
Abstract
In the setting of extended life expectancy, cardiovascular diseases are predicted to increase in the following decades in terms of incidence and prevalence representing a medical, scientific, and economic challenge for clinicians, investigators, and health systems.
A strong correlation between age and cardiovascular disorders is broadly accepted, although its nature remains elusive as it is still questioned whether age-associated defects are due to a chronic wear-and-tear process or simply due to an accumulation in a stochastic fashion.
The cardiovascular apparatus can be affected in all its different components (muscle, vessels, conduction system, and valves) by a number of non-mutually exclusive molecular and cellular mechanisms (inflammation, dyslipidemia, apoptosis, calcification, uncontrolled fibrosis, dysfunctional genetic program reactivation, protein misfolding, and neurohormonal dysregulation). In some cases, these physiological ploys have beneficial effects in the short run, but they are of deleterious impact chronically and they ultimately characterize the various morphofunctional phenotypes of the elderly heart. Independently of primary and ancillary driving mechanisms of damage, older patients will tend to suffer from an end-stage condition called heart failure. Unfortunately, the management of this condition is based on supportive, symptomatic, and judicious medical management since the elderly are often underrepresented in clinical trials.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Ageing
- Vascular dysfunction
- Atrial fibrillation
- Valve disease
- Myocardial ischemia
- Amyloidosis
- Hypertension
- Heart failure
1 Epidemiology
According to the World Health Organization, cardiovascular diseases (CVD) are the leading causes of morbidity and mortality in industrialized nations, and they are expected to become the first cause of mortality also in low- and middle-income countries in the near future.
Incidence of CVD has reached pandemic proportions in light of combined social changes and medical improvements occurred in the last decades, resulting in a significant demographic shift that will lead by 2040 to a 22% prevalence of subjects older than 65 [1]. Owing to numerous preventive, diagnostic, and therapeutic means implemented on a global scale, life expectancy has dramatically increased over the course of the last 50 years, resulting in a 16.5 year gain in life expectancy within this timeframe and reaching a life expectancy of 71.9 years worldwide.
Increased life expectancy has, particularly in high-income areas, expanded the terminal phase of life which is characterized by a series of different disabilities affecting the quality of life itself where non-mutually exclusive CVD lead to a final stage of cardiac impairment called heart failure [2]. Concomitantly, the clinical picture and therefore the general population outlook is heterogeneous with non-cardiovascular diseases also playing a role. Together with intrinsic risk factors, environmental ones such as nutrition [3], alcohol abuse [4], lack of physical exercise [5], and pollution [6] can determine the morphofunctional phenotype of cardiac impairment.
A recent population study in the UK showed that, despite a reduction in the annual incidence of heart failure diagnosis (−7%), its prevalence increased (+12%) in light of adequate public health policies and effective therapeutic strategies [7].
These numbers are translated into a public healthcare system challenge with an annual expenditure of nearly $125,000 per heart failure patient, a sum that is doomed to increase in the near future [8].
2 Features
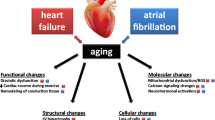
With ageing, the cardiovascular system undergoes a series of structural and functional changes dictated by molecular and cellular mechanisms affecting different anatomical structures such as the myocardium, the valve apparatus, the conduction system, and the vasculature as shown in Fig. 2.1.

Age-dependent changes to cardiovascular tissues. Molecular, cellular, and tissue changes occurring with time ultimately altering morphology and function of the heart and vessels. (North BJ, Sinclair DA, The Intersection between Aging and Cardiovascular Disease, Circulation Research 2012;110:1097–1108)
The indisputable evidence connecting CVD with age notwithstanding, it still remains elusive whether ageing per se should be considered as an independent risk factor, a process, or rather an epiphenomenon of accumulated stochastic molecular events exhausting compensatory defensive response and ultimately yielding to reduced homeostasis and therefore morbid conditions.
3 Arterial Ageing
Over time, significant peripheral and coronary vascular changes occur leading to the two most common forms of CVD observed in the elderly: arterial hypertension and coronary artery disease (CAD). A key early step in the development of these diseases is represented by endothelial dysfunction where the bioavailability of nitric oxide (NO)—a key protective factor with vasodilatory, anti-adhesion, and anti-aggregation properties—is drastically reduced.
