
Abstract
Precursor lymphoid neoplasm consists of mainly B lymphoblastic leukemia/lymphoma (B-ALL) and T lymphoblastic leukemia/lymphoma (T-ALL). B-ALL has characteristic molecular genetic profiles, including cytogenetic and molecular changes, such as BCR-ABL1 fusion and KMT2A/MLL translocations. B-ALL with different recurrent chromosomal abnormalities has been recognized as B-ALL subtypes since they demonstrate distinct clinical or prognostic features, including two new entities, B-ALL with intrachromosomal amplification of chromosome 21 (iAMP21) and BCR-ABL1-like B-ALL. Molecular genetic studies on B-ALL have extended our understanding of the genetic landscape of B-ALL, and the gene mutations or aberrations involved in various key pathways have been illustrated. T-ALL has recurrent cytogenetic changes such as translocations involving TR loci and characteristic molecular alterations involved in different pathways including NOTCH1 activating mutations and loss of CDKN2A/2B locus. A subtype of T-ALL, early T-cell precursor lymphoblastic leukemia (ETP-ALL), demonstrates different molecular genetic profiles from other T-ALL, harboring gene mutations more often associated with acute myeloid leukemia. Furthermore, molecular methodologies recommended in initial B-ALL and T-ALL workup and minimal residual disease (MRD) detection have been discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- B lymphoblastic leukemia
- T lymphoblastic leukemia
- Early T-cell precursor lymphoblastic leukemia
- Cytogenetics
- Molecular genetics
- Molecular pathology
- Molecular diagnostics
List of Frequently Asked Questions
-
1.
What are the recommendations for initial molecular genetic workup of B lymphoblastic leukemia/lymphoma (B-ALL)?
-
2.
What are the recommendations for initial molecular genetic workup of T lymphoblastic leukemia/lymphoma (T-ALL)?
-
3.
What are the most common cytogenetic abnormalities in B-ALL? How are these changes integrated into the subclassification of B-ALL?
-
4.
Why BCR-ABL1 fusion and KMT2A (MLL) translocations are important in B-ALL subtyping?
-
5.
What genetic alterations are associated with TKI resistance and relapse of B-ALL with BCR-ABL1?
-
6.
What are the cytogenetic features of KMT2A/MLL translocation?
-
7.
What are the prognostic implications of the chromosomal numerical abnormalities in B-ALL?
-
8.
What is the significance of TP53 mutations in hypodiploid B-ALL?
-
9.
What are some other rare cytogenetic changes associated with poor prognosis in B-ALL?
-
10.
What are the clinicopathological features of B-ALL with t(12;21)/ETV6-RUNX1?
-
11.
How is B-ALL with intrachromosomal amplification of chromosome 21 (iAMP21) diagnosed and what are its clinical features?
-
12.
What is BCR-ABL1-like B-ALL, and how can the diagnosis be made?
-
13.
What are the specific molecular genetic changes in BCR-ABL1-like B-ALL?
-
14.
What is the mutational landscape of B-ALL?
-
15.
What is the significance of IKZF1 mutations in B-ALL?
-
16.
What is the significance of CRLF2 alterations in B-ALL?
-
17.
What is the significance of PAX5 alterations in B-ALL?
-
18.
What is the significance of CREBBP mutations in B-ALL?
-
19.
What is the significance of ERG mutations in B-ALL?
-
20.
What are the molecular methods to assess ALL minimal residual disease (MRD)?
-
21.
What are the molecular genetic changes in T lymphoblastic leukemia (T-ALL)?
-
22.
What are the molecular genetic changes in early T-cell precursor lymphoblastic leukemia (ETP-ALL)?
Frequently Asked Questions
-
1.
What are the recommendations for initial molecular genetic workup of B lymphoblastic leukemia/lymphoma (B-ALL)?
-
The most recently published guidelines by College of American Pathologists and American Society of Hematology (CAP/ASH) recommend the below initial molecular genetic workup for B-ALL [1]:
-
Conventional cytogenetic analysis (i.e., karyotype), appropriate molecular genetic testing, and/or fluorescence in situ hybridization (FISH) testing should be performed.
-
Testing for t(12;21)(p13.2;q22.1); ETV6-RUNX1, t(9;22)(q34.1;q11.2); BCR-ABL1, KMT2A (MLL) translocation, iAMP21, and trisomy 4 and 10 is recommended for pediatric cases with suspected or confirmed B-ALL.
-
Testing for t(9;22)(q34.1;q11.2); BCR-ABL1 is recommended for adult cases with suspected or confirmed B-ALL. Testing for KMT2A (MLL) translocations may be performed.
-
Mutational analysis for selected genes that influence diagnosis, prognosis, and/or therapeutic management, which include, but are not limited to, PAX5, JAK1, JAK2, and/or IKZF1, is recommended for B-ALL. Testing for overexpression of CRLF2 may also be performed.
-
-
National Comprehensive Cancer Network (NCCN) guidelines have similar recommendations of karyotype and appropriate FISH, as well as reverse transcription polymerase chain reaction (RT-PCR) for BCR-ABL1 product (i.e., p190 vs. p210) in BCR-ABL1-positive B-ALL. When BCR-ABL1 is negative, NCCN guidelines encourage testing for gene mutations associated with BCR-ABL1-like B-ALL including gene fusions of ABL1, ABL2, CRLF2, CSF1R, EPOR, JAK2, or PDGFRB and mutations of FLT3, IL7R, SH2B3, JAK1, JAK3, and JAK2 (in combination with CRLF2 gene fusions). Additional optional tests include genomic assessment (comparative genomic hybridization array, CGH array) in cases of aneuploidy or failed karyotype [2].
-
-
2.
What are the recommendations for initial molecular genetic workup of T lymphoblastic leukemia/lymphoma (T-ALL)?
CAP/ASH and NCCN guidelines recommend the below testing [3]:
-
Conventional cytogenetic analysis.
-
Mutational analysis for selected genes that influence diagnosis, prognosis, and/or therapeutic management, such as NOTCH1 and/or FBXW7.
-
-
3.
What are the most common cytogenetic abnormalities in B-ALL? How are these changes integrated into the subclassification of B-ALL?
-
B-ALL is a heterogeneous disease that is associated with a plethora of chromosomal abnormalities, involving both numerical and structural alterations, such as hyperdiploidy, hypodiploidy, translocation, and intrachromosomal amplification. Approximately 75% of B-ALL cases have recurrent chromosomal changes detectable by conventional cytogenetic analysis [4], many of which have impacts on prognosis and are used for risk stratification on some treatment protocols [4,5,6] (Table 15.1, Fig. 15.1).
