
Abstract
Traditionally, stem cells are grown in two-dimensional vessels, although this poses a major challenge in the clinical-grade production of a large number of stem cells and their derivatives to fulfill the dose requirements in clinical trials (at a scale of ~109–1010 cells). The cultivation of microcarriers in stirred bioreactors has been recognized as among the most promising strategies for the rapid scale-up of stem cell-derived therapeutics under controlled conditions. In addition to large-scale production, microcarriers have been applied in cell delivery systems for in vivo transplantation, to enhance cell survival and engraftment. ‘Microcarrier’ is a term used to refer to microspheres that support cells in mammalian cell culture, in which cells grow as monolayers on the surface of the particles. Microcarriers are spherical particles with a size ranging between 100 and 200 µm. Due to their small size, microcarriers have a wide variety of applications, one of which is cell culture and tissue engineering. This study proposes a novel model system for the in vitro study of cell proliferation ability on microcarriers.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Introduction
Microspheres are spherical particles between 0.1 and 200 µm in size (Sahil et al., 2011). Due to its small size, microspheres can be categorized as a microparticle, microcarrier, and microcapsule. It has a wide variety of applications, e.g. in drug delivery systems, cell culture and tissue engineering, protein immobilization, and gene delivery. In each application, there are different terms used to indicate the particular use of microspheres specifically. The term microparticle is commonly used in the drug delivery system where a consistent and predictable particle surface area is important (Nikam et al., 2012). The microsphere that can entrap cells in the inner compartment is called a microcapsule (Tan et al., 2010). Meanwhile, the microsphere is used to support cells in mammalian cell culture which grow as monolayers on the surface of the microsphere is called a microcarrier (Brun-Graeppi et al., 2011; Tan et al., 2010). In this article, the term microcarrier is used as it is applied to cultivate and propagate mammalian cells.
4.1.1 Principle
The microcarrier cell culture system served two significant purposes. First, mass production of certain bioproducts, such as recombinant proteins, hormones, and vaccines, whereby animal cells are routinely cultured in a bioreactor to meet industrial demand (van der Velden-de Groot, 1995) and in a clinical trial stage (Goh et al., 2013). The second purpose is to serve as the delivery of cultured cells, and the transplantation of biodegradable microcarriers loaded with cultured cells into the body (Seland et al., 2011). Therefore, materials with appropriate degradation rates are beneficial in the microcarrier cell culture system to minimize the effect of a toxic degradation product. Formerly, in cell culture work, anchorage-dependent cells are typically activated on the wall of roller bottles or unagitated vessels, such as tissue culture flasks (White & Ades, 1990). This system is perfectly suited for research and lab scale. Moving to the industrial scale, these systems were no longer relevant due to limitations in culture space, cell yield, control in culture condition, and sterility. Particularly, industrial production includes the production of large quantities of mammalian cells and their bioproduct. Microcarrier is one of the most established technological platforms for industrial production to increase productivity (Chu & Robinson, 2001). Microcarrier acts as a substrate for cells to attach and is cultured in suspension in a bioreactor. There are various types of bioreactors used by microcarriers to grow mammalian cells, such as a stirred tank and a fluidized bed bioreactor.
Cell culture using microcarrier beads as a three-dimensional (3D) substrate was first introduced by Wezel (1967). This 3D cell culture has undergone extensive modifications to optimize conditions for cell propagation and simulating in vitro and in vivo conditions (Overstreet et al., 2003). Under proper conditions, the cells attach and spread to the carrier beads and gradually expand into a confluent monolayer (van der Velden-de Groot, 1995).
Three different types of polycaprolactone (PCL) based microcarriers that have been developed in previous research (Samsudin et al., 2017) (gelatin-coated PCL microspheres, UV/O3-treated PCL, and untreated PCL microcarrier) were further tested to support the attachment and growth of rat amniotic fluid stem cells (AFSC).
4.2 Objective of Experiment
This study was set to determine the biocompatibility of the newly developed microcarrier, as well as to investigate the ability of AFSCs to maintain pluripotency after being cultivated in microcarrier culture.
