
Abstract
Surgical resection and adjuvant radiation are the mainstays of treatment for patients with chordoma. However, these local therapies are easily exhausted during the treatment of chordoma of the axial skeleton in patients with advanced, recurrent, or metastatic disease. There is currently no approved standard of care first-line systemic therapy for the systemic treatment of chordoma. Conventional cytotoxic chemotherapy has been shown to have few benefits for the treatment of chordoma. Therefore, efforts to improve and expand systemic treatment options have focused on the identifying and targeting genetic, epigenetic, cellular signaling, and immunologic characteristics of chordoma cells. In this chapter, we review preclinical and clinical data from the use of molecularly targeted therapies for the treatment of chordoma, including those targeted against receptor tyrosine kinases, intracellular signaling pathways, the cell cycle, and epigenetic regulation of gene control. Finally, we detail ongoing efforts to use immunotherapy strategies to treat patients with chordoma.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Surgical resection and adjuvant radiation can lead to long-term disease control in many patients with chordoma [1]. However, gross total resection is not always technically feasible, and subtotal resection is associated with disease recurrence, possible tumor seeding, and development of metastatic disease [2, 3]. Five-year conditional disease-specific survival (DSS) for patients with localized chordoma is 83%; however, 5-year conditional DSS is only 71% for those with metastatic disease [4]. The use of cytotoxic chemotherapy has been proposed and tested as an option for the treatment of metastatic chordoma; however, there has been little evidence to date supporting its clinical efficacy [5]. Furthermore, the rarity of chordoma precludes the study of systemic therapies using large-scale, prospective trials. Nonetheless, data from genomic, epigenomic, and transcriptomic analyses, along with the study of the chordoma tumor-immune microenvironment, have identified a number of promising new treatment targets.
Cytotoxic Chemotherapy
Chordoma is considered unresponsive to cytotoxic chemotherapy , and there is no prospective data to support its clinical use. To date the only available clinical trial data is derived from a small phase II, prospective study by Chugh et al. studying this use of cytotoxic chemotherapy in patients with advanced chordoma. In this trial, the authors found that a regimen of the topoisomerase I inhibitor, rubitecan, in 15 patients with advanced chordoma was associated with moderate toxicity and little clinical benefit; only one patient had an objective response to treatment [6].
Multi-agent regimens have been described in a handful of case reports with varying degrees of reported success. In their report, Fleming et al. described complete remission in two patients with dedifferentiated sacral chordoma treated with neoadjuvant radiation, sacrectomy, and adjuvant systemic therapy. One patient was treated with ifosfamide, and the other was treated with a six-drug regimen including cisplatin , etoposide, vincristine, dacarbazine, cyclophosphamide, and doxorubicin [7]. Similarly, in a retrospective review of six pediatric patients with clival chordomas, Dhall et al. found the use of adjuvant ifosfamide and etoposide (VP-16) resulted in disease stability in two patients with a median follow-up of 9 years from diagnosis [8]. Ceruso et al. similarly documented both clinical and radiographic responses in a patient with advanced sacral chordoma that was treated with oral cyclophosphamide and prednisone given on a metronomic schedule (continuous administration of low doses of the active drugs) [9].
Despite these isolated case reports, there are no high-quality data to suggest that cytotoxic chemotherapy is an effective treatment for either conventional and chondroid chordoma. Thus, development of targeted and immune-based therapies has been a major focus of chordoma research.
Molecular-Targeted Therapies: Receptor Tyrosine Kinases and Cell Signaling Pathways
Given the lack of demonstrated efficacy for conventional cytotoxic chemotherapy, there has been a drive to identify the key molecular changes associated with chordoma oncogenesis. Identification of the mutated or overexpressed genes and dysregulated molecular pathways has provided opportunities for the use of targeted therapies in the treatment of chordoma [10, 11]. Key changes that have been associated with chordoma tumorigenesis include increased expression and activation of receptor tyrosine kinases (RTKs) and alterations in both the downstream effectors of these RTKs and the negative regulators of RTK signaling [12, 13]. While no single, dominant molecular pathway has been implicated in the development of chordoma, analysis of patient chordoma samples has noted increased expression of the following RTKs : epidermal growth factor receptor (EGFR), tyrosine-protein kinase KIT (KIT), platelet-derived growth factor receptors A and B (PDGFRA and PDGFRB), and vascular endothelial growth factor receptor 2 (VEGFR2) [12, 14, 15].
Perhaps consistent with the relatively indolent growth pattern of chordomas, histopathology studies have found chordomas to have a relatively low burden of somatic mutations. Analysis of 37 chordoma tumor exomes or genomes revealed a median of 21 coding substitutions and 4 insertion-deletion mutations (indels) per case [16]. In spite of the relatively low mutation burden, alterations in a small number of pathways have been consistently identified. These include changes in the phosphoinositide-3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway. Noted changes include activating mutations in the PIK3CA gene and truncations or deletions in its negative regulator – phosphatase and tensin homolog on chromosome 10 (PTEN). Alterations in genes encoding regulators of the cell cycle have also been identified, including mutations in cyclin D/cyclin-dependent kinases 4 and 6 (CDK4/6), retinoblastoma (RB), and the Janus kinase (JAK)/signal transducer activator of transcription (STAT) pathways. All have been postulated to be involved in tumorigenesis [16,17,18,19].
