
Abstract
Oil paint is a dynamic system that undergoes chemical alteration on several time and length scales. At the short term, curing reactions are necessary for oil to dry properly. At longer time scales, a wide variety of other chemical processes can negatively affect the visual appearance or mechanical properties of historical artistic paint systems. The development of chemical imaging methods capable of covering length scales continuously from the millimetric to micro- or even nanoscale is key in understanding the chemical composition of a painting and the historical changes thereof. Such imaging methods can help in assessing to which extent the original painting’s composition may have been modified by chemical degradation processes. Processes that occur in the highly heterogeneous mixtures of binders, pigments, additives, alteration products and possibly later repainting and restoration treatments. Establishing the precise biography of the painting contributes to evaluate its authenticity. New modalities and novel methods of microchemical imaging provide access to previously unexplored length scales, are capable of better differentiation between the various oil paint components (original composition or later addition), and allow performing faster analysis to produce higher definition images. In this review, we report on recent methodological developments and future prospects to determine oil paints composition using microchemical imaging at the micro- and nanoscale.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
For centuries, oil paint has been one of the most important artistic media. It was used as the medium of choice for some of the world’s most recognized artworks, such as Leonardo da Vinci’s Mona Lisa, Rembrandt’s The Night Watch, and Vincent van Gogh’s Starry Night. Despite their static appearance, oil paintings are in fact dynamic systems that undergo a wide variety of chemical changes. A fresh oil paint undergoes various chemical reactions, resulting in a dry and mechanically stable paint system. However, at longer time scales a multitude of chemical reactions take place that may (severely) deteriorate the painting’s visual appearance or mechanical stability. Since these degradative processes are chemically complex and hard to predict, they pose a monumental challenge to art curators and conservators worldwide. By modifying the painting chemical composition, they may also impact the authentication evaluation which usually aims at identifying which materials were used, in which parts of the paintings, when applied and above all by whom (main artist, copyist, conservators, or even forgers!)
Past studies have shown that forgers have tried various strategies to speed up the drying of the oil paint, such as accelerated ageing conditions or the use of additives to create a hard paint film (Blumenroth et al. 2019; Breek and Froentjes 1975). Access to suitable analytical techniques and an understanding of the drying processes of oil paint are important factors in unravelling these frauds. The use of pigments that have come onto the market after the dating of the painting is a good marker for authenticity issues, but often the pallet of the painting is not characteristic enough and needs further investigation into the material and its context (Craddock 2009; Sloggett 2019). Focusing on past conservation treatments, conservators are confronted with paint retouches which can be very difficult to distinguish from the original paint (Noble et al. 2018). Paint morphologies, trace elements, layer build-ups, degradation components and their distribution can help identify these later additions to the painting. In all cases, knowledge of various chemical alteration and degradation processes is needed to address authenticate specific sections of oil paintings, or oil paintings as a whole (Doménech-Carbó et al. 2012; Tanasa et al. 2020).
The most basic description of oil paint is that of a mixture of a drying oil binder (triacylglycerol with a high degree of polyunsaturated fatty acid chains) and one or more organic or inorganic pigments, mixed in a ratio such that it is workable by the artist. Upon application of the paint, the drying oil binder is exposed to oxygen and the double bonds of the unsaturated fatty acids start to undergo autoxidation and form cross-links (van den Berg et al. 1999). This process is called the curing of the oil paint and eventually leads to a 3D-polymerized network that holds the pigment particles in place. As the curing process tends to take multiple days or even weeks, driers or other additives may be added to speed up the solidification of the oil paint.
On a longer timescale, the ongoing autoxidation and hydrolysis start to break down the polymerized oil binder network to form carboxylic acids—either still covalently bonded to the oil network or present as free or metal-bound dicarboxylic or monocarboxylic fatty acids. Previous studies have shown that—for a mature oil paint—the degree of carboxylic acid formation due to autoxidation is in the order of one acid group per triacylglycerol, while hydrolysis increases the free fraction of saturated fatty acids from less than 1% for fresh oils to over 90% for some historical oil paints (Baij et al. 2019; van den Berg et al. 1999). Like the binding medium, many pigments also undergo certain alteration and degradation processes. Some pigments react with saturated free fatty acids to form metal soaps (Heeren et al. 1999; Van der Weerd et al. 2004). Although this process is not necessarily disadvantageous for an oil paint (Cotte et al. 2017a), in some cases, the metal soaps agglomerate, forming brittle domains that may occupy a larger volume than the separate starting materials. In such cases, metal soap formation has been shown to lead to conservation issues such as paint layer delamination and the formation of protrusions (Ebert et al. 2011; Heeren et al. 1999; Keune and Boon 2007; Van Loon et al. 2019; Noble et al. 2002; Osmond et al. 2013; Van der Weerd et al. 2004). Another phenomenon that affects many oil paintings is the light-induced transformation of pigments into polymorphs or oxidation products (Van Der Snickt et al. 2009; Trentelman et al. 1996). As these polymorphs and oxidation products may have a different color than the original pigment material, these processes can lead to discoloration of the painting over time and therefore loss of the original artistic intention.
Aged oil paint leads to the formation of systems that are chemically complex and spatially highly heterogeneous. This heterogeneity shows up on multiple length scales: both in the plane of the painting as well as in-depth, due to the multi-layered build-up of oil paintings. A paint stratigraphy typically consists of several chemically distinct paint layers, each 10–100 μm thick, containing pigment particles with dimensions of about 0.05–10 μm that are embedded in a medium of organic binder and various micrometric and submicrometric domains of organic and inorganic alteration and degradation products. The existence of these various distinct scales of chemical heterogeneity means that information on composition and degradation processes can also be found on various length scales, ranging from tens of micrometers to nanometers. Given these length scales, there is a strong interest in analyses using high-resolution microchemical imaging (techniques hereafter denoted with the prefix μ, e.g., μ-XRF). Due to the multiscale nature of oil paints, the scale of analysis needs to be chosen such that relevant details can be spatially resolved while the total sampled area (or volume) provides a representative chemical overview.
For the purpose of performing microchemical analysis on oil paints, microsamples can sometimes be obtained from historical works of art. Microsamples typically have dimensions in the order of a few hundred micrometers and are mostly taken by conservators from small pre-existing cracks, other surface damages, or from the edge, so as not to leave any marks that can be distinguished by the naked eye. An alternative and complementary approach is to mimic oil paint composition and degradation phenomena by ageing (naturally or artificially) paint mock-ups. This approach allows to control the type and extent of ageing, paint composition, and has less strict limitations when it comes to sampling, but may not always accurately represent the chemistry that occurs in actual historical works of art.
What measurement modality will be applied for the study of either a historical microsample or a sample obtained from a paint mock-up strongly depends on the specific chemical question. For instance, an investigation of oxidative discoloration of an inorganic pigment may require that a measurement can provide reliable chemical contrast between the different oxidation states of the affected element, while a study of delamination in saponified zinc white (ZnO) paints requires a measurement that can provide chemical contrast between metal carboxylates and one or more of the organic and inorganic reactants. If the migration of a reactant or a degradation product is of interest, it may be insightful to resolve chemical distributions throughout one or multiple paint layers, directly around the pigment, or in the vicinity of a possible catalytically active compound. Depending on what distribution is being studied, a spatial resolution may be required in the order of tens of micrometers (multilayer gradients), a few micrometers (gradients within layers), or tens to hundreds of nanometers (distributions around pigment particles or particles of possibly catalytically active compounds). The study of all the various aspects of oil paint degradation requires an extensive methodological toolbox with a wide variety of contrasting mechanisms and the ability to perform analysis at multiple length scales. Due to the complexity of oil paints and their degradation phenomena, answering chemical questions typically requires the use of multiple methods from this toolbox.
Over the past few decades, rapid advances have been made in the development of spectroscopic, spectrometric, and diffraction tools that are capable of characterizing and identifying organic and inorganic compounds in terms of molecular fragments, functional groups, elemental composition, coordination chemistry, defect chemistry, crystalline structure, and physical texture. Developments in focusing optics and sources for mid-IR to hard X-ray photons and charged particles have led to the availability of microanalytical tools that can perform these chemical characterizations and identifications on the micro- or even nanoscale. As the various developments progress, the time necessary to perform chemical microanalysis at single points decreases further and further. Reduced measurement times have made it possible to implement almost all spectroscopic, spectrometric and diffraction modalities as microchemical imaging tools; allowing to resolve—in two or even three dimensions—the spatial distributions of specific chemical species at micrometric or nanometric spatial resolution. Microchemical imaging is therefore particularly useful for detecting trends and patterns in chemical distributions, which can inform on the underlying chemistry of migration and degradation processes. Especially for the study of systems that are as chemically complex and spatially heterogeneous as oil paints, such trends and patterns may only be detected through the analysis of many spatial points and the use of some form of microchemical imaging is essential to collect the required information.
The first part of this chapter will review the state-of-the-art applications of microchemical imaging methodologies to the study of oil paint composition and degradation. As there may be some ambiguity with regards to the term microchemical imaging, in this review it is defined as any technique that can probe compound-specific spectroscopic transitions, diffraction patterns, or mass fragments, and resolve their distributions at a sub-10 μm lateral resolution over a certain area or volume of interest. The following four sections will discuss recent advances in microchemical imaging instrumentation, covering optics, detectors, and light sources, and go over what types of chemical information can be obtained and at what length scales analysis can currently be performed. This review is structured along the working principles of the discussed analytical tools; separating infrared-, UV-Vis-, X-ray-, and charged particle-based methods. The second part of this chapter will critically discuss the capabilities and limitations of the current methodological toolbox and formulate perspectives on future research endeavors in oil paint studies. Such perspectives are illustrated by identifying four general research objectives that address the technological limitations of the microchemical imaging techniques reviewed previously: improved retrieval of spatial information, improved retrieval of chemical information, addressing the limited statistical relevance of analysis on paint microsamples, and integration of computational methods in the processing of microchemical data.
2 Infrared-based Methods
Starting at the lower-end of the energy range, infrared-based analysis is primarily used for the characterization of the organic phase of paint systems. Especially in the mid-infrared (mid-IR) range (2.5–25 μm, 400–4000 cm−1), most functional groups exhibit fundamental vibrational modes. Since each fundamental mode has a narrow corresponding absorption feature, mid-IR absorption spectroscopy has a very high chemical specificity. This property has made mid-IR spectroscopy one of the most widely applied methods for the study of oil paint degradation. In the wider context of paint analysis, mid-IR spectroscopy has been used extensively to identify and analyze pigments, additives, binding medium (Meilunas et al. 1990; Shearer et al. 1983), metal carboxylates (Cotte et al. 2007; Gabrieli et al. 2017; Henderson et al. 2019; Keune and Boon 2007; Mass et al. 2013a; Mazzeo et al. 2008; Noble et al. 2002; Osmond 2014; Osmond et al. 2012; Pouyet et al. 2015; Romano et al. 2020; Van der Weerd et al. 2004), as well as environmentally-induced inorganic pigment degradation products (Mass et al. 2013a; Pouyet et al. 2015). Here, we will cover the state-of-the-art of two approaches to microscale mid-IR analysis of oil paints: μ-FTIR and AFM-IR.
2.1 μ-FTIR
-
A detailed description of the μ-FTIR technique and its various measuring modalities can be found in ref. (Salzer and Siesler 2009).
Fourier-transform infrared microscopy (μ-FTIR) is a technique in which an FTIR spectrometer is coupled to a microscope so as to obtain full mid-IR absorption spectra either on single points (using a single-element detector) or on many points at once (using array detectors). Although the high chemical specificity and sensitivity to the organic phase makes μ-FTIR a suitable technique for studies of oil paint degradation, its use of mid-IR radiation inherently comes with restrictions in terms of spatial resolution. Even in the most ideal optical set-up, the spatial resolution of conventional far-field μ-FTIR is limited by the diffraction limit according to:
Where d is the minimum distance between adjacent features where both features can still be resolved, λ is the wavelength of the used electromagnetic radiation, and NA is the numerical aperture of the optical system. In transmission μ-FTIR experiment, lenses with numerical apertures of 0.3–0.8 are most common, meaning features smaller than 10 μm can typically not be resolved. Obviously, for oil paint samples with a characteristically high degree of heterogeneity, this puts significant limits on the types of phenomena that can be studied.
As a means to improve the applicability of μ-FTIR for studies of oil paint composition and degradation, one instrumental development has gained particular popularity. Attenuated total reflectance (ATR) is a measurement geometry that is somewhat comparable to specular reflectance (see Fig. 12.1), but makes use of a high refractive index crystal that is pressed onto the sample. Rather than reflecting directly on the sample surface, the mid-IR beam reflects internally on the crystal surface pressed onto the sample. In the process of reflecting internally on the crystal-sample interface, parts of the electromagnetic wave penetrate several micrometers into the sample (known as an evanescent wave). The reflected light will therefore be partially absorbed by the sample, as would be the case in a specular reflectance geometry. As an ATR crystal, germanium is typically used. Besides being IR-transparent, germanium possesses a refractive index of around 4.0 in the mid-IR spectral range. Due to the proportionality between numerical aperture and refractive index, substantial improvements can be made in terms of spatial resolution. Although this value depends on the wavelength, detector, and other optics used in the set-up, the spatial resolution of μ-ATR-FTIR with a germanium ATR crystal is thought to be in the order of several μm.

The three common measurement geometries in μ-FTIR using a conventional (reflection) Schwarzschild objective. From left to right: transmission, specular reflectance and ATR. The red arrows indicate the direction of the mid-IR beam
Two of the earliest applications of μ-ATR-FTIR for microchemical studies of oil paint were reported by Spring et al. (2008) and Mazzeo et al. (2007). Both on historical cross-sections and model samples they show the ability to identify and map various metal carboxylate species. More recently, Gabrieli et al. (2017) showed how μ-ATR-FTIR allows to resolve the morphology of zinc soap agglomerates and their formation relative to particles of the jellifying agent aluminum stearate. Maps of ionomeric zinc carboxylates, zinc soaps, aluminum stearate, and corresponding FTIR spectra are shown in Fig. 12.2.

μ-FTIR data recorded in ATR geometry on a sample obtained from Jackson Pollock’s Alchemy. The four FTIR spectra were recorded on locations indicated by numbers 1–4 in the heatmaps, representing the distributions of ionomeric zinc carboxylates (1590 cm−1 broad), crystalline zinc carboxylates (1540 cm−1), and aluminum stearate (1588 cm−1). The black scale bars correspond to 10 μm. Reprinted with permission from ref. (Gabrieli et al. 2017)
For studies in which a high signal-to-noise ratio (SNR) is of primary concern, the use of synchrotron-generated IR beams is a development that has gained particular attention the last 10–15 years. Although mid-IR synchrotron beams are not necessarily brighter than the regular Globar IR sources, their low divergence make that the actual photon flux delivered to the focal volume is substantially higher (Cotte et al. 2007). While the most significant benefits are obtained when working in a confocal configuration with diffraction-limit-sized apertures and single-element mercury cadmium telluride (MCT) detectors (Cotte et al. 2007; Levenson et al. 2006; Osmond 2014; Osmond et al. 2012; Pouyet et al. 2015; Schiering et al. 2000), synchrotron radiation has also been used as an IR source for studies of oil paint degradation in external reflection and transmission geometries (see Fig. 12.1) using 2D focal plane array (FPA) detectors (Henderson et al. 2019; Kidder et al. 1997; Mass et al. 2013a).
Another development in the use of μ-FTIR applied to studies of oil paint is the use of chemical derivatization of the sample surface to separate otherwise overlapping absorption features. This approach was first reported in the context of degraded oil paint by Zumbühl et al. (Zumbühl et al. 2014), who demonstrate how exposure of polished cross-sections to gaseous sulfur tetrafluoride (SF4) can for instance help in distinguishing absorption features coming from non-hydrolyzed triglyceride esters with those coming from free fatty acids and metal carboxylates. In addition, it has been shown that differences in the sensitivity of different metal carboxylates to SF4 exposure allows to readily differentiate metal oxalates from other metal carboxylate species.
2.2 AFM-IR
-
A detailed description of the AFM-IR technique can be found in ref. (Dazzi et al. 2005), while a schematic illustration of the technique is shown in Fig. 12.3.

Schematic representation of the AFM-IR technique. A pulsed mid-IR laser is focused onto the sample and directed to coincide with the apex of the AFM cantilever. Absorption of mid-IR pulses by the sample induces transient or resonant oscillations of the AFM cantilever that are monitored by the deflection of a red laser
A lot of methodological research has been conducted over the years to combine the powerful capabilities of mid-IR microspectroscopy with the ability to perform analysis on length scales smaller than the diffraction limit. A near-field approach that couples atomic force microscopy with mid-IR absorption spectroscopy (AFM-IR) has been demonstrated to be especially powerful (Dazzi et al. 2005; Dazzi and Prater 2017). AFM-IR makes use of an AFM cantilever to probe local thermal expansion induced by a pulsed mid-infrared laser (see Fig. 12.3). Due to the nanometric radius of the apex of the AFM cantilever, field exaltation induced by the tip, and the fast dissipation of thermal energy induced by the laser pulses, lateral resolutions in the order of several tens of nanometers can readily be obtained.
Two studies have been published so far that involve the use of AFM-IR for the study of oil paints. Morsch et al. (2017) demonstrate how AFM-IR can inform on the photocatalytic activity of titanium white (TiO2), showing that radical oxygenation of the oil binder is induced in the direct vicinity of nanometric TiO2 pigment crystallites. Ma et al. (2019) studied the effects of aluminum stearate on the formation of zinc soaps in naturally aged paint-outs. Resolving the distributions of zinc carboxylates around a micrometric aluminum stearate agglomerate informs on how it acts as a local source of free fatty acids, hereby fostering the growth of zinc stearate crystallites.
As Ma et al. studied the same systems as Gabrieli et al. (2017), but used AFM-IR instead of μ-FTIR, it is interesting to note the different types of information that can be obtained at different length scales. A false-color image showing the distribution of crystalline zinc soaps and glycerol esters around an aluminum stearate agglomerate is shown in Fig. 12.4. It is clear from resolving the zinc soap distributions at the nanoscale that the formation of zinc soap crystallites at the rim of the aluminum stearate mass only constitutes a small fraction of all saponification. Moreover, it can be seen that the distribution of the zinc soap crystallites does not exhibit a clear (anti-) correlation with the distribution of non-hydrolyzed triglyceride esters. Both observations imply an important role for the diffusion of free fatty acids in the saponification of oil paints; something that could not be concluded from the distributions resolved at micrometric resolution (Fig. 12.2). On the other hand, the very small field of view of the AFM-IR maps means that certain trends and patterns cannot be observed. Whereas it is clear from Fig. 12.2 that in the Alchemy sample zinc soap formation is fostered as much as 20 μm away from the aluminum stearate mass, but not beyond, the occurrence of this phenomenon in the naturally aged paint-outs could not be confirmed due to the much smaller scale of analysis.

False-color AFM-IR images obtained on a thin section of a naturally aged titanium white/zinc white paint-out. Colors were assigned as follows: crystalline zinc carboxylates (1540 cm−1, red), ionomeric zinc carboxylates plus aluminum stearate (1590 cm−1, green), glycerol esters (1742 cm−1, blue). The yellow rectangle on the left indicates where the high-resolution map on the right was recorded. Reprinted from (Ma et al. 2019)
It is important to note that AFM-IR is still heavily under development, with different instrumental approaches rapidly succeeding one another. Comparing the two examples mentioned in the previous paragraph, the approach taken by Morsch et al. involves an optical parametric oscillator (OPO) light source pulsed at 1 kHz, which induces a transient resonance in the AFM cantilever, while Ma et al. operate the AFM-IR with a high-frequency heterodyne detection scheme and a quantum cascade laser (QCL) light source (Dazzi et al. 2015). Moreover, Morsch et al. report the operation of the AFM in contact mode, working on paint films cut straight from the substrate, while Ma et al. operate in tapping mode, working on samples cut with an ultramicrotome to a thickness of around 200 nm. Although high-frequency heterodyne detection and samples prepared as thin sections are thought to provide significant benefit in terms of measurement speed and spatial resolution, a lot remains as of yet unknown about the exact effects of these parameters on the analysis of degraded paint samples.
3 Methods Based on UV and Visible Light
Moving up in energy from the infrared, one reaches the visible and UV regions of the electromagnetic spectrum. In this particular spectral region, two spectroscopic principles have been most widely applied in studies of oil paint: Raman scattering and photoluminescence (PL).
3.1 PL Microimaging
-
More detailed descriptions of the various aspects of PL microspectroscopy and microimaging specifically applied to cultural heritage samples can be found in refs. (Thoury et al. 2011, Nevin et al. 2014)
PL is a process in which a compound absorbs a photon of a given energy (excitation) and then emits a photon of a lower energy (emission). Although the macroscopic use of PL for the study of heritage objects has been reported as far back as the late 1920s and early 1930s (Rorimer 1929, 1931), it has only been over the last 10 years that a wide range of PL-based methods have started to become widely used as microanalytical tools for studies of oil paint degradation. In most common modern PL microimaging set-ups, a sample area of interest is illuminated with a defocused UV beam, the emitted PL is magnified by an optical objective, and full-field images are captured through one or more emission bandpass filters. This multispectral approach has become commonplace, because PL emission spectra of most materials of interest exhibit relatively broad bands that can often be selectively imaged even with relatively broad emission bandpass filters (see Fig. 12.5). Due to the use of visible and UV photons, PL microimaging has the ability to routinely achieve submicrometric spatial resolutions. Chemical specificity can be achieved along no less than three dimensions (absorption wavelength, emission wavelength, and emission lifetime).

