
Abstract
Finger millet (Eleusine coracana subsp. coracana) is a cereal food crop for millions of resource-poor people of Africa and Indian subcontinent. It is climate-resilient nutri-cereal which survives under drought and marginal fertile soil. Owing to its ability to grow under diverse agroclimatic conditions, it is a model crop to study the genomic factors responsible for tolerance to abiotic stresses and the same knowledge can be transferred to improve other cereal crops. Finger millet can give nutritional security since it is rich in iron, methionine, and calcium. It is a recommended diet for diabetic patients because of its low glycemic index, high fiber content and lowers cholesterol level. In spite of this, genomic resources developed for this crop were very meager. Fortunately, two genomes of finger millet varieties namely ML365 and PR202 have been sequenced and made available to the scientific community. These genome sequencing efforts will strengthen millet scientists for allele discovery, development of DNA markers, genetic mapping, and identification of candidate genes for disease resistance, drought tolerance and other agronomically important traits, which will boost the finger millet production in near future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
6.1 Introduction
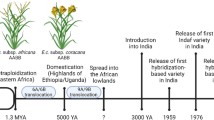
Finger millet (Eleusine coracana subsp. coracana) belongs to the family Poaceae and is one of the neglected and underutilized cereal crops. The grain is nutritious but can be further improved into a nutritious “super cereal” that alleviates malnutrition, especially in women and pre-school children in most countries of south-east Asia and Africa. Finger millet is a more nutritious food grain crop than other cereals (Bhandari et al. 2004; Dida and Devos 2006) in terms of protein (7.3g/100 g) (Malleshi and Klopfenstein 1998), dietary fiber (15–20%) (Chethan and Malleshi 2007) and calcium content (344 mg/100 g) (Gopalan et al. 1971). Furthermore, finger millet is cultivated in semi-arid areas. There are predictions indicating its production will be adversely affected by climate change and it will become a risk to livelihoods of millions of people who are depending on this crop. Hence, development of climate-smart finger millet varieties through innovative technologies is very essential to sustain finger millet cultivation. Initially, finger millet improvement was carried out by conventional breeding methodologies. Such breeding efforts across the globe have led to release of many varieties possessing improved yield and other desirable traits. However, the speed of crop improvement strategy in this crop is very slow as compared to other major cereal crops. This arduous nature could be attributed to slow release of variability, due to polyploidy and difficulty in hybridization owing to small florets in finger millet. The polyploid nature of the crop demands greater shuffling of genes for reaching higher genetic potential. However, these bottlenecks could be delimited with the availability of versatile genetic and genomic resources.
6.2 Complexity of Finger Millet Genome
The cultivated Finger millet (Eleusine coracana subsp. coracana) is an allotetraploid with “AABB” genomes with estimated genome size of ~1.5 billion bases (Dida et al. 2007; Hiremath and Salimath 1991; Mysore and Baird 1997). The E. coracana (2n = 4x = 36) exhibits morphological similarity to both E. indica (2n = 18) and E. africana (2n = 36). Based on the cytological, biochemical and molecular evidences E. indica has been “AA” genome progenitor of cultivated E. coracana and E. africana (Hilu 1988, 1995; Mallikharjun et al. 2005; Werth et al. 1994) species. Both the tetraploid species of finger millet viz., E. africana and E. coracana are genetically related and had a greater advantage of gene flow between them, which is indicating the origin of E. coracana from E. africana through selections and with mutations toward larger grain type (Chennaveeraiah and Hiremath 1974; Hilu and Wet 1976). Further, the detailed study of diploid (E. indica and E. floccifolia) species with ribosomal DNA (rDNA) in comparison with tetraploid species (E. africana) suggested that, the two diploid species might be the donors of two genomes to E. africana (Bisht and Mukai 2001, 2000). But the possibility of E. floccifolia to be as B genome donor was disproved based on nuclear internal transcribed spacers (ITS) and plasmid trnT–trnF sequences (Neves et al. 2005). Kinship was estimated between E. indica and E. tristachya, and between E. floccifolia and E. jaegeri based on biochemical and genetic evidences between them (Hilu and Johnson 1992; Hiremath and Chennaveeraiah 1982; Hiremath and Salimath 1991; Liu et al. 2011). Genetic characterization of E. africana, E. coracana and E. kigeziensis, based on plastid phylogeny indicated the presence of a common ancestor with E. indica—E. tristachya clade, which represents a source for the maternal parents. However, with the recent whole genome and transcriptome sequencing of Eleusine species confirmed that E. indica is the maternal parent of Eleusine coracana and Eleusine africana (Zhang et al. 2019a, b).The exploration studies in defining the source of paternal parent (contributor of BB genome) of three tetraploids indicated the extinction of the actual/probable donor of BB genome and they are not available in any of the germplasm resources available across the globe (Liu et al. 2014).
