
Abstract
Duchenne muscular dystrophy (DMD) is a severe, progressive, genetic muscle wasting disorder arising from the absence of the membrane stabilizing protein, dystrophin, that renders muscle fibers susceptible to damage and degeneration. Since the discovery of the dystrophin gene, research efforts have focused on the development of regenerative gene- and cell-based therapies for DMD, although many obstacles need to be overcome before they can be considered for clinical application. The development of adjunct therapies that can slow the pathologic progression, preserve muscle mass, enhance muscle regeneration, and promote muscle growth, is therefore essential. Rehabilitation through physical exercise or muscle contraction protocols may help attenuate muscle weakness and dysfunction in DMD, with evidence supporting rehabilitation as an adjunct treatment to gene- and cell-mediated therapies. This chapter summarizes the current state of research for DMD therapy and explores the potential for combined regenerative and rehabilitation therapies to improve outcomes for DMD patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Introduction
Duchenne muscular dystrophy (DMD) is a severe and progressive muscle wasting disorder caused by mutations in the dystrophin gene resulting in the absence of the membrane stabilizing protein, dystrophin. Loss of dystrophin renders muscle fibers fragile and susceptible to membrane tears that facilitate an influx of Ca2+ that can activate inflammatory and muscle degenerative pathways. Although considerable efforts are being directed to the development of gene- and cell-based therapies for DMD, these techniques are far from perfect, and many obstacles need to be overcome in order for them to advance to clinical application. In the interim, it is essential that alternative therapies also be developed, including research directed to slowing the pathologic progression, preserving muscle mass, enhancing muscle regeneration, and promoting muscle growth. This chapter will highlight the current state of research for regenerative medicine (e.g., gene-, cell-, and pharmacologic therapies), the current state of research for rehabilitation (e.g., exercise modalities), and explore the potential for combined regenerative and rehabilitation therapies to improve outcomes for DMD patients.
4.2 Duchenne Muscular Dystrophy: A Difficult Disease to Treat
DMD is a debilitating X-linked genetic disorder affecting 1:3500–6000 males worldwide, caused by mutations in the dystrophin (DMD) gene that result in an absence of the dystrophin protein. In striated muscle, the dystrophin protein is a component of the dystrophin-glycoprotein complex (DGC), a multimeric protein complex located at the sarcolemma of striated muscle fibers which links the intracellular actin cytoskeleton to the extracellular matrix (ECM) to transmit the forces generated by muscle contraction (Ervasti and Campbell 1993). The loss of dystrophin destabilizes this link, causing failure of the DGC to assemble at the sarcolemma (Ohlendieck and Campbell 1991). The loss of integrity leads to a progressive loss of muscle mass and function and early lethality in DMD patients. The current gold-standard treatment for most DMD patients is administration of corticosteroids that can slow the pathologic progression and prolong ambulation (Manzur et al. 2004), but does not cure the disease.
DMD is not an easy disease to treat or cure. Within affected patients, all muscle fibers and stem cells within the body contain a defective DMD gene. Therefore, a vast volume of tissue requires restorative, lifelong treatment. This is further complicated by the fact that the dystrophin protein is expressed locally (i.e., it does not circulate) and must therefore be delivered into every muscle cell, or at least a significant proportion of muscle fibers. Furthermore, although DMD is a monogenic disorder, over 1800 different mutations have been reported within the DMD gene that result in DMD, including nonsense and missense mutations, duplications, and deletions. It is therefore unlikely that a single therapy will be effective for all patients. Clinical trials have also demonstrated that the therapeutic dystrophin protein may be identified as “foreign” by the immune system of some patients, resulting in immune-mediated destruction of the therapeutic protein. Dystrophin is a large, complex protein, originating from the largest known gene in the human genome, and it is therefore a difficult gene to reintroduce into the body. Therapeutic approaches must therefore consider factors such as: the route of delivery; the carrying capacity of vector systems; the ideal timing for administration; the requirement for patient-specific versus generic therapies; and the potential for an immune response to both the delivery vehicle and the therapeutic protein. Efforts to identify a cure, via gene, cell, pharmacologic, and/or physical therapies remain ongoing and are discussed in detail.
4.3 Gene Therapies for DMD
The development of dystrophin restoration therapies for DMD has received much attention since the discovery of the causative mutation in dystrophin over 30 years ago (Hoffman et al. 1987a). This discovery was a key milestone in the development of a potential cure for DMD, with patient advocates and research communities working on the assumption that once the underlying genetic defect was identified, a cure would soon follow. This proved not to be the case, with the development of gene replacement therapies for DMD facing many hurdles. However, over in the most recent 10–15 years, significant progress has been made toward developing both viral and non-viral gene correction/replacement strategies, with a small number being approved for conditional use in DMD patients.
4.3.1 Gene Correction Strategies
4.3.1.1 Stop Codon Readthrough
Stop codon readthrough refers to the process by which various drugs facilitate the continuation of mRNA translation to restore protein expression. The process, originally referred to as “phenotypic suppression” was first observed for the aminoglycoside antibiotics in bacteria and yeast (Palmer et al. 1979; Singh et al. 1979). Within the population, it is estimated that approximately 10–15% of all DMD cases arise from nonsense mutations, in which mutations result in the introduction of a premature stop codon (Aartsma-Rus et al. 2006). Therefore, a significant proportion of DMD patients would benefit from stop codon readthrough-based therapy. Gentamicin, an aminoglycoside antibiotic, was the first such drug tested for treating DMD, and was shown to restore dystrophin expression in DMD myoblasts in vitro and restore sarcolemmal dystrophin expression with some functional benefit in mdx dystrophic mice (Barton-Davis et al. 1999). Several clinical trials have assessed the potential for gentamicin to increase dystrophin protein expression and improve pathology in DMD patients (Dunant et al. 2003; Malik et al. 2010; Wagner et al. 2001; Politano et al. 2003). Of these, the most comprehensive showed increases in dystrophin protein expression up to 15% of normal levels, with reduced serum creatine kinase (CK) levels and stabilization of muscle strength after 6 months of intravenous administration (Malik et al. 2010). Based on the success of these studies, more potent drugs were developed with the potential to drive stop codon readthrough, the most successful being ataluren (PTC142, Translarna®). Ataluren received approval from the European Medicines Agency (EMA) in 2014 for the treatment of ambulant patients aged 5 years and older with DMD resulting from a nonsense mutation in the DMD gene. Compared with gentamicin which requires intravenous administration, ataluren is orally bioavailable and therefore repeat administration is more feasible. The therapeutic potential of ataluren remains under investigation in clinical trials, particularly to determine efficacy in non-ambulatory patients. Early studies suggest that earlier intervention with ataluren leads to the best outcomes (Ruggiero et al. 2018).
4.3.1.2 Exon Skipping
The concept of exon skipping was first proposed in the mid-to-late 1990s, as a method to reduce the severity of some genetic mutations. A large majority of DMD-causative mutations arise from missense mutations, in which a mutation induces a shift in the mRNA reading frame of the DMD gene, resulting in a lack of dystrophin protein production. Exon skipping aims to correct the reading frame by inducing the “skipping” of mutation-containing exons during pre-mRNA splicing using antisense oligonucleotides (AONs) that interfere with the splicing of targeted exons. Therefore, skipping over exons containing these frame-shift mutations facilitates the restoration of a smaller, but functional, dystrophin protein that can reduce disease severity. While mutations have been identified throughout the DMD gene, 70% of DMD patients have a mutation in a “hot-spot” in the central genomic region, between exons 45 and 53 (Den Dunnen et al. 1989; Koenig et al. 1989; Nobile et al. 1997). Therefore, the majority of exon skipping strategies have to date focused on this region of the gene, with the aim of treating as large a percentage of the DMD patient population as possible with a small number of drugs.
The most studied tools for exon skipping include the AONs, the phosphorodiamidate morpholino oligomers (PMOs), and the peptide conjugated PMOs (PPMOs). Exon skipping with AONs was first shown in the early 2000s to restore dystrophin protein expression in human myotubes in vitro and reduce disease severity in mdx mice in vivo (van Deutekom et al. 2001; Lu et al. 2003), but initial clinical trials demonstrated variable benefit and FDA approval was subsequently denied in 2016 (Flanigan et al. 2014; Goemans et al. 2011, 2016, 2018; Voit et al. 2014). Subsequent testing of a morpholino-based drug showed levels of dystrophin protein restoration between 10 and 50% and improved skeletal muscle function in the mdx mouse (Alter et al. 2006). This was followed by significant improvements in DMD patient ambulation and respiration in clinical trials (Mendell et al. 2013), leading to FDA approval in 2016 of the first exon skipping therapy for DMD patients amenable to exon 51 skipping, eteplirsen. Since then, a further three drugs have been approved by the FDA for treatment of DMD patients amenable to exon 53 (golodirsen, Vyondys 53®, approved in 2019; viltolarsen, Viltepso®, approved in 2020) and exon 45 skipping (casimersen, Amondys 45®, approved in 2021). Exon skipping is at the forefront of DMD therapeutics, with significant promise to improve the lives of many DMD patients.
4.3.1.3 Gene Editing
The most recent strategy for gene correction in DMD utilizes clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene editing technology. CRISPR/Cas9-mediated genomic editing was first demonstrated to correct the dystrophin mutation in the mdx mouse in 2014 (Long et al. 2014). Injection of guide RNAs (sgRNA) and the Cas9 enzyme into mdx mouse zygotes, to correct the mutation in the germ line, restored dystrophin protein expression 17–83% in skeletal muscles and the hearts of treated mice (Long et al. 2014). Studies are currently focussed on optimizing the therapeutic potential for CRISPR/Cas9 gene editing to correct dystrophin mutations in postnatal mice (Xu et al. 2016; Long et al. 2016). Investigations are currently focussed on combining gene editing technology with adeno-associated viral (AAV) vector delivery.
4.3.2 Gene Replacement Strategies
4.3.2.1 Delivery of Plasmid DNA
Early methods attempted to reintroduce the DMD gene by direct injection of a plasmid containing the DMD gene into the skeletal muscles of mdx mice. While this restored dystrophin expression in injected muscles, efficiency was only around one percent due to the limited ability of skeletal muscle fibers to take up the large DMD gene (Acsadi et al. 1991; Danko et al. 1993). Subsequent attempts utilized adjunct methods to improve uptake of the plasmid DNA, including electroporation, chemical induction of injury, liposome encapsulation, copolymer administration, and co-administration of hyaluronidase (Wells 1993; Yanagihara et al. 1996; Baranov et al. 1999; Vilquin et al. 2001; Danialou et al. 2002; Gollins et al. 2003; Murakami et al. 2003; Ferrer et al. 2004; Molnar et al. 2004; Richard et al. 2005; Wong et al. 2005). While these methods enabled delivery of the full-length dystrophin cDNA to the muscle, the process was highly inefficient and not amenable to systemic gene restoration. More recently, groups have demonstrated the potential for systemic delivery of plasmid DNA to reach multiple muscles, including the diaphragm (Liu et al. 2001; Zhang et al. 2004, 2010). While exciting, these methods are invasive and therefore currently limited in their potential for clinical translation.
4.3.2.2 Viral-Mediated Gene Therapy
Viral-mediated gene delivery strategies utilizing vectors and viruses carrying genetic material into cells, are at the forefront of DMD gene therapies. While some viral vectors contain large genetic constructs, the carrying capacity of most vectors is a limiting factor. The DMD gene, at 14 kb, is the largest gene in the human genome and therefore presents significant challenges for gene delivery systems, with most viral vectors being too small to carry such a large gene. However, in 1990, a patient with very mild Becker muscular dystrophy (BMD, a dystrophinopathy like DMD, but with a milder phenotype) was identified with a mutation that resulted in deletion of 46% of the DMD gene, demonstrating that large portions of the gene could be removed yet continue to make a reasonably functional protein (England et al. 1990). Subsequent mutagenesis studies revealed a series of dystrophin mini- and micro-genes that were small enough to be packaged into even the smallest of viral vectors (Harper et al. 2002).
4.3.2.2.1 Adenoviral Vectors
The first viral vectors to deliver dystrophin to mdx mice were based on the adenoviruses. The adenoviruses have a double stranded (ds)DNA genome with a 35 kb capacity, and therefore the capacity to carry the entire DMD gene. First-generation adenoviral vectors were generated by removal of the E1 and/or E3 viral genes, resulting in a carrying capacity of approximately 8 kb. These vectors successfully delivered a miniaturized DMD gene to mdx mice via intramuscular injection (Ragot et al. 1993; Vincent et al. 1993). However, the presence of the remaining viral genes limited the potential for these vectors to be delivered systemically due to risk of an immune response. The newer generation vectors, termed “gutted” adenoviral vectors, lack all viral genes and therefore have the capacity to deliver the full-length DMD gene (Hartigan-O’Connor et al. 2002). Gutted adenoviral vectors have effectively delivered the full-length dystrophin protein to skeletal muscle after direct intramuscular injection into both adult and newborn mice, resulting in stable protein expression and functional improvements up to a year after injection (Dudley et al. 2004; Gilbert et al. 2001, 2003). Unfortunately, the use of these vectors is limited by the ongoing risk of an immune response, and therefore not suitable for systemic administration and potential clinical application.
4.3.2.2.2 Lentiviral Vectors
Vectors based on lentiviruses, which have an RNA genome with an 8 kb capacity, have also been utilized for delivery of dystrophin mini-genes. Early studies demonstrated that injection of lentiviral vectors containing a dystrophin mini-gene could confer almost lifelong restoration of dystrophin protein expression and functional improvement when subcutaneously injected into a newborn mouse; proposed to be a result of in vivo targeting of the muscle stem cells (Kobinger et al. 2003; Kimura et al. 2010). This benefit was reduced when injected intramuscularly into an adult mouse (Kobinger et al. 2003). As lentiviral vectors have the capacity to integrate into the genome, they possess the ability to confer lifelong protection by integrating into the DNA of muscle stem cells, enabling restoration of dystrophin protein into muscle fibers as a consequence of injury-repair. However, this comes with a significant risk of insertional mutagenesis, and so in vivo systemic delivery of lentiviral vectors to treat skeletal muscle is not therapeutically viable. Instead, the use of lentiviral vectors is widespread in the treatment of muscle stem cells, facilitating autologous muscle stem cell transplantation.
