampicin treatment for tuberculosis. Infect Genet Evol 74. https://doi.org/10.1016/j.meegid.2019.103937
Groisman EA, Parra-Lopez C, Salcedo M, Lipps CJ, Heffron F (1992) Resistance to host antimicrobial peptides is necessary for Salmonella virulence. Proc Natl Acad Sci U S A 89(24):11939. https://doi.org/10.1073/pnas.89.24.11939
Groo A-C, Matougui N, Umerska A, Saulnier P (2018) Reverse micelle-lipid nanocapsules: a novel strategy for drug delivery of the plectasin derivate AP138 antimicrobial peptide. Int J Nanomedicine 13:7565–7574. https://doi.org/10.2147/ijn.s180040
Gross M (2013) Antibiotics in crisis. Curr Biol 23(24):R1063–R1065. https://doi.org/10.1016/j.cub.2013.11.057
Gudmundsson G, Magnusson K, Chowdhary B, Johansson M, Andersson L, Boman H (1995) Structure of the gene for porcine peptide antibiotic PR-39, a cathelin gene family member: comparative mapping of the locus for the human peptide antibiotic FALL-39. Proc Natl Acad Sci USA 92(15):7085–7089. https://doi.org/10.1073/2Fpnas.92.15.7085
Gupta S, Jain A, Chakraborty M, Sahni JK, Ali J, Dang S (2013) Oral delivery of therapeutic proteins and peptides: a review on recent developments. Drug Deliv 20(6):237–246. https://doi.org/10.3109/10717544.2013.819611
Han D, Sherman S, Filocamo S, Steckl AJ (2017) Long-term antimicrobial effect of nisin released from electrospun triaxial fiber membranes. Acta Biomater 53:242–249. https://doi.org/10.1016/j.actbio.2017.02.029
Hancock LE, Perego M (2004) The enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J Bacteriol 186(17):5629. https://doi.org/10.1128/jb.186.17.5629-5639.2004
Hancock REW (2001) Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect Dis 1(3):156–164. https://doi.org/10.1016/s1473-3099(01)00092-5
Hancock REW, Haney EF, Gill EE (2016) The immunology of host defence peptides: beyond antimicrobial activity. Nat Rev Immunol 16(5):321–334. https://doi.org/10.1038/nri.2016.29
Hancock REW, Rozek A (2002) Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol Lett 206:143–149. https://doi.org/10.1111/j.1574-6968.2002.tb11000.x
Handwerger S, Skoble J (1995) Identification of chromosomal mobile element conferring high-level vancomycin resistance in Enterococcus faecium. Antimicrob Agents Chemother 39(11):2446. https://doi.org/10.1128/aac.39.11.2446
Hart BL (2011) Behavioural defences in animals against pathogens and parasites: parallels with the pillars of medicine in humans. Philos Trans Royal Soc Lond Ser B Biol Sci 366(1583):3406–3417. https://doi.org/10.1098/2Frstb.2011.0092
Hassel C, Hafner C, Soberg R, Adelmann J, Haywood R (2002) Using Chinese medicine to understand medicinal herb quality: an alternative to biomedical approaches? J Agric Food Human Values Soc 19(4):337–347. https://doi.org/10.1023/A:1021144102102
Hayes D, Jensen H (2003) Lessons from the Danish Ban on Feed-Grade Antibiotics. IDEAS Working Paper Series from RePEc. https://doi.org/10.22004/ag.econ.93711
Hecht S, Huc I (2007) Foldamers: structure, properties and applications. Wiley. https://doi.org/10.1093/ww/9780199540884.013.u42774
Heilborn DJ, Margareta Frohm N, Gunnar K, Günther W, Ole S, Niels B, Mona S-B (2003) The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J Investig Dermatol 120(3):379. https://doi.org/10.1046/j.1523-1747.2003.12069.x
Helenius A, Fries E, Kartenbeck J (1977) Reconstitution of Semliki forest virus membrane. J Cell Biol 75(3):866–880. https://doi.org/10.1083/jcb.75.3.866
Hirakura Y, Kobayashi S, Matsuzaki K (2002) Specific interactions of the antimicrobial peptide cyclic β-sheet tachyplesin I with lipopolysaccharides. Biochim Biophys Acta Biomembr 1562(1):32–36. https://doi.org/10.1016/s0005-2736(02)00358-9
Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC (1997) Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother 40(1):135–136. https://doi.org/10.1093/jac/40.1.135
Hollmann A, Martinez M, Maturana P, Semorile LC, Maffia PC (2018) Antimicrobial peptides: interaction with model and biological membranes and synergism with chemical antibiotics. Front Chem 6:204–204. https://doi.org/10.3389/fchem.2018.00204
Holmes EC (2011) Plague's progress: the Black Death was one of the most devastating pandemics in human history. The first complete genome sequence of the causative Yersinia pestis bacterium provides a fresh perspective on plague evolution.(GENOMICS)(Report). Nature 478(7370):465. https://doi.org/10.1038/478465a
Homaeigohar S, Boccaccini AR (2020) Antibacterial biohybrid nanofibers for wound dressings. Acta Biomater 107:25–49. https://doi.org/10.1016/j.actbio.2020.02.022
Hu J, Peidaee P, Elshagmani E, Istivan T, Pirogova E (2013) The effects of synthetic azurocidin peptide analogue on staphylococcus aureus bacterium. In: 13th IEEE International Conference on BioInformatics and BioEngineering, pp 1–4. https://doi.org/10.1109/BIBE.2013.6701685
Huang Y, Gui S (2018) Factors affecting the structure of lyotropic liquid crystals and the correlation between structure and drug diffusion. RSC Adv 8:6978–6987. https://doi.org/10.1039/C7RA12008G
Hultmark D, Engström Å, Bennich H, Kapur R, Boman HG (1982) Insect immunity: isolation and structure of cecropin D and four minor antibacterial components from cecropia pupae. Eur J Biochem 127(1):207–217. https://doi.org/10.1111/j.1432-1033.1982.tb06857.x
Istivan TS, Pirogova E, Gan E, Almansour NM, Coloe PJ, Cosic I (2011) Biological effects of a de novo designed myxoma virus peptide analogue: evaluation of cytotoxicity on tumor cells. PLoS One 6(9). https://doi.org/10.1371/journal.pone.0024809
Jeevanandam J, Barhoum A, Chan YS, Dufresne A, Danquah MK (2018) Review on nanoparticles and nanostructured materials: history, sources, toxicity and regulations. Beilstein J Nanotechnol 9:1050–1074. https://doi.org/10.3762/2Fbjnano.9.98
Jenssen H, Fjell CD, Cherkasov A, Hancock REW (2008) QSAR modeling and computer-aided design of antimicrobial peptides. J Pept Sci 14(1):110–114. https://doi.org/10.1002/psc.908
Jenssen H, Gutteberg TJ, Lejon T (2005) Modelling of anti-HSV activity of lactoferricin analogues using amino acid descriptors. J Pept Sci 11(2):97–103. https://doi.org/10.1002/psc.908