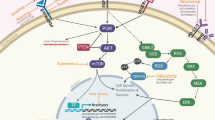
Numerous preclinical studies support the use of targeted small molecule inhibitors in chordomas harboring alterations in these pathways among others. Clinical implementation of these therapies has been attempted with the use of several tyrosine kinase inhibitors (TKIs). TKIs that have been previously described in the preclinical or clinical setting include the PDGFR inhibitor imatinib; the KIT inhibitor dasatinib; the EGFR/ human epidermal growth factor receptor 2 (HER2) inhibitors erlotinib, lapatinib, and gefitinib ; and the VEGFR inhibitors sorafenib, pazopanib, and sunitinib. Other inhibitors that have been described include the anti-EGFR antibody cetuximab and the PI3K/Akt/mTOR pathway inhibitors temsirolimus, sirolimus, and everolimus (Fig. 16.1) [10]. These pathways present targets for molecular therapies that have been evaluated in preclinical and, in some cases, clinical studies [10, 20]. A summary of phase II trials of TKIs for the treatment of chordoma is presented in Table 16.1.

Selected molecular-targeted therapies against RTK signaling pathways for the treatment of chordoma
PDGFR
Imatinib, a multi-kinase inhibitor with activity against BCR-ABL, KIT, and PDGFR, was the first molecular-targeted therapy to show clinical benefit in the treatment of chordoma. A phase II study of patients with advanced chordoma expressing platelet-derived growth factor β (PDGFB)/PDGF receptor β (PDGFRB) treated 56 patients with 800 mg/day of imatinib. While only one partial response (PR) was seen by at 6-month follow-up (ORR, 2%), 35 patients (70%) had stable disease (SD) for an overall clinical benefit rate of 64%. The median progression-free survival (PFS) and overall survival (OS) were 9.2 and 34.9 months, respectively [21]. Multiple other studies, including two other clinical trials, have investigated the use of imatinib, either as monotherapy or in combination with chemotherapy, in patients with chordoma [10, 22, 23].
Dasatinib, another PDGFR inhibitor that also has activity against Src, was studied in a phase II trial involving patients with locally advanced or metastatic sarcomas. Across the 32 included patients, median PFS was 6 months and OS was 43% and 18% at 2 and 5 years, respectively [24].
mTOR
Multiple clinical case reports have found mTOR inhibitors to have clinically significant activity against chordoma [25,26,27]. The combination of the mTOR inhibitor everolimus and imatinib was recently examined in a phase II clinical trial by Stacchiotti and colleagues. The examined cohort comprised 43 patients with advanced or metastatic chordoma that had progressed following initial surgical, radiation, or medical treatment. Of note, 13 patients had previously been treated with imatinib. While the antitumor effect was modest, with an ORR of 22.5%, 6 of 30 chemotherapy-naïve patients (20%) and 3 of 13 patients (23%) previously treated with imatinib demonstrated a partial response, as defined by Choi criteria [28]. Molecular characterization of this subgroup found that these patients had tumors with high levels of mTOR phosphorylation (present in ≥60% of tumor cells). Similar phosphorylation levels were not observed in the tumors from patients who did not benefit from treatment with everolimus and imatinib [29].
VEGFR
VEGF protein expression is detected in chordoma samples [15, 30, 31]. Accordingly, multiple anti-VEGF therapies have been investigated for their clinical utility in patients with advanced or unresectable chordoma . Agents that have been previously examined include the small molecule VEGF inhibitors sorafenib [32], pazopanib [23, 33, 34], and sunitinib [23], as well as the humanized anti-VEGF-A antibody, bevacizumab [35], and thalidomide, which modulates VEGF expression [36, 37].
Of these, the small molecule inhibitors have been most popular, and to date there have been four phase II clinical trials examining the clinical utility of anti-VEGF TKIs in patients with chordoma [38,39,40,41]. Two trials [38, 39] evaluated the use of sorafenib, a VEGFR1/2/3 and PDGFRB inhibitor [42]. One of these trials, the phase II Angionext trial, included 26 patients with advanced chordoma who were treated with sorafenib at a dose of 800 mg/day. This treatment was associated with a 9-month PFS of 72.9% [39]. Sunitinib was similarly examined in a trial of advanced non-GIST soft tissue sarcomas, including nine patients with chordoma. Patients were treated with 37.5 mg of daily sunitinib and examinations at 16 and 24 weeks demonstrated stable disease in 44% and 22% of chordoma patients, respectively [40], as defined by RECIST criteria [43]. Finally, a single-arm , phase II nonrandomized trial of apatinib, a VEGFR-2 inhibitor, was performed at a single institution [41, 44]. The results suggested it may have mild efficacy on disease control. Although only one patient had a partial response by RECIST criteria, median PFS was 18 months, 1-year OS was 88%, and 63% of patients had local disease control at 6 months.
EGFR
The genes encoding EGFR and its ligands are highly expressed in patient-derived chordoma samples. Additionally, comparison of gene expression in fetal nucleus pulposus tissue and chordoma samples demonstrates significantly higher EGFR protein expression in tumor sample [17, 45]. Inhibition of EGFR and other human epidermal growth receptor family members has therefore been an attractive strategy for the treatment of chordoma. Case reports have detailed the use of small molecule inhibitors of EGFR, including erlotinib and gefitinib, as well as the anti-EGFR chimeric monoclonal antibody, cetuximab. These case reports have suggested anti-EGFR treatments to have clinical benefit both in terms of tumor response and improvement in neurologic symptoms secondary to chordoma mass effect [35, 46,47,48,49,50,51,52].
