
Abstract
Leprosy results from infection with Mycobacterium leprae, an unculturable pathogen with an exceptionally long generation time that has afflicted human populations for millennia. The history of leprosy has been documented by various civilizations, and its global spread deduced from the anthropological record, although scientific proof for the latter is often lacking. Considerable insight into the biology, genetics, and evolution of the leprosy bacillus has been obtained from genomics. M. leprae has undergone extensive reductive evolution, losing DNA from its genome, half of which is now occupied by pseudogenes. Comparative genomics of four different strains from India, Brazil, Thailand, and the USA revealed remarkable conservation of the ~3.27-megabase genome (99.995% identity) yet uncovered 215 polymorphic sites, mainly single-nucleotide polymorphisms (SNP), and a handful of new pseudogenes. Mapping these polymorphisms in a large panel of strains defined 16 SNP subtypes that showed strong geographical associations and helped retrace the evolution of M. leprae and the dissemination of leprosy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Background
Leprosy, a chronic, dermatological, and neurological disease, results from infection with the unculturable pathogen Mycobacterium leprae [1]. The disease is curable yet remains a public health problem even though there is no known ubiquitous reservoir for transmission of M. leprae other than human beings. Thanks to the massive implementation in the 1980s of multidrug therapy (MDT) by the World Health Organization, over 14 million patients have been cured and the incidence of leprosy has declined considerably. Nonetheless, an average of 250,000–300,000 new cases have been reported annually during the last 5 years throughout the world [2]. Many of these cases occur in children, thereby indicating that the chain of transmission remains, albeit weakened; this highlights the need for sensitive and reliable epidemiological methods to detect M. leprae and to monitor its spread both locally and globally. Genome sequencing has proved a particularly powerful means of understanding the biology and genetics of the leprosy bacillus, and comparative genomics has uncovered polymorphisms that can serve as the basis for developing molecular epidemiological tools. Such tools have started to find application and are helping us to understand how M. leprae has evolved.
2 History
Leprosy is often described as an “ancient” disease, but what does that really mean? Certainly, in the context of anatomically modern humans and the civilizations they established in the past 5000–10,000 years, the disease may appear ancient, but then one should recall that, from an evolutionary standpoint, where millions of years are the norm, the species Homo sapiens itself actually appeared quite recently. Various lines of evidence, based on haploid markers from mitochondrial DNA and the Y chromosome, together with the archeological, anthropological, and linguistic records, indicate that the cradle of mankind was East Africa. After initially spreading within Africa, humans migrated to the Near East about 60,000 years ago, before spreading to southern Asia and Australasia, Europe, and Central Asia, until finally reaching the Americas via the Bering Strait about 14,000–20,000 years ago [3]. Now, when we consider that M. leprae and the tubercle bacillus Mycobacterium tuberculosis are thought to have diverged from their most recent common ancestor 66 million years ago [4], the term “ancient” assumes a different meaning.
There are very old historical reports indicating that leprosy most likely existed in human populations in Egypt [5], India [6], and China [7], although the disease may often have been confused with other dermatological conditions and infections [8]. The oldest written records of leprosy are from the ancient Indian texts in the Sushruta Samhita around 600 BCE, which accurately describes the characteristic features and diagnosis of leprosy, and even the traditional treatment with chaulmoogra oil [6]. Skeletal remains provide a more reliable indicator, as the deformities characteristic of leprosy can often be readily identified. For instance, the “clawed hand” and enlarged or degraded maxillofacial regions can often be discerned on well-preserved skeletons [9]. Examples of the latter from 200 BCE are from the Dakhleh Oasis in Egypt [10]. The oldest leprosy skeletal remains found, from Balathal in India, were estimated to be 4000 years old by radiocarbon dating [11]. Such remains are relatively common in Europe, where leprosy was endemic in medieval times, but have not been found in the pre-Colombian Americas [12].