Schematic representation of the three main approaches to PL microanalysis. The bottom left shows a high-resolution emission spectrum retrieved from a hyperspectral map recorded on a zinc white oil paint microsample that was excited at 280 nm. The figures shown in grey frames represent full-field images recorded through emission band pass filters of which the spectral ranges are indicated in the full emission spectrum with grey rectangles. The figures shown in blue frames represent full-field images taken through one emission bandpass filter at different time delays. NB: PL images are for illustration purposes only and have not necessarily been obtained using the indicated parameters
Two research themes have particularly benefited from the contribution of microchemical imaging by PL: the study of semiconductor pigments and that of metal soap formation. Semiconductor pigments are often brightly luminescing under suitable excitation and exhibit emission properties informative both about their characteristic band gap and intrinsic crystal defects. The hypothesized relation between crystal defects and the (photo-) catalytic activity of semiconductor pigments such as zinc white and cadmium yellow (CdS) make the identification of these defects of especially high interest in the context of oil paint degradation (Comelli et al. 2019; Drouilly et al. 2013; French et al. 2001; Hageraats et al. 2019b; Kurtz et al. 2005). As was discussed in Sect. 12.2.2, metal soap formation is usually studied using techniques based on absorption in the mid-IR, where metal carboxylates have multiple distinct and characteristic absorption features. However, due to the intrinsically limited spatial resolution of conventional μ-FTIR, there is significant interest to complement μ-FTIR observations of metal soaps with PL microimaging, which works with ultraviolet and visible light and can therefore produce images at submicrometric spatial resolution.
Notable recent additions to the PL microimaging toolbox include methods based on excitation in the deep-UV regime (deep-UV PL) and methods based on resolving the emission dynamics over time (time-resolved PL, see Fig. 12.5). Deep-UV excited PL microspectroscopy and microimaging have been shown particularly useful for studies of the brightly luminescing semiconductor pigment zinc white and its zinc carboxylate alteration products. With its characteristic band gap emission centered around 380 nm and the various defect-related emissions at longer wavelengths (Bertrand et al. 2013a; Zhang et al. 2019), the PL-based study of degraded zinc white oil paints has been shown to benefit from excitation around 280 nm. As zinc white does not luminescence at energies higher than its bandgap, excitation at 280 nm opens up a significant spectral region (280–380 nm) without spectral interference from the brightly luminescing zinc white pigment. To illustrate this principle, Fig. 12.5 shows the average PL emission spectrum of a degraded zinc white oil paint microsample, recorded with an excitation energy of 280 nm. Whereas the spectral region above 380 nm is dominated by zinc white emission, the spectrum shows a clear low-intensity emission band around 340 nm which is thought to be emitted by crystalline zinc soaps (Hageraats et al. 2019a; Van Loon et al. 2019; Thoury et al. 2019). According to a previous evaluation of the optical system used in these studies, this method provides chemically specific analysis of zinc white degradation products close to the diffraction limit of about 150 nm (Bertrand et al. 2013a).
Particularly interesting results have been reported using time-resolved PL microspectroscopy and microimaging (also known as fluorescence lifetime imaging or FLIM) to study of the semiconductor pigments cadmium yellow and zinc white. Besides differing in emission wavelength, band gap and defect-related emissions can also be distinguished based on their emission lifetime, with band gap emission typically occurring in the ps or ns regime, and defect-related emission typically occurring in the μs regime. This principle has been exploited by Comelli et al. (2017) and Artesani et al. (2019) in studies combining multispectral PL microimaging with time-gated detection to aid the identification of pigments in chemically complex historical samples. It has been shown that the measurement of PL lifetimes can inform on the chemical interaction of the reactive pigment with the organic binding medium (Artesani et al. 2017, 2018). However, the retrieval of such information in the context of a microchemical imaging study has not yet been reported.
3.2 Raman Microspectroscopy
-
A more detailed account of Raman (micro-)spectroscopy applied to studies of oil paint degradation and authentication can be found in Chap. 13 . This section therefore presents only a concise summary of the technique and its applications in microscopic studies of oil paint degradation.
Raman scattering is an inelastic scattering phenomenon in which a scattered visible or near-IR photon loses an amount of energy corresponding to a vibrational transition in the scattering molecule. Following this principle, Raman scattering (like mid-IR absorption) can therefore be used to probe vibrational transitions. Raman spectroscopy experiments are performed by focusing a laser (with a wavelength of typically 500–800 nm) onto the sample and recording a high-resolution spectrum of the scattered light (Opilik et al. 2013). Besides an elastic scattering peak and possible broad luminescence bands, Raman spectra contain a series of narrow peaks with a small energy offset relative to the laser energy. The offset of each peak then corresponds to the energies of certain vibrational transitions that are expressed in terms of a Raman shift.
Within studies of oil paint, Raman scattering has mostly been applied in microspectroscopy experiments where only one or several spatial points are analyzed (Chen-Wiegart et al. 2017; Higuchi et al. 1997; Keune et al. 2013; Mahon et al. 2020; Monico et al. 2011, 2013, 2014; Otero et al. 2014; Spring and Grout 2002; Trentelman et al. 1996). In some studies, where resolving spatial distributions is of crucial importance, raster scans are performed (Cato et al. 2017; Lau et al. 2008; Monico et al. 2015b, 2020a; Ropret et al. 2010). This yields hyperspectral Raman spectroscopy maps with micrometric spatial resolution, from which specific features can be integrated to obtain chemically specific distribution images on the microscale. Monico et al. (2015b, 2020a) show how this approach can be used to map out different types of chrome yellows and to detect poorly crystalline, hexagonal CdS and some of its degradation products. Cato et al. (2017) show how mapping a specific vibration of the blue S•3− radical anion chromophore over an ultramarine oil paint surface reveals a local decrease in the chromophore concentration in optically altered regions: implying that the oxidation of sulfur chromophores may have caused ultramarine discoloration.
4 Methods Based on X-Rays
The X-ray regime is very broad in terms of energy range and provides a variety of analytical methods to study oil paint degradation. Photons on the lower end of this energy range—spanning roughly from 100 eV to 1 keV—are collectively referred to as soft X-rays, while photons on the upper end of the energy range (>5 keV) are typically referred to as hard X-rays. The intermediate range (1–5 keV) is sometimes colloquially called the tender X-ray regime. Applications in each energy range largely depend on the penetration depth in the sample of interest. Generally speaking, the higher the X-ray photon energy, the greater the attenuation depth, and the heavier the element, the shorter the attenuation depth.
All the X-ray based studies reviewed in this following section were conducted at synchrotron facilities. Synchrotron-based light sources offer low-divergence beams of an extremely high brightness that can be over 10 orders of magnitude greater than that of conventional X-ray tube sources. These properties make them particularly suitable for use with tunable monochromators and focusing optics. Although the high energy (and therefore short wavelength) of X-rays makes that the diffraction limit at most energies is subnanometric, their focusing is generally technically challenging due to the small difference in refractive index between air (or vacuum) and condensed matter and the very small critical angle for total external reflection (Ice et al. 2011). Still, a number of techniques were developed over the years that address these physical limitations in different ways. The four most commonly applied focusing optics for microscopic X-ray experiments are Kirkpatrick-Baez (KB) mirrors (Kirkpatrick and Baez 1948), Fresnel zone plates (FZP) (Rösner et al. 2018), compound refractive lenses (CRL), and X-ray capillary optics (illustrated in Fig. 12.6). Depending on the type of optics, design, X-ray energy, and beam divergence of the beamline, beam diameters can be obtained in the order of tens of micrometers (capillary optics) down to several nanometers (FZP).

Schematic representations of the four main X-ray focusing optics
-
A detailed review of X-ray focusing optics can be found in ref. (Ice et al. 2011), while an overview of applications of synchrotron techniques for the study of ancient materials can be found in ref. (Bertrand et al. 2012).
For microchemical imaging applications, X-rays are primarily used to probe core-level transitions and to perform crystal diffraction. Probing core-level transitions, chemically-specific information can be obtained by making use of either of two dependencies: the element-dependency of core-level transition energies, or the dependency of the density-of-states (DOS) of core-level transitions to the oxidation state and to the coordination environment of the target atom. X-ray fluorescence spectrometry (XRF) uses this element-dependency of core-level transition energies, whereas the dependency of the DOS of core-level transitions to the oxidation state and coordination environment is exploited in X-ray absorption spectroscopies (XAS). While XRF is especially useful for elemental mapping, XAS has its main applications in determining oxidation states, coordination numbers, and the bond lengths for the first few coordination shells. Microfocused X-ray beams can also be used for crystal diffraction experiments (X-ray diffraction, XRD) to determine the crystalline phases present in a sample of interest.
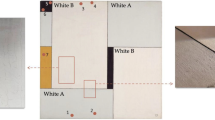
The principles and state-of-the-art of μ-XRF, μ-XAS, and μ-XRD as applied to oil paint research are discussed in the following three sub-sections. In addition, to demonstrate the capabilities and complementarity of these three techniques, Figs. 12.8, 12.9, and 12.11 show the X-ray based chemical microimaging of an extraordinarily thick (~3 mm) stratigraphy of historical white oil paints, taken from the window frame of a historical home (De Witte Roos) in Delft, The Netherlands (see Fig. 12.7). These wooden frames have been repainted regularly over the past three centuries, as can be seen from the ~50 yellowish/whitish paint layers composing the stratigraphy (Fig. 12.7b. c). This sample therefore presents the recording of most of the commonly used white pigments over the past ~300 years. Besides, it can provide relevant information about their long-term stability and degradation. Therefore, its analysis established a record biography of a painting fragment. After embedding the paint fragment in resin, it was prepared as a cross-section (Fig. 12.7b) and as a microtomed 15 μm thin section (Fig. 12.7c).

(a) De Witte Roos in Delft. (b and c) Optical microscopy image of a paint fragment prepared as a polished cross-section and as a thin section, respectively. The most ancient (yellowish) paint layers are at the bottom while the most recent (whiter) layers are at the top of the section
4.1 μ-XRF
-
A description of the principle of XRF can be found in ref. (Mantler and Schreiner 2000), while a more detailed account of the instrumental aspects of modern μ-XRF instrumentation can be found in ref. (Cotte et al. 2017b).
The ability of μ-XRF to map elemental distributions makes the technique particularly suitable to map pigment distributions in oil paint cross-sections. μ-XRF usually requires microfocused synchrotron-generated X-ray beams, making this technique less accessible than its electron-based counterpart (SEM-EDX) which is routinely used for mapping pigments. μ-XRF is mostly applied to studies of oil paint systems when sensitivity to certain (low concentration) elements is of concern or when other forms of synchrotron-based X-ray microanalysis are involved and μ-XRF can be performed for parallel characterization of the sample at the same location using the same instrumentation and set-up (Chen-Wiegart et al. 2017; Cotte et al. 2008; Cotte and Susini 2009; Van Der Snickt et al. 2009, 2012). Some research findings that were obtained through μ-XRF include the role played by chlorine contamination in the blackening of cinnabar (HgS) (Cotte et al. 2008; Cotte and Susini 2009; Radepont et al. 2011), the formation of a cadmium-sulfur degradation compound on the surface of cadmium yellow paints (Van Der Snickt et al. 2009, 2012), and the disintegration of lead tin yellow to form lead soap aggregates (Chen-Wiegart et al. 2017). Combined with macro- X-ray imaging techniques on the entire painting itself (see Chap. 11), μXRF can be used to distinguish originally paint layers from repainted layers in micro-samples, and help in reorienting and optimizing the conservation strategy, as recently done during the restoration of van Eyck’s renowned Ghent Altarpiece (Van der Snickt et al. 2017).
State-of-the-art applications of μ-XRF to studies of oil paint include the simultaneous recording of μ-XRF and μ-XRD by making use of fluorescence and transmission geometry respectively (Cotte et al. 2008; Van Der Snickt et al. 2012), and the ability to perform hard X-ray μ-XRF at spatial resolutions as high as 200 nm (Casadio and Rose 2013; Cotte et al. 2008). More routinely, beam sizes in the order of one to a few μm are being used, with μ-XRF experiments typically being combined with μ-XAS—enabled by the tunability of synchrotron-generated X-ray beams and the shared ability to record data in fluorescence geometry.
The application of the μ-XRF technique to the Delft sample is shown in Fig. 12.8. Two maps were acquired at 7.2 keV (above the Fe K-edge), one covering the older, yellowish paint layers and one covering the more recent, whiter, upper paint layers. The elemental maps show that in the older paint layers, lead is the main element, with some layers containing calcium, barium, and sulfur. The finding of lead in the older paint layers is in line with the common use of lead white in that time period, and based on similar knowledge of pigment use in modern times, the finding of titanium in the upper paint layers can for instance be hypothesized to be due to the pigment titanium white. However, in these cases, μ-XRF data cannot provide a definitive identification, for which one relies on techniques with a higher chemical specificity. This is also the case for a number of other μ-XRF observations—such as the finding of barium and titanium in some of the oldest paint layers and the presence of many thin layers of sulfur—for which more precise identification is necessary in order to determine their origin.

μ-XRF data obtained on a cross-section of the Delft sample. (a) Optical microscopy image, with the red and black rectangles indicating the recent and ancient paint layers respectively. (b) μ-XRF maps of Si, S, Ca, Ti, Ba, and Pb. (c) Fits to the raw data using known energies of K, L, and M emission lines. Data was obtained at ID21, ESRF (Cotte et al. 2017b), using an X-ray beam tuned to 7.2 keV and focused down to 0.5 × 1.0 μm using a Kirkpatrick-Baez mirror system (see Fig. 12.6).
4.2 μ-XAS
-
Detailed information about the methodological and theoretical principles behind XAS can be found in refs. (Penner-Hahn 1999) and (Rehr and Albers 2000).
μ-XAS refers to a set of techniques that aim to extract chemical information from the spectral structure around and above elemental absorption edges. Since the collection of X-ray absorption spectra requires extensive tunability and high monochromaticity of the X-ray beam, it is still today essentially a synchrotron-based technique, particularly for heterogenous complex systems as encountered in paint composition and alteration studies. XAS is performed in two distinct energy ranges: X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). XANES focuses on the energy range close to the absorption edge—containing the discrete orbital-to-orbital transition features—and is particularly useful for probing oxidation states and identifying compounds. EXAFS focuses on the higher energy range that corresponds to core-shell-to-continuum transitions. It can be used to extract precise quantitative measurements of the average coordination numbers and bond lengths. As EXAFS has as of yet not been shown in a microchemical imaging application in the context of oil paint degradation studies, this section will be focused on μ-XANES, while the prospects of the capabilities of μ-EXAFS imaging are discussed in Sect. 12.6.
The capability of XANES to probe oxidation states of elements makes μ-XANES a particularly powerful technique to study (mostly) environmentally induced degradation processes of inorganic pigments on the microscale. Some notable examples of results obtained through μ-XANES on degradation oil paint systems include the identification of cinnabar degradation products (Cotte and Susini, 2009), the identification of oxidative cadmium yellow degradation products (Van Der Snickt et al. 2009, 2012), the elucidation of the various factors that affect the reduction of lead chromate (Monico et al. 2011, 2015a, 2016), and the characterization of the degradation-migration pathways of realgar (As4S4), orpiment (As2S3), and emerald green (3Cu(AsO2)2·Cu(CH3COO)2) (Keune et al. 2015, 2016). Besides degradation studies, μ-XANES can also be used to distinguish different qualities. It was used for example to identify a marker for the preparation method (in particular heat treatment) of ultramarine pigment from lapis lazuli in historical paints (Gambardella et al. 2020).
One of the primary goals of μ-XANES analysis on oil paints is to resolve spatial distributions of the different components, in particular degradation products relative to the original pigment and possible other reactants. Two main approaches are currently taken, depending on the chemical species under study and the available instrumentation. The first approach is sometimes called chemical state (or speciation) mapping and works particularly well when absorption edges exhibit markedly different spectral features. Chemical state mapping involves the recording of a small number (2–4) of XRF maps at different excitation energies (see also examples below). When the excitation energies are chosen so as to correspond to XANES absorption features that are specific for certain compounds or oxidation states, chemical maps can be reconstructed that specifically show the distribution of one compound or the other (Monico et al. 2015a, 2016; Pouyet et al. 2015; Radepont et al. 2011; Van Der Snickt et al. 2009, 2012).
The second approach is based on recent developments in detector technology and involves the recording of hyperspectral μ-XANES maps and subsequent fitting with reference spectra. This approach is particularly useful when the X-ray absorption behavior of the species of interest is more subtly different, or when complex mixtures of compounds are present. Using single-element X-ray detectors, hyperspectral maps can only be recorded by raster scanning the same area many times for different excitation energies. With conventional detector technology, this approach is only feasible for low-definition images. One demonstrated solution involves the use of the recently developed new-generation Maia detector: an annular X-ray detector with a much larger solid angle that enables faster raster scanning of samples and has been used to record hyperspectral XANES maps to study chrome yellow degradation (Monico et al. 2015a). The second solution is to employ a full-field imaging approach using scintillators and very sensitive CCD or CMOS cameras. Megapixel hyperspectral images can be recorded at once, circumventing time-costly raster scanning and the need for tightly focused X-ray beams. This principle has been demonstrated for the study of the degradation of cadmium yellow and for the characterization of sulfur chemistry in ultramarine pigments as a function of their post-synthesis treatments, mentioned above (Gambardella et al. 2020; Pouyet et al. 2015).
The application of the μ-XANES technique to the Delft sample is shown in Fig. 12.9. The goal of these experiments was to determine more precisely the identity of the sulfur species, in particular in the many thin layers observed using μ-XRF (see Fig. 12.8). First, a μ-XRF map was acquired at 2.55 keV (i.e., above the S K-edge and Pb M5-edge) to localize lead and the thin sulfur layers. 51 XANES spectra were then acquired at the maximum XRF emission intensity of each of these S layers. These spectra could be grouped into four main classes and averaged (Fig. 12.9c). They reveal different sulfur species, ranging from reduced sulfur (sulfide, S2−, here preliminary attributed to ZnS), to more oxidized forms (sulfite and sulfate, S4+ and S6+). The reference spectra of three sulfates (PbSO4, CaSO4.2H2O and BaSO4) show that they all have a maximum absorption at a similar energy position (around 2.482 keV), but differ by their fine structure. Comparing the four experimental XANES with these five reference spectra, experimental spectrum 1 can be hypothesized to have been recorded on a mixture of CaSO4⋅2H2O and a sulfite, spectrum 2 on a mixture of PbSO4 and a sulfite, spectrum 3 on BaSO4, and spectrum 4 on a mixture of ZnS and BaSO4. Based on these spectra, four energies were chosen to map these different species: Sa = 2.4736 keV (maximum absorption for ZnS), Sb = 2.477 keV (maximum absorption for sulfite), Sc = 2.481 keV (shoulder in the PbSO4 spectrum) and Sd = 2.4822 keV (maximum absorption for all sulfates). From these maps and the experimental S K-edge XANES spectra, it could be concluded that the top paint layers contain sulfur in the forms of ZnS and BaSO4, suggesting the use of the white pigment lithopone. Furthermore, μ-XANES analyses of the thin sulfur-rich layers reveal a mixture of sulfites and sulfates. The sulfates in the layers found among lead-pigmented layers were found to most closely resemble anglesite (PbSO4), whereas those found on top of the calcium-rich layer more closely resemble gypsum (CaSO4⋅2H2O). Although these compounds have been used as paint components in the past (anglesite as Flemish white and gypsum as an extender), here, their mixture with sulfites and their systematic distribution as thin layers alternated with lead white layers suggests a degradative origin; likely due to prolonged exposure to atmospheric pollutants, such as SO2.