Another small molecule EGFR inhibitor, lapatinib, has also been evaluated in a phase II clinical trial. In this trial, lapatinib, a dual EGFR and HER2 inhibitor, was administered to 18 patients with locally advanced or metastatic, EGFR-expressing chordoma at a dose of 1500 mg/day (mean dose intensity of 1282 mg/day). Six of the 18 patients (33%) experienced partial disease response (ORR 33.3%), and seven others had stable disease by RECIST criteria with a minimum follow-up of 6 months. Median PFS in the cohort was 8 months, and the overall clinical benefit rate was reported at 22% [53]. Based upon these promising initial results, Stacchiotti et al. have begun a phase II, single-arm trial of the second-generation EGFR TKI, afatinib. The trial is currently enrolling patients with locally advanced or metastatic EGFR-expressing chordoma (NCT03083678).
CDK4/6
Progression through the cell cycle is tightly controlled by a large network of proteins, and dysregulation of this cycle has been implicated in the tumorigenesis of multiple malignancies, including chordoma [54]. One key molecule is p16, encoded by the gene cyclin-dependent kinase inhibitor 2A (CDKN2A). p16 negatively regulates CDK4/6 , which is responsible for stimulating cells to progress through the G1/S transition of the cell cycle [55]. CDK4/6 are often overactivated in chordoma samples due to loss of p16, resulting in increased cellular proliferation [56, 57]. Inhibition or downregulation of CDK4/6 may therefore represent a potential therapeutic target. Both strategies have proven effective in vitro, with CDK4/6 inhibition in p16-deleted cells resulting in decreased cellular proliferation and repression of other oncogenic properties [57, 58]. The CDK4/6 inhibitor used in these studies – palbociclib – is currently approved for the treatment of breast cancer [59] and is undergoing evaluation of its efficacy in a phase II study of patients with advanced or metastatic chordoma (NCT03110744).
Epigenetic Therapies
In the past decade, epigenetic regulation of chromatin structure and gene expression has been increasingly recognized as an important driver in tumorigenesis [60]. Control of chromatin packing dynamics is therefore another potential target for novel chordoma therapies. Missense and nonsense mutations in SMARCB1 , a member of the ATP-dependent SWI/SNF chromatin-remodeling complex, have been documented in chordoma samples [18]. Low expression of the SMARCB1 protein is additionally associated with poor prognosis in skull base chordomas [61]. SMARCB1 and the SWI/SNF complex play key roles in the epigenetic regulation of cell cycle progression and multiple signaling pathways [62]. Inactivation of SMARCB1, through either loss of protein expression or mutational inactivation, leads to increased activity of enhancer of zeste 2 (EZH2), a histone-lysine N-methyltransferase that forms the catalytic subunit of the polycomb repressive complex 2 (PRC2) [63, 64]. Increased PRC2 activity amplifies chromatin methylation, notably in regions encoding genes crucial to cell survival, proliferation, and invasion [65].
Curiosity about the efficacy of EZH2 inhibitors served as the basis of a phase I trial of tazemetostat, a selective EZH2 inhibitor [66]. In this trial of 46 patients with relapsed or refractory INI1-negative tumors, two children were being treated for chordoma. Though the chordoma cohort was too small to reach definitive conclusions, one of the chordoma patients was forced to withdraw from the study after experiencing worsening pain from her sacral mass. However, her tumor was found to be stable by RECIST 1.1 criteria at the time of discontinuation, and she experienced complete response of her pulmonary metastases. After withdrawing, she received radiation to the sacral mass, and radiographic follow-up at 4 months demonstrated persistent remission of her distant pulmonary metastases, suggesting that tazemetostat may induce an antitumor immune response [67]. This remains speculative though, and a phase II clinical trial is currently underway, which exams tazemetostat for the treatment of adult patients with SMARCB1-negative tumors, including poorly differentiated chordoma (NCT02601950).
Immunotherapy
The immune system and its impact on the development and treatment of cancer has been studied since the nineteenth century [68]. Work over the previous three decades has led to a more complete understanding of the role the immune system plays in the detection and elimination of neoplastic cells. This process of immune surveillance is regulated by multiple factors, including T cell immune checkpoints, molecular interactions that negatively regulate T cell function. For example, the programmed cell death-ligand 1 (PD-L1) found on cancer cell membranes binds to the programmed cell death protein 1 (PD-1) on CD8+ effector T cells resulting in CD8+ T cell anergy [69]. Disruption of this anergy-inducing interaction has therefore been a focus of novel immunotherapies and has stimulated the development of drugs that alter immunosurveillance by inhibiting these immune checkpoints. These checkpoint inhibitors are rapidly becoming a key aspect of chemotherapy as they have been successfully used to treat patients with cancers in which chemotherapy had been ineffective. Their use in the treatment of chordoma is therefore also the subject of great interest and study [5, 70].
The FDA has approved use of multiple antibodies that target components of two key immune checkpoint pathways: the binding of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) to CD80/CD86 and the binding of PD-1 and PD-L1. These inhibitors include the anti-CTLA-4 antibody ipilimumab, the anti-PD-1 antibodies nivolumab and pembrolizumab, and the anti-PD-L1 antibodies atezolizumab, avelumab, and durvalumab [70]. Preclinical data has shown that chordoma cell lines express both PD-1 and PD-L1 at the gene and protein levels [71]. The chordoma tumor immune microenvironment may serve to increase the growth and invasive potential of chordoma cells. For instance, expression of the PD-L1 gene and protein is upregulated by exposure to pro-inflammatory cytokines such as IFN-γ [71]. Exposure to TNF-α also increases PD-L1 gene expression and upregulates expression of genes associated with the epithelial-mesenchymal transition, PI3K/AKT signaling, pro-angiogenesis, and anti-apoptotic pathways (Fig. 16.2) [72]. This suggests that chordomas may activate T cell checkpoints and decrease the activity of T cells in response to an otherwise immune stimulating environment.