3.1 Arterial Ageing: Arterial Hypertension
Starting from the age of 50, arterial hypertension in elderly patients is principally characterized by isolated, elevated systolic pressure with a progressive decline in diastolic pressure, intuitively leading to widened pulse pressure and an increased pulse wave velocity (PWV). Such condition is primarily driven by concomitant endothelial dysfunction and central arterial stiffness, which are intimately interconnected within a self-sustaining vicious cycle of hemodynamic load, endothelial activation, inflammation, and persistent damage. Endothelial dysfunction includes reduced vasodilatatory capability determined by reduced NO bioavailability [9], impaired endothelial-dependent responsiveness to prostaglandins [10], increased levels of the vasoconstrictor endothelin-1, and systemic molecular signature changes towards pro-inflammatory molecules, i.e. TNF-alpha, IL-6. Such changes are associated with altered vascular homeostasis, favouring a prooxidant and pro-inflammatory milieu with a tendency to cardiovascular adverse events [11]. On the other hand, arterial stiffness is due to altered proteolytic activities by metalloproteinases (MMPs), cathepsins, and neutrophil elastase as well as an age-related increased production of TGF-β favouring abnormal elastin fragmentation, fibers calcification, augmented collagen deposition, endothelial senescence, and tissue invasion by inflammatory cells and smooth muscle cells overgrowth [12]. This process is macroscopically translated into luminal enlargement and wall thickening as well as wall stiffening with reduced arterial distensibility yielding an increased PWV.
In order to compensate these changes and preserve sufficient peripheral perfusion, the myocardium undergoes a series of cellular, structural, and functional ploys which in the long run predispose to irreversible chronic cardiac dysfunction. As shown in Fig. 2.2, increased aortic impedance and ventricular loading are counteracted by increased wall tension featuring augmented wall thickness and prolonged systole.

Conceptual model of arterial ageing and arterial decline. Herein are reported the intertwined, compensating mechanisms which will eventually lead to the well-characterized vascular dysfunction of the elderly (Lakatta EG. So! What’s Ageing? Is Cardiovascular Ageing a Disease? Journal of Molecular and Cellular Cardiology;83:1–13)
A longer systolic interval is possible when time from diastole is borrowed. In order to preserve contractile activity, this physiological compromise causes an incomplete relaxation and thus forces the heart to increase its cavities as well as filling pressures.
3.2 Arterial Ageing: Coronary Artery Disease
Among CVD, CAD is the most common cause of morbidity and mortality in elderly patients responsible for nearly one-fifth of death cases and representing the first aetiology of heart failure [13] as illustrated in Fig. 2.3.

The vicious cycle of myocardial ischemia—Ischemic damage, independently of magnitude and extension, leads to ventricular dysfunction which is temporarily compensated by a number of physiological ploys (autonomic, neurohormonal, etc.) preserving myocardial function. Over time, these ploys exhaust their compensatory capabilities yielding to a decompensated myocardium. (Remme WJ, Overview of the relationship between ischemia and congestive heart failure, Clinical Cardiology 2000;23 (Suppl. IV):1–13)
In comparison to the general population, elderly patients are usually more difficult to diagnose in light of the concomitant comorbidities and of atypical clinical presentation; this still holds true despite this subpopulation being more frequently affected and with extended involvement of the coronary tree.
Pathogenesis of CAD is based on a plethora of interconnected factors determining atherosclerosis. Despite being initially considered solely as a cholesterol storage disease, it is now accepted that inflammation plays a central role in atherosclerotic plaque formation, progression, and rupture. Initiation sites are usually localized in peculiar sectors where laminar blood flow is disturbed (branches); lack of luminal elastin coupled with proteoglycans exposure favours subendothelial low-density lipoproteins’ (LDL) deposition. In the presence of inflammatory cells, LDLs are a vulnerable substrate to a number of posttranslational modifications as well as to oxidative stress caused by myeloperoxidase or lypoxygenases release. LDL oxidation triggers endothelial adhesion molecules overexpression (CCL5, CXCR3, CCL2, CCR5, CCR2, CXCR1), promoting further cellular invasion. Over time, plaques evolve from fatty streaks where T-cells and monocytes-derived foam cells load are predominant towards more complex histological lesions [14,15,16].