Jenssen H, Lejon T, Hilpert K, Fjell CD, Cherkasov A, Hancock REW (2007) Evaluating different descriptors for model design of antimicrobial peptides with enhanced activity toward P. aeruginosa. Chem Biol Drug Des 70(2):134–142. https://doi.org/10.1111/j.1747-0285.2007.00543.x
Jerse AE, Sharma ND, Simms AN, Crow ET, Snyder LA, Shafer WM (2003) A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect Immun 71(10):5576. https://doi.org/10.1128/2FIAI.71.10.5576-5582.2003
Jiang M, Zhang H, Pfeifer BA (2013) The logic, experimental steps, and potential of heterologous natural product biosynthesis featuring the complex antibiotic erythromycin A produced through E. coli. J Vis Exp 71:e4346. https://doi.org/10.3791/4346
Jochum FD, Theato P (2013) Temperature- and light-responsive smart polymer materials. Chem Soc Rev 42(17):7468–7483. https://doi.org/10.1039/C2CS35191A
Jolivet-Gougeon A, Bonnaure-Mallet M (2014) Biofilms as a mechanism of bacterial resistance. Drug Discov Today Technol 11(1):49–56. https://doi.org/10.1016/j.ddtec.2014.02.003
Juretić D, Simunić J (2019) Design of α-helical antimicrobial peptides with a high selectivity index. Expert Opin Drug Discovery 14(10):1053–1063. https://doi.org/10.1080/17460441.2019.1642322
Kasapis S (2006) Definition and applications of the network glass transition temperature. Food Hydrocoll 20(2–3):218–228. https://doi.org/10.1016/j.foodhyd.2005.02.020
Kasapis S (2008) Recent advances and future challenges in the explanation and exploitation of the network glass transition of high sugar/biopolymer mixtures. Crit Rev Food Sci Nutr 48(2):185–203. https://doi.org/10.1080/10408390701286025
Khan I, Saeed K, Khan I (2019) Nanoparticles: properties, applications and toxicities. Arab J Chem 12(7):908–931. https://doi.org/10.1016/j.arabjc.2017.05.011
Kier B, Anderson J, Andersen N (2015) Disulfide-mediated beta-strand dimers: hyperstable beta-sheets lacking tertiary interactions and turns. J Am Chem Soc 137(16):5363–5371. https://doi.org/10.1021/ja5117809
Kirchhelle C (2018) Pharming animals: a global history of antibiotics in food production (1935–2017). Palgrave Commun 4(1):96. https://doi.org/10.1057/s41599-018-0152-2
Koczulla R, Degenfeld G, Kupatt C, Krötz F, Zahler S, Gloe T, Bieber K, Unterberger P, Zaiou M, Lebherz C, Karl A, Raake P, Pfosser A, Boekstegers P, Welsch U, Hiemstra P, Vogelmeier C, Gallo R, Clauss M, Bals R (2003) An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest 111:1665–1672. https://doi.org/10.1172/jci17545
Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA (1983) Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm 15(1):25–35. https://doi.org/10.1016/0378-5173(83)90064-9
Kościuczuk EM, Lisowski P, Jarczak J, Strzałkowska N, Jóźwik A, Horbańczuk J, Krzyżewski J, Zwierzchowski L, Bagnicka E (2012) Cathelicidins: family of antimicrobial peptides. A review. Mol Biol Rep 39(12):10957–10970. https://doi.org/10.1007/s11033-012-1997-x
Koynova R, Tenchov B (2015) Recent progress in liposome production, relevance to drug delivery and nanomedicine. Recent Patents Nanotechnol 9. https://doi.org/10.2174/187221050902150819151721
Kulkarni N, Muddapur U (2014) Biosynthesis of metal nanoparticles: a review. J Nanotechnol 2014:510246. https://doi.org/10.1155/2014/510246
Ladokhin AS, White SH (1999) Folding of amphipathic α-helices on membranes: energetics of helix formation by melittin. J Mol Biol 285(4):1363–1369. https://doi.org/10.1006/jmbi.1998.2346
Lai Y, Gallo RL (2009) AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol 30(3):131–141. https://doi.org/10.1016/2Fj.it.2008.12.003
Landers TF, Cohen B, Wittum TE, Larson EL (2012) A review of antibiotic use in food animals: perspective, policy, and potential. Public Health Rep (Washington, D.C. : 1974) 127(1):4–22. 10.1177%2F003335491212700103
Laporte DC, Rosenthal KS, Storm DR (1977) Inhibition of Escherichia coli growth and respiration by polymyxin B covalently attached to agarose beads. Biochemistry 16(8):1642–1648. https://doi.org/10.1021/bi00627a019
Lasic DD (1995) Mechanisms of liposome formation. J Liposome Res 5(3):431–441. https://doi.org/10.3109/08982109509010233
Lau YE, Rozek A, Scott MG, Goosney DL, Davidson DJ, Hancock REW (2005) Interaction and cellular localization of the human host defense peptide LL-37 with lung epithelial cells. Infect Immun 73(1):583–591. https://doi.org/10.1128/iai.73.1.583-591.2005
Lazăr V, Chifiriuc M (2010) Architecture and physiology of microbial biofilms. Roum Arch Microbiol Immunol 69:95–107. https://doi.org/10.1016/b978-0-12-381045-8.00008-4
Lazzaro BP, Zasloff M, Rolff J (2020) Antimicrobial peptides: application informed by evolution. Science 368(6490):eaau5480. https://doi.org/10.1126/science.aau5480
Leader B, Baca QJ, Golan DE (2008) Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov 7(1):21–39. https://doi.org/10.1038/nrd2399
Lederberg J (2000) Infectious history. Science (Washington) 288(5464):287–293. https://doi.org/10.1126/science.288.5464.287
Lee B, Park J, Ryu M, Kim S, Joo M, Yeom J-H, Kim S, Park Y, Lee K, Bae J (2017) Antimicrobial peptide-loaded gold nanoparticle-DNA aptamer conjugates as highly effective antibacterial therapeutics against Vibrio vulnificus. Sci Rep 7(1):13572. https://doi.org/10.1038/s41598-017-14127-z
Lee H-T, Lee C-C, Yang J-R, Lai JZC, Chang KY (2015) A large-scale structural classification of antimicrobial peptides. BioMed Res Inter 2015. https://doi.org/10.1155/2015/475062
Lehrer RI, Ganz T (1999) Antimicrobial peptides in mammalian and insect host defence. Curr Opin Immunol 11(1):23–27. https://doi.org/10.1016/s0952-7915(99)80005-3
Lei J, Sun L, Huang S, Zhu C, Li P, He J, Mackey V, Coy DH, He Q (2019) The antimicrobial peptides and their potential clinical applications. Am J Transl Res 11(7):3919–3931
Lester W (1972) Rifampin: a semisynthetic derivative of rifamycin-A prototype for the future. Annu Rev Microbiol 26(1):85–102. https://doi.org/10.1146/annurev.mi.26.100172.000505
Levine DP (2006) Vancomycin: a history. Clin Infect Dis 42(Supplement_1):S5–S12. https://doi.org/10.1086/491709
Li J, Mooney D (2016) Designing hydrogels for controlled drug delivery. Nat Rev Mater 1(12). https://doi.org/10.1038/natrevmats.2016.71
Li L-H, Ju T-C, Hsieh C-Y, Dong W-C, Chen W-T, Hua K-F, Chen W-J (2017) A synthetic cationic antimicrobial peptide inhibits inflammatory response and the NLRP3 inflammasome by neutralizing LPS and ATP. PLoS One 12(7):e0182057. https://doi.org/10.1371/journal.pone.0182057