It is thought that the Phoenicians from the eastern Mediterranean region were responsible for the dissemination of leprosy around the Mediterranean Basin from 1500 to 300 BCE and that Alexander the Great’s soldiers brought leprosy back from India about 325 BCE. In turn, the expansion of the Roman Empire is thought to have resulted in the introduction of leprosy into France, Germany, and the Iberian Peninsula. After the collapse of the Roman Empire, other invaders such as the Barbarians and the Saracens may have disseminated the disease, which reached the British Isles and Scandinavia, from where the Vikings likely served as carriers [13]. Leprosy was certainly endemic in Europe before the Crusades began in 1095 CE and started to decline gradually from the thirteenth century onward, although the reasons for this are unknown. Norway was one of the last European countries to eliminate leprosy, and it was there, in 1873, that Armauer Hansen made his seminal discovery of the leprosy bacillus in biopsies from fishermen in Bergen (editor note: last autochthonous Italian case is reported in 2010). One way of retracing the history of leprosy is to examine the genomes of strains of M. leprae, both ancient and modern, and to identify polymorphisms that have been vertically transmitted for use as molecular epidemiological markers.
3 Genomics
Since M. leprae has never been successfully cultured in the laboratory and has a generation time of 14 days, one of the longest known for a bacterium, it has proved extremely challenging to perform microbiological or genetic research with this pathogen. An important breakthrough came when it was found that the nine-banded armadillo Dasypus novemcinctus was naturally susceptible to infection [14]. Thus, for the first time, it became possible to obtain sufficient bacilli to perform biomedical research, such as vaccine development, with the leprosy bacillus. Once large numbers of bacteria become available, it was a fairly simple matter to extract their DNA in order to carry out whole-genome sequencing. The first M. leprae genome to be completely sequenced was that of the TN strain, originally isolated from a patient in Tamil Nadu, India.
The genome sequence of the TN strain of M. leprae contains 3,268,212 base pairs (bp) and has average G + C content of 57.8% [15]. The leprosy bacillus thus has the smallest and most A + T-rich genome of any known mycobacterium; for comparison, the close relatives M. tuberculosis and Mycobacterium marinum have genomes of 4,411,532 and 6,636,827 bp, respectively, and G + C content of 65.9% [16, 17]. Bioinformatic analysis uncovered 1614 genes coding for proteins in the TN genome and a further 50 that encode stable RNAs. These account for a mere 49.5% of the genome, with the remainder occupied by pseudogenes, i.e., inactive reading frames that still have functional counterparts in other mycobacteria. Initially, 1116 pseudogenes were found [15], but this figure rose to 1310 when other mycobacterial genome sequences became available for comparison [4].
The reductive evolution undergone by M. leprae, in which DNA was deleted from the genome and pseudogenes accumulated, provides a general explanation for its unusually slow growth, although no specific defect could be identified to account for this. Loss of DNA can be explained by homologous recombination events involving dispersed repeats, of which four families have been named [18]. This proposal is supported by the presence of single copies of these repeats at sites in the genome where synteny with other mycobacterial genomes breaks down as the gene order changes abruptly. None of these repeated elements contain open reading frames, although they do show some properties of transposable elements [18]. The presence of some of these repetitive sequences within pseudogenes suggests that they were once capable of undergoing transposition, but as will emerge below, this is no longer the case.
Analysis of the genome sequence has improved our understanding of the physiology, pathogenesis, and genetics of M. leprae and is underpinning the development of better diagnostics and molecular epidemiological tools for monitoring disease transmission.
4 Comparative Genomics of M. leprae Strains
To identify polymorphic DNA markers that could be used as the basis of a molecular epidemiological test for leprosy, the genomes of three other strains were sequenced, namely, Br4923, from Brazil; Thai-53, from Thailand; and NHDP63, from the USA. Astonishingly, when the four genome sequences were compared, they were found to share 99.995% identity and to display near-perfect collinearity and size. There was no evidence for gene deletions, insertions, or translocations, with all of the dispersed repeats occupying the same positions in all cases [19, 20]. This four-way comparison revealed only 215 polymorphic sites, including single-nucleotide polymorphisms (SNPs) and small insertion-deletion events (indels). Five new pseudogenes were uncovered, three in strain Thai-53, and one each in strains Br4923 and NHDP63.