μ-XANES data obtained on the Delft sample. (a) Elemental map of Pb as a reference for the distribution of lead pigments (acquired at 2.55 keV, above Pb M5-edge). (b) XRF S-K maps recorded at the four absorption features identified in (c). (c) The bottom four spectra show averages of S K-edge XANES acquired at 51 points, represented as colored dots in (b) (#1–4), while the top five spectra shown S K-edge XANES from five reference compounds (grey). Data was obtained at ID21, ESRF, using an X-ray beam focused down to 0.5 μm × 1.0 μm using a Kirkpatrick-Baez mirror system (see Fig. 12.6).
4.3 μ-XRD
-
A comprehensive review of both the instrumental and theoretical aspects of XRD and μ-XRD can be found in refs. (Giacovazzo et al. 2009; Gonzalez et al. 2020). The application of μ-XRD to cultural heritage in general has been reviewed previously (Gonzalez et al. 2020).
When scattered elastically off a material with crystalline properties, X-rays form diffraction patterns that are highly characteristic of the chemical make-up of the material and its various crystalline phases. X-ray diffractograms are recorded by irradiating a crystalline sample with a monochromatic X-ray beam and recording the intensity of reflected X-rays as a function of the outgoing angle. According to Bragg’s law, positive interference occurs for specific angles θ, under the condition that the following relation holds:
Here, d is the spacing between certain crystallographic planes, λ is the wavelength of the X-ray beam, and n is an integer that corresponds to the order of the interference maximum. X-ray diffractograms typically consist of many (>10) narrow peaks, making the technique highly chemically specific for the analysis of crystalline compounds.
Common applications of μ-XRD in studies of oil paint include the identification and mapping of crystalline metal carboxylates and inorganic degradation products (Cotte et al. 2008; Radepont et al. 2011; Salvadó et al. 2009; Simoen et al. 2019; Van Der Snickt et al. 2009, 2012), and the characterization of certain pigments (Cotte et al. 2008; Gonzalez et al. 2017b; Monico et al. 2013; Welcomme et al. 2007). In currently reported μ-XRD analyses of oil paint, both mapping and characterization experiments are performed in transmission geometry. For mapping, the use of a transmission geometry (as compared to reflection geometry) reduces the on-sample lateral spot size of microfocused X-ray beams and prevents shadowing of Debye-Scherrer rings, especially at small diffraction angles (Welcomme et al. 2007).
One approach to recording transmission geometry μ-XRD maps is currently shown to be particularly useful for applications in oil paint analysis. In this approach, the sample is raster scanned under an X-ray microbeam and diffraction patterns are imaged in full-field using a scintillator in combination with a 2D CCD (Cotte and Susini 2009; Gonzalez et al. 2017b; Radepont et al. 2011; Van Der Snickt et al. 2009, 2012). Compared to the more conventional way of recording powder X-ray diffractograms, in which an angular range is scanned using a single-element X-ray detector, the full-field approach is substantially faster, allowing many points to be analyzed in a relatively short period of time. Some of the previously referenced papers show how it allows the imaging of oxidation products of cadmium yellow (Cotte and Susini 2009; Van Der Snickt et al. 2009), lead sulfates, copper oxalates, and copper hydroxyl chlorides (Mantler and Schreiner 2000), different grades of lead white (Welcomme et al. 2007), and degradation products of vermillion (Cotte and Susini 2009).
A second approach that is particularly noteworthy in discussing the state-of-the-art of μ-XRD analysis of oil paints is X-ray diffraction tomography. Reported first in this context by Vanmeert et al. (2015), the technique is used to preserve the depth information that is normally lost when recording diffraction data in transmission. As reported by Vanmeert et al. and later by Price et al. (2019), it works by repeatedly scanning a line of the microsample with a microfocused X-ray beam and recording diffractograms in transmission with a 2D scintillator-CMOS or hybrid photon counting (HPC) detector system. After each repetition the sample is rotated by a few degrees, such that a rotation of 180° or 360° is reached after about 100 successive rotational steps. After reconstruction with a filtered back-projection (FBP) or maximum-likelihood expectation-maximization (MLEM) algorithm, a virtual 2D cross-section is obtained with a full X-ray diffractogram for each pixel. The spatial resolution of this technique is determined by the diameter of the X-ray beam and the number of rotational steps and is in the order of one to a few μm (Vanmeert et al. 2015; Price et al. 2019). A comparison of 2D XRD tomograms and a regular μ-XRD map recorded on a pustular mass from a painting by Vincent van Gogh is shown in Fig. 12.10. Here, it can be seen that voids in the distribution of crystalline material have formed in the pustular mass—something that cannot be observed in the regular 2D μ-XRD map.

2D μ-XRD mapping (a) versus 2D XRD tomography (b) of a pustular mass sampled from Vincent van Gogh’s Wheat Stack Under a Cloudy Sky (Kröller-Müller Museum). The two 2D tomograms show identical virtual cross-sections with the green color in the bottom tomogram representing a plumbonacrite phase. The white arrows indicate the voids in the distribution of crystalline material. Reprinted from ref. (Vanmeert et al. 2015)
The application of the μ-XRD technique to the Delft sample is shown in Fig. 12.11. The diffraction data was recorded on a microtomed 15 μm thin section in a transmission geometry (Fig. 12.7c) in order to have good control of the probed volume and optimize the absorption and diffraction intensity. Having access to full X-ray diffractograms for each pixel, it is now possible to unambiguously identify all crystalline compounds present throughout the 50+ different paint layers. Hydrocerussite and cerussite are both detected in the lead-pigmented layers in various ratios, indicating the presence of lead white produced using different manufacturing processes or having undergone different post-synthesis treatments (Gonzalez et al. 2017b). In the more recently applied paint layers towards the top of the sample, zincite (ZnO) is detected, indicating the use of the pigment zinc white, introduced in the first half of the nineteenth century. Here, alternating zinc white and lead white layers show the gradual progression in the use of different white pigments. TiO2 (rutile) is the main component of five recent layers, separated by one ZnS/BaSO4 layer, indicating the use of the modern pigment titanium white and lithopone respectively. In addition, a lead titanate (PbTiO3) phase was observed in some of the upper titanium white paint layers. The use of lead titanate as a highly durable pigment for use in protective paint has been proposed in the 1930s, but has never found widespread use, reportedly due to its high cost of production (Robertson 1936). In terms of (possible) degradation products, anglesite could now be identified with a high degree of certainty, and its distribution mapped with respect to the many lead white paint layers. Gypsum is also confirmed at the surface of the early calcite layers.

μ-XRD data obtained on the thin section of the Delft sample. (a) False color image showing the distribution of different crystalline phases, calculated by integrating the XRD intensity at specific diffraction peaks, indicated by colored asterisks in (b). (b) Diffractograms obtained by averaging XRD patterns over pixels exhibiting corresponding colors in the phase maps. Data was obtained at ID13, ESRF, using an X-ray beam tuned to 13 keV and focused down to 1.5 μm × 2.2 μm using a compound refractive lens (see Fig. 12.6).
Comparing the information obtained from μ-XRD with the information obtained using μ-XRF and μ-XANES, it is clear that μ-XRD is particularly powerful for unambiguously identifying pigments and crystalline degradation products. On the other hand, only μ-XANES appears to be able to show the presence of sulfite species in the bottom lead white layers, most likely because this phase is (mostly) amorphous. Overall, for studies of oil paint, the choice of (X-ray) based imaging method strongly depends on the research question, the sample composition, as well as on constraints with regards to sample preparation.
5 Methods Based on Charged Particle Beams
The two main principles involving charged particles beams that are applied to the study of paint are electron scattering and secondary ion generation. Electron scattering forms the basis for electron microscopy and the commonly integrated energy dispersive X-ray spectrometry, while secondary ion generation forms the basis for secondary ion mass spectrometry (SIMS). In the following sections three different techniques will be discussed: scanning electron microscopy energy-dispersive X-ray spectrometry (SEM-EDX), transmission electron microscopy (TEM), and imaging SIMS.
5.1 SEM-EDX
-
A comprehensive overview of the instrumental aspects of SEM-EDX can be found in ref. (Goldstein et al. 2003).
SEM-EDX works with a tightly focused electron beam that is scattered off the analyzed sample and subsequently detected using an electron-sensitive detector. For imaging purposes, the most common approach is to detect the elastically backscattered electrons—analogous to the use of reflection geometry in optical microscopy. As compared to photon-based imaging methods, SEM has a clear advantage in terms of spatial resolution. While the diffraction limit of IR, visible, and UV photons and the physically limited focusability of X-rays pose substantial challenges for photon-based imaging at the nanoscale, electron beams can readily be focused down to only a few nanometers to obtain images of comparable spatial resolution.
Besides scattering elastically on the sample surface and inelastically generating secondary electrons, the focused electron beam also induces the ejection of core electrons from the measurement volume. This process is similar to the X-ray-induced core electron ejection that forms the basis for XRF and likewise results in the emission of characteristic X-rays. In this respect, the elemental mapping capabilities of SEM-EDX are comparable to the capabilities of μ-XRF, with the clear advantage that SEM-EDX does not require synchrotron-generated X-rays. A conscious choice may be made for either one, depending on whether there is an interest in low-concentration elements and whether the study of a sample is already planned to involve the use of synchrotron-generated microfocused X-ray beams (see Sects. 12.4.2 and 12.4.3).
Due to the unique combination of high-resolution imaging and elemental mapping capabilities and the fact that mature lab-based instrumentation has been around for several decades (Goldstein et al. 2003), SEM-EDX has evolved to become one of the primary methods of oil paint microanalysis. Common ways by which SEM-EDX analysis can inform on oil paint composition and degradation is through identifying the elements in degradation-related surface crusts (Van Loon et al. 2011; Mass et al. 2013b; Spring and Grout 2002), visualizing and identifying aggregates and protrusions of degradation products (Gabrieli et al. 2017; Helwig et al. 2014; Keune et al. 2011; Osmond et al. 2013; Romano et al. 2020; Van der Weerd et al. 2004), studying early signs of paint delamination (Helwig et al. 2014; Van Loon et al. 2019; Rogala et al. 2010), and resolving surface texture for studies of water sensitivity, blanching, and ultramarine disease (Burnstock et al. 2006; de la Rie et al. 2017; Mills et al. 2008). Two examples that show the value of the high-resolution imaging capabilities of SEM and the elemental mapping capabilities of EDX are shown in Fig. 12.12. a and b show SEM backscatter images of lead soap aggregates in two distinct stages of development, demonstrating how contrast in electron density can inform on migration and remineralization phenomena in degraded oil paints. Figure 12.12c, d show an example of a 2D EDX raster scan and corresponding SEM backscatter image, recorded on a whole cross-section taken from an oil painting suffering from cracking. Lead white and zinc white can be identified in the top and middle layer respectively, which—due to some discontinuity—are both in contact with a carbon black layer that is painted on top, but cannot be observed clearly using SEM-EDX.

SEM backscatter images of lead soap aggregates in early (a) and later (b) stages of development. (c) SEM backscatter image of a cross-section a painting whose upper layers have suffered from cracking. The white rectangle indicates the region that was scanned using EDX. (d) Overlay of SEM backscatter image and false color EDX map. (a) and (b) reproduced with permission from ref. (Keune et al. 2011). (c) and (d) Reprinted from ref. (Rogala et al. 2010) with the permission of Taylor and Francis Ltd.