One more complexity of finger millet genome is transposable elements. Though transposable elements are the major components of eukaryotic genomes but their integration in the genome plays a vital role in genome evolution and duplication. Based on ML365 genome study (Hittalmani et al. 2017), ~49.92% of finger millet genome is repetitive comprising of various retroelements (35.56%), unclassified repeats (9.73%) and DNA transposons (4.48%). Previous study prior to whole genome publication also indicated the abundance of repeats in finger millet genome based on DNA reassociation kinetics (Gupta and Ranjekar 1981). This repetitive nature of the finger millet genome is attributed to larger lengths of interspersed DNA repeats as reported in pearl millet (Gupta and Ranjekar 1981; Wimpee and Rawson 1979). This was one of the major problems in assembling genome to chromosome/pseudomolecule level. Next-generation sequencing (NGS) technology particularly second-generation (Illumina and Ion Proton sequencers) can provide genome sequence in a short period of time (weeks to months) with reduced cost per genome. Although, assembly and annotation are technically challenging due to short read nature of second-generation sequencing. Many crop plants with polyploidy genomes with high repetitive DNA have seen a thrust in their crop improvement programs after the inception of genomic studies providing reference genome assemblies. The later developments in third-generation sequencers like Oxford Nanopore and Pacific Biosystems SMRT sequencing have immensely helped to resolve the repetitive DNA structures of several genomes including finger millet. One such effort was attempted in finger millet by Hatakeyama and coworkers to develop a better assembly of PR202 variety using diverse sequencing technologies. They have used novel multiple hybrid genome assembly workflow coupled with whole genome optical mapping using the BionanoIrys system (Hatakeyama et al. 2018).
Taking into consideration of the facts of genome sequencing and its challenges, attempt in sequencing finger millet genome has been done by two individual groups (Hittalmani et al. 2017; Hatakeyama et al. 2018), which was a leap in developing genomic resources for finger millet. Further, very recent reports of high throughput genotyping and marker trait studies by several researchers will make finger millet a resource-rich crop for further genomics-assisted breeding programs. The current chapter focuses on discussing the available genome assemblies of finger millet.
6.3 ML365 and PR202 Genomes
Recent advances made in NGS technologies facilitated whole genome sequencing of many orphan crops and largescale sequence-based genotyping. Much awaited whole genome sequence of ML365, a drought-tolerant finger millet variety was released in 2017 (Hittalmani et al. 2017), followed by whole genome hybrid assembly release for PR202 variety (Hatakeyama et al. 2018).
ML365 is developed through recombination breeding of Indaf-5 × IE1012 and released in 2008 as a short duration, drought-tolerant and neck blast resistant variety (Gandhi et al. 2012). The PR202(Godavari) is a pureline selection from a landrace of Mettachodi ragi of Araku valley released in 1976 as a drought tolerant and blast susceptible variety (https://www.dhan.org/smallmillets/docs/report/Compendium_of_Released_Varieties_in_Small_millets.pdf), where as PR202 is also used as national check by All India Co-ordinated Research Project (AICRP) on Small Millets for multilocation yield-evaluation trials.
Comparative studies of these genomes is a large-scale approach to understand the similarities and difference of the crop at genome level in multiple perspectives. The important highlights of comparative studies are enumerated in this chapter.