The natural history of CAD is strongly affected by age: in fact, CAD is more commonly diagnosed in aged individuals and a positive direct correlation between age and number and size of lesions is present. As later discussed, compensatory myocardial hypertrophy represents an additional metabolic challenge for coronary circulation. With prolonged systolic time, reduced luminal diameter, and decreased vascular density, chronic and/or acute ischemic damage can occur more frequently.
Specific histological features distinguish atherosclerotic lesions that are more prone to rupture and thus cause acute coronary syndromes. Such lesions are usually larger, presenting a bigger necrotic core (cell debris and cholesterol crystals) and a protective fibrous cap invaded by pro-inflammatory cells and less smooth muscle cells [17].
3.3 Arterial Ageing: Cardiac Microvascular Disease
Microcirculation is defined as blood vessels with a diameter inferior to 100 mm and its role is particularly important in regulating tissue perfusion and cell function in response to the release of a multitude of dilating and constricting factors [18].
Even in the absence of image-detectable and clinically significant coronary lesions, dysfunctional microcirculation can dramatically change patients’ prognosis. Microcirculation can be affected in a wide range of chronic and acute conditions (ischemia, hypertension, diabetes, obesity, tobacco use, renal impairment, and with age per se). As previously mentioned, senescent endothelium as well as smooth muscle cells, in low calibre arterioles, tend to lose their capability to regulate vascular resistance and match energy request with blood flow, ultimately affecting cardiac performance itself and increasing the chances of myocardial ischemia. In fact, myocardial oxygen extraction tends already to maximal capabilities in resting conditions, and therefore its delivery heavily relies on blood flow. In case of increased oxygen demand, a proportional increase in coronary blood flow must be matched with metabolic requests [19, 20].
4 Myocardial Hypertrophy
Pathological myocardial hypertrophy as frequently observed in the elderly is an irreversible process governed by different yet intertwined molecular pathways in contrast to the physiological reversible hypertrophy observed in other conditions such as in athletes. Hypertrophy in the failing ageing heart is characterized by cellular loss, partially compensated with survived hypertrophic cardiomyocytes, and due to the aforementioned ischemic burden, progressively replaced with nonfunctional fibroblasts.
Such structural changes can be regarded as a maladaptation of cardiomyocytes to mechanical and chemical stress events, reactivating the so-called “fetal gene program” by promoting chromatin remodelling, transcriptional, and posttranscriptional upregulation for specific genes transcription factors such as the myocyte enhancer factor 2 (MEF2A-C), erythroid transcription factor (GATA4).
Upon reactivation, a series of common fetal isoforms genes are accessed for transcription such as muscle creatine kinase (Ckm), alpha myosin chain (Myh-6), myosin light chain (Myl1), and Troponins C and I (Tnnc1, Tnni3). It is now widely accepted that the aberrant expression of fetal genes in the postnatal heart involved in contractility, calcium handling, and myocardial energetics has only a temporary beneficial effect as it bears a negative prognostic significance [21].
5 Amyloidosis
An additional, yet underestimated, cause of myocardial hypertrophy is represented by amyloidosis caused by the extracellular deposition of insoluble fibrils. This is an autoptic finding; in nearly 20% of subjects older than 80, it is the amyloid deposition within the myocardium.
Amyloid plaques are stable, extracellular aggregates derived from proteinaceous by-products yielding to histological changes and organ dysfunction. All amyloidoses have a common pathogenic mechanism consisting of erroneous protein folding. In fact, each protein sequence undergoes a number of quality control systems in order to acquire a correct tridimensional structure and therefore fulfil the physiological function, localization, and interactions. In the unfortunate, yet not uncommon, case of combined adverse events (genetic mutation, protein overproduction, age per se, concomitant comorbidities, iatrogenic factors), proteins can acquire aberrant conformations. Once a single protein is abnormally arranged (monomer) and has overwhelmed additional cellular control systems, it aggregates into larger and more complex structures called oligomers, and further on into fibers and finally deposit into stable and pathognomonic structures called amyloid plaques [22].
With the term cardiac amyloidosis, we engulf a heterogenous group of medical conditions affecting the heart muscle which can have variable degrees of severity, prevalence, and evolution.