The chordoma tumor immune microenvironment. (a) In response to pro-inflammatory signals and cytokines such as INF-γ, chordoma cells increase expression of PD-L1. (b) Higher levels of PDL-1 expression lead to activation of T cell immune checkpoints through binding to PD-1 receptors on the T cell surface. (c) Activation of T cell immune checkpoints induces T cell anergy to tumor-specific antigens and apoptosis of effector T cells. (d) Brachyury vaccine strategies use epitopes from brachyury-derived tumor antigens to induce production and activation of brachyury antigen-specific CD8+ T cells. These T cells have an enhanced ability to recognize and possibly destroy chordoma cells. Clinical trials of brachyury vaccines have also incorporated the use of radiation with the goal of enhancing the anti-tumor immune response
Analysis of nine human chordoma tissue samples showed that 94.9% of the samples expressed PD-L1 as measured in a tissue microarray, and samples from patients with metastatic chordoma had higher expression of PD-L1 than those from patients with non-metastatic disease [73]. Infiltrating T lymphocytes (TILs) have also been examined. One study showing that 22% of TILs found in chordoma samples expressed PD-1, and a second observed detectable CTLA-4 on the majority of TILs in chordoma patient samples [74, 75]. Furthermore, treatment with avelumab, an anti-PD-L1 antibody, increased the immune-mediated killing of human-derived chordoma cell lines in vitro [76]. Taken together, these studies serve as rationale for targeting the immune system in the treatment of chordoma.
Recent case reports have shown that treatment with anti-PD-1 antibodies, such as pembrolizumab or nivolumab, led to reduction in tumor sizes and subjective clinical improvement. Notably, these improvements were observed even in patients with chordoma that was refractory to multiple earlier lines of therapy [77]. Both phase I and II clinical trials evaluating the use of anti-PD-1 antibodies alone, in conjunction with radiation or in combination with an inhibitor of another immune checkpoint, lymphocyte-activation gene 3 (LAG-3) , are recruiting patients with chordoma (NCT03173950, NCT02989636, and NCT03623854).
Despite this, recent data has suggested that only a subset of patients with chordoma may be amenable to immunotherapy with checkpoint inhibitors. In order to be eliminated by the host immune system, tumor cells must not only downregulate anergy-inducing checkpoint molecules, but they must also actively express peptides novel peptides demonstrating them to be “non-self.” This presentation is done through the interaction of human leukocyte antigen (HLA) class 1-peptide complexes on tumor cells and surface receptors on CD8+ effector T cells. Recently, Patel et al. [78] examined samples for surgically treated chordoma patients and found that 40% or more of chordoma samples may not express HLA class 1/peptide complexes. However, in vitro work has found proton- and photon-based radiation to increase HLA expression [79]. Consequently, paired radiotherapy and checkpoint inhibitor administration may improve tumor cell killing. This is currently undergoing investigation in a phase I trial (NCT02989636).
Brachyury
Brachyury is a transcription factor encoded by the T-box transcription factor T gene (TBXT) located on the long arm of chromosome 6 [80]. Brachyury has a conserved role in development [81], and its expression has been established as a diagnostic marker in chordoma [82, 83]. Brachyury is highly expressed in patient chordoma samples, with the rare exception of a subset of dedifferentiated chordomas [84]. Chromosome 6 duplication and TBXT copy number gain have both been observed in chordoma cell lines and patient samples. The increased somatic copy number gain of TBXT has in turn been correlated with increased brachyury expression [85,86,87,88].
Preclinical studies have shown that brachyury is involved in numerous critical cellular pathways. These include pathways regulating cell growth, apoptosis, the epithelial-to-mesenchymal transition, and cellular differentiation [89,90,91,92]. In vivo experimental models have been used to demonstrate that disruption of brachyury expression or activity can prevent the formation of chordoma tumors or decrease the size of existing tumors [90, 92].
Based upon this preclinical evidence, brachyury has also become as an attractive target for systemic therapies. One such therapy is the chordoma Brachyury vaccine, originally described by Hamilton et al. in a murine model of metastatic colorectal adenocarcinoma [93]. The group found that treating mice transplanted with MC38 cells using a Saccharomyces cerevisiae-based brachyury vaccine produced robust brachyury-specific CD4+ and CD8+ T cell responses with minimal toxicity. Importantly, treated mice also showed a significant reduction in their lung metastasis burden. Based upon this, a phase I clinical trial of a yeast-based cancer Brachyury vaccine was launched in patients with chordoma or other advanced solid tumors (Fig. 16.2) [94]. The vaccine was well tolerated and induced the development or enhancement of brachyury-specific CD4+ and/or CD8+ T cells in 55% of patients. Of the ten patients with chordoma, none had evidence of disease progression at 5-month evaluation. Among these patients, the median PFS was 8.3 months, though only two patients had disease response (one with partial response and one with mixed response). Of note, both patients demonstrating disease response had received radiation treatment prior to their vaccination. This radiotherapy may have upregulated HLA class 1/brachyury complex expression, enhancing the “non-self” signal provided by the chordoma cells. Based upon this, two phase II trials have been initiated looking at concomitant treatment with radiotherapy and a Brachyury vaccine. One trial is examining patients with locally advanced, unresectable chordoma using concomitant radiotherapy and vaccination with the yeast-based Brachyury vaccine (NCT02383498) [95]. The second (NCT03595228) is using a poxvirus-based Brachyury vaccine (BN-Brachyury vaccine) with radiation in patients with chordoma. In this second study, the vaccine consists of two recombinant poxvirus vectors delivered in tandem as a prime-boost strategy. These viruses are modified to express brachyury as well as viral molecules known to increase immune cell activation [96].