Liston SD, McMahon SA, Le Bas A, Suits MDL, Naismith JH, Whitfield C (2018) Periplasmic depolymerase provides insight into ABC transporter-dependent secretion of bacterial capsular polysaccharides. Proc Natl Acad Sci U S A 115(21):E4870–E4879. https://doi.org/10.1073/pnas.1801336115
Liu D, Yang F, Xiong F, Gu N (2016) The smart drug delivery system and its clinical potential. Theranostics 6(9):1306–1323. https://doi.org/10.7150/thno.14858
Liu F, Myers AG (2016) Development of a platform for the discovery and practical synthesis of new tetracycline antibiotics. Curr Opin Chem Biol 32:48–57. https://doi.org/10.1016/j.cbpa.2016.03.011
Lobanovska M, Pilla G (2017) Penicillin's discovery and antibiotic resistance: lessons for the future? Yale J Biol Med 90(1):135–145. https://doi.org/10.2217/fmb.15.78
Lu J, Xu H, Xia J, Ma J, Xu J, Li Y, Feng J (2020) D- and unnatural amino acid substituted antimicrobial peptides with improved proteolytic resistance and their proteolytic degradation characteristics. Front Microbiol 11(2869). https://doi.org/10.3389/fmicb.2020.563030
Lung O, Kuo L, Wolfner MF (2001) Drosophila males transfer antibacterial proteins from their accessory gland and ejaculatory duct to their mates. J Insect Physiol 47(6):617–622. https://doi.org/10.1016/s0022-1910(00)00151-7
Magana M, Pushpanathan M, Santos AL, Leanse L, Fernandez M, Ioannidis A, Giulianotti MA, Apidianakis Y, Bradfute S, Ferguson AL, Cherkasov A, Seleem MN, Pinilla C, de la Fuente-Nunez C, Lazaridis T, Dai T, Houghten RA, Hancock REW, Tegos GP (2020) The value of antimicrobial peptides in the age of resistance. Lancet Infect Dis. https://doi.org/10.1016/s1473-3099(20)30327-3
Maggi N, Vigevani A, Gallo GG, Pasqualucci CR (1968) Acetyl migration in rifampicin. J Med Chem 11(5):936. https://doi.org/10.1021/jm00311a004
Mahalakshmi R, Balaram P (2006) Non-protein amino acids in the design of secondary structure scaffolds. Methods Mol Biol (Clifton, N.J.) 340:71–94. https://doi.org/10.1385/1-59745-116-9:71
Makowski M, Silva ÍC, do Amaral CP, Gonçalves S, Santos NC (2019) Advances in lipid and metal nanoparticles for antimicrobial peptide delivery. Pharmaceutics 11(11):588. https://doi.org/10.3390/pharmaceutics11110588
Malheiros PS, Cuccovia IM, Franco BDGM (2016) Inhibition of Listeria monocytogenes in vitro and in goat milk by liposomal nanovesicles containing bacteriocins produced by Lactobacillus sakei subsp. sakei 2a. Food Control 63:158–164. https://doi.org/10.1016/2Fj.foodcont.2015.11.037
Manikkath J, Parekh HS, Mutalik S (2020) Surface-engineered nanoliposomes with lipidated and non-lipidated peptide-dendrimeric scaffold for efficient transdermal delivery of a therapeutic agent: development, characterization, toxicological and preclinical performance analyses. Eur J Pharm Biopharm 156:97–113. https://doi.org/10.1016/j.ejpb.2020.09.001
Marshall J (1946) Penicillin. Nature 158(4022):772. https://doi.org/10.1038/158772a0
Martin KW, Ernst E (2003) Herbal medicines for treatment of bacterial infections: a review of controlled clinical trials. J Antimicrob Chemother 51(2):241–246. https://doi.org/10.1093/jac/dkg087
McBride SM, Sonenshein AL (2011) The dlt operon confers resistance to cationic antimicrobial peptides in Clostridium difficile. Microbiology (Reading, England) 157(Pt 5):1457–1465. https://doi.org/10.1099/2Fmic.0.045997-0
McIntosh J, Fildes P, Parker HB, Bulloch W, Sequeira JH (1912) Neosalvarsan. Lancet 180(4637):82–83. https://doi.org/10.1016/S0140-6736(01)41235-9
Mecke A, Lee D-K, Ramamoorthy A, Orr BG, Banaszak Holl MM (2005) Membrane thinning due to antimicrobial peptide binding: an atomic force microscopy study of MSI-78 in lipid bilayers. Biophys J 89(6):4043–4050. https://doi.org/10.1529/biophysj.105.062596
Menon R (2014) A review on various reasons for teeth discolouration. Res J Pharm Technol 7(7):815–820. https://doi.org/10.5958/0974-360X
Mika JT, Moiset G, Cirac AD, Feliu L, Bardají E, Planas M, Sengupta D, Marrink SJ, Poolman B (2011) Structural basis for the enhanced activity of cyclic antimicrobial peptides: the case of BPC194. BBA-Biomembranes 1808(9):2197–2205. https://doi.org/10.1016/j.bbamem.2011.05.001
Mirski T, Niemcewicz M, Bartoszcze M, Gryko R, Michalski A (2018) Utilisation of peptides against microbial infections – a review. Ann Agric Env Med 25(2):205–210. https://doi.org/10.26444/aaem/74471
Mishra A, Choi J, Moon E, Baek K (2018) Tryptophan-rich and proline-rich antimicrobial peptides. Molecules 23(4). https://doi.org/10.3390/molecules23040815
Misra R, Rudnick-Glick S, Adler-Abramovich L (2021) From folding to assembly: functional supramolecular architectures of peptides comprised of non-canonical amino acids. Macromol Biosci:2100090. https://doi.org/10.1002/mabi.202170020
Moghimi SM, Porter CJH, Muir IS, Illum L, Davis SS (1991) Non-phagocytic uptake of intravenously injected microspheres in rat spleen: influence of particle size and hydrophilic coating. Biochem Biophys Res Commun 177(2):861–866. https://doi.org/10.1016/0006-291x(91)91869-e
Mohanty S, Jena P, Mehta R, Pati R, Banerjee B, Patil S, Sonawane A (2013) Cationic antimicrobial peptides and biogenic silver nanoparticles kill mycobacteria without eliciting DNA damage and cytotoxicity in mouse macrophages. Antimicrob Agents Chemother 57(8):3688–3698. https://doi.org/10.1128/aac.02475-12
Mojsoska B, Jenssen H (2015) Peptides and peptidomimetics for antimicrobial drug design. Pharmaceuticals 8:366–415. https://doi.org/10.3390/2Fph8030366
Mok H, Jeong H, Kim S-J, Chung BH (2012) Indocyanine green encapsulated nanogels for hyaluronidase activatable and selective near infrared imaging of tumors and lymph nodes. Chem Commun (Camb) 48(69):8628. https://doi.org/10.1039/C2CC33555G
Monteiro N, Martins A, Reis RL, Neves NM (2014) Liposomes in tissue engineering and regenerative medicine. J R Soc Interface 11(101):20140459–20140459. https://doi.org/10.1098/rsif.2014.0459
Mootz HD, Marahiel MA (1997) The tyrocidine biosynthesis operon of Bacillus brevis: complete nucleotide sequence and biochemical characterization of functional internal adenylation domains. J Bacteriol 179(21):6843. https://doi.org/10.1128/jb.179.21.6843-6850.1997