In light of the massive gene decay and extensive DNA loss undergone by M. leprae, the exceptional conservation of the genomes of these strains was truly unexpected, even more so when their widely different geographical origins are taken into account. Furthermore, it has been estimated from the number of nonsynonymous substitutions per site occurring in the pseudogenes that a single pseudogenization event occurred in the leprosy bacillus in the last 10–20 million years [4]. Given this vast timeframe, it is truly puzzling that so little diversity is observed within either the genes or the pseudogenes. This may indicate that the emergence of M. leprae as a human pathogen may have been due to a recent event, such as passing through an evolutionary bottleneck or introduction from another host. The long generation time is another factor that might contribute to genetic homogeneity. The leprosy bacillus is an excellent example of a genetically monomorphic pathogen [21], and in principle, all cases of leprosy stem from the initial infection with a single clone.
5 Strain Typing and Molecular Epidemiology
The exact route of transmission of M. leprae remains obscure, but early identification of infectious leprosy cases is critical in order to implement MDT rapidly and thus prevent disease progression, disabilities, and further contagion. After diagnosis, molecular typing of strains can help to improve understanding of the transmission and dynamics of the disease. Different molecular epidemiological tools are being developed to trace the possible sources of infection, to differentiate cases of relapse from reinfection, and to probe possible links between human and environmental reservoirs. One should recall that phenotypic methods of drug susceptibility testing (DST) of M. leprae, such as the mouse footpad model [22], have now been almost completely replaced by molecular methods [23] that use the polymerase chain reaction (PCR) to amplify regions of the gene encoding the drug target [24, 25]. Ideally, molecular DST and strain typing tests will eventually be combined.
Most genotyping methods take advantage of polymorphic sites in DNA, which exist in several forms. In addition to the SNPs and indels described above, there are variable number tandem repeats (VNTR) that can be exploited. VNTRs are sometimes called short tandem repeats (STRs) , or microsatellites (repeat length 2–5 bp) and minisatellites (repeat length 6–50 bp). The paradigm VNTR in mycobacteria is the mycobacterial interspersed repetitive unit (MIRU), a tandemly arranged minisatellite that is a major source of diversity in the genomes of tubercle bacilli and some other mycobacteria. MIRUs serve as the basis of a robust PCR-based typing system that exploits differences in their copy number [26,27,28]. Unlike in M. tuberculosis, none of the 20 MIRU loci in M. leprae contain tandem repeats [18], so this target sequence could not be used.
Instead, VNTRs consisting of repeat units of 1, 2, 3, 6, 12, 18, 21, and 27 bp have been screened and exploited for typing purposes [29, 30]. While VNTRs provide a better dynamic range for typing, they may be error prone due to the homoplasies frequently associated with such minisatellites [31]. To be successful in molecular epidemiology, the loci targeted must behave in a reproducible, stable, and discriminatory manner. Some of the limitations associated with VNTR typing can be overcome by interrogating multiple loci simultaneously, a technique known as multilocus VNTR analysis .
SNPs can be restricted to a single strain, and are therefore relatively uninformative, or be transmitted vertically and thus present in multiple strains, in which case they are more informative markers. The first SNP typing system was based upon three informative SNPs and used to screen ~400 different strains from 28 different countries across the world. This approach revealed only four combinations (SNP types 1–4) but with strong geographical associations [19]. Subsequently, a further 84 informative markers (78 SNPs and 6 indels) were discovered among the 215 polymorphic sites found by comparative genomics with four strains of the leprosy bacillus. Interrogation of these 84 sites enabled M. leprae strains to be classified into 16 different SNP genotypes (1A–4P), again displaying a phylogeographical relationship [20]. Figure 1.1 provides an algorithm of how SNP typing can be performed.

