
Abstract
During the past two decades, high-throughput transcriptome sequencing has developed into a well-established and widely used research method. Transcriptomics experiments allow researchers to classify transcriptional behavior, concentrate on a subset of specific target genes and transcripts, or simultaneously profile thousands of genes to create a global image of cell function. Besides, the simultaneous maturation of bioinformatics tools are playing a crucial role in the analysis of generated transcriptomic data for novel discovery. Finger millet (Eleusine coracana L.), an essential species of C4 which is known for its resistance to stress and dietary significance is a nutritional and health security crop as well due to extraordinarily high levels of calcium (Ca). Ca being considered the most important macronutrient in the diet, is needed in relatively large quantities to maintain a healthy body. Therefore, analysis of transcriptome/RNA-seq data generated via different high-throughput sequencing platform and their integration through systems biology offered new opportunities to decode the molecular mechanisms of entire finger millet systems in different conditions for identification of key genes involved in drought stress, growth and development, disease resistance, synthesis of bioactive compounds, proteins, carbohydrate and, Ca accumulation and so on. This book chapter highlights the principles of finger millet transcriptome analysis from data generation to data analysis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Finger millet
- High-throughput sequencing technologies
- Transcriptome analysis
- Bioinformatics
- Systems biology
8.1 Introduction
Finger millet (Eleusine coracana L.) a nutritionally important crop has recently gained a lot of attention due to its nutritional components. The finger millet (FM; also known as ragi) ranks fourth after the Sorghum (Sorghum bicolor), pearl millet (Pennisetum glaucum), and foxtail millet (Setaria italica) worldwide (Kumar et al. 2016). It is an important food crop, which is widely cultivated in arid and semi-arid regions of the world, particularly in eastern Africa; India and the rest of Asia including Sri Lanka and China (Belton and Taylor 2002; Kumar et al. 2016).
Despite being an excellent source of nutrients and minerals with the well-documented health benefits FM, also possess some anti-nutrients (commonly referred to as phytochemicals). The slow digestibility of FM is supplying energy during the day. The plant itself is diaphoretic, diuretic and vermifuge, and its leaf juice was offered to women during childbirth (Antony and Chandra 1998; Vadivoo et al. 1998; Subba Rao and Muralikrishna 2002; Fernandez et al. 2003; Ćujić et al. 2016; Kumar et al. 2018). It has also been used for various diseases, including leprosy, liver disease, pleurisy, pneumonia and smallpox, as a folk remedy. Finger millet has a high fiber content that tends to reduce constipation, high cholesterol, diabetes and intestinal cancer. Besides, it is also suggested that the risk of diabetes and gastrointestinal tract disorders may be effectively minimized with daily intake of FM in diet (Gibson and Helme 2001; Mooser and Carr 2001; Scalbert et al. 2005; Lattimer and Haub 2010; Schatzkin et al. 2007; Fardet et al. 2008).
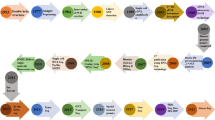
Despite the fact that many essential nutritional features are available in finger millet grains, research work has largely been ignored to exploit the tremendous potential of this crop for solving the problems associated with protein-energy malnutrition and nutritional deficiencies (Murtaza et al. 2014; Kumar et al. 2018). This may be attributed to the fact that before and during the green revolution, staple crops such as wheat, rice and maize gained a great deal of research attention in order to improve food production. Moreover, the availability of very limited genomic knowledge in past years has further limited finger millet crops improvement programs. However, but, the first time the transcriptome (Rahman et al. 2014; Kumar et al. 2015a, b) and genome data (Hittalmani et al. 2017; Antony et al. 2018) of finger millet is now available due to advances in omics science and technology. After that many papers are now available addressing the complexity of transcriptome data and reported some essential genes/proteins associated with different biological processes and its nutritional potential (Lanham-New 2008; Singh et al. 2014; Chinchole et al. 2017; Kokane et al. 2018; Gupta et al. 2018; Avashthi et al. 2018, 2020; Parvathi et al. 2019). These available data hold enormous information related to millet breeding program. There is a need of time to explore these data and generate new transcriptome data for comparative analysis and mining of novel information through computational and experimental approaches. The chapter highlights the pipeline of finger millet transcriptome analysis from data generation to analysis, integration and deposition of data in public repository for the benefit of the scientific society (Fig. 8.1).