Conclusions
Treatment of chordoma after exhaustion of local therapy options remains challenging. Chordoma has no established standard of care therapy with conventional cytotoxic chemotherapy proving ineffective. Large randomized trials of systemic therapy are difficult to conduct owing to the rarity of chordoma, the heterogeneous nature of the disease, and the varying rates of progression observed across patients. Nevertheless, an enhanced understanding of the molecular drivers and tumor immune microenvironment in chordoma has opened the door for new, targeted treatment paradigms which may prove useful in long-term control or remission for those with advanced disease. Early results have been promising, but continued study is necessary, and the ultimate solution may rely on establishment of patient-specific regimens dictated by each tumor’s unique genetic fingerprint.
Abbreviations
- CDK4/6:
-
Cyclin-dependent kinases 4 and 6
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- DSS:
-
Disease-specific survival
- EGFR:
-
Epidermal growth factor receptor
- EZH2:
-
Enhancer of zeste 2
- FDA:
-
Food and Drug Administration
- HER2:
-
Human epidermal growth factor receptor 2
- JAK:
-
Janus kinase
- KIT:
-
Tyrosine-protein kinase KIT
- LAG-3:
-
Lymphocyte-activation gene 3
- mTOR:
-
Mammalian target of rapamycin
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PD1:
-
Programmed cell death protein 1
- PDGFB:
-
Platelet-derived growth factor β
- PDGFR:
-
Platelet-derived growth factor receptor
- PDGFRA:
-
Platelet-derived growth factor receptor ɑ
- PDGFRB:
-
Platelet-derived growth factor receptor β
- PD-L1:
-
Program cell death ligand 1
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphoinositide-3-kinase (PI3K)
- PR:
-
Partial response
- PRC2:
-
Polycomb repressive complex 2
- PTEN:
-
Phosphatase and tensin homolog deleted on chromosome 10
- RB:
-
Retinoblastoma
- RTK(s):
-
Receptor tyrosine kinase(s)
- SD:
-
Stable disease
- SMARCB1:
-
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1
- STAT:
-
Signal transducer activator of transcription
- SWI/SNF:
-
Switch/sucrose non-fermentable
- TBXT:
-
T-box transcription factor T
- TCR:
-
T cell receptor
- TKI(s):
-
Tyrosine kinase inhibitor(s)
- VEGFR:
-
Vascular endothelial growth factor receptor
- VP-16 :
-
Etoposide
References
Catton C, O’Sullivan B, Bell R, Laperriere N, Cummings B, Fornasier V, Wunder J. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41:67–72.
Hulen CA, Temple HT, Fox WP, Sama AA, Green BA, Eismont FJ. Oncologic and functional outcome following sacrectomy for sacral chordoma. J Bone Jt Surg. 2006;88:1532–9.
Chambers PW, Schwinn CP. Chordoma: a clinicopathologic study of metastasis. Am J Clin Pathol. 1979;72:765–76.
Huang J-F, Chen D, Zheng X-Q, Lin J-L, Wang X-Y, Wu A-M. Conditional survival and changing risk profile in patients with chordoma: a population-based longitudinal cohort study. J Orthop Surg Res. 2019;14:181.
Colia V, Stacchiotti S. Medical treatment of advanced chordomas. Eur J Cancer. 2017;83:220–8.
Chugh R, Dunn R, Zalupski MM, Biermann JS, Sondak VK, Mace JR, Leu KM, Chandler WF, Baker LH. Phase II study of 9-nitro-Camptothecin in patients with advanced chordoma or soft tissue sarcoma. J Clin Oncol. 2005;23:3597–604.
Fleming GF, Heimann PS, Stephens JK, Simon MA, Ferguson MK, Benjamin RS, Samuels BL. Dedifferentiated chordoma: response to aggressive chemotherapy in two cases. Cancer. 1993;72:714–8.
Dhall G, Traverso M, Finlay JL, Shane L, Gonzalez-Gomez I, Jubran R. The role of chemotherapy in pediatric clival chordomas. J Neuro-Oncol. 2011;103:657–62.
Spalato Ceruso M, Napolitano A, Silletta M, Vincenzi B. Unexpected benefit from an ‘old’ metronomic chemotherapy regimen in advanced chordoma. BMJ Case Rep. 2019;12:e228728.
Meng T, Jin J, Jiang C, Huang R, Yin H, Song D, Cheng L. Molecular targeted therapy in the treatment of chordoma: a systematic review. Front Oncol. 2019;9:30.
Ozair MZ, Shah PP, Mathios D, Lim M, Moss NS. New prospects for molecular targets for chordomas. Neurosurg Clin N Am. 2020;31:289–300.
Dewaele B, Maggiani F, Floris G, Ampe M, Vanspauwen V, Wozniak A, Debiec-Rychter M, Sciot R. Frequent activation of EGFR in advanced chordomas. Clin Sarcoma Res. 2011;1:4.
D’Agati G, Cabello EM, Frontzek K, Rushing EJ, Klemm R, Robinson MD, White RM, Mosimann C, Burger A. Active receptor tyrosine kinases, but not Brachyury, are sufficient to trigger chordoma in zebrafish. Dis Model Mech. 2019;12:dmm039545.
Tamborini E, Miselli F, Negri T, et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin Cancer Res. 2006;12:6920–8.
Akhavan-Sigari R, Gaab MR, Rohde V, Brandis A, Tezval H, Abili M, von Eckardstein K, Ostertag H. Expression of vascular endothelial growth factor receptor 2 (VEGFR-2), inducible nitric oxide synthase (iNOS), and Ki-M1P in skull base chordoma: a series of 145 tumors. Neurosurg Rev. 2014;37:79–88.