SNP typing scheme for M. leprae. Using the first three SNPs shown on the left (SNP14676, 1642879, and 2935693), a strain may first be typed into one of the four SNP types (1–4) and then subtyped using three or four markers shown at the right per SNP type to give the 16 subtypes (A–P). Yellow cells denote identity to the base present in the TN reference strain (subtype 1A), while dark green indicates identity to the base present in the Brazilian strain Br4923 at this position (subtype 4P). # These SNPs are the same as those used for typing into types 1–4 (left side of the figure)
While the SNP-based typing system is both very robust and highly reliable for classifying strains over broader geographical regions, at present, short-range transmission studies, within a district for example, are difficult with this typing system due to too little variation. Combining SNP and multilocus VNTR typing appears to be the method of choice for future typing studies of M. leprae by merging the phylogeographic component imparted by SNP subtypes with the dynamic branching provided by VNTRs.
6 Phylogeography
In the past decade, it has become increasingly apparent that one can retrace the evolutionary history of peoples and their migrations by studying the genotypes of the pathogens they carry. In this regard, disease-causing bacteria such as Helicobacter pylori [21, 32, 33], Yersinia pestis [34], and M. leprae have served as valuable tools. Conversely, understanding the structure of human populations and their immunogenetics can also provide insight into the evolution of pathogens [35].
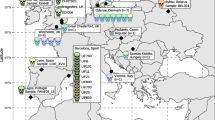
Phylogenetic studies of M. leprae have allowed us to infer relationships between different strains and to attempt to retrace their history. There is a fairly strict correlation between the geographical origin of the leprosy patient and the SNP profile: types 1A–D occur predominantly in Asia, the Pacific region, and East Africa; types 3I–M in Europe, North Africa, and the Americas; and types 4 N–P in West Africa, the Caribbean region, and South America. M. leprae belonging to SNP types 2E–H appears to be the rarest, although this may be due to undersampling, and has only been detected in Ethiopia, Malawi, Nepal/North India, and New Caledonia (Fig. 1.2). Applying the principles of maximum parsimony, two plausible evolutionary schemes arise. In the first scheme, which we consider more probable, M. leprae of SNP type 2, present in the region of East Africa/Near East, preceded SNP type 1, which migrated eastward, and SNP type 3, which disseminated westward in human populations, before SNP type 3 gave rise to type 4. In the second scenario, type 1 was the progenitor of type 2, with SNP types 3 and 4 following in numerical order. Comparison of what is known of human migrations with the phylogeny of M. leprae is highly informative and reveals general agreement with the historical record and several contradictions.

Global dissemination of M. leprae from SNP typing. Pillars are located on the country of origin of the M. leprae sample and color coded according to the scheme for the 16 SNP subtypes. The thickness of each pillar corresponds to the number of samples tested (1–5, thin; 6–29, intermediate; >30, broad). Gray arrows indicate the routes of migration taken by early humans with the estimated time of migration in years [3, 36]. Dashed line indicates the location of the Silk Road in the first century, and * denotes result obtained from ancient DNA. (Reproduced with permission from [20])
The proposition that leprosy originated in the Indian subcontinent and was introduced into Europe by Greek soldiers returning from the Indian campaign of Alexander the Great [8] is not compatible with the phylogeny of M. leprae since SNP type 1 predominates in India, whereas SNP type 3 afflicted European populations. From India, leprosy is thought to have moved to China, in about 500 BC, and then to Japan, reaching Pacific islands such as New Caledonia as recently as the nineteenth century. Once again, this is partly contradicted by the results of M. leprae genotyping, as leprosy appears to have been introduced into Asia by two different routes. The first of these, which is historically consistent, is the southern one associated with the SNP type 1 strains present in the Indian subcontinent, Indonesia, and the Philippines. The second is a more northerly route beginning in the eastern Mediterranean region and extending via Turkey and Iran to China, and hence to Korea and Japan. The strains encountered along this route are mainly of SNP subtype 3K [20], and the Silk Road appears to have been a likely means of transport and disease transmission (Fig. 1.2). The Plague or Black Death, caused by Yersinia pestis [34], is thought to have reached Europe in the fourteenth century from China via the Silk Road, being carried by humans and their fleas. With respect to leprosy, the opposite route of transmission may have been traveled, with the disease originating in Europe or the Near/Middle East and then spreading to the Far East.
