
Abstract
A 31-year-old female admitted in a respiratory high dependency care unit with acute respiratory failure and acute pulmonary edema, showing hypopnea (9 breaths/min), drowsiness (GCs: 11), hypoxemia (PaO2/FiO2: 145 mmHg), hypercapnia and a mixed acidosis was successfully treated by non-invasive ventilation (CPAP with PEEP 5 cmH2O). Beside the increase in extravascular lung water, iatrogenic factors such as hyponatremia and hyposmolarity could make the therapeutic management more difficult.
Corrado Mollica and Giovacchino Pedicelli are retired.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Extra-vascular lung water (EVLW)
- Hypocapnia
- Hyposmolarity
- Hyponatremia
- Continuous positive air pressure (CPAP)
1 Introduction
As shown by Bone and Balk 1988 [1], a non-invasive respiratory care unit is a cost-effective solution in acute respiratory failure treatment of patients whose primary need consists in monitoring or assistance with weaning, and are hemodynamically stable.
In Italy, respiratory high dependency care units (RHDCUs) provide an intermediate level of care between the intensive care unit (ICU) and the general ward for patients with single organ respiratory failure, who do not need ICU admission. Although Italian RHDCUs are mainly devoted to the monitoring and treatment of acute and chronic respiratory failure by non-invasive ventilation (NIV), they also help weaning from invasive mechanical ventilation. In 1984, an 8 beds RHDCU at the S. Camillo-Forlanini Hospital, Rome (Italy) was instituted. In this short paper, we report the arguments that were discussed by RHDCU’s medical staff in 1990 and that allowed to recognize the kind of edema and to choose a proper treatment, in terms of both medical and ventilatory therapy.
Clinical Case
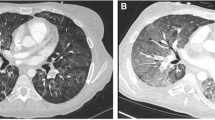
A 31-year-old female, coming from another Hospital, where she had been admitted for drug intoxication (benzodiazepines) that she had taken to commit suicide (Lorazepam 1 mg >20 tablets; Trihexyphenidyl Hydrochloride 2 mg >15 tablets); Bromperidol tablets (unspecified number), and treated with Flumazenil 1 mg/10 mL IV, diuretics, and fluid therapy (crystalloid), was admitted to the emergency department (ED) of our Hospital. At admittance she was afebrile and tachypneic—respiratory rate (RR) equal to 30 breaths/min—showing dyspnoea, light deterioration of mental status: Glasgow Coma score (GCs: 13). She was mydriatic with pupil poorly reacting to light and accommodation; moreover, auscultation of the lungs highlighted decreased breath sounds at the right base and diffuse crackling rales. No lower extremity edema. The electrocardiogram showed sinus rhythm rate 100 beats/min. Blood pressure was 130/90. A chest radiograph (CXR) revealed “widespread ground-glass thickening of the lung parenchyma with relative sparing of the lower right half. The pleural sinuses appear free. The mediastinal image is morphologically normal with moderate widening of the vascular pedicle. CRX compatible with ARDS in early stage” (Fig. 33.1). The arterial blood gas (ABG) showed hypoxemia (PaO2: 59.2 mmHg; Standard PaO2: 42 mmHg) at inspiratory oxygen fraction (FiO2): 21%, (PaO2/FiO2 or P/F = 280 mmHg), hypocapnia with (acute) respiratory