4.3 Methodology
4.3.1 Cell Line
The cell line was kindly provided by Dr. Norshariza Nordin, Genetic and Regenerative Medicine Research Centre, Department of Obstetrics and Gynecology, Faculty of Medicine and Health Sciences, University Putra Malaysia. Amniotic fluid was collected from time mated Sprague Dawley rats as described in Mun-Fun et al. (2015).
4.3.2 Media Preparation
Two types of media were prepared. Essential stem media (ESM) for the cultivation of the AFSC and spontaneous stem cell (EBM) differentiation in a biocompatibility study. The basic media component (Table 4.1) was prepared and kept at 4 °C until further used. Mixing was conducted under a sterile condition in a biosafety hood.
4.3.3 Embryonic Stem Media (ESM)
ESM was prepared by mixing basic media with 15% FBS and 10 ng/mL of rat LIF and made to a total volume of 50 mL. It is best to prepare the ES media fresh before use.
4.3.4 Embryoid Bodies Media (EBM)
EBM was prepared by combining the basic media with 15% FBS and made to a total volume of 50 mL. It is best to prepare the EB media fresh before use.
4.3.5 Cell Propagation in a 2D Cultured Flask
4.3.5.1 Thawing of Cryopreserved Cells
The method suggested by Freshney (2010) was closely followed. The DEMEM media supplemented by 15% FBS was prepared accordingly before the thawing process. A vial of a cryopreserved cell (1 mL) from −150° condition was warmed in a 37 °C water bath. Once the cryopreserved media starts to melt, DMEM medium (1 mL) was added into the vial and the mixture was mixed using a micropipette. The mixture was then transferred into a 15 mL centrifuge tube and centrifuged at 800 × g for 5 min at 25 °C. The supernatant was discarded and 1 mL of fresh media containing 10% FBS was used to resuspend the cell pellet. About 4 mL of fresh medium were added to the homogeneous cell and the cells were counted. Cells were made to \( 1\times 1 0^{ 5} \) cells/mL concentration seeded into 25 cm2 T-flask containing media with the volume that was summed up to a total of 5 mL. The flask containing cells was then incubated in the CO2 incubator supplied with 5% CO2 at 37 °C.
4.3.5.2 Cell Counting
The concentration of cells in the suspension was determined using a haemacytometer with the aid of trypan blue. About 20 mL of cell suspension were mixed with an equal volume of trypan blue dye. The microliters of the mixture were placed on the haemacytometer and allowed to spread by capillary action. Cells were counted under an inverted microscope and the concentration of cells (cells/mL) was calculated using Eq. (4.1).
c = cell concentration (cells/mL)
n = number of cells
v = volume counted (mL).
Standard Heamacytometer used to have the depth of chamber of 1 mm and the area of the central grid is 1 mm2. Therefore v = 0.1 mm3. The formula then becomes
or
If the cells were too concentrated, the cell suspension can be diluted, and the dilution factor was added to the calculation as follows:
4.3.5.3 Sub-culture of Cells
Media supplemented with 10% FBS was prepared according to the number and size of the flask to be used. Once the cells reached confluency or the media was exhausted, the cells were sub-cultured using a method described by Butler (2004). The spent media from confluent monolayer cells was carefully removed from the flask and the surface of the flask was washed with 2 mL of PBS to remove remaining FBS. One mL of Accutase (an enzyme with proteolytic and collagenolytic activity for detachment of cells from the flask’s surface) was added to the flask and incubated in a CO2 incubator for 5 min for detachment. About 4 mL of culture with 10% FBS was added to the flask, 1 mL of culture with 10% FBS was added to the flask. Cell detachment was monitored at 5 min intervals with a microscope to ensure the cell has been detached. To stop the reaction of the proteolytic enzyme, 1 mL of culture with 10% FBS was added to the flask. The cell’s mixture was then removed into a 15-mL tube and centrifuged at 800 × g for 5 min. The supernatant was discarded and the pellet was resuspended in 5 mL of media. Cell concentration was determined and was made to \( 1 \times 10^{5} \) cells/mL concentration seeded into a T-flask containing media. The flask containing the cells was then incubated in a CO2 incubator supplied with 5% CO2 at 37 °C.