Nothing is known of the history of leprosy in sub-Saharan Africa except that the disease was present prior to the colonial era [8, 13]. The phylogeny of M. leprae suggests that the disease was most likely introduced into West Africa by infected explorers, traders, or colonialists of European or north African descent, rather than by migrants from East Africa, as M. leprae of SNP type 4 is endemic in West Africa and much closer to type 3 than to type 2. West and southern Africa are believed to have been settled by migrants from the eastern part of the continent before the arrival of humans in the Eurasian regions [3, 37]. It seems unlikely that early humans brought leprosy into West Africa unless clonal replacement has since occurred. From West Africa, leprosy was then introduced by the slave trade from the seventeenth century onward into the Caribbean region, Venezuela, Brazil, and other parts of South America, as isolates of M. leprae with the same SNP type, 4N or O, are found there as in West Africa, which is consistent with the history of slavery. Strains of SNP subtype 4P are restricted to South America or the Caribbean, and they must have branched off during the last 400 years, since the introduction of the ancestral strain from West Africa, which was likely of SNP type 4 N or O.
It appears improbable that leprosy was introduced into the Americas by early humans via the Bering Strait; instead, it seems more likely that immigrants from Europe brought the disease, as most of the M. leprae strains found in North, Central, and South America have the 3I genotype associated with European leprosy cases. In the eighteenth and nineteenth centuries, when the Mid-Western states of the USA were settled by Scandinavian immigrants, many cases of leprosy were recorded at a time when a major epidemic was underway in Norway [8]. Further support for the “European origin” hypothesis is provided by the finding that wild armadillos from Louisiana, USA, which are naturally infected with M. leprae, also harbor the SNP type 3 strain, indicating that they were contaminated from human sources of European origin [19]. Immigrants from France and Spain primarily were the main settlers in the state of Louisiana.
It is also noteworthy that, on islands such as the French West Indies and New Caledonia, there is much greater SNP variety of M. leprae (Fig. 1.2), reflecting the passage of, and settlement by, successive human populations. It is known that migrations by sea rather than over land or via coastal routes lead to more diversity in humans through intermixing, and a greater range of diversity is also apparent among their pathogens.
7 Paleomicrobiology
Insight into ancient leprosy can be gained from the study of skeletons with the telltale signs of leprosy described above. Although this is a technically challenging approach, fascinating results have been obtained with skeletal remains from a number of settings. Studying these “extinct” cases not only enables comparisons with extant strains of M. leprae but also provides information for countries where leprosy has long been eradicated.
In the first report, two archeological cases of leprosy from Medieval England were studied using ancient DNA methods and PCR. The highly characteristic repetitive DNA was used for confirmation of the identity M. leprae, and three VNTR loci were also amplified [37]. In subsequent work, SNP typing was undertaken with specimens from the UK, Turkey, Egypt, Denmark, and Croatia. The M. leprae present in these samples all exhibited an SNP type 3 profile, and in some cases, further typing was possible, revealing the presence of subtypes 3I, 3K, and 3M [20, 38]. The oldest specimen examined to date, roughly 1500 years old, came from an Egyptian skeleton from Dakhleh Oasis [39], a region close to the proposed origin of M. leprae and Homo sapiens. The ancient DNA analysis indicated that M. leprae of SNP type 3 was involved. Finally, the most recent study was with a sample from Uzbekistan, of a similar age to that from Egypt, and this too revealed that the causative strain of M. leprae exhibited an SNP type 3 profile [40]. This is consistent with the fact that extant strains of M. leprae from Iran, a country nearby, are often of type 3K [20]. With improvements in ancient DNA methodologies and the massively parallel sequencing capabilities offered by next-generation sequencing technologies, it is not inconceivable that we will be able to use skeletal remains to generate a draft genome sequence for an “extinct” strain of M. leprae , as was done recently for the Neanderthal genome [41].