6.3.1 Sequencing Platforms, Data Pre-processing and Assembly
The ML365 whole genome sequencing (WGS) was performed by Illumina and SOLiD sequencing chemistries. Whereas, PR202 genome was sequenced by combination of both second and third generation sequencing technologies like Illumina and PacBio, respectively. In addition, genome optical mapping was carried out for PR202 on a BionanoIrys® system (Bionano Genomics). The WGS of ML365 and PR202 assemblies used both pair-end and mate-pair library preparation workflow. In paired-end sequencing, sequencing will be done from both the 5ʹ and 3ʹ ends producing both forward and reverse orientation of sequence reads. Mate pair sequencing involves long insert paired-end libraries, which are later circularized, fragmented and ligated to another set of adapters and sequencing them in pair-end sequencing chemistry. Once the raw sequence is generated from the sequencer, analysis starts with quality check and pre-processing of raw sequence reads, which are scanned for low base quality, adapter contamination, and duplicate reads. Quality scores measures the probability of incorrect base calling. There are many tools which have been developed for read quality control and pre-processing including FastQC, PRINSEQ, Trimmomatic, Cutadapt, FastX and many more (Table 6.1). Currently, for pre-processing of PR202 raw sequence Trimmomatic was used (Bolger et al. 2014). Trimmomatic is a java based tool that facilitate to remove adapters and trim reads based on quality. FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) is another popular tool used for pre-processing of raw fastq/fasta files of ML365.
6.3.2 Genome Assembly, Scaffolding and Hybrid Assembly
Genome assembly refers to aligning DNA sequence to reconstruct the correct order of original sequence. The reference-based and de novo-based are the two methodologies opted for assembling high-quality sequence reads into contigs. ML365 genome assembly is the first de novo assembly published for finger millet and SOAPdenovo assembler was used for the same, where it is specially designed for Illumina sequences and optimized for large genomes like plant and animal.
Assembling a genome has many computational limits one among them is long tandem repeats, resolving them using short reads is highly difficult which can be accomplished using long reads generated from third-generation sequencing technology, the main challenge of using this technique alone is limitation in accuracy and higher error rates comparatively to short read sequencing. Hybrid assembly is a method that uses data from different sequencing technologies to achieve accuracy. PR202 was assembled using hybrid assembly technique. Platanus is a massive parallel shotgun de novo assembler used for assembling Illumina data and DBG2OLC is a hybrid assembler used for assembling PacBio CLR long reads along with Illumina contigs as anchor points. Pilon is a widely used assembly polish software, used in improving the accuracy of PR202 through an internal reassembly process by correcting sequence errors, mis-assemblies and filling gaps.
Scaffolding is a reconstruction of genome sequence from contigs. SSPACE is a stand-alone scaffolding program used in both ML365 and PR202 assemblies, where contigs were assessed for order, distance and orientation to find a link and combine into scaffolds. Contigs are always of different length, if part of the terminal sequence of one contig fragment overlaps with other reads of contigs it will be combined to make single scaffold and gaps are included as ‘N’ (any nucleotide base) where varied sequence ends are present, so contigs are continuous genomic sequences whereas scaffold contains contigs and gaps (represented as N). As gaps are very commonly generated through scaffolding process, gap filling is a part of assembly process. The GapCloser and Gmclose tools were used in ML365 and PR202, respectively for closing the gaps. GapCloser is a SOAPdenovo module designed to close the gaps using pair relationship of short reads. Gmcloser uses pre-assembled contigs and measures likelihood ratios to improve accuracy and efficiency. Gmcloser was used on super scaffolds of PR202. Super scaffold of PR202 includes assembly from BNG contigs generated from BioNanoIrys®System through optical mapping. Optical mapping is a comprehensive method to understand the genomic structure and structural variation through genome-wide restriction map, it has been widely used to improve the de novo assemblies of eukaryotic as well as prokaryotic genomes. BioNanoIrys®System was used for optical mapping of PR202. Optical mapping was followed by IrysView for de novo assembly for the same. IrysSolve was used for generating contigs from BNG contigs and hybrid scaffolding using BNG contigs as well as hybrid assembly from DBG2OLC assembler.