4.3.2.2.3 Adeno-Associated Viral Vectors
The adeno-associated viruses (AAVs) are small, DNA parvoviruses with a single stranded (ss)DNA genome with a 5 kb capacity. Although vectors based on AAV are limited to the delivery of only small dystrophin micro-genes, these vectors have shown the most promise for viral-mediated DMD gene therapy. There are many different AAV capsid serotypes, with AAV1, 6, 7, 8, 9, and 10 having a high tropism for striated muscle, effectively transducing both skeletal and cardiac muscle after systemic administration (Muraine et al. 2020; Zincarelli et al. 2008). Delivery of different mini- and micro-dystrophin genes with various AAV serotypes, restored dystrophin protein expression and ameliorated disease in skeletal and cardiac muscle (of mice) after systemic administration (Fabb et al. 2002; Lai et al. 2005; Liu et al. 2005; Wang et al. 2000; Watchko et al. 2002; Yoshimura et al. 2004; Yue et al. 2003). Systemic delivery of a micro-dystrophin gene with an AAV6 vector rescued the pathology and improve lifespan in the severely dystrophic dystrophin/utrophin double knockout mouse (Gregorevic et al. 2006). This was the first evidence that AAV-mediated micro-dystrophin gene therapy might be a viable treatment for DMD. Early clinical trials with an optimized AAV vector and micro-dystrophin gene failed to restore dystrophin protein expression and improve pathology, which was attributed to patients developing an immune response to the therapeutic dystrophin protein (Mendell et al. 2010). Both the AAV vectors and dystrophin micro-genes underwent significant optimization and three different clinical trials were underway as of 2021, with Sarepta, Pfizer, and Solid Biosciences, demonstrating therapeutic potential of AAV-micro-dystrophin gene therapy for DMD.
While AAV-mediated micro-dystrophin gene therapy is progressing through clinical trials, researchers are investigating the therapeutic potential of combining the AAV-mediated delivery system with exon skipping and CRISPR technologies, to enable systemic in vivo gene editing. In vivo gene editing was first reported by two groups in 2016 and 2017, using AAV9 and AAV6 to systemically deliver the CRISPR/Cas9 machinery to muscles of dystrophic mice (Bengtsson et al. 2017; Tabebordbar et al. 2016). These studies showed improved functional outcomes (Bengtsson et al. 2017) and correction in both muscle stem cells and differentiated muscle fibers of dystrophic mice (Tabebordbar et al. 2016). Subsequent studies have aimed to improve the efficacy of this methodology using self-complementary (sc)AAV vectors (Zhang et al. 2020), testing the smaller Streptococcus aureus Cas9 to enable packaging into a single AAV vector (Zhang et al. 2021), and utilizing a Pax7nGFP;Ai9 dual reporter to specifically correct mutations within muscle stem cells via CRISPR/Cas9 delivery using AAV9 in vivo, thus conferring lifelong disease correction (Kwon et al. 2020). Together, this combination of viral delivery of gene editing tools provides a powerful and exciting toolkit for DMD gene therapy.
4.4 Cell-Mediated Therapies for DMD
While gene therapies should eventually provide a cure for DMD, not all patients may benefit, depending on the progression of the disease. For example, older DMD patients will likely present with significant replacement of their skeletal muscle fibers with fat and fibrotic tissue, which not only limit the amount of muscle able to be corrected but serve as physical barriers for some treatments. Cell-mediated therapies with potential to replace skeletal muscle fibers will be similarly important in the treatment of DMD, especially for older patients. Although cell therapies for DMD have not advanced as far as gene therapies, several advances have been made over the past 10–15 years.
4.4.1 Myoblast/Muscle Stem Cell Transplantation
Myoblasts were the initial choice for muscle cell transplantation to restore muscle due to their ease of isolation, and demonstrated potential when injected into the muscles of mdx mice (Partridge et al. 1989). Unfortunately, subsequent clinical trials identified only 10% dystrophin-positive fibers in muscles of immunosuppressed DMD patients despite multiple cell injections. These disappointing results were attributed to the poor survival of the injected cells in vivo, limited migration from the injection sites, and an inability to participate in long-term regeneration as the muscle stem cell population was not restored (Mendell et al. 1995). Myoblasts therefore have limited therapeutic potential as a cell population for treating DMD.
The ability to successfully isolate muscle stem cells from skeletal muscle, first characterized in a Pax3-GFP knock-in mouse (Montarras et al. 2005), was a significant advance in the mid-2000s. Injection of freshly isolated cells into muscles of mdx mice resulted in significant engraftment and improvements in muscle function (Cerletti et al. 2008), as well as successful engraftment into the muscle stem cell niche (Montarras et al. 2005; Sacco et al. 2008), demonstrating potential for long-term therapeutic benefit. While these early studies were encouraging, it became apparent that the myogenic potential and survival/migration of these cells were severely reduced if the cells were cultured prior to injection (Montarras et al. 2005). Studies have since focused on developing strategies to improve the survival and engraftment of these cells, most of which have attempted to mimic the conditions within the muscle stem cell niche to promote a more quiescent, self-renewal state. Both culture substrate rigidity and oxygen levels have been demonstrated to be important in the maintenance of “stemness” in cultured muscle stem cells to enhance the efficacy of myoblast transplantation in vivo (Duguez et al. 2012; Gilbert et al. 2010; Liu et al. 2012). Investigations are ongoing to identify the “ideal” culture conditions to expand muscle stem cells for transplantation.
While allogenic transplants have been successful in immunocompetent mice (Vilquin et al. 1995), autologous transplantations are preferred because they limit the possibility of immune responses to the transplanted cells. Advances in the development of induced pluripotent stem cells (iPSCs) and their differentiation into myogenic cells have led to further developments. Isolation and differentiation of patient-derived iPS cells into myogenic progenitors have been successful in vitro and genetic correction of both these and patient-derived myoblasts has been shown feasible using lentiviral, transposon, and CRISPR-based technologies (Filareto et al. 2013; Ifuku et al. 2018; Kazuki et al. 2010; Li et al. 2015; Quenneville et al. 2007; Young et al. 2016). Myoblast transplants remain a viable option for targeted replacement of lost muscle tissue in DMD patients.
4.4.2 Mesoangioblasts and Pericytes
While myoblast transplantation continues to be optimized in preclinical and clinical studies, one major drawback is that myoblasts are not suitable for systemic delivery, meaning their use is limited to local, intramuscular delivery. As DMD patients will require dystrophin restoration in the entire musculature, including the diaphragm, this reduces their potential as a standalone therapy. Mesoangioblasts, and a related cell population, termed “pericytes,” are alternative cell populations which became attractive candidates for muscle cell transplantation due to their myogenic potential and suitability for systemic delivery (Dellavalle et al. 2007; Minasi et al. 2002). Mesoangioblasts engraft efficiently into skeletal muscle, restore dystrophin expression, and improve functional and histological parameters in dystrophic mice and dogs when delivered either intramuscularly or systemically (Berry et al. 2007; Sampaolesi et al. 2006). Based on these encouraging studies, an initial clinical trial tested the efficacy of intraarterial delivery of human leukocyte antigen (HLA)-matched donor mesoangioblasts in five DMD patients, demonstrating delivery of the cells to be reasonably safe and well-tolerated, but with little or no restoration of dystrophin expression. This was attributed to the advanced disease progression in the patients, the impact of steroid therapy on the cells (reduced extravasation), and an insufficient number of injected cells (Cossu et al. 2015). The therapeutic potential of these cells to treat DMD remains under investigation. The potential for autologous transplantation has been demonstrated using PiggyBac transposons to correct the genetic defect in mesoangioblasts from dystrophic SCID-mdx mice. This resulted in 11–44% restoration of dystrophin expression after transplantation back into the mice which was stable for up to 5 months (Iyer et al. 2018). Other studies turned to differentiating iPS cells into mesoangioblasts for transplantation, with and without lentiviral transduction to restore genetic insufficiencies, in mouse models of other muscular dystrophies (Tedesco et al. 2012; Gerli et al. 2014). These blood-vessel associated progenitors hold promise for DMD, and their therapeutic potential continues to be investigated.
4.5 Pharmacological Therapies for DMD
Although gene- and/or cell-mediated therapies have significant potential to eventually cure DMD, the success of these approaches will likely be reduced in patients with a more advanced pathology. Therefore, pharmacologic interventions that can delay the disease progression by tackling different aspects of the dystrophic pathology, remain crucial treatments for DMD patients.
4.5.1 Targeting Myostatin
Myostatin signaling is a potent negative regulator of skeletal muscle mass. Inhibition of the myostatin signaling pathway has proved promising in preclinical studies in dystrophic mice, with antibody treatments increasing muscle mass and force production, and decreasing fibrosis (Morine et al. 2010; Murphy et al. 2010b; Pistilli et al. 2011; Nakatani et al. 2008; Parsons et al. 2006; Bogdanovich et al. 2002). Various strategies to inhibit myostatin signaling have since moved into clinical trials. AAV-mediated overexpression of the myostatin inhibitor, follistatin, using AAV1-FS344 increased muscle mass and strength in dystrophic mice and non-human primates (Haidet et al. 2008; Kota et al. 2009), and was subsequently proven safe and efficacious in BMD patients, with improved outcomes in the six-minute walk test (Mendell et al. 2015). AAV1-FS344 has subsequently progressed to Phase I/II trials with intramuscular injections in DMD patients, although the results have yet to be reported.
Antibody-directed myostatin inhibition also attenuated muscle atrophy in mouse models of muscle atrophy, including cachexia, disuse, sarcopenia, and muscular dystrophy (Murphy et al. 2010a, b, 2011a, b), leading to the development of anti-myostatin antibodies (MYO-029, PF-06252616) which had variable success in clinical trials (Krivickas et al. 2009; Singh et al. 2016; Wagner et al. 2008). Fusion of a soluble activin receptor inhibitor to IgG (ActRIIB-IgG), which reduces signaling through the TGF-β pathway, increased muscle mass in injected muscles of dystrophic mice with reduced off-target effects (Pearsall et al. 2019), and was shown safe in healthy volunteers in clinical trials (Glasser et al. 2018). Disappointingly, despite myostatin inhibition showing great promise in preclinical models, clinical trials have not progressed due to a lack of demonstrated efficacy (reviewed in Wagner 2020). The reasons for the lack of efficacy in patients are unclear but are thought to be attributed to differences in myostatin expression in mice relative to humans, a lack of improvement in muscle function despite increases in mass, and potential interference between corticosteroid therapy and myostatin inhibition (Rybalka et al. 2020).
4.5.2 Reducing Inflammation and Fibrosis
4.5.2.1 HDAC Inhibition
Compounds that inhibit class I and/or II histone deacetylases (HDACs) have been shown to ameliorate dystrophic pathology in the mdx mouse (Colussi et al. 2008; Johnson et al. 2013; Minetti et al. 2006; Vianello et al. 2014). In preclinical studies using two murine models of DMD, the HDAC inhibitor, givinostat, increased muscle mass and muscle fiber size, reduced fat and collagen deposition, and improved fatigue resistance after oral administration (Consalvi et al. 2013; Licandro et al. 2021). High dose administration of givinostat improved muscle function and pathology to a greater extent than conventional steroid (glucocorticoid) therapy in severely dystrophic mice (Licandro et al. 2021). Givinostat is in clinical trials to test efficacy in DMD patients. Oral administration of givinostat to ambulant DMD boys aged 7–11, who were already receiving corticosteroid treatment, for greater than 1 year, reduced fibrosis, necrosis, and fatty tissue deposition in muscle biopsies, showing the drug to be safe for long-term administration and effective in delaying aspects of the pathology (Bettica et al. 2016). Givinostat is a promising intervention for DMD patients and Phase III trials are ongoing with results expected in 2022.
4.5.2.2 NF-κB Inhibition
Signaling via the nuclear factor kappa B (NF-κB) pathway is tightly linked to changes in skeletal muscle mass. Transgenic mice with chronically elevated NF-κB signaling have severe wasting of limb and trunk muscles and conversely, NF-κB inhibition protects against wasting (Cai et al. 2004; Mourkioti et al. 2006). Increased NF-κB signaling is linked to disease progression in DMD patients and in mouse models of DMD, and targeted inhibition of NF-κB is an attractive therapeutic option for skeletal muscle atrophy. Preclinical studies in dystrophic mice showed inhibition of NF-κB improved skeletal muscle structure and function (Hammers et al. 2016). Edasalonexent (CAT-1004) is an oral NF-κB inhibitor under investigation for treatment of DMD patients, shown to effectively inhibit NF-κB signaling and to be well-tolerated in Phase I safety studies in healthy adults and pediatric DMD patients (Donovan et al. 2017; Finanger et al. 2019).
Another promising NF-κB inhibitor, VBP15, is in development by ReveraGen BioPharma as an alternative to current glucocorticoids for DMD. In mdx mice, VBP15 produced consistent improvements in muscle inflammation and function, with increased fore- and hind-limb grip strength and improved ex vivo contractile properties in the extensor digitorum longus (EDL) muscle (Heier et al. 2013). Importantly, VBP15 has been proposed to inhibit NF-κB signaling to a greater extent than traditional steroid therapy, with reduced off-target effects (Conklin et al. 2018; Hoffman et al. 2018). Phase I and Phase IIa studies demonstrate VBP15 to be well-tolerated with benefits to motor function in young DMD patients (Smith et al. 2020; Mavroudis et al. 2019). Phase III trials are currently underway.