TEM-EELS images of chrome yellow before (top row) and after aging (bottom row) under UV-Vis light. The numbers in the top right of each map show the estimated fraction of each compound in the analyzed sample. Reprinted from ref. (Tan et al. 2013) with the permission of Wiley-VCH
One alternative approach to EDX for elemental mapping using the SEM platform is wavelength-dispersive X-ray spectrometry (WDS). This approach is based on the wavelength-dependent diffraction angle of X-rays on a single crystal and actually predates the EDX approach. Although WDS is much slower than EDX due to the mechanical scanning of diffraction angles, its spectral resolution and quantitative capabilities are superior. One example of how SEM-WDS can be used for studies of oil paint degradation was reported by Geldof et al. (2019). Here, WDS is chosen over EDX for the characterization of chrome yellows (PbCr1-xSxO4) in terms of the stoichiometric factor x, due to the strong spectral overlap of the sulfur Kα and lead Mα emission lines.
5.2 TEM
-
A detailed account of the instrumental aspects of TEM and the associated sample preparation can be found in ref. (Williams and Carter 2009).
TEM is based on the imaging of transmitted electrons either by scanning a focused electron beam (scanning transmission electron microscopy, STEM) or by employing a full-field configuration with a 2D electron detector array. Due to complex interplay between higher accelerating voltages (shorter electron wavelength), lower electron densities, and different scattering and absorption processes of the detected electrons, TEM has a spatial resolution that is about an order of magnitude better than conventional SEM. Despite this interesting capability, TEM is much less widely applied than SEM-EDX for the study of oil paint. This can mostly be traced back to one primary reason; since TEM is based on the transmission of electrons, samples need to be extremely thin. Depending on the material, a sample thickness of no more than a few hundred nanometers is required.
In the paint studies, TEM was initially used as an alternative to SEM-EDX for pigment identification (Barba et al. 1995; Papillon et al. 1987; San Andres et al. 1997). In these cases, the reason to choose TEM over SEM-EDX is the ability to perform electron diffraction; providing direct structural information that allows unambiguous identification of paint components. A TEM modality that is of particular interest for studies of oil paint degradation is electron energy loss spectroscopy (EELS). EELS measures the loss in energy of transmitted electrons using an electron spectrometer. These energy losses tend to correspond to core-level electronic transitions in a way that is conceptually similar to XANES. Similar transitions are probed; providing information about the oxidation state and coordination environment of certain elements. Combining STEM and EELS, Casadio et al. (2011) were able to elucidate the chemical process leading to the darkening of zinc yellow (K2O·4ZnCrO4·3H2O) oil paints. Here, EELS works by recording electron losses in resonance with Cr L-edge transitions, providing highly localized information about the oxidation state of chromium. Monico et al. (2011) and Tan et al. (2013) show how the extraordinarily high lateral resolution of TEM-EELS allows to visualize the core-shell structure of degraded chrome yellow particles—identifying up to three different phases based on O K-edge and Cr L2,3-edge speciation. Taken from Tan et al. distribution maps of these three phases obtained through fitting reference spectra to the hyperspectral maps are shown in Fig. 12.12. Comparing the pigment before and after light-induced aging reveals that the PbCrO4 core is reduced to a Cr2O3 shell. Given the nanometric thickness of the reduced shell structure, it is evident that the ability to perform chemically specific analysis at the nanoscale is an important asset in the microchemical imaging toolbox.
The combination of STEM and EDX was demonstrated by Vandivere et al. (2019a, b) to obtain very detailed information on pigmentation in Vermeer’s The Girl with a Pearl Earring. Here, preparation of samples as 200 nm thin sections by means of a focused ion beam (FIB) was shown to drastically improve the spatial resolution of the obtained elemental maps. STEM-EDX was also shown by Van Driel et al. (2018) to be able to perform mapping and identification of different types of inorganic surface coatings and non-stoichiometric surfaces on titanium whites. The ability to resolve coatings and the stoichiometry of titanium white surfaces has previously been hypothesized to be related to the pigment’s photocatalytic properties (Cushing et al. 2017; van Driel et al. 2016; Pan et al. 2013; Zhao et al. 2017), again showing the utility of chemical imaging at the nanoscale for the study of oil paint composition.
Another particularly noteworthy application of TEM to the study of oil paint was recently published by Hermans et al. (2018). They report on the use of TEM to produce 3D reconstructions of nanometric zinc soap agglomerates in degraded zinc white oil paint model systems. Similar to most other tomography methods, transmission images are recorded under a series of tilt angles. From the obtained set of projections, a 3D reconstruction can algorithmically be retrieved. Reconstructions of a single agglomerate show a lamellar structure with a high degree of disorder. The crystalline domains are in the order of several tens of nanometers, with many domains measuring no more than a few unit cells.
5.3 Imaging SIMS
-
A detailed account of the various aspects of imaging SIMS and some other imaging mass spectrometry techniques can be found in ref. 136. (Massonnet and Heeren 2019).
Moving from electrons to ions, imaging SIMS is the last technique to be discussed in this review. It is unique in the context of all other discussed methods, as it derives its chemical specificity from the mass of atoms and molecules. SIMS uses a primary ion beam to generate secondary ions on the surface of a sample. These primary ions are metallic ions or ionic clusters of gallium, indium, gold, or bismuth, while the secondary positive or negative ions consist of sample material (or fragments thereof) and are separated based on their weight-to-charge ratio m/z. Analysis of m/z in SIMS is typically done using either a time-of-flight (TOF) or quadrupole mass analyzer (Dawson 1975; Massonnet and Heeren 2019; Niehuis et al. 1987). Due to the limited ability of these primary ions or ionic clusters to penetrate into the sample and the limited ability of secondary ions to escape from the sample, SIMS is very much a surface technique, probing only the first 1–2 nm. Spatially resolved SIMS analysis is enabled by the use of a microfocused ion beam which is raster scanned over the sample surface. For imaging applications, the ion beam is slightly defocused, yielding beam diameters ranging from several hundreds of nanometers to several micrometers.
In the context of oil paint studies, SIMS has a broad chemical sensitivity that can perform chemically specific analysis of low- and high-Z elements (Baij et al. 2019; Boon et al. 2001; Keune et al. 2009; Keune and Boon 2004, 2007; Voras et al. 2015), common moieties such as sulfates or carbonates (Boon et al. 2001), lead chloride alteration products (Keune et al. 2009), as well as fatty acids and their salts such as lead and zinc soaps (Baij et al. 2019; Boon et al. 2001; Keune et al. 2009; Keune and Boon 2004, 2005, 2007; Noun et al. 2016; Richardin et al. 2011; Sanyova et al. 2011). One recent study that shows some of the wider capabilities of imaging SIMS enabled by using Bi3+ clusters was reported by Voras et al. (Voras et al. 2015). Here, as a follow-up to two previous studies of Matisse’s Le Bonheur de Vivre (Mass et al. 2013a, b), the authors demonstrate how imaging SIMS can perform chemically specific analysis of the intact cadmium yellow pigment, its various oxidation products, and amino acid fragments, while also resolving elemental distributions. The demonstration of such analyses at an estimated spatial resolution of 7.5 μm shows that modern imaging SIMS instrumentation can retrieve a wide range of information that may otherwise have required XRD, EDX, XANES, and FTIR analyses.
6 Future Prospects
Sections 12.2, 12.3, 12.4 and 12.5 reviewed the state-of-the-art of microchemical imaging currently applied to the study of oil paint composition and degradation. The aim of this final section is to critically discuss the capabilities and limitations of the current methodological toolbox and formulate perspectives on future research endeavors in oil paint studies. Such perspectives are illustrated by identifying four general research objectives that address the technological limitations of the microchemical imaging techniques reviewed previously: improved retrieval of spatial information, improved retrieval of chemical information, addressing the limited statistical relevance of analysis on paint microsamples, and integration of computational methods in the processing of microchemical data.
6.1 Improved Retrieval of Spatial Information
The spatial information captured in a chemical image is hard to define formally and has aspects far beyond the scope of this chapter (Yu and Winkler 2013), but here the discussion will be limited to the two aspects of spatial information that are considered most relevant for the study of highly heterogeneous degraded oil paint samples: spatial resolution and definition (Bertrand et al. 2013b). Here, spatial resolution is defined as the dimensions of the smallest detail that can still be resolved, while definition is defined as the total number of spatial points that can be analyzed in a reasonable timeframe. An improvement in spatial resolution reduces the smallest scale at which chemical information can be gathered, whereas an increase in the total number of analyzed spatial points allows access to different length scales at once and can increase the statistical relevance of the obtained information (Bertrand et al. 2013b). Here, a perspective is formulated on how certain technological developments and statistical approaches could lead to improvements in the retrieval of spatial information of degraded oil paint samples at various length scales through microchemical imaging.
6.1.1 Improvements in Spatial Resolution
In studies of oil paint, different chemical questions need to be addressed at different length scales. For instance, while the origin of metal soap-related delamination issues can typically be identified through chemical imaging on a length scale in the order of 10 μm (Van Loon et al. 2019), some fundamental, chemically relevant properties of oil paints only show up on length scales in the order of tens to hundreds of nanometers (Bertrand et al. 2013a; Casadio and Rose 2013). Especially in studies that aim to specifically address the earliest signs of degradation (e.g., microfissure formation) or the chemical interactions between submicrometric pigment particles and the binding medium, crucial information is expected to be found in the nanometric reaction volume at the pigment-medium interface. The following section discusses some technologies that are capable of significant improvements in spatial resolution, but has so far only been applied sparsely (or not at all) in studies of oil paint.
As was discussed in Chap. 2, Sect. 2.1, μ-FTIR is a technique that has proven very powerful for the study of oil paint composition, but also suffers from intrinsic limitations in terms of spatial resolution. AFM-IR circumvents these intrinsic limitations by employing an ultrafine mechanical probe, reaching a spatial resolution in the order of tens of nanometers (see Chap. 2, Sect. 2.2). Despite its demonstrated potential for oil paint studies (Ma et al. 2019; Morsch et al. 2017), preliminary experiments by the authors have revealed that the mechanical heterogeneity of cured oil paint samples severely hampers the routine application of the technique. Furthermore, AFM-IR is a relatively young technique which is still under rapid development. For these reasons, further studies are required to fully elucidate the compatibility of this technique with mechanically heterogeneous samples and to explore the positive and negative effects of the various measurement modalities that already have been developed or are currently under development. A recent demonstration of how specific preparations of mechanically heterogeneous cultural heritage samples can facilitate compatibility with AFM-IR was reported by Reynaud et al. (2020).
In Sect. 12.3.2, some previously reported applications of Raman microspectroscopy were discussed. The Raman scattering modality was shown to be very useful in complementing other means of analysis, providing chemical specificity for compounds that are challenging to discriminate otherwise. The spatial resolution of the technique is determined by the focal volume of the excitation laser and is in the order of 1–2 μm. Significant improvements to this resolution can be realized by making use of wide-field Raman imaging or tip-enhanced Raman scattering (TERS). Wide-field Raman imaging is a technique that is operationally similar to PL microimaging (see Chap. 2, Sect. 2.3), but instead of broadband emission filters, employs tunable narrow-band emission filters (Schaeberle et al. 1999). As the technique works according to a full-field configuration and records images in the visible or near-IR range, a submicrometric spatial resolution can readily be achieved (Schaeberle et al. 1999). Moreover, this full-field approach means that millions—instead of hundreds—of spatial points can be analyzed in a reasonable timeframe. Wide-field Raman imaging therefore also addresses the focus of Sect. 12.6.1.2: increasing the number of analyzed spatial points.
TERS is based on the phenomenon of surface-enhanced Raman scattering (SERS) (Campion and Kambhampati 1998). SERS is induced when the probed molecule is in contact with a nanostructured metallic surface and can locally enhance the Raman signal by several orders of magnitude. The SERS phenomenon is particularly useful as the unenhanced Raman process has an extremely low cross-section, which prohibits analysis of low-concentration compounds and necessitates the use of intense laser irradiation—posing a substantial risk in terms of radiation damage. Using a gold-coated AFM tip to induce the SERS effect, it is possible to record Raman spectra well below the diffraction limit, improving further upon the diffraction-limited spatial resolution of wide-field Raman microimaging (Anderson 2000). This technique does not employ the mechanical detection scheme used for AFM-IR, so the compatibility issues with mechanically heterogeneous cured oil paint samples are not expected to show up as strongly.
As described in Sect. 12.4, methods in the X-ray range are widely applied in oil paint studies. Microscopic X-ray based methodologies provide access to information on redox chemistry, coordination chemistry, while allowing discrimination between compounds of high chemical similarity. Besides beam divergence, measurement volume, and sample thickness, the spatial resolution of X-ray based techniques is for a large part limited by the X-ray focusing optics. Recent developments in synchrotron technology and X-ray focusing optics have led to more routine application of both soft and hard X-ray nanoprobes with sizes in the order of tens of nanometers (Sakdinawat and Attwood 2010). Moreover, similar focusing optics can be applied as imaging optics in full-field X-ray transmission experiments. The two main techniques associated with X-ray nanoprobes and full-field X-ray nanoimaging are called scanning transmission X-ray microscopy (STXM) and transmission X-ray microscopy (TXM) respectively. Due to the tunability of synchrotron-generated X-ray beams, STXM and TXM can be operated in XANES contrasting mode and are therefore for instance foreseen to have the ability to provide crucial information on the early stages of the formation and migration of oxidation and reduction products.
6.1.2 Increasing the Number of Analyzed Spatial Points
The second point of focus in this section is the projected increase in the total number of spatial points that can be analyzed in a reasonable timeframe. The retrieval of chemically specific information at a single spatial point may take anywhere between a few milliseconds (e.g., multispectral PL microimaging) to several minutes (e.g., Raman microspectroscopy), meaning that the total amount of spatial information obtained in a reasonable experimental timeframe may be anywhere between several tens and several millions of spatial points. Especially when studying highly heterogeneous samples, the information obtained from the analysis of several tens of points may hold very little statistical relevance. Moreover, when analyzing certain radiation sensitive compounds, long exposure times may cause radiation damage, which often changes the outcome of the measurement and can prevent subsequent chemical analysis on the same sample. As oil paint samples exhibit a high degree of heterogeneity and can contain materials prone to photo-induced reactions, technological developments aimed at speeding up microchemical analysis are considered crucial. Here, a perspective is proposed on the implementation of both instrumental and statistical methods to increase the total number of analyzed spatial points in the same timeframe.
The first approach is an instrumental one and has been developed specifically to speed up X-ray absorption spectroscopy analysis. All of the currently reviewed studies that involve the use of μ-XAS record spectra by scanning the energy of the excitation beam with a monochromator and recording either a raster-scanned or full-field image. Energy-dispersive XAS uses an elliptically bent polychromator to select a broad energy range that covers an entire absorption edge at once, while simultaneously focusing the beam to micrometric dimensions (Couves et al. 1990). As the transmitted divergent beam is energy-dispersed, a linear detector array placed some distance behind the sample allows to record a full absorption spectrum at once. Due to the extremely high brightness of third-generation synchrotron beamlines, it has recently been shown that full X-ray absorption spectra can be recorded using only a single 100-ps electron bunch (Pascarelli et al. 2016). Besides its applications for time-resolved XAS studies (Gervais et al. 2013b), the technique has specific potential for dramatically reducing the exposure time needed to record full hyperspectral X-ray absorption maps on thin sections of oil paint samples.
The second approach is based on the XANES/XRF speciation mapping method described in Sect. 12.4.2 (Pouyet et al. 2015; Radepont et al. 2011; Van Der Snickt et al. 2009, 2012) and the previously discussed multispectral PL microimaging method (Bertrand et al. 2013a; Comelli et al. 2017; Hageraats et al. 2019a; Van Loon et al. 2019; Thoury et al. 2011, 2019), but is foreseen to be similarly applicable to any other technique that relies on scanning an excitation or emission energy. Both XANES/XRF speciation mapping and multispectral PL microimaging essentially compromise the spectral resolution from hundreds of spectral points to just a few, in order to drastically reduce the measurement time per point. The rationale behind reducing the number of spectral points is (1) the notion that the intrinsic width of absorption and emission features tends to exceed the spectral resolution of most spectrometers and (2) the notion that, within a typically probed spectral range, many spectral features provide redundant information about the same compound. In the previously referenced examples on XANES/XRF speciation mapping and multispectral PL microimaging, energies were chosen either based on visual inspection of XANES spectra or simply based on the availability of spectral bandpass filters. It is postulated here that studies of oil paint could greatly benefit from the development of statistical models (for instance based on discriminant analysis) that determine from high-resolution spectra the number and energy of spectral points required to obtain certain threshold fractions of information. In this way, maps recorded in a multispectral manner could approach the chemical specificity of hyperspectral maps while significantly lowering the analysis time and radiation dose.
-
A detailed account of the theoretical background of this approach applied to as well as an example on cerium speciation in a paleontological sample was recently reported by Cohen et al. (2020).
Just like a full spectrum contains many redundant spectral points, the signal in any data set can be said to manifest a certain level of redundancy with respect to the noise level. That is, the information content in a data set with a high signal-to-noise ratio may be approximately equal to a similar data set with a low signal-to-noise ratio, even though the latter took less time to record. Therefore, the third method for increasing the number of analyzed spatial points is based on a critical evaluation of the signal-to-noise ratio (SNR) that is required to discriminate between different compounds. It is here proposed to make use of the principle of zeta-scores, which measure the distance between two data points xi and xj normalized to the expected standard deviation σ in this distance if the two data points were recorded on exactly the same sample (Analytical Methods Committee 2016):
Recognizing that the SNR of a spectrum R can be calculated by taking the ratio between its mean value μ and its standard deviation, the zeta-score equation for spectra can be rewritten as:
A rough estimate of the SNR required to determine the chemical identity of a sample based on some spectrum X and two reference spectra R1 and R2 could then be made as follows:
Where ‖R1 − R2‖ is the Cartesian distance between the two reference spectra, Rmin is the reference spectrum with the lowest SNR, and ζ is the zeta-score, which sets the required level of confidence for the identification based on spectrum X and reference spectra R1 and R2. For instance, for a 95% confidence level, ζ needs to be set to 1.96. This expression for SNRX can be generalized to n reference spectra simply by finding the set of two reference spectra for which the zeta score is minimal (according to Eq. 12.4) and plugging the values for Ri and Rj into Eq. 12.5.
6.2 Improved Retrieval of Chemical Information
Besides the instrumental and statistical methods to improve the retrieval of spatial information for a given analytical approach, there is a continuous interest to improve upon the ability of microchemical imaging techniques to discriminate between different chemical species. Here, seven methodologies are briefly discussed that are meant to improve upon the chemical specificity of PL microimaging, μ-XANES, and SIMS, and extend the analytical capabilities of SEM.
6.2.1 Semi-hyperspectral Total Synchronous PL Microimaging
In Sect. 12.3.1 a number of PL-based microimaging and microspectroscopy methods were described that have so far been shown to be powerful analytical tools for the study of semiconductor pigments and zinc carboxylate degradation products. Still, the authors identify a recurring issue with PL-based methodologies in terms of chemical specificity, which limits the number of possible applications in oil paint studies. This issue is thought to be related primarily to the width of PL emission bands at room temperature and the large dynamic range of photoluminescence quantum yields of different paint components. This makes that emission bands of interest are often obscured either by emission bands with a similar energy, or even by emission bands with a very different energy, but a much higher quantum yield. In order to improve upon the chemical specificity of PL-based microimaging, there may be a strong interest in exploiting both the absorption and emission behavior of compounds by implementing total synchronous PL approaches in PL microanalysis methodologies. Total synchronous PL spectroscopy works by recording 2D absorption-emission spectra and has demonstrated potential in the classification of organic paint media and the characterization of differently produced lead whites (Gonzalez et al. 2017a; Nevin et al. 2009), but has so far not been demonstrated for microanalytical purposes.
It is proposed here to extend the multispectral PL microimaging methodology with a tunable narrow-band excitation source to obtain high-resolution spectral information without compromising on spatial resolution. Such a semi-hyperspectral total synchronous PL microimaging set-up could be realized by performing the excitation using a monochromatized tunable xenon arc lamp and recording full-field images for a large series of excitation wavelengths. Repeating the series through a number of emission band pass filters then yields a total synchronous PL dataset with the ability to reach diffraction-limited lateral resolutions. Of course, in order for total synchronous PL microimages to be translated into chemically specific maps of organic compounds, comprehensive (standardized) studies into the PL properties of binders, pigments, additives, and degradation products are first required. Moreover, spectral unfolding (see Sect. 12.6.4) will foster the ability to map compounds with strongly overlapping absorption and/or emission features.
6.2.2 Site-Selective and High-Energy Resolution μ-XANES
Within the current state-of-the-art, μ-XANES on oil paint microsamples is often recorded in a fluorescence geometry. Detection of the X-ray fluorescence signal typically occurs using energy-dispersive silicon drift or high purity germanium (HPGe) detectors, providing for each excitation energy an X-ray fluorescence spectrum with low energy resolution (< 130 eV). By either recording all signal, irrespective of energy (total fluorescence mode), or binning the emission signal falling only in a detector channel region over the element emission lines of interest, a measurement is obtained of the local X-ray absorption at that specific excitation energy. It must be stressed that the low spectral resolution of energy-dispersive detectors has an effect on the spectral resolution of X-ray absorption spectra and consequently also has an effect on the chemical specificity of the technique.
This effect is best described by considering a resonant X-ray emission spectroscopy (RXES) plane, such as the one shown in Fig. 12.14. An RXES plane shows a high-resolution X-ray emission spectrum for each X-ray excitation energy—exhibiting multiple narrow resonant emission bands whose energies and intensities depend strongly on the excitation energy. In case of negligible molecular motion, the resonant emission bands follow diagonal lines each characterized by a definite energy transfer corresponding to ω − ω′: the difference between the excitation and emission energies. When recording XANES with a low-energy-resolution detector, the whole RXES plane is effectively projected onto a single energy channel, significantly broadening absorption features and often losing important spectral and therefore chemical information. Looking for instance at the RXES plane shown in Fig. 12.14, there are clearly two (or even three) separate peaks in the region of incident photon energies between 4560 and 4567 eV, which would all be observed as one using a low-resolution energy-dispersive detector (Marchenko et al. 2011).

