
Abstract
Diseases of the heart and skeletal muscle represent a major cause of mortality and morbidity in the United States. Many, if not most, are refractory to contemporary medical approaches, which rarely address the root pathogenesis. Regenerative medicine is a specialized field focused on replacing or repairing damaged tissue, which is otherwise incapable of self-healing. For more than four decades, stem and progenitor cells have been recognized for their potential as therapeutic products for regenerative medicine. Despite the long history of scientific inquiry, no cell therapy product has received regulatory approval for regenerative medicine applications in the United States. Recent initiatives focused on understanding the mechanistic basis of cell therapy have fundamentally redirected the trajectory of their development. In doing so, a class of extracellular vesicles called exosomes have emerged as next-generation therapeutic candidates for regenerative medicine. In this chapter, we discuss the difficulties with the commercial development of cell therapy products and the promise of exosomes as next-generation therapeutics. The field of regenerative medicine is ever-changing, and the latest technological advances continue to define our path forward.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
7.1 Diseases of the Heart and Skeletal Muscle
All vertebrates have specialized muscle cells called myocytes which form the functional apparatus of the heart and skeletal muscle. Although myocytes from both organs share similarities such as contracting to generate force to perform their functional duties, crucial differences distinguish the two. Cardiomyocytes beat spontaneously and are electromechanically coupled to form the myocardium—the contractile tissue of the heart. In contrast, skeletal myocytes do not beat spontaneously (motor neurons trigger contraction in response to efferent input) and skeletal myocytes are electrically isolated from each other. Another distinguishing feature of heart and skeletal muscle is the intrinsic ability to repair in response to injury or disease. Unlike the adult heart, which lacks meaningful regenerative capabilities, skeletal muscle exhibits a robust regenerative response primarily driven by resident stem cells. Notwithstanding such ability, certain injuries overwhelm the innate regenerative capacity, and some chronic skeletal muscle conditions impair the reparative machinery, which consequently manifests as muscle dysfunction. Diseases of the heart and skeletal muscle, for which a paucity of effective therapies often exist, can lead to long-lasting physical inability and poor quality of life. In this chapter, we discuss the most prevalent forms of degenerative or traumatic heart and skeletal muscle disease (see Fig. 7.1 for a schematic overview), avenues for regenerative cell therapy, challenges in the translation of therapeutic products to the clinic, and the promise of novel cell-based approaches. We conclude with a look at future perspectives in regenerative medicine.

Diseases of the heart and skeletal muscle. Illustration of shared pathology in the heart and skeletal muscle, and targets for cell-based regenerative therapies. Some schematics were adapted from Servier Medical Art
7.1.1 Heart Failure with Reduced Ejection Fraction
Heart failure (HF) is a disorder in which exercise tolerance is reduced and pulmonary congestion develops. HF affects more than five million people in the United States, with cases expected to climb to eight million within the next decade (Mozaffarian et al. 2016). Based on ejection fraction (EF), that is, a gross measure of the heart’s pumping efficiency, HF can be broadly classified into two groups: (1) HF with reduced EF (HFrEF) and (2) HF with preserved EF (HFpEF) (Ponikowski et al. 2016; Savarese and Lund 2017). Approximately half of HF cases in the USA are due to HFrEF and the remainder to HFpEF. In this section, we summarize the etiology of HFrEF and provide an outlook of clinical outcomes in patients with HFrEF.
HFrEF is often an evolution of the heart’s failed adaptation to ischemic injury: thrombosis of a coronary artery reduces blood and oxygen supply to the myocardium resulting in necrosis—a process commonly known as myocardial infarction (Marbán 2018a). Prompt restoration of blood flow is crucial to minimize infarct size and thus has become a staple in the first-line treatment of myocardial ischemia. However, despite successful recanalization of the blocked artery, persistent ischemic regions in the myocardium can remain, leading to HF and poor clinical outcomes (Basalay et al. 2020). In humans, clear evidence of irreversible myocardial damage can be seen in as little as 24 h post-infarction. The damaged myocardium undergoes a dynamic remodeling process, which unfavorably reshapes the heart (Heusch et al. 2014), manifesting as ventricular dilation and infarct expansion (Pfeffer and Braunwald 1990). Independent of the death of living heart tissue, myocardial remodeling contributes to the development of ischemic heart disease and progression to HF, a stage at which prognosis is generally considered poor. The Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) study which enrolled 20,118 patients with HFrEF reported three dismal statistics: (1) a 60–90 day mortality of 9.8%, (2) a rehospitalization rate of 29.9%, and (3) an in-hospital mortality of 3.9% (Savarese and Lund 2017). Despite significant advancements in the field, standard-of-care for HFrEF mostly relies on symptom relief and slowing of disease progression by a variety of neurohormonal blockers and other agents acting indirectly on the heart.
7.1.1.1 Current Clinical Practice in HFrEF
In HFrEF, both pharmacological and non-pharmacological interventions are in current clinical practice and have been demonstrated to extend life. Pharmacological agents such as angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), beta-adrenergic blockers, and angiotensin receptor-neprilysin inhibitors (ARNI) have been associated with improvements in clinically meaningful outcomes in patients with HFrEF (Yancy et al. 2013). Thus, a patient with HFrEF is typically on a cocktail of various types of agents, titrated to maximal doses tolerated (Yancy et al. 2017). Non-pharmacologic interventions include: (a) implantable cardioverter defibrillator to prevent sudden death due to ventricular tachyarrhythmias, (b) cardiac resynchronization therapy to improve myocardial mechanics, and, in the most extreme cases, (c) organ transplantation (Yancy et al. 2013). Despite pharmacological and non-pharmacological interventions, the 5-year mortality rate for symptomatic HFrEF patients approaches 50–75% (Shah et al. 2017). This gloomy statistic highlights the desperate need for the development of novel therapies.
A major limitation of therapeutic approaches to HFrEF is that none addresses the root physiologic cause of the HF: loss of functional myocardium. This is where regenerative approaches may, in principle, prove helpful.
7.1.2 Heart Failure with Preserved Ejection Fraction
The other form of HF, HFpEF, already constitutes ~50% of all HF admissions and is increasing in prevalence (Gladden et al. 2018). Despite having a normal EF, HFpEF patients exhibit classical HF symptoms including breathlessness and exertional intolerance. In HFpEF, the myocardium tends to be thicker than normal, rather than dilating and thinning as seen in HFrEF (Borlaug 2014). Clinical diagnosis of HFpEF requires a patient to present with normal EF and diastolic dysfunction. However, recent research suggests HFpEF is a multifactorial systemic disease involving hypertension, metabolic disorders, and inflammation (Soni et al. 2020).
Regardless of comorbidities, the main cause of functional deterioration is diastolic dysfunction, which is clinically defined as an impaired ability of the ventricle to fill with blood to an appropriate preload volume at a physiological pulmonary venous pressure (Borlaug et al. 2013). When other heart conditions such as endocardial or pericardial disease can be ruled out, diastolic dysfunction is the direct result of increased myocardial stiffness which can be regulated by both the extracellular matrix and the cardiomyocyte (Borlaug and Paulus 2011). In the most fundamental sense, deposition of matricellular proteins such as collagen contributes to increases in extracellular matrix stiffness. However, changes inside the cardiomyocyte can be a little more complex. The sarcomeric protein titin has been identified as the primary culprit: up to 80% of passive stiffness can be explained by this protein alone—especially when the sarcomere length is within the physiological range (Borlaug and Paulus 2011). Mechanistically, the more compliant titin isoform, N2BA, shifts to the less compliant, that is, more stiff, isoform N2B (Borlaug and Paulus 2011). Additionally, passive tension of the cardiomyocyte can be increased by posttranslational phosphorylation of titin by protein kinases A and G (Borlaug and Paulus 2011). Clinical outcomes from the OPTIMIZE-HF study, which enrolled 21,149 HFpEF patients, reported nearly identical statistics compared to HFrEF: (1) a 60–90 day mortality of 9.5%, (2) a rehospitalization rate of 29.2%, and (3) in-hospital mortality of 2.9% (Savarese and Lund 2017). Thus, while HFpEF is fundamentally distinct from HFrEF, HFpEF patients experience a correspondingly poor prognosis.