Moravej H, Moravej Z, Yazdanparast M, Heiat M, Mirhosseini A, Moghaddam MM, Mirnejad R (2018) Antimicrobial peptides: features, action, and their resistance mechanisms in bacteria. Microbial Drug Resist (Larchmont, N.Y.) 24(6):747–767. https://doi.org/10.3389/fmicb.2020.593215
Moritz M, Geszke-Moritz M (2015) Recent developments in application of polymeric nanoparticles as drug carriers. Adv Clin Exp Med 24(5):749–758. https://doi.org/10.17219/acem/31802
Motta I, Gohlke A, Adrien V, Li F, Gardavot H, Rothman JE, Pincet F (2015) Formation of giant unilamellar proteo-liposomes by Osmotic Shock. Langmuir 31(25):7091–7099. https://doi.org/10.1021/2Facs.langmuir.5b01173
Muheem A, Shakeel F, Jahangir MA, Anwar M, Mallick N, Jain GK, Warsi MH, Ahmad FJ (2016) A review on the strategies for oral delivery of proteins and peptides and their clinical perspectives. Saudi Pharm J 24(4):413–428. https://doi.org/10.1016/j.jsps.2014.06.004
Muñoz-Carrillo JL, Contreras-Cordero JF, Gutiérrez-Coronado O, Villalobos-Gutiérrez PT, Ramos-Gracia LG, Hernández-Reyes VE (2018) Cytokine profiling plays a crucial role in activating immune system to clear infectious pathogens. In: Immune response activation and immunomodulation. IntechOpen. https://doi.org/10.5772/intechopen.80843
Murray J, Schraufnagel D, Hopewell P (2015) Treatment of tuberculosis: a historical perspective. Ann Am Thorac Soc 12(12):1749–1759. https://doi.org/10.1513/annalsats.201509-632ps
Nagaoka I, Hirota S, Niyonsaba F, Hirata M, Adachi Y, Tamura H, Heumann D (2001) Cathelicidin family of antibacterial peptides CAP18 and CAP11 inhibit the expression of TNF-α by blocking the binding of LPS to CD14(+) cells. J Immunol 167(6):3329–3338. https://doi.org/10.4049/jimmunol.167.6.3329
Nakayama J, Cao Y, Horii T, Sakuda S, Akkermans ADL, De Vos WM, Nagasawa H (2001) Gelatinase biosynthesis-activating pheromone: a peptide lactone that mediates a quorum sensing in enterococcus faecalis. Mol Microbiol 41(1):145–154. https://doi.org/10.1046/j.1365-2958.2001.02486.x
Nassar N, Whitehead F, Istivan T, Shanks R, Kasapis S (2021) Manipulation of the glass transition properties of a high-solid system made of acrylic acid-N,N′-methylenebisacrylamide copolymer grafted on hydroxypropyl methyl cellulose. Int J Mol Sci 22(5):2682. https://doi.org/10.3390/ijms22052682
Nature PG (1937) Prontosil in puerperal infections. Nature 140(3537):284. https://doi.org/10.1136/bmj.2.4005.695
Nature PG (1938) Prontosil. Nature 142(3594):533. https://doi.org/10.1038/142533a0
Nawrocki KL, Crispell EK, McBride SM (2014) Antimicrobial peptide resistance mechanisms of gram-positive bacteria. Antibiotics (Basel) 3(4):461–492. https://doi.org/10.3390/antibiotics3040461
Nekkanti V, Venkatesan N, Betageri G (2015) Proliposomes for oral delivery: progress and challenges. Curr Pharm Biotechnol 16. https://doi.org/10.2174/1389201016666150118134256
Nelson M, Levy SB (2011) The history of the tetracyclines. Ann NY Acad Sci 1241(1):17–32. https://doi.org/10.1111/j.1749-6632.2011.06354.x
Nelson ML, Dinardo A, Hochberg J, Armelagos GJ (2010) Brief communication: mass spectroscopic characterization of tetracycline in the skeletal remains of an ancient population from Sudanese Nubia 350–550 CE. Am J Phys Anthropol 143(1):151–154. https://doi.org/10.1002/ajpa.21340
Neuhaus FC, Baddiley J (2003) A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol Mol Biol Rev 67(4):686–723. https://doi.org/10.1128/mmbr.67.4.686-723.2003
Newman DJ, Cragg GM (2007) Natural products as sources of new drugs over the last 25 years. J Nat Prod 70(3):461–477. https://doi.org/10.1021/np068054v
Newton BA (1956) The properties and mode of action of the polymyxins. Bacteriol Rev 20(1):14–27. https://doi.org/10.1128/br.20.1.14-27.1956
Niikura K, Matsunaga T, Suzuki T, Kobayashi S, Yamaguchi H, Orba Y, Kawaguchi A, Hasegawa H, Kajino K, Ninomiya T, Ijiro K, Sawa H (2013) Gold nanoparticles as a vaccine platform: influence of size and shape on immunological responses in vitro and in vivo. ACS Nano 7(5):3926–3938. https://doi.org/10.1021/nn3057005
Niyonsaba F, Ushio H, Nakano N, Ng W, Sayama K, Hashimoto K, Nagaoka I, Okumura K, Ogawa H (2007) Antimicrobial peptides human β-defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J Investig Dermatol 127(3):594–604. https://doi.org/10.1038/sj.jid.5700599
Nusrath Unissa A, Hanna LE (2017) Molecular mechanisms of action, resistance, detection to the first-line anti tuberculosis drugs: rifampicin and pyrazinamide in the post whole genome sequencing era. Tuberculosis 105:96–107. https://doi.org/10.1016/j.tube.2017.04.008
Nwabuife JC, Madhaorao Pant A, Govender T (2021) Liposomal delivery systems and their applications against Staphylococcus aureus and Methicillin-resistant Staphylococcus aureus. Adv Drug Del Rev:113861. https://doi.org/10.1016/j.addr.2021.113861
Nwodo UU, Green E, Okoh AI (2012) Bacterial exopolysaccharides: functionality and prospects. Int J Mol Sci 13(11):14002–14015. https://doi.org/10.3390/ijms131114002
Oesper RE (1954) Gerhard Domagk and chemotherapy. J Chem Educ 31(4):188. https://doi.org/10.1021/ed031p188
Oh N, Park J-H (2014) Surface chemistry of gold nanoparticles mediates their exocytosis in macrophages. ACS Nano 8(6):6232–6241. https://doi.org/10.1021/nn501668a
Oku N, Macdonald RC (1983) Formation of giant liposomes from lipids in chaotropic ion solutions. Biochim Biophys Acta Biomembr 734(1):54–61. https://doi.org/10.1016/0005-2736(83)90074-3
Osaki T, Omotezako M, Nagayama R, Hirata M, Iwanaga S, Kasahara J, Hattori J, Ito I, Sugiyama H, Kawabata S (1999) Horseshoe crab hemocyte-derived antimicrobial polypeptides, tachystatins, with sequence similarity to spider neurotoxins. J Biol Chem 274(37):26172–26178. https://doi.org/10.1074/jbc.274.37.26172
Otsuka H, Nagasaki Y, Kataoka K (2003) PEGylated nanoparticles for biological and pharmaceutical applications. Adv Drug Deliv Rev 55(3):403–419. https://doi.org/10.1016/S0169-409X(02)00226-0
Pal I, Bhattacharyya D, Kar RK, Zarena D, Bhunia A, Atreya HS (2019) A peptide-nanoparticle system with improved efficacy against multidrug resistant bacteria. Sci Rep 9(1):4485. https://doi.org/10.1038/s41598-019-41005-7