4.5.2.3 Inhibition of Collagen
Halofuginone (HT-100) had potent anti-fibrotic properties in multiple mouse models of disease with fibrosis and was tested in dystrophic mice to determine its efficacy for attenuating fibrosis in skeletal muscle. In mdx mice, halofuginone administration inhibited muscle fibrosis and improved muscle histopathology and strength (Turgeman et al. 2008). In addition, halofuginone has been shown to have direct effects on muscle cells, enhancing cell survival and myoblast fusion in both primary and C2C12 myoblasts (Bodanovsky et al. 2014; Roffe et al. 2010). HT-100 was in clinical trials to test safety and efficacy in DMD patients, but extended Phase II studies to study long-term impacts of HT-100 administration were terminated in 2016 after the unexpected death of a patient. No further updates have been provided.
4.5.2.4 Sodium/Proton Exchanger Type 1 (NHE-1) Inhibition
In the skeletal muscles and hearts of patients and mouse models of DMD, membrane tears caused by mechanical stress during contraction can lead to Ca2+ influx and consequent increase in [Na+]. Rimeporide, an NHE-1 inhibitor, has potent anti-inflammatory and anti-fibrotic effects in both skeletal and cardiac muscles in mdx mice (Porte-Thome et al. 2015), and demonstrated improved cardiac function in dystrophic dogs (Ghaleh et al. 2020). Phase 1b studies found Rimeporide to be well-tolerated and positive indications as a cardioprotective treatment in DMD patients (Previtali et al. 2020). Further clinical development is underway.
4.5.3 Upregulation of Utrophin
Since dystrophin gene replacement therapies may induce immune responses in some DMD patients, a possible safer alternative could be the therapeutic upregulation of the related protein, utrophin. In the mdx mouse, upregulation of utrophin can compensate for the loss of dystrophin (Tinsley et al. 1996, 1998). This was the basis of the development of the first orally bioavailable small molecule upregulator of utrophin, SMT-C1100, which was shown to reduce inflammation and fibrosis and improve force production in muscles of sedentary and exercised mdx mice (Tinsley et al. 2011). Phase Ia and Phase Ib trials confirmed SMT-C1100 to be safe and well-tolerated in healthy adults and in DMD patients (Ricotti et al. 2016; Tinsley et al. 2015). However, drug blood plasma concentrations were found to be lower in DMD patients than in healthy adults when administered at the same dose; an effect attributed to differences in diet and other disease-related factors. Absorption was subsequently improved in patients on a controlled diet (Muntoni et al. 2019). Unfortunately, development of SMT-C1100 was discontinued in 2018 due to it not meeting primary and secondary endpoints at the conclusion of the Phase II trial (Babbs et al. 2020). Preclinical development is continuing on a second-generation compound, SMT022357, which improved the dystrophic phenotype in mdx mice along with improved absorption, distribution, metabolism, and excretion profiles compared to SMT-C1100 (Babbs et al. 2020; Guiraud et al. 2015).
4.5.4 Improving Membrane Stability
As calcium ion (Ca2+) influx is thought to be a primary initiator of skeletal and cardiac muscle cell degeneration in DMD, compounds able to seal damaged membranes have therapeutic potential. Poloxamer-188 (P-188) is a non-ionic tri-block copolymer that can act as a membrane sealant after different types of injury. P-188 was cardioprotective after systemic administration in both mouse and dog models of muscular dystrophy. One- or 2-week treatments improved left ventricle function and promoted survival in mice after challenge with cardiac stimulants and reduced myocardial fibrosis and left ventricle remodeling in dystrophic dogs after chronic infusion for 8 weeks (Spurney et al. 2011; Townsend et al. 2010; Yasuda et al. 2005). In addition, in vitro studies showed improvements in dystrophic skeletal muscle after P-188 treatment (Spurney et al. 2011; Ng et al. 2008). P-188 (Carmeseal-MD™) improved respiratory and cardiac function in dystrophic mice (Markham et al. 2015), although some studies have reported less success with respect to ameliorating contraction-induced injury in muscles of mdx mice (Terry et al. 2014). Although most studies in animal models report positive benefits, studies examining the safety and efficacy of P-188 for DMD, are warranted.
4.6 Physical Therapies
Skeletal muscle is comprised of functionally diverse fibers that can differ in their size, metabolism, and contractility; with extremes being classically referred to as “slow oxidative” or “fast glycolytic” (Egan and Zierath 2013). Based on myosin heavy chain (MyHC) protein isoforms, which largely dictate the rate of force development, shortening velocity and the rate of cross-bridge cycling, slow oxidative (type I) fibers are typically small with a high oxidative capacity and fatigue resistance compared with fast glycolytic (type II) fibers that are typically larger, reliant on glycolysis and highly fatigable (Egan and Zierath 2013). Subtypes of the fast fibers (type IIa, type IIx) vary in their reliance on oxidative and glycolytic metabolism. Most mammalian muscles are usually comprised of different proportions of these four main fiber types (type I, type IIa, type IIb, and type IIx), although muscle fibers can exist along a continuum, with subtypes exhibiting different variations of the main attributes, especially metabolic features, as revealed by single fiber proteomics (Schiaffino et al. 2020).
Muscle fibers can exhibit remarkable plasticity, capable of altering their intrinsic structural, functional, metabolic, and molecular properties in response to changes in loading, contractile activity, and circulating hormones (Pette and Vrbova 1999; Blaauw et al. 2013; Lynch 2017; Schiaffino and Reggiani 2011). This plasticity was first demonstrated through pioneering nerve cross-reinnervation studies in cats, which revealed when fast muscles were innervated by a slow nerve, the muscle transformed from a fast (glycolytic) to a slower, more oxidative phenotype and contracted more slowly. When slow muscles were innervated by a fast nerve, the muscle transformed from an oxidative to a more glycolytic phenotype and contracted more quickly. Such phenotypic changes were attributed to the specific impulse patterns delivered to the muscle via the motor neuron (Buller et al. 1960). Muscular contractions through physical activity (exercise) can be an effective stimulus to induce adaptations in muscle if exercise duration and intensity are sufficient. Endurance exercise (e.g., running, cycling) can increase muscle oxidative capacity and fatigue resistance, while resistance exercise (e.g., lifting weights) increases fiber size (hypertrophy) and strength (Egan and Zierath 2013).
In DMD and well-characterized murine models of the disease linked to the genetic loss of the protein dystrophin, fast muscle fibers are more susceptible to contraction-mediated damage and pathological progression than slow muscle fibers, which are resistant to injury and relatively spared (Webster et al. 1988). Although a cure for DMD will eventually come from the corrective gene therapies described earlier, limitations of delivery systems, gene carrying capacity, dissemination efficiency, expression persistence, and immunological tolerance, all pose significant obstacles for clinical application. There remains an urgent and unmet clinical need for therapies that can ameliorate the pathology, preserve and protect dystrophic muscles from damage. Physical modalities such as exercise have many localized and systemic health benefits, and therefore may ultimately serve as adjuvant therapies for any gene- or cell-based approaches.
Much of our understanding as to whether physical activity and exercise training interventions can improve quality of life in DMD patients has come from exercise studies conducted in appropriate mouse models, particularly mdx dystrophic mice. These studies have examined: (1) whether exercise exacerbates the dystrophic pathology, typically from high-intensity, involuntary exercise protocols; or (2) the therapeutic potential of low-intensity, exercise protocols (voluntary and involuntary) to attenuate the dystrophic pathology (Grange and Call 2007). Several exercise protocols in mdx mice have demonstrated beneficial adaptations, with some of these low-intensity exercises having translational relevance for DMD.
4.6.1 Involuntary Exercise
It is generally accepted that low-intensity, low-weight bearing exercise promote beneficial adaptations for the dystrophic pathology, whereas exercises involving potentially injurious lengthening (i.e., eccentric) contractions may aggravate the pathology (Markert et al. 2012). Several models of exercise training have been developed and utilized in preclinical studies that have improved our understanding of the therapeutic potential of exercise for muscular dystrophy (reviewed in Hyzewicz et al. 2015; Markert et al. 2011).
4.6.1.1 Treadmill Running
Treadmill exercise training in healthy mice promotes adaptations similar to endurance exercise in humans. One main advantage of treadmill exercise is that it allows researchers to precisely control the training workload (e.g., frequency, intensity, duration) to interrogate specific muscle adaptations to submaximal or maximal workloads. Most studies have demonstrated detrimental effects of treadmill exercise training in mdx mice (reviewed in Hyzewicz et al. 2015), especially since, in most cases, exercise intensity was matched to levels achieved by otherwise healthy wild-type mice. When performed at lower intensities, treadmill running has been shown to promote beneficial adaptations to the dystrophic pathology, including decreasing intramuscular collagen deposition (Fernandes et al. 2019; Gaiad et al. 2017), reducing serum creatine kinase levels (Hall et al. 2007), and increasing activity of antioxidant enzymes (Fernandes et al. 2019).
4.6.1.2 Swimming
Swimming exercise training has been widely used in mdx mice to examine acute responses and training adaptations to endurance exercise. Swimming activity recruits muscle groups throughout body and can be used to monitor adaptations in the heart, diaphragm, and limb skeletal muscles. Endurance swimming (2 h/day, 5 days/week, for 10–20 weeks) improved the functional capacity of hindlimb muscles (in mdx mice) through adaptations arising from an increased proportion of oxidative fibers and reducing muscle fatigue (Lynch et al. 1993; Hayes et al. 1993). Favorable adaptations to low-intensity, swim exercise could also be achieved in older mdx mice (Hayes and Williams 1998), at a time when the progressive pathology more closely mimics that of DMD. Swim training was shown to improve the dystrophic pathology when combined with pharmacologic (Hayes and Williams 1997) and cell-based interventions (Bouchentouf et al. 2006). These and many other confirmative studies support the contention that low-intensity exercise, when performed alone or in combination with other interventions, may have therapeutic potential for muscle wasting conditions, including the muscular dystrophies.
4.6.1.3 Electrical Stimulation
Despite physical activity and muscular contraction, especially endurance exercise training, having many beneficial effects on muscle health, the sad reality is that many patients with neuromuscular diseases are simply unable to exercise, especially boys with DMD who are often confined to a wheelchair before their teens. Therefore, protocols of muscle contractions that can mimic the benefits of exercise are under investigation to determine whether such interventions can attenuate the loss of muscle health and function and potentially improve quality of life for patients.
Electrical stimulation to induce muscular contractions can provide an exercise-like stimulus that has been shown to induce beneficial muscle adaptations and clinical outcomes in adults with advanced progressive disease (Jones et al. 2016). Electrical stimulation at varying frequencies can be used to induce concentric, isometric, and/or eccentric muscle contractions, and several variations have been examined in preclinical mouse models and in DMD patients. Isometric contractions generate force or torque without a change in muscle length or joint angle. Repeated bouts of isometric contractions induced by percutaneous stimulation of the peroneal nerve in mdx mice improved aspects of the dystrophic pathology, including increased force production, satellite cell number, and myofiber hypertrophy, while reducing fibrosis and injury susceptibility (Lindsay et al. 2019).
Chronic low-frequency stimulation (LFS) mimics the electrical discharge pattern of slow motor neurons innervating slow muscles and induces downstream molecular signaling pathways that promote transcription of slow, more oxidative fiber-specific genes (Pette and Vrbova 1999). The resultant fast-to-slow adaptations include increased oxidative metabolism and mitochondrial biogenesis concurrent with fiber transitions in the type IIb > type IId/x > type IIa > type I direction (Leeuw and Pette 1993; Termin et al. 1989). While LFS can challenge a muscle to its full adaptive potential, it can do so efficiently and typically in the absence of injury and regeneration (Pette and Vrbova 1999). Therefore, LFS is an ideal model for investigating the therapeutic potential of promoting a slower, more oxidative muscle phenotype to ameliorate the dystrophic pathology. Initial studies of LFS in dystrophic mice were not conducted on mouse models of DMD (Dangain and Vrbova 1989; Luthert et al. 1980; Reichmann et al. 1981, 1983). Nonetheless, these studies showed that LFS exerted beneficial effects on laminin-deficient muscles in C57BL/6J-dy2j (dy/dy) mice, including improved strength (Dangain and Vrbova 1989; Luthert et al. 1980), and normalized enzyme activities (Reichmann et al. 1981, 1983).
More recently, we and others have evaluated the therapeutic merit of LFS in well-characterized mouse models of DMD. In dystrophin-deficient mdx mice, LFS (10 Hz, 12 h/day, 7 days/week, 28 days) was sufficient to induce remodeling of mitochondrial respiratory chain complexes, enhanced fiber respiration, and conferred protection from eccentric contraction-mediated damage (Hardee et al. 2021). However, this adaptive remodeling was attenuated or abrogated in dystrophic muscles lacking both dystrophin and utrophin (i.e., dko mice), highlighting a role for utrophin in the adaptations of dystrophic skeletal muscles. Using an alternate approach via transcutaneous surface stimulation, others have found that neuromuscular electrical stimulation (NMES) to mdx mice improved muscle-derived stem cell (MDSC) engraftment sufficient to enhance muscle strength, and, in combination with MDSC transplantation, improve recovery from fatigue (Distefano et al. 2013). Collectively, these findings highlight the therapeutic potential of LFS to ameliorate the dystrophic pathology and protect from contraction-induced injury with important implications for DMD and related muscle disorders.