7.1.2.1 Current Clinical Practice in HFpEF
Given the principal distinction between HFpEF and HFrEF, it is not surprising the management of HFpEF should differ from that of HFrEF. In support of this notion, clinical trials testing the efficacy of agents commonly used for HFrEF, such as ACE inhibitors and ARBs, failed to decrease morbidity and mortality in HFpEF (Ma et al. 2020). Given the lack of clinical trials reporting positive results, HFpEF treatment remains largely focused on managing associated conditions such as hypertension and acute decompensations due to pulmonary edema. Like HFrEF, the 5-year mortality rate for HFpEF is 50–75% (Shah et al. 2017), further driving the need for medical interventions to meet dire demand.
In terms of therapeutic options, the situation in HFpEF is worse than that in HFrEF, in that no drugs or devices have been shown to increase survival. Our best current understanding implicates fibrosis and inflammation in the pathogenesis of HFpEF, so targeting those processes may eventually turn out to be more fruitful. Here, cell therapies may have a niche, not by replacing lost myocardium but rather by virtue of anti-fibrotic and anti-inflammatory effects. Such pleiotropic effects of cell therapy are discussed further below.
7.1.3 Duchenne Muscular Dystrophy
For an overview of Duchenne muscular dystrophy (DMD), we turn the reader to Chap. 4 where DMD has been reviewed in detail; here we cover regenerative approaches only. Initially identified as a disease of skeletal muscle, DMD is now recognized also as a disease of the heart. As heart function deteriorates in DMD patients, clinical signs of HF develop. Improved respiratory care has prolonged life such that HF is now often the terminal process in DMD (Buddhe et al. 2018). As part of disease management for the DMD patient, regular heart evaluations have become standard (Birnkrant et al. 2018), but little can be done clinically in terms of improving heart function in DMD. The goal is to identify early myocardial changes and initiate therapy to favorably affect adverse myocardial remodeling, and thus improving clinical outcomes and quality of life (Buddhe et al. 2018). Given a lack of therapies for DMD-related cardiomyopathy, the 2018 DMD Care Considerations have endorsed following traditional HF treatment strategies, such as those highlighted in Sect. 7.1.1.1. Unfortunately, with the lack of information regarding the use of these therapies in patients with DMD, specific recommendations for their use in DMD-related cardiomyopathy remains challenging. And, in most cases, the degree of respiratory insufficiency and muscular weakness commonly observed in patients with DMD, who have developed severe myocardial dysfunction, is thought to be a contraindication to transplantation (Connuck et al. 2008; Buddhe et al. 2018). Therefore, novel interventions that improve both heart and skeletal muscle health are desperately needed.
Other than exon-skipping therapies and corticosteroids, which appear not to improve the heart in DMD, no therapies address the root pathophysiological causes of the muscle dysfunction.
7.1.4 Volumetric Muscle Loss
In the field of regenerative medicine, much of the clinical and basic research experience to date has been focused on diseases of the heart. Owing to its ability to self-repair after nontraumatic injury, skeletal muscle has received much less attention in the realm of regenerative medicine. Nevertheless, certain disorders of skeletal muscle may represent viable targets. One such condition is volumetric muscle loss (VML). Because VML has been covered in detail in Chap. 6, the discussion here will be focused on regenerative approaches in the context of cell-based therapy. In VML, suboptimal, if any, endogenous muscle regeneration occurs, which is thought to be the result of three principal defects to the skeletal muscle repair machinery (Greising et al. 2019). First, resident muscle stem cells (satellite cells) no longer populate the affected area, which are strictly required for skeletal myogenesis (Caldwell et al. 1990; Lepper et al. 2011). Second, the muscle extracellular matrix, which provides a three-dimensional scaffold for cell migration and tissue organization, becomes disrupted (Caldwell et al. 1990). And third, a prolonged wave of inflammation stimulates the deposition of fibrotic scar tissue (Greising et al. 2017; Aguilar et al. 2018). Without successful regenerative medicine interventions, the VML-injured limb, even if salvaged, may have little function and thus is later amputated.
7.2 Cell Therapy for Regenerative Medicine
Regenerative medicine is a specialized branch of medicine which seeks to repair or replace tissues and organs damaged by trauma or disease. The recognition that stem or progenitor cells can be instructed to differentiate into numerous cell types quickly garnered the attention of researchers and has since become a conceptually attractive approach to repair damaged tissue. In principle, cell therapy can be used for a broad spectrum of acquired and genetic disorders by targeting the primary and/or secondary disease features. Nevertheless, the amount of hype in this area far exceeds that of translationally viable approaches to date, such that, despite four decades of preclinical and clinical efforts, no cell therapy products have been approved for regenerative indications.
7.2.1 From Discovery to Clinical Translation
The process of translating findings, beginning from discovery-level science to preclinical development, to clinical evaluation is instrumental in the effort to advance medical science. In this section, we discuss the dedication of decades of scientific inquiry that has led to what is now an era of superlative possibilities (but generally disappointing realities).
7.2.1.1 Heart Failure with Reduced Ejection Fraction
The first efforts in regenerative cardiology were founded on the earlier finding that autologous skeletal muscle-derived myoblasts could engraft and proliferate when transplanted directly into the heart (Voronov 1975). As a reminder, skeletal myocytes (unlike cardiomyocytes) do not couple in syncytium, nor do they spontaneously contract. Even so, the hypothesis was that the transplanted cells would stimulate the generation of new contractile tissue within the host myocardium. The development of skeletal myoblasts for cardiac regeneration started with small animal models (Koh et al. 1993), advanced to more clinically relevant models (Taylor et al. 1998), and ultimately finished with in-human clinical trials (Hare et al. 2008). Although signs of efficacy were evident, efforts were later abandoned after it was discovered that transplanted myoblasts were arrhythmogenic in patients with HFrEF (Marbán 2018a).
The next cell type evaluated for cardiac regeneration was unselected bone marrow-derived cells, which were shown to drive repair in preclinical studies (Orlic et al. 2001; Jackson et al. 2001). While early phase clinical testing demonstrated improvements in EF (Meyer et al. 2006), the study of bone marrow cells was short-lived insofar as larger randomized, placebo-controlled clinical trials were unable to reproduce earlier findings (Traverse et al. 2012; Sürder et al. 2013). A second-generation of cell products began experimental evaluation as the trials of bone marrow cells were concluding. Notably, bone marrow-derived mesenchymal stem cells (MSCs) (Wang et al. 2016) were tested for cardiac regeneration owing to success in other disease models, in which they had already been evaluated previously. More specifically, antigen-selected mesenchymal precursor cells (Psaltis et al. 2010) and cardiopoietic cells (autologous MSCs reinforced in vitro) (Behfar et al. 2008) have reached clinical testing for heart failure, but have been met with a modest level of improvement. In the search for alternative strategies, recruiting and augmenting innate regenerative responses in the heart by cardiac progenitor cells seemed particularly attractive. In this context, cardiac progenitor cells were evaluated due to their potential to engraft, proliferate, and differentiate into new viable myocardium. Cells isolated from hearts by the selection of the c-kit surface antigen were proposed to regenerate the myocardium by these canonical mechanisms (Beltrami et al. 2003), which eventually motivated the SCIPIO trial using autologous c-kit-selected heart cells (Bolli et al. 2011). Unfortunately, the background science supporting this trial was later called into question (Van Berlo et al. 2014), numerous basic studies were deemed fraudulent, and the SCIPIO trial itself was later retracted (Lancet 2019).