RXES plane of CH3I recorded close to the I L3-edge. Reprinted from ref. (Marchenko et al. 2011) with the permission of AIP Publishing
Methods have therefore been proposed to improve the spectral resolution of fluorescence detection systems, in order to record X-ray absorption spectra for specific X-ray emission energies (Glatzel and Bergmann 2005). Glatzel et al. (2002) reported on a setup in which—using a Rowland geometry wavelength-dispersed spectrometer—the X-ray emission energy of different Kβ lines could be selectively probed. It was shown that with this site-selective approach it is possible to selectively probe different iron species in mixed-valence Prussian blue. A demonstration of how this approach can be made to work with X-ray nanoprobes was reported by Cotte et al. (2011), showing significant improvements on the spectral resolution of Sb L3-edge XANES using a compact double-crystal wavelength-dispersive detection system operated in a confocal configuration. Due to the high chemical heterogeneity of oil paint samples, site-selective and high-resolution μ-XANES are anticipated to provide increased chemical specificity in studies of pigment degradation that involve complex redox chemistry. For imaging purposes, it must be noted that the wavelength-dispersed detection scheme requires a higher radiation dose to be deposited onto the sample as compared to regular μ-XAS. Therefore, there is an elevated risk of radiation damage in the analyzed sample (Bertrand et al. 2015; Monico et al. 2020b).
6.2.3 Energy-Dispersive μ-EXAFS
EXAFS is conceptually similar to XANES (see Sect. 12.4.2), but rather than focusing on discrete near-edge transitions, information is retrieved from the oscillations that appear at higher energies corresponding to core-shell-to-continuum transition. These oscillations are interference patterns of ejected electrons that are scattered off atoms in the first few coordination shells; corresponding spectra can be transformed and fitted to retrieve information about a compound’s coordination number and bond lengths. μ-EXAFS has been applied previously to studies of oil paint degradation, providing unique insights into the discoloration of smalt and the blackening of copper resinate (Cartechini et al. 2008; Cianchetta et al. 2012; Robinet et al. 2011). These μ-EXAFS studies all make use of point analysis, due to the fact that experiments were performed in fluorescence geometry with stepwise scanning of the excitation energy, leading to measurement times per spectrum in the order of tens of minutes.
As was discussed in Sect. 12.6.1.2, the measurement time of μ-XAS measurements can be reduced by several orders of magnitude by making use of a transmission geometry in combination with an energy-dispersive detection scheme. The fact that the elliptically bent polychromator crystals can disperse a range of X-ray energies that is much wider than a typical XANES make this approach also highly suitable—and in fact primarily designed—for EXAFS analysis (Couves et al. 1990; Pascarelli et al. 2016). Given the previously reported relationship between changes in coordination chemistry and pigment discoloration (Cianchetta et al. 2012; Couves et al. 1990; Robinet et al. 2011), the implementation of an energy-dispersive detection scheme in the EXAFS regime is anticipated to provide unique information on the precise chemical processes that underlie the discoloration of certain pigments.
6.2.4 X-Ray Raman Scattering
A limitation of XAS techniques for light elements (such as carbon, oxygen or nitrogen) is the high attenuation of X-rays which requires working on TEM-class (∼100 nm) thin samples. Yet, the preparation necessary to obtain samples that thin may not be compatible with mechanically heterogeneous cured oil pains. An alternative is the excitation of the same transitions (such as C, O or N K edges) using the energy loss in the sample resulting from inelastic scattering processes in the hard X-ray domain; a modality called X-ray Raman scattering (XRS). Using such an approach on historical materials, Gueriau et al. (2017) could for instance differentiate chemical compositions among carbon black pigments. Apart from the study of carbonaceous pigments, XRS is thought to be particularly promising to gain a deeper understanding of the transformation of the organic binder in oil paints.
On chemically heterogenous samples, a micrometric beam (typically 50 μm) can be raster-scanned across the sample, with XRS spectral information being recorded for each position. With this approach, chemical speciation maps can be collected on heterogeneous samples without the hassle and limitations of preparing thin sections. Due to the low attenuation of hard X-rays in most materials, XRS also lends itself as a contrasting method in 3D imaging techniques. To demonstrate this ability, Sahle et al. (2017a, b) have shown that the detected (scattered) X-ray beams can be resolved on a pixelated detector. The point-to-point correlation between voxels illuminated along the beam path in the depth of the sample allows to attain 3D reconstructed volumes of chemical speciation of carbon, oxygen and other elements in organic materials following the direct tomography method (Georgiou et al. 2019).
Currently, the limited spatial resolution of XRS-based methodologies prohibits them from providing spatially resolved chemical information on oil paints at resolutions better than a few tens of micrometers. Still, currently reported values are not due to fundamental physical limitations, but rather to the very low cross-section of the XRS process, which makes it challenging to obtain a sufficient signal-to-noise ratio from small measurement volumes while keeping radiation damage below acceptable thresholds. It is anticipated that improved detection systems with higher solid angles of detection can eventually bring the spatial resolution to ∼10 μm or below. Here, it must be noted that, due to the low cross-section of the XRS process, a high photon flux is often required to obtain sufficient signal. Exposure of organic materials to such high photon fluxes has been shown to induce radiation damage that is observable with the human eye (Gueriau et al. 2017).
6.2.5 SEM-Raman & Electron Backscatter Diffraction
One of the primary limitations of SEM—even when equipped with an EDX system—is its limited chemical specificity. For this reason, many research efforts have been focused on combining scanning electron microscopes with various other measurement modalities. Here, based on their prospective applications in studies of oil paint, two of such hyphenated techniques are discussed: SEM-Raman and electron backscatter diffraction (EBSD). SEM-Raman has been developed very recently to introduce the analytical capabilities of Raman microspectroscopy into a SEM—hereby circumventing the need to transfer the sample between microscopes and allowing easy correlation between high-resolution electron backscatter images and Raman scattering data. It works by leading the beam of an excitation laser into the chamber, focusing it onto the sample, and directing the scattered light to a spectrometer using collection optics and an optical fiber (Wille et al. 2014). The technique has already been shown to work well for the identification and characterization of pigments obtained from an ancient cave painting from Grottes de la Garenne. In the context of oil paint, the technique can be expected to provide quick identification of pigments and degradation products in and around the degradation features observed using SEM.
EBSD is a more established technique that combines SEM electron backscatter imaging with the exceptional chemical specificity of crystal diffraction (Schwartz et al. 2009). Instead of using X-rays, EBSD makes use of the wave properties of electrons. By firing the electron beam at a crystalline sample at a large angle and imaging the scattered electrons using a scintillator-CCD/CMOS detection system, complex (Kikuchi) patterns are observed that are representative of the local crystal structure. For highly heterogeneous samples, EBSD can be used to identify crystalline phases with dimensions in the order of tens of nanometers. Its application for the study of historical artworks has already been demonstrated by Berrie et al. (2016), who used EBSD to identify various pigments in a microscopic cross-section. Just like SEM-Raman, the technique can be envisioned to provide direct and accurate chemical identification of compounds found in features resolved using SEM backscatter imaging.
6.2.6 Imaging MALDI Mass Spectrometry
In Sect. 12.5.3 the imaging applications of secondary ion mass spectrometry (SIMS) were discussed. Using a focused beam of ions or small ionic clusters, secondary ions can be generated locally on the sample surface. Chemical maps are then obtained by scanning the beam over an area of interest, and integrating the signal coming from certain chemical species. Imaging MALDI mass spectrometry is a technique that is conceptually similar, but uses matrix-assisted laser desorption (MALDI) as an ionization mechanism (Sabatini et al. 2016). By using a focused laser beam, a spatial resolution in the order of 25 μm can be obtained. Despite this compromise on spatial resolution as compared to imaging SIMS, the MALDI ionization method is extremely soft and is therefore useful for analysis of large molecules, such as proteins. For this reason, MALDI mass spectrometry has for instance been used in identification and characterization studies of proteinaceous binders and organic dyes (Kuckova et al. 2007; Sabatini et al. 2016). In the future, imaging MALDI mass spectrometry may for instance be applied to map distributions of more complex organic original and degradation products. In this regard, the method would be unique within the conservation scientist’s microchemical imaging toolbox.
6.3 Addressing the Limited Statistical Relevance of Analysis on Paint Microsamples
One fundamental problem that is inherent to the analysis of oil paint microsamples is the limited statistical relevance of the sample in the context of the object as a whole. Moreover, microchemical imaging of paint microsamples conducted in a reflection or fluorescence geometry is often only sensitive to the uppermost micrometers (e.g., μ-ATR-FTIR, μ-XANES) or uppermost nanometers (e.g., AFM-IR, imaging SIMS) of the sample. The authors identify two main perspectives for improving the statistical relevance of the analysis of oil paint microsamples: three-dimensional microchemical imaging techniques to resolve chemical distributions throughout entire microsamples and object-based sub-surface microchemical imaging to perform cross-sectional microanalysis without the need for invasive sampling.
6.3.1 Three-Dimensional Microchemical Imaging
Within the current state-of-the-art of microchemical imaging of oil paints, the focus lies primarily on two-dimensional imaging of paint cross-sections, fragments, or pigment powders. Still, extending microchemical analysis into the third dimension is foreseen to strongly increase the amount of information that can be obtained from a single microsample, thereby decreasing the risk of false positives that are inherent to interpretations of data sets with limited spatial representation. Depending on the purpose of the study, there are two general groups of 3D imaging techniques that may see applications for the microchemical study of oil paints: those relying solely on material density and those relying on compound-specific X-ray absorption or emission transitions.
The principle of material density-based 3D imaging applied to oil paints was first demonstrated in a series of three studies by Ferreira et al. (2009, 2011) and Gervais et al. (2013a), who show how X-ray absorption tomography can help in determining the porosity of oil paints. Here, the use of tomography—instead of a 2D imaging method—allows to obtain much better statistics on overall porosity, but also permits to calculate accurate porosity metrics as a function of depth. Instead of imaging the absorption of X-rays to detect differences in density, the coherence of synchrotron-generated X-rays also allows to detect density differences based on differences in the phase shift. This principle is used by ptychographic X-ray computed tomography and is particularly useful for imaging small differences in density (Dierolf et al. 2010).
An alternative to X-ray tomography for 3D imaging has also been developed that eliminates the need for synchrotron radiation and is based on a SEM instead. Serial block-face scanning electron microscopy (SBFSEM) is a method in which an ultramicrotome is introduced into the SEM experimental chamber to cut many consecutive thin (∼30 nm) slices off the surface of the sample (Denk and Horstmann 2004). After each cut, the surface is imaged to obtain a stack of images that can be reconstructed to a 3D volume with voxel sizes in the order of 20 × 20 × 30 nm. Although no applications have yet been demonstrated on oil paint, multiple reports have been published of SBFSEM applied to distinguish the various phases in coatings (Chen et al. 2013, 2014; Hughes et al. 2014).
In all these studies, no direct chemical information could be obtained. In an effort to retrieve chemical information from SBFSEM or single-energy X-ray tomography data, processing algorithms have recently been developed that can quantitatively assess local attenuation lengths and electron densities (Andres et al. 2008; Li et al. 2019; Maldanis et al. 2020; Reynaud et al. 2020). In studies of oil paint, this could allow coupling microscopic observation of morphological features with phase identification at micrometric to nanometric resolutions. The collection of billions of voxels means that statistical processing can be performed, which can produce a more complete description of samples and can help to identify previously unidentified phases in complex heterogenous degraded oil paint samples (Li et al. 2019).
The second way to obtain microchemical information in 3D is by making use of the chemical information embedded in elemental absorption edges or by collecting the XRF signal. The principle of using XAS contrast in X-ray absorption tomography has for instance been demonstrated on composite electrodes (Meirer et al. 2011), where rotational tomography series were repeated for 154 X-ray energies, covering the full Ni K-edge. The principle of using XRF contrast in X-ray absorption tomography has recently been demonstrated on catalyst particles (Bossers et al. 2020). Here, consecutive XRF images are recorded by raster scanning a nanofocused X-ray beam and recording the full XRF spectrum per sample position using a fast annular X-ray detector.
In the context of oil paint, three-dimensional reconstructions of elemental distributions or their oxidation states are foreseen to provide invaluable, statistically sound information on the presence and migration of original and degradative species. Moreover, with spatial resolutions being reported in the order of tens of nanometers (Meirer et al. 2011), these techniques can provide a unique view to a variety of interface effects and early stages of degradation.
6.3.2 Object-Based Sub-surface Microchemical Imaging
Technologies that possess the ability to analyze paintings as a whole are commonly referred to as macroscopic techniques and include for instance visible-NIR hyperspectral reflectance imaging, macroscopic XRF (MA-XRF) and macroscopic X-ray powder diffraction (MA-XRPD) (Cucci et al. 2016; Dik et al. 2008; Vanmeert et al. 2018). Although such techniques have proven exceptionally powerful for studying artistic composition, underpaintings, and pigmentation, the ability to resolve chemical distributions in-depth and/or on the microscale is still underdeveloped. One particularly promising research objective is therefore to bridge the current gap between macroscopic and microscopic methodologies, by developing object-based sub-surface microchemical imaging methodologies.
One of the primary advantages of performing analysis on microsamples is the ability to analyze paint layers in cross-section. In order to bring chemical microanalysis to whole objects, the ability to probe underneath the paint surface is therefore a crucial point of focus. Here, we will discuss four techniques that have a proven ability to probe samples in-depth and rely on contrasting methods that could be exploited for the study of oil paint sub-surface composition: confocal XRF/XAS, pump-probe microscopy, and Compton scattering tomography.
Confocal XRF makes use of a set of two perpendicularly placed X-ray focusing optics so as to only detect X-ray fluorescence from the intersection between the two foci (Kanngießer et al. 2003). Most commonly, polycapillary optics are used, providing lateral and axial resolutions in the order of tens of micrometers. With the ability to discriminate between elements, this technique is capable of distinguishing different paint layers and identifying with some degree of certainty the pigmentation in each layer. Although these capabilities can for instance provide virtual cross-sectional elemental distributions (Alfeld and Broekaert 2013; Kanngießer et al. 2012), the limited chemical specificity of XRF and the low spatial resolution provided by polycapillary optics make conventional confocal XRF largely unsuitable for studies of oil paint degradation. Instead, it is proposed to make use of the tunability of synchrotron radiation and use the confocal X-ray excitation-detection scheme in conjunction with XAS. Confocal XAS follows the same principle as confocal XRF, but measures the total fluorescence yield as a function of the excitation energy at certain absorption edges (Denecke et al. 2009). In order to combine the higher chemical specificity of XAS with a spatial resolution suitable for microanalytical studies of oil paint sub-surface layers, a KB mirror can be used to focus the excitation beam, while collimating channel array (CCA) optics can be used to define the measurement volume. This combination has been shown to provide a lateral resolution of 2.0 μm and a depth resolution of 2.5 μm (Sun et al. 2008), thereby making the confocal XAS technique a potentially powerful tool for noninvasive subsurface microchemical imaging of oil paintings.
Pump-probe microscopy is a technique that originates from biology and exploits de-excitation dynamics as a means to achieve chemical contrast in the overlapping excitation volume of two pulsed light sources (Helmchen and Denk 2005). Due to the use of a confocal configuration, pump-probe microscopy is a suitable technique to obtain virtual cross-sections. This principle has been demonstrated by Villafana et al. (2014), where the different absorption spectra and de-excitation dynamics of ultramarine and quinacridone red are used to compose cross-sectional distributions maps of both pigments in mock-ups and a fourteenth century painting. As was shown by Yu et al. (2019) in a pump-probe microscopy study on vermillion powders and a historical cross-section, the technique can also be used to study pigment degradation. It is foreseen that such principles could be similarly applied in object-based studies.
Compton scattering tomography is a technique that is highly promising for applications in subsurface imaging of oil paintings, but has not been demonstrated in practice yet (Norton 1994). Recently, Guerrero Prado et al. (2017) provided the numerical support for the feasibility of this technique for the three-dimensional electron density imaging of flat objects. The proposed approach to Compton scattering tomography does not require relative rotations of the object with respect to the imaging set-up and can be performed even when the object is supported by an electronically dense material. In combination with the processing algorithms discussed briefly in Sect. 12.6.3.1, it ought to be possible to use this technique to obtain sub-surface microchemical information on oil paintings.
6.4 Integrating Computational Methodologies in the Processing of Microchemical Data
In the current state-of-the-art of microchemical analysis of oil paint degradation, the overwhelming majority of all chemical information is still retrieved through manual data analysis by means of the expert eye. However, with the current and prospective technological developments discussed in all foregoing sections, there is a strong tendency for microchemical images to consist of more and more spectral and/or spatial points. With all this extra information, relying solely on the expert eye will often mean missing patterns or compounds that computer algorithms can identify. Following the rapid increase in readily available computational power, more and more methods are becoming available for the retrieval of chemical information through algorithmic processing (Gendrin et al. 2008). Due to the complex nature of degraded oil paints and the increasing volume of data sets, the authors identify a significant opportunity for the integration of computational methodologies for the improved retrieval of chemical information from microanalytical data.
For the study of samples that are chemically as complex and heterogeneous as mature oil paint samples, particularly useful computational methodologies are those involved in dimensionality reduction. Given a large microspectroscopic dataset, dimensionality reduction algorithms aim to find so-called latent variables: variables that reflect certain chemical parameters, but are hidden in the dataset. In this way, a series or map of spectra with several hundreds of spectral variables (e.g., excitation or emission energies) can be reduced to less than 10 variables—each of which can be interpreted chemically. Common examples of such algorithms include principal component analysis (PCA), partial least squares (PLS), non-negative matrix factorization (NMF), and singular value decomposition (SVD). As a follow-up, each spectrum in the dataset can be expressed (i.e., modeled) as a linear combination of the calculated latent variables, returning chemically relevant weight matrices. A more detailed review of methods to perform dimensionality reduction to obtain chemical maps can be found in (Guerrero Prado et al. 2017).
As has been expressed previously, the application of such approaches to microspectroscopic datasets obtained in oil paint degradation studies has been rather rare. The most noteworthy applications have been reported very recently and include the work by Romano et al. (2020) and Gambardella et al. (2020). Although the microspectroscopic data sets were recorded using μ-FTIR and full-field μ-XANES respectively, in both cases NMF is used to retrieve the latent variables. Romano et al. use the weight matrices retrieved through modeling of the μ-FTIR data cube using NMF factors to reconstruct chemical phase maps of cadmium red paint, zinc white paint, and a gel-like zinc carboxylate protrusion. Similarly, Gambardella et al. visualize the distributions of two distinct chemical phases in ultramarine pigments by color-coding the difference between the NMF weight matrices of the first and third factor.
These examples illustrate two instances in which the different chemical phases cannot directly be identified by mapping single spectral points or single integrated absorption features, but are rather hidden in the complex sets of data. For instance, looking at the result reported by Romano et al. the pigments zinc white and cadmium red have no significant absorption in the mid-IR range themselves, but the two paints could still be distinguished computationally; likely due to different chemical interactions with the infrared-active oil binder. Considering the chemical contrast within ultramarines reported by Gambardella et al. and its origin in subtle spectral differences in S K-edge XANES, it is hard to imagine a manual approach to yield the same chemical information as the reported computational procedure.
Even without the previously described dimensionality reduction algorithms, there are still significant improvements that can be made in the retrieval of information from microchemical imaging datasets by making use of spectral unfolding. Spectral unfolding is based on the notion that most spectra are essentially made up of sums of probability distributions, such as Gaussians, Lorentzians, or Voigt profiles. These probability distributions reflect instrumental peak broadening, statistical variance in molecular structure, and—especially for rapid transitions—natural broadening. In chemically complex samples, there tends to be a high degree of overlap between adjacent probability distributions, often obscuring the precise peak locations of spectral bands. As the adjacency of spectral bands may for instance reflect the occurrence of relevant chemical transitions or the presence of one compound in two or more structural configurations, it is often favorable to be able to analyze the separate spectral contributions. Spectral unfolding is an exclusively computational endeavor that fits a model consisting of sums of probability distributions to raw spectral data.
This procedure is currently routinely applied to XRF data obtained with energy-dispersive X-ray detectors, where emission energies are known and peak widths can be approximated by knowing the spectral resolution of the detector. For this particular purpose, the PyMCA software has been developed at the European Synchrotron Radiation Facility (ESRF) (Solé et al. 2007), and has been used extensively among the μ-XRF studies discussed in part I of this chapter (see for instance Fig. 12.8). Still, in oil paint studies involving μ-FTIR or PL microspectroscopy—where spectral overlap similarly obscures chemically relevant information—spectral unfolding is not yet commonly applied. Explanations as to why spectral unfolding has so far been limited to XRF studies include lack of prior knowledge about the location and width of spectral features and complex and unpredictable baselines.
7 Conclusion
In part I of this chapter, we reviewed the current state-of-the-art of microchemical imaging studies on oil pain tcomposition. Based on the specific characteristics of oil paint samples, techniques have been applied that are based on the absorption of mid-IR radiation, the emission of photoluminescence, the absorption, reflection, and emission of X-rays, the scattering and absorption of electrons, and the mass-specific detection of secondary ions. By either employing tightly focused beams of electromagnetic radiation or charged particles, or full-field detection in combination with dedicated microscopy objectives, spatially resolved analysis can be performed at the micro- or even nanoscale. The current toolbox that makes up the state-of-the-art has for instance been applied to the identification and localization of (different grades of) original and degraded pigments and later added paint layers. By combining multiple microchemical imaging modalities, it is often possible to obtain the information necessary to elucidate the entire painting life history.
From the review of over a hundred reports on the microchemical imaging of oil paints, one can conclude that the conservation science community has shown an eagerness to adapt new developments, such as synchrotron-generated micro- and nanobeams, full-field X-ray imaging detectors, XRD tomography, and AFM-based infrared nanospectroscopy. Still, due to the complexity of oil paints and the current challenges associated both with object-based and sample-based microanalysis, even current state-of-the-art technology comes with substantial limitations in terms of the chemical information that can be obtained and the statistical relevance thereof. Ways in which these limitations can be addressed were discussed in Sect. 12.6.
In this sectioncond part, it was proposed that by making use of certain recent instrumental developments, smart statistical evaluations of data requirements, and subsequent computational data analysis, some of these limitations could potentially be tackled. The authors foresee particularly promising prospects for nanoscale chemical imaging methodologies (see Sect. 12.6.1.1), object-based sub-surface microchemical imaging techniques (see Sect. 12.6.3.2), and data dimensionality reduction approaches for mapping chemical contrast in complex, heterogeneous samples (see Sect. 12.6.4). A summary of all methodologies discussed in the first and second part of this chapter is presented in Fig. 12.15. In this respect, the specific properties of ancient painting materials (which are inherently heterogeneous, aged, and unique) not only constitute a set of constraints on analytical studies concerning the identification of their composition and the understanding of degradation pathways, they also constitute an important source of inspiration, encouraging the optimization and development of analytical techniques sensitive to small chemical contrasts at high spectral and spatial resolution, and their coupling to advanced statistical processing of data, for the heritage science field and far beyond.