4.6.1.4 LFS in DMD Patients
From a clinical perspective, there was considerable interest in LFS as a therapy for DMD during the 1970s through to the early 1990s, but after the discovery of dystrophin in 1987, unsurprisingly the field focused on addressing the dystrophic pathophysiology through molecular biochemical approaches (Hoffman et al. 1987a–c). These early studies found that electrical stimulation reduced the rate of deterioration of ankle dorsiflexors and quadriceps muscles in boys with DMD (Scott et al. 1990; Zupan 1992; Zupan et al. 1993, 1995), provided that the stimulation was performed before the patients were not severely disabled (Scott et al. 1986). Similar findings related to safety, practicality, and improved muscular strength and endurance have been reported in facioscapulohumeral muscular dystrophy (FSHD) and myotonic dystrophy type 1 patients (Colson et al. 2010; Cudia et al. 2016). While the initial studies on DMD patients were encouraging (e.g., some showing preserved strength), they were largely preliminary in nature (with few patients and studies of limited duration) with a lack of rigorous scientific and statistical clarity (Lynch 2017). Nonetheless, there remains a dearth of information on the application of such a well-described and utilized intervention like LFS (with existing applications in rehabilitation medicine and physical therapy) to ameliorate aspects of the dystrophic pathology.
4.6.2 Voluntary Exercise
4.6.2.1 Wheel Running
Providing access to running wheels in cages is one approach to elicit low-intensity endurance training adaptations in rodents. While this model permits normal physiological patterns of motor unit recruitment, it is dependent on intrinsic, voluntary physical activity and therefore total work performed can vary dependent on the animal’s volition based on age and disease severity (Wineinger et al. 1998). Voluntary wheel running in mdx mice has been shown to promote favorable adaptations in plasma CK levels (Carter et al. 1995), improve force production (Baltgalvis et al. 2012; Call et al. 2010; Carter et al. 1995; Dupont-Versteegden et al. 1994; Hayes and Williams 1996; Hourde et al. 2013), decrease injury susceptibility (Hourde et al. 2013; Delacroix et al. 2018), improve fatigue resistance (Baltgalvis et al. 2012; Hayes and Williams 1996; Wineinger et al. 1998), and alter myofiber size and type (Delacroix et al. 2018; Hayes and Williams 1996; Landisch et al. 2008). Mechanisms mediating these functional outcomes were identified, such as increased utrophin protein expression (Gordon et al. 2014) and muscle oxidative capacity (Baltgalvis et al. 2012). A recent study combining voluntary wheel running and micro-dystrophin gene therapy reported improved running capacity, increased muscle contractile properties, protection from eccentric contraction-induced injury, and enhanced mitochondrial respiration (Hamm et al. 2021). Thus, exercise may be a complementary intervention for enhancing the efficacy of micro-dystrophin gene therapies. Further studies are warranted to examine the efficacy of combined interventions, including investigations of different aged mice and disease severities before this might be considered for potential clinical application.
While it is generally accepted that low-intensity exercise may be more beneficial for attenuating the dystrophic process, resisted wheel running exercise may also induce favorable adaptations such as enhanced muscle growth through myofiber hypertrophy, improved regeneration, and force production. Indeed, voluntary wheel running with progressive resistance in mdx mice was shown to improve grip strength and increase specific force of the soleus muscle (Call et al. 2010). Overall, these studies support the contentions that: (1) voluntary exercise is not detrimental to dystrophic pathology in mice; and (2) dystrophic skeletal muscles retain the adaptive potential to respond to muscular contractions, with favorable, clinically relevant outcomes.
4.6.2.2 Exercise in DMD Patients
4.6.2.2.1 Resistance Exercise
Initial studies examining physical training interventions in DMD patients utilized resistance-type exercise. At the medical conference for Muscular Dystrophy Associations of America in 1952, Abramson and Rogoff (Abramson and Rogoff 1952) first reported in 27 patients with muscular dystrophy that active, assisted active, and resistive exercises (3 days/week, 7 months) led to improvements (n = 13) and/or no changes (n = 13) in the manual muscle chart. Based on these encouraging findings, Hoberman (Hoberman 1955) studied ten children with progressive muscular dystrophy. The patients performed 4 months of physical medicine and rehabilitation tests, instruction and training. While no changes in muscle strength were observed, there were improvements in performance of activities of daily living, vital capacity, and endurance (Hoberman 1955). Vignos and Watkins (1966) studied 24 patients with muscular dystrophy (14 DMD, 6 limb-girdle, and 4 facioscapulohumeral), who performed home-based resistance exercise program for 12 months. In the year leading up to the study, DMD patients exhibited declines in muscle strength. Unexercised controls continued to exhibit a decline in muscle strength, while the exercised group maintained or improved slightly from their initial muscle strength (Vignos and Watkins 1966). Most of the improvements in muscle strength occurred within the first 4 months of the exercise program and the degree of improvement was related to the initial strength of the exercised muscle; i.e., a stronger muscle improved more, while weaker muscles improved less. Unfortunately, functional improvements were not permanent and only occurred in 7/52 patients at 4 months and 1/52 patients at 12 months (Vignos and Watkins 1966). To determine if submaximal exercise could improve strength, four DMD patients performed unilateral isokinetic exercise of the quadriceps (4–5 days/week, 6 months). The authors reported a modest, though not statistically significant, increase in strength during the 6-month period, which was maintained for 3 months after the cessation of training (de Lateur and Giaconi 1979). More recently, Lott et al. (2021) reported that a 12-week in-home, remotely-supervised, mild-moderate intensity resistance isometric leg exercise program was safe, feasible, and increased strength (knee extension, knee flexion) and function (descending step) in ambulatory boys with DMD. Overall, these studies indicate that resistance-type exercise programs can improve aspects of activities of daily living and functional performance. It is noted and that the exercise program should be started earlier in the disease trajectory when muscles are most functional. Importantly, it also highlighted that type of regimen was feasible (e.g., no ill events reported) and did not cause deleterious effects on muscle strength in the patients. Regardless, exercise should be prescribed cautiously and the therapeutic merit of any resistance training for patients with neuromuscular diseases must be assessed against the risk for overwork and potential for exacerbating the pathology.
4.6.2.2.2 Endurance Exercise
More recently, the “No Use is Disuse” study was the first randomized control trial in ambulant and wheelchair-dependent DMD boys that examined whether low-intensity physical training through assisted cycling training using the arms and legs (5 days/week, 24 weeks) could improve muscle endurance and functional capacity (Jansen et al. 2010). The authors found all participants could complete the training protocol (except one) with no serious adverse events reported. Importantly, the primary outcome of total Motor Function Measure remained stable with physical training, whereas it decreased in the control group (Jansen et al. 2013). However, no improvements in the Assisted 6-Min Cycling Test were observed. These findings suggest that assisted bicycle training of the legs and arms was feasible and safe for both ambulant and wheelchair-dependent DMD patients and that physical training helped maintain functional capacity.
Others have demonstrated improvements in clinically relevant outcomes with different types of endurance exercise modalities. Compared to range of motion exercises alone, upper extremity training with an arm ergometer (40 min/session, 3 days/week, 8 weeks) was more effective in preserving and improving the functional level of early-stage DMD patients (Alemdaroglu et al. 2015). Overall, the studies performed to date demonstrate that dystrophic skeletal muscles retain the capacity to adapt favorably to exercise training and this can attenuate the functional decline with disease progression. However, as highlighted in a recent Cochrane Review of exercise in muscle diseases (Voet et al. 2019), the evidence regarding endurance and resistance exercise training interventions in muscle diseases remains uncertain, and more research with robust methodology and greater numbers of participants are still required.
4.7 Conclusions
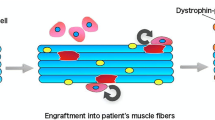
Improving quality of life for DMD patients through exercise requires activities that can improve function in all muscles of the body, ideally including the heart and respiratory muscles. Exercise has the capacity to improve or maintain physical function, body composition, and overall quality of life for patients. While endurance and resistance exercise training can individually promote health benefits, the muscle adaptive responses are unique to the stimulus/intervention provided. Exercise interventions could attenuate muscle weakness and dysfunction in DMD, and current international guidelines recommend regular submaximal exercise activities for boys with DMD (Bushby et al. 2010). Although studies support rehabilitation as an adjunct treatment to gene- and cell-mediated therapies for DMD patients (see Fig. 4.1), current recommendations are based on theories, practical experience of the practitioners, and knowledge gained from animal studies. Randomized, controlled trials are warranted to investigate the therapeutic merit of adjunct rehabilitation in conjunction with other interventions based on the benefits observed in preclinical models and DMD patients.

Therapeutic combinations for DMD. Striated muscle pathology in DMD arises from a loss of the dystrophin protein causing membrane instability, contractile and metabolic dysfunction, and muscle wasting and weakness, ultimately resulting in premature death. Regenerative gene, cell, and/or pharmacologic therapies have varying efficacy for increasing dystrophin/utrophin protein expression to improve membrane stability and physical function. In addition, rehabilitative therapies including physical exercise and muscle contraction protocols elicited by electrical stimulation (e.g., neuromuscular electrical stimulation, functional electrical stimulation, or low-frequency stimulation) have also shown promise for improving skeletal muscle pathology by potentially driving a slow muscle phenotype that improves mitochondrial function, oxidative capacity, and contractile performance. Combinations of regenerative and rehabilitative therapies have significant potential to improve quality of life and survival in DMD patients. Created with BioRender.com
References
Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT (2006) Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34(2):135–144. https://doi.org/10.1002/mus.20586
Abramson AS, Rogoff J (1952) An approach to the rehabilitation of children with muscular dystrophy. Physical treatment in muscular dystrophy. Paper presented at the 1st and 2nd medical conferences, Muscular Dystrophy Association of America, New York
Acsadi G, Dickson G, Love DR, Jani A, Walsh FS, Gurusinghe A, Wolff JA, Davies KE (1991) Human dystrophin expression in mdx mice after intramuscular injection of DNA constructs. Nature 352(6338):815–818. https://doi.org/10.1038/352815a0
Alemdaroglu I, Karaduman A, Yilmaz OT, Topaloglu H (2015) Different types of upper extremity exercise training in Duchenne muscular dystrophy: effects on functional performance, strength, endurance, and ambulation. Muscle Nerve 51(5):697–705. https://doi.org/10.1002/mus.24451
Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, Partridge TA, Lu QL (2006) Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med 12(2):175–177. https://doi.org/10.1038/nm1345
Babbs A, Berg A, Chatzopoulou M, Davies KE, Davies SG, Edwards B, Elsey DJ, Emer E, Figuccia ALA, Fletcher AM, Guiraud S, Harriman S, Moir L, Robinson N, Rowley JA, Russell AJ, Squire SE, Thomson JE, Tinsley JM, Wilson FX, Wynne GM (2020) Synthesis of SMT022357 enantiomers and in vivo evaluation in a Duchenne muscular dystrophy mouse model. Tetrahedron 76(2):130819. https://doi.org/10.1016/j.tet.2019.130819
Baltgalvis KA, Call JA, Cochrane GD, Laker RC, Yan Z, Lowe DA (2012) Exercise training improves plantar flexor muscle function in mdx mice. Med Sci Sports Exerc 44(9):1671–1679. https://doi.org/10.1249/MSS.0b013e31825703f0
Baranov A, Glazkov P, Kiselev A, Ostapenko O, Mikhailov V, Ivaschenko T, Sabetsky V, Baranov V (1999) Local and distant transfection of mdx muscle fibers with dystrophin and LacZ genes delivered in vivo by synthetic microspheres. Gene Ther 6(8):1406–1414. https://doi.org/10.1038/sj.gt.3300954
Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL (1999) Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest 104(4):375–381. https://doi.org/10.1172/JCI7866
Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, Hauschka SD, Chamberlain JR, Chamberlain JS (2017) Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun 8:14454. https://doi.org/10.1038/ncomms14454
Berry SE, Liu J, Chaney EJ, Kaufman SJ (2007) Multipotential mesoangioblast stem cell therapy in the mdx/utrn-/- mouse model for Duchenne muscular dystrophy. Regen Med 2(3):275–288. https://doi.org/10.2217/17460751.2.3.275
Bettica P, Petrini S, D’Oria V, D’Amico A, Catteruccia M, Pane M, Sivo S, Magri F, Brajkovic S, Messina S, Vita GL, Gatti B, Moggio M, Puri PL, Rocchetti M, De Nicolao G, Vita G, Comi GP, Bertini E, Mercuri E (2016) Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul Disord 26(10):643–649. https://doi.org/10.1016/j.nmd.2016.07.002
Blaauw B, Schiaffino S, Reggiani C (2013) Mechanisms modulating skeletal muscle phenotype. Compr Physiol 3(4):1645–1687. https://doi.org/10.1002/cphy.c130009
Bodanovsky A, Guttman N, Barzilai-Tutsch H, Genin O, Levy O, Pines M, Halevy O (2014) Halofuginone improves muscle-cell survival in muscular dystrophies. Biochim Biophys Acta 1843(7):1339–1347. https://doi.org/10.1016/j.bbamcr.2014.03.025
Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana TS (2002) Functional improvement of dystrophic muscle by myostatin blockade. Nature 420(6914):418–421. https://doi.org/10.1038/nature01154
Bouchentouf M, Benabdallah BF, Mills P, Tremblay JP (2006) Exercise improves the success of myoblast transplantation in mdx mice. Neuromuscul Disord 16(8):518–529. https://doi.org/10.1016/j.nmd.2006.06.003
Buller AJ, Eccles JC, Eccles RM (1960) Interactions between motoneurones and muscles in respect of the characteristic speeds of their responses. J Physiol 150:417–439. https://doi.org/10.1113/jphysiol.1960.sp006395
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C, Group DMDCCW (2010) Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 9(2):177–189. https://doi.org/10.1016/S1474-4422(09)70272-8
Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE (2004) IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119(2):285–298. https://doi.org/10.1016/j.cell.2004.09.027
Call JA, McKeehen JN, Novotny SA, Lowe DA (2010) Progressive resistance voluntary wheel running in the mdx mouse. Muscle Nerve 42(6):871–880. https://doi.org/10.1002/mus.21764
Carter GT, Wineinger MA, Walsh SA, Horasek SJ, Abresch RT, Fowler WM Jr (1995) Effect of voluntary wheel-running exercise on muscles of the mdx mouse. Neuromuscul Disord 5(4):323–332. https://doi.org/10.1016/0960-8966(94)00063-f
Cerletti M, Jurga S, Witczak CA, Hirshman MF, Shadrach JL, Goodyear LJ, Wagers AJ (2008) Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell 134(1):37–47. https://doi.org/10.1016/j.cell.2008.05.049
Colson SS, Benchortane M, Tanant V, Faghan JP, Fournier-Mehouas M, Benaim C, Desnuelle C, Sacconi S (2010) Neuromuscular electrical stimulation training: a safe and effective treatment for facioscapulohumeral muscular dystrophy patients. Arch Phys Med Rehabil 91(5):697–702. https://doi.org/10.1016/j.apmr.2010.01.019
Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S, Ragone G, Pescatori M, Zaccagnini G, Antonini A, Minetti G, Martelli F, Piaggio G, Gallinari P, Steinkuhler C, Clementi E, Dell’Aversana C, Altucci L, Mai A, Capogrossi MC, Puri PL, Gaetano C (2008) HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci U S A 105(49):19183–19187. https://doi.org/10.1073/pnas.0805514105
Conklin LS, Damsker JM, Hoffman EP, Jusko WJ, Mavroudis PD, Schwartz BD, Mengle-Gaw LJ, Smith EC, Mah JK, Guglieri M, Nevo Y, Kuntz N, McDonald CM, Tulinius M, Ryan MM, Webster R, Castro D, Finkel RS, Smith AL, Morgenroth LP, Arrieta A, Shimony M, Jaros M, Shale P, McCall JM, Hathout Y, Nagaraju K, van den Anker J, Ward LM, Ahmet A, Cornish MR, Clemens PR (2018) Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug. Pharmacol Res 136:140–150. https://doi.org/10.1016/j.phrs.2018.09.007
Consalvi S, Mozzetta C, Bettica P, Germani M, Fiorentini F, Del Bene F, Rocchetti M, Leoni F, Monzani V, Mascagni P, Puri PL, Saccone V (2013) Preclinical studies in the mdx mouse model of Duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med 19:79–87. https://doi.org/10.2119/molmed.2013.00011
Cossu G, Previtali SC, Napolitano S, Cicalese MP, Tedesco FS, Nicastro F, Noviello M, Roostalu U, Natali Sora MG, Scarlato M, De Pellegrin M, Godi C, Giuliani S, Ciotti F, Tonlorenzi R, Lorenzetti I, Rivellini C, Benedetti S, Gatti R, Marktel S, Mazzi B, Tettamanti A, Ragazzi M, Imro MA, Marano G, Ambrosi A, Fiori R, Sormani MP, Bonini C, Venturini M, Politi LS, Torrente Y, Ciceri F (2015) Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol Med 7(12):1513–1528. https://doi.org/10.15252/emmm.201505636
Cudia P, Weis L, Baba A, Kiper P, Marcante A, Rossi S, Angelini C, Piccione F (2016) Effects of functional electrical stimulation lower extremity training in myotonic dystrophy type I: a pilot controlled study. Am J Phys Med Rehabil 95(11):809–817. https://doi.org/10.1097/PHM.0000000000000497
Dangain J, Vrbova G (1989) Long term effect of low frequency chronic electrical stimulation on the fast hind limb muscles of dystrophic mice. J Neurol Neurosurg Psychiatry 52(12):1382–1389. https://doi.org/10.1136/jnnp.52.12.1382
Danialou G, Comtois AS, Dudley RW, Nalbantoglu J, Gilbert R, Karpati G, Jones DH, Petrof BJ (2002) Ultrasound increases plasmid-mediated gene transfer to dystrophic muscles without collateral damage. Mol Ther 6(5):687–693
Danko I, Fritz JD, Latendresse JS, Herweijer H, Schultz E, Wolff JA (1993) Dystrophin expression improves myofiber survival in mdx muscle following intramuscular plasmid DNA injection. Hum Mol Genet 2(12):2055–2061. https://doi.org/10.1093/hmg/2.12.2055
Delacroix C, Hyzewicz J, Lemaitre M, Friguet B, Li Z, Klein A, Furling D, Agbulut O, Ferry A (2018) Improvement of dystrophic muscle fragility by short-term voluntary exercise through activation of calcineurin pathway in mdx mice. Am J Pathol 188(11):2662–2673. https://doi.org/10.1016/j.ajpath.2018.07.015
de Lateur BJ, Giaconi RM (1979) Effect on maximal strength of submaximal exercise in Duchenne muscular dystrophy. Am J Phys Med 58(1):26–36
Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R, Li S, Belicchi M, Peretti G, Chamberlain JS, Wright WE, Torrente Y, Ferrari S, Bianco P, Cossu G (2007) Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol 9(3):255–267. https://doi.org/10.1038/ncb1542
Den Dunnen JT, Grootscholten PM, Bakker E, Blonden LA, Ginjaar HB, Wapenaar MC, van Paassen HM, van Broeckhoven C, Pearson PL, van Ommen GJ (1989) Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet 45(6):835–847
Distefano G, Ferrari RJ, Weiss C, Deasy BM, Boninger ML, Fitzgerald GK, Huard J, Ambrosio F (2013) Neuromuscular electrical stimulation as a method to maximize the beneficial effects of muscle stem cells transplanted into dystrophic skeletal muscle. PLoS One 8(3):e54922. https://doi.org/10.1371/journal.pone.0054922
Donovan JM, Zimmer M, Offman E, Grant T, Jirousek M (2017) A novel NF-kappaB inhibitor, edasalonexent (CAT-1004), in development as a disease-modifying treatment for patients with Duchenne muscular dystrophy: phase 1 safety, pharmacokinetics, and pharmacodynamics in adult subjects. J Clin Pharmacol 57(5):627–639. https://doi.org/10.1002/jcph.842
Dudley RW, Lu Y, Gilbert R, Matecki S, Nalbantoglu J, Petrof BJ, Karpati G (2004) Sustained improvement of muscle function one year after full-length dystrophin gene transfer into mdx mice by a gutted helper-dependent adenoviral vector. Hum Gene Ther 15(2):145–156. https://doi.org/10.1089/104303404772679959
Duguez S, Duddy WJ, Gnocchi V, Bowe J, Dadgar S, Partridge TA (2012) Atmospheric oxygen tension slows myoblast proliferation via mitochondrial activation. PLoS One 7(8):e43853. https://doi.org/10.1371/journal.pone.0043853
Dunant P, Walter MC, Karpati G, Lochmuller H (2003) Gentamicin fails to increase dystrophin expression in dystrophin-deficient muscle. Muscle Nerve 27(5):624–627. https://doi.org/10.1002/mus.10341
Dupont-Versteegden EE, McCarter RJ, Katz MS (1994) Voluntary exercise decreases progression of muscular dystrophy in diaphragm of mdx mice. J Appl Physiol (1985) 77(4):1736–1741. https://doi.org/10.1152/jappl.1994.77.4.1736
Egan B, Zierath JR (2013) Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17(2):162–184. https://doi.org/10.1016/j.cmet.2012.12.012
England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, Bulman DE, Harris JB, Davies KE (1990) Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 343(6254):180–182. https://doi.org/10.1038/343180a0
Ervasti JM, Campbell KP (1993) A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol 122(4):809–823. https://doi.org/10.1083/jcb.122.4.809
Fabb SA, Wells DJ, Serpente P, Dickson G (2002) Adeno-associated virus vector gene transfer and sarcolemmal expression of a 144 kDa micro-dystrophin effectively restores the dystrophin-associated protein complex and inhibits myofibre degeneration in nude/mdx mice. Hum Mol Genet 11(7):733–741. https://doi.org/10.1093/hmg/11.7.733
Fernandes DC, Cardoso-Nascimento JJA, Garcia BCC, Costa KB, Rocha-Vieira E, Oliveira MX, Machado ASD, Santos AP, Gaiad TP (2019) Low intensity training improves redox status and reduces collagen fibers on dystrophic muscle. J Exerc Rehabil 15(2):213–223. https://doi.org/10.12965/jer.1938060.030
Ferrer A, Foster H, Wells KE, Dickson G, Wells DJ (2004) Long-term expression of full-length human dystrophin in transgenic mdx mice expressing internally deleted human dystrophins. Gene Ther 11(11):884–893. https://doi.org/10.1038/sj.gt.3302242
Filareto A, Parker S, Darabi R, Borges L, Iacovino M, Schaaf T, Mayerhofer T, Chamberlain JS, Ervasti JM, McIvor RS, Kyba M, Perlingeiro RC (2013) An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat Commun 4:1549. https://doi.org/10.1038/ncomms2550
Finanger E, Vandenborne K, Finkel RS, Lee Sweeney H, Tennekoon G, Yum S, Mancini M, Bista P, Nichols A, Liu H, Fretzen A, Donovan JM (2019) Phase 1 study of edasalonexent (CAT-1004), an oral NF-kappaB inhibitor, in pediatric patients with Duchenne muscular dystrophy. J Neuromuscul Dis 6(1):43–54. https://doi.org/10.3233/JND-180341
Flanigan KM, Voit T, Rosales XQ, Servais L, Kraus JE, Wardell C, Morgan A, Dorricott S, Nakielny J, Quarcoo N, Liefaard L, Drury T, Campion G, Wright P (2014) Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne muscular dystrophy: results of a double-blind randomized clinical trial. Neuromuscul Disord 24(1):16–24. https://doi.org/10.1016/j.nmd.2013.09.004
Gaiad TP, Oliveira MX, Lobo AR Jr, Liborio LR, Pinto PAF, Fernandes DC, Santos AP, Ambrosio CE, Machado ASD (2017) Low-intensity training provokes adaptive extracellular matrix turnover of a muscular dystrophy model. J Exerc Rehabil 13(6):693–703. https://doi.org/10.12965/jer.1735094.547