At about the same time many researchers were evaluating the efficacy of bone marrow cells and autologous MSCs (circ. 2006), a population of cardiac progenitor cells, called cardiosphere-derived cells (CDCs) was first described (Smith et al. 2007). CDCs do not require antigenic selection and were confirmed to be of intrinsic cardiac origin Marbán and Liao (2022). Early preclinical work in animal models of HFrEF demonstrated CDCs exhibit profound regenerative bioactivity in mice, rats, and pigs, and in some experiments CDCs outperformed MSCs (Malliaras and Marbán 2011). In light of the remarkable preclinical safety and efficacy data demonstrated by CDCs, a first in-human clinical trial (CADUCEUS) of autologous CDCs began in 2009. The results from CADUCEUS showed a reduction in infarct size and an increase in viable myocardium, as evidenced by cardiac MRI at 6 and 12 months following treatment (Makkar et al. 2012). While CADUCEUS was in process, basic studies made it clear that transplanted CDCs left behind lasting structural and functional benefits but did not linger in the heart for more than 3–4 weeks, leading to a shift from the autologous paradigm (cells grown from a given patient and transplanted back into that same patient) to an allogeneic (unrelated donor) paradigm. Following the CADUCEUS trial, two additional trials tested the efficacy of allogeneic CDCs—such that a cell product can be manufactured and stored for on-demand access. The first trial (ALLSTAR), while showing a favorable safety profile, did not meet the primary efficacy endpoint. Nevertheless, reductions in left ventricular volumes and circulating natriuretic peptides demonstrated disease-modifying bioactivity (Makkar et al. 2020). The second trial (DYNAMIC) tested CDCs in patients with advanced HFrEF, which reported improvements in EF, end-systolic volume, and quality of life score at 6 months. The improvements in EF and quality of life score remained significant at the 12-month follow-up (Chakravarty et al. 2020).
At the time of writing, only two cell types appear to be on a path for active commercial development as cell-based therapies with the ultimate goal of product registration for cardiac indications: allogeneic mesenchymal precursor cells are being evaluated in a phase III study for HFrEF (DREAM HF-1 trial), and allogeneic CDCs are being developed for the cardiomyopathy (and the skeletal myopathy) associated with DMD (HOPE-2 trial). More and more, the realization that cell therapy exerts indirect benefits has led to changes in treatment strategies, with a shift toward systemic (typically intravenous) delivery and repeat dosing in the case of chronic illnesses such as DMD (Marbán 2018a). Both of these innovations are first reflected in the design of the HOPE-2 trial, which is discussed more extensively below.
7.2.1.2 Heart Failure with Preserved Ejection Fraction
Given the bleak outlook in the search of effective therapeutic strategies for HFpEF, and recent mechanistic insights implicating inflammation, fibrosis, and vascular dysfunction, some researchers have turned to cell therapy for answers. To date, only a limited number of cell therapy studies have been performed in preclinical models of HFpEF—most notably CDCs—for which the evidence at present indicates favorable therapeutic bioactivity. In the first published study of cell therapy in HFpEF, CDCs normalized myocardial relaxation and diastolic pressure while improving survival in a preclinical rat model (Gallet et al. 2016). These benefits occurred despite the continued presence of hypertension and myocardial hypertrophy. Serendipitously, CDCs reversed myocardial inflammation and fibrosis—two critical pathological features of HFpEF. Subsequent preclinical studies revealed cardiac electrical abnormalities underlie sudden death in HFpEF (Cho et al. 2017, 2018b) which could be reversed by CDC treatment (Cho et al. 2018a).
With the favorable safety profile and hints of efficacy of CDCs in human patients with HFrEF, and preclinical data supporting efficacy in HFpEF, the REGRESS-HFpEF trial was initiated. The trial is currently testing allogenic CDCs using catheter-based intracoronary infusions in patients with HFpEF. The only other clinical trial using cell therapy for HFpEF is CELLpEF, which is assessing the safety and efficacy of transendocardial CD34+ cells. If the preliminary evidence from either of these two clinical trials is promising, it may lay the foundation for future studies exploring the efficacy of novel cell therapies for HFpEF. Nevertheless, the conceptual innovations of systemic delivery and repeat dosing (Rogers et al. 2019b, 2020b; Aminzadeh et al. 2018; Reich et al. 2016) have not yet reached clinical testing in the case of HFpEF, such that any benefit seen in the ongoing trials may be a lower limit estimate of likely therapeutic value.
7.2.1.3 Duchenne Muscular Dystrophy
As in the initial efforts of cell therapy for cardiac regeneration, transplantation of skeletal myoblasts was also evaluated as a therapeutic solution in DMD. Here, the underlying hypothesis was that transplanted myoblasts with a non-mutated dystrophin gene would engraft, proliferate, differentiate, and form new myofibers, which would also express functional dystrophin. This notion was supported by pilot studies in the mdx mouse, a widely studied preclinical model of DMD, which showed transplanted myoblasts (with a non-mutated copy of the dystrophin gene) sufficed to regenerate dystrophic skeletal muscle (Law et al. 1988; Partridge et al. 1989). Success of these early myoblast transfer studies motivated in-human clinical testing in DMD patients. However, despite preclinical efficacy, the majority of early phase clinical trials failed to demonstrate any functional improvements nor any evidence of dystrophin-positive myofibers (Mendell et al. 1995; Gussoni et al. 1997; Karpati et al. 1993). Poor survival and insufficient migration of the transplanted myoblasts, and the possibility of immune rejection were offered as explanations for the lack of efficacy (Skuk and Tremblay 2014). However, nearly a decade of technical and conceptual innovations in trial design, transplantation techniques, and immunosuppression strategies have provided insight to improve outcomes. Consequently, the next-generation of clinical trials demonstrated donor-derived dystrophin-positive myofibers (Skuk et al. 2004, 2006; 2007). Still, there remains much room for improvement. These latest trials illustrate the importance of trial design based on foundations supported by basic studies.
Other cell types have also been evaluated, based on known mechanism of action, for efficacy in DMD. One such example is mesangioblasts, which not only have a demonstrated ability to differentiate into several mesodermal lineages including skeletal muscle but also associate with the vascular endothelium to enable extravasation from the circulation after systemic infusion (Sampaolesi et al. 2003; Minasi et al. 2002). In preclinical studies, mesoangioblasts exert therapeutic bioactivity in murine models of muscular dystrophy, including DMD (Sampaolesi et al. 2006; Berry et al. 2007; Diaz-Manera et al. 2010). Moreover, genetically corrected mesoangioblasts, delivered systemically to the golden retriever muscular dystrophy (GRMD) dog model, stimulated dystrophin expression in up to 50% of myofibers from studied hind-limb muscles, leading to improved muscle structure and function (Sampaolesi et al. 2006). As a result, a phase I/IIa trial (EudraCT) delivering HLA-matched mesoangioblasts to five DMD patients was launched (Cossu et al. 2015). Unfortunately, the hope for mesoangioblasts as a therapeutic candidate for DMD has faded due to poor donor cell engraftment and dystrophin expression (Biressi et al. 2020).
The potential of MSCs to give rise to myogenic stem cells (a population distinct from satellite cells) (Liu et al. 2007; De Bari et al. 2003) supported their evaluation in DMD. MSCs from diverse sources such as human adipose tissue (Vieira et al. 2012) and human dental pulp (Kerkis et al. 2008) engraft and express dystrophin to varying degrees in GRMD dogs. In one pediatric and two adult Becker muscular dystrophy patients, intravenous administration of human umbilical cord-derived MSCs (UC-MSCs) were studied (Li et al. 2015). No improvements in the histology of muscle biopsies were reported; however, gait improvements were observed during the clinical examination of the pediatric patient. A subsequent in-human study (NCT02484560) recruited nine DMD patients to receive allogeneic UC-MSCs delivered in a combination of intramuscular and systemic injections. Despite pulmonary function being improved in all patients, variable dystrophin expression was reported, and limb muscle strength was not different between pre- and post-treatment assessments, though most treated patients had a reduction in circulating creatine kinase levels (Dai et al. 2018).