Graphical summary of the microchemical imaging methods currently employed for the study of oil paint and their recent and prospective developments
References
Alfeld, M., Broekaert, J.A.C.: Mobile depth profiling and sub-surface imaging techniques for historical paintings—a review. Spectrochim. Acta Part B Atom. Spectrosc. 88, 211–230 (2013). https://doi.org/10.1016/j.sab.2013.07.009
Analytical Methods Committee. (2016). z-Scores and other scores in chemical proficiency testing-their meanings, and some common misconceptions. Anal. Methods. 8, 5553–5555. https://doi.org/10.1039/C6AY90078J
Anderson, M.S.: Locally enhanced Raman spectroscopy with an atomic force microscope. Appl. Phys. Lett. 76, 3130–3132 (2000). https://doi.org/10.1063/1.126546
Andres, B., Köthe, U., Helmstaedter, M., Denk, W., Hamprecht, F.A.: Segmentation of SBFSEM Volume Data of Neural Tissue by Hierarchical Classification. Pattern Recognit, pp. 142–152. Springer Berlin Heidelberg, Berlin/Heidelberg (2008). https://doi.org/10.1007/978-3-540-69321-5_15
Artesani, A., Gherardi, F., Nevin, A., Valentini, G., Comelli, D.: A photoluminescence study of the changes induced in the zinc white pigment by formation of zinc complexes. Materials (Basel). 10, 340 (2017). https://doi.org/10.3390/ma10040340
Artesani, A., Gherardi, F., Mosca, S., Alberti, R., Nevin, A., Toniolo, L., et al.: On the photoluminescence changes induced by ageing processes on zinc white paints. Microchem. J. 139, 467–474 (2018). https://doi.org/10.1016/j.microc.2018.03.032
Artesani, A., Ghirardello, M., Mosca, S., Nevin, A., Valentini, G., Comelli, D.: Combined photoluminescence and Raman microscopy for the identification of modern pigments: explanatory examples on cross-sections from Russian avant-Garde paintings. Herit. Sci. 7, 17 (2019). https://doi.org/10.1186/s40494-019-0258-x
Baij, L., Chassouant, L., Hermans, J.J., Keune, K., Iedema, P.D.: The concentration and origins of carboxylic acid groups in oil paint. RSC Adv. 9, 35559–35564 (2019). https://doi.org/10.1039/C9RA06776K
Barba, C., Andrés, M.S., Peinado, J., Báez, M.I., Baldonedo, J.L.: A note on the characterization of paint layers by transmission electron microscopy. Stud. Conserv. 40, 194–200 (1995). https://doi.org/10.1179/sic.1995.40.3.194
Berrie, B.H., Leona, M., McLaughlin, R.: Unusual pigments found in a painting by Giotto (c. 1266-1337) reveal diversity of materials used by medieval artists. Herit. Sci. 4, 1 (2016). https://doi.org/10.1186/s40494-016-0070-9
Bertrand, L., Cotte, M., Stampanoni, M., Thoury, M., Marone, F., Schöder, S.: Development and trends in synchrotron studies of ancient and historical materials. Phys. Rep. 519, 51–96 (2012). https://doi.org/10.1016/j.physrep.2012.03.003
Bertrand, L., Réfrégiers, M., Berrie, B., Échard, J.-P., Thoury, M.: A multiscalar photoluminescence approach to discriminate among semiconducting historical zinc white pigments. Analyst. 138, 4463 (2013a). https://doi.org/10.1039/c3an36874b
Bertrand, L., Thoury, M., Anheim, E.: Ancient materials specificities for their synchrotron examination and insights into their epistemological implications. J. Cult. Herit. 14, 277–289 (2013b). https://doi.org/10.1016/j.culher.2012.09.003
Bertrand, L., Schöeder, S., Anglos, D., Breese, M.B.H., Janssens, K., Moini, M., et al.: Mitigation strategies for radiation damage in the analysis of ancient materials. TrAC Trends Anal. Chem. 66, 128–145 (2015). https://doi.org/10.1016/j.trac.2014.10.005
Blumenroth, D., Dietz, S., Müller, W., Zumbühl, S., Caseri, W., Heydenreich, G.: Inside the Forger’s oven: identification of drying products in oil paints during and after accelerated drying with increased temperatures. In: Conservation of Modern Oil Paintings, pp. 437–450. Springer International Publishing, Cham (2019). https://doi.org/10.1007/978-3-030-19254-9_34
Boon, J.J., Keune, K., Van der Weerd, J., Geldof, M., Van Asperen de Boer, J.: Imaging microspectroscopic, secondary ion Mass spectrometric and electron microscopic studies on Discoloured and partially Discoloured smalt in cross-sections of 16th century paintings. Chimia (Aarau). 55, 952–960 (2001)
Bossers, K.W., Valadian, R., Zanoni, S., Smeets, R., Friederichs, N., Garrevoet, J., et al.: Correlated X-ray Ptychography and fluorescence Nano-tomography on the fragmentation behavior of an individual catalyst particle during the early stages of olefin polymerization. J. Am. Chem. Soc. 142, 3691–3695 (2020). https://doi.org/10.1021/jacs.9b13485
Breek, R., Froentjes, W.: Application of pyrolysis gas chromatography on some of Van Meegeren’s faked Vermeers and Pieter de Hooghs. Stud. Conserv. 20, 183 (1975). https://doi.org/10.2307/1505738
Burnstock, A., van den Berg, K.J., de Groot, S., Wijnberg, L.: An investigation of water-sensitive oil paints in twentieth-century paintings. In: Modern Paints Uncovered, pp. 177–188. Getty Conservation Institute, Los Angeles (2006)
Campion, A., Kambhampati, P.: Surface-enhanced Raman scattering. Chem. Soc. Rev. 27, 241 (1998). https://doi.org/10.1039/a827241z
Cartechini, L., Miliani, C., Brunetti, B.G., Sgamellotti, A., Altavilla, C., Ciliberto, E., et al.: X-ray absorption investigations of copper resinate blackening in a XV century Italian painting. Appl. Phys. A Mater. Sci. Process. 92, 243–250 (2008). https://doi.org/10.1007/s00339-008-4498-y
Casadio, F., Rose, V.: High-resolution fluorescence mapping of impurities in historical zinc oxide pigments: hard X-ray nanoprobe applications to the paints of Pablo Picasso. Appl. Phys. A Mater. Sci. Process. 111, 1–8 (2013). https://doi.org/10.1007/s00339-012-7534-x
Casadio, F., Xie, S., Rukes, S.C., Myers, B., Gray, K.A., Warta, R., et al.: Electron energy loss spectroscopy elucidates the elusive darkening of zinc potassium chromate in Georges Seurat’s A Sunday on La Grande Jatte—1884. Anal. Bioanal. Chem. 399, 2909–2920 (2011). https://doi.org/10.1007/s00216-010-4264-9
Cato, E., Scherrer, N., Ferreira, E.S.B.: Raman mapping of the S 3 − chromophore in degraded ultramarine blue paints. J. Raman Spectrosc. 48, 1789–1798 (2017). https://doi.org/10.1002/jrs.5256
Chen, B., Guizar-Sicairos, M., Xiong, G., Shemilt, L., Diaz, A., Nutter, J., et al.: Three-dimensional structure analysis and percolation properties of a barrier marine coating. Sci. Rep. 3, 1177 (2013). https://doi.org/10.1038/srep01177
Chen, B., Hashimoto, T., Vergeer, F., Burgess, A., Thompson, G., Robinson, I.: Three-dimensional analysis of the spatial distribution of iron oxide particles in a decorative coating by electron microscopic imaging. Prog. Org. Coat. 77, 1069–1072 (2014). https://doi.org/10.1016/j.porgcoat.2014.03.005
Chen-Wiegart, Y.K., Catalano, J., Williams, G.J., Murphy, A., Yao, Y., Zumbulyadis, N., et al.: Elemental and molecular segregation in oil paintings due to Lead soap degradation. Sci. Rep. 7, 11656 (2017). https://doi.org/10.1038/s41598-017-11525-1
Cianchetta, I., Colantoni, I., Talarico, F., D’Acapito, F., Trapananti, A., Maurizio, C., et al.: Discoloration of the smalt pigment: experimental studies and ab initio calculations. J. Anal. At. Spectrom. 27, 1941–1948 (2012). https://doi.org/10.1039/c2ja30132f
Cohen, S.X., Webb, S.M., Gueriau, P., Curis, E., Bertrand, L.: Robust framework and software implementation for fast speciation mapping. J. Synchrotron Radiat. 27, 1049–1058 (2020). https://doi.org/10.1107/S1600577520005822
Comelli, D., Artesani, A., Nevin, A., Mosca, S., Gonzalez, V., Eveno, M., et al.: Time-resolved photoluminescence microscopy for the analysis of semiconductor-based paint layers. Materials (Basel). 10, 1335 (2017). https://doi.org/10.3390/ma10111335
Comelli, D., MacLennan, D., Ghirardello, M., Phenix, A., Schmidt Patterson, C., Khanjian, H., et al.: Degradation of cadmium yellow paint: new evidence from photoluminescence studies of trap states in Picasso’s femme ( Époque des “demoiselles d’Avignon”). Anal. Chem. 91, 3421–3428 (2019). https://doi.org/10.1021/acs.analchem.8b04914
Cotte, M., Susini, J.: Watching ancient paintings through synchrotron-based X-ray microscopes. MRS Bull. 34, 403–405 (2009). https://doi.org/10.1557/mrs2009.115
Cotte, M., Checroun, E., Susini, J., Walter, P.: Micro-analytical study of interactions between oil and lead compounds in paintings. Appl. Phys. A Mater. Sci. Process. 89, 841–848 (2007). https://doi.org/10.1007/s00339-007-4213-4
Cotte, M., Susini, J., Solé, V.A., Taniguchi, Y., Chillida, J., Checroun, E., et al.: Applications of synchrotron-based micro-imaging techniques to the chemical analysis of ancient paintings. J. Anal. At. Spectrom. 23, 820–828 (2008). https://doi.org/10.1039/b801358f
Cotte, M., Szlachetko, J., Lahlil, S., Salomé, M., Solé, V.A., Biron, I., et al.: Coupling a wavelength dispersive spectrometer with a synchrotron-based X-ray microscope: a winning combination for micro-X-ray fluorescence and micro-XANES analyses of complex artistic materials. J. Anal. At. Spectrom. 26, 1051–1059 (2011). https://doi.org/10.1039/c0ja00217h
Cotte, M., Checroun, E., De Nolf, W., Taniguchi, Y., De Viguerie, L., Burghammer, M., et al.: Lead soaps in paintings: friends or foes? Stud. Conserv. 62, 2–23 (2017a). https://doi.org/10.1080/00393630.2016.1232529
Cotte, M., Pouyet, E., Salomé, M., Rivard, C., De Nolf, W., Castillo-Michel, H., et al.: The ID21 X-ray and infrared microscopy beamline at the ESRF: status and recent applications to artistic materials. J. Anal. At. Spectrom. 32, 477–493 (2017b). https://doi.org/10.1039/C6JA00356G
Couves, J.W., Thomas, J.M., Catlow, C.R.A., Greaves, G.N., Baker, G., Dent, A.J.: In situ studies of the dehydration of zeolitic catalysts by time-resolved energy-dispersive x-ray absorption spectroscopy. J. Phys. Chem. 94, 6517–6519 (1990). https://doi.org/10.1021/j100380a001
Craddock, P. (ed.): Scientific Investigation of Copies, Fakes and Forgeries. Butterworth-Heinemann, Oxford (2009)
Cucci, C., Delaney, J.K., Picollo, M.: Reflectance hyperspectral imaging for investigation of works of art: old master paintings and illuminated manuscripts. Acc. Chem. Res. 49, 2070–2079 (2016). https://doi.org/10.1021/acs.accounts.6b00048
Cushing, S.K., Meng, F., Zhang, J., Ding, B., Chen, C.K., Chen, C.-J., et al.: Effects of defects on photocatalytic activity of hydrogen-treated titanium oxide Nanobelts. ACS Catal. 7, 1742–1748 (2017). https://doi.org/10.1021/acscatal.6b02177
Dawson, P.H.: Quadrupoles for secondary ion mass spectrometry. Int. J. Mass Spectrom. 17, 447–467 (1975). https://doi.org/10.1016/0020-7381(75)80018-0
Dazzi, A., Prater, C.B.: AFM-IR: technology and applications in nanoscale infrared spectroscopy and chemical imaging. Chem. Rev. 117, 5146–5173 (2017). https://doi.org/10.1021/acs.chemrev.6b00448
Dazzi, A., Prazeres, R., Glotin, F., Ortega, J.M.: Local infrared microspectroscopy with subwavelength spatial resolution with an atomic force microscope tip used as a photothermal sensor. Opt. Lett. 30, 2388 (2005). https://doi.org/10.1364/OL.30.002388
Dazzi, A., Saunier, J., Kjoller, K., Yagoubi, N.: Resonance enhanced AFM-IR: a new powerful way to characterize blooming on polymers used in medical devices. Int. J. Pharm. 484, 109–114 (2015). https://doi.org/10.1016/j.ijpharm.2015.02.046
de la Rie, E.R., Michelin, A., Ngako, M., Del Federico, E., Del Grosso, C.: Photo-catalytic degradation of binding media of ultramarine blue containing paint layers: a new perspective on the phenomenon of “ultramarine disease” in paintings. Polym. Degrad. Stab. 144, 43–52 (2017). https://doi.org/10.1016/j.polymdegradstab.2017.08.002
Denecke, M.A., Brendebach, B., De Nolf, W., Falkenberg, G., Janssens, K., Simon, R.: Spatially resolved micro-X-ray fluorescence and micro-X-ray absorption fine structure study of a fractured granite bore core following a radiotracer experiment. Spectrochim. Acta Part B Atom. Spectrosc. 64, 791–795 (2009). https://doi.org/10.1016/j.sab.2009.05.025
Denk, W., Horstmann, H.: Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biol. 2, 1900–1909 (2004). https://doi.org/10.1371/journal.pbio.0020329
Dierolf, M., Menzel, A., Thibault, P., Schneider, P., Kewish, C.M., Wepf, R., et al.: Ptychographic X-ray computed tomography at the nanoscale. Nature. 467, 436–439 (2010). https://doi.org/10.1038/nature09419
Dik, J., Janssens, K., Van Der Snickt, G., van der Loeff, L., Rickers, K., Cotte, M.: Visualization of a lost painting by Vincent van Gogh using synchrotron radiation based X-ray fluorescence elemental mapping. Anal. Chem. 80, 6436–6442 (2008). https://doi.org/10.1021/ac800965g
Doménech-Carbó, M.T., Edwards, H.G.M., Doménech-Carbó, A., del Hoyo-Meléndez, J.M., de la Cruz-Cañizares, J.: An authentication case study: Antonio Palomino versus Vicente Guillo paintings in the vaulted ceiling of the Sant Joan del Mercat church (Valencia, Spain). J. Raman Spectrosc. 43, 1250–1259 (2012). https://doi.org/10.1002/jrs.3168
Drouilly, C., Krafft, J.-M., Averseng, F., Lauron-Pernot, H., Bazer-Bachi, D., Chizallet, C., et al.: Origins of the deactivation process in the conversion of methylbutynol on zinc oxide monitored by operando DRIFTS. Catal. Today. 205, 67–75 (2013). https://doi.org/10.1016/j.cattod.2012.08.011
Ebert, B., Macmillan, A.S., Singer, B.W., Grimaldi, N.: Analysis and conservation treatment of vietnamese paintings. In: ICOM-CC 16th Triennial Conference Lisbon 19–23 September 2011 Preprints (2011)
Ferreira, E.S.B., Boon, J.J., van der Horst, J., Scherrer, N.C., Marone, F., Stampanoni, M.: 3D synchrotron x-ray microtomography of paint samples. In: Pezzati, L., Salimbeni, R. (eds.) SPIE 7391, O3A Optics for Arts, Architecture, and Archaeology II, p. 73910L (2009). https://doi.org/10.1117/12.837366
Ferreira, E.S.B., Boon, J.J., Stampanoni, M., Marone, F.: Study of the mechanism of formation of calcium soaps in early 20th century easel paintings with correlative 2D and 3D microscopy. In: ICOM Lisbon 2011 Preprints 16th Triennial Conference Lisbon, 19–23 September 2011. ICOM (2011)
French, S.A., Sokol, A.A., Bromley, S.T., Catlow, C.R.A., Rogers, S.C., King, F., et al.: From CO2 to methanol by hybrid QM/MM embedding this work was supported by EU Esprit IV project 25047. S.A.F. is grateful to ICI and Synetix for funding. K. Waugh, L. Whitmore, S. Cristol, and P. Sushko are thanked for their helpful insights. QM/MM=quantum. Angew. Chem. Int. Ed. 40, 4437–4440 (2001). https://doi.org/10.1002/1521-3773(20011203)40:23<4437::AID-ANIE4437>3.0.CO;2-L
Gabrieli, F., Rosi, F., Vichi, A., Cartechini, L., Pensabene Buemi, L., Kazarian, S.G., et al.: Revealing the nature and distribution of metal carboxylates in Jackson Pollock’s Alchemy (1947) by micro-attenuated Total reflection FT-IR spectroscopic imaging. Anal. Chem. 89, 1283–1289 (2017). https://doi.org/10.1021/acs.analchem.6b04065
Gambardella, A.A., Cotte, M., de Nolf, W., Schnetz, K., Erdmann, R., van Elsas, R., et al.: Sulfur K-edge micro- and full-field XANES identify marker for preparation method of ultramarine pigment from lapis lazuli in historical paints. Sci. Adv. 6, eaay8782 (2020). https://doi.org/10.1126/sciadv.aay8782
Geldof, M., van der Werf, I.D., Haswell, R.: The examination of Van Gogh’s chrome yellow pigments in ‘Field with irises near Arles’ using quantitative SEM–WDX. Herit. Sci. 7, 100 (2019). https://doi.org/10.1186/s40494-019-0341-3
Gendrin, C., Roggo, Y., Collet, C.: Pharmaceutical applications of vibrational chemical imaging and chemometrics: a review. J. Pharm. Biomed. Anal. 48, 533–553 (2008). https://doi.org/10.1016/j.jpba.2008.08.014
Georgiou, R., Gueriau, P., Sahle, C.J., Bernard, S., Mirone, A., Garrouste, R., et al.: Carbon speciation in organic fossils using 2D to 3D x-ray Raman multispectral imaging. Sci. Adv. 5, eaaw5019 (2019). https://doi.org/10.1126/sciadv.aaw5019
Gervais, C., Boon, J.J., Marone, F., Ferreira, E.S.B.: Characterization of porosity in a 19th century painting ground by synchrotron radiation X-ray tomography. Appl. Phys. A Mater. Sci. Process. 111, 31–38 (2013a). https://doi.org/10.1007/s00339-012-7533-y
Gervais, C., Languille, M.-A., Reguer, S., Gillet, M., Vicenzi, E.P., Chagnot, S., et al.: “Live” Prussian blue fading by time-resolved X-ray absorption spectroscopy. Appl. Phys. A Mater. Sci. Process. 111, 15–22 (2013b). https://doi.org/10.1007/s00339-013-7581-y
Giacovazzo, C., et al.: Fundamentals of crystallography. Oxford University Press, Oxford/New York (2009). https://doi.org/10.1093/acprof:oso/9780199573653.001.0001
Glatzel, P., Bergmann, U.: High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes—electronic and structural information. Coord. Chem. Rev. 249, 65–95 (2005). https://doi.org/10.1016/j.ccr.2004.04.011
Glatzel, P., Jacquamet, L., Bergmann, U., de Groot, F.M.F., Cramer, S.P.: Site-selective EXAFS in mixed-valence compounds using high-resolution fluorescence detection: a study of iron in Prussian blue. Inorg. Chem. 41, 3121–3127 (2002). https://doi.org/10.1021/ic010709m
Goldstein, J.I., Newbury, D.E., Echlin, P., Joy, D.C., Lyman, C.E., Lifshin, E., et al.: Scanning Electron Microscopy and X-Ray Microanalysis. Springer US, Boston (2003). https://doi.org/10.1007/978-1-4615-0215-9
Gonzalez, V., Gourier, D., Calligaro, T., Toussaint, K., Wallez, G., Menu, M.: Revealing the origin and history of Lead-white pigments by their photoluminescence properties. Anal. Chem. 89, 2909–2918 (2017a). https://doi.org/10.1021/acs.analchem.6b04195
Gonzalez, V., Wallez, G., Calligaro, T., Cotte, M., De Nolf, W., Eveno, M., et al.: Synchrotron-based high angle resolution and high lateral resolution X-ray diffraction: revealing Lead white pigment qualities in old masters paintings. Anal. Chem. 89, 13203–13211 (2017b). https://doi.org/10.1021/acs.analchem.7b02949
Gonzalez, V., Cotte, M., Vanmeert, F., Nolf, W., Janssens, K.: X-ray diffraction mapping for cultural heritage science: a review of experimental configurations and applications. Chem. Eur. J. 26, 1703–1719 (2020). https://doi.org/10.1002/chem.201903284
Gueriau, P., Rueff, J.-P., Bernard, S., Kaddissy, J.A., Goler, S., Sahle, C.J., et al.: Noninvasive synchrotron-based X-ray Raman scattering discriminates carbonaceous compounds in ancient and historical materials. Anal. Chem. 89, 10819–10826 (2017). https://doi.org/10.1021/acs.analchem.7b02202
Guerrero Prado, P., Nguyen, M.K., Dumas, L., Cohen, S.X.: Three-dimensional imaging of flat natural and cultural heritage objects by a Compton scattering modality. J. Electron. Imaging. 26, 011026 (2017). https://doi.org/10.1117/1.JEI.26.1.011026
Hageraats, S., Keune, K., Réfrégiers, M., van Loon, A., Berrie, B., Thoury, M.: Synchrotron deep-UV photoluminescence imaging for the submicrometer analysis of chemically altered zinc white oil paints. Anal. Chem. 91, 14887–14895 (2019a). https://doi.org/10.1021/acs.analchem.9b02443
Hageraats, S., Keune, K., Thoury, M., Hoppe, R.: A synchrotron photoluminescence microscopy study into the use and degradation of zinc white in ‘the woodcutters’ by Bart van der. In: Conservation of Modern Oil Paintings, pp. 275–288. Springer International Publishing, Cham (2019b). https://doi.org/10.1007/978-3-030-19254-9_21