-

Frequency of cytogenetic and molecular genetic abnormalities in pediatric B-ALL (a) and adult B-ALL (b)
-
The cytogenetic changes have also been integrated into the 2017 revision of WHO classification of tumors of hematopoietic and lymphoid tissues [7]. They have been categorized as such because the recurrent cytogenetic changes are associated with distinctive clinical or phenotypic properties, have important prognostic implications, demonstrate other biologically distinct features, and are generally mutually exclusive with other entities. The subclassification of acute lymphoblastic leukemia/lymphoma includes the below entities:
-
B-lymphoblastic leukemia/lymphoma, not otherwise specified (NOS)
-
B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities
-
B-lymphoblastic leukemia/lymphoma with t(9;22)(q34.1;q11.2); BCR-ABL1
-
B-lymphoblastic leukemia/lymphoma with t(v;11q23.3); KMT2A rearranged
-
B-lymphoblastic leukemia/lymphoma with t(12;21)(p13.2;q22.1); ETV6-RUNX1
-
B-lymphoblastic leukemia/lymphoma with hyperdiploidy
-
B-lymphoblastic leukemia/lymphoma with hypodiploidy
-
B-lymphoblastic leukemia/lymphoma with t(5;14)(q31.1;q32.3); IL3-IGH
-
B-lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3); TCF3-PBX1
-
Provisional: B-lymphoblastic leukemia/lymphoma, BCR-ABL1-like
-
Provisional: B-lymphoblastic leukemia/lymphoma with iAMP21
-
-
4.
Why BCR-ABL1 fusion and KMT2A ( MLL ) translocations are important in B-ALL subtyping?
-
BCR-ABL1 fusion (also known as t(9;22) translocation, Philadelphia chromosome, and Ph chromosome) and KMT2A/MLL translocations are associated with an increased risk of disease relapse or worse prognosis.
-
The clinical outcome with conventional chemotherapy in the patient group with BCR-ABL1 is extremely poor. However, tyrosine kinase inhibitors (TKIs) such as imatinib mesylate, in combination with intensive chemotherapy, have been used successfully, although primary or secondary drug resistance and high rates of relapse are problematic [9].
-
Rearrangements involving the KMT2A/MLL gene and partner genes are associated with poor prognosis. There is also a high frequency of central nervous system involvement at diagnosis.
-
-
5.
What genetic alterations are associated with TKI resistance and relapse of B-ALL with BCR-ABL1 ?
-
Point mutations within the ABL1 kinase domain and alternative signaling pathways mediated by the Src family kinase are implicated in the mechanism of resistance to TKI therapy [10, 11].
-
ABL1 mutations (frequently T315I, Y253F/H, E255K/V, M351T, G250E, F359C/V, H396R/P, M244V, E355G, F317L, M237I, Q252H/R, D276G, L248V, F486S, etc.) are the major contributors to the TKI resistance [12], for which new TKIs have been developed to bypass the signaling pathways or to bind to alternative sites, including bosutinib, dasatinib, nilotinib, and ponatinib. They have shown great improvement on the clinical response in certain patients [13].
-
Clonal evolution and secondary gene aberrations such as deletions or mutations of IKZF1 or other genes are found to be significantly associated with the resistance and relapse [14, 15].
-
-
6.
What are the cytogenetic features of KMT2A/MLL translocation?
-
The mixed-lineage leukemia 1 (MLL1) gene (now renamed lysine [K]-specific MethylTransferase 2A or KMT2A) has more than 100 different partner genes described.
-
Most cases show MLL1 fusions with one of the six common partner genes: AFF1/AF4 [t(4,11)], MLLT3/AF9 [t(9,11)], MLLT1/ENL [t(11,19)(q23,p13.3)], AF10 [t(10,11)], ELL [t(11,19)(q23,p13.1)], and AF6 [t(6,11)] [16, 17].
-
The translocation may be missed by conventional karyotyping, and FISH with a KMT2A/MLL break-apart probe can be performed. PCR may be used to identify major translocation partners.
-
-
7.
What are the prognostic implications of the chromosomal numerical abnormalities in B-ALL?
-
Conventional cytogenetic analysis can identify changes in chromosome number. Numerical abnormalities may involve the whole chromosome set, resulting in ploidy changes or the gain or loss of individual chromosomes (aneuploidy).
-
Chromosomal numerical changes have prognostic significance.
-
High hyperdiploidy (51–65 chromosomes) is an independent indicator in childhood ALL with favorable prognosis [18].
-
Hypodiploidy (<46 chromosomes, with some people suggesting a stricter criteria with <44 chromosomes [19]) is a poor prognostic indicator.
-
Hypodiploidy can be further classified into different categories: high hypodiploidy (40–43 chromosomes), low hypodiploidy (33–39 chromosomes), and near haploidy (23–29 chromosomes) [20]. The patient has progressively poor prognosis with decreasing chromosome numbers. Near-haploidy and low-hypoploidy B-ALL patients have extremely poor prognosis [21]. Near-diploid (44–45 chromosomes) B-ALL is usually not included in hypodiploid B-ALL.
-
B-ALL cases with hypodiploid karyotype may appear to be hyperdiploid by conventional karyotyping, when the hypodiploid cells undergo endoreduplication. The set of chromosomes is often doubled, which allows a distinction between hypodiploid ALL with doubled chromosome set and hyperdiploid B-ALL [7, 22]. SNP microarray, FISH , and DNA content flow cytometry may be helpful in differentiating the ploidy level (see case study #2).
-
-
8.
What is the significance of TP53 mutations in B-ALL, particularly hypodiploid B-ALL?
-
TP53 deletions and mutations are initially found in 2–4% of pediatric patients [23] and 8% of adult patients [24] at initial diagnosis of B-ALL. Next-generation sequencing (NGS) data revealed that overall TP53 mutations were present in up to 16% of B-ALL cases [25,26,27].
-
TP53 alterations, often germline as seen in Li-Fraumeni syndrome [27], are present in almost all cases of B-ALL with low hypodiploidy [28].
-
TP53 germline mutations are associated with early relapse and poor overall survival in pediatric and adult B-ALL [24, 29,30,31].
-
TP53 alterations are associated with alterations of the lymphoid transcription factor IKZF2 and the tumor suppressor gene loci CDKN2A and CDKN2B [28].