Light chain immunoglobulins can affect the heart in 50% of cases depending on the type of cell dyscrasia and median age of presentation can vary for the same reason. Of note, heart failure represents the worst prognostic factor in these patients [23].
Transthyretin, whether wild-type or mutated, is the biological precursor of systemic senile and systemic familial amyloidosis, respectively. In the absence of mutations, the mean age of presentation is around 75 years and cardiac involvement prevalence increases with age. Nowadays, nearly 100 different transthyretin mutations have been reported, each with their peculiar physicochemical properties determining their noxious properties and natural progression. Cardiac involvement greatly varies in terms of age presentation and concomitant nervous involvement based on the type of mutation [24].
Other proteins of cardiac and noncardiac origins can deposit within the myocardium such as Atrial Natriuretic Peptide giving rise to isolated atrial amyloidosis, serum amyloid A to amyloid A amyloidosis, and β2-microglobulin which tends to deposit in patients with long-standing dialytic treatment [25].
At early stages when typical signs of prominent cardiac involvement such as thickened myocardial walls with reduced electrical voltages on ECG tracings are not apparent yet, cardiac amyloidosis can represent a challenging diagnosis. While endomyocardial biopsy represents the definitive mean for confirming the diagnostic hypothesis [26, 27], Technetium 99 m pyrophosphate (Tc 99 m PYP) cardiac imaging has emerged as a highly sensitive and specific technique for detecting ATTR cardiac amyloidosis and capable of distinguishing it from AL cardiac amyloidosis [28].
6 Atrial Fibrillation
AF is already the most commonly occurring dysrhythmia with a lifetime risk for AF of around 25%, indicating that one out of four women or men over age 40 will experience AF.
The estimated global prevalence of AF of 33 million in 2010 is expected to double by 2050 because of population ageing, the rising prevalence of cardiometabolic risk factors, and the improved survival from cardiovascular events [29, 30]. Importantly, prevalence, both of the global incidence and the age-adjusted mortality rates, is also rising [29]. The most important modifiable risk factors, particularly elevated blood pressure and obesity, explain about 50% of the population’s attributable risk for AF development.
While AF is associated with a five-fold increase in the risk of ischemic stroke and up to 20% of all strokes are attributable to AF, the lack of temporal relationship between arrhythmia episodes and adverse outcomes has questioned the causal role of AF in the development of stroke.
Characterized by the presence of rapid, irregular, and fibrillatory waves that vary in magnitude, shape, and timing, possibly affecting hemodynamic cardiac performance itself [31], atrial fibrillation usually develops in the context of a diseased left atrium due to the hemodynamical challenges of high filling pressures, as in hypertension and/or heart failure with preserved or reduced ejection and fraction, and altered histological substrates that predispose to arrhythmic event or increased vulnerability. Indeed, on top of structural abnormalities, AF normally requires a trigger event of cardiac or noncardiac origin such as autonomic tone change, neurohormonal activation, inflammation, or other stimuli.
7 Valvular Diseases
Cardiac valve diseases are of remarkable importance in the general population; over the last decades, they have changed aetiology and demographic patterns transitioning from prevalent rheumatologic sequelae of the adult to a typical degenerative process of the elderly with increasing prevalence starting from the sixth and seventh decade of life.
As shown in Fig. 2.4, heart valve diseases of any severity rapidly increase from 0.5% before the age of 55 and they reach at least a 12% prevalence in subjects older than 75, with mitral regurgitation being the most common (from 10.9% to 7.1%) form followed by aortic stenosis (4.6–2.8%), aortic regurgitation (2–1.7%), and mitral stenosis (0.2%) [32].

Prevalence of valve heart disease per age. Both aortic and mitral valves undergo a series of herein described histological changes that are clinically translated into stenosis and insufficiency. Prevalence increases over the course of three decades from nearly 1% (45–54 years of age) to 12% (>75 years of age) representing a common finding in this population. (Nkomo VT, Gardin JM, Skelton TN, Gottidiner JC, Scott GC, Enrriquez-Sarano M. Burden of valvular heart diseases: a population-based study, Lancet. 2006;368: 1005–1011)
Each type of valve disease challenges the myocardium with peculiar combinations of pressure and/or volume overload determining temporary compensatory responses, ultimately exhausting the morphofunctional reserve of the heart itself and becoming a significant determinant of mortality.