alkalosis (PaCO2: 29.6 mmHg; pH: 7.50; HCO3−: 23.3 mmol/L).Footnote 1 Oxygen arterial saturation (SaO2): 92.8%, with alveolar-arterial oxygen gradient (DA-aO2): 54 mmHg (expected DA-aO2 for age: 11.8 mmHg).Footnote 2 Electrolytes: hyponatraemia (Na+: 129 mmol/L) with plasmatic hyposmolality (Osm: 255 mOsm/kg) (n.v. = 285–305 mOsm/kg), hypokalaemia (K+: 3.1 mmol/L), Cl−: 98 mmol/L. Other values: Urea: 90 mg/dL, Glicemia: 83 mg/dL, Red Blood Cells (RBC): 3,940,000/mm3; White Blood Cells (WBC): 11,600/mm3; Hb: 12.9 g%, Ht: 37%. Acute Physiology And Chronic Health Evaluation (APACHE) II score: 10 [2] (Table 33.1). The trans-cutaneous pulse oximetry saturation (SpO2) on 100% oxygen delivered for 20 min by a non-rebreather mask was equal to 96% (Rossier test). Therapy was started with loop diuretics and fluid infusions. Twenty-four hours after, the drowsiness worsened (GCs: 12); at chest, radiograph showed a dense right lower lobe consolidation. The lung examination showed inspiratory crackles, rhonchi and silence in lower (base) of the right lung. Worsening in ABG values (P/F = 222 mmHg), hyponatraemia (Na+: 122 mmol/L) with plasmatic hyposmolality (244 mOsm/kg) and negative water balance (urine output: 1 L within the last 24 h), both furosemide (Lasix®) 40 mg × 4/24 h, Albumine (Behring®) 200 g/L (20% 50 mL) and saline 0.9% NaCl (≈308 mOsm/L) in I.V. slow infusion were administered; the latter in order to counterbalance the hypotensive effect of diuretics. Aerosolized Ambroxol fl (Mucosolvan®) was added to antibiotics, corticosteroids, digoxin already on line. Oxygen therapy (O2T) via Ventimask, with FiO2 equal to 28% was set up. Forty-eight hours after admittance, further worsening in neural status (GCs: 11), hemodynamics (HR: 130 b/min; PA: 90/60), increasing in WBC: 16,000/mm3, and ABG values occurred: PaO2: 30.4 mmHg, PaCO2: 56.4 mmHg pH: 7.27 (FiO2: 21%) (P/F = 145 mmHg) (APACHE II score: 19). CXR showed “widespread lung parenchyma consolidation with confluent patches morphology and moderate asymmetric volumetric reduction of both lungs. Widened vascular peduncle. Pleural sinuses free from effusion. The finding is compatible with diagnosis of ARDS in the acute phase” (Fig. 33.2).

CRX at hospital admittance

CRX at RHDCU admittance
It was necessary to start mechanical ventilation (MV) by NIV in RHDCU. NIV was performed by Bird 8.400 ST® (Bird Products Palm Springs, CA, USA) via face mask (Respironics® Murrysville, PA, USA) in continuous positive air pressure (CPAP) mode (ventilator) with positive end-expiratory pressure (PEEP) equal to 5 cmH2O and an FiO2 (30%) able to obtain a SaO2 >90% and to reduce RR <25/m. In this kind of ventilator (in CPAP mode) neither backup respiratory rate, nor controlled air-leaks system was present (Table 33.1). During NIV, on-line measurements recorded included heart rate (HR) and SpO2 (with a finger probe). At 10 h pm, after ABG measurement, the patient was disconnected and treated with a FiO2 equal to 35%. In the following days a reduction in FiO2 value was gradually done, thanks to an improvement in neural status and in chest infiltration (Fig. 33.4). Four days after, O2T was definitively suspended, and patient was discharged and entrusted to a psychiatric institution.