Pal I, Brahmkhatri VP, Bera S, Bhattacharyya D, Quirishi Y, Bhunia A, Atreya HS (2016) Enhanced stability and activity of an antimicrobial peptide in conjugation with silver nanoparticle. J Colloid Interface Sci 483:385–393. https://doi.org/10.1016/j.jcis.2016.08.043
Pang Y, Lu J, Wang Y, Song Y, Wang S, Zhao Y (2013) Study of the rifampin monoresistance mechanism in Mycobacterium tuberculosis. Antimicrob Agents Chemother 57(2):893–900. https://doi.org/10.1128/AAC.01024-12
Panigrahi S, Kundu S, Ghosh S, Nath S, Pal T (2004) General method of synthesis for metal nanoparticles. J Nanopart Res 6(4):411–414. https://doi.org/10.1007/s11051-004-6575-2
Patil YP, Jadhav S (2014) Novel methods for liposome preparation. Chem Phys Lipids 177:8–18. https://doi.org/10.1016/j.chemphyslip.2013.10.011
Patra JK, Das G, Fraceto LF, Campos EVR, Rodriguez-Torres MDP, Acosta-Torres LS, Diaz-Torres LA, Grillo R, Swamy MK, Sharma S, Habtemariam S, Shin H-S (2018) Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnol 16(1):71–71. https://doi.org/10.1186/s12951-018-0392-8
Pavlin M, Bregar VB (2012) Stability of nanoparticle suspensions in different biologically relevant media. Dig J Nanomater Bios 7(4)
Pence MA, Rooijakkers SHM, Cogen AL, Cole JN, Hollands A, Gallo RL, Nizet V (2010) Streptococcal inhibitor of complement promotes innate immune resistance phenotypes of invasive M1T1 group a streptococcus. J Innate Immun 2(6):587–595. https://doi.org/10.1159/000317672
Peng S, Li L, Hu Y, Srinivasan M, Cheng F, Chen J, Ramakrishna S (2015) Fabrication of spinel one-dimensional architectures by single-spinneret electrospinning for energy storage applications. ACS Nano 9(2):1945–1954. https://doi.org/10.1021/nn506851x
Peppas NA, Korsmeyer RW (1986) Dynamically swelling hydrogels in controlled release applications. Prop Appl 3
Peppas NA, Sahlin JJ (1989) A simple equation for the description of solute release. III Coupling of diffusion and relaxation. Int J Pharm 57(2):169–172. https://doi.org/10.1016/0378-5173(89)90306-2
Perron GG, Zasloff M, Bell G (2006) Experimental evolution of resistance to an antimicrobial peptide. Proc Biol Sci 273(1583):251–256. https://doi.org/10.1098/2Frspb.2005.3301
Peschel A (2002) How do bacteria resist human antimicrobial peptides? Trends Microbiol 10:179–186. https://doi.org/10.1016/S0966-842X(02)02333-8
Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Götz F (1999) Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274(13):8405–8410. https://doi.org/10.1074/jbc.274.13.8405
Petrin D, Delgaty K, Bhatt R, Garber G (1998) Clinical and microbiological aspects of Trichomonas vaginalis. Clin Microbiol Rev 11(2):300–317. https://doi.org/10.1128/CMR.11.2.300
Pfalzgraff A, Brandenburg K, Weindl G (2018) Antimicrobial peptides and their therapeutic potential for bacterial skin infections and wounds. Front Pharmacol 9(281). https://doi.org/10.3389/fphar.2018.00281
Phoenix DA, Dennison SR, Harris F (2013) Antimicrobial peptides. Wiley-VCH, Weinheim. https://doi.org/10.1002/9783527652853
Pidgeon C, McNeely S, Schmidt T, Johnson JE (1987) Multilayered vesicles prepared by reverse-phase evaporation: liposome structure and optimum solute entrapment. Biochemistry 26(1):17–29. https://doi.org/10.1021/bi00375a004
Piepenbrink H (1986) Two examples of biogenous dead bone decomposition and their consequences for taphonomic interpretation. J Archaeol Sci 13(5):417–430. https://doi.org/10.1016/0305-4403(86)90012-9
Pinilla CMB, Lopes NA, Brandelli A (2021) Lipid-based nanostructures for the delivery of natural antimicrobials. Molecules 26(12):3587. https://doi.org/10.3390/molecules26123587
Pirogova E, Akay M, Cosic I (2009) Investigating the interaction between oncogene and tumor suppressor protein. IEEE Trans Inf Technol Biomed 13(1):10–15. https://doi.org/10.1109/titb.2008.2003338
Pirogova E, Istivan T, Gan E, Cosic I (2011) Advances in methods for therapeutic peptide discovery, design and development. Curr Pharm Biotechnol 12(8):1117–1127. https://doi.org/10.1109/titb.2008.2003338
Poblete H, Agarwal A, Thomas SS, Bohne C, Ravichandran R, Phopase J, Comer J, Alarcon EI (2016) New insights into peptide–silver nanoparticle interaction: deciphering the role of cysteine and lysine in the peptide sequence. Langmuir 32(1):265–273. https://doi.org/10.1021/acs.langmuir.5b03601
Podolsky SH (2018) The evolving response to antibiotic resistance (1945–2018). Palgrave Commun 4(1):124. https://doi.org/10.1057/s41599-018-0181-x
Pokorny A, Almeida PFF (2004) Kinetics of dye efflux and lipid flip-flop induced by δ-Lysin in phosphatidylcholine vesicles and the mechanism of graded release by amphipathic, α-helical peptides. Biochemistry 43(27):8846–8857. https://doi.org/10.1021/bi0497087
Powers J-PS, Hancock REW (2003) The relationship between peptide structure and antibacterial activity. Peptides 24(11):1681–1691. https://doi.org/10.1016/j.peptides.2003.08.023
Pushpanathan M, Gunasekaran P, Rajendhran J (2013) Antimicrobial peptides: versatile biological properties. Int J Pept 2013:675391. https://doi.org/10.1155/2013/675391
Qiao M, Ying G-G, Singer AC, Zhu Y-G (2018) Review of antibiotic resistance in China and its environment. Environ Int 110:160–172. https://doi.org/10.1016/j.envint.2017.10.016
Qiu L, Qiao M, Chen Q, Tian C, Long M, Wang M, Li Z, Hu W, Li G, Cheng L, Cheng L, Hu H, Zhao X, Chen D (2014) Enhanced effect of pH-sensitive mixed copolymer micelles for overcoming multidrug resistance of doxorubicin. Biomaterials 35(37):9877–9887. https://doi.org/10.1016/j.biomaterials.2014.08.008
Rai M, Yadav A, Gade A (2009) Silver nanoparticles as a new generation of antimicrobials. Biotechnol Adv 27(1):76–83. https://doi.org/10.1016/j.biotechadv.2008.09.002
Rai NK, Tripathi K, Sharma D, Shukla VK (2005) Apoptosis: a basic physiologic process in wound healing. Int J Low Extrem Wounds 4(3):138–144. https://doi.org/10.1177/1534734605280018
Rashid R, Veleba M, Kline KA (2016) Focal targeting of the bacterial envelope by antimicrobial peptides. Front Cell Dev Biol 4(55). https://doi.org/10.3389/fcell.2016.00055
Ravdin JI (1995) Amebiasis. Clin Infect Dis 20(6):1453–1466. https://doi.org/10.1093/clinids/20.6.1453
Rebstock MC, Crooks HM, Controulis J, Bartz QR (1949) Chloramphenicol (Chloromycetin).1 IV.1a Chemical studies. J Am Chem Soc 71(7):2458–2462. https://doi.org/10.1021/ja01175a065