4.3.6 Microcarrier Spinner Vessel Culture
4.3.6.1 Microcarriers Preparation
PCL microcarriers were sterilized using 70% ethanol rather than the standard autoclaving method, since the heat may cause the biopolymer to melt. PCL microcarriers (3 g/mL) were washed in Ca2+, Mg2+-free PBS. Once settled, the supernatant was decanted and was replaced by 70% (v/v) ethanol in distilled water. Microcarriers were washed twice with ethanol solution and then incubated overnight in 70% (v/v) ethanol. The ethanol solution was removed and microcarriers were rinsed three times in sterile Ca2+and Mg2+-free PBS (50 mL/g microcarrier) and once in culture medium (20–50 mL/g microcarriers) before use.
4.3.6.2 Spinner Vessel Culture
Cells were grown using a 500 mL spinner vessel with 200 mL working volume. Before cell inoculation, the inner surface of the spinner vessel was coated with 5% silicon oil in ethyl acetate to prevent microcarriers from attaching to the inner surface. After sterilization by standard autoclaving, the vessel was transferred to a biosafety cabinet. The culture medium (150 mL) supplemented by the desired concentration of fetal bovine serum (FBS) was transferred aseptically into the spinner vessel. This was followed by inoculation of 30 mL of culture medium containing suspended microcarriers, and 20 mL of culture medium supplemented by the desired concentration of FBS containing cells at a concentration of \( 1. 5\times 1 0^{ 5} \) cells/mL. The spinner vessel was then transferred to the humidified CO2 incubator and agitated at low speed (30 rpm) for the first two hours and then continued at the desired agitation. Cell sampling was taken at 8 h–interval to determine cell growth. The cell growth on the newly developed biodegradable microcarrier was observed and the growth kinetic was calculated based on
4.3.6.3 Sampling and Cell Counting
One mL of microcarriers culture was aseptically pipetted out from the spinner flask culture and placed in a 15 mL tube. The microcarriers could settle and the supernatant was discarded. Microcarriers were washed twice with PBS before treatment with Accutase and the tube was incubated in a CO2 incubator for 15 min at 37 °C. After 15 min the mixture was gently flushed to detach the immobilized cells. The concentration of cells in the suspension was determined using a haemacytometer with the aid of trypan blue. Twenty mL of cell suspension was mixed with an equal volume of trypan blue dye. Ten microlitres of the mixture were placed on the haemacytometer and allowed to spread by capillary action. Cells were counted under an inverted microscope and the concentration of cells (cells/mL) was calculated using Eq. (4.2).
4.4 Result and Discussion
This study investigated the ability of the developed microcarriers to support rat amniotic fluid stem cell (AFSC), a primary cell line. AFSC was first isolated in 2007 by De Coppi et al. (2007) (as cited in Mun-Fun et al., 2015, p. 89) with high differentiation capacities that enable cells to develop into three primary germ layers linages (ectoderm, mesoderm, and endoderm). Under proper conditions, cells can be differentiated into dopaminergic neuron using a directed monolayer differentiation protocol which is potentially useful for Parkinson’s disease treatment (Mun-Fun et al., 2015)
In this study, several AFSC responses upon interaction with microcarriers were determined. This includes AFSC adherence and proliferation during expansion on microcarriers, as well as their ability to retain cell shape and organization through enzymatic retrieval from microcarriers. The ability of the cells to retain their properties is a crucial aspect in maintaining the differentiation potential of the stem cell. The expansion of AFSCs in 3D cultures has been previously studied by Liu (2004). The result shows an improvement in cell yield when a microcarrier-based spinner flask culture system was used to scale up AFSC cell expansion.
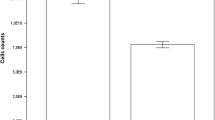
Figure 4.1 shows the growth performance of AFSC on gelatin-coated PCL microcarrier, UV/O3-treated PCL microcarrier, and untreated PCL microcarrier (control). Meanwhile, Table 4.2 shows the results of the calculated maximum cell number, growth rate, and the doubling time of the three microcarriers used. All experiments were carried out at a seeding concentration of \( 1. 5\times 1 0^{ 5} \) cells/mL and microcarrier concentration of 3 g/L. The number of cells that adhered to the microcarrier was calculated every 12 h for 5 days. Cell growth kinetics for gelatin-coated PCL microcarrier exhibited the highest final yield of \( 1. 6 5\times 1 0^{ 6} \) cell/mL (11-fold expansion) on the second day of cultivation. The culture showed fast growth (doubling time 27.75 h) before a lag phase at 36 h before the exponential phase (Fig. 4.1). The number of cells started to decrease 70 h later due to nutrient depletion and accumulation of waste in the culture media. Slow growth with a doubling time of 55.91 h was observed in UV/O3 culture with UV/O3-treated PCL. The maximum yield obtained was very low \( ( 4. 2 5\times 1 0^{ 5} {\text{cells}}/{\text{mL }} \)) compared to the gelatin-coated PCL microcarrier culture. This low yield could be attributed to the incompatibility of the surface as AFSC is a type of primary cell which may have low plating efficiency.