6.3.3 Comparison of Genome Statistics
Total genome size was estimated in M365 and PR202 finger millet varieties. Propidium iodide was used in staining the nuclei and suspension of stained nuclei were analyzed using a flow cytometer using Pisum sativum and Lycopersicon esculentum as an internal standard for ML365 and PR202, respectively. The assembly generated for ML365 includes Illumina and SOLiD sequencing technologies with mate pair and pair-end reads. Total assembly generated for ML365 is 1196 Mb covering 82.31% of the estimated genome size, which includes 525,759 scaffolds/contigs with average length of 2.2 Kb representing N50 of 23.73 Kb (Table 6.2). PR202 assembly was generated from combination of short reads from Illumina with mate pair and pair-end reads and PacBio long reads. Total assembled genome size is 1189 Mb covering 78.20% representing 28,22,919 contigs in 1,897 hybrid scaffolds with average contig length of 464.72 bp and average hybrid scaffold length of 626.66 Kb representing N50 of 1.4 Kb for contig and 2.6 Mb for hybrid scaffold. The highest GC content of land plants have been found in grasses (Poaceae). GC content of monocots varies between 33.6 and 48.9% (Šmarda et al. 2014), finger millet being monocot belonging to Poaceae family, the GC content of the ML365 assembly was 40.98% and GC content of PR202 assembly estimated to be 44.76% (Table 6.2).
6.3.4 Validation of Genome Completeness and Comparison of Gene Annotation
Assessing the completeness of the assembly for its quality, contiguity and correctness is most important. CEGMA (Core Eukaryotic Genes Mapping Approach) was used in ML365. CEGMA uses highly conserved orthologous genes across eukaryotic species. As per the analysis, around 94.35% of core eukaryotic genes (CEG) were present in the ML365 genome. Whereas BUSCO (Benchmarking Universal Single-Copy Orthologs) which assesses the assembly through single-copy ortholog from OrthoDB database was used in PR202. Around 96.5% of the universal single-copy genes were identified indicating that quality of the PR202 genome was good. BUSCO is an improved version of CEGMA. Both the assemblies possessed good quality for further downstream annotation. Annotation refers to identification of relevant features of genome sequence. It is divided into two types structural and functional annotation; structural annotation identifies the gene locations which includes exons, introns, UTRs, etc., whereas functional annotation refers to assigning the function to genes which it encodes like physiological function, cellular function, biochemical and metabolic activity so on. Structural gene annotation of ML365 genome was carried out by AUGUSTUS (tool widely used in eukaryotic gene prediction based on generalized hidden Markov model) with RNA-seq evidence using Zea mays as a reference model. This enabled prediction of 85,243 genes in ML365 genes and gene ontology (GO) annotation was done with Viridiplantae protein sequences retrieved from UniProt database. The PR202 hybrid assembly was used for gene prediction using MAKER tool using Oryza sativa as a model species and 62,348 genes were identified. RNA-seq data generated from young leaves were mapped to the hybrid assembly of PR202 using STAR aligner showed 95.9% mapping, which strongly supported assembly may contain ~95% predicted gene set through MAKER.
Owing to finger millet’s ability to grow under diverse agro-climatic conditions, it is a model crop to study the genomics of drought tolerance as compared to other crops like rice, sorghum, maize, foxtail millet. In ML365, 2,866 drought tolerant genes have been identified based on RNA sequencing experiment followed by gene annotation. In addition, 11,125 genes in ML365 genome are known to harbor transcription factor (TFs) distributed across 56 various families. The most widely distributed TFs are bHLH, MYB, FAR1, WRKY, NAC, MYB related, B3, ERF, bZIP, HD-ZIP, C2H2, C3H, G2- like, TALE, GRAS, ARF, M-type, Trihelix, GATA, WOX, LBD, HSF, MIKC, S1Fa-like, HB other, CPP, and YABBY. This strongly validates the inherent nature of drought tolerance character of the finger millet.
6.3.5 Comparative Analysis of Functional Classification of Proteins
Total proteins predicted from both the assembly were subjected for their functional classification using reverse position specific BLAST (RPS-BLAST) program on KOG (EuKaryotic Orthologous Groups) database with e-value 0.001 to obtain specific hit using WebMGA. The RPS-BLAST searches protein query against the conserved domain database (CDD) collected from many source databases either in standalone or using web server. Functional domains were classified into three main classes viz.., information storage and processing, cellular process and signaling, and metabolism. There is one more class which is poorly characterized which includes 2 sub class falls on general and unknown function classification, remaining 23 classes out of 25 falls on main class functional groups (Fig. 6.1a). All predicted genes in ML365 (85,243) were subjected to RPS-BLAST to predict the domain, out of them only 36,866 were predicted to have specific hit and found in 3,519 conserved protein domain family (Pfam). In case of PR202, total number of genes predicted is 62,348 and all of them were used in RPS-BLAST. 37,711 were found to have specific hit into 3677 Conserved Protein Domain Family. Comparative Pfam domain analysis between ML365 and PR202 showed that 30,460 ORFs (open reading frames) were common, 7,251 ORFs were unique to PR202 and 6,406 ORFs were unique to ML365 (Fig. 6.1b).