Mitral regurgitation provokes left ventricular cavities enlargement without compensatory wall hypertrophy; on the other hand, aortic insufficiency predisposes to ventricular enlargement coupled to hypertrophy. Patients with aortic stenosis have hypertrophic hearts without cavities dilatation, whereas mitral stenosis presents with significant left atrial enlargement leaving the left ventricle unaffected.
Mitral insufficiency can be divided into two distinct nosocomial entities: a primary degeneration of the valve called organic and a function regurgitation due to altered surrounding structures. Organic mitral valve degeneration can have two major pathogenic mechanisms: myxomatous degeneration or fibroelastic deficiency. The former is believed to be driven by myofibroblast activation secreting MMPs and altering extracellular matrix turnover as well as TGF-β sustaining myofibroblast proliferation and differentiation. The latter is still not well-understood, but it is characterized by an age-dependent impairment of connective tissue fiber synthesis. Myxomatous mitral valves feature redundant prolapsing tissue, leaflet, and annulus calcification as well as annular dilatation in contrast to the other form which usually tends to tether chordae and eventually rupture them [33, 34]. Patients may remain symptomatic over the vast majority of the natural history of the disease until an irreversible stage characterized by preserved systolic function with pulmonary hypertension or atrial fibrillation begins.
Concerning aortic stenosis, the sclerotic process (with or without stenosis) represents a classic echocardiographic finding of the elderly being detectable in nearly half of subjects older than 85 [35]. Aortic valve calcification is believed to progress with the same cellular and molecular mechanisms of atherosclerosis; nevertheless, such hypothesis remains open in light of the lack of benefit from statin therapy.
After a long asymptomatic phase where the myocardium can counteract flow obstruction with compensatory hypertrophy, patients with severe aortic stenosis rapidly complain of angina, syncope, and fatigue and mortality rate abruptly rises to 25% per year [36].
8 Therapy
Medical therapeutic mainstays of the various cardiovascular conditions rely on judicious evaluation in the setting of polymorbic patients who most likely require multidrug therapy. In addition, renal function should be regularly monitored in order to correctly provide dose medication and to avoid expectable, adverse, and overdosing effects. Unfortunately, the elderly population is underrepresented in the vast majority of clinical trials in spite of the unmet need for tailored medical therapy for this subpopulation. The above-mentioned complex clinical scenario poses an additional challenge in the decision making process since classic, clinical hard endpoints should be coupled with the quality of life and frailty scoring systems. In fact, life expectancy might not be perceived as a pivotal determinant by senior patients who are more concerned in preserving their daily activity independence.
References
Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123:933–44.
Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20.
Miller V, Mente A, Dehghan M, et al. Fruit, vegetable, and legume intake, and cardiovascular disease and deaths in 18 countries (PURE): a prospective cohort study. Lancet. 2017;390:2037–49.
Whitman IR, Agarwal V, Nah G, et al. Alcohol abuse and cardiac disease. J Am Coll Cardiol. 2017;69:13–24.
Lear SA, Hu W, Rangarajan S, et al. The effect of physical activity on mortality and cardiovascular disease in 130,000 people from 17 high-income, middle-income, and low-income countries: the PURE study. Lancet. 2017;390:2643–54.
Aung N, Sanghvi MM, Zemrak F, et al. Association between ambient air pollution and cardiac morpho-functional phenotypes: insights from the UK Biobank Population Imaging Study. Circulation. 2018;138:2175–86.
Conrad N, Judge A, Tran J, et al. Temporal trends and patterns in heart failure incidence: a population-based study of 4 million individuals. Lancet. 2018;391:572–80.
Lesyuk W, Kriza C, Kolominsky-Rabas P. Cost-of-illness studies in heart failure: a systematic review 2004–2016. BMC Cardiovasc Disord. 2018;18:74.
Donato AJ, Eskurza I, Silver AE, et al. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ Res. 2007;100:1659–66.
Singh N, Prasad S, Singer DR, MacAllister RJ. Ageing is associated with impairment of nitric oxide and prostanoid dilator pathways in the human forearm. Clin Sci (Lond). 2002;102:595–600.