Growth kinetics of rat amniotic stem cell (AFSC) on different microcarriers in stirred spinner flasks: (x) UV/O3 PCL, (●) gelatin immobilized, (▲) untreated PCL
According to Hwang and coworkers (as cited in Amelia & Mohd Ridzuan, 2015, p. 18153), cells have different requirements for attachment depending on the cell’s specificity. Therefore, incorporation of gelatin into PCL microcarrier surface may cater to the needs of AFSC as gelatin contains many reactive groups that allow binding to cell integrin leading to better attachment and growth. In contrast, UV/O3-treated- PCL microcarrier offers only a charged surface. Besides, the immobilization of cells, particularly stem cells and primary cells, may enable the control of the behavior of the cells, including its differentiation potential. For example, Yang et al. (2003) showed that stem cells cultured on gelatin-coated microsphere maintain ex vivo expansion of stem cells while preserving their differentiation potential (Yang et al., 2003).
It is also important to establish a stable propagation of AFSC without differentiation during cultivation. According to Mun-Fun et al. (2015), AFSC is capable to differentiate into three types of germ layers (ectoderm, mesoderm, and endoderm). As such, spontaneous differentiation may give rise to a variety of phenotypes that may lead to the formation of teratomas (consisting of three tissue from the germ layers) at the transplantation site (Hentze et al., 2009). In a study by Murray and Edgar (2001), Leukemia Inhibitor Factor (LIF) was added to the culture medium to inhibit the differentiation of epiblast cell mouse embryonic stem cells into visceral and parietal (endoderm lineage). Therefore, it inhibits the formation of embryonic bodies during cultivation (Murray & Edgar, 2001).
Figure 4.2 shows the morphology of AFSC cells on untreated PCL, UV/O3-treated PCL, and gelatin-coated PCL observed using phase contrast microscope and SEM at 60 h of cultivation. No aggregates were observed for all cultures for the first 3 days of cultivation, but cell bridges start to form in UV/O3-treated PCL and gelatin-coated PCL microcarrier cultures.

Micrograph of AFSC at 96 h on (a) untreated PCL (b) UV/O3 treated PCL (c) gelatin coated PCL visualized using an inverted phase contrast microscope (100 x amplification) Scale bar: 100 µm. SEM image of (d) AFSC on UV/O3 treated PCL and (e) AFSC on gelatin-coated PCL (1000 x amplification) Scale bar: 100 µm
4.4.1 Spontaneous Differentiation of AFSC
The main goal of studying spontaneous differentiation of AFSC was to confirm that AFSC cells cultured for 5 days under stirred conditions using microcarriers retained their pluripotency. The functional pluripotency was determined by the ability of AFSC to differentiate spontaneously to form the good quality of embryonic bodies (EBs) (Itskovitz-Eldor et al., 2000). According to Mun-Fun et al. (2015), the formation of EBs is one of the principal tests to determine the “stemness” of the stem cell. In this phase of the study, a hanging drop method was applied in which the collected cells post trypsinization from UV/O3-treated PCL and gelatin-coated PCL were resuspended in EB medium (without LIF), aliquoted to 20 µl drops containing 4000 cells, and placed on the underside of the petri dish lid. Trypsinized cells from untreated PCL microcarriers were not included in the formation of EB due to the insufficient number of cells to perform the spontaneous differentiation. Figure 4.3 illustrates the flow of AFSC cultivation on PCL microcarrier (UV/O3-treated and gelatin-coated) and 2D culture (T-flask) as a positive control. Following is the spontaneous differentiation of the AFSC to the EB produced by the hanging drop method. Figures 4.3 (d and e) shows the unsuccessful spontaneous differentiation using different strains of AFSC into EB as a reference. Cells tend to be attached to the culture flask rather than to form cell aggregates (EBs).