Comparison of functional classification of KOGs (Eukaryotic Orthologous Groups) between ML365 and PR202
6.3.6 Clustering of Gene Families
Predicted genes of ML365 (85,243) and PR202 (62,348) were clustered using OrthoVenn2 tool. Gene repertoire of these varieties formed a 24,445 core homologous clusters consisting of 75,459 genes (41,906 from ML365 and 33,553 from PR202). The gene ontology (GO) annotation of these core homologous genes showed that majority of genes belonging to response to osmotic stress, oxidative stress and lipid catabolic process (Fig. 6.2a). Remaining 43,337genes of ML365 were unique which formed 3,113 clusters (18,651 genes are 2 copies—paralogs) and 24,686 single-copy genes. Tricarbolic acid cycle, glycolytic process, DNA integration, RNA-mediated transposition and fatty acid beta-oxidation were the major gene ontology annotations of paralogs genes in ML365 (Fig. 6.2b). Similarly, 4,115 paralog clusters comprising of 11,026 genes and 17,769 single-copy genes were identified in PR202 genome. Majority of these paralogs have a gene ontology function in plasma membrane, protein glycosylation response to oxidative stress and osmotic stress (Fig. 6.2c).

Comparative gene family analysis of ML365 and PR202
6.4 Current Status of Finger Millet Production
Over the last two decades area and production of finger millet is in declining phase due to replacement of finger millet by other competitive crops. Finger millet varieties have been developed mostly through selection or hybridization followed by selection. Attempts have been made in realizing high yield potential using the worldwide genetic resources. The maximum yield potential of 5000–5500 kg/ha have been achieved and is almost reaching stagnation. In three decades, finger millet productivity in India has increased considerably and is the highest (1500 kg/ha) among all the millets including sorghum and pearl millet. This is largely due to the development and spread of high yielding and blast-resistant varieties by exploiting African germplasm. But newly developed and currently cultivated varieties are relatively constrained by biotic and abiotic stresses. Therefore, there is an urgent need to enhance tolerance to biotic and abiotic stresses in order to stabilize productivity, extend their adaptation and tailor them to suit changing climate. In the coming years climate change, water scarcity, increasing world population, rising food prices, and other socio-economic impacts are expected to generate a great threat to agriculture and food security worldwide, especially for the poorest people who live in arid and sub-arid regions. The crisis can be challenged by sustainable food production through development of high-yielding cultivars in finger millet. Breeding of finger millet with genetic and genomic studies aided by recent high throughput genotyping platforms may be helpful to develop cultivars/varieties with desired features/traits of interest.
6.5 Production Constraints in Finger Millet
Finger millet production is severely affected by both biotic and abiotic stresses (Saha et al. 2016). Fungal blast is a major disease caused by Magnaporthe grisea (anamorph Pyricularia grisea), affecting growth and yield of finger millet. The fungus mostly infects young leaf and causes leaf blast, whereas under highly favorable conditions, neck and finger blasts are also formed at flowering (Babu et al. 2013). This disease has been identified as the highest priority constraint to finger millet production in Eastern Africa, and India since most of the genotypes are highly susceptible. The disease affects the crop at all growth stages however, neck blast and finger blast are the most destructive forms of disease. The blast fungus enters and causes the breakdown of parenchymatous, sclerenchymatous, and vascular tissues of the neck region, thereby inhibiting the flow of nutrients into the grains (Rath and Mishra 1975). Subsequently, grain formation is partially or totally inhibited and the infected spikelets will be shorter than healthy spikelets, which affects the grain formation (Ekwamu 1991; Rath and Mishra 1975). Till now there are no reports on the molecular characterization and mapping of blast resistant genes in finger millet. So, there is a need to identify the molecular markers linked to the blast resistance for their further introgression into locally well-adapted germplasm.