-
-
9.
What are some other rare cytogenetic changes associated with poor prognosis in B-ALL?
-
B-ALL with rearrangement of IGH locus occurs in less than 5% of the cases and confers poor prognosis [32]. The most common partner gene is cytokine receptor-like factor 2 (CRLF2) located at chromosome X, and other partner genes can be inhibitor of DNA binding 4 (ID4), EPOR, CCAAT/enhancer-binding protein (CEBP) family members, BCL2, and the LIM domain homeobox 4 (LHX4) [33].
-
Some other genetic changes that are associated with poor prognosis include the very rare t(17;19)/E2A-HLF translocation [34], abnormal 17p, and loss of 13q [5], as well as complex karyotype with five or more abnormalities in adult B-ALL patients [35].
-
-
10.
What should be considered when testing for t(12;21)/ ETV6-RUNX1 in B-ALL?
-
Cytogenetic testing for t(12;21)/ETV6-RUNX1 is important since B-ALL with the t(12;21)/ETV6-RUNX1 (>90% are pediatric cases) has a very favorable prognosis.
-
Abnormality of t(12;21)/ETV6-RUNX1 is usually cryptic by conventional karyotyping but detectable by FISH or PCR.
-
FISH for t(12;21) can actually detect copy number changes of chromosome 21 in B-ALL with iAMP21.
-
-
11.
How is B-ALL with intrachromosomal amplification of chromosome 21 (iAMP21) diagnosed and what are its clinical features?
-
iAMP21 is amplification of a large but variable region of chromosome 21.
-
iAMP21 can be detected by FISH with a RUNX1 probe that reveals extra signals (five or more copies per interphase nucleus or three or more copies on a single abnormal chromosome 21 in metaphase FISH).
-
The abnormality can be detected by conventional karyotyping analysis with finding of the absence of a second normal copy of chromosome 21, which may not always be present [36].
-
B-ALL with iAMP21 is present in about 2% of pediatric B-ALL, mostly in older children and adolescents (median age 9 years), but is uncommon in adults. The patients are characterized by lower white blood cell and blast cell counts, older age, the French-American-British classification (FAB) L1 morphology, and common B-lymphoblast immunophenotype with a subset showing aberrant myeloid-associated antigen expression [36].
-
B-ALL with iAMP21 patients demonstrated a consistently poor prognosis with worse event-free survival and overall survival when treated with standard-risk chemotherapy regimens [37]. It is justified to assign such patients in the very-high-risk group and treat them with more intensive chemotherapy. The clinical outcome has been significantly improved with more aggressive therapy [21, 38].
-
B-ALL with iAMP21 is extremely rare in adults, and its prognostic effect in adult group is undetermined.
-
-
12.
What is BCR-ABL1 -like B-ALL, and how can the diagnosis be made?
-
BCR-ABL1-like ALL is a subgroup of BCR-ABL1-negative B-ALL exhibiting similar gene expression profile to that of B-ALL with BCR-ABL1 rearrangement [39, 40]. The patients have similar poor prognosis and high risk for relapse to BCR-ABL1-positive B-ALL [41, 42]. Patients in this group, particularly those with translocations involving tyrosine kinases, show improved clinical outcome with remarkable responses to TKI therapy [43, 44] or JAK inhibitors such as ruxolitinib [45].
-
BCR-ABL1-like ALL demonstrates a number of different genetic alterations: translocation involving cytokine receptor genes such as CRLF2 (leading to CRLF2 overexpression), translocations involving tyrosine kinases (other than BCR-ABL1), and activating mutations or deletions of critical genes (ABL1, JAK2, etc.) leading to activation of the Ras or JAK-STAT pathway [43]. The gene fusions and mutations include ABL1, ABL2, CRLF2, CSF1R, EPOR, JAK1, JAK2, JAK3, PDGFRB, EBF1, FLT3, IL7R, NIRK3, and SH2B3 genes [39].
-
There is no consensus regarding the approach to screen and diagnose BCR-ABL1-like B-ALL. It is difficult to screen for every case for all possible fusions and mutations and not practically feasible due to inaccessible genetic testing (such as gene expression profiling assays) to most labs and excessive cost.
-
It has been reported that 50% of BCR-ABL1-like B-ALL cases harbor CRLF2 rearrangements. Flow cytometry may be used to detect CRLF2 overexpression, and a subsequent FISH study can be performed to confirm CRLF2 rearrangement.
-
NGS -based targeted RNA sequencing is widely available and can be used to identify a broad variety of gene fusions including those related to BCR-ABL1-like B-ALL.
-
Other less readily available methodologies to identify BCR-ABL1-like B-ALL include gene expression profiling assay, low-density gene expression arrays, RT-PCR, and FISH for known translocations.
-
Different centers around the world use different approaches to screen and confirm BCR-ABL1-like B-ALL. Some groups in Europe use multiplex PCR or commercially available targeted RNA sequencing kits, while others use a FISH for primary screening. Some group in the USA uses low-density microarray (LDA) as the screening approach. Some others use comprehensive RNA sequencing [46].
-
Although there has been no standard guideline established for BCR-ABL1-like ALL diagnosis at initial workup of B-ALL, different algorithms have been proposed by different authors. Below is an algorithm modified from several literatures [46, 47] (Fig. 15.2).
-
-
13.
What are the specific molecular genetic changes in BCR-ABL1 -like B-ALL?
-
ABL1-like rearrangements involving ABL1, ABL2, CSF1R, and PDGFRB.
-
JAK2 or EPOR rearrangements.
-
CRLF2 rearrangements (often with JAK gene mutations and activation of JAK-STAT signaling).
-
Ras signaling pathway gene mutations.
-
Uncommon kinase alterations including NTRK3, PTK2B TYK2, etc. [43].
-
They usually have a high frequency of IKZF1 deletion (~70%), CRLF2 overexpression (~50%), and JAK mutations (~30%) [43, 48].
-
See Fig. 15.3 for the breakdown of the molecular genetic alterations [43, 49].
-

Proposed flowchart for the initial BCR-ABL1-like ALL workup
-
14.
What is the mutational landscape of B-ALL?
-
Genome-wide genetic profiling studies on B-ALL have extended our understanding of the genetic landscape of B-ALL in children and young adults over the past decade. Mutations involved in various key pathways are found in different subtypes of B-ALL.