Airways pressure (cmH2O) and waveform in spontaneous breathing (S.B.) without PEEP (ZEEP) and in CPAP with PEEP at 5 cmH2O. (From: Ventrella F. (2015) [46]; Courtesy of the author)
2 Discussion
2.1 Formation and Resolution of Edema
Acute pulmonary edema (APE) is caused by an excess of fluid in the lungs due to an elevated vascular pressure or to a more permeable membrane that is not matched by an adequate lymph clearance rate. It can be defined as the abnormal increase in the amount of extra-vascular lung water (EVLW). In an un-anesthetized sheep, Erdman et al. (1975) showed that EVLW content, measured post-mortem, does not change significantly until microvascular hydrostatic pressure is more than doubled, indicating a large safety factor that normally protects the lungs against fluid accumulation [3]. Nevertheless if the trans-capillary protein osmotic pressure decreases, along with lymphatic removal incapacity, pulmonary edema can develop at a lower than usual level of net filtration pressure [3]. Lung lymphatics remove edema fluid in either hydrostatic or increased permeability lung edema, but they cannot entirely compensate for an increase in intrans-vascular fluid flux or an impaired alveolar fluid clearance (AFC) [4]. The mechanism for the resolution of alveolar edema is provided by the active ion transport across the alveolar epithelium that creates an osmotic gradient that drives AFC [5]. Both type I and type II alveolar cells are involved in transepithelial ion transport. The primary driving force for alveolar fluid clearance is the active transport of sodium from the alveolar space to the interstitium by alveolar epithelial type II cells. The transport of sodium ions is the most important driver for the generation of the osmotic gradient [6]. The system of active ion-driven alveolar fluid reabsorption is the primary mechanism that removes alveolar edema fluid under both physiologic and pathological conditions, such as hypoxia [7]. If pulmonary hydrostatic pressures are elevated, the rate of AFC is also reduced. ‘In vivo’ studies conducted on animals, showed that net AFC was reduced under clinically relevant pathologic conditions [7]. Alveolar fluid clearance driven by active epithelial Na+ and secondary Cl− ions absorption counteracts edema formation in the intact lung. In left heart disease, lung edema was previously attributed to passive fluid filtration across an intact alveolo-capillary barrier. Conversely, it has been demonstrated that a major part of cardiogenic edema results from an active epithelial secretion of Cl− and secondary fluid flux into the alveolar space [8]. As the inhibition of (Na+)–(K+)–(Cl−)-cotransporters blocks alveolar fluid secretion, it has been identified as a unique therapeutic target in cardiogenic lung edema. “This evolving concept may not be uniquely restricted to cardiogenic pulmonary edema, but it could also apply to other forms of hydrostatic lung edema” [8].
2.2 Detection Methods
Criteria generally used to define hydrostatic pulmonary edema include central venous pressure (CVP) ≥14 mmHg, pulmonary arterial wedge pressure (PAWP) ≥18 mmHg, or a cardiac ejection fraction ≤45% by echocardiogram, radionuclide, or contrast ventriculography, and/or positive physical findings, including a third heart sound and jugular venous distension [9].
2.2.1 Radiological Aspects
Patients with pulmonary edema are likely to present many upholding causes (cardiogenic as opposed to non-cardiogenic). Different hemodynamic conditions and changes of the extravascular protein osmotic forces may be the main factors underlying the radiographic patterns in the various types of pulmonary edema. Conventional chest radiograph aspects can help orienting the diagnosis: as opposed to chronic cardiac failure, where the distribution of flow is usually “inverted” (base-to-apex redistribution), in overload edema, the distribution of pulmonary blood flow and pulmonary regional edema is “balanced” (homogeneous), along the horizontal axis (central), with increased pulmonary blood volume; heart size and vascular pedicle width (VPW) are enlarged;Footnote 3 lung volume is normal or increased; not commune are septal lines and air bronchogram, while peribronchial cuffs and pleural effusions are very commune findings [10].
As to the case in point, in the absence of echocardiogram, CVP, PAWP, or brain natriuretic peptide and troponin-T values, the only chest radiograph—using portable, supine CXR—did not offer an accurate evaluation for distinguishing causes of pulmonary edema.
There are explanations for the limited diagnostic accuracy of the chest radiograph.
Edema may not be visible until the amount of lung water increases by 30% [11]. Moreover, VPW, which could provide a clue in assessing patients’ intravascular volume status, varies depending on the position; the supine position, as in our case, can increase the VPW by nearly 20% compared to the upright position [11].