Nevertheless, the daunting search for novel therapeutic candidates for DMD continues. Because of their ability to favorably target inflammation, fibrosis, and cardiomyogenesis, CDCs represent a logical candidate to antagonize the pathophysiology of DMD. We first evaluated the therapeutic bioactivity of intramyocardially delivered CDCs to impact on cardiomyopathy in mdx mice (Aminzadeh et al. 2018). Based on insights discovered from earlier work in HFrEF, the hope was that CDCs would target the secondary pathology in the hearts of mdx mice—that is, inflammation, fibrosis, and necrosis. Indeed, not only did CDCs reduce myocardial inflammation and fibrosis they also stimulated cardiomyogenesis by way of increased cardiomyocyte proliferation. A salient finding from this study was the discovery that CDCs were also bioactive in the skeletal muscle of mdx mice—a finding that has paved the way for their use in other skeletal muscle indications. Consequently, this work motivated a phase I/IIa clinical trial (HOPE-Duchenne) to deliver CDCs into the coronary circulation, and a follow-on preclinical study to deliver CDCs intravenously to mdx mice (Rogers et al. 2019b). The HOPE-Duchenne trial demonstrated a favorable safety profile for CDCs with indications of disease-modifying bioactivity, while the preclinical study demonstrated profound disease-modifying bioactivity of CDCs when delivered intravenously to mdx mice. The results from both preclinical and clinical studies motivated a second phase II clinical trial (HOPE-2) to repeatedly deliver CDCs intravenously to patients with advanced-stage DMD. Data reported from this clinical trial showed significant improvements in both cardiac and skeletal muscle parameters (Marbán et al. 2020; McDonald et al. 2022), supporting both the insights discovered from the preclinical work and the adoption of a systemic delivery with repeat dosing paradigm.
7.2.1.4 Volumetric Muscle Loss
Despite decades of research, VML has remained largely refractory to many experimental strategies—all designed to recover lost muscle tissue and function. Although skeletal myoblasts were initially considered a promising cell source for VML treatment, several limitations preclude their clinical translation. Such limitations include low abundance within muscle, challenges related to purification, poor engraftment posttransplantation, and a meager self-renewal and differentiation capacity (Ding et al. 2017; Pantelic and Larkin 2018). Because of these limitations, MSCs represent an alternative cell type with a demonstrated ability to accelerate the repair of injured animal skeletal muscle. Compared to skeletal myoblasts, MSCs can be found in greater abundance and can be harvested by minimally invasive processes such as bone marrow extraction or lipoaspiration (Mori et al. 2015; Berebichez-Fridman and Montero-Olvera 2018). By themselves, MSCs may be a suitable cell type for the treatment of VML. Unfortunately, MSCs are also susceptible to low engraftment potential and a lack of lineage-specific differentiation. The discovery of microenvironmental cues that direct MSCs toward a myogenic phenotype will be instrumental to harness the therapeutic potential of MSCs. Future studies should delineate whether MSCs work canonically or by noncanonical mechanisms such as paracrine signaling to repair damaged skeletal muscle (Shayan and Huang 2020).
We have learned a great deal regarding the appropriateness of CDCs in skeletal muscle regeneration from our experience with mdx mice. Our basic studies have followed a logical sequence starting with the discovery of regenerative bioactivity in mdx skeletal muscle, to the exploration of the regenerative mechanisms, and the adoption to pathologically similar skeletal muscle injuries such as VML. For example, in dystrophic skeletal muscle, CDCs (by way of paracrine actions) regulate inflammation by modulating the macrophage phenotype, reducing fibrotic scar size, and coaxing dysfunctional satellite cells into forming de novo myofibers (Rogers et al. 2019a, b). The congruency of these actions in DMD and VML reasonably motivated us to test the efficacy of CDCs for the VML indication. Our initial experience with CDCs in a mouse model of VML has been quite promising (Rogers et al. 2021), where CDCs: (1) improve recovery of muscle function, (2) partially restore lost muscle volume and mass, (3) increase the number of innervated myofibers, and (4) stimulate the endogenous repair machinery. If proven successful, CDCs may be a therapeutic candidate for in-human clinical testing for trauma patients with associated VML.
7.3 Current Challenges and Novel Approaches
The field of cell therapy has not been short of novel ideas, some of which have been substantiated by considerable preclinical testing to establish concepts that define the current state of regenerative medicine. Unfortunately, to date, no cell product has received FDA approval for regenerative applications. How is this possible, despite developments produced from decades of preclinical and clinical research? Several obstacles to the translation of cell products are discussed in detail below (Marbán 2018a).
7.3.1 Barriers to Product Approval
Manufacturing Challenges
Nearly four decades of preclinical research have shown ample evidence for the efficacy of cell therapy in regenerative applications. Despite this fact, cells are fragile living entities that can be often difficult to manufacture and handle, which precludes the implementation of conventional manufacturing protocols (Dodson and Levine 2015). In academic settings, where cellular therapeutic candidates generally originate, little consideration is given to regulatory compliance or scalability. Early phase in-human studies generally require only a modest degree of manufacturing process optimization. Further, reproducibility of the cell product, scalability, and cost are secondary concerns as these studies are primarily designed to treat only a small cohort of patients. The progress beyond phase I clinical trials of cell therapies is dismal, in large part owing to pitfalls that lie in the progression that follows. When therapeutic products advance to further development, manufacturing priorities shift toward the cost of goods sold, process development, and quality control. Herein lies a significant barrier for cell products: in contrast to small molecules, cells are living entities with dynamic and adapting responses to environmental conditions. Therefore, a continuous, yet fundamental, challenge is maintaining therapeutic efficacy, especially for cell types that are susceptible to senescence after several cycles of cell division (Campbell et al. 2015). Another important consideration is the optimization of manufacturing processes. For example, seemingly subtle changes such as a switch from monolayer culture to suspension culture can introduce phenotypic changes that can unfavorably skew the final product (Karnieli 2015). It is easy to envision, in extreme cases, therapeutic candidates may lose their disease-modifying properties as they drift further away from the intended formulation (Szymczak et al. 2011; Marbán 2018a).
Reproducibility and Efficacy
Product consistency is a major concern for commercialization. In order to be compliant with regulatory standards, a commercial product must be comparable in identity, potency, and composition from year to year. Quality control measures must be employed at integral steps in the manufacturing process to ensure product stability of master cell banks and those destined for product manufacturing. Since no allogeneic cell therapy products for regenerative medicine have been approved in the United States, no standard operating procedures exist detailing how this should be done. An ever-present concern in commercial product development is diminished efficacy, which warrants the development and validation of assays that reliably measure product potency. In the context of cell therapy, this can be quite challenging as cell therapy is often multifactorial with diverse targets, making it difficult to identify one molecule or pathway as being strictly required for a global view of therapeutic efficacy. Both in vivo and in vitro models are met with unique challenges that often preclude their utility as potency assays. Mechanistic dissection is a seemingly obvious route for identifying key factors of efficacy which, in turn, may reveal markers for potency assay development (Marbán 2018a).
Safety
One critical concern in cell therapy is safety. Generally speaking, such concerns can be categorized as either: (a) related to how the cells are delivered or (b) as intrinsic to the cells themselves. Cell delivery methods can vary from being relatively harmless to exceptionally invasive, which (of course) tracks accordingly with an escalating risk. Although intravenous infusion is the least invasive method used clinically, it has been considered to be inferior if the goal is targeting specific tissues such as the heart or skeletal muscle—due to the absence of direct administration to the organ itself. In light of mounting evidence that cells can exert long-distance effects by way of secreted exosomes (as reviewed in the next section), intravenous delivery is poised for a resurgence.