Tarpey PS, Behjati S, Young MD, et al. The driver landscape of sporadic chordoma. Nat Commun. 2017;8:890.
Tamborini E, Virdis E, Negri T, et al. Analysis of receptor tyrosine kinases (RTKs) and downstream pathways in chordomas. Neuro-Oncology. 2010;12:776–89.
Choy E, MacConaill LE, Cote GM, Le LP, Shen JK, Nielsen GP, Iafrate AJ, Garraway LA, Hornicek FJ, Duan Z. Genotyping cancer-associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PLoS One. 2014;9:e101283.
Wang L, Zehir A, Nafa K, et al. Genomic aberrations frequently alter chromatin regulatory genes in chordoma. Genes Chromosom Cancer. 2016;55:591–600.
Foundation C. Therapeutic targets in chordoma. 2020. https://www.chordomafoundation.org/targets. Accessed 1 Sept 2020.
Stacchiotti S, Longhi A, Ferraresi V, et al. Phase II study of Imatinib in advanced chordoma. J Clin Oncol. 2012;30:914–20.
Adenis A, Ray-Coquard I, Italiano A, et al. A dose-escalating phase I of imatinib mesylate with fixed dose of metronomic cyclophosphamide in targeted olid tumours. Br J Cancer. 2013;109:2574–8.
Lipplaa A, Dijkstra S, Gelderblom H. Efficacy of pazopanib and sunitinib in advanced axial chordoma: a single reference centre case series. Clin Sarcoma Res. 2016;6:19.
Schuetze SM, Bolejack V, Choy E, et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer. 2017;123:90–7.
Stacchiotti S, Marrari A, Tamborini E, et al. Response to imatinib plus sirolimus in advanced chordoma. Ann Oncol. 2009;20:1886–94.
Ricci-Vitiani L, Runci D, D’Alessandris QG, et al. Chemotherapy of skull base chordoma tailored on responsiveness of patient-derived tumor cells to rapamycin. Neoplasia. 2013;15:773–82.
Lebellec L, Chauffert B, Blay J-Y, et al. Advanced chordoma treated by first-line molecular targeted therapies: outcomes and prognostic factors. A retrospective study of the French Sarcoma Group (GSF/GETO) and the Association des Neuro-Oncologues d’Expression Française (ANOCEF). Eur J Cancer. 2017;79:119–28.
Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, Chen LL, Podoloff DA, Benjamin RS. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with Imatinib Mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–9.
Stacchiotti S, Morosi C, Lo Vullo S, et al. Imatinib and everolimus in patients with progressing advanced chordoma: a phase 2 clinical study. Cancer. 2018;124:4056–63.
Li X, Ji Z, Ma Y, Qiu X, Fan Q, Ma B. Expression of hypoxia-inducible factor-1α, vascular endothelial growth factor and matrix metalloproteinase-2 in sacral chordomas. Oncol Lett. 2012;3:1268–74.
Tauziède-Espariat A, Bresson D, Polivka M, et al. Prognostic and therapeutic markers in chordomas: a study of 287 tumors. J Neuropathol Exp Neurol. 2016;75:111–20.
Eroukhmanoff J, Castinetti F, Penel N, Salas S. Auto-immune thyroid dysfunction induced by tyrosine kinase inhibitors in a patient with recurrent chordoma. BMC Cancer. 2016;16:679.
Ribeiro MFSA, de Sousa MC, Hanna SA, Maldaun MVC, Kurimori CO, de Lima LGCA, Mattedi RL, Munhoz RR. Tumor reduction with Pazopanib in a patient with recurrent lumbar chordoma. Case Rep Oncol Med. 2018;2018:1–7.
Stein J, Milhem M, Vaena D. Clinical outcomes and toxicities of pazopanib administered orally in crushed form: case reports and review of the literature. J Oncol Pharm Pract. 2020;26:232–5.
Asklund T, Sandström M, Shahidi S, Riklund K, Henriksson R. Durable stabilization of three chordoma cases by bevacizumab and erlotinib. Acta Oncol (Madr). 2014;53:980–4.
Sherbet GV. Therapeutic potential of thalidomide and its analogues in the treatment of cancer. Anticancer Res. 2015;35:5767–72.
Chay WY, Teo M, Sittampalam K, Toh HC. Effective use of thalidomide in the treatment of recurrent metastatic chordoma. J Clin Oncol. 2011;29:e477–80.
Bompas E, Le Cesne A, Tresch-Bruneel E, et al. Sorafenib in patients with locally advanced and metastatic chordomas: a phase II trial of the French sarcoma group (GSF/GETO). Ann Oncol. 2015;26:2168–73.
Lebellec L, Bertucci F, Tresch-Bruneel E, et al. Circulating vascular endothelial growth factor (VEGF) as predictive factor of progression-free survival in patients with advanced chordoma receiving sorafenib: an analysis from a phase II trial of the french sarcoma group (GSF/GETO). Oncotarget. 2016;7:73984–94.
George S, Merriam P, Maki RG, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J Clin Oncol. 2009;27:3154–60.
Liu C, Jia Q, Wei H, et al. Apatinib in patients with advanced chordoma: a single-arm, single-centre, phase 2 study. Lancet Oncol. 2020;21:1244–52.
Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129–40.
Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92:205–16.
Liao Z, Li F, Zhang C, et al. Phase II trial of VEGFR2 inhibitor apatinib for metastatic sarcoma: focus on efficacy and safety. Exp Mol Med. 2019;51:1–11.
Chen H, Zhang K, Lu J, Wu G, Yang H, Chen K. Comprehensive analysis of mRNA-lncRNA co-expression profile revealing crucial role of imprinted gene cluster DLK1-MEG3 in chordoma. Oncotarget. 2017;8:112623–35.