Camici GG, Sudano I, Noll G, Tanner FC, Luscher TF. Molecular pathways of aging and hypertension. Curr Opin Nephrol Hypertens. 2009;18:134–7.
Chung HY, Sung B, Jung KJ, Zou Y, Yu BP. The molecular inflammatory process in aging. Antioxid Redox Signal. 2006;8:572–81.
Remme WJ. Overview of the relationship between ischemia and congestive heart failure. Clin Cardiol. 2000;23:IV4–8.
Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–8.
Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74.
Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–22.
Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–8.
Gutterman DD, Chabowski DS, Kadlec AO, et al. The human microcirculation: regulation of flow and beyond. Circ Res. 2016;118:157–72.
Taqueti VR, Di Carli MF. Coronary microvascular disease pathogenic mechanisms and therapeutic options: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72:2625–41.
Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830–40.
Dirkx E, da Costa Martins PA, De Windt LJ. Regulation of fetal gene expression in heart failure. Biochim Biophys Acta. 2013;1832:2414–24.
Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–96.
Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-chain) cardiac amyloidosis: a review of diagnosis and therapy. J Am Coll Cardiol. 2016;68:1323–41.
Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12:91–102.
Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1:e000364.
Rapezzi C, Lorenzini M, Longhi S, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20:117–24.
Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203–12.
Castano A, Haq M, Narotsky DL, et al. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol. 2016;1:880–9.
Chugh SS, Havmoeller R, Narayanan K, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014;129:837–47.
Schnabel RB, Yin X, Gona P, et al. 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet. 2015;386:154–62.
Falk RH. Atrial fibrillation. N Engl J Med. 2001;344:1067–78.
Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368:1005–11.
Levine RA, Hagege AA, Judge DP, et al. Mitral valve disease--morphology and mechanisms. Nat Rev Cardiol. 2015;12:689–710.
Anyanwu AC, Adams DH. Etiologic classification of degenerative mitral valve disease: Barlow’s disease and fibroelastic deficiency. Semin Thorac Cardiovasc Surg. 2007;19:90–6.
Supino PG, Borer JS, Preibisz J, Bornstein A. The epidemiology of valvular heart disease: a growing public health problem. Heart Fail Clin. 2006;2:379–93.
Ross J Jr, Braunwald E. Aortic stenosis. Circulation. 1968;38:61–7.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Ethics declarations
Prof. Ruschitzka has not received personal payments by pharmaceutical companies or device manufacturers in the last 3 years (remuneration for the time spent in activities, such as participation as steering committee member of clinical trials and member of the Pfizer Research Award selection committee in Switzerland, were made directly to the University of Zurich). The Department of Cardiology (University Hospital of Zurich/University of Zurich) reports research-, educational-, and/or travel grants from Abbott, Amgen, Astra Zeneca, Bayer, Berlin Heart, B. Braun, Biosense Webster, Biosensors Europe AG, Biotronik, BMS, Boehringer Ingelheim, Boston Scientific, Bracco, Cardinal Health Switzerland, Corteria, Daiichi, Diatools AG, Edwards Lifesciences, Guidant Europe NV (BS), Hamilton Health Sciences, Kaneka Corporation, Kantar, Labormedizinisches Zentrum, Medtronic, MSD, Mundipharma Medical Company, Novartis, Novo Nordisk, Orion, Pfizer, Quintiles Switzerland Sarl, Sahajanand IN, Sanofi, Sarstedt AG, Servier, SIS Medical, SSS International Clinical Research, Terumo Deutschland, Trama Solutions, V-Wave, Vascular Medical, Vifor, Wissens Plus, and ZOLL. The research and educational grants do not impact on Prof. Ruschitzka’s personal remuneration.
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Luciani, M., Ruschitzka, F., Camici, G.G. (2022). Cardiovascular Ageing. In: Pape, HC., Kates, S.L., Hierholzer, C., Bischoff-Ferrari, H.A. (eds) Senior Trauma Patients . Springer, Cham. https://doi.org/10.1007/978-3-030-91483-7_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-91483-7_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-91482-0
Online ISBN: 978-3-030-91483-7
eBook Packages: MedicineMedicine (R0)