Naturally, the risk of neoplasm formation is pervasive with any proliferating transplanted cell. In the extreme, pluripotent cells are known to form tumors in vivo. As such, pre-differentiation and removal of residual pluripotent cells are critical precautions one must consider prior to clinical application (Fox et al. 2014). However, allogeneic adult stem and progenitor cells are likely cleared by the patient’s immune system over time (Malliaras et al. 2012), which may fortuitously confer an added level of safety. On the other hand, expedient immune clearance may undermine the ability of the allogeneic cells to exert their paracrine effects, potentially limiting their efficacy (Schu et al. 2012). A related concern with allogeneic cells is the risk for sensitization to foreign antigens, which may subvert the efficacy of repeat dosing protocols (Schu et al. 2012). In cases of progressive underlying pathology such as DMD, wherein multiple sequential doses over time may be needed to sustain efficacy, sensitization to allogeneic cells may be problematic. In theory, however, crossmatching donor compatibility with the recipient may mitigate the potential concern of sensitization, if such a concern proves to be true (Al-Daccak and Charron 2015; Marbán 2018a). Another approach involves premedication before infusion to avoid immune sensitization and/or infusion-related hypersensitivity reactions.
Lastly, and unique to cardiac applications of cell therapy, is the possibility of elevated electrical disturbances, that is, the potential for arrhythmogenesis. As previously discussed, the heart functions as an electromechanical syncytium where efficient coupling is required to facilitate the conduction of electrical impulses and the subsequent transmission of mechanical force. Transplanted cells may disrupt the propagation of electrical impulses in two ways: (1) by creating uncoupled or even poorly coupled clusters of heart tissue which create roadblocks for conduction and (2) by electrically coupling to the adjacent myocardium and eliciting local activation and repolarization (Smith et al. 2008). Using skeletal myoblasts as an example, their transplantation into the myocardium worsens arrhythmia by the first mechanism (Miyagawa et al. 2017). Alternatively, transplanting immature cardiomyocytes derived from pluripotent cells triggers arrhythmias by the second mechanism, creating regional substrates that facilitate reentrant circuits (Smith et al. 2008). Because of this, non-integrating adult heart cells, which typically work by secreting paracrine factors (such as exosomes), may offer a safety advantage over engrafting cell types (Marbán 2018a).
7.3.2 A Novel Paradigm in Regenerative Medicine
The development of cell therapies, to date, has not been without its challenges. To succeed in the clinic, their development must be focused on the underlying biological mechanisms that govern therapeutic efficacy. In this context, deep mechanistic insights are now placing cells themselves in the backseat and shifting attention to the cells’ paracrine functions. For example, the development of CDCs for HF (and now skeletal muscle disease and injury) illustrates how mechanistic discoveries can influence the translational trajectory of cell therapies. Such paradigm shifts are only possible by questioning accepted dogma and following the data (Marbán 2018a).
The Promise of Exosomes
It is widely known now that many adult stem and progenitor cells, such as MSCs and CDCs, function primarily by paracrine mechanisms. One such paracrine function is the secretion of extracellular vesicles, including exosomes, which are lipid bilayer nanoparticles laden with rich repertoires of bioactive molecules such as nucleic acids, proteins, and lipids. Exosomes are synthesized by the ceramide-requiring endolysosomal pathway, where cell membrane invaginations (endosomes) fuse with products of the Golgi apparatus to create multivesicular bodies (Ibrahim and Marbán 2016). When multivesicular bodies fuse to the intracellular face of the cell membrane, they release their contents (including exosomes) into the extracellular space. Neighboring or distant cells internalize the exosomes which can reshape the transcriptomic landscape and greatly influence cell behavior.
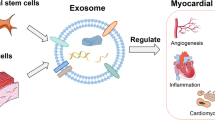
Several lines of evidence (in both heart and skeletal muscle disease) now demonstrate blocking exosome production disrupts CDC bioactivity, whereas CDC-derived exosomes (CDCEXO) suffice to reproduce the benefits of the parent CDCs (Ibrahim et al. 2014; Rogers et al. 2019b). Thus, exosomes secretion is a necessary step for CDC efficacy (Fig. 7.2). Many of the effects of exosomes can be explained by their RNA cargo, specifically non-coding RNAs (ncRNAs) (Rogers et al. 2020a). Among the several species of ncRNAs, miRs and Y RNA fragments are abundantly present in CDCEXO and (generally speaking) regulate gene expression in the recipient cell (Rogers et al. 2020a). These discoveries have shaped the emerging concept that the mechanism of action of CDCEXO requires the parent cells to secrete exosomes which act as vehicles to transfer bioactive cargo into recipient cells; thus, inducing the transcriptomic and phenotypic changes that underlie the benefits of cell therapy. Given the fact exosomes are bioactive themselves, they may be an attractive next-generation cell-free therapeutic candidate (Marbán 2018a).

Mechanisms of action underlying CDCs’ bioactivity. CDCs secrete exosomes which target key pathological mechanisms of heart and skeletal muscle disease. Adapted from Akhmerov and Marbán (2020)
The question then becomes: will exosomes entirely replace cells as therapeutic products? The answer is not so trivial, but with all things considered—probably not. Therapeutic cells have been in development for decades, in contrast to exosomes, which have been tested in only a handful of trials in humans (Marbán 2018b). This may be in large part due to the fact exogenously delivered exosomes are still poorly understood in terms of optimal delivery methods, their tissue distribution profile, and mechanisms of bioactivity. The challenge is identifying situations where cells would be preferable to exosomes. Acute myocardial infarction, for example, may be one indication where cells may outperform exosomes. Here, the rationale is when both products are directly infused into the coronary circulation (Gallet et al. 2017), cells become lodged within the capillaries and may function as sustained-release factories for exosomes, whereas exosomes (being up to 1000 times smaller) quickly pass through the coronary vessels to the systemic circulation thus diluting their bioactivity (Marbán 2018a).
Notwithstanding, therapeutic exosomes offer the potential to circumvent the main limitations of cell therapy. For example, some advantages of exosomes include product stability (Akers et al. 2016) and immune tolerance (i.e., human CDCEXO are therapeutic in non-immunosuppressed mice (Rogers et al. 2019b), rats (Cambier et al. 2017), and pigs (Gallet et al. 2017; Dawkins et al. 2022), even after multiple doses (Aminzadeh et al. 2018)). Further, exosome dosing strategies are not limited by microvascular plugging, and their efficacy can be enhanced by multiple approaches such as engineering of the producer cells (Conlan et al. 2017). Given the role of exosomes is clear, focus has now shifted to identifying the factors within exosomes that are responsible for mediating their disease-modifying bioactivity in any given indication. For example, miR-146a, miR-181b, and a Y RNA fragment have been shown to be central mediators of cardioprotection in acute myocardial infarction (Ibrahim et al. 2014; de Couto et al. 2017; Cambier et al. 2017); and miR-148a is required for CDCEXO-mediated myogenesis in mdx skeletal muscle (Rogers et al. 2019b). Two important implications can be borne from the identification of defined factors themselves: (1) the factors themselves, individually or in combination, may be developed as new therapeutic candidates and (2) the potential to use mechanistic discoveries to aid the development of cell therapies toward the goal of enhanced efficacy. For example, the development of potency assays based on mechanistic insights may offer the quantification of the defined factors in exosomes secreted by prospective cell candidate. Thus, the search for mechanistic discoveries, instead of simply drifting science away from cells completely and toward exosomes and their defined factors, should lead to the development of approaches to improve the quality, reproducibility, and efficacy of cell therapy (Marbán 2018a).