Integrated approach for decoding the finger millet transcriptome for novel discovery
8.2 Overview of High-Throughput Transcriptome Sequencing Platform
The pyrosequencing-based 454 system by Roche, the sequencing-by-synthesis-based GA/HiSeq/MiSeq devices from Illumina and the sequencing-by-ligation SOLiD system, Pacific Biosciences and Oxford Nanopore are currently the most widely used sequencing platform, and some others are under development (Wolf 2013; Klepikova et al. 2016; Pathak et al. 2018b). These sequencing platforms will be utilized for finger millet transcriptome sequencing as per objective of the research projects (Kumar et al. 2015a, b). The sequencing technology and its estimated experimental cost are mentioned in Table 8.1 (Pathak et al. 2018a, b).
8.3 Experimental Set-Up for Data Generation
As we know that finger millet is one of the agriculturally important crop which holds immense potential for improving human life and prevent from several diseases due to its nutraceuticals properties. Therefore, transcriptome sequencing and its analysis is necessary for identifying key components involved in different biological processes for several purposes. Before, going to the experimental setup for data generation, several points’ need to be addressed which are discussed in the following sections.
8.3.1 Purpose of Transcriptome Sequencing
The starting point for each experiment is to identify its basic aims and determine its viability with respect to the budget and the technique available. For a transcriptome sequencing experiment, questions like, why we need transcriptome data, what type of genes will be characterized or identified and why, what are the future applications of identified genes, etc., need to be addressed.
8.3.2 Statistical Design
It is one of the important steps before going for transcriptome sequencing. Here, we will focus on biological replication and statistical model selection. It is a common practice in many biological investigations. In order to integrate biological replication, the statistical treatment of transcriptome data has moved from single sample studies to more complex statistical designs, such as generalized linear regression models. In case of transcriptome analysis, it will help in sample collection and analysis of data for comparative analysis. For example collections of samples in different time interval as compared to control plants followed by sequencing and analysis. This will help in data analysis for fruitful results.
8.3.3 Choice of Tissue and Time
The abundance of transcript and isoform identity is radically different across tissues, and they change drastically not only throughout embryological development, but also over the entire life of an organism. Therefore, it is important to understand, which tissue and at which, physiological level is most likely to observe difference related to the problem at hand.
8.3.4 Collection of Sample
Sample collections will be done carefully because RNA degrades rapidly. Therefore, after cutting put sample immediately in ice box, generally it a good job for preserving RNA for some time at room temperature, then stored at −80 °C for further utilization and their isolation.
8.3.5 RNA Extraction and Their Quality Assessments
RNA extraction has to be suited to the focal RNA species: during normal mRNA extraction, tiny RNA molecules (<200 bp) will be destroyed following traditional LiCl precipitation or commercially available kits. For small transcripts, such as micro-RNAs, different extraction protocols will be needed. The RNA integrity assessment is a crucial first step for obtaining significant measurements of gene expression.
8.3.6 cDNA Synthesis
Until sequencing, most sequencing platforms usually require RNA to be converted to cDNA. The reverse transcriptase enzymatic reaction may either be prepared by hybridizing an oligo-dT primer onto the mRNA template of poly-A tail or by use of random hexamer primers. It can easily be done by following standard lab protocol or protocol provided through kit.
8.3.7 Library Preparation
Library preparation is an important step and it is platform specific. Library can be prepared as per objective of the research project. During library preparation, cDNA is broken into smaller pieces, which then act as a sequencing template. The cDNA pieces are partly sequenced from one end while a single-end approach is used, where paired-end sequencing read short sequences from both ends. For initial transcriptome assembly and isoform identification, paired-end sequencing may be helpful, but one should be careful that the insert size should not be too big (usually <300 bp), else the small size fraction of transcripts would be lost. On the other hand, too short sizes of insert can give adapter contamination, which may need trimming or removal of reads, leading to complicated analysis.