Heeren, R.M.A., Boon, J.J., Noble, P., Wadum, J.: Integrating imaging FTIR and secondary ion mass spectrometry for the analysis of embedded paint cross-sections. In: ICOM-CC Triennial Meeting, (12th), Lyon, 29 August-3 September 1999 Preprint, pp. 228–233 (1999)
Helmchen, F., Denk, W.: Deep tissue two-photon microscopy. Nat. Methods. 2, 932–940 (2005). https://doi.org/10.1038/nmeth818
Helwig K, Poulin J, Corbeil M-C, Moffatt E, Duguay D. Conservation issues in several twentieth-century Canadian oil paintings: the role of zinc carboxylate reaction products. In: van den Berg KJ, Burnstock A, de Keijzer M, Krueger J, Learner T, de Tagle A, et al.. Issues in Contemporary Oil Paint, Cham: Springer International Publishing; 2014, p. 167–184. doi: https://doi.org/10.1007/978-3-319-10100-2_11
Henderson, E.J., Helwig, K., Read, S., Rosendahl, S.M.: Infrared chemical mapping of degradation products in cross-sections from paintings and painted objects. Herit. Sci. 7, 71 (2019). https://doi.org/10.1186/s40494-019-0313-7
Hermans, J., Osmond, G., van Loon, A., Iedema, P., Chapman, R., Drennan, J., et al.: Electron microscopy imaging of zinc soaps nucleation in oil paint. Microsc. Microanal. 24, 318–322 (2018). https://doi.org/10.1017/S1431927618000387
Higuchi, S., Hamada, T., Gohshi, Y.: Examination of the photochemical curing and degradation of oil paints by laser Raman spectroscopy. Appl. Spectrosc. 51, 1218–1223 (1997). https://doi.org/10.1366/0003702971941782
Hughes, A.E., Trinchi, A., Chen, F.F., Yang, Y.S., Cole, I.S., Sellaiyan, S., et al.: The application of multiscale quasi 4D CT to the study of SrCrO4 distributions and the development of porous networks in epoxy-based primer coatings. Prog. Org. Coat. 77, 1946–1956 (2014). https://doi.org/10.1016/j.porgcoat.2014.07.001
Ice, G.E., Budai, J.D., Pang, J.W.L.: The race to X-ray microbeam and Nanobeam science. Science (80-). 334, 1234–1239 (2011). https://doi.org/10.1126/science.1202366
Kanngießer, B., Malzer, W., Reiche, I.: A new 3D micro X-ray fluorescence analysis set-up – first archaeometric applications. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact Mater. Atoms. 211, 259–264 (2003). https://doi.org/10.1016/S0168-583X(03)01321-1
Kanngießer, B., Malzer, W., Mantouvalou, I., Sokaras, D., Karydas, A.G.: A deep view in cultural heritage—confocal micro X-ray spectroscopy for depth resolved elemental analysis. Appl. Phys. A Mater. Sci. Process. 106, 325–338 (2012). https://doi.org/10.1007/s00339-011-6698-0
Keune, K., Boon, J.J.: Imaging secondary ion Mass spectrometry of a paint cross section taken from an early Netherlandish painting by Rogier van der Weyden. Anal. Chem. 76, 1374–1385 (2004). https://doi.org/10.1021/ac035201a
Keune, K., Boon, J.J.: Analytical imaging studies clarifying the process of the darkening of vermilion in paintings. Anal. Chem. 77, 4742–4750 (2005). https://doi.org/10.1021/ac048158f
Keune, K., Boon, J.J.: Analytical imaging studies of cross-sections of paintings affected by Lead soap aggregate formation. Stud. Conserv. 52, 161–176 (2007). https://doi.org/10.1179/sic.2007.52.3.161
Keune, K., Hoogland, F., Boon, J.J., Peggie, D., Higgitt, C.: Evaluation of the “added value” of SIMS: a mass spectrometric and spectroscopic study of an unusual Naples yellow oil paint reconstruction. Int. J. Mass Spectrom. 284, 22–34 (2009). https://doi.org/10.1016/j.ijms.2008.10.016
Keune, K., van Loon, A., Boon, J.J.: SEM backscattered-electron images of paint cross sections as information source for the presence of the Lead white pigment and Lead-related degradation and migration phenomena in oil paintings. Microsc. Microanal. 17, 696–701 (2011). https://doi.org/10.1017/S1431927610094444
Keune, K., Boon, J.J., Boitelle, R., Shimadzu, Y.: Degradation of Emerald green in oil paint and its contribution to the rapid change in colour of the Descente des vaches (1834–1835) painted by Théodore Rousseau. Stud. Conserv. 58, 199–210 (2013). https://doi.org/10.1179/2047058412Y.0000000063
Keune, K., Mass, J., Meirer, F., Pottasch, C., van Loon, A., Hull, A., et al.: Tracking the transformation and transport of arsenic sulfide pigments in paints: synchrotron-based X-ray micro-analyses. J. Anal. At. Spectrom. 30, 813–827 (2015). https://doi.org/10.1039/C4JA00424H
Keune, K., Mass, J., Mehta, A., Church, J., Meirer, F.: Analytical imaging studies of the migration of degraded orpiment, realgar, and emerald green pigments in historic paintings and related conservation issues. Herit. Sci. 4, 10 (2016). https://doi.org/10.1186/s40494-016-0078-1
Kidder, L.H., Levin, I.W., Lewis, E.N., Kleiman, V.D., Heilweil, E.J.: Mercury cadmium telluride focal-plane array detection for mid-infrared Fourier-transform spectroscopic imaging. Opt. Lett. 22, 742–744 (1997). https://doi.org/10.1364/OL.22.000742
Kirkpatrick, P., Baez, A.V.: Formation of optical images by X-rays. J. Opt. Soc. Am. 38, 766–774 (1948). https://doi.org/10.1364/JOSA.38.000766
Kuckova, S., Hynek, R., Kodicek, M.: Identification of proteinaceous binders used in artworks by MALDI-TOF mass spectrometry. Anal. Bioanal. Chem. 388, 201–206 (2007). https://doi.org/10.1007/s00216-007-1206-2
Kurtz, M., Strunk, J., Hinrichsen, O., Muhler, M., Fink, K., Meyer, B., et al.: Active sites on oxide surfaces: ZnO-catalyzed synthesis of methanol from CO and H 2. Angew. Chem. Int. Ed. 44, 2790–2794 (2005). https://doi.org/10.1002/anie.200462374
Lau, D., Villis, C., Furman, S., Livett, M.: Multispectral and hyperspectral image analysis of elemental and micro-Raman maps of cross-sections from a 16th century painting. Anal. Chim. Acta. 610, 15–24 (2008). https://doi.org/10.1016/j.aca.2007.12.043
Levenson, E., Lerch, P., Martin, M.C.: Infrared imaging: synchrotrons vs. arrays, resolution vs. speed. Infrared Phys. Technol. 49, 45–52 (2006). https://doi.org/10.1016/j.infrared.2006.01.026
Li, J., Guériau, P., Bellato, M., King, A., Robbiola, L., Thoury, M., et al.: Synchrotron-based phase mapping in corroded metals: insights from early Copper-Base artifacts. Anal. Chem. 91, 1815–1825 (2019). https://doi.org/10.1021/acs.analchem.8b02744
Ma, X., Beltran, V., Ramer, G., Pavlidis, G., Parkinson, D.Y., Thoury, M., et al.: Revealing the distribution of metal carboxylates in oil paint from the micro- to nanoscale. Angew. Chem. Int. Ed. 58, 11652–11656 (2019). https://doi.org/10.1002/anie.201903553
Mahon, D., Centeno, S.A., Iacono, M., Carό, F., Stege, H., Obermeier, A.: Johannes Vermeer’s mistress and maid: new discoveries cast light on changes to the composition and the discoloration of some paint passages. Herit. Sci. 8, 30 (2020). https://doi.org/10.1186/s40494-020-00375-2
Maldanis, L., Hickman-Lewis, K., Verezhak, M., Gueriau, P., Guizar-Sicairos, M., Jaqueto, P., et al.: Nanoscale 3D quantitative imaging of 1.88 Ga Gunflint microfossils reveals novel insights into taphonomic and biogenic characters. Sci. Rep. 10, 8163 (2020). https://doi.org/10.1038/s41598-020-65176-w
Mantler, M., Schreiner, M.: X-ray fluorescence spectrometry in art and archaeology. X-Ray Spectrom. 29, 3–17 (2000). https://doi.org/10.1002/(SICI)1097-4539(200001/02)29:1<3::AID-XRS398>3.0.CO;2-O
Marchenko, T., Journel, L., Marin, T., Guillemin, R., Carniato, S., Žitnik, M., et al.: Resonant inelastic x-ray scattering at the limit of subfemtosecond natural lifetime. J. Chem. Phys. 134, 144308 (2011). https://doi.org/10.1063/1.3575514
Mass, J., Sedlmair, J., Patterson, C.S., Carson, D., Buckley, B., Hirschmugl, C.: SR-FTIR imaging of the altered cadmium sulfide yellow paints in Henri Matisse’s Le Bonheur de vivre (1905–6) – examination of visually distinct degradation regions. Analyst. 138, 6032 (2013a). https://doi.org/10.1039/c3an00892d
Mass, J.L., Opila, R., Buckley, B., Cotte, M., Church, J., Mehta, A.: The photodegradation of cadmium yellow paints in Henri Matisse’s le Bonheur de vivre (1905–1906). Appl. Phys. A Mater. Sci. Process. (2013b). https://doi.org/10.1007/s00339-012-7418-0
Massonnet, P., Heeren, R.M.A.: A concise tutorial review of TOF-SIMS based molecular and cellular imaging. J. Anal. At. Spectrom. 34, 2217–2228 (2019). https://doi.org/10.1039/C9JA00164F
Mazzeo, R., Joseph, E., Prati, S., Millemaggi, A.: Attenuated Total reflection–Fourier transform infrared microspectroscopic mapping for the characterisation of paint cross-sections. Anal. Chim. Acta. 599, 107–117 (2007). https://doi.org/10.1016/j.aca.2007.07.076
Mazzeo, R., Prati, S., Quaranta, M., Joseph, E., Kendix, E., Galeotti, M.: Attenuated total reflection micro FTIR characterisation of pigment–binder interaction in reconstructed paint films. Anal. Bioanal. Chem. 392, 65–76 (2008). https://doi.org/10.1007/s00216-008-2126-5
Meilunas, R.J., Bentsen, J.G., Steinberg, A.: Analysis of aged paint binders by FTIR spectroscopy. Stud. Conserv. 35, 33–51 (1990). https://doi.org/10.1179/sic.1990.35.1.33
Meirer, F., Cabana, J., Liu, Y., Mehta, A., Andrews, J.C., Pianetta, P.: Three-dimensional imaging of chemical phase transformations at the nanoscale with full-field transmission X-ray microscopy. J. Synchrotron Radiat. 18, 773–781 (2011). https://doi.org/10.1107/S0909049511019364
Mills, L., Burnstock, A., Duarte, F., De Groot, S., Margens, L., Bisschoff, M., et al.: Water sensitivity of modern artists’ oil paints. In: ICOM Preprint International Council Museums-Committee for Conservation 15th Triennial Conference New Delhi, pp. 651–659. Allied Publishers PVT, New Delhi (2008)
Monico, L., Van der Snickt, G., Janssens, K., De Nolf, W., Miliani, C., Verbeeck, J., et al.: Degradation process of Lead chromate in paintings by Vincent van Gogh studied by means of synchrotron X-ray Spectromicroscopy and related methods. 1. Artificially aged model samples. Anal. Chem. 83, 1214–1223 (2011). https://doi.org/10.1021/ac102424h
Monico, L., Janssens, K., Miliani, C., Brunetti, B.G., Vagnini, M., Vanmeert, F., et al.: Degradation process of Lead chromate in paintings by Vincent van Gogh studied by means of Spectromicroscopic methods. 3. Synthesis, characterization, and detection of different crystal forms of the chrome yellow pigment. Anal. Chem. 85, 851–859 (2013). https://doi.org/10.1021/ac302158b
Monico, L., Janssens, K., Hendriks, E., Brunetti, B.G., Miliani, C.: Raman study of different crystalline forms of PbCrO 4 and PbCr 1−x S x O 4 solid solutions for the noninvasive identification of chrome yellows in paintings: a focus on works by Vincent van Gogh. J. Raman Spectrosc. 45, 1034–1045 (2014). https://doi.org/10.1002/jrs.4548
Monico, L., Janssens, K., Alfeld, M., Cotte, M., Vanmeert, F., Ryan, C.G., et al.: Full spectral XANES imaging using the Maia detector array as a new tool for the study of the alteration process of chrome yellow pigments in paintings by Vincent van Gogh. J. Anal. At. Spectrom. 30, 613–626 (2015a). https://doi.org/10.1039/C4JA00419A
Monico, L., Janssens, K., Hendriks, E., Vanmeert, F., der Snickt, G., Cotte, M., et al.: Evidence for degradation of the chrome yellows in van Gogh’s sunflowers : a study using noninvasive in situ methods and synchrotron-radiation-based X-ray techniques. Angew. Chem. Int. Ed. 54, 13923–13927 (2015b). https://doi.org/10.1002/anie.201505840
Monico, L., Janssens, K., Cotte, M., Sorace, L., Vanmeert, F., Brunetti, B.G., et al.: Chromium speciation methods and infrared spectroscopy for studying the chemical reactivity of lead chromate-based pigments in oil medium. Microchem. J. 124, 272–282 (2016). https://doi.org/10.1016/j.microc.2015.08.028
Monico, L., Cartechini, L., Rosi, F., Chieli, A., Grazia, C., De Meyer, S., et al.: Probing the chemistry of CdS paints in the scream by in situ noninvasive spectroscopies and synchrotron radiation x-ray techniques. Sci. Adv. 6, eaay3514 (2020a). https://doi.org/10.1126/sciadv.aay3514
Monico, L., Cotte, M., Vanmeert, F., Amidani, L., Janssens, K., Nuyts, G., et al.: Damages induced by synchrotron radiation-based X-ray microanalysis in chrome yellow paints and related Cr-compounds: assessment, quantification, and mitigation strategies. Anal. Chem. 92, 14164–14173 (2020b). https://doi.org/10.1021/acs.analchem.0c03251
Morsch, S., van Driel, B.A., van den Berg, K.J., Dik, J.: Investigating the photocatalytic degradation of oil paint using ATR-IR and AFM-IR. ACS Appl. Mater. Interfaces. 9, 10169–10179 (2017). https://doi.org/10.1021/acsami.7b00638
Nevin, A., Cesaratto, A., Bellei, S., D’Andrea, C., Toniolo, L., Valentini, G., Comelli, D.: Time-resolved photoluminescence spectroscopy and imaging: New approaches to the analysis of cultural heritage and its degradation. Sensors. 14(4), 6338–6355 (2014). https://doi.org/10.3390/s140406338
Nevin, A., Comelli, D., Valentini, G., Cubeddu, R.: Total synchronous fluorescence spectroscopy combined with multivariate analysis: method for the classification of selected resins, oils, and protein-based media used in paintings. Anal. Chem. 81, 1784–1791 (2009). https://doi.org/10.1021/ac8019152
Niehuis, E., Heller, T., Feld, H., Benninghoven, A.: Design and performance of a reflectron based time-of-flight secondary ion mass spectrometer with electrodynamic primary ion mass separation. J. Vac. Sci. Technol. A Vac. Surf. Film. 5, 1243–1246 (1987). https://doi.org/10.1116/1.574781
Noble, P., Boon, J.J., Wadum, J.: Dissolution, aggregation, and protrusion: Lead soap formation in 17th century grounds and paint layers. Art Matters. 1, 46–61 (2002)
Noble, P., Van Duijn, E., Hermens, E., Keune, K., Van Loon, A., Smelt, S., et al.: An exceptional commission. Rijksmus Bull. 66, 308–345 (2018). https://doi.org/10.52476/trb.9762
Norton, S.J.: Compton scattering tomography. J. Appl. Phys. 76, 2007–2015 (1994). https://doi.org/10.1063/1.357668
Noun, M., Van Elslande, E., Touboul, D., Glanville, H., Bucklow, S., Walter, P., et al.: High mass and spatial resolution mass spectrometry imaging of Nicolas Poussin painting cross section by cluster TOF-SIMS. J. Mass Spectrom. 51, 1196–1210 (2016). https://doi.org/10.1002/jms.3885
Opilik, L., Schmid, T., Zenobi, R.: Modern Raman imaging: vibrational spectroscopy on the micrometer and nanometer scales. Annu. Rev. Anal. Chem. 6, 379–398 (2013). https://doi.org/10.1146/annurev-anchem-062012-092646
Osmond, G.: Zinc white and the influence of paint composition for stability in oil based media. In: Issues in Contemporary Oil Paint, pp. 263–281. Springer International Publishing, Cham (2014). https://doi.org/10.1007/978-3-319-10100-2_18
Osmond, G., Boon, J.J., Puskar, L., Drennan, J.: Metal stearate distributions in modern artists’ oil paints: surface and cross-sectional investigation of reference paint films using conventional and synchrotron infrared microspectroscopy. Appl. Spectrosc. 66, 1136–1144 (2012). https://doi.org/10.1366/12-06659
Osmond, G., Ebert, B., Drennan, J.: Zinc oxide-centred deterioration in 20th century Vietnamese paintings by Nguyễ Trọng Kiệm (1933–1991). AICCM Bull. 34, 4–14 (2013). https://doi.org/10.1179/bac.2013.34.1.002
Otero, V., Sanches, D., Montagner, C., Vilarigues, M., Carlyle, L., Lopes, J.A., et al.: Characterisation of metal carboxylates by Raman and infrared spectroscopy in works of art. J. Raman Spectrosc. 45, 1197–1206 (2014). https://doi.org/10.1002/jrs.4520
Pan, X., Yang, M.-Q., Fu, X., Zhang, N., Xu, Y.-J.: Defective TiO2 with oxygen vacancies: synthesis, properties and photocatalytic applications. Nanoscale. 5, 3601–3614 (2013). https://doi.org/10.1039/c3nr00476g
Papillon, M., Lefevre, R., Lahanier, C., Duval, A., Rioux, J.: Analyses de pigments blancs appliquées à l’étude chronologique des peintures de chevalet—blanc de titane. In: Grimstad, K. (ed.) Committee for Conservation; 8th Triennial Meeting. Getty Conservation Institute, Los Angeles (1987)
Pascarelli, S., Mathon, O., Mairs, T., Kantor, I., Agostini, G., Strohm, C., et al.: The time-resolved and extreme-conditions XAS (TEXAS) facility at the European Synchrotron Radiation Facility: the energy-dispersive X-ray absorption spectroscopy beamline ID24. J. Synchrotron Radiat. 23, 353–368 (2016). https://doi.org/10.1107/S160057751501783X
Penner-Hahn, J.E.: X-ray absorption spectroscopy in coordination chemistry. Coord. Chem. Rev. 190–192, 1101–1123 (1999). https://doi.org/10.1016/S0010-8545(99)00160-5
Pouyet, E., Cotte, M., Fayard, B., Salomé, M., Meirer, F., Mehta, A., et al.: 2D X-ray and FTIR micro-analysis of the degradation of cadmium yellow pigment in paintings of Henri Matisse. Appl. Phys. A Mater. Sci. Process. 121, 967–980 (2015). https://doi.org/10.1007/s00339-015-9239-4
Price, S.W.T., Van Loon, A., Keune, K., Parsons, A.D., Murray, C., Beale, A.M., et al.: Unravelling the spatial dependency of the complex solid-state chemistry of Pb in a paint micro-sample from Rembrandt’s Homer using XRD-CT. Chem. Commun. 55, 1931–1934 (2019). https://doi.org/10.1039/C8CC09705D
Radepont, M., de Nolf, W., Janssens, K., Van der Snickt, G., Coquinot, Y., Klaassen, L., et al.: The use of microscopic X-ray diffraction for the study of HgS and its degradation products corderoite (α-Hg3S2Cl2), kenhsuite (γ-Hg3S2Cl2) and calomel (Hg2Cl2) in historical paintings. J. Anal. At. Spectrom. 26, 959–968 (2011). https://doi.org/10.1039/c0ja00260g
Rehr, J.J., Albers, R.C.: Theoretical approaches to x-ray absorption fine structure. Rev. Mod. Phys. 72, 621–654 (2000). https://doi.org/10.1103/RevModPhys.72.621
Reynaud, C., Thoury, M., Dazzi, A., Latour, G., Scheel, M., Li, J., et al.: In-place molecular preservation of cellulose in 5,000-year-old archaeological textiles. Proc. Natl. Acad. Sci. 117, 19670–19676 (2020). https://doi.org/10.1073/pnas.2004139117
Richardin, P., Mazel, V., Walter, P., Laprévote, O., Brunelle, A.: Identification of different copper green pigments in renaissance paintings by cluster-TOF-SIMS imaging analysis. J. Am. Soc. Mass Spectrom. 22, 1729–1736 (2011). https://doi.org/10.1007/s13361-011-0171-3
Robertson, D.W.: Lead Titanate. Ind. Eng. Chem. 28, 216–218 (1936). https://doi.org/10.1021/ie50314a017
Robinet, L., Spring, M., Pagès-Camagna, S., Vantelon, D., Trcera, N.: Investigation of the discoloration of smalt pigment in historic paintings by micro-X-ray absorption spectroscopy at the Co K-Edge. Anal. Chem. 83, 5145–5152 (2011). https://doi.org/10.1021/ac200184f
Rogala, D., Lake, S., Maines, C., Mecklenburg, M.: Condition problems related to zinc oxide Underlayers: examination of selected abstract expressionist paintings from the collection of the Hirshhorn Museum and Sculpture Garden, Smithsonian Institution. J. Am. Inst. Conserv. 49, 96–113 (2010). https://doi.org/10.1179/019713610804489937
Romano, C., Lam, T., Newsome, G.A., Taillon, J.A., Little, N., Tsang, J.: Characterization of zinc carboxylates in an oil paint test panel. Stud. Conserv. 65, 14–27 (2020). https://doi.org/10.1080/00393630.2019.1666467
Ropret, P., Miliani, C., Centeno, S.A., Tavzes, Č., Rosi, F.: Advances in Raman mapping of works of art. J. Raman Spectrosc. 41, 1462–1467 (2010). https://doi.org/10.1002/jrs.2733
Rorimer, J.: Marble sculpture and the ultra-violet ray. Metrop. Mus. Art Bull. 24, 185–186 (1929)
Rorimer, J.J.: Ultra-Violet Rays and Their Use in the Examination of Works of Art. Metropolitan Museum of Art, New York (1931)