Gerli MF, Maffioletti SM, Millet Q, Tedesco FS (2014) Transplantation of induced pluripotent stem cell-derived mesoangioblast-like myogenic progenitors in mouse models of muscle regeneration. J Vis Exp 83:e50532. https://doi.org/10.3791/50532
Ghaleh B, Barthelemy I, Wojcik J, Sambin L, Bize A, Hittinger L, Tran TD, Thome FP, Blot S, Su JB (2020) Protective effects of rimeporide on left ventricular function in golden retriever muscular dystrophy dogs. Int J Cardiol 312:89–95. https://doi.org/10.1016/j.ijcard.2020.03.031
Gilbert R, Nalbantoglu J, Howell JM, Davies L, Fletcher S, Amalfitano A, Petrof BJ, Kamen A, Massie B, Karpati G (2001) Dystrophin expression in muscle following gene transfer with a fully deleted (“gutted”) adenovirus is markedly improved by trans-acting adenoviral gene products. Hum Gene Ther 12(14):1741–1755. https://doi.org/10.1089/104303401750476249
Gilbert R, Dudley RW, Liu AB, Petrof BJ, Nalbantoglu J, Karpati G (2003) Prolonged dystrophin expression and functional correction of mdx mouse muscle following gene transfer with a helper-dependent (gutted) adenovirus-encoding murine dystrophin. Hum Mol Genet 12(11):1287–1299. https://doi.org/10.1093/hmg/ddg141
Gilbert PM, Havenstrite KL, Magnusson KE, Sacco A, Leonardi NA, Kraft P, Nguyen NK, Thrun S, Lutolf MP, Blau HM (2010) Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 329(5995):1078–1081. https://doi.org/10.1126/science.1191035
Glasser CE, Gartner MR, Wilson D, Miller B, Sherman ML, Attie KM (2018) Locally acting ACE-083 increases muscle volume in healthy volunteers. Muscle Nerve 57(6):921–926. https://doi.org/10.1002/mus.26113
Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, Holling T, Janson AA, Platenburg GJ, Sipkens JA, Sitsen JM, Aartsma-Rus A, van Ommen GJ, Buyse G, Darin N, Verschuuren JJ, Campion GV, de Kimpe SJ, van Deutekom JC (2011) Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med 364(16):1513–1522. https://doi.org/10.1056/NEJMoa1011367
Goemans NM, Tulinius M, van den Hauwe M, Kroksmark AK, Buyse G, Wilson RJ, van Deutekom JC, de Kimpe SJ, Lourbakos A, Campion G (2016) Long-term efficacy, safety, and pharmacokinetics of drisapersen in Duchenne muscular dystrophy: results from an open-label extension study. PLoS One 11(9):e0161955. https://doi.org/10.1371/journal.pone.0161955
Goemans N, Mercuri E, Belousova E, Komaki H, Dubrovsky A, McDonald CM, Kraus JE, Lourbakos A, Lin Z, Campion G, Wang SX, Campbell C, Group DIs (2018) A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul Disord 28(1):4–15. https://doi.org/10.1016/j.nmd.2017.10.004
Gollins H, McMahon J, Wells KE, Wells DJ (2003) High-efficiency plasmid gene transfer into dystrophic muscle. Gene Ther 10(6):504–512. https://doi.org/10.1038/sj.gt.3301927
Gordon BS, Lowe DA, Kostek MC (2014) Exercise increases utrophin protein expression in the mdx mouse model of Duchenne muscular dystrophy. Muscle Nerve 49(6):915–918. https://doi.org/10.1002/mus.24151
Grange RW, Call JA (2007) Recommendations to define exercise prescription for Duchenne muscular dystrophy. Exerc Sport Sci Rev 35(1):12–17. https://doi.org/10.1249/01.jes.0000240020.84630.9d
Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, Finn E, Adams ME, Froehner SC, Murry CE, Chamberlain JS (2006) rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med 12(7):787–789. https://doi.org/10.1038/nm1439
Guiraud S, Squire SE, Edwards B, Chen H, Burns DT, Shah N, Babbs A, Davies SG, Wynne GM, Russell AJ, Elsey D, Wilson FX, Tinsley JM, Davies KE (2015) Second-generation compound for the modulation of utrophin in the therapy of DMD. Hum Mol Genet 24(15):4212–4224. https://doi.org/10.1093/hmg/ddv154
Haidet AM, Rizo L, Handy C, Umapathi P, Eagle A, Shilling C, Boue D, Martin PT, Sahenk Z, Mendell JR, Kaspar BK (2008) Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci U S A 105(11):4318–4322. https://doi.org/10.1073/pnas.0709144105
Hall JE, Kaczor JJ, Hettinga BP, Isfort RJ, Tarnopolsky MA (2007) Effects of a CRF2R agonist and exercise on mdx and wildtype skeletal muscle. Muscle Nerve 36(3):336–341. https://doi.org/10.1002/mus.20820
Hamm SE, Fathalikhani DD, Bukovec KE, Addington AK, Zhang H, Perry JB, McMillan RP, Lawlor MW, Prom MJ, Vanden Avond MA, Kumar SN, Coleman KE, Dupont JB, Mack DL, Brown DA, Morris CA, Gonzalez JP, Grange RW (2021) Voluntary wheel running complements microdystrophin gene therapy to improve muscle function in mdx mice. Mol Ther Methods Clin Dev 21:144–160. https://doi.org/10.1016/j.omtm.2021.02.024
Hammers DW, Sleeper MM, Forbes SC, Coker CC, Jirousek MR, Zimmer M, Walter GA, Sweeney HL (2016) Disease-modifying effects of orally bioavailable NF-kappaB inhibitors in dystrophin-deficient muscle. JCI Insight 1(21):e90341. https://doi.org/10.1172/jci.insight.90341
Hardee JP, Martins KJB, Miotto PM, Ryall JG, Gehrig SM, Reljic B, Naim T, Chung JD, Trieu J, Swiderski K, Philp AM, Philp A, Watt MJ, Stroud DA, Koopman R, Steinberg GR, Lynch GS (2021) Metabolic remodeling of dystrophic skeletal muscle reveals biological roles for dystrophin and utrophin in adaptation and plasticity. Mol Metab 45:101157. https://doi.org/10.1016/j.molmet.2020.101157
Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, Chamberlain JS (2002) Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med 8(3):253–261. https://doi.org/10.1038/nm0302-253
Hartigan-O’Connor D, Barjot C, Crawford R, Chamberlain JS (2002) Efficient rescue of gutted adenovirus genomes allows rapid production of concentrated stocks without negative selection. Hum Gene Ther 13(4):519–531. https://doi.org/10.1089/10430340252809810
Hayes A, Williams DA (1996) Beneficial effects of voluntary wheel running on the properties of dystrophic mouse muscle. J Appl Physiol (1985) 80(2):670–679. https://doi.org/10.1152/jappl.1996.80.2.670
Hayes A, Williams DA (1997) Contractile properties of clenbuterol-treated mdx muscle are enhanced by low-intensity swimming. J Appl Physiol (1985) 82(2):435–439. https://doi.org/10.1152/jappl.1997.82.2.435
Hayes A, Williams DA (1998) Contractile function and low-intensity exercise effects of old dystrophic (mdx) mice. Am J Physiol 274(4):C1138–C1144. https://doi.org/10.1152/ajpcell.1998.274.4.C1138
Hayes A, Lynch GS, Williams DA (1993) The effects of endurance exercise on dystrophic mdx mice. I Contractile and histochemical properties of intact muscles. Proc Biol Sci 253(1336):19–25. https://doi.org/10.1098/rspb.1993.0077
Heier CR, Damsker JM, Yu Q, Dillingham BC, Huynh T, Van der Meulen JH, Sali A, Miller BK, Phadke A, Scheffer L, Quinn J, Tatem K, Jordan S, Dadgar S, Rodriguez OC, Albanese C, Calhoun M, Gordish-Dressman H, Jaiswal JK, Connor EM, McCall JM, Hoffman EP, Reeves EK, Nagaraju K (2013) VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med 5(10):1569–1585. https://doi.org/10.1002/emmm.201302621
Hoberman M (1955) Physical medicine and rehabilitation: its value and limitations in progressive muscular dystrophy. Am J Phys Med 34(1):109–115
Hoffman EP, Brown RH Jr, Kunkel LM (1987a) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51(6):919–928. https://doi.org/10.1016/0092-8674(87)90579-4
Hoffman EP, Knudson CM, Campbell KP, Kunkel LM (1987b) Subcellular fractionation of dystrophin to the triads of skeletal muscle. Nature 330(6150):754–758. https://doi.org/10.1038/330754a0
Hoffman EP, Monaco AP, Feener CC, Kunkel LM (1987c) Conservation of the Duchenne muscular dystrophy gene in mice and humans. Science 238(4825):347–350. https://doi.org/10.1126/science.3659917
Hoffman EP, Riddle V, Siegler MA, Dickerson D, Backonja M, Kramer WG, Nagaraju K, Gordish-Dressman H, Damsker JM, McCall JM (2018) Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes. Steroids 134:43–52. https://doi.org/10.1016/j.steroids.2018.02.010
Hourde C, Joanne P, Medja F, Mougenot N, Jacquet A, Mouisel E, Pannerec A, Hatem S, Butler-Browne G, Agbulut O, Ferry A (2013) Voluntary physical activity protects from susceptibility to skeletal muscle contraction-induced injury but worsens heart function in mdx mice. Am J Pathol 182(5):1509–1518. https://doi.org/10.1016/j.ajpath.2013.01.020
Hyzewicz J, Ruegg UT, Takeda S (2015) Comparison of experimental protocols of physical exercise for mdx mice and Duchenne muscular dystrophy patients. J Neuromuscul Dis 2(4):325–342. https://doi.org/10.3233/JND-150106
Ifuku M, Iwabuchi KA, Tanaka M, Lung MSY, Hotta A (2018) Restoration of dystrophin protein expression by exon skipping utilizing CRISPR-Cas9 in myoblasts derived from DMD patient iPS cells. Methods Mol Biol 1828:191–217. https://doi.org/10.1007/978-1-4939-8651-4_12
Iyer PS, Mavoungou LO, Ronzoni F, Zemla J, Schmid-Siegert E, Antonini S, Neff LA, Dorchies OM, Jaconi M, Lekka M, Messina G, Mermod N (2018) Autologous cell therapy approach for Duchenne muscular dystrophy using PiggyBac transposons and mesoangioblasts. Mol Ther 26(4):1093–1108. https://doi.org/10.1016/j.ymthe.2018.01.021
Jansen M, de Groot IJ, van Alfen N, Geurts A (2010) Physical training in boys with Duchenne muscular dystrophy: the protocol of the no use is disuse study. BMC Pediatr 10:55. https://doi.org/10.1186/1471-2431-10-55
Jansen M, van Alfen N, Geurts AC, de Groot IJ (2013) Assisted bicycle training delays functional deterioration in boys with Duchenne muscular dystrophy: the randomized controlled trial “no use is disuse”. Neurorehabil Neural Repair 27(9):816–827. https://doi.org/10.1177/1545968313496326
Johnson NM, Farr GH 3rd, Maves L (2013) The HDAC inhibitor TSA ameliorates a zebrafish model of Duchenne muscular dystrophy. PLoS Curr 5. https://doi.org/10.1371/currents.md.8273cf41db10e2d15dd3ab827cb4b027
Jones S, Man WD, Gao W, Higginson IJ, Wilcock A, Maddocks M (2016) Neuromuscular electrical stimulation for muscle weakness in adults with advanced disease. Cochrane Database Syst Rev 10:CD009419. https://doi.org/10.1002/14651858.CD009419.pub3
Kazuki Y, Hiratsuka M, Takiguchi M, Osaki M, Kajitani N, Hoshiya H, Hiramatsu K, Yoshino T, Kazuki K, Ishihara C, Takehara S, Higaki K, Nakagawa M, Takahashi K, Yamanaka S, Oshimura M (2010) Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol Ther 18(2):386–393. https://doi.org/10.1038/mt.2009.274
Kimura E, Li S, Gregorevic P, Fall BM, Chamberlain JS (2010) Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol Ther 18(1):206–213. https://doi.org/10.1038/mt.2009.253
Kobinger GP, Louboutin JP, Barton ER, Sweeney HL, Wilson JM (2003) Correction of the dystrophic phenotype by in vivo targeting of muscle progenitor cells. Hum Gene Ther 14(15):1441–1449. https://doi.org/10.1089/104303403769211655
Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, Meng G, Muller CR, Lindlof M, Kaariainen H, de la Chapellet A, Kiuru A, Savontaus ML, Gilgenkrantz H, Recan D, Chelly J, Kaplan JC, Covone AE, Archidiacono N, Romeo G, Liechti-Gailati S, Schneider V, Braga S, Moser H, Darras BT, Murphy P, Francke U, Chen JD, Morgan G, Denton M, Greenberg CR, Wrogemann K, Blonden LA, van Paassen MB, van Ommen GJ, Kunkel LM (1989) The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 45(4):498–506
Kota J, Handy CR, Haidet AM, Montgomery CL, Eagle A, Rodino-Klapac LR, Tucker D, Shilling CJ, Therlfall WR, Walker CM, Weisbrode SE, Janssen PM, Clark KR, Sahenk Z, Mendell JR, Kaspar BK (2009) Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci Transl Med 1(6):6ra15. https://doi.org/10.1126/scitranslmed.3000112
Krivickas LS, Walsh R, Amato AA (2009) Single muscle fiber contractile properties in adults with muscular dystrophy treated with MYO-029. Muscle Nerve 39(1):3–9. https://doi.org/10.1002/mus.21200
Kwon JB, Ettyreddy AR, Vankara A, Bohning JD, Devlin G, Hauschka SD, Asokan A, Gersbach CA (2020) In vivo gene editing of muscle stem cells with adeno-associated viral vectors in a mouse model of Duchenne muscular dystrophy. Mol Ther Methods Clin Dev 19:320–329. https://doi.org/10.1016/j.omtm.2020.09.016
Lai Y, Yue Y, Liu M, Ghosh A, Engelhardt JF, Chamberlain JS, Duan D (2005) Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol 23(11):1435–1439. https://doi.org/10.1038/nbt1153