References
Britton WJ, Lockwood DN. Leprosy. Lancet. 2004;363(9416):1209–19.
Anon. Global leprosy situation. Wkly Epidemiol Rec. 2010;85:337–48.
Cavalli-Sforza LL, Feldman MW. The application of molecular genetic approaches to the study of human evolution. Nat Genet. 2003;33(Suppl):266–75.
Gomez-Valero L, Rocha EP, Latorre A, Silva FJ. Reconstructing the ancestor of Mycobacterium leprae: the dynamics of gene loss and genome reduction. Genome Res. 2007;17(8):1178–85.
Hulse EV. Leprosy and ancient Egypt. Lancet. 1972;2(7785):1024–5.
Dharmendra. History of spread and decline of leprosy. 2nd ed. New Delhi: Ministry of Health; 1967.
Skinsnes OK, Chang PH. Understanding of leprosy in ancient China. Int J Lepr Other Mycobact Dis. 1985;53(2):289–307.
Browne SG. The history of leprosy. In: Hastings RC, editor. Leprosy. Edinburgh: Churchill Livingstone; 1985. p. 1–14.
Scollard DM, Skinsnes OK. Oropharyngeal leprosy in art, history, and medicine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87(4):463–70.
Dzierzykray-Rogalski T. Paleopathology of the Ptolemaic inhabitants of the Dakhleh oasis (Egypt). J Hum Evol. 1980;9:71–4.
Robbins G, Tripathy VM, Misra VN, et al. Ancient skeletal evidence for leprosy in India (2000 B.C.). PLoS One. 2009;4(5):e5669.
Ortner DJ. Infectious diseases: tuberculosis and leprosy. In: Ortner DJ, editor. The identification of pathological conditions in human skeletal remains. London: Academic; 2003. p. 227–72.
Sansarricq H. Histoire de la lèpre. In: Sansarricq H, editor. La lèpre. Paris: Ellipses; 1995. p. 22–32.
Kirchheimer WF, Storrs EE. Attempts to establish the armadillo (Dasypus novemcinctus Linn.) as a model for the study of leprosy. I. Report of lepromatoid leprosy in an experimentally infected armadillo. Int J Lepr Other Mycobact Dis. 1971;39(3):693–702.
Cole ST, Eiglmeier K, Parkhill J, et al. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–11.
Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–44.
Stinear TP, Seemann T, Harrison PF, et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008;18(5):729–41.
Cole ST, Supply P, Honoré N. Repetitive sequences in Mycobacterium leprae and their impact on genome plasticity. Lepr Rev. 2001;72:449–61.
Monot M, Honore N, Garnier T, et al. On the origin of leprosy. Science. 2005;308(5724):1040–2.
Monot M, Honore N, Garnier T, et al. Comparative genomic and phylogeographic analysis of Mycobacterium leprae. Nat Genet. 2009;41(12):1282–9.
Achtman M. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol. 2008;62:53–70.
Shepard CC, Congdon CC. Increased growth of Mycobacterium leprae in thymectomized-irradiated mice after foot pad inoculation. Int J Lepr Other Mycobact Dis. 1968;36(2):224–7.
Honore N, Cole ST. Molecular basis of rifampin resistance in Mycobacterium leprae. Antimicrob Agents Chemother. 1993;37(3):414–8.
Maeda S, Matsuoka M, Nakata N, et al. Multidrug resistant Mycobacterium leprae from patients with leprosy. Antimicrob Agents Chemother. 2001;45(12):3635–9.