Major abiotic stresses such as deficiencies of nutrients [nitrogen (N), phosphorus (P), and zinc (Zn)], drought, and salinity also seem to affect the growth and yield of finger millet. According to a recent study, N deficiency decreased the tiller number in finger millet (Goron and Raizada 2015). Low P stress also affected the growth and biomass of finger millet seedlings in glass house conditions (Ramakrishnan et al. 2017). Zinc deficiency resulted in stunted growth, delayed seed maturity, appearance of chlorosis, shortened internodes and petioles, and malformed leaves (Yamunarani et al. 2016). Drought is also one of the major abiotic constraints of finger millet production. Drought stress caused wilting and leaf rolling and resulted in the reduction of leaf solute potential and chlorophyll content with the induction of many drought stress-responsive genes when compared to control condition (Parvathi et al. 2013). Salinity also reduced the water content, plant height, leaf expansion, finger length and width, grain weight, and delayed the flowering (Anjaneyulu et al. 2014). In addition to these, the other concerns are poor understanding of the Finger millet genome biology, non-availability of microsatellite markers and single nucleotide polymorphisms (SNPs). Non-availability of an appropriate bi-parental mapping population for traits of interest leading to non-availability of genetic linkage maps, limiting the application of translational genomics and marker-assisted selection (MAS). Similarly, the non-availability of physical maps till date has limited the deployment of genome-wide association study (GWAS) and genomic selection (GS) strategy in crop improvement programs and limited knowledge about sequence diversity between cultivated and wild species in finger millet has stalled the prospective of genomic assisted breeding.
6.6 Future Perspectives
Indian agriculture is always a gambling with monsoon and this uncertainty could be minimized by growing drought-tolerant crops like finger millet and other millet crops. Finger millet is right now occupying an important position as a ‘Nutri-cereal’ rather than as a coarse cereal due to its potential use in combating malnutrition and hidden hunger worldwide. Finger millet is rich in iron, methionine, and calcium and can give a nutritional security to mitigate malnourishment in the country. Finger millet grain contains ten times more calcium than any other cereals, such calcium transport and accumulation-related genes have been identified through ML365 genome sequencing study. With profound nutritional significance now a day's finger millet is popularizing as a trendy food among diet-conscious people to maintain healthy lifestyle and to prevent lifestyle disorders, chronic and non-communicable diseases. Finger millet is a recommended food for diabetic patients because of its low glycemic index (slow releasing of sugar to the blood), high fiber and cholesterol-lowering ability. These inherent properties make finger millet an ideal model for studying genomics and a plausible source for gene mining for complex traits. Molecular breeding has witnessed its importance as a promising tool for imparting stress tolerance in economically important plants, however, until now the progress is limited in finger millet mainly due to lack of appropriate genomic resources. High throughput sequencing platforms have enabled to generate more of genomic resources is less possible time in neglected/overlooked crops like finger millet. The recent release of draft genomes may aid to develop high-resolution studies, namely, forward and reverse genetics, functional genomics, and proteomics studies in finger millet. With the availability of the high-quality genome sequence of E. coracana, it will be possible to identify new targets of selection and its use in genomic selection coupled with prediction approaches. In addition, these advances will not only enable us to overcome the challenges of understanding large and complex finger millet genome but will also help to understand the regulation of genes at transcriptional, post-transcriptional, epigenetic level. This will speed up the breeding process and will allow cumulative improvement for yield, disease resistance and nutritional quality. Development of ‘ideotype breeding’ in finger millet may also be possible in the future by incorporating various agronomically important traits into the genome of a single finger millet genotype/cultivar. Thus, utilization of current advances in molecular breeding and with advanced well-defined genome assemblies will have an impact in improving the present scenario of research in finger millet. The new genomic resource is expected to enrich the finger millet research in many domains including dissection of key traits involved in nutrient enrichment and drought tolerance using GWAS, genetic diversity analysis based on SNP and functional genomics studies. Overall, the recently released WGS of finger millet is expected to augment the finger millet research for its breeding and improvement.