8.3.8 Sequencing Strategy and Platform Selection
Illumina HiSeq, IonTorrent, Pacific Biosciences are the most commonly used sequencing platform right now and others are under development. For platform selection, cost per base pair, error rate and error profiles, total output and read length are the relevant parameters to note. Where there is a trade-off among read length and total output, for transcriptome data, the latter seems more significant. In de novo assembly, longer reads are very helpful and paired-end reads also work equally well. In the end, what matters is the number of appropriately aligned reads per gene, which defines the precision of the calculation of gene expression.
8.4 Bioinformatics for Data Analysis
Bioinformatics is an interdisciplinary science because it is made from combination of several scientific disciplines, i.e. plant science, animal science, chemical science, physical science, pharmaceutical science, mathematical and statistical science supported by computer science and information technology as support system. We can’t think about transcriptome analysis without bioinformatics. It is playing vital role in dissecting the complexity of transcriptome data for identification of key genes/proteins involved in various biological processes and their expression with respect to time in different tissues. A strong computational skill is required for handling of the transcriptome data and softwares. Several important tools used for transcriptome analysis are highlighted in Table 8.2.
8.4.1 Computational Resources and Programming Skills
During transcriptome sequencing, a big amount of data will be generated. Therefore, we need good computational resources in terms of data storage and analysis, because during data analysis other files such as assembled file, ban, sam, etc., will be generated in the form of results, which also take big space in the computer. Besides, good programming skills are necessary for fetching of key sequences, their annotation and analysis. R, Perl and python are the most demanding language right now in this area. Good command in UNIX/Linux operating systems is must for transcriptome analysis.
8.4.1.1 Understanding of File Format
At the time of transcriptome sequencing and analysis, various files will be generated. The knowledge of these files formats such as fastq, fasta, sam, bam, vcf, etc., is necessary to understand data and results.
8.4.2 Quality Analysis of Generated Data
After transcriptome sequencing, the generated data will be analyzed using bioinformatics tools such as fastqc to determine the quality of generated data. If some error has occurred in data, the software like trimmomatic, cut-adapter, etc., will be run to remove low quality reads from the data. Further, fastqc will be run to evaluate the quality of data after trimming.
8.4.3 Method for Data Assembly
It is a computational method to reconstruct longer sequence (e.g. a transcript) from sort sequence reads. Basically, two methods are available for the assembly of transcriptome data, i.e. de novo assembly and reference-based assembly. In finger millet, the first de novo assembled transcriptome was published in the year 2013.
8.4.3.1 De novo Assembly
De novo assembly uses more computational resources as compared to reference-based assembly. A computer may contain at least 8 cores and 256 GB of RAM to allow assembly within a suitable time frame. An assembled transcript facilitates gene expression studies and annotations for novel discovery. Usually, de novo assembly is more challenging and less accurate than reference-based assembly.
8.4.3.2 Reference Based or Genome-Guided Assembly
Here, the genome of a target organism (if available) or closely related organism will be taken as reference. The transcriptome/RNA-seq reads will be mapped on the reference genome to construct the longer leads from small reads for further investigation. This is considered as more accurate method for assembly of transcriptome data.
8.4.4 Annotation of Assembled Transcript
Annotation is a very common term in Bioinformatics. It is a process to find out biologically important regions in sequences, its expression with respect to particular condition and time, and their involvement/role in different biological processes. The obtained information will be further utilized for validation and other research program.
8.4.4.1 Identification of Differentially Expressed Genes (DEGs)
Identification of differentially expressed genes (DEGs) from transcriptome data is one of the key steps in transcriptome assembly and annotation. If an observed difference or expression level among two experimental conditions is statistically important, a gene is declared as differentially expressed. Bioinformatics analysis of transcriptome data play vital role in investigation of differentially expressed genes. These genes will be further annotated and validated for various uses including crop improvement program through molecular breeding and genetic engineering approaches.