Trapani D, Conforti F, De Pas T. EGFR inhibition in a pretreated sacral chordoma: a role for Erlotinib? Case report and a brief review of literature. Transl Med @ UniSa. 2017;16:30–3.
Houessinon A, Boone M, Constans J-M, Toussaint P, Chauffert B. Sustained response of a Clivus chordoma to Erlotinib after Imatinib failure. Case Rep Oncol. 2015;8:25–9.
Singhal N, Kotasek D, Parnis FX. Response to erlotinib in a patient with treatment refractory chordoma. Anti-Cancer Drugs. 2009;20:953–5.
Launay SG, Chetaille B, Medina F, Perrot D, Nazarian S, Guiramand J, Moureau-Zabotto L, Bertucci F. Efficacy of epidermal growth factor receptor targeting in advanced chordoma: case report and literature review. BMC Cancer. 2011;11:423.
Linden O, Ehinger M, Kinhult S, Relander T, Lundgren L, Engellau J, Kjellén E. Cetuximab and EGFR inhibitors in patients with neurological symptoms due to chordoma. J Clin Oncol. 2012;30:10024.
Hof H, Welzel T, Debus J. Effectiveness of Cetuximab/Gefitinib in the therapy of a sacral chordoma. Oncol Res Treat. 2006;29:572–4.
Lindèn O, Stenberg L, Kjellén E. Regression of cervical spinal cord compression in a patient with chordoma following treatment with cetuximab and gefitinib. Acta Oncol (Madr). 2009;48:158–9.
Stacchiotti S, Tamborini E, Lo Vullo S, et al. Phase II study on lapatinib in advanced EGFR-positive chordoma. Ann Oncol. 2013;24:1931–6.
Gordon E, Ravicz J, Liu S, Chawla S, Hall F. Cell cycle checkpoint control: the cyclin G1/Mdm2/p53 axis emerges as a strategic target for broad-spectrum cancer gene therapy - a review of molecular mechanisms for oncologists. Mol Clin Oncol. 2018;9:115–34.
Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–38.
Yakkioui Y, Temel Y, Creytens D, Jahanshahi A, Fleischeuer R, Santegoeds RGC, Van Overbeeke JJ. A comparison of cell-cycle markers in skull base and sacral chordomas. World Neurosurg. 2014;82:e311–8.
von Witzleben A, Goerttler LT, Marienfeld R, et al. Preclinical characterization of novel chordoma cell systems and their targeting by pharmocological inhibitors of the CDK4/6 cell-cycle pathway. Cancer Res. 2015;75:3823–31.
Liu T, Shen JK, Choy E, Zhang Y, Mankin HJ, Hornicek FJ, Duan Z. CDK4 expression in chordoma: a potential therapeutic target. J Orthop Res. 2018;36:1581–9.
Serra F, Lapidari P, Quaquarini E, Tagliaferri B, Sottotetti F, Palumbo R. Palbociclib in metastatic breast cancer: current evidence and real-life data. Drugs Context. 2019;8:1–16.
Ahuja N, Sharma AR, Baylin SB. Epigenetic therapeutics: a new weapon in the war against cancer. Annu Rev Med. 2016;67:73–89.
Li M, Zhai Y, Bai J, Wang S, Gao H, Li C, Gui S, Du J, Zhang Y. SNF5 as a prognostic factor in skull base chordoma. J Neuro-Oncol. 2018;137:139–46.
Kohashi K, Oda Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. 2017;108:547–52.
Kim KH, Roberts CWM. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. 2014;207:365–72.
Geller JI, Roth JJ, Biegel JA. Biology and treatment of Rhabdoid tumor. Crit Rev Oncog. 2015;20:199–216.
Gan L, Yang Y, Li Q, Feng Y, Liu T, Guo W. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res. 2018;6:10.
Chi S, Fouladi M, Shukla N, et al. Abstract A175: phase 1 study of the EZH2 inhibitor, tazemetostat, in children with relapsed or refractory INI1-negative tumors including rhabdoid tumors, epithelioid sarcoma, chordoma, and synovial sarcoma. Mol Cancer Ther. 2018;17:A175.
Gounder MM, Zhu G, Roshal L, Lis E, Daigle SR, Blakemore SJ, Michaud NR, Hameed M, Hollmann TJ. Immunologic correlates of the abscopal effect in a SMARCB1/INI1-negative poorly differentiated Chordoma after EZH2 inhibition and radiotherapy. Clin Cancer Res. 2019;25:2064–71.
Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. 2019;27:S87–97.
Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, Wang X. The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol. 2019; https://doi.org/10.3389/fimmu.2019.02298.
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–68.
Mathios D, Ruzevick J, Jackson CM, et al. PD-1, PD-L1, PD-L2 expression in the chordoma microenvironment. J Neuro-Oncol. 2015;121:251–9.
Gulluoglu S, Tuysuz EC, Sahin M, Yaltirik CK, Kuskucu A, Ozkan F, Dalan AB, Sahin F, Ture U, Bayrak OF. The role of TNF-α in chordoma progression and inflammatory pathways. Cell Oncol. 2019;42:663–77.
Feng Y, Shen J, Gao Y, Liao Y, Cote G, Choy E, Chebib I, Mankin H, Hornicek F, Duan Z. Expression of programmed cell death ligand 1 (PD-L1) and prevalence of tumor-infiltrating lymphocytes (TILs) in chordoma. Oncotarget. 2015;6:11139–49.
Liang WS, Millis SZ, Gatalica Z, Reddy SK, Little A, Van Tine BA. Identification of actionable targets in chordomas using a multiplatform molecular analysis, and response with targeted therapy. J Clin Oncol. 2015;33:e22215.