References
Anjaneyulu E, Reddy PS, Sunita MS, Kishor PBK, Meriga B (2014) Salt tolerance and activity of antioxidative enzymes of transgenic finger millet overexpressing a vacuolar H+-pyrophosphatase gene (SbVPPase) from Sorghum bicolor. J Plant Physiol 171(10):789–798
Babu TK, Thakur R, Upadhyaya H, Reddy PN, Sharma R, Girish A, Sarma N (2013) Resistance to blast (Magnaporthe grisea) in a mini-core collection of finger millet germplasm. Eur J Plant Pathol 135(2):299–311
Bhandari B, Gyawali S, Rijal D, Sthapit B, Shrestha P (2004) Value addition and market promotion in finger millet crop: LI-BIRD’s experiences. In: Proceedings on 24th national summer crops workshop, Nepal Agricultural Research Council, Lalitpur, Nepal, pp 28–30
Bisht M, Mukai Y (2001) Genomic in situ hybridization identifies genome donor of finger millet (Eleusine coracana). Theor Appl Genet 102(6–7):825–832
Bisht MS, Mukai Y (2000) Mapping of rDNA on the chromosomes of Eleusine species by fluorescence in situ hybridization. Genes Genet Syst 75(6):343–348
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Chennaveeraiah M, Hiremath S (1974) Genome analysis of Eleusine coracana (L.) Gaertn Euphytica 23(3):489–495
Chethan S, Malleshi N (2007) Finger millet polyphenols: Characterization and their nutraceutical potential. Amer J Food Technol 2(7):582–592
Dida MM, Devos KM (2006) Finger millet. In: Kole C (ed) Genome mapping and molecular breeding in plants, Volume 1: cereals and millets. Springer, pp 333–343
Dida MM, Ramakrishnan S, Bennetzen JL, Gale MD, Devos KM (2007) The genetic map of finger millet, Eleusine coracana. Theor Appl Genet 114(2):321–332
Ekwamu A (1991) Influence of head blast infection on seed germination and yield components of finger millet (Eleusine coracana L. Gaertn). Intl J Pest Manag 37(2):122–123
Gandhi RV, Shailaja H, Babu NN (2012) Performance of new ragi variety ML 365 by adopting production technology in southern Karnataka India. Intl J Plant Sci (muzaffarnagar) 7(1):176–180
Gopalan C, Rama Sastri B, Balasubramanian S (1971) Nutritive values of indian foods. National Institute of Nutrition (ICMR), Hyderabad (India)
Goron TL, Raizada MN (2015) Genetic diversity and genomic resources available for the small millet crops to accelerate a New Green Revolution. Front in Plant Sci 6:157
Gupta VS, Ranjekar P (1981) DNA sequence organization in finger millet (Eleusine coracana). J Biosci (bangalore) 3(4):417–430
Hatakeyama M, Aluri S, Balachadran MT, Sivarajan SR, Patrignani A, Grüter S, Poveda L, Shimizu-Inatsugi R, Baeten J, Francoijs K-J (2018) Multiple hybrid de novo genome assembly of finger millet, an orphan allotetraploid crop. DNA Res 25(1):39–47
Hilu K, Johnson J (1992) Ribosomal DNA variation in finger millet and wild species of Eleusine (Poaceae). Theor Appl Genet 83(6–7):895–902
Hilu K, Wet JD (1976) Racial evolution in Eleusine coracana ssp. coracana (finger millet). Amer J Bot 63 (10):1311–1318
Hilu KW (1988) Identification of the “A” genome of finger millet using chloroplast DNA. Genetics 118(1):163–167
Hilu KW (1995) Evolution of finger millet: evidence from random amplified polymorphic DNA. Genome 38(2):232–238
Hiremath S, Chennaveeraiah M (1982) Cytogenetical studies in wild and cultivated species of Eleusine (Gramineae). Caryologia 35(1):57–69
Hiremath S, Salimath S (1991) Quantitative nuclear DNA changes in Eleusine (Gramineae). Plant Syst Evol 178(3):225–233
Hittalmani S, Mahesh H, Shirke MD, Biradar H, Uday G, Aruna Y, Lohithaswa H, Mohanrao A (2017) Genome and transcriptome sequence of finger millet (Eleusine coracana (L.) Gaertn.) provides insights into drought tolerance and nutraceutical properties. BMC Genomics 18(1):465