8.4.4.2 Gene Name Assignment
The identified DEGs, i.e. up-regulated and down-regulated sequences will be subjected for gene name assignment based on the available information in databases through computational prediction. Designation of gene name on identified sequences is one of the key steps in characterization of genes and construction of their relationship with known sequences of the related organisms through multiple sequence alignment and building of phylogenetic tree.
8.4.4.3 Gene Set Enrichment Analysis
A set of up-regulated and down-regulated sequences will be subjected to enrichment analysis in terms of gene ontology, i.e. molecular function, biological process and cellular components. Based on gene set enrichment analysis, we will annotate the function of genes identified via transcriptome analysis.
8.4.4.4 Pathway Analysis
Pathway analysis is now being done with bioinformatics applications or web resources that accept and interpret various data from omics (Cirillo et al. 2017). It has become an essential tool for determination of candidate genes involved in different pathways. These studies have promoted an interactive assessment of the genes, their role, regulation or association.
8.4.5 Variant Calling
Development of molecular markers within the putatively functional genomic elements of transcribed DNA is one of the key applications of transcriptomic data. Many tools are available for variant calling, such as the extremely versatile GATK pipeline. Generally, partially overlapping sets of variants will call, as they take various statistical approaches and vary in which parts of the data are used.
8.4.6 Systems Biology for Data Integration and Novel Discovery
Systems biology has emerged in recent years as an effective approach for understanding crop plant systems to improve food production and how their dietary components support our health and avoid diseases, as well as for studying the bioactive molecules that are involved in these impacts (Pathak and Singh 2020). Computational methods and statistical models allow for a broad study of the response of key genes and their role in improving the nutritional content and influence of plant products on human health. This has contributed to the recognition of several essential genes and proteins involved in the growth and nutrition of plants, as well as the discovery of bioactive molecules linked with human, animal and plant health (Kumar et al. 2015a, b, 2018; Pathak et al. 2017a, b; Pathak et al. 2018a; Rana et al. 2020; Pathak and Singh 2020).
8.4.6.1 Pathway Modeling and Simulation Analysis
It is a powerful approach to study the behavior of different genes involved in pathway at different amount/concentration with respect to time. The obtained information from transcriptome analysis and information already available in literatures will be used to build model pathway using systems biology graphical notation (SBGN). The different types of SBGN symbols are available for representing gene, protein, transcription factor, metabolite, etc. (Pathak et al. 2013). We can use these symbols to model pathway. Further, kinetic rate equation will be generated for each species in the model to simulate its dynamic behavior for identification of key components involved in regulation of different molecular mechanisms in the biological systems. A pathway of molecular interactions between calcium exchangers and sensors in different tissues involved in the regulation of calcium transport and their accumulation in seeds of finger millet has been proposed through transcriptomics and systems biology approaches (Fig. 8.2) (Kokane et al. 2018).

Figure depicted the molecular mechanism of transport and accumulation of calcium in finger millet seeds
8.4.6.2 Network Generation and Analysis
The key genes or proteins obtained from transcriptome analysis will be utilized for network generation and analysis for identification of hubs from the large set of genes. It is a powerful approach that emerged in recent years in the area of biological sciences and made a new discipline called network biology. With the help of network biology approaches, we can sort key genes as hub from large set of genes based on topological parameters for further validation and implementation in the crop improvement program. The modeled pathway will be imported as network via network visualization tools for their analysis. Dehydroascorbate reductase (DHAR) was identified as a key gene regulating different biological processes in Finger millet through network analysis (Fig. 8.3) (Avashthi et al. 2020).

Identification of hub genes involved in different biological processes in finger millet through integrated transcriptome data analysis
8.4.7 Validation
The results generated from above analysis, i.e. transcriptome sequencing, analysis and their integration with systems biology will be validated through experimental approaches for further implementation in finger millet research program with respect to resistance to abiotic and biotic stresses, food and nutritional security.