-
The mutated genes include transcriptional factors promoting early lymphoid cell development, e.g., PAX5, IKZF1, EBF1, ETV6, and LMO2, which were detected in ~40% of B-ALL cases [50], and other genes including tumor suppressor genes and cell cycle regulators (e.g., TP53, RB1, CDKN2A/CDKN2B), cytokine receptor (e.g., CRLF2, RPOR), kinase (e.g., ABL1, ABL2, CSF1R, JAK2, PDGFRB), Ras signaling pathway (e.g., KRAS, NF1, NRAS, PTPN11), lymphoid signaling (e.g., BTLA, CD200), and epigenetic modification (e.g., EZH2, CREBBP, SETD2, MLL2, NSD2) [43, 50].
-
Among them, some genetic alterations are found to be associated with adverse or favorable clinical outcome [51] such as IKZF1, CREBBP, and ERG mutations or alterations (see Questions 15–19).
-
-
15.
What is the significance of IKZF1 mutations in B-ALL?
-
Mutations of transcription factors involved in early lymphoid development are considered a hallmark of B-ALL genetic changes. The transcription factors include IKZF1, PAX5, EBF1, ETV6, LMO2, etc.
-
IKZF1 mutation is one of the most frequent genetic aberrations in B-ALL. IKZF1 gene encodes the Ikaros transcription factor that is an important regulator of normal lymphoid development and differentiation [52, 53].
-
IKZF1 gene mutation is observed in high-risk B-ALL, including approximately 80% of BCR-ABL1-positive B-ALL cases and 70% of BCR-ABL1-like B-ALL cases [54, 55].
-
IKZF1 mutations are often deletions and rarely point mutations [56, 57]. Most deletions are monoallelic and involve exons 3–6, which encode the N-terminal zinc finger DNA-binding domain [56]. The deletions result in dominant negative form of the Ikaros protein that inhibits the function of wild-type Ikaros. It has been shown that induction of mutant, dominant negative Ikaros in early pre-B cells arrests the cell differentiation, suggesting that loss of Ikaros activity contributes to B-ALL leukemogenesis and IKZF1 mutations are likely driver mutations [58].
-
Multiple studies support that IKZF1 mutation/deletion is an independent indicator of B-ALL unfavorable clinical outcome including chemotherapy resistance and higher risk for relapse [15, 43, 59,60,61].
-
Different methodologies including single-nucleotide polymorphism (SNP) microarray, transcriptional profiling, sequencing, and CGH can be used to detect IKZF1 mutation/deletion [40, 62].
-
-
16.
What is the significance of CRLF2 alterations in B-ALL?
-
CRLF2 alterations are found in approximately 8% of pediatric B-ALL patients, and more than 50% of patients with Down syndrome-associated B-ALL [63].
-
CRLF2 alterations are commonly gene rearrangement with immunoglobulin heavy chain locus resulting in IGH-CRLF2 fusion gene, less often interstitial deletions resulting in P2RY8-CRLF2 fusion gene, and rarely can be point mutations [48, 64].
-
These changes usually result in overexpression of CRLF2 (therefore can be analyzed by flow cytometry). CRLF2 alterations are associated with constitutive activation in the JAK-2 pathway such as JAK-STAT, PI3K/mTOR, and BCL-2 transduction [65].
-
The alterations are often found in high-risk B-ALL [48], although the prognostic significance of CRLF2 deregulation in B-ALL remains controversial [64].
-
-
17.
What is the significance of PAX5 alterations in B-ALL?
-
Alterations of PAX5 have been found in ~30% of B-ALL cases [50].
-
The alterations include acquired mutations, rearrangements involving various partner genes such as ETV6 and JAK2, and germline mutations [50, 66, 67].
-
Unlike IKZF1, PAX5 alterations do not appear to impact clinical outcomes; however, the PAX5 mutations may be driver mutations in B-ALL leukemogenesis and play a role in susceptibility to B-ALL [67, 68].
-
Sequencing is usually the methodology to detect PAX5 mutations [50].
-
-
18.
What is the significance of CREBBP mutations in B-ALL?
-
Deletions and mutations of CREBBP, which encodes the transcriptional coactivators and acetyltransferase CREB binding protein, are found in 18% of relapsed pediatric B-ALL patients, but less than 1% at diagnosis in those who did not relapse [69], suggesting CREBBP gene mutations are associated with relapse of the disease.
-
The mutations result in loss of function of CREBBP. In one study, CREBBP mutations were associated with hyperdiploid B-ALL relapse. Up to 60% of high-hyperdiploid relapse cases show CREBBP mutation, altering the clinical outcome in the favorable B-ALL group [70]. It might be a marker that can be integrated into risk stratification system after large cohort study.
-
-
19.
What is the significance of ERG mutations in B-ALL?
-
Several studies have identified a subgroup of pediatric B-ALL patients, comprising 3–5% of B-ALL cases, with monoallelic deletion of ERG gene, which encodes an ETS-domain-containing transcription factor [71, 72].
-
The deletions result in an aberrant ERG protein that functions as a competitive inhibitor of wild-type ERG [73].
-
The ERG deletion and other known classifying genetic lesions are mutually exclusive, suggesting that B-ALL with ERG deletion may be a distinct subtype.
-
These patients generally have excellent prognosis, despite an association with frequent IKZF1 deletions, which is different from BCR-ABL1-positive and BCR-ABL1-like B-ALL cases [72]. Whether or not the ERG mutations function as a negative regulator under IKZF1 mutated status needs to be explored.
-
-
20.
What are the molecular methods to assess ALL minimal residual disease (MRD)?
-
The most frequently used molecular methods to assess ALL MRD are 1) antigen-receptor (immunoglobulin/IG and T-cell receptor/TCR) gene rearrangement analysis, 2) real-time quantitative polymerase chain reaction (RQ-PCR) to detect known fusion genes such as BCR-ABL1, and 3) NGS-based assay to detect clonal immunoglobulin heavy-chain gene or T-cell receptor gene rearrangements [2].
-
RQ-PCR-based IG and TCR gene rearrangement analyses and RQ-PCR-based fusion gene assay can detect MRD at a level of 1 x 10−5 sensitivity [74], and NGS-based MRD assay to detect clone-specific IG or TCR index sequence can reach a sensitivity level of 1 × 10−6, comparing to a sensitivity of 1 × 10−4 cells by using six-color flow cytometry [2, 74].
-
About 5–10% ALL cases do not carry clonal IG or TR gene rearrangements.