Liu Q, Jiang B, Wen J, Peterson PM (2014) Low-copy nuclear gene and McGISH resolves polyploid history of Eleusine coracana and morphological character evolution in Eleusine. Turk J Bot 38(1):1–12
Liu Q, Triplett JK, Wen J, Peterson PM (2011) Allotetraploid origin and divergence in Eleusine (Chloridoideae, Poaceae): evidence from low-copy nuclear gene phylogenies and a plastid gene chronogram. Ann Bot 108(7):1287–1298
Malleshi N, Klopfenstein C (1998) Nutrient composition, amino acid and vitamin contents of malted sorghum, pearl millet, finger millet and their rootlets. Intl J Food Sci Nutr 49(6):415–422
Mallikharjun DR, Hiremath SC, Rao SR, Kumar A, Sheelavanthmath SS (2005) Genome interrelationship in the genus Eleusine (Poaceae) as revealed through heteroploid crosses. Caryologia 58(4):300–307
Mysore K, Baird V (1997) Nuclear DNA content in species of Eleusine (Gramineae): a critical re-evaluation using laser flow cytometry. Plant Syst Evol 207(1):1–11
Neves SS, Swire-Clark G, Hilu KW, Baird WV (2005) Phylogeny of Eleusine (Poaceae: Chloridoideae) based on nuclear ITS and plastid trnT–trnF sequences. Mol Phylogenetics Evol 35(2):395–419
Parvathi M, Nataraja KN, Yashoda B, Ramegowda H, Mamrutha H, Rama N (2013) Expression analysis of stress responsive pathway genes linked to drought hardiness in an adapted crop, finger millet (Eleusine coracana). J Plant Biochem Biotechnol 22(2):193–201
Ramakrishnan M, Ceasar SA, Vinod K, Duraipandiyan V, Ajeesh Krishna T, Upadhyaya HD, Al-Dhabi N, Ignacimuthu S (2017) Identification of putative QTLs for seedling stage phosphorus starvation response in finger millet (Eleusine coracana L. Gaertn.) by association mapping and cross species synteny analysis. PLoS One 12(8):e0183261
Rath G, Mishra D (1975) Nature of losses due to neck blast infection in ragi. Sci Cult 41(7):322–323
Saha D, Gowda MC, Arya L, Verma M, Bansal KC (2016) Genetic and genomic resources of small millets. Crit Rev Plant Sci 35(1):56–79
Šmarda P, Bureš P, Horová L, Leitch IJ, Mucina L, Pacini E, Tichý L, Grulich V, Rotreklová O (2014) Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc Natl Acad Sci USA 111(39):E4096–E4102
Werth CR, Hilu KW, Langner CA (1994) Isozymes of Eleusine (Gramineae) and the origin of finger millet. Amer J Bot 81(9):1186–1197
Wimpee CF, Rawson JR (1979) Characterization of the nuclear genome of pearl millet. Biochim Biophys Acta (BBA)-Nucleic Acids Protein Synthesis 562(2):192–206
Yamunarani R, Govind G, Ramegowda V, Thammegowda HV, Guligowda SA (2016) Genetic diversity for grain Zn concentration in finger millet genotypes: potential for improving human Zn nutrition. Crop J 4(3):229–234
Zhang H, Hall N, Goertzen LR, Bi B, Chen CY, Peatman E, Lowe EK, Patel J, McElroy JS (2019a) Development of a goosegrass (Eleusine indica) draft genome and application to weed science research. Pest Manage Sci 75(10):2776–2784
Zhang H, Hall N, Goertzen LR, Chen CY, Peatman E, Patel J, McElroy JS (2019b) Transcriptome analysis reveals unique relationships among Eleusine species and heritage of Eleusine coracana. G3: Genes Genomes Genetics 9(6):2029–2036
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mahesh, H.B., Manasa, K.G., Raghavendra, N.R., Shirke, M.D., Hittalmani, S. (2022). The Complete Genome Sequence of Finger Millet. In: Kumar, A., Sood, S., Babu, B.K., Gupta, S.M., Rao, B.D. (eds) The Finger Millet Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-031-00868-9_6
Download citation
DOI: https://doi.org/10.1007/978-3-031-00868-9_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-00867-2
Online ISBN: 978-3-031-00868-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)