8.4.8 Submission of Generated Data in International Data Repository
Advances in sequencing platforms generated huge amount of sequencing data in daily basis right now. Storage and management of these data is a challenging task for bioinformatician. Bioinformatics has tremendous potential in management and analysis of omics data. A lot of database, i.e. primary, composite, secondary, structural and specialized databases are available on internet for management of biological data. Right now Sequence Read Archive, commonly known as SRA (https://www.ncbi.nlm.nih.gov/sra) hosted at National Center for Biotechnology Information (NCBI) is a major resource for submission of transcriptome data. Data related to finger millet transcriptome are also available at SRA for further analysis and their integration with newly sequenced data for novel discovery. A list of finger millet transcriptome sequencing data available in public domains is highlighted in Table 8.3.
8.5 Application of Transcriptome Sequencing and Data Analysis in Breeding and Improvement of Finger Millet
The availability of finger millet transcriptome sequencing data, re-sequencing approaches and computational resources has led to a new era of breeding, as they make it easier to research the genotype and its relationship to the phenotype, particularly for complex traits. It aids in the discovery of new genes and regulatory sequences, as well as discovery of molecular markers. Besides, breeders will learn about the molecular basis of complex traits via expression studies. They also make it possible to find markers that are related to genes and QTLs. The generated information from the data analysis will be further utilized in the development of smart finger millet crops through molecular breeding or biotechnological approaches for ensuring nutritional security. The identified key candidate’s genes will be also useful in improving nutritional quality, drought resistance, heat resistance, etc., to the other cereal crops, i.e. wheat, rice, maize, etc., via genetic engineering method.
8.6 Conclusion
As per our traditional knowledge finger millet seeds is considered a powerhouse of nutritionally important compound as biochemical factory for ensuring nutritional security. The present chapter highlights the key information related to finger millet transcriptome data analysis for identification of key genes/proteins involved in different biological processes, i.e. calcium accumulation, synthesis of secondary metabolites useful in human health and other information. This will be helpful for the readers for understanding the methodology of transcriptome data analysis and their outcomes, which will be further utilized in crop improvement program for food and nutritional security.
References
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data
Antony Ceasar S, Maharajan T, Ajeesh Krishna TP, Ramakrishnan M, Victor Roch G, Satish L, Ignacimuthu S (2018) Finger millet [Eleusine coracana (L.) Gaertn.] improvement: current status and future interventions of whole genome sequence. Front Plant Sci 9:1054
Antony U, Chandra TS (1998) Antinutrient reduction and enhancement in protein, starch, and mineral availability in fermented flour of finger millet (Eleusine coracana). J Agri Food Chem 46(7):2578–2582
Avashthi H, Pathak RK, Gaur VS, Singh S, Gupta VK, Ramteke PW, Kumar A (2020) Comparative analysis of ROS-scavenging gene families in finger millet, rice, sorghum, and foxtail millet revealed potential targets for antioxidant activity and drought tolerance improvement. NetMAHIB 9(1):33
Avashthi H, Pathak RK, Pandey N, Arora S, Mishra AK, Gupta VK, Ramteke PW, Kumar A (2018) Transcriptome-wide identification of genes involved in Ascorbate–Glutathione cycle (Halliwell–Asada pathway) and related pathway for elucidating its role in antioxidative potential in finger millet (Eleusine coracana (L.)). 3 Biotech 8(12):499