Rösner, B., Koch, F., Döring, F., Bosgra, J., Guzenko, V.A., Kirk, E., et al.: Exploiting atomic layer deposition for fabricating sub-10 nm X-ray lenses. Microelectron. Eng. 191, 91–96 (2018). https://doi.org/10.1016/j.mee.2018.01.033
Sabatini, F., Lluveras-Tenorio, A., Degano, I., Kuckova, S., Krizova, I., Colombini, M.P.: A matrix-assisted laser desorption/ionization time-of-flight Mass spectrometry method for the identification of Anthraquinones: the case of Historical Lakes. J. Am. Soc. Mass Spectrom. 27 (2016). https://doi.org/10.1007/s13361-016-1471-4
Sahle, C.J., Mirone, A., Vincent, T., Kallonen, A., Huotari, S.: Improving the spatial and statistical accuracy in X-ray Raman scattering based direct tomography. J. Synchrotron Radiat. 24, 476–481 (2017a). https://doi.org/10.1107/S1600577517000169
Sahle, C.J., Rosa, A.D., Rossi, M., Cerantola, V., Spiekermann, G., Petitgirard, S., et al.: Direct tomography imaging for inelastic X-ray scattering experiments at high pressure. J. Synchrotron Radiat. 24, 269–275 (2017b). https://doi.org/10.1107/S1600577516017100
Sakdinawat, A., Attwood, D.: Nanoscale X-ray imaging. Nat. Photonics. 4, 840–848 (2010). https://doi.org/10.1038/nphoton.2010.267
Salvadó, N., Butí, S., Nicholson, J., Emerich, H., Labrador, A., Pradell, T.: Identification of reaction compounds in micrometric layers from gothic paintings using combined SR-XRD and SR-FTIR. Talanta. 79, 419–428 (2009). https://doi.org/10.1016/j.talanta.2009.04.005
Salzer, R.W., Siesler, H. (eds.): Infrared and Raman Spectroscopic Imaging. John Wiley & Sons, Ltd, Weinheim (2009)
San Andres, M., Baez, M.I., Baldonedo, J.L., Barba, C.: Transmission electron microscopy applied to the study of works of art: sample preparation methodology and possible techniques. J. Microsc. 188, 42–50 (1997). https://doi.org/10.1046/j.1365-2818.1997.2460804.x
Sanyova, J., Cersoy, S., Richardin, P., Laprévote, O., Walter, P., Brunelle, A.: Unexpected materials in a Rembrandt painting characterized by high spatial resolution cluster-TOF-SIMS imaging. Anal. Chem. 83, 753–760 (2011). https://doi.org/10.1021/ac1017748
Schaeberle, M.D., Morris, H.R., Ii, J.F.T., Treado, P.J.: Peer reviewed: Raman chemical imaging spectroscopy. Anal. Chem. 71, 175A–181A (1999). https://doi.org/10.1021/ac990251u
Schiering, D.W., Tague, T.J., Reffner, J.A., Vogel, S.H.: A dual confocal aperturing microscope for IR microspectrometry. Analusis. 28, 46–52 (2000). https://doi.org/10.1051/analusis:2000280046
Schwartz, A.J., Kumar, M., Adams, B.L., Field, D.P. (eds.): Electron Backscatter Diffraction in Materials Science. Springer US, Boston (2009). https://doi.org/10.1007/978-0-387-88136-2
Shearer, J.C., Peters, D.C., Hoepfner, G., Newton, T.: FTIR in the service of art conservation. Anal. Chem. 55, 874A–880A (1983). https://doi.org/10.1021/ac00259a002
Simoen, J., De Meyer, S., Vanmeert, F., de Keyser, N., Avranovich, E., Van der Snickt, G., et al.: Combined micro- and macro scale X-ray powder diffraction mapping of degraded orpiment paint in a 17th century still life painting by Martinus Nellius. Herit. Sci. 7, 83 (2019). https://doi.org/10.1186/s40494-019-0324-4
Sloggett, R.: Unmasking art forgery: scientific approaches. In: The Palgrave Handbook on Art Crime, pp. 381–406. Palgrave Macmillan UK, London (2019). https://doi.org/10.1057/978-1-137-54405-6_19
Solé, V.A., Papillon, E., Cotte, M., Walter, P., Susini, J.: A multiplatform code for the analysis of energy-dispersive X-ray fluorescence spectra. Spectrochim. Acta Part B Atom. Spectrosc. 62, 63–68 (2007). https://doi.org/10.1016/j.sab.2006.12.002
Spring, M., Grout, R.: The blackening of Vermillion: an analytical study of the process in paintings. Natl. Gall Tech. Bull. 23, 50–61 (2002)
Spring, M., Ricci, C., Peggie, D.A., Kazarian, S.G.: ATR-FTIR imaging for the analysis of organic materials in paint cross sections: case studies on paint samples from the National Gallery, London. Anal. Bioanal. Chem. 392, 37–45 (2008). https://doi.org/10.1007/s00216-008-2092-y
Sun, T., Ding, X., Liu, Z., Zhu, G., Li, Y., Wei, X., et al.: Characterization of a confocal three-dimensional micro X-ray fluorescence facility based on polycapillary X-ray optics and Kirkpatrick–Baez mirrors. Spectrochim. Acta Part B Atom. Spectrosc. 63, 76–80 (2008). https://doi.org/10.1016/j.sab.2007.11.003
Tan, H., Tian, H., Verbeeck, J., Monico, L., Janssens, K., Van Tendeloo, G.: Nanoscale investigation of the degradation mechanism of a historical chrome yellow paint by quantitative electron energy loss spectroscopy mapping of chromium species. Angew. Chem. Int. Ed. 52, 11360–11363 (2013). https://doi.org/10.1002/anie.201305753
Tanasa, P.O., Sandu, I., Vasilache, V., Sandu, I.G., Negru, I.C., Sandu, A.V.: Authentication of a painting by Nicolae Grigorescu using modern multi-analytical methods. Appl. Sci. 10, 3558 (2020). https://doi.org/10.3390/app10103558
Thoury, M., Echard, J.-P., Réfrégiers, M., Berrie, B., Nevin, A., Jamme, F., et al.: Synchrotron UV−visible multispectral luminescence microimaging of historical samples. Anal. Chem. 83, 1737–1745 (2011). https://doi.org/10.1021/ac102986h
Thoury, M., Van Loon, A., Keune, K., Hermans, J.J., Réfrégiers, M., Berrie, B.H.: Photoluminescence micro-imaging sheds new light on the development of metal soaps in oil paintings. In: Casadio, F., Keune, K., Noble, P., Van Loon, A., Hendriks, E., Centeno, S., et al. (eds.) Metal Soaps in Art, pp. 211–225 (2019). https://doi.org/10.1007/978-3-319-90617-1_12
Trentelman, K., Stodulski, L., Pavlosky, M.: Characterization of Pararealgar and other light-induced transformation products from Realgar by Raman microspectroscopy. Anal. Chem. 68, 1755–1761 (1996). https://doi.org/10.1021/ac951097o
van den Berg, J.D.J., van den Berg, K.J., Boon, J.J.: Chemical changes in curing and ageing oil paints. In: ICOM Committee for Conservation, 12th Triennial Meeting. Lyon 29 August – 3 September 1999. James & James (Science Publishers) Ltd, Lyon (1999)
Van Der Snickt, G., Dik, J., Cotte, M., Janssens, K., Jaroszewicz, J., De Nolf, W., et al.: Characterization of a degraded cadmium yellow (CdS) pigment in an oil painting by means of synchrotron radiation based X-ray techniques. Anal. Chem. 81, 2600–2610 (2009). https://doi.org/10.1021/ac802518z
Van Der Snickt, G., Janssens, K., Dik, J., De Nolf, W., Vanmeert, F., Jaroszewicz, J., et al.: Combined use of synchrotron radiation based micro-X-ray fluorescence, micro-X-ray diffraction, micro-X-ray absorption near-edge, and micro-Fourier transform infrared spectroscopies for revealing an alternative degradation pathway of the pigment cadmium ye. Anal. Chem. 84, 10221–10228 (2012). https://doi.org/10.1021/ac3015627
Van der Snickt, G., Dubois, H., Sanyova, J., Legrand, S., Coudray, A., Glaude, C., et al.: Large-area elemental imaging reveals Van Eyck’s original paint layers on the Ghent altarpiece (1432), Rescoping its conservation treatment. Angew. Chem. Int. Ed. 56, 4797–4801 (2017). https://doi.org/10.1002/anie.201700707
Van der Weerd, J., Geldof, M., Van der Loeff, L., Heeren, R., Boon, J.J.: Zinc soap aggregate formation in ‘falling leaves’ (Les Alyscamps)’ by Vincent van Gogh. Zeitschrift Für Kunsttechnologie Und Konserv. 17, 407–416 (2004)
van Driel, B.A., Kooyman, P.J., van den Berg, K.J., Schmidt-Ott, A., Dik, J.: A quick assessment of the photocatalytic activity of TiO2 pigments — from lab to conservation studio! Microchem. J. 126, 162–171 (2016). https://doi.org/10.1016/j.microc.2015.11.048
van Driel, B., Artesani, A., van den Berg, K.J., Dik, J., Mosca, S., Rossenaar, B., et al.: New insights into the complex photoluminescence behaviour of titanium white pigments. Dyes Pigments. 155, 14–22 (2018). https://doi.org/10.1016/j.dyepig.2018.03.012
Van Loon, A., Noble, P., Boon, J.J.: White hazes and surface crusts in Rembrandt’s homer and related paintings. In: ICOM-CC 16th Triennial Conference Lisbon 19–23 September 2011 Preprint. ICOM (2011)
Van Loon, A., Hoppe, R., Keune, K., Hermans, J.J., Diependaal, H., Bisschoff, M., et al.: Paint delamination as a result of zinc soap formation in an early Mondrian painting. In: Casadio, F., Keune, K., Noble, P., Van Loon, A., Hendriks, E., Centeno, S., et al. (eds.) Metal Soaps in Art, pp. 359–373. Springer, Houten (2019). https://doi.org/10.1007/978-3-319-90617-1_21
Vandivere, A., van Loon, A., Callewaert, T., Haswell, R., Proaño Gaibor, A.N., van Keulen, H., et al.: Fading into the background: the dark space surrounding Vermeer’s girl with a pearl earring. Herit. Sci. 7, 69 (2019a). https://doi.org/10.1186/s40494-019-0311-9
Vandivere, A., van Loon, A., Dooley, K.A., Haswell, R., Erdmann, R.G., Leonhardt, E., et al.: Revealing the painterly technique beneath the surface of Vermeer’s girl with a pearl earring using macro- and microscale imaging. Herit. Sci. 7, 64 (2019b). https://doi.org/10.1186/s40494-019-0308-4
Vanmeert, F., Van der Snickt, G., Janssens, K.: Plumbonacrite identified by X-ray powder diffraction tomography as a missing link during degradation of red Lead in a van Gogh painting. Angew. Chem. Int. Ed. 54, 3607–3610 (2015). https://doi.org/10.1002/anie.201411691
Vanmeert, F., De Nolf, W., De Meyer, S., Dik, J., Janssens, K.: Macroscopic X-ray powder diffraction scanning, a new method for highly selective chemical imaging of works of art: instrument optimization. Anal. Chem. 90, 6436–6444 (2018). https://doi.org/10.1021/acs.analchem.8b00240
Villafana, T.E., Brown, W.P., Delaney, J.K., Palmer, M., Warren, W.S., Fischer, M.C.: Femtosecond pump-probe microscopy generates virtual cross-sections in historic artwork. Proc. Natl. Acad. Sci. 111, 1708–1713 (2014). https://doi.org/10.1073/pnas.1317230111
Voras, Z.E., DeGhetaldi, K., Wiggins, M.B., Buckley, B., Baade, B., Mass, J.L., et al.: ToF–SIMS imaging of molecular-level alteration mechanisms in Le Bonheur de vivre by Henri Matisse. Appl. Phys. A Mater. Sci. Process. 121, 1015–1030 (2015). https://doi.org/10.1007/s00339-015-9508-2
Welcomme, E., Walter, P., Bleuet, P., Hodeau, J.-L., Dooryhee, E., Martinetto, P., et al.: Classification of lead white pigments using synchrotron radiation micro X-ray diffraction. Appl. Phys. A Mater. Sci. Process. 89, 825–832 (2007). https://doi.org/10.1007/s00339-007-4217-0
Wille, G., Bourrat, X., Maubec, N., Lahfid, A.: Raman-in-SEM, a multimodal and multiscale analytical tool: performance for materials and expertise. Micron. 67, 50–64 (2014). https://doi.org/10.1016/j.micron.2014.06.008
Williams, D.B., Carter, C.B.: Transmission Electron Microscopy. Springer US, Boston (2009). https://doi.org/10.1007/978-0-387-76501-3
Yu, H., Winkler, S.: Image complexity and spatial information. In: 2013 Fifth International Workshop on Quality of Multimedia Experience, pp. 12–17. IEEE (2013)
Yu, J., Warren, W.S., Fischer, M.C.: Visualization of vermilion degradation using pump-probe microscopy. Sci. Adv. 5, eaaw3136 (2019). https://doi.org/10.1126/sciadv.aaw3136
Zhang, M., Averseng, F., Haque, F., Borghetti, P., Krafft, J.-M., Baptiste, B., et al.: Defect-related multicolour emissions in ZnO smoke: from violet, over green to yellow. Nanoscale. 11, 5102–5115 (2019). https://doi.org/10.1039/C8NR09998G
Zhao, H., Pan, F., Li, Y.: A review on the effects of TiO2 surface point defects on CO2 photoreduction with H2O. J. Mater. 3, 17–32 (2017). https://doi.org/10.1016/j.jmat.2016.12.001
Zumbühl, S., Scherrer, N.C., Eggenberger, U.: Derivatization technique to increase the spectral selectivity of two-dimensional Fourier transform infrared focal plane Array imaging: analysis of binder composition in aged oil and tempera paint. Appl. Spectrosc. 68, 458–465 (2014). https://doi.org/10.1366/13-07280
Acknowledgements
The authors would like to express their gratitude to The Bennink Foundation for funding this research, to Edwin Verweij for providing the sample from the window frame of De Witte Roos in Delft, and to Dawn Rogala, Frederik Vanmeert, and Xiao Ma for providing figures for and/or proofreading the examples of SEM-EDX, XRD tomography and AFM-IR. They also warmly thank ESRF for granting in-house beamtime and Manfred Burghammer and Wout de Nolf for their support in data acquisition and data processing. SH, MT, LB and KK acknowledge support from the France–Netherlands Van Gogh program (Partenariat Hubert Curien MEAE/MESRI and NUFFIC).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Glossary
Glossary
Concept/abbreviation | Description |
Absorption edge | Distinct step in the absorption spectrum of a compound, corresponding to element-specific electronic transitions from a core-level orbital to other core-level orbitals, valence-level orbitals, and continuum states. |
AFM | Atomic force microscopy |
Annular detector | A ring-shaped detector primarily used to collect signal from a large solid angle, while permitting transmission of an excitation beam through the center. |
ATR | Attenuated total reflectance |
Band gap | Property of a semiconductor: the gap between the highest occupied and lowest unoccupied energy levels. The bandgap strongly influences the ability of the semiconductor to absorb (or emit) ultraviolet, visible, and /or infrared light. |
CCD detector | Charge-coupled device: a 2D photodetector array used primarily for the detection of ultraviolet and visible light. See also: CMOS detector. |
Chemical contrast | Ambiguous term, here defined as: differences in the signal recorded by a measurement modality from different chemical compounds. |
Chemical specificity | Ambiguous term, here defined as: the ability of a measurement modality to distinguish different chemical compounds. A modality with a high chemical specificity is capable of recording signal from specific compounds without interference from any others. |
CMOS detector | Complementary metal oxide semiconductor: a 2D photodetector array used primarily for the detection of ultraviolet and visible light. Similar to, but operated differently than CCD detectors. |
Compton scattering | An inelastic scattering effect in which energy is transferred from an X-ray photon to an electron. |
Core-level transition | An electronic transition in which the lower energy level corresponds to a tightly bound core-orbital. Core-level transition energies are diagnostic of certain elements. |
Cross-section (of a process) | The probability that a certain process will take place. Primarily used in the context of scattering, absorption, and emission processes. |
Energy-dispersive detection | Ambiguous term. In the context of SEM-EDX: a means of detecting photon energy directly by probing the amount of ionization the photon produces in a detector material. In the context of energy-dispersive XAS: a detection method in which the energies in a polychromatic X-ray beam are spatially dispersed and detected separately on a detector array. See also: wavelength-dispersive detection. |
EXAFS | Extended X-ray absorption fine structure |
Far-field | An approach in optics in which the distance between the sample and the collection or focusing elements is far greater than the wavelength of the photons being used. |
FTIR | Fourier-transform infrared |
Full-field imaging | An approach to imaging in which a whole region-of-interest is projected onto a 2D detector array at once. |
Hyperspectral | An approach to spectral imaging in which full spectra are collected for each pixel. |
Length scale | Ambiguous term. Imaging at a certain length scale is here defined to be done with a spatial resolution and field of view that allows to resolve physical features whose dimensions lie in a certain range. |
Measurement modality | Ambiguous term, here defined as: the physical principle according to which an analytical measurement is performed. |
Metal soap | A salt consisting of metal ions and fatty acids. Often found in oil paints as a result of pigment degradation. |
Method | Ambiguous term, defined in the context of microchemical imaging as: a physical or computational tool that can be used to prepare or analyze a sample or dataset. |
Methodology | Ambiguous term, defined in the context of microchemical imaging as: the overarching approach by which analysis is performed; includes sample preparation, instrumental analysis, and data treatment. |
Multispectral | An approach to spectral imaging in which absorption, emission, or scattering is probed at a small number of energies (typically 5-15) for each pixel. |
Near-field | An approach in optics in which the distance between the sample and the collection or focusing elements is smaller than the wavelength of the photons being used. |
Numerical aperture | A metric of optical elements that measures the angular range over which light is accepted or emitted. See also: solid angle |
PL | Photoluminescence |
Polymorph | A compound that exists in a particular crystal structure, but can exist in one or more others. |
Pump-probe | An approach to spectroscopy in which one light source (the pump) promotes electrons from one energy level to another and a second light source (the probe) is used to measure the population or depopulation of certain energy levels. |
RXES | Resonant X-ray emission spectroscopy, sometimes referred to as resonant inelastic X-ray scattering (RIXS) |
Saponification | The process of fatty acids being converted into (metal) soaps. |
SEM-EDX | Scanning electron microscopy-energy-dispersive X-ray spectrometry. See also: energy-dispersive detection. |
Semiconductor pigment | An inorganic pigment that consists of crystallites that possess semiconducting properties, such as a bandgap and the ability to luminesce. Due to its bandgap structure, small changes in the composition of the pigment can strongly affect its color or luminescing properties. |
SIMS | Secondary ion mass spectrometry |
SNR | Signal-to-noise ratio |
Solid angle | The angular range over which an optical element or detector accepts incoming photons. |
STEM | Scanning transmission electron microscopy |
Synchrotron | A large cyclic particle accelerator that can be used as a source of high-intensity and tunable X-rays, infrared, visible, and ultraviolet radiation. |
TEM | Transmission electron microscopy |
Tomography | A collection of techniques that are aimed at reconstructing sections through a sample. |
Voxel | The three-dimensional equivalent of a pixel. |
Wavelength-dispersive detection | A means to detect a spectrum in which different wavelengths are separated based on some form of diffraction and directed towards a 2D detector array. |
XANES | X-ray absorption near-edge structure |
XAS | X-ray absorption spectroscopy |
XRD | X-ray diffraction |
XRF | X-ray fluorescence |
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hageraats, S., Thoury, M., Cotte, M., Bertrand, L., Janssens, K., Keune, K. (2022). Microchemical Imaging of Oil Paint Composition and Degradation: State-of-the-Art and Future Prospects. In: Colombini, M.P., Degano, I., Nevin, A. (eds) Analytical Chemistry for the Study of Paintings and the Detection of Forgeries. Cultural Heritage Science. Springer, Cham. https://doi.org/10.1007/978-3-030-86865-9_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-86865-9_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-86864-2
Online ISBN: 978-3-030-86865-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)