Illustrative flow of cultivation of AFSC in spinner vessel on UV/O3 and gelatin coated PCL microcarrier and T-flask culture as a control. The formation of EBs using hanging drop method from UV/O3 (b) and gelatin coated (c) were compared to control (a). Figure (d) and (e) unsuccessful spontaneous differentiation to form EB. Scale bar: 100 µm
EBs were analyzed based on smooth boundaries, size, and occurrence of the cavitation process (Kim et al., 2011). AFSC cultured on microcarriers (Figs. 4.3b and 4.3c) was able to form EBs with a size range between 100 and 300 µm. This is a significant measure to ensure the quality and successful differentiation into the three cell lineages (Messana et al., 2008).
Besides, the formation of a cavity in the middle of EB is another measure of good EBs and its formation is significantly related to the initiation of EBs to differentiate into three germs layers (Rodda et al., 2002). According to Itskovitz-Eldor et al. (2000), stem cells are true pluripotent because when they are capable to differentiate into all embryonic lineages. Thus, the formation of post-microcarrier EBs (3D) cultivation has confirmed that pluripotency of AFSC is being preserved; which is comparable to EBs of AFSC culture in 2D culture (T-flask).
4.5 Conclusion
This study explores the development of functional surface biodegradable PCL microcarrier and the use of PCL microcarrier in cell and tissue culture and regenerative medicine. The result shows that the gelatin-coated PCL microcarrier is capable to support the growth and proliferation of AFSC. The growth of AFSC was assisted only by the gelatin-coated PCL microcarrier as they are the primary type of cells with low plating efficiency that require supplementary growth factor to attach and proliferate in vitro. As a result of easy and safe accessibility, abundant cell numbers, and lack of ethical concerns, AFSC has emerged as an attractive source of stem cells for basic research and clinical applications. Compared to 2D cultures, AFSCs grown in 3D microenvironments of gelatin microcarrier had stable proliferation with a significantly higher expansion fold, suggesting that the gelatin-coated PCL microcarrier is an effective 3D support for anchorage-dependent AFSC.
References
Amelia, A. K., & Mohd Ridzuan, A. (2015). A review of cell adhesion studies for biomedical and biological applications. International Journal of Molecular Sciences, 16, 18149–18184.
Brun-Graeppi, A. K. A. S., Richard, C., Bessodes, M., Scherman, D., & Merten, D. W. (2011). Cell microcarrier and microcapsule of stimuli-responsive polymers. Journal of Controlled Release, 149(3), 209–224.
Butler, M. (2004). The basic animal cell culture and technology. Oxford University Press.
Chu, L., & Robinson, K. R. (2001). Industrial choice for protein production by large scale cell culture. Biochemical Engineering, 12, 180–187.
Freshney, R. I. (2010). Culture of animal cells: A manual of basic technique (6th ed.). Wiley-Blackwell.
Goh, T. K.-P., Zhang, Z.-Y., Chen, A. K.-L., Reuveny, S., Choolani, M., Chan, J. K. Y., & Oh, S. K.-W. (2013). Microcarrier culture for efficient expansion and osteogenic differentiation of human fetal mesenchymal stem cells. BioResearch Open Access, 2(2), 84–97. https://doi.org/10.1089/biores.2013.0001.
Hentze, H., Soong, P. L., Wang, S. T., Phillips, B. W., Putti, T. C., & Dunn, N. R. (2009). Teratoma formation by human embryonic stem cells: Evaluation of essential parameters for future safety studies. Stem Cell Research, 2(3), 198–210.
Itskovitz-Eldor, J., Schuldiner, M., Karsenti, D., Eden, A., Yanuka, O., Amit, M., Soreq, H., & Benvenisty, N. (2000). Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Molecular Medicine, 6(2), 88–95.
Kim, J. M., Moon, S.-H., Lee, S. G., Cho, Y. J., Hong, K. S., Lee, J. H., & Chung, H.-M. (2011). Assessment of differentiation aspects by the morphological classification of embryoid bodies derived from human embryonic stem cells. Stem Cells and Development, 20(11), 1925–1935.