He G, Liu X, Pan X, Ma Y, Liu X. Cytotoxic T lymphocyte antigen-4 (CTLA-4) expression in chordoma and tumor-infiltrating lymphocytes (TILs) predicts prognosis of spinal chordoma. Clin Transl Oncol. 2020;22:2324–32.
Fujii R, Friedman ER, Richards J, Tsang KY, Heery CR, Schlom J, Hodge JW. Enhanced killing of chordoma cells by antibody-dependent cell-mediated cytotoxicity employing the novel anti-PD-L1 antibody avelumab. Oncotarget. 2016;7:33498–511.
Migliorini D, Mach N, Aguiar D, Vernet R, Landis BN, Becker M, McKee T, Dutoit V, Dietrich P-Y. First report of clinical responses to immunotherapy in 3 relapsing cases of chordoma after failure of standard therapies. Onco Targets Ther. 2017;6:e1338235.
Patel SS, Nota SP, Sabbatino F, Nielsen GP, Deshpande V, Wang X, Ferrone S, Schwab JH. Defective HLA class I expression and patterns of lymphocyte infiltration in chordoma tumors. Clin Orthop Relat Res. 2020; https://doi.org/10.1097/CORR.0000000000001587.
Gameiro SR, Malamas AS, Bernstein MB, et al. Tumor cells surviving exposure to proton or photon radiation share a common immunogenic modulation signature, rendering them more sensitive to T cell–mediated killing. Int J Radiat Oncol. 2016;95:120–30.
Edwards YH, Putt W, Lekoape KM, Stott D, Fox M, Hopkinson DA, Sowden J. The human homolog T of the mouse T(Brachyury) gene; gene structure, cDNA sequence, and assignment to chromosome 6q27. Genome Res. 1996;6:226–33.
Le Gouar M, Guillou A, Vervoort M. Expression of a SoxB and a Wnt2/13 gene during the development of the mollusc Patella vulgata. Dev Genes Evol. 2004;214:250–6.
Karpathiou G, Dumollard JM, Dridi M, Dal Col P, Barral F-G, Boutonnat J, Peoc’h M. Chordomas: a review with emphasis on their pathophysiology, pathology, molecular biology, and genetics. Pathol - Res Pract. 2020;216:153089.
Schwab JH, Boland PJ, Agaram NP, et al. Chordoma and chondrosarcoma gene profile: implications for immunotherapy. Cancer Immunol Immunother. 2009;58:339–49.
Vujovic S, Henderson S, Presneau N, Odell E, Jacques T, Tirabosco R, Boshoff C, Flanagan A. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209:157–65.
Presneau N, Shalaby A, Ye H, et al. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: a genetic and functional-based study. J Pathol. 2011;223:327–35.
Hallor KH, Staaf J, Jönsson G, et al. Frequent deletion of the CDKN2A locus in chordoma: analysis of chromosomal imbalances using array comparative genomic hybridisation. Br J Cancer. 2008;98:434–42.
Rinner B, Froehlich EV, Buerger K, et al. Establishment and detailed functional and molecular genetic characterisation of a novel sacral chordoma cell line, MUG-Chor1. Int J Oncol. 2011;40:443–51.
Otani R, Mukasa A, Shin M, Omata M, Takayanagi S, Tanaka S, Ueki K, Saito N. Brachyury gene copy number gain and activation of the PI3K/Akt pathway: association with upregulation of oncogenic Brachyury expression in skull base chordoma. J Neurosurg. 2018;128:1428–37.
Nelson AC, Pillay N, Henderson S, et al. An integrated functional genomics approach identifies the regulatory network directed by brachyury ( T ) in chordoma. J Pathol. 2012;228:274–85.
Shah SR, David JM, Tippens ND, et al. Brachyury-YAP regulatory Axis drives Stemness and growth in cancer. Cell Rep. 2017;21:495–507.
Jäger D, Lechel A, Tharehalli U, Seeling C, Möller P, Barth TFE, Mellert K. U-CH17P, -M and -S, a new cell culture system for tumor diversity and progression in chordoma. Int J Cancer. 2018;142:1369–78.
Sharifnia T, Wawer MJ, Chen T, et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat Med. 2019;25:292–300.
Hamilton DH, Litzinger MT, Jales A, Huang B, Fernando RI, Hodge JW, Ardiani A, Apelian D, Schlom J, Palena C. Immunological targeting of tumor cells undergoing an epithelial-mesenchymal transition via a recombinant brachyury-yeast vaccine. Oncotarget. 2013;4:1777–90.
Heery CR, Singh BH, Rauckhorst M, et al. Phase I trial of a yeast-based therapeutic cancer vaccine (GI-6301) targeting the transcription factor Brachyury. Cancer Immunol Res. 2015;3:1248–56.
Menon H, Ramapriyan R, Cushman TR, et al. Role of radiation therapy in modulation of the tumor stroma and microenvironment. Front Immunol. 2019;10:193.
García-Arriaza J, Esteban M. Enhancing poxvirus vectors vaccine immunogenicity. Hum Vaccin Immunother. 2014;10:2235–44.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zabransky, D.J., Pennington, Z., Meyer, C. (2021). Systemic Therapy, Trials, and Future Directions for Chordoma of the Spine. In: Sciubba, D.M., Schwab, J.H. (eds) Chordoma of the Spine. Springer, Cham. https://doi.org/10.1007/978-3-030-76201-8_16
Download citation
DOI: https://doi.org/10.1007/978-3-030-76201-8_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-76200-1
Online ISBN: 978-3-030-76201-8
eBook Packages: MedicineMedicine (R0)