-
Examples of fusion genes that can be used in RQ-PCR to detect MRD include BCR-ABL1, ETV6-RUNX1, KMT2A/MLL rearrangement, and TCF3-PBX1 in B-ALL and TAL1 deletions (SIL/TAL1) in T-ALL. The detection of these potential MRD markers should be performed at diagnosis and monitored throughout the course of disease.
-
Novel technologies and applications are under further investigation and validation. Examples are droplet digital PCR [75, 76] and deep sequencing NGS [77].
-
Table 15.2 is the comparison of different methods commonly used to detect ALL MRD [74, 78,79,80,81].
-

Breakdown of kinase alterations in children, adolescents, and young adults BCR-ABL1-like ALL
-
21.
What are the molecular genetic changes in T lymphoblastic leukemia (T-ALL)?
-
Rearrangements between the regulatory region of a T-cell receptor locus (alpha and delta TR loci at 14q11.2, beta locus at 7q35, and gamma locus at 7p14-15) and an oncogenic transcription factor (such as TAL1 at 1p32, TLX1 at 10q24, MYC at 8q24.1, LMO1 at 11p15, LMO2 at 11p13, and LYL1 at 19p13) are also common [7].
-
Rearrangements of MLLT10, KMT2A, ABL1, or NIP98 with a variety of different partner genes result in deregulated expression of the transcription factor [82].
-
Most of the translocations can only be detected by molecular genetic studies, but not by conventional karyotyping. For example, TAL1 is fused to SIL/STIL as a result of a cryptic interstitial deletion at chromosome 1p32 [7].
-
NOTCH1 activating mutations and loss of CDKN2A locus occur in over 70% of the T-ALL cases. NOTCH1 pathway activation (NOTCH1 mutations, FBXW7 mutations (resulting in increased half-life of NOTCH1) or, rarely, NOTCH1 rearrangement) is present in >50% of T-ALL cases [3]. CDKN2A/2B mutations and deletion of chromosome 9p that result in inactivation of CDKN2A/2B occur in >50% of the T-ALL cases [83].
-
Genome-wide sequencing has revealed a broad spectrum of mutations and copy number alterations of genes involving JAK-STAT (IL7R, JAK1, JAK3, DNM2), RAS (NRAS, KRAS, and NF1), PI3K-AKT (PTEN, AKT1, PIK3CA, PIK3CD), epigenetic regulators (PHF6, SUZ12, EZH2, KDM6A), transcription factors and regulators (ETV6, GATA3, RUNX1, LEF1, WT1, BCL11B), and translation regulators (CNOT3, RPL5, RPL10) [83, 84].
-
-
22.
What are the molecular genetic changes in early T-cell precursor lymphoblastic leukemia (ETP-ALL)?
-
The mutational landscape of ETP-ALL includes alterations in genes involved in cytokine and RAS signaling (e.g., NRAS, KRAS, FLT3, and JAK1), epigenetic regulation (e.g., EZH2, DNMT3A, and SUZ12), and hematopoietic development (e.g., ETV6, RUNX1, and IKZF1) [85].
-
The mutation profile of ETP-ALL is more similar to that of myeloid leukemias, with high frequency mutations of FLT3, the RAS family of genes, DNMT3A, IDH1, and IDH2 [7].
-
The incidence of activating NOTCH1 mutations is lower in ETP-ALL (15%) than that of T-ALL (higher than 50%) [85].
-
ETP-ALL shares some genomic and epigenomic features with T/myeloid mixed phenotype acute leukemia, with frequent biallelic WT1 alterations, and alterations in other several transcription factors (ETV6, RUNX1, CEBPA) and signaling pathways (JAK-STAT, FLT3, RAS) [84].
-
Case Presentations
Case 1
Learning Objective
-
Review CAP/ASH and NCCN guidelines for initial ALL workup recommendations.
-
KMT2A/MLL translocation may be missed by conventional karyotyping.
-
B-ALL with KMT2A/MLL translocation cases can show peculiar immunophenotype.
Case History
A 34-year-old female complained of vague symptoms including dyspnea, headache, and fatigue. She was diagnosed with COVID-19 in 2020 and started developing new symptoms after that. She was found to have an elevated white blood cell count of 15.9 K/μL and circulating immature mononuclear cells in the peripheral blood.
Laboratory Findings
Blood cell counts: WBC 9.39 K/μL; hemoglobin 7.9 g/dL; platelets 332 K/μL
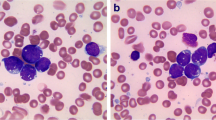
Peripheral blood smear showed 3% circulating blasts. The bone marrow was hypercellular with sheets of blasts (Fig. 15.4a, b). By immunohistochemical staining, the blasts were positive for CD34, CD19, and PAX5 and negative for TdT, CD117, CD3, CD20, and myeloperoxidase (Fig. 15.4c–f).

The bone marrow biopsy and aspirate smear showed sheets of blasts (a, b). By immunohistochemical staining, the blasts were positive for CD19 (c), PAX5 (d), and CD34 (e) and negative for TdT (F)
Flow cytometric analysis demonstrated a population of blasts expressing dim CD45, CD19, CD34, CD38, and cytoplasmic CD79a and lacking cytoplasmic CD3, CD10, CD20, CD22, CD117, myeloperoxidase, and terminal deoxynucleotidyl transferase (TdT).
Genetic Study
Chromosome analysis revealed a normal karyotype: 46,XX [10]. FISH study for BCR-ABL1 was negative. NGS analysis (targeted gene DNA sequencing and RNA sequencing) detected KMT2A (MLL)/MLLT1 (ENL) fusion. Multiple variants of unknown significance (VUS) were also detected,.
Final Diagnosis
B lymphoblastic leukemia/lymphoma, with KMT2A-MLLT1 rearrangement
Discussion
CAP/ASH and NCCN guidelines recommend initial molecular genetic workup for B-ALL to include conventional cytogenetic analysis (i.e., karyotype), appropriate molecular genetic testing, and/or FISH testing. FISH for KMT2A/MLL translocation is optional. However, conventional cytogenetic analysis may miss the KMT2A/MLL aberrancy. Some commercially available NGS-based mutational assays include both DNA sequencing and RNA sequencing and can detect certain gene rearrangements. In this case, the KMT2A -MLLT1 translocation was detected by NGS-based mutational analysis.
Additionally, B-ALL with KMT2A rearrangements may show immunophenotypic variation different from the other B-ALL cases, usually with more mature phenotype, positive for CD19, negative for CD10 and CD24, and variable expression of CD34 and TdT [86]. In this case, the immunophenotype was unusual, with negative CD10 and TdT.