Reinholz J, Landfester K, Mailänder V (2018) The challenges of oral drug delivery via nanocarriers. Drug Deliv 25(1):1694–1705. 10.1080%2F10717544.2018.1501119
Ren X, Ying P, Yang Z, Shang M, Hou H, Gao F (2015) Foaming-assisted electrospinning of large-pore mesoporous ZnO nanofibers with tailored structures and enhanced photocatalytic activity. RSC Adv 5(21):16361–16367. https://doi.org/10.1039/C4RA15421E
Reynolds P (1989) Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis 8(11):943–950. https://doi.org/10.1007/bf01967563
Rideau E, Wurm FR, Landfester K (2018) Giant polymersomes from non-assisted film hydration of phosphate-based block copolymers. Polym Chem 9(44):5385–5394. https://doi.org/10.1039/C8PY00992A
Robertsen HL, Musiol-Kroll EM (2019) Actinomycete-derived polyketides as a source of antibiotics and lead structures for the development of new antimicrobial drugs. Antibiotics 8(4). https://doi.org/10.3390/2Fantibiotics8040157
Rodriguez N, Pincet F, Cribier S (2005) Giant vesicles formed by gentle hydration and electroformation: a comparison by fluorescence microscopy. Colloids Surf B: Biointerfaces 42(2):125–130. https://doi.org/10.1016/j.colsurfb.2005.01.010
Romberg B, Hennink WE, Storm G (2007) Sheddable coatings for long-circulating nanoparticles. Pharm Res 25(1):55–71. https://doi.org/10.1007/2Fs11095-007-9348-7
Rughani RV, Schneider JP (2008) Molecular design of β-hairpin peptides for material construction. MRS Bull 33(5):530–535. https://doi.org/10.1557/mrs2008.106
Saar-Dover R, Bitler A, Nezer R, Shmuel-Galia L, Firon A, Shimoni E, Trieu-Cuot P, Shai Y (2012) D-alanylation of lipoteichoic acids confers resistance to cationic peptides in group B streptococcus by increasing the cell wall density. PLoS Pathog 8(9):e1002891. https://doi.org/10.1371/journal.ppat.1002891
Saghazadeh S, Rinoldi C, Schot M, Kashaf SS, Sharifi F, Jalilian E, Nuutila K, Giatsidis G, Mostafalu P, Derakhshandeh H, Yue K, Swieszkowski W, Memic A, Tamayol A, Khademhosseini A (2018) Drug delivery systems and materials for wound healing applications. Adv Drug Deliv Rev 127:138–166. https://doi.org/10.1016/j.addr.2018.04.008
Salopek B, Krasic D, Filipovic S (1992) Measurement and application of zeta-potential. Rud Geol Naft Zb 4(1):147. https://doi.org/10.1007/springerreference_67936
Samuelson J (1999) Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother 43(7):1533–1541. https://doi.org/10.1128/AAC.43.7.1533
Sánchez-López E, Gomes D, Esteruelas G, Bonilla L, Lopez-Machado AL, Galindo R, Cano A, Espina M, Ettcheto M, Camins A, Silva AM, Durazzo A, Santini A, Garcia ML, Souto EB (2020) Metal-based nanoparticles as antimicrobial agents: an overview. Nanomaterials (Basel, Switzerland) 10(2):292. https://doi.org/10.3390/nano10020292
Sanson N, Rieger J (2010) Synthesis of nanogels/microgels by conventional and controlled radical crosslinking copolymerization. Polym Chem 1(7):965–977. https://doi.org/10.1039/C0PY00010H
Schaller M, Braunsdorf C, Mailänder-Sanchez D, Jäckel A, Müller J, Borelli C (2015) Comparison of user-friendliness and treatment cost of Loceryl® vs. Ciclopoli® – a patient's perspective. Mycoses 58(10):632–636. https://doi.org/10.1111/myc.12371
Schild HG (1992) Poly(N-isopropylacrylamide): experiment, theory and application. Prog Polym Sci 17(2):163–249. https://doi.org/10.1016/0079-6700(92)90023-R
Schmidtchen A, Frick IM, Andersson E, Tapper H, Björck L (2002) Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol 46(1):157–168. https://doi.org/10.1046/j.1365-2958.2002.03146.x
Schmidtchen A, Frick IM, Björck L (2001) Dermatan sulphate is released by proteinases of common pathogenic bacteria and inactivates antibacterial α-defensin. Mol Microbiol 39(3):708–713. https://doi.org/10.1046/j.1365-2958.2001.02251.x
Scorciapino MA, Serra I, Manzo G, Rinaldi AC (2017) Antimicrobial dendrimeric peptides: structure, activity and new therapeutic applications. Int J Mol Sci 18(3). https://doi.org/10.3390/ijms18030542
Scott MG, Davidson DJ, Gold MR, Bowdish D, Hancock REW (2002) The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J Immunol 169(7):3883–3891. https://doi.org/10.4049/jimmunol.169.7.3883
Scott MG, Dullaghan E, Mookherjee N, Glavas N, Waldbrook M, Thompson A, Wang A, Lee K, Doria S, Hamill P, Yu JJ, Li Y, Donini O, Guarna MM, Finlay BB, North JR, Hancock REW (2007) An anti-infective peptide that selectively modulates the innate immune response. Nat Biotechnol 25(4):465–472. https://doi.org/10.1038/nbt1288
Scovil ER (1912) Salvarsan. Am J Nurs 12(5):387–390
Sengupta S, Chattopadhyay M, Grossart H-P (2013) The multifaceted roles of antibiotics and antibiotic resistance in nature. Front Microbiol 4(47). https://doi.org/10.3389/2Ffmicb.2013.00047
Sensi P (1975) Recent progress in the chemistry and biochemistry of rifamycins. Pure Appl Chem ( Chimie Pure et Appliquee) 41(1–2):15–29. https://doi.org/10.1351/pac197541010015
Sensi P (1983) History of the development of rifampin. Rev Infect Dis 5(Supplement_3):S402–S406. https://doi.org/10.1093/clinids/5.supplement_3.s402
Shafer WM, Qu XD, Waring AJ, Lehrer RI (1998) Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc Natl Acad Sci U S A 95(4):1829. https://doi.org/10.1073/pnas.95.4.1829
Shahriar SMS, Mondal J, Hasan MN, Revuri V, Lee DY, Lee Y-K (2019) Electrospinning nanofibers for therapeutics delivery. Nano 9(4):532. https://doi.org/10.3390/nano9040532
Shai Y (2002) Mode of action of membrane active antimicrobial peptides. N Y 66:236–248
Shasmitha R (2014) Tetracycline in Discolouration of teeth. Res J Pharm Technol 7(6):712–714. https://doi.org/10.5958/0974-360X
Shinn DLS (1962) Metronidazole in acute ulcerative gingivitis. Lancet 279(7240):1191–1191. https://doi.org/10.1016/S0140-6736(62)92243-2
Shohda K, Takahashi K, Suyama A (2015) A method of gentle hydration to prepare oil-free giant unilamellar vesicles that can confine enzymatic reactions. Biochem Biophys Rep 3:76–82. https://doi.org/10.1016/j.bbrep.2015.07.005
Shurkin J (2014) News feature: animals that self-medicate. Proc Natl Acad Sci U S A 111(49):17339–17341. https://doi.org/10.1073/pnas.1419966111