Landisch RM, Kosir AM, Nelson SA, Baltgalvis KA, Lowe DA (2008) Adaptive and nonadaptive responses to voluntary wheel running by mdx mice. Muscle Nerve 38(4):1290–1303. https://doi.org/10.1002/mus.21141
Leeuw T, Pette D (1993) Coordinate changes in the expression of troponin subunit and myosin heavy-chain isoforms during fast-to-slow transition of low-frequency-stimulated rabbit muscle. Eur J Biochem 213(3):1039–1046. https://doi.org/10.1111/j.1432-1033.1993.tb17851.x
Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H, Yamamoto T, Yamanaka S, Hotta A (2015) Precise correction of the dystrophin gene in Duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep 4(1):143–154. https://doi.org/10.1016/j.stemcr.2014.10.013
Licandro SA, Crippa L, Pomarico R, Perego R, Fossati G, Leoni F, Steinkuhler C (2021) The pan HDAC inhibitor Givinostat improves muscle function and histological parameters in two Duchenne muscular dystrophy murine models expressing different haplotypes of the LTBP4 gene. Skelet Muscle 11(1):19. https://doi.org/10.1186/s13395-021-00273-6
Lindsay A, Larson AA, Verma M, Ervasti JM, Lowe DA (2019) Isometric resistance training increases strength and alters histopathology of dystrophin-deficient mouse skeletal muscle. J Appl Physiol (1985) 126(2):363–375. https://doi.org/10.1152/japplphysiol.00948.2018
Liu F, Nishikawa M, Clemens PR, Huang L (2001) Transfer of full-length Dmd to the diaphragm muscle of Dmd(mdx/mdx) mice through systemic administration of plasmid DNA. Mol Ther 4(1):45–51. https://doi.org/10.1006/mthe.2001.0419
Liu M, Yue Y, Harper SQ, Grange RW, Chamberlain JS, Duan D (2005) Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol Ther 11(2):245–256. https://doi.org/10.1016/j.ymthe.2004.09.013
Liu W, Wen Y, Bi P, Lai X, Liu XS, Liu X, Kuang S (2012) Hypoxia promotes satellite cell self-renewal and enhances the efficiency of myoblast transplantation. Development 139(16):2857–2865. https://doi.org/10.1242/dev.079665
Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN (2014) Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 345(6201):1184–1188. https://doi.org/10.1126/science.1254445
Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351(6271):400–403. https://doi.org/10.1126/science.aad5725
Lott DJ, Taivassalo T, Cooke KD, Park H, Moslemi Z, Batra A, Forbes SC, Byrne BJ, Walter GA, Vandenborne K (2021) Safety, feasibility, and efficacy of strengthening exercise in Duchenne muscular dystrophy. Muscle Nerve 63(3):320–326. https://doi.org/10.1002/mus.27137
Lu QL, Mann CJ, Lou F, Bou-Gharios G, Morris GE, Xue SA, Fletcher S, Partridge TA, Wilton SD (2003) Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med 9(8):1009–1014. https://doi.org/10.1038/nm897
Luthert P, Vrbova G, Ward KM (1980) Effects of slow frequency electrical stimulation on muscles of dystrophic mice. J Neurol Neurosurg Psychiatry 43(9):803–809. https://doi.org/10.1136/jnnp.43.9.803
Lynch GS (2017) Therapeutic potential of skeletal muscle plasticity and slow muscle programming for muscular dystrophy and related muscle conditions. In: Sakuma K (ed) The plasticity of skeletal muscle. Springer, Singapore, pp 277–292
Lynch GS, Hayes A, Lam MH, Williams DA (1993) The effects of endurance exercise on dystrophic mdx mice. II Contractile properties of skinned muscle fibres. Proc Biol Sci 253(1336):27–33. https://doi.org/10.1098/rspb.1993.0078
Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, Lewis S, Shilling CJ, Kota J, Serrano-Munuera C, Hayes J, Mahan JD, Campbell KJ, Banwell B, Dasouki M, Watts V, Sivakumar K, Bien-Willner R, Flanigan KM, Sahenk Z, Barohn RJ, Walker CM, Mendell JR (2010) Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol 67(6):771–780. https://doi.org/10.1002/ana.22024
Manzur AY, Kuntzer T, Pike M, Swan A (2004) Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev (2):CD003725. https://doi.org/10.1002/14651858.CD003725.pub2
Markert CD, Ambrosio F, Call JA, Grange RW (2011) Exercise and Duchenne muscular dystrophy: toward evidence-based exercise prescription. Muscle Nerve 43(4):464–478. https://doi.org/10.1002/mus.21987
Markert CD, Case LE, Carter GT, Furlong PA, Grange RW (2012) Exercise and Duchenne muscular dystrophy: where we have been and where we need to go. Muscle Nerve 45(5):746–751. https://doi.org/10.1002/mus.23244
Markham BE, Kernodle S, Nemzek J, Wilkinson JE, Sigler R (2015) Chronic Dosing with Membrane Sealant Poloxamer 188 NF Improves Respiratory Dysfunction in Dystrophic Mdx and Mdx/Utrophin-/- Mice. PLoS One 10(8):e0134832. https://doi.org/10.1371/journal.pone.0134832
Mavroudis PD, van den Anker J, Conklin LS, Damsker JM, Hoffman EP, Nagaraju K, Clemens PR, Jusko WJ (2019) Population pharmacokinetics of vamorolone (VBP15) in healthy men and boys with Duchenne muscular dystrophy. J Clin Pharmacol 59(7):979–988. https://doi.org/10.1002/jcph.1388
Mendell JR, Kissel JT, Amato AA, King W, Signore L, Prior TW, Sahenk Z, Benson S, McAndrew PE, Rice R et al (1995) Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N Engl J Med 333(13):832–838. https://doi.org/10.1056/NEJM199509283331303
Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, Bowles D, Gray S, Li C, Galloway G, Malik V, Coley B, Clark KR, Li J, Xiao X, Samulski J, McPhee SW, Samulski RJ, Walker CM (2010) Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med 363(15):1429–1437. https://doi.org/10.1056/NEJMoa1000228
Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, Alfano L, Gomez AM, Lewis S, Kota J, Malik V, Shontz K, Walker CM, Flanigan KM, Corridore M, Kean JR, Allen HD, Shilling C, Melia KR, Sazani P, Saoud JB, Kaye EM, Eteplirsen Study Group (2013) Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 74(5):637–647. https://doi.org/10.1002/ana.23982
Mendell JR, Sahenk Z, Malik V, Gomez AM, Flanigan KM, Lowes LP, Alfano LN, Berry K, Meadows E, Lewis S, Braun L, Shontz K, Rouhana M, Clark KR, Rosales XQ, Al-Zaidy S, Govoni A, Rodino-Klapac LR, Hogan MJ, Kaspar BK (2015) A phase 1/2a follistatin gene therapy trial for Becker muscular dystrophy. Mol Ther 23(1):192–201. https://doi.org/10.1038/mt.2014.200
Minasi MG, Riminucci M, De Angelis L, Borello U, Berarducci B, Innocenzi A, Caprioli A, Sirabella D, Baiocchi M, De Maria R, Boratto R, Jaffredo T, Broccoli V, Bianco P, Cossu G (2002) The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 129(11):2773–2783
Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, Fortuni S, Straino S, Sampaolesi M, Di Padova M, Illi B, Gallinari P, Steinkuhler C, Capogrossi MC, Sartorelli V, Bottinelli R, Gaetano C, Puri PL (2006) Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med 12(10):1147–1150. https://doi.org/10.1038/nm1479
Molnar MJ, Gilbert R, Lu Y, Liu AB, Guo A, Larochelle N, Orlopp K, Lochmuller H, Petrof BJ, Nalbantoglu J, Karpati G (2004) Factors influencing the efficacy, longevity, and safety of electroporation-assisted plasmid-based gene transfer into mouse muscles. Mol Ther 10(3):447–455. https://doi.org/10.1016/j.ymthe.2004.06.642
Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M (2005) Direct isolation of satellite cells for skeletal muscle regeneration. Science 309(5743):2064–2067. https://doi.org/10.1126/science.1114758
Morine KJ, Bish LT, Selsby JT, Gazzara JA, Pendrak K, Sleeper MM, Barton ER, Lee SJ, Sweeney HL (2010) Activin IIB receptor blockade attenuates dystrophic pathology in a mouse model of Duchenne muscular dystrophy. Muscle Nerve 42(5):722–730. https://doi.org/10.1002/mus.21743
Mourkioti F, Kratsios P, Luedde T, Song YH, Delafontaine P, Adami R, Parente V, Bottinelli R, Pasparakis M, Rosenthal N (2006) Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest 116(11):2945–2954. https://doi.org/10.1172/JCI28721
Muntoni F, Tejura B, Spinty S, Roper H, Hughes I, Layton G, Davies KE, Harriman S, Tinsley J (2019) A phase 1b trial to assess the pharmacokinetics of ezutromid in pediatric Duchenne muscular dystrophy patients on a balanced diet. Clin Pharmacol Drug Dev 8(7):922–933. https://doi.org/10.1002/cpdd.642
Muraine L, Bensalah M, Dhiab J, Cordova G, Arandel L, Marhic A, Chapart M, Vasseur S, Benkhelifa-Ziyyat S, Bigot A, Butler-Browne G, Mouly V, Negroni E, Trollet C (2020) Transduction efficiency of adeno-associated virus serotypes after local injection in mouse and human skeletal muscle. Hum Gene Ther 31(3–4):233–240. https://doi.org/10.1089/hum.2019.173
Murakami T, Nishi T, Kimura E, Goto T, Maeda Y, Ushio Y, Uchino M, Sunada Y (2003) Full-length dystrophin cDNA transfer into skeletal muscle of adult mdx mice by electroporation. Muscle Nerve 27(2):237–241. https://doi.org/10.1002/mus.10283
Murphy KT, Koopman R, Naim T, Leger B, Trieu J, Ibebunjo C, Lynch GS (2010a) Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J 24(11):4433–4442. https://doi.org/10.1096/fj.10-159608
Murphy KT, Ryall JG, Snell SM, Nair L, Koopman R, Krasney PA, Ibebunjo C, Holden KS, Loria PM, Salatto CT, Lynch GS (2010b) Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx mice. Am J Pathol 176(5):2425–2434. https://doi.org/10.2353/ajpath.2010.090932
Murphy KT, Chee A, Gleeson BG, Naim T, Swiderski K, Koopman R, Lynch GS (2011a) Antibody-directed myostatin inhibition enhances muscle mass and function in tumor-bearing mice. Am J Physiol Regul Integr Comp Physiol 301(3):R716–R726. https://doi.org/10.1152/ajpregu.00121.2011
Murphy KT, Cobani V, Ryall JG, Ibebunjo C, Lynch GS (2011b) Acute antibody-directed myostatin inhibition attenuates disuse muscle atrophy and weakness in mice. J Appl Physiol (1985) 110(4):1065–1072. https://doi.org/10.1152/japplphysiol.01183.2010
Nakatani M, Takehara Y, Sugino H, Matsumoto M, Hashimoto O, Hasegawa Y, Murakami T, Uezumi A, Takeda S, Noji S, Sunada Y, Tsuchida K (2008) Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J 22(2):477–487. https://doi.org/10.1096/fj.07-8673com
Ng R, Metzger JM, Claflin DR, Faulkner JA (2008) Poloxamer 188 reduces the contraction-induced force decline in lumbrical muscles from mdx mice. Am J Physiol Cell Physiol 295(1):C146–C150. https://doi.org/10.1152/ajpcell.00017.2008
Nobile C, Marchi J, Nigro V, Roberts RG, Danieli GA (1997) Exon-intron organization of the human dystrophin gene. Genomics 45(2):421–424. https://doi.org/10.1006/geno.1997.4911
Ohlendieck K, Campbell KP (1991) Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol 115(6):1685–1694. https://doi.org/10.1083/jcb.115.6.1685
Palmer E, Wilhelm JM, Sherman F (1979) Phenotypic suppression of nonsense mutants in yeast by aminoglycoside antibiotics. Nature 277(5692):148–150. https://doi.org/10.1038/277148a0
Parsons SA, Millay DP, Sargent MA, McNally EM, Molkentin JD (2006) Age-dependent effect of myostatin blockade on disease severity in a murine model of limb-girdle muscular dystrophy. Am J Pathol 168(6):1975–1985. https://doi.org/10.2353/ajpath.2006.051316
Partridge TA, Morgan JE, Coulton GR, Hoffman EP, Kunkel LM (1989) Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature 337(6203):176–179. https://doi.org/10.1038/337176a0
Pearsall RS, Davies MV, Cannell M, Li J, Widrick J, Mulivor AW, Wallner S, Troy ME, Spaits M, Liharska K, Sako D, Castonguay R, Keates S, Grinberg AV, Suragani R, Kumar R (2019) Follistatin-based ligand trap ACE-083 induces localized hypertrophy of skeletal muscle with functional improvement in models of neuromuscular disease. Sci Rep 9(1):11392. https://doi.org/10.1038/s41598-019-47818-w
Pette D, Vrbova G (1999) What does chronic electrical stimulation teach us about muscle plasticity? Muscle Nerve 22(6):666–677. https://doi.org/10.1002/(sici)1097-4598(199906)22:6<666::aid-mus3>3.0.co;2-z
Pistilli EE, Bogdanovich S, Goncalves MD, Ahima RS, Lachey J, Seehra J, Khurana T (2011) Targeting the activin type IIB receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am J Pathol 178(3):1287–1297. https://doi.org/10.1016/j.ajpath.2010.11.071
Politano L, Nigro G, Nigro V, Piluso G, Papparella S, Paciello O, Comi LI (2003) Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol 22(1):15–21
Porte-Thome F, Nagaraju K, Yu Q, Tatem K, Bkaily G, Scholz W, Slade A, Bot N, Kant C (2015) Development of Rimeporide, a sodium-hydrogen exchanger (NHE-1) inhibitor, for patients with Duchenne muscular dystrophy. Neuromuscul Disord 25:S259–S260. Abstract.