Case 2 (Courtesy of Dr. Peter Papenhausen at LabCorp)
Learning Objective
-
Hypodiploidy in B-ALL can present as pseudo-hyperdiploidy.
-
SNP microarray may be helpful in differentiating the ploidy level.
Case History
A 10-year-old boy complained of fatigue and bone pain. He was found to have an elevated WBC count, anemia, and thrombocytopenia. A bone marrow aspiration was performed.
Laboratory Findings
Blood cell counts: WBC 17.5 K/μL, 60% blasts
The bone marrow aspirate smears show hypercellular marrow with greater than 90% blasts.
Flow cytometric analysis demonstrated a population of blasts (97% of total events) positive for CD45 (dim), CD19, CD10, CD22, CD34, CD38, HLA-DR, TdT, CD52, CD58, and CD123 and negative for CD2, CD3, CD4, CD5, CD7, CD8, CD11c, CD13, CD15, CD20, CD23, CD25, CD35, CD41, CD61, CD64, CD79b, CD235a, sIgM, cIgM, MPO, kappa light chain, and lambda light chain.
Genetic Study
Chromosome analysis revealed an abnormal karyotype: 52,XY,+X,+Y,+14,+14,+21,+21[cp20] (Fig. 15.5).

Chromosome analysis showed a karyotype with tetrasomies, which actually evolved from a hypodiploid karyotype
FISH studies demonstrated no ETV6(TEL)/RUNX1, MLL, BCR/ABL1, or TCF3 gene rearrangements, but 95% of nuclei showed four copies of the RUNX1 (AML1) gene locus on chromosome 21q22.
SNP microarray analysis was performed and demonstrated near-haploid clone (26 chromosomes with two copies of chromosomes 14 and 21 and single copies of all other chromosomes).
Final Diagnosis
B lymphoblastic leukemia/lymphoma , with hypodiploidy
Discussion
The conventional cytogenetics in this case showed a karyotype appearing to be hyperdiploid. However, tetrasomy 14 and 21 suggested that the karyotype might actually have evolved from hypodiploid karyotype, with chromosomes doubled from a near-haploid karyotype. SNP microarray, FISH, and DNA content flow cytometry may be helpful in differentiating the ploidy level.
B-ALL cases with hypodiploid karyotype may appear to be hyperdiploid by conventional karyotyping, when the hypodiploid cells undergo endoreduplication. The set of chromosomes is often doubled (so-called hypotriploidy, or very low tetraploid chromosome set). This results in a typical pattern of two or four chromosomes, which allows a distinction between hypodiploid ALL with doubled chromosome set and hyperdiploid B-ALL [7, 22]. Hypodiploidy is a poor prognostic indicator, while hyperdiploidy is a favorable prognostic indicator in pediatric B-ALL patients. Importantly, the subsequent therapy selection for these two subtypes is different. Current recommendations for pediatric B-ALL patients with near haploid are to proceed to allogeneic stem cell transplant at the first complete remission.
Case 3
Learning Objective
-
Diagnosis of BCR-ABL1-like B-ALL requires molecular genetic workup.
Case History
A 46-year-old female presented with progressing weakness and fatigue. Initial CBC showed leukocytosis. Peripheral blood smear showed 90% circulating blasts. CT scan showed axillary adenopathy.
Laboratory Findings
Blood cell counts: WBC 112 K/μL; hemoglobin 9.2 g/dL; platelets 103 K/μL.
The bone marrow was hypercellular with sheets of blasts (85%).
Flow cytometric analysis demonstrated a population of lymphoblasts (88% of total events) expressing dim CD45, CD19, CD10 (bright), CD22 (dim), CD58, CD200, cytoplasmic CD79a, CD33 (dim), CD99 (dim), CD34, and TdT and lacking cytoplasmic CD3, CD117, and myeloperoxidase.
Genetic Study
Chromosome analysis revealed an abnormal karyotype: 46,XX [20]. FISH for BCR-ABL1 translocation was negative.
NGS analysis (targeted gene DNA sequencing and RNA sequencing) detected JAK2 R683S mutation and IGH-CRLF2 rearrangement. Multiple VUS was also detected, including CHEK2 L236P, HDAC4 P64A, IKZF1 F145L, KMT2A A53V TNFRSF14 T160A, and TSC1 K587R.
Final Diagnosis
B lymphoblastic leukemia/lymphoma, BCR-ABL1-like
Follow-Up
The patient received hyper-CVAD chemotherapy (hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone), and a repeat bone marrow biopsy showed residual disease. The treatment was switched to augmented hyper-CVAD (plus PEG-asparaginase with higher dose of vincristine). Follow-up bone marrow biopsy showed evidence of minimal residual disease. She was then treated with blinatumomab and POMP (6-mercaptopurine (Purinethol), vincristine (Oncovin), methotrexate, and prednisone) maintenance. Unfortunately, the disease relapsed after 3 years at the last follow-up.
Discussion
This is an example of BCR-ABL1-negative B-ALL, with findings of JAK2 mutation and IGH-CRLF2 rearrangement by molecular study, consistent with BCR-ABL1-like B-ALL. JAK2 mutation and coexistent IGH-CRLF2 rearrangement (therefore high level expression of CRLF2) occur in 47% of BCR-ABL1-like B-ALL [43].
Currently there is no consensus as to the approach to diagnose BCR-ABL1-like B-ALL, and it is somehow challenging in the practice to screen and confirm this subtype of B-ALL. Mutational analysis for selected genes including the genes frequently found in BCR-ABL1-like B-ALL is recommended in BCR-ABL1-negative B-ALL. In the labs where there are resources, certain diagnostic algorithm may be adopted to be more cost-effective, such as performing FISH and CRLF2 flow cytometry analysis before mutational testing. It is clinically significant to diagnose BCR-ABL1-like B-ALL since this subtype of B-ALL shows poor prognosis and high relapse rate and, more importantly, may be treated with TKI.
Case 4
Learning Objective
-
The molecular profiling study can assist in the diagnosis of ETP-ALL .
Case History
A 70-year-old male presented with weakness and falls and CT scan of abdomen and pelvic areas showed lymphadenopathy and splenomegaly.
Laboratory Findings
Blood cell counts: WBC 14.58 K/μL; hemoglobin 9.8 g/dL; platelets 33 K/μL.