7.4 Outlook and Future Perspectives
Within the last decade, the deconstruction of cell therapy into its mechanistic underpinnings has shifted focus away from autologous cell therapy to allogeneic applications and onwards, to exosomes and their contents as next-generation therapies. The discovery that exosomes are key mediators stemmed from the recognition that both autologous and allogeneic cell sources resulted in equivalent benefits that persisted long after the original cells were no longer detectable. Mining of the exosomal cargo, in turn, has implicated specific ncRNA species as defined factors worthy of scientific inquiry. The commercialization of therapeutic exosome products is in its infancy, and their trajectory toward clinical use remains to be determined. Despite the shift away from cells toward exosomes and defined factors, cell therapies still hold potential for application in well-defined indications.
References
Aguilar CA, Greising SM, Watts A, Goldman SM, Peragallo C, Zook C, Larouche J, Corona BT (2018) Multiscale analysis of a regenerative therapy for treatment of volumetric muscle loss injury. Cell Death Dis 4(1):1–11
Akers JC, Ramakrishnan V, Yang I, Hua W, Mao Y, Carter BS, Chen CC (2016) Optimizing preservation of extracellular vesicular miRNAs derived from clinical cerebrospinal fluid. Cancer Biomark 17(2):125–132
Akhmerov A, Marbán E (2020) COVID-19 and the heart. Circ Res 126(10):1443–1455
Al-Daccak R, Charron D (2015) Allogenic benefit in stem cell therapy: cardiac repair and regeneration. Tissue Antigens 86(3):155–162
Aminzadeh MA, Rogers RG, Fournier M, Tobin RE, Guan X, Childers MK, Andres AM, Taylor DJ, Ibrahim A, Ding X, Torrente A, Goldhaber JI, Lewis MI, Gottlieb RA, Victor RA, Marbán E (2018) Exosome-mediated benefits of cell therapy in mouse and human models of Duchenne muscular dystrophy. Stem Cell Rep 10(3):942–955
Basalay M, Yellon D, Davidson S (2020) Targeting myocardial ischaemic injury in the absence of reperfusion. Basic Res Cardiol 115(6):1–16
Behfar A, Faustino RS, Arrell DK, Dzeja PP, Perez-Terzic C, Terzic A (2008) Guided stem cell cardiopoiesis: discovery and translation. J Mol Cell Cardiol 45(4):523–529
Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114(6):763–776
Berebichez-Fridman R, Montero-Olvera PR (2018) Sources and clinical applications of mesenchymal stem cells: state-of-the-art review. Sultan Qaboos Univ Med J 18(3):e264
Berry SE, Liu J, Chaney EJ, Kaufman SJ (2007) Multipotential mesoangioblast stem cell therapy in the mdx/utrn-/-mouse model for Duchenne muscular dystrophy. Regen Med 2(3):275–288
Biressi S, Filareto A, Rando TA (2020) Stem cell therapy for muscular dystrophies. J Clin Invest 130(11):5652–5664
Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, Case LE, Cripe L, Hadjiyannakis S, Olson AK (2018) Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 17(4):347–361
Bolli R, Chugh AR, D’Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T (2011) Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. The Lancet 378(9806):1847–1857
Borlaug BA (2014) The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 11(9):507–515
Borlaug BA, Paulus WJ (2011) Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J 32(6):670–679
Borlaug BA, Redfield MM, Melenovsky V, Kane GC, Karon BL, Jacobsen SJ, Rodeheffer RJ (2013) Longitudinal changes in left ventricular stiffness: a community-based study. Circ Heart Fail 6(5):944–952
Buddhe S, Cripe L, Friedland-Little J, Kertesz N, Eghtesady P, Finder J, Hor K, Judge DP, Kinnett K, McNally EM (2018) Cardiac management of the patient with Duchenne muscular dystrophy. Pediatrics 142(suppl 2):S72–S81
Caldwell C, Mattey D, Weller R (1990) Role of the basement membrane in the regeneration of skeletal muscle. Neuropathol Appl Neurobiol 16(3):225–238
Cambier L, de Couto G, Ibrahim A, Echavez AK, Valle J, Liu W, Kreke M, Smith RR, Marbán L, Marbán E (2017) Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. EMBO Mol Med 9(3):337–352
Campbell A, Brieva T, Raviv L, Rowley J, Niss K, Brandwein H, Oh S, Karnieli O (2015) Concise review: process development considerations for cell therapy. Stem Cells Transl Med 4(10):1155–1163
Chakravarty T, Henry TD, Kittleson M, Lima J, Siegel RJ, Slipczuk L, Pogoda JM, Smith RR, Malliaras K, Marbán L, Ascheim DD, Marbán E, Makkar RR (2020) Allogeneic cardiosphere-derived cells for the treatment of heart failure with reduced ejection fraction: results of the Dilated cardiomYopathy iNtervention with Allogeneic Myocardially-regeneratIve Cells (DYNAMIC) trial. EuroIntervention 16:e293–e300
Cho JH, Zhang R, Kilfoil PJ, Gallet R, de Couto G, Bresee C, Goldhaber JI, Marbán E, Cingolani E (2017) Delayed repolarization underlies ventricular arrhythmias in rats with heart failure and preserved ejection fraction. Circulation 136(21):2037–2050
Cho JH, Kilfoil PJ, Zhang R, Solymani RE, Bresee C, Kang EM, Luther K, Rogers RG, de Couto G, Goldhaber JI, Marbán E, Cingolani E (2018a) Reverse electrical remodeling in rats with heart failure and preserved ejection fraction. JCI Insight 3(19)
Cho JH, Zhang R, Aynaszyan S, Holm K, Goldhaber JI, Marbán E, Cingolani E (2018b) Ventricular arrhythmias underlie sudden death in rats with heart failure and preserved ejection fraction. Circ Arrhythm Electrophysiol 11(8):e006452
Conlan RS, Pisano S, Oliveira MI, Ferrari M, Pinto IM (2017) Exosomes as reconfigurable therapeutic systems. Trends Mol Med 23(7):636–650
Connuck DM, Sleeper LA, Colan SD, Cox GF, Towbin JA, Lowe AM, Wilkinson JD, Orav EJ, Cuniberti L, Salbert BA (2008) Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J 155(6):998–1005
Cossu G, Previtali SC, Napolitano S, Cicalese MP, Tedesco FS, Nicastro F, Noviello M, Roostalu U, Natali Sora MG, Scarlato M (2015) Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol Med 7(12):1513–1528
Dai A, Baspinar O, Yeşilyurt A, Sun E, Aydemir Çİ, Öztel ON, Capkan DU, Pinarli F, Agar A, Karaöz E (2018) Efficacy of stem cell therapy in ambulatory and nonambulatory children with Duchenne muscular dystrophy—phase I–II. Degener Neurol Neuromuscul Dis 8:63
Dawkins JF, Ehdaie A, Rogers R, Soetkamp D, Valle J, Holm K et al (2022) Biological substrate modification suppresses ventricular arrhythmias in a porcine model of chronic ischaemic cardiomyopathy. Eur Heart J
De Bari C, Dell’Accio F, Vandenabeele F, Vermeesch JR, Raymackers J-M, Luyten FP (2003) Skeletal muscle repair by adult human mesenchymal stem cells from synovial membrane. J Cell Biol 160(6):909–918
de Couto G, Gallet R, Cambier L, Jaghatspanyan E, Makkar N, Dawkins JF, Berman BP, Marbán E (2017) Exosomal microRNA transfer into macrophages mediates cellular postconditioning. Circulation 136(2):200–214
Diaz-Manera J, Touvier T, Dellavalle A, Tonlorenzi R, Tedesco F, Messina G, Meregalli M, Navarro C, Perani L, Bonfanti C (2010) Partial dysferlin reconstitution by adult murine mesoangioblasts is sufficient for full functional recovery in a murine model of dysferlinopathy. Cell Death Dis 1(8):e61–e61
Ding S, Wang F, Liu Y, Li S, Zhou G, Hu P (2017) Characterization and isolation of highly purified porcine satellite cells. Cell Death Dis 3(1):1–11
Dodson BP, Levine AD (2015) Challenges in the translation and commercialization of cell therapies. BMC Biotechnol 15(1):1–15