Siepmann J, Kranz H, Bodmeier R, Peppas NA (1999) HPMC-matrices for controlled drug delivery: a new model combining diffusion, swelling, and dissolution mechanisms and predicting the release kinetics. Pharm Res 16(11):1748–1756. https://doi.org/10.1023/a:1018914301328
Sieprawska-Lupa M, Mydel P, Krawczyk K, Wójcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, Shafer W, McAleese F, Foster T, Travis J, Potempa J (2005) Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother 48:4673–4679. https://doi.org/10.1128/aac.48.12.4673-4679.2004
Sivalanka G, Rai S, Alex A (2016) Antibiotic resistance: emergence and solutions. Manipal J Pharm Sci 2(2):26–30. https://doi.org/10.1016/c2015-0-00104-6
Soni G, Yadav KS (2016) Nanogels as potential nanomedicine carrier for treatment of cancer: a mini review of the state of the art. Saudi Pharm J 24(2):133–139. https://doi.org/10.1016/j.jsps.2014.04.001
Soni KS, Desale SS, Bronich TK (2016) Nanogels: an overview of properties, biomedical applications and obstacles to clinical translation. J Control Release 240:109–126. https://doi.org/10.1016/j.jconrel.2015.11.009
Sousa MGC, Maximiano MR, Costa RA, Rezende TMB, Franco OL (2020) Nanofibers as drug-delivery systems for infection control in dentistry. Expert Opin Drug Deliv 17(7):919–930. https://doi.org/10.1080/17425247.2020.1762564
Sousa MGC, Rezende TMB, Franco OL (2021) Nanofibers as drug-delivery systems for antimicrobial peptides. Drug Discov Today 26(8):2064–2074. https://doi.org/10.1016/j.drudis.2021.03.008
Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J (2008) The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis 46(2):155–164. https://doi.org/10.1086/524891
Spinney L (2019) How pandemics shape social evolution. Nature 574(7778):324–326. https://doi.org/10.1038/d41586-019-03048-8
Sprigg K, Pietrangeli C (2019) Bacterial antibiotic resistance: on the cusp of a post-antibiotic world. Curr Treat Options Infect Dis 11(1):42–57. https://doi.org/10.1007/s40506-019-0181-4
Stallmann H, Faber C, Amerongen A, Wuisman P (2006) Antimicrobial peptides: review of their application in musculoskeletal infections. Inj Int J Care Inj 37(2):34–40. https://doi.org/10.1016/j.injury.2006.04.007
Stansly PG, Schlosser ME (1947) Studies on polymyxin: isolation and identification of bacillus polymyxa and differentiation of polymyxin from certain known antibiotics. J Bacteriol 54(5):549. https://doi.org/10.1128/jb.54.5.549-556.1947
Stanwell-Smith R (2007) From the golden age of antibiotics to the era of the superbug: the lessons of antibiotic resistance. Health Hyg 28(1):11–12. https://doi.org/10.1016/j.puhe.2006.09.005
Steinberg DA, Hurst MA, Fujii CA, Kung AH, Ho JF, Cheng FC, Loury DJ, Fiddes JC (1997) Protegrin-1: a broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob Agents Chemother 41(8):1738–1742. https://doi.org/10.1128/aac.41.8.1738
Steiner H, Hultmark D, Bennich EÅH, Boman HG (1981) Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 292(5820):246. https://doi.org/10.1038/292246a0
Stetefeld J, McKenna SA, Patel TR (2016) Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophys Rev 8(4):409–427. https://doi.org/10.1007/s12551-016-0218-6
Stockwell V, Duffy B (2012) Use of antibiotics in plant agriculture. Rev Sci Tech 31(1):199–210. https://doi.org/10.20506/rst.31.1.2104
Storm DR, Rosenthal KS, Swanson PE (1977) Polymyxin and related peptide antibiotics. Annu Rev Biochem 46:723. https://doi.org/10.1146/annurev.bi.46.070177.003451
Strebhardt K, Ullrich A (2008) Paul Ehrlich's magic bullet concept: 100 years of progress. Nat Rev Cancer 8(6):473–480. https://doi.org/10.1038/nrc2394
Strömstedt AA, Ringstad L, Schmidtchen A, Malmsten M (2010) Interaction between amphiphilic peptides and phospholipid membranes. Curr Opin Colloid Interface Sci 15(6):467–478. https://doi.org/10.1016/j.cocis.2010.05.006
Sun C, Lee JSH, Zhang M (2008) Magnetic nanoparticles in MR imaging and drug delivery. Adv Drug Deliv Rev 60(11):1252–1265. https://doi.org/10.1016/j.addr.2008.03.018
Sun L, Klein EY, Laxminarayan R (2012) Seasonality and temporal correlation between community antibiotic use and resistance in the United States. Clin Infect Dis 55(5):687. https://doi.org/10.1093/cid/cis509
Sun Y, Shang D (2015) Inhibitory effects of antimicrobial peptides on lipopolysaccharide-induced inflammation. Mediat Inflamm 2015:167572–167572. https://doi.org/10.1155/2015/167572
Swann JP (1983) The search for synthetic penicillin during world war II. Br J Hist Sci 16(2):154–190. https://doi.org/10.1017/s0007087400026789
Swenson RM (1988) Plagues, history, and AIDS. Am Sch 57(2):183–200. https://doi.org/10.1093/swra/24.2.88-a
Taboureau O, Olsen OH, Nielsen JD, Raventos D, Mygind PH, Kristensen HH (2006) Design of novispirin antimicrobial peptides by quantitative structure-activity relationship. Chem Biol Drug Des 68(1):48–57. https://doi.org/10.1111/j.1747-0285.2006.00405.x
Tan SY, Tatsumura Y (2015) Alexander Fleming (1881–1955): discoverer of penicillin. Singapore Med J 56(7):366–367. https://doi.org/10.11622/2Fsmedj.2015105
Territo MC, Ganz T, Selsted ME, Lehrer R (1989) Monocyte-chemotactic activity of defensins from human neutrophils. J Clin Invest 84(6):2017–2020. https://doi.org/10.1172/jci114394
Thapa RK, Diep DB, Tønnesen HH (2021) Nanomedicine-based antimicrobial peptide delivery for bacterial infections: recent advances and future prospects. J Pharm Investig 51(4):377–398. https://doi.org/10.1007/s40005-021-00525-z
Thurlow LR, Thomas VC, Narayanan S, Olson S, Fleming SD, Hancock LE (2010) Gelatinase contributes to the pathogenesis of endocarditis caused by enterococcus faecalis. Infect Immun 78(11):4936. https://doi.org/10.1128/iai.01118-09
Tian X, Sun F, Zhou XR, Luo SZ, Chen L (2015) Role of peptide self-assembly in antimicrobial peptides. J Pept Sci 21(7):530–539. https://doi.org/10.1002/psc.2788
Topcu B, Gultekinoglu M, Timur SS, Eroglu I, Ulubayram K, Eroglu H (2021) Current approaches and future prospects of nanofibers: a special focus on antimicrobial drug delivery. J Drug Target 29(6):563–575. https://doi.org/10.1080/1061186X.2020.1867991
Tossi A, Sandri L, Giangaspero A (2000) Amphipathic, alpha -helical antimicrobial peptides. Biopolymers (Peptide Science) 55(1):4–30. https://doi.org/10.1002/1097-0282(2000)55:1%3C4::aid-bip30%3E3.0.co;2-m