Liu, Q. (2004). Tissue engineering. In D. Shi (Ed.), Biomaterials and tissue engineering. Springer-Verlag Berlin Heidelberg.
Messana, J. M., Hwang, N. S., Coburn, J., Elisseeff, J. H., & Zhang, Z. (2008). Size of the embryoid body influences chondrogenesis of mouse embryonic stem cells. Journal of Tissue Engineering and Regenerative Medicine, 2, 499–506.
Mun-Fun, H., Ferdaos, N., Hamzah, S. N., Ridzuan, N., Hisham, N. A., Abdullah, S., & Nordin, N. (2015). Rat full term amniotic fluid harbors highly potent stem cells. Research in Veterinary Science, 102, 89–99.
Murray, P., & Edgar, D. (2001). The regulation of embryonic stem cell differentiation by leukaemia inhibitor factor (LIF). Differentiation, 68(4–5), 227–234.
Nikam, V. K., Gudsoorkar, V. R., Hiremath, S. N., Dolas, R. T., & Kashid, V. A. (2012). Microspheres-A novel grug delivery system: an overview. International Journal of Pharmaceutical Chemical, 1(1), 113–128.
Overstreet, M., Sohrabi, A., Polotsky, A., Hungerford, D., & Frondoza, C. G. (2003). Collagen microcarrier spinner culture promotes osteoblast proliferation and synthesis of matrix proteins. Vitro Cellular and Developmental Biology, 39, 228–234.
Rodda, S. J., Kavanagh, S. J., Rathjen, J., & Rathjen, P. D. (2002). Embryonic stem cell differentiation and the analysis of mammalian development. International Journal of Developmental Biology, 46, 449–458.
Sahil, K., Akanksha, M., Premjeet, S., Bilandi, A., & Kapoor, B. (2011). Microsphere: A review. International Journal of Research in Pharmacy and Chemistry, 1(4), 1184–1198.
Samsudin, N., Hashim, Y. Z. H-Y., Arifin, M. A., Mel, A., Salleh, H. M., Spoyan, I., & Jimat, D. N. (2017). Optimization of ultraviolet ozone treatment process for improvement of polycaprolactone (PCL) microcarrier performance. Cytotech. https://doi.org/10.1007/s10616-017-0071-x.
Seland, H., Gustafson, C.-J., Johnson, H., Junker, J. P. E., & Kratz, G. (2011). Transplantation of acellular dermis and keratinocytes cultured on porous biodegradable microcarriers into full-thickness skin injuries on athymic rats. Burns: Journal of the International Society for Burn Injuries, 37(1), 99–108.
Tan, H., Wu, J., Huang, D., & Gao, C. (2010). The design of biodegradable microcarriers for induced cell aggregation. Macromolecular Bioscience, 10(2), 156–163.
van der Velden-de Groot, C. a. (1995). Microcarrier technology, present status and perspective. Cytotechnology, 18(1–2), 51–6.
Wezel, V. (1967). Growth of cell-strains and primary cells on micro-carriers in homogeneous culture. Nature, 216, 64–65.
White, L. A., & Ades, E. W. (1990). Growth of vero E-6 cells on microcarriers in a cell bioreactor. Journal of Clinical Microbiology, 28(2), 283–286.
Yang, Y., Porte, M., Marmey, P., El Hai, A. J., Amedee, J., & Baquey, C. (2003). Covalent bonding of collagen on poly(L-lactic acid) by gamma irradiation. Nuclear Instrumentation Methods, 207, 165–174.
Acknowledgements
The authors are grateful to the Ministry of Higher Education Malaysia for their research grant (PRGS 11-001-0001) under the Prototype Research Grant Scheme (PRGS) and the Department of Biotechnology Engineering, International Islamic University Malaysia for their support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Samsudin, N., Hashim, Y.Z.HY., Salleh, H.M., Ariffin, A. (2021). Proliferation of Rat Amniotic Stem Cell (AFSC) on Modified Surface Microcarrier. In: Amid, A. (eds) Multifaceted Protocols in Biotechnology, Volume 2. Springer, Cham. https://doi.org/10.1007/978-3-030-75579-9_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-75579-9_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-75578-2
Online ISBN: 978-3-030-75579-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)