Fox IJ, Daley GQ, Goldman SA, Huard J, Kamp TJ, Trucco M (2014) Use of differentiated pluripotent stem cells in replacement therapy for treating disease. Science 345(6199)
Gallet R, de Couto G, Simsolo E, Valle J, Sun B, Liu W, Tseliou E, Zile MR, Marbán E (2016) Cardiosphere-derived cells reverse heart failure with preserved ejection fraction in rats by decreasing fibrosis and inflammation. JACC Basic Transl Sci 1(1):14–28
Gallet R, Dawkins J, Valle J, Simsolo E, De Couto G, Middleton R, Tseliou E, Luthringer D, Kreke M, Smith RR, Marbán L, Ghaleh B, Marbán E (2017) Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur Heart J 38(3):201–211
Gladden JD, Chaanine AH, Redfield MM (2018) Heart failure with preserved ejection fraction. Annu Rev Med 69:65–79
Greising SM, Rivera JC, Goldman SM, Watts A, Aguilar CA, Corona BT (2017) Unwavering pathobiology of volumetric muscle loss injury. Sci Rep 7(1):1–14
Greising SM, Corona BT, McGann C, Frankum JK, Warren GL (2019) Therapeutic approaches for volumetric muscle loss injury: a systematic review and meta-analysis. Tissue Eng Part B Rev 25(6):510–525
Gussoni E, Blau HM, Kunkel LM (1997) The fate of individual myoblasts after transplantation into muscles of DMD patients. Nat Med 3(9):970–977
Hare J, Traverse J, Henry T (2008) The myoblast autologous grafting in ischemic cardiomyopathy (MAGIC) trial: first randomized placebo controlled study of myoblast transplantation. Circulation 117:1189–1200
Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, Opie L (2014) Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 383(9932):1933–1943
Ibrahim A, Marbán E (2016) Exosomes: fundamental biology and roles in cardiovascular physiology. Annu Rev Physiol 78:67–83
Ibrahim A, Cheng K, Marbán E (2014) Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep 2(5):606–619
Jackson KA, Majka SM, Wang H, Pocius J, Hartley CJ, Majesky MW, Entman ML, Michael LH, Hirschi KK, Goodell MA (2001) Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J Clin Invest 107(11):1395–1402
Karnieli O (2015) Cell therapy: early process development and optimization of the manufacturing process are critical to ensure viability of the product, quality, consistency and cost efficiency. J Commerc Biotechnol 21(1)
Karpati G, Ajdukovic D, Arnold D, Gledhill RB, Guttmann R, Holland P, Koch PA, Shoubridge E, Spence D, Vanasse M (1993) Myoblast transfer in Duchenne muscular dystrophy. Ann Neurol 34(1):8–17
Kerkis I, Ambrosio CE, Kerkis A, Martins DS, Zucconi E, Fonseca SA, Cabral RM, Maranduba CM, Gaiad TP, Morini AC (2008) Early transplantation of human immature dental pulp stem cells from baby teeth to golden retriever muscular dystrophy (GRMD) dogs: local or systemic? J Transl Med 6(1):1–13
Koh GY, Klug MG, Soonpaa MH, Field LJ (1993) Differentiation and long-term survival of C2C12 myoblast grafts in heart. J Clin Invest 92(3):1548–1554
Lancet T (2019) Retraction—Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet (London, England) 393(10176):1084
Law PK, Goodwin TG, Wang MG (1988) Normal myoblast injections provide genetic treatment for murine dystrophy. Muscle Nerve 11(6):525–533
Lepper C, Partridge TA, Fan C-M (2011) An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 138(17):3639–3646
Li P, Cui K, Zhang B, Wang Z, Shen Y, Wang X, Zhang J, Tong F, Li S (2015) Transplantation of human umbilical cord-derived mesenchymal stems cells for the treatment of Becker muscular dystrophy in affected pedigree members. Int J Mol Med 35(4):1051–1057
Liu Y, Yan X, Sun Z, Chen B, Han Q, Li J, Zhao RC (2007) Flk-1+ adipose-derived mesenchymal stem cells differentiate into skeletal muscle satellite cells and ameliorate muscular dystrophy in mdx mice. Stem Cells Dev 16(5):695–706
Ma C, Luo H, Fan L, Liu X, Gao C (2020) Heart failure with preserved ejection fraction: an update on pathophysiology, diagnosis, treatment, and prognosis. Braz J Med Biol Res 53(7)
Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marbán L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marbán E (2012) Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. The Lancet 379(9819):895–904
Marbán E, Liao K (2022) On the cellular origin of cardiosphere-derived cells (CDCs). Basic Res Cardiol 117(1):1–4
Makkar RR, Kereiakes DJ, Aguirre F, Kowalchuk G, Chakravarty T, Malliaras K, Francis GS, Povsic TJ, Schatz R, Traverse JH, Pogoda JM, Smith RR, Marbán L, Ascheim DD, Ostovaneh MR, Lima J, Demaria A, Marbán E, Henry TD (2020) Intracoronary ALLogeneic heart STem cells to Achieve myocardial Regeneration (ALLSTAR): a randomized, placebo-controlled, double-blinded trial. Eur Heart J 41(36):3451–3458
Malliaras K, Marbán E (2011) Cardiac cell therapy: where we’ve been, where we are, and where we should be headed. Br Med Bull 98(1):161–185
Malliaras K, Li T-S, Luthringer D, Terrovitis J, Cheng K, Chakravarty T, Galang G, Zhang Y, Schoenhoff F, Van Eyk J, Marbán L, Marbán E (2012) Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation 125(1):100–112
Marbán E (2018a) A mechanistic roadmap for the clinical application of cardiac cell therapies. Nature biomedical engineering 2(6):353–361
Marbán E (2018b) The secret life of exosomes: what bees can teach us about next-generation therapeutics. J Am Coll Cardiol 71(2):193–200
Marbán L, Rogy S, McDonald C, Eagle M, Finkel R, Tian C, Taylor M, Janas J, Harmelink M, Varadhachary A, Hor K, Mayer OH, Furlong P, Committee H-S (2020) HOPE-2 one-year results show clinically relevant improvements in upper limb & cardiac function in patients with later stage Duchenne muscular dystrophy. Neuromuscul Disord 30:S168–S169
McDonald CM, Marbán E, Hendrix S, Hogan N, Smith RR, Eagle M et al (2022) Repeated intravenous cardiosphere-derived cell therapy in late-stage Duchenne muscular dystrophy (HOPE-2): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 399(10329):1049–1058
Mendell JR, Kissel JT, Amato AA, King W, Signore L, Prior TW, Sahenk Z, Benson S, McAndrew PE, Rice R (1995) Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N Engl J Med 333(13):832–838
Meyer GP, Wollert KC, Lotz J, Steffens J, Lippolt P, Fichtner S, Hecker H, Schaefer A, Arseniev L, Hertenstein B (2006) Intracoronary bone marrow cell transfer after myocardial infarction. Circulation 113(10):1287–1294
Minasi MG, Riminucci M, De Angelis L, Borello U, Berarducci B, Innocenzi A, Caprioli A, Sirabella D, Baiocchi M, De Maria R (2002) The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 129(11):2773–2783
Miyagawa S, Domae K, Yoshikawa Y, Fukushima S, Nakamura T, Saito A, Sakata Y, Hamada S, Toda K, Pak K (2017) Phase I clinical trial of autologous stem cell–sheet transplantation therapy for treating cardiomyopathy. J Am Heart Assoc 6(4):e003918
Mori R, Kamei N, Okawa S, Nakabayashi A, Yokota K, Higashi Y, Ochi M (2015) Promotion of skeletal muscle repair in a rat skeletal muscle injury model by local injection of human adipose tissue-derived regenerative cells. J Tissue Eng Regen Med 9(10):1150–1160
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, De Ferranti S, Després J-P, Fullerton HJ (2016) Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation 133(4):e38–e360
Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM (2001) Bone marrow cells regenerate infarcted myocardium. Nature 410(6829):701–705