Beier S, Thiel T, Münch T, Scholz U, Mascher M (2017) MISA-web: a web server for microsatellite prediction. Bioinformatics 33(16):2583–2585
Belton PS, Taylor JR (eds) (2002) Pseudocereals and less common cereals: grain properties and utilization potential. Springer Science and Business Media
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Chinchole M, Pathak RK, Singh UM, Kumar A (2017) Molecular characterization of EcCIPK24 gene of finger millet (Eleusine coracana) for investigating its regulatory role in calcium transport. 3 Biotech 7(4):267
Cirillo E, Parnell LD, Evelo CT (2017) A review of pathway-based analysis tools that visualize genetic variants. Fron Genet 8:174
Conesa A, Götz S, Blast2GO (2008) A comprehensive suite for functional analysis in plant genomics. Inter J Plant Genom
Ćujić N, Šavikin K, Janković T, Pljevljakušić D, Zdunić G, Ibrić S (2016) Optimization of polyphenols extraction from dried chokeberry using maceration as traditional technique. Food Chem 194:135–142
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158
Fardet A, Rock E, Remesy C (2008) Is the in vitro antioxidant potential of whole-grain cereals and cereal products well reflected in vivo? J Cereal Sci 48:258–276
Fernandez DR, Vanderjagt DJ, Millson M, Huang YS, Chuang LT, Pastuszyn A, Glew RH (2003) Fatty acid, amino acid and trace mineral composition of Eleusine coracana (Pwana) seeds from northern Nigeria. Plant Foods Hum Nutr 58(3):1
Funahashi A, Morohashi M, Kitano H, Tanimura N (2003) Cell Designer: a process diagram editor for gene-regulatory and biochemical networks. Biosilico 1(5):159–162
Gibson SJ, Helme RD (2001) Age-related differences in pain perception and report. Clin Geriatr Med 17(3):433–456
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech 7:644–652
Gupta S, Pathak RK, Gupta SM, Gaur VS, Singh NK, Kumar A (2018) Identification and molecular characterization of Dof transcription factor gene family preferentially expressed in developing spikes of Eleusine coracana L. 3 Biotech 8(2):82
Hittalmani S, Mahesh HB, Shirke MD, Biradar H, Uday G, Aruna YR, Lohithaswa HC, Mohanrao A (2017) Genome and transcriptome sequence of finger millet (Eleusine coracana (L.) Gaertn.) provides insights into drought tolerance and nutraceutical properties. BMC genom 18(1):465
Klepikova AV, Kasianov AS, Gerasimov ES, Logacheva MD, Penin AA (2016) A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. The Plant J 88(6):1058–1070
Kokane SB, Pathak RK, Singh M, Kumar A (2018) The role of tripartite interaction of calcium sensors and transporters in the accumulation of calcium in finger millet grain. Biol Plant 62(2):325–334
Kumar A, Gaur VS, Goel A, Gupta AK (2015a) De novo assembly and characterization of developing spikes transcriptome of finger millet (Eleusine coracana): a minor crop having nutraceutical properties. Plant Mol Biol Report 33(4):905–922
Kumar A, Metwal M, Kaur S, Gupta AK, Puranik S, Singh S, Singh M, Gupta S, Babu BK, Sood S, Yadav R (2016) Nutraceutical value of finger millet [Eleusine coracana (L.) Gaertn.], and their improvement using omics approaches. Front Plant Sci 7:934
Kumar A, Pathak RK, Gayen A, Gupta S, Singh M, Lata C, Sharma H, Roy JK, Gupta SM (2018) Systems biology of seeds: decoding the secret of biochemical seed factories for nutritional security. 3 Biotech 8(11):460
Kumar A, Pathak RK, Gupta SM, Gaur VS, Pandey D (2015b) Systems biology for smart crops and agricultural innovation: filling the gaps between genotype and phenotype for complex traits linked with robust agricultural productivity and sustainability. Omics: a j integ bio 19(10):581–601.