Williams DL, Gillis TP. Molecular detection of drug resistance in Mycobacterium leprae. Lepr Rev. 2004;75(2):118–30.
Allix-Beguec C, Fauville-Dufaux M, Supply P. Three-year population-based evaluation of standardized mycobacterial interspersed repetitive-unit-variable-number tandem-repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2008;46(4):1398–406.
Allix-Beguec C, Harmsen D, Weniger T, Supply P, Niemann S. Evaluation and strategy for use of MIRU-VNTRplus, a multifunctional database for online analysis of genotyping data and phylogenetic identification of Mycobacterium tuberculosis complex isolates. J Clin Microbiol. 2008;46(8):2692–9.
Mazars E, Lesjean S, Banuls AL, et al. High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci U S A. 2001;98(4):1901–6.
Groathouse NA, Rivoire B, Kim H, et al. Multiple polymorphic loci for molecular typing of strains of Mycobacterium leprae. J Clin Microbiol. 2004;42(4):1666–72.
Matsuoka M, Maeda S, Kai M, et al. Mycobacterium leprae typing by genomic diversity and global distribution of genotypes. Int J Lepr Other Mycobact Dis. 2000;68(2):121–8.
Filliol I, Motiwala AS, Cavatore M, et al. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J Bacteriol. 2006;188(2):759–72.
Falush D, Wirth T, Linz B, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299(5612):1582–5.
Wirth T, Wang X, Linz B, et al. Distinguishing human ethnic groups by means of sequences from Helicobacter pylori: lessons from Ladakh. Proc Natl Acad Sci U S A. 2004;101(14):4746–51.
Achtman M, Morelli G, Zhu P, et al. Microevolution and history of the plague bacillus, Yersinia pestis. Proc Natl Acad Sci U S A. 2004;101(51):17837–42.
Wong SH, Gochhait S, Malhotra D, et al. Leprosy and the adaptation of human toll-like receptor 1. PLoS Pathog. 2010;6:e1000979.
Underhill PA, Shen P, Lin AA, et al. Y chromosome sequence variation and the history of human populations. Nat Genet. 2000;26:358–61.
Taylor GM, Watson CL, Lockwood DNJ, Mays SA. Variable nucleotide tandem repeat (VNTR) typing of two cases of lepromatous leprosy from the archaeological record. J Archaeol Sci. 2006;33:1569–79.
Watson CL, Lockwood DN. Single nucleotide polymorphism analysis of European archaeological Mycobacterium leprae DNA. PLoS One. 2009;4(10):e7547.
Molto JE. Leprosy in Roman period skeletons from Kellis 2, Dakhleh, Egypt. In: Roberts CA, Lewis ME, Manchester K, editors. The past and present of leprosy: archaeological, historical, paleopathological and clinical approaches, BAR international series, vol. 1054. Oxford: Archaeopress; 2002. p. 179–92.
Taylor GM, Blau S, Mays S, et al. Genotyping of Mycobacterium leprae amplified from an archaeological case of lepromatous leprosy from Central Asia. J Archaeol Sci. 2009;36:2408–14.
Green RE, Krause J, Briggs AW, et al. A draft sequence of the neandertal genome. Science. 2010;328(5979):710–22.
Acknowledgments
We thank all the patients and participants who contributed to this work, particularly Philippe Busso, Nadine Honoré, and Marc Monot. Financial support was generously provided by the Foundation Raoul Follereau, and the National Institutes of Health, National Institute of Allergy and Infectious Diseases (grant RO1-AI47197-01A1).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cole, S.T., Singh, P. (2022). History and Phylogeography of Leprosy. In: Nunzi, E., Massone, C., Portaels, F. (eds) Leprosy and Buruli Ulcer. Springer, Cham. https://doi.org/10.1007/978-3-030-89704-8_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-89704-8_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-89703-1
Online ISBN: 978-3-030-89704-8
eBook Packages: MedicineMedicine (R0)