Previtali SC, Gidaro T, Diaz-Manera J, Zambon A, Carnesecchi S, Roux-Lombard P, Spitali P, Signorelli M, Szigyarto CA, Johansson C, Gray J, Labolle D, Porte Thome F, Pitchforth J, Domingos J, Muntoni F (2020) Rimeporide as a first-in-class NHE-1 inhibitor: results of a phase Ib trial in young patients with Duchenne Muscular Dystrophy. Pharmacol Res 159:104999. https://doi.org/10.1016/j.phrs.2020.104999
Quenneville SP, Chapdelaine P, Skuk D, Paradis M, Goulet M, Rousseau J, Xiao X, Garcia L, Tremblay JP (2007) Autologous transplantation of muscle precursor cells modified with a lentivirus for muscular dystrophy: human cells and primate models. Mol Ther 15(2):431–438. https://doi.org/10.1038/sj.mt.6300047
Ragot T, Vincent N, Chafey P, Vigne E, Gilgenkrantz H, Couton D, Cartaud J, Briand P, Kaplan JC, Perricaudet M et al (1993) Efficient adenovirus-mediated transfer of a human minidystrophin gene to skeletal muscle of mdx mice. Nature 361(6413):647–650. https://doi.org/10.1038/361647a0
Reichmann H, Pette D, Vrbova G (1981) Effects of low frequency electrical stimulation on enzyme and isozyme patterns of dystrophic mouse muscle. FEBS Lett 128(1):55–58. https://doi.org/10.1016/0014-5793(81)81078-2
Reichmann H, Pette D, Vrbova G (1983) Electrostimulation-induced changes in the enzyme activity pattern of dystrophic mouse muscles. Acta Histochem Suppl 28:125–127
Richard P, Bossard F, Desigaux L, Lanctin C, Bello-Roufai M, Pitard B (2005) Amphiphilic block copolymers promote gene delivery in vivo to pathological skeletal muscles. Hum Gene Ther 16(11):1318–1324. https://doi.org/10.1089/hum.2005.16.1318
Ricotti V, Spinty S, Roper H, Hughes I, Tejura B, Robinson N, Layton G, Davies K, Muntoni F, Tinsley J (2016) Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to pediatric patients with Duchenne muscular dystrophy. PLoS One 11(4):e0152840. https://doi.org/10.1371/journal.pone.0152840
Roffe S, Hagai Y, Pines M, Halevy O (2010) Halofuginone inhibits Smad3 phosphorylation via the PI3K/Akt and MAPK/ERK pathways in muscle cells: effect on myotube fusion. Exp Cell Res 316(6):1061–1069. https://doi.org/10.1016/j.yexcr.2010.01.003
Ruggiero L, Iodice R, Esposito M, Dubbioso R, Tozza S, Vitale F, Santoro L, Manganelli F (2018) One-year follow up of three Italian patients with Duchenne muscular dystrophy treated with ataluren: is earlier better? Ther Adv Neurol Disord 11:1756286418809588. https://doi.org/10.1177/1756286418809588
Rybalka E, Timpani CA, Debruin DA, Bagaric RM, Campelj DG, Hayes A (2020) The failed clinical story of myostatin inhibitors against Duchenne muscular dystrophy: exploring the biology behind the battle. Cell 9(12). https://doi.org/10.3390/cells9122657
Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM (2008) Self-renewal and expansion of single transplanted muscle stem cells. Nature 456(7221):502–506. https://doi.org/10.1038/nature07384
Sampaolesi M, Blot S, D’Antona G, Granger N, Tonlorenzi R, Innocenzi A, Mognol P, Thibaud JL, Galvez BG, Barthelemy I, Perani L, Mantero S, Guttinger M, Pansarasa O, Rinaldi C, Cusella De Angelis MG, Torrente Y, Bordignon C, Bottinelli R, Cossu G (2006) Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444(7119):574–579. https://doi.org/10.1038/nature05282
Schiaffino S, Reggiani C (2011) Fiber types in mammalian skeletal muscles. Physiol Rev 91(4):1447–1531. https://doi.org/10.1152/physrev.00031.2010
Schiaffino S, Reggiani C, Murgia M (2020) Fiber type diversity in skeletal muscle explored by mass spectrometry-based single fiber proteomics. Histol Histopathol 35(3):239–246. https://doi.org/10.14670/HH-18-170
Scott OM, Vrbova G, Hyde SA, Dubowitz V (1986) Responses of muscles of patients with Duchenne muscular dystrophy to chronic electrical stimulation. J Neurol Neurosurg Psychiatry 49(12):1427–1434. https://doi.org/10.1136/jnnp.49.12.1427
Scott OM, Hyde SA, Vrbova G, Dubowitz V (1990) Therapeutic possibilities of chronic low frequency electrical stimulation in children with Duchenne muscular dystrophy. J Neurol Sci 95(2):171–182. https://doi.org/10.1016/0022-510x(90)90240-n
Singh A, Ursic D, Davies J (1979) Phenotypic suppression and misreading Saccharomyces cerevisiae. Nature 277(5692):146–148. https://doi.org/10.1038/277146a0
Singh P, Rong H, Gordi T, Bosley J, Bhattacharya I (2016) Translational pharmacokinetic/pharmacodynamic analysis of MYO-029 antibody for muscular dystrophy. Clin Transl Sci 9(6):302–310. https://doi.org/10.1111/cts.12420
Smith EC, Conklin LS, Hoffman EP, Clemens PR, Mah JK, Finkel RS, Guglieri M, Tulinius M, Nevo Y, Ryan MM, Webster R, Castro D, Kuntz NL, Kerchner L, Morgenroth LP, Arrieta A, Shimony M, Jaros M, Shale P, Gordish-Dressman H, Hagerty L, Dang UJ, Damsker JM, Schwartz BD, Mengle-Gaw LJ, McDonald CM, Cinrg VBP, Investigators D (2020) Efficacy and safety of vamorolone in Duchenne muscular dystrophy: an 18-month interim analysis of a non-randomized open-label extension study. PLoS Med 17(9):e1003222. https://doi.org/10.1371/journal.pmed.1003222
Spurney CF, Guerron AD, Yu Q, Sali A, van der Meulen JH, Hoffman EP, Nagaraju K (2011) Membrane sealant Poloxamer P188 protects against isoproterenol induced cardiomyopathy in dystrophin deficient mice. BMC Cardiovasc Disord 11:20. https://doi.org/10.1186/1471-2261-11-20
Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, Church GM, Wagers AJ (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351(6271):407–411. https://doi.org/10.1126/science.aad5177
Tedesco FS, Gerli MF, Perani L, Benedetti S, Ungaro F, Cassano M, Antonini S, Tagliafico E, Artusi V, Longa E, Tonlorenzi R, Ragazzi M, Calderazzi G, Hoshiya H, Cappellari O, Mora M, Schoser B, Schneiderat P, Oshimura M, Bottinelli R, Sampaolesi M, Torrente Y, Broccoli V, Cossu G (2012) Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci Transl Med 4(140):140ra189. https://doi.org/10.1126/scitranslmed.3003541
Termin A, Staron RS, Pette D (1989) Changes in myosin heavy chain isoforms during chronic low-frequency stimulation of rat fast hindlimb muscles. A single-fiber study. Eur J Biochem 186(3):749–754. https://doi.org/10.1111/j.1432-1033.1989.tb15269.x
Terry RL, Kaneb HM, Wells DJ (2014) Poloxamer [corrected] 188 has a deleterious effect on dystrophic skeletal muscle function. PLoS One 9(3):e91221. https://doi.org/10.1371/journal.pone.0091221
Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE (1996) Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 384(6607):349–353. https://doi.org/10.1038/384349a0
Tinsley J, Deconinck N, Fisher R, Kahn D, Phelps S, Gillis JM, Davies K (1998) Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med 4(12):1441–1444. https://doi.org/10.1038/4033
Tinsley JM, Fairclough RJ, Storer R, Wilkes FJ, Potter AC, Squire SE, Powell DS, Cozzoli A, Capogrosso RF, Lambert A, Wilson FX, Wren SP, De Luca A, Davies KE (2011) Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One 6(5):e19189. https://doi.org/10.1371/journal.pone.0019189
Tinsley J, Robinson N, Davies KE (2015) Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to healthy male adult volunteers. J Clin Pharmacol 55(6):698–707. https://doi.org/10.1002/jcph.468
Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN, Metzger JM (2010) Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest 120(4):1140–1150. https://doi.org/10.1172/JCI41329
Turgeman T, Hagai Y, Huebner K, Jassal DS, Anderson JE, Genin O, Nagler A, Halevy O, Pines M (2008) Prevention of muscle fibrosis and improvement in muscle performance in the mdx mouse by halofuginone. Neuromuscul Disord 18(11):857–868. https://doi.org/10.1016/j.nmd.2008.06.386
van Deutekom JC, Bremmer-Bout M, Janson AA, Ginjaar IB, Baas F, den Dunnen JT, van Ommen GJ (2001) Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet 10(15):1547–1554. https://doi.org/10.1093/hmg/10.15.1547
Vianello S, Consolaro F, Bich C, Cancela JM, Roulot M, Lanchec E, Touboul D, Brunelle A, Israel M, Benoit E, de la Porte S (2014) Low doses of arginine butyrate derivatives improve dystrophic phenotype and restore membrane integrity in DMD models. FASEB J 28(6):2603–2619. https://doi.org/10.1096/fj.13-244798
Vignos PJ Jr, Watkins MP (1966) The effect of exercise in muscular dystrophy. JAMA 197(11):843–848
Vilquin JT, Wagner E, Kinoshita I, Roy R, Tremblay JP (1995) Successful histocompatible myoblast transplantation in dystrophin-deficient mdx mouse despite the production of antibodies against dystrophin. J Cell Biol 131(4):975–988. https://doi.org/10.1083/jcb.131.4.975
Vilquin JT, Kennel PF, Paturneau-Jouas M, Chapdelaine P, Boissel N, Delaere P, Tremblay JP, Scherman D, Fiszman MY, Schwartz K (2001) Electrotransfer of naked DNA in the skeletal muscles of animal models of muscular dystrophies. Gene Ther 8(14):1097–1107. https://doi.org/10.1038/sj.gt.3301484
Vincent N, Ragot T, Gilgenkrantz H, Couton D, Chafey P, Gregoire A, Briand P, Kaplan JC, Kahn A, Perricaudet M (1993) Long-term correction of mouse dystrophic degeneration by adenovirus-mediated transfer of a minidystrophin gene. Nat Genet 5(2):130–134. https://doi.org/10.1038/ng1093-130
Voet NB, van der Kooi EL, van Engelen BG, Geurts AC (2019) Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst Rev 12:CD003907. https://doi.org/10.1002/14651858.CD003907.pub5
Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, Campion G, De Kimpe SJ, Eagle M, Guglieri M, Hood S, Liefaard L, Lourbakos A, Morgan A, Nakielny J, Quarcoo N, Ricotti V, Rolfe K, Servais L, Wardell C, Wilson R, Wright P, Kraus JE (2014) Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol 13(10):987–996. https://doi.org/10.1016/S1474-4422(14)70195-4
Wagner KR (2020) The elusive promise of myostatin inhibition for muscular dystrophy. Curr Opin Neurol 33(5):621–628. https://doi.org/10.1097/WCO.0000000000000853
Wagner KR, Hamed S, Hadley DW, Gropman AL, Burstein AH, Escolar DM, Hoffman EP, Fischbeck KH (2001) Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol 49(6):706–711
Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, Flanigan KM, Pestronk A, Tawil R, Wolfe GI, Eagle M, Florence JM, King WM, Pandya S, Straub V, Juneau P, Meyers K, Csimma C, Araujo T, Allen R, Parsons SA, Wozney JM, Lavallie ER, Mendell JR (2008) A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 63(5):561–571. https://doi.org/10.1002/ana.21338
Wang B, Li J, Xiao X (2000) Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci U S A 97(25):13714–13719. https://doi.org/10.1073/pnas.240335297
Watchko J, O’Day T, Wang B, Zhou L, Tang Y, Li J, Xiao X (2002) Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum Gene Ther 13(12):1451–1460. https://doi.org/10.1089/10430340260185085
Webster C, Silberstein L, Hays AP, Blau HM (1988) Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 52(4):503–513. https://doi.org/10.1016/0092-8674(88)90463-1
Wells DJ (1993) Improved gene transfer by direct plasmid injection associated with regeneration in mouse skeletal muscle. FEBS Lett 332(1–2):179–182. https://doi.org/10.1016/0014-5793(93)80508-r
Wineinger MA, Abresch RT, Walsh SA, Carter GT (1998) Effects of aging and voluntary exercise on the function of dystrophic muscle from mdx mice. Am J Phys Med Rehabil 77(1):20–27. https://doi.org/10.1097/00002060-199801000-00004
Wong SH, Lowes KN, Quigley AF, Marotta R, Kita M, Byrne E, Kornberg AJ, Cook MJ, Kapsa RM (2005) DNA electroporation in vivo targets mature fibres in dystrophic mdx muscle. Neuromuscul Disord 15(9-10):630–641. https://doi.org/10.1016/j.nmd.2005.04.008
Xu L, Park KH, Zhao L, Xu J, El Refaey M, Gao Y, Zhu H, Ma J, Han R (2016) CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther 24(3):564–569. https://doi.org/10.1038/mt.2015.192
Yanagihara I, Inui K, Dickson G, Turner G, Piper T, Kaneda Y, Okada S (1996) Expression of full-length human dystrophin cDNA in mdx mouse muscle by HVJ-liposome injection. Gene Ther 3(6):549–553
Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM (2005) Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 436(7053):1025–1029. https://doi.org/10.1038/nature03844
Yoshimura M, Sakamoto M, Ikemoto M, Mochizuki Y, Yuasa K, Miyagoe-Suzuki Y, Takeda S (2004) AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol Ther 10(5):821–828. https://doi.org/10.1016/j.ymthe.2004.07.025
Young CS, Hicks MR, Ermolova NV, Nakano H, Jan M, Younesi S, Karumbayaram S, Kumagai-Cresse C, Wang D, Zack JA, Kohn DB, Nakano A, Nelson SF, Miceli MC, Spencer MJ, Pyle AD (2016) A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 18(4):533–540. https://doi.org/10.1016/j.stem.2016.01.021
Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan D (2003) Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation 108(13):1626–1632. https://doi.org/10.1161/01.CIR.0000089371.11664.27
Zhang G, Ludtke JJ, Thioudellet C, Kleinpeter P, Antoniou M, Herweijer H, Braun S, Wolff JA (2004) Intraarterial delivery of naked plasmid DNA expressing full-length mouse dystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther 15(8):770–782. https://doi.org/10.1089/1043034041648408
Zhang G, Wooddell CI, Hegge JO, Griffin JB, Huss T, Braun S, Wolff JA (2010) Functional efficacy of dystrophin expression from plasmids delivered to mdx mice by hydrodynamic limb vein injection. Hum Gene Ther 21(2):221–237. https://doi.org/10.1089/hum.2009.133
Zhang Y, Li H, Min YL, Sanchez-Ortiz E, Huang J, Mireault AA, Shelton JM, Kim J, Mammen PPA, Bassel-Duby R, Olson EN (2020) Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci Adv 6(8):eaay6812. https://doi.org/10.1126/sciadv.aay6812
Zhang Y, Nishiyama T, Li H, Huang J, Atmanli A, Sanchez-Ortiz E, Wang Z, Mireault AA, Mammen PPA, Bassel-Duby R, Olson EN (2021) A consolidated AAV system for single-cut CRISPR correction of a common Duchenne muscular dystrophy mutation. Mol Ther Methods Clin Dev 22:122–132. https://doi.org/10.1016/j.omtm.2021.05.014
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE (2008) Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16(6):1073–1080. https://doi.org/10.1038/mt.2008.76
Zupan A (1992) Long-term electrical stimulation of muscles in children with Duchenne and Becker muscular dystrophy. Muscle Nerve 15(3):362–367. https://doi.org/10.1002/mus.880150316
Zupan A, Gregoric M, Valencic V, Vandot S (1993) Effects of electrical stimulation on muscles of children with Duchenne and Becker muscular dystrophy. Neuropediatrics 24(4):189–192. https://doi.org/10.1055/s-2008-1071537
Zupan A, Gregoric M, Valencic V (1995) Long-lasting effects of electrical stimulation upon muscles of patients suffering from progressive muscular dystrophy. Clin Rehabil 9(2):102–109
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Swiderski, K., Hardee, J.P., Lynch, G.S. (2022). Regenerative Rehabilitation for Duchenne Muscular Dystrophy. In: Greising, S.M., Call, J.A. (eds) Regenerative Rehabilitation. Physiology in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-95884-8_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-95884-8_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-95883-1
Online ISBN: 978-3-030-95884-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)