Peripheral blood shows circulating blasts (73%). The bone marrow was 60% cellular with 80% blasts (Fig. 15.6a).

The bone marrow aspirate smear showed increased medium-sized lymphoblasts (a, 1000×). Flow cytometry analysis demonstrated characteristic immunophenotype (CD45dim+, CD34+, CD117 partial+, cytoplasmic CD3+, CD7+, CD33+, TdT dim+, CD1a-, CD5-, CD8-, and cytoplasmic myeloperoxidase-) (b)
Flow cytometric analysis demonstrated a population of lymphoblasts (70% of total events) expressing dim CD45, CD34, CD117 (partial), cytoplasmic CD3, CD7, CD13, CD33, CD56, and TdT (dim) and negative for CD1a, CD5, CD4, CD8, CD2, HLA-DR, CD19, CD10, CD20, CD79a, cytoplasmic CD22, cytoplasmic CD79a, and cytoplasmic myeloperoxidase (Fig. 15.6b).
Genetic Study
NGS analysis (targeted gene DNA sequencing) detected the following mutations: NRAS G12D, VAF 38.8%; DNMT3A R882H, VAF 37.25%; DNMT3A E30A, VAF 50.59%; RAD21, D276Vfs*15, VAF 20%; NOTCH1 Q2184*, VAF 8.11%; and ETV6 G91D, VAF 39.94%.
Final Diagnosis
Early T-cell precursor lymphoblastic leukemia
Follow-Up
The patient was treated with hyper-CVAD and intrathecal methotrexate and cytarabine. Two months later, repeat peripheral blood testing showed recurrent/persistent disease. He was given comfort measures and deceased 3 months after the initial diagnosis.
Discussion
By definition, ETP-ALL cases show characteristic immunophenotype. The T lymphoblasts express CD7 but lack CD1a and CD8 and are positive for one or more of the myeloid/stem cell markers (CD34, CD117, HLA-DR, CD13, CD33, CD11b, and CD65). CD5 is either negative or positive in < 75% of the blasts [7]. Different from the classic T lymphoblastic leukemia, the molecular mutation profile is similar to T/myeloid mixed phenotype acute leukemia. Besides the mutation in genes usually associated with T-ALL such as NOTCH1, there can be mutations frequently seen in myeloid neoplasms, like DNMT3A, RAS and ETV6 mutations, as what we see in this case.
In this case, findings from flow cytometry, together with the molecular study results, are consistent with the final diagnosis of ETP-ALL. ETP-ALL is derived from cells committed to the T-cell lineage but retain the potential for myeloid/dendritic cell differentiation. The clinical outcomes of adult ETP-ALL to standard chemotherapy are suboptimal, and different clinical regimen and management including molecular targeting therapy may be pursued [87].
References
de Haas V, et al. Initial diagnostic work-up of acute leukemia: ASCO clinical practice guideline endorsement of the College of American Pathologists and American Society of hematology guideline. J Clin Oncol. 2019;37(3):239–53.
National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines) acute lymphoblastic leukemia (Version 2.2020). 2020 [cited 2020 Nov 19]; Available from: https://www.nccn.org/professionals/physician_gls/pdf/all.pdf.
Harris MH, et al. Genetic testing in the diagnosis and biology of acute leukemia. Am J Clin Pathol. 2019;152(3):322–46.
Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122(10):3407–15.
Moorman AV, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11(5):429–38.
Pui CH, Jeha S. New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat Rev Drug Discov. 2007;6(2):149–65.
Swerdlow S, et al., editors. World Health Organization classification of tumours of haematopoietic and lymphoid tissues, World Health Organization classification of tumours. Revised 4th ed. Lyon: International Agency for Research on Cancer; 2017.
Mrozek K, Harper DP, Aplan PD. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Hematol Oncol Clin North Am. 2009;23(5):991–1010. v
Liu-Dumlao T, et al. Philadelphia-positive acute lymphoblastic leukemia: current treatment options. Curr Oncol Rep. 2012;14(5):387–94.
Branford S, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99(9):3472–5.
Soverini S, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA working party on chronic myeloid leukemia. Clin Cancer Res. 2006;12(24):7374–9.
Shah NP, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117–25.
Bitencourt R, Zalcberg I, Louro ID. Imatinib resistance: a review of alternative inhibitors in chronic myeloid leukemia. Rev Bras Hematol Hemoter. 2011;33(6):470–5.
Wetzler M, et al. Additional cytogenetic abnormalities in adults with Philadelphia chromosome-positive acute lymphoblastic leukaemia: a study of the Cancer and Leukaemia group B. Br J Haematol. 2004;124(3):275–88.
Martinelli G, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol. 2009;27(31):5202–7.
Meyer C, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013;27(11):2165–76.
Winters AC, Bernt KM. MLL-rearranged leukemias-an update on science and clinical approaches. Front Pediatr. 2017;5:4.
Harrison CJ. Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia. Br J Haematol. 2009;144(2):147–56.
Nachman JB, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110(4):1112–5.
Harrison CJ, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol. 2004;125(5):552–9.
Safavi S, et al. Loss of chromosomes is the primary event in near-haploid and low-hypodiploid acute lymphoblastic leukemia. Leukemia. 2013;27(1):248–50.
Charrin C, et al. A report from the LALA-94 and LALA-SA groups on hypodiploidy with 30 to 39 chromosomes and near-triploidy: 2 possible expressions of a sole entity conferring poor prognosis in adult acute lymphoblastic leukemia (ALL). Blood. 2004;104(8):2444–51.
Zhang J, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood. 2011;118(11):3080–7.
Chiaretti S, et al. TP53 mutations are frequent in adult acute lymphoblastic leukemia cases negative for recurrent fusion genes and correlate with poor response to induction therapy. Haematologica. 2013;98(5):e59–61.
Stengel A, et al. TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood. 2014;124(2):251–8.
Muhlbacher V, et al. Acute lymphoblastic leukemia with low hypodiploid/near triploid karyotype is a specific clinical entity and exhibits a very high TP53 mutation frequency of 93%. Genes Chromosomes Cancer. 2014;53(6):524–36.
Holmfeldt L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–52.
Comeaux EQ, Mullighan CG. TP53 mutations in hypodiploid acute lymphoblastic leukemia. Cold Spring Harb Perspect Med. 2017;7(3):a026286.
Qian M, et al. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J Clin Oncol. 2018;36(6):591–9.