Pantelic MN, Larkin LM (2018) Stem cells for skeletal muscle tissue engineering. Tissue Eng Part B Rev 24(5):373–391
Partridge TA, Morgan J, Coulton G, Hoffman E, Kunkel L (1989) Conversion of mdx myofibres from dystrophin-negative to-positive by injection of normal myoblasts. Nature 337(6203):176–179
Pfeffer MA, Braunwald E (1990) Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 81(4):1161–1172
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González-Juanatey JR, Harjola V-P, Jankowska EA (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 37(27):2129–2200
Psaltis P, Paton S, See F, Arthur A, Martin S, Itescu S, Worthley S, Gronthos S, Zannettino A (2010) Enrichment for STRO-1 expression enhances the cardiovascular paracrine activity of human bone marrow-derived mesenchymal cell populations. J Cell Physiol 223(2):530–540
Reich H, Tseliou E, de Couto G, Angert D, Valle J, Kubota Y, Luthringer D, Mirocha J, Sun B, Smith RR (2016) Repeated transplantation of allogeneic cardiosphere-derived cells boosts therapeutic benefits without immune sensitization in a rat model of myocardial infarction. J Heart Lung Transplant 35(11):1348–1357
Rogers RG, de Couto G, Liu W, Sanchez L, Marbán E (2019a) Cardiosphere-derived cell exosomes modulate mdx macrophage phenotype and alter their secretome. FASEB J 33(S1):lb611–lb611
Rogers RG, Fournier M, Sanchez L, Ibrahim AG, Aminzadeh MA, Lewis MI, Marbán E (2019b) Disease-modifying bioactivity of intravenous cardiosphere-derived cells and exosomes in mdx mice. JCI Insight 4(7)
Rogers RG, Ciullo A, Marbán E, Ibrahim AG (2020a) Extracellular vesicles as therapeutic agents for cardiac fibrosis. Front Physiol 11:479
Rogers RG, Fournier M, Lee Y, Jones K, Sanchez L, Marbán E (2020b) Repeated administration of cardiosphere-derived cells preserves exercise capacity and muscle function for at least one year in mdx mice. Neuromuscul Disord 30:S107–S108
Rogers RG, Li L, Peck K, Sanchez L, Liu W, Ciullo A, Alfaro J, Rannou A, Fournier M, Lee Y, Marbán E (2021) Cardiosphere-derived cells, with and without a biological scaffold, stimulate myogenesis and recovery of muscle function in mice with volumetric muscle loss. Biomaterials 274:120852
Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D’Antona G, Pellegrino MA, Barresi R, Bresolin N, De Angelis MGC, Campbell KP (2003) Cell therapy of α-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science 301(5632):487–492
Sampaolesi M, Blot S, D’Antona G, Granger N, Tonlorenzi R, Innocenzi A, Mognol P, Thibaud J-L, Galvez BG, Barthélémy I (2006) Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444(7119):574–579
Savarese G, Lund LH (2017) Global public health burden of heart failure. Card Fail Rev 3(1):7
Schu S, Nosov M, O’Flynn L, Shaw G, Treacy O, Barry F, Murphy M, O’Brien T, Ritter T (2012) Immunogenicity of allogeneic mesenchymal stem cells. J Cell Mol Med 16(9):2094–2103
Shah KS, Xu H, Matsouaka RA, Bhatt DL, Heidenreich PA, Hernandez AF, Devore AD, Yancy CW, Fonarow GC (2017) Heart failure with preserved, borderline, and reduced ejection fraction: 5-year outcomes. J Am Coll Cardiol 70(20):2476–2486
Shayan M, Huang NF (2020) Pre-clinical cell therapeutic approaches for repair of volumetric muscle loss. Bioengineering 7(3):97
Skuk D, Tremblay JP (2014) Clarifying misconceptions about myoblast transplantation in myology. Mol Ther 22(5):897–898
Skuk D, Roy B, Goulet M, Chapdelaine P, Bouchard J-P, Roy R, Dugré FJ, Lachance J-G, Deschênes L, Senay H (2004) Dystrophin expression in myofibers of Duchenne muscular dystrophy patients following intramuscular injections of normal myogenic cells. Mol Ther 9(3):475–482
Skuk D, Goulet M, Roy B, Chapdelaine P, Bouchard J-P, Roy R, Dugré FJ, Sylvain M, Lachance J-G, Deschênes L (2006) Dystrophin expression in muscles of Duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J Neuropathol Exp Neurol 65(4):371–386
Skuk D, Goulet M, Roy B, Piette V, Côté CH, Chapdelaine P, Hogrel J-Y, Paradis M, Bouchard J-P, Sylvain M (2007) First test of a “high-density injection” protocol for myogenic cell transplantation throughout large volumes of muscles in a Duchenne muscular dystrophy patient: eighteen months follow-up. Neuromuscul Disord 17(1):38–46
Smith R, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marbán E (2007) Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 115(7):896–908
Smith RR, Barile L, Messina E, Marbán E (2008) Stem cells in the heart: what’s the buzz all about? Part 2: Arrhythmic risks and clinical studies. Heart Rhythm 5(6):880–887
Soni M, Ferrell B, Wikholm C, Wilson LT (2020) Stem cell therapies and treatment advances for heart failure with preserved ejection fraction. Georgetown Med Rev 4(1):12344
Sürder D, Manka R, Lo Cicero V, Moccetti T, Rufibach K, Soncin S, Turchetto L, Radrizzani M, Astori G, Schwitter J (2013) Intracoronary injection of bone marrow-derived mononuclear cells early or late after acute myocardial infarction: effects on global left ventricular function. Circulation 127(19):1968–1979
Szymczak MM, Friedman RL, Uppoor R, Yacobi A (2011) Detection, measurement, and control in pharma manufacturing. Pharm Technol 35:70–76
Taylor DA, Atkins BZ, Hungspreugs P, Jones TR, Reedy MC, Hutcheson KA, Glower DD, Kraus WE (1998) Regenerating functional myocardium: improved performance after skeletal myoblast transplantation. Nat Med 4(8):929–933
Traverse JH, Henry TD, Pepine CJ, Willerson JT, Zhao DX, Ellis SG, Forder JR, Anderson RD, Hatzopoulos AK, Penn MS (2012) Effect of the use and timing of bone marrow mononuclear cell delivery on left ventricular function after acute myocardial infarction: the TIME randomized trial. JAMA 308(22):2380–2389
Van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin S-CJ, Middleton RC, Marbán E, Molkentin JD (2014) C-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509(7500):337–341
Vieira N, Valadares M, Zucconi E, Secco M, Junior CB, Brandalise V, Assoni A, Gomes J, Landini V, Andrade T (2012) Human adipose-derived mesenchymal stromal cells injected systemically into GRMD dogs without immunosuppression are able to reach the host muscle and express human dystrophin. Cell Transplant 21(7):1407–1417
Voronov R (1975) Experimental study of the regenerative potentialities of the cardiac and somatic musculatures. Arkh Anat Gistol Embriol 69(9):35–40
Wang L-T, Ting C-H, Yen M-L, Liu K-J, Sytwu H-K, Wu KK, Yen BL (2016) Human mesenchymal stem cells (MSCs) for treatment towards immune-and inflammation-mediated diseases: review of current clinical trials. J Biomed Sci 23(1):1–13
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL (2013) 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 62(16):e147–e239
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM (2017) 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 70(6):776–803
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Rogers, R.G., Marbán, E. (2022). Novel Cell-Based Therapeutics for Diseases of the Heart and Skeletal Muscle. In: Greising, S.M., Call, J.A. (eds) Regenerative Rehabilitation. Physiology in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-95884-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-95884-8_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-95883-1
Online ISBN: 978-3-030-95884-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)