Tracey C, Lindsay FL, John MK, Mariana FW, Linda P (1995) Cost of mating in Drosophila melanogaster females is mediated by male accessory gland products. Nature 373(6511):241. https://doi.org/10.1038/373241a0
Truskewycz A, Truong VK, Ball AS, Houshyar S, Nassar N, Yin H, Murdoch BJ, Cole I (2021) Fluorescent magnesium hydroxide nanosheet bandages with tailored properties for biocompatible antimicrobial wound dressings and pH monitoring. ACS Appl Mater Interfaces 13(24):27904–27919. https://doi.org/10.1021/acsami.1c05908
Vanessa MDC, Christine EK, Lindsay K, Mariya M, Wilson WLS, Carsten S, Duane F, Grant Z, Fabrice C, Regis D, Golding GB, Hendrik NP, Gerard DW (2011) Antibiotic resistance is ancient. Nature 477(7365):457. https://doi.org/10.1038/nature10388
Venter H, Henningsen ML, Begg SL (2017) Antimicrobial resistance in healthcare, agriculture and the environment: the biochemistry behind the headlines. Essays Biochem 61(1):1–10. https://doi.org/10.1042/ebc20160053
Ventola CL (2015) The antibiotic resistance crisis: part 1: causes and threats. Pharmacol Ther 40(4):277–283
Wainwright M (1989a) Moulds in ancient and more recent medicine. Mycologist 3(1):21–23. https://doi.org/10.1016/S0269-915X(89)80010-2
Wainwright M (2002) Fleming's unfinished. Perspect Biol Med 45(4):529. https://doi.org/10.1353/pbm.2002.0065
Wakabayashi H, Oda H, Yamauchi K, Abe F (2014) Lactoferrin for prevention of common viral infections. J Infect Chemother 20(11):666–671. https://doi.org/10.1016/j.jiac.2014.08.003
Waksman SA (1948) Antibiotics. Biol Rev 23(4):452–487. https://doi.org/10.1111/j.1469-185x.1948.tb00567.x
Waksman SA, Schatz A, Reynolds DM (1946) Production of antibiotic substances by actinomycetes. Ann N Y Acad Sci 48(2):73–86. https://doi.org/10.1111/j.1749-6632.2010.05861.x
Wang G, Li X, Wang Z (2016) APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 44(D1):D1087. https://doi.org/10.1093/nar/gkv1278
WHO (2014) Antibiotic resistance a worry, spreading fast. WHO, Mumbai
Wierup M (2001) The Swedish experience of the 1986 year ban of antimicrobial growth promoters, with special reference to animal health, disease prevention, productivity, and usage of antimicrobials. Microb Drug Resist (Larchmont, N.Y.) 7(2):183. https://doi.org/10.1089/10766290152045066
Wray SK (2012) Plagues in world history. J World Hist 23(3):682–685. https://doi.org/10.1353/jwh.2012.0074
Wright AJ (1999) The penicillins. Mayo Clin Proc 74(3):290–307. https://doi.org/10.4065/74.3.290
Wu Q, Patočka J, Kuča K (2018) Insect antimicrobial peptides, a mini review. Toxins 10(11). https://doi.org/10.3390/toxins10110461
Xia X, Cheng L, Zhang S, Wang L, Hu J (2018) The role of natural antimicrobial peptides during infection and chronic inflammation. Antonie Van Leeuwenhoek 111(1):5–26. https://doi.org/10.1007/s10482-017-0929-0
Xu X, Yang L, Xu X, Wang X, Chen X, Liang Q, Zeng J, Jing X (2005) Ultrafine medicated fibers electrospun from W/O emulsions. J Control Release 108(1):33–42. https://doi.org/10.1016/j.jconrel.2005.07.021
Yadav S, Sharma AK, Kumar P (2020) Nanoscale self-assembly for therapeutic delivery. Front Bioeng Biotechnol 8(127). https://doi.org/10.3389/fbioe.2020.00127
Yan S, Song W (2014) Photo-transformation of pharmaceutically active compounds in the aqueous environment: a review. Environ Sci Processes Impacts 16(4):697–720. https://doi.org/10.1039/c3em00502j
Yasumoto T, Satake M (1998) Bioactive compounds from marine microalgae. CHIMIA Int J Chem 52(1–2):63–68
Yeaman MR, Yount NY (2003) Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 55(1):27–55. https://doi.org/10.1124/pr.55.1.2
Yu B, Lee RJ, Lee LJ (2009) Microfluidic methods for production of liposomes. Methods Enzymol 465:129–141. https://doi.org/10.1016/s0076-6879(09)65007-2
Yu H, Park J-Y, Kwon C, Hong S-C, Park K-M, Chang P-S (2018) An overview of nanotechnology in food science: preparative methods, practical applications, and safety. J Chem 2018:1–10. https://doi.org/10.1155/2018/5427978
Zaat JOM, Mank TG, Assendelft WJJ (1997) A systematic review on the treatment of giardiasis. Tropical Med Int Health 2(1):63–82. https://doi.org/10.1046/j.1365-3156.1997.d01-132.x
Zanetti M (2004) Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol 75(1):39–48. https://doi.org/10.1189/jlb.0403147
Zasloff M (1987) Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci U S A 84(15):5449–5453. https://doi.org/10.1073/pnas.84.15.5449
Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415(6870):389. https://doi.org/10.1038/415389a
Zeng Q, Jiang X, Yu A, Lu M (2007) Growth mechanisms of silver nanoparticles: a molecular dynamics study. Nanotechnology 18:035708. https://doi.org/10.1088/0957-4484/18/3/035708
Zhang D, Lin L, Luo Z, Yan C, Zhang X (2011) Occurrence of selected antibiotics in Jiulongjiang River in various seasons, South China. J Environ Monit 13(7):1953–1960. https://doi.org/10.1039/C0EM00765J
Zhang H, Zhai Y, Wang J, Zhai G (2016) New progress and prospects: the application of nanogel in drug delivery. Mater Sci Eng C 60:560–568. https://doi.org/10.1016/j.msec.2015.11.041
Zhang L-J, Gallo RL (2016) Antimicrobial peptides. Curr Biol 26(1):R14–R19. https://doi.org/10.1016/j.cub.2015.11.017
Zhang L, Cao Z, Li Y, Ella-Menye J-R, Bai T, Jiang S (2012) Softer Zwitterionic Nanogels for longer circulation and lower splenic accumulation. ACS Nano 6(8):6681–6686. https://doi.org/10.1021/nn301159a
Zhao Z, Wang Y, Han J, Wang K, Yang D, Yang Y, Du Q, Song Y, Yin X (2014) Self-assembled micelles of amphiphilic poly(L-phenylalanine)-b-poly(L-serine) polypeptides for tumor-targeted delivery. Int J Nanomedicine 9:5849. https://doi.org/10.2147/IJN.S73111
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Nassar, N., Kasapis, S., Pyreddy, S., Istivan, T. (2022). The History of Antibiotics Illumes the Future of Antimicrobial Peptides Administered Through Nanosystems. In: Kumar, V., Shriram, V., Shukla, R., Gosavi, S. (eds) Nano-Strategies for Addressing Antimicrobial Resistance. Nanotechnology in the Life Sciences. Springer, Cham. https://doi.org/10.1007/978-3-031-10220-2_1
Download citation
DOI: https://doi.org/10.1007/978-3-031-10220-2_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-10219-6
Online ISBN: 978-3-031-10220-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)