Salmoiraghi S, et al. Mutations of TP53 gene in adult acute lymphoblastic leukemia at diagnosis do not affect the achievement of hematologic response but correlate with early relapse and very poor survival. Haematologica. 2016;101(6):e245–8.
Hof J, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185–93.
Dyer MJ, et al. Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: rare clinical curios or potent genetic drivers? Blood. 2010;115(8):1490–9.
Ghazavi F, et al. Molecular basis and clinical significance of genetic aberrations in B-cell precursor acute lymphoblastic leukemia. Exp Hematol. 2015;43(8):640–53.
de Boer J, et al. The E2A-HLF oncogenic fusion protein acts through Lmo2 and Bcl-2 to immortalize hematopoietic progenitors. Leukemia. 2011;25(2):321–30.
Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012;26(3):123–35.
Johnson RC, et al. Cytogenetic variation of B-lymphoblastic leukemia with intrachromosomal amplification of chromosome 21 (iAMP21): a multi-institutional series review. Am J Clin Pathol. 2015;144(1):103–12.
Moorman AV, et al. Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21). Blood. 2007;109(6):2327–30.
Harrison CJ. Blood spotlight on iAMP21 acute lymphoblastic leukemia (ALL), a high-risk pediatric disease. Blood. 2015;125(9):1383–6.
Den Boer ML, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125–34.
Mullighan CG, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–80.
Boer JM, et al. Expression profiling of adult acute lymphoblastic leukemia identifies a BCR-ABL1-like subgroup characterized by high non-response and relapse rates. Haematologica. 2015;100(7):e261–4.
Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica. 2016;101(4):407–16.
Roberts KG, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15.
Weston BW, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. J Clin Oncol. 2013;31(25):e413–6.
Ding YY, et al. Clinical efficacy of ruxolitinib and chemotherapy in a child with Philadelphia chromosome-like acute lymphoblastic leukemia with GOLGA5-JAK2 fusion and induction failure. Haematologica. 2018;103(9):e427–31.
Frisch A, Ofran Y. How I diagnose and manage Philadelphia chromosome-like acute lymphoblastic leukemia. Haematologica. 2019;104(11):2135–43.
Herold T, Gokbuget N. Philadelphia-like acute lymphoblastic leukemia in adults. Curr Oncol Rep. 2017;19(5):31.
Harvey RC, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116(23):4874–84.
Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015;12(6):344–57.
Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–64.
Liu YF, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173–83.
Rebollo A, Schmitt C. Ikaros, Aiolos and Helios: transcription regulators and lymphoid malignancies. Immunol Cell Biol. 2003;81(3):171–5.
Georgopoulos K, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79(1):143–56.
Ensor HM, et al. Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood. 2011;117(7):2129–36.
Hertzberg L, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the international BFM study group. Blood. 2010;115(5):1006–17.
Mullighan CG, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–4.
Olsson L, Johansson B. Ikaros and leukaemia. Br J Haematol. 2015;169(4):479–91.
Joshi I, et al. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol. 2014;15(3):294–304.
Yao QM, et al. Prognostic impact of IKZF1 deletion in adults with common B-cell acute lymphoblastic leukemia. BMC Cancer. 2016;16(1):269.
Boer JM, et al. Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia. 2016;30(1):32–8.
Clappier E, et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia. 2015;29(11):2154–61.
Stanulla M, Cave H, Moorman AV. IKZF1 deletions in pediatric acute lymphoblastic leukemia: still a poor prognostic marker? Blood. 2020;135(4):252–60.
Buitenkamp TD, et al. Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia. 2012;26(10):2204–11.
Zhou Y, et al. Advances in B-lymphoblastic leukemia: cytogenetic and genomic lesions. Ann Diagn Pathol. 2016;23:43–50.
Tasian SK, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012;120(4):833–42.
Nebral K, et al. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23(1):134–43.
Shah S, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45(10):1226–31.
Auer F, et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia. 2014;28(5):1136–8.
Mullighan CG, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471(7337):235–9.
Inthal A, et al. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia. 2012;26(8):1797–803.
Zaliova M, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia. 2014;28(1):182–5.
Clappier E, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2014;28(1):70–7.
Mullighan CG. The molecular genetic makeup of acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2012;2012:389–96.
Della Starza I, et al. Minimal residual disease in acute lymphoblastic leukemia: technical and clinical advances. Front Oncol. 2019;9:726.
Jennings LJ, et al. Detection and quantification of BCR-ABL1 fusion transcripts by droplet digital PCR. J Mol Diagn. 2014;16(2):174–9.
Coccaro N, et al. Droplet digital PCR is a robust tool for monitoring minimal residual disease in adult Philadelphia-positive acute lymphoblastic leukemia. J Mol Diagn. 2018;20(4):474–82.
Faham M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120(26):5173–80.
van Dongen JJ, et al. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. 2015;125(26):3996–4009.
Bruggemann M, et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia. 2019;33(9):2241–53.
Kim IS. Minimal residual disease in acute lymphoblastic leukemia: technical aspects and implications for clinical interpretation. Blood Res. 2020;55(S1):S19–26.
Reyes-Barron C, et al. Next-generation sequencing for minimal residual disease surveillance in acute lymphoblastic leukemia: an update. Crit Rev Oncog. 2017;22(5–6):559–67.
Liu Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–8.
Girardi T, et al. The genetics and molecular biology of T-ALL. Blood. 2017;129(9):1113–23.
Kimura S, Mullighan CG. Molecular markers in ALL: clinical implications. Best Pract Res Clin Haematol. 2020;33(3):101193.
Fransecky L, et al. Silencing of GATA3 defines a novel stem cell-like subgroup of ETP-ALL. J Hematol Oncol. 2016;9(1):95.
Sajaroff EO, et al. B-cell acute lymphoblastic leukemia with mature phenotype and MLL rearrangement: report of five new cases and review of the literature. Leuk Lymphoma. 2016;57(10):2289–97.
Castaneda Puglianini O, Papadantonakis N. Early precursor T-cell acute lymphoblastic leukemia: current paradigms and evolving concepts. Adv Hematol. 2020;11:2040620720929475.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zhang, X. (2021). Precursor Lymphoid Neoplasms. In: Ding, Y., Zhang, L. (eds) Practical Oncologic Molecular Pathology. Practical Anatomic Pathology. Springer, Cham. https://doi.org/10.1007/978-3-030-73227-1_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-73227-1_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-73226-4
Online ISBN: 978-3-030-73227-1
eBook Packages: MedicineMedicine (R0)