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357
Lanham-New SA (2008) Importance of calcium, vitamin D and vitamin K for osteoporosis prevention and treatment: symposium on ‘diet and bone health.’ Proc Nutr Soc 67(2):163–176
Lattimer JM, Haub MD (2010) Effects of dietary fiber and its components on metabolic health. Nutrients 2(12):1266–1289
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17(1):10–12
McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40(10):4288–4297
Mooser V, Carr A (2001) Antiretroviral therapy-associated hyperlipidaemia in HIV disease. Curr Opin Lipid 12(3):313–319
Murtaza N, Baboota RK, Jagtap S, Singh DP, Khare P, Sarma SM, Podili K, Alagesan S, Chandra TS, Bhutani KK, Boparai RK (2014) Finger millet bran supplementation alleviates obesity-induced oxidative stress, inflammation and gut microbial derangements in high-fat diet-fed mice. Br J Nutr 112(9):1447–1458
Parvathi MS, Nataraja KN, Reddy YN, Naika MB, Gowda MC (2019) Transcriptome analysis of finger millet (Eleusine coracana (L.) Gaertn.) reveals unique drought responsive genes. J Genet 98(2):46
Pathak RK, Baunthiyal M, Pandey D, Kumar A (2018b) Augmentation of crop productivity through interventions of omics technologies in India: challenges and opportunities. 3 Biotech 8(11):1–28
Pathak RK, Baunthiyal M, Pandey N, Pandey D, Kumar A (2017a) Modeling of the jasmonate signaling pathway in Arabidopsis thaliana with respect to pathophysiology of Alternaria blight in Brassica. Sci Rep 7(1):1–2
Pathak RK, Baunthiyal M, Shukla R, Pandey D, Taj G, Kumar A (2017b) In Silico Identification of mimicking molecules as defense inducers triggering jasmonic acid mediated immunity against alternaria blight disease in Brassica species. Front Plant Sci 8:609
Pathak RK, Gupta A, Shukla R, Baunthiyal M (2018b) Identification of new drug-like compounds from millets as Xanthine oxidoreductase inhibitors for treatment of Hyperuricemia: a molecular docking and simulation study. Comput Bio Chem 76:32–41
Pathak RK, Singh DB (2020) Systems Biology Approaches for Food and Health. In: Advances in agri-food biotechnology. Springer, Singapore, pp 409–426
Pathak RK, Taj G, Pandey D, Arora S, Kumar A (2013) Modeling of the MAPK machinery activation in response to various abiotic and biotic stresses in plants by a system biology approach. Bioinformation 9(9):443
Rahman H, Jagadeeshselvam N, Valarmathi R, Sachin B, Sasikala R, Senthil N, Sudhakar D, Robin S, Muthurajan R (2014) Transcriptome analysis of salinity responsiveness in contrasting genotypes of finger millet (Eleusine coracana L.) through RNA-sequencing. Plant Mol Biol 85(4–5):485–503
Rana G, Pathak RK, Shukla R, Baunthiyal M (2020) In silico identification of mimicking molecule (s) triggering von Willebrand factor in human: a molecular drug target for regulating coagulation pathway. J Biom Struct Dyn 38(1):124–136
Scalbert A, Manach C, Morand C, Rémésy C, Jiménez L (2005) Dietary polyphenols and the prevention of diseases. Critical Rev Food Sci Nutr 45(4):287–306
Schatzkin A, Mouw T, Park Y, Subar AF, Kipnis V, Hollenbeck A, Leitzmann MF, Thompson FE (2007) Dietary fiber and whole-grain consumption in relation to colorectal cancer in the NIH-AARP Diet and Health Study. Am J Clin Nutr 85(5):1353–1360
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504
Singh UM, Chandra M, Shankhdhar SC, Kumar A (2014) Transcriptome wide identification and validation of calcium sensor gene family in the developing spikes of finger millet genotypes for elucidating its role in grain calcium accumulation. PLoS ONE 9(8):e103963
Subba Rao MV, Muralikrishna G (2002) Evaluation of the antioxidant properties of free and bound phenolic acids from native and malted finger millet (Ragi, Eleusine coracana Indaf-15). J Agri Food Chem 50(4):889–892
Thiel T, Michalek W, Varshney R, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). TAG 106(3):411–22
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Prot 7(3):562–578
Vadivoo AS, Joseph R, Ganesan NM (1998) Genetic variability and diversity for protein and calcium contents in finger millet (Eleusine coracana (L.) Gaertn) in relation to grain color. Plant Foods Human Nutri 52(4):353–64
Wolf JB (2013) Principles of transcriptome analysis and gene expression quantification: an RNA-seq tutorial. Mol Ecol Resour 13(4):559–572
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Pathak, R.K., Singh, D.B., Pandey, D., Gaur, V.S., Kumar, A. (2022). Finger Millet Transcriptome Analysis Using High Throughput Sequencing Technologies. In: Kumar, A., Sood, S., Babu, B.K., Gupta, S.M., Rao, B.D. (eds) The Finger Millet Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-031-00868-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-031-00868-9_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-00867-2
Online ISBN: 978-3-031-00868-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)