Recognizing and differentiating acute hydrostatic pulmonary edema from acute respiratory distress syndrome (ARDS) is always a tricky task in the early stages. This is due to the fact that ARDS pts are not necessarily always affected by edema, as it happens, for instance, when low hydrostatic pressures in the lungs, or other factors in the Starling equation, alleviate the increased trans-vascular transport of fluid. Conversely, over-hydration by aggressive fluid therapy can affect the lungs, thus resulting in pulmonary edema but without increased permeability. This is the reason why over-hydration is hard to recognize at the bedside and hence to differentiate from ARDS. A case in point, which mirrors our present one, is that of pts infused with crystalloid fluids, in that over-hydration is likely to lower plasma colloid osmotic pressure. As a consequence of this—and even in the absence of increased permeability—we witness a lowering of the threshold value of hydrostatic pressure (i.e. pulmonary capillary wedge pressure) above which interstitial edema and thus the alveolar flooding develop [12]. Therefore, the distinction of hydrostatic from permeability pulmonary edema is difficult, especially when using portable, supine CXRs; nevertheless “in supine, mechanically ventilated patients, measurements of high-resolution computed tomography and VPW correlate with pulmonary artery occlusion pressure” [13]. Although the absence of clinical signs of congestive heart failure along with the hemodynamically stable situation could suggest to rule out an acute cardiogenic pulmonary edema, it is well known that APE caused by excessive infusions of blood, blood products and fluids is included among the cardiogenic types of edema, because the overload edema too, is caused by increased hydrostatic pressure [14]. As a result, the diagnosis of hydrostatic pulmonary edema usually relies on clinical information and response to treatment: if oxygenation and radiograph finding improve rapidly with diuresis, this favours hydrostatic edema [14]. As to the case in point, despite diuretic therapy, neither oxygenation nor radiograph improved in the first 24 h after admission. Quite the contrary, the clinical and functional status worsened considerably, in accordance with a CXR finding, also compatible with diagnosis of ARDS in the acute phase.
2.2.2 Acid Base Findings
Extravasation of fluid into the alveoli prevents oxygen from being absorbed into the bloodstream and neutralizes surfactant’ s lubricating properties. The lungs become less compliant and the effort of breathing increases, causing dyspnea and CO2 retention. The hypoxemia that occurs in alveolar flooding is more severe than in interstitial pulmonary edema—which causes ventilation-perfusion (Va/Q) mismatch—and is caused by right-to-left shunting of blood [14].
In our case, as opposed to radiological aspects of ARDS, neither aetiology (fluid overload) nor P/F value at admittance and in the first 24 h induced to a diagnosis of a (even mild) ARDS [15].
Chest radiogram, hyponatraemia with plasmatic hyposmolality and the presence of negative water balance, oriented towards over-hydration acute pulmonary edema; the history established that in 48 h before admission the patient was really resuscitated with an unspecified number of liters of crystalloid (Saline and Normosol M), the last one at hyperosmotic set up (Osm: 390 mOsm/L) that can cause interstitial fluid withdrawn into the bloodstream. Moreover, loop diuretics (and thiazides) were previously administered, which could be the cause of the electrolyte imbalance (hypokalaemia, hyponatraemia and hypochloraemic alkalosis) [16]. An “inappropriate” anti-diuretic hormone (ADH) syndrome (hyponatremia with hypervolemia) is described in situations in which ADH secretion may be increased as postoperative ADH release due to barbiturates or transient “idiopathic” hyponatremia (secondary to diuretics, especially thiazides) [16]. Furthermore it cannot be ruled out that the drowsiness was also caused by over-hydration of the central nervous system, that may occur in the presence of concentrations of plasma sodium equal to or below 120 mEq/L [16]. Hypocapnia (with respiratory alkalosis) is generally caused by hypoxemia; as to the case in point, fluid overload could be a contributing factor in leading to hypocapnia, via juxta-capillary receptors (J-receptors, or pulmonary C-fiber receptors) that are involved in events which cause a decrease in oxygenation, responding by an increase in respiration [17]. Hypocapnia in APE is often associated with the rise in serum lactate produced by a marked reduction of splanchnic and muscle blood flow associated with a high degree of hypoxemia [18, 19]. This compensatory hyperventilation is relatively slow and is not complete for 12–24 h [20]. In our case acute respiratory alkalosis (PaCO2: 29.6 mmHg, pH: 7.50) at admission was not associated to metabolic acidosisFootnote 4 [21]. In the absence of serum lactate measure and despite anion gap (AG) value was normal (10.7 mEq/L; nv: 8–12 mEq/L) at admission, we cannot rule out the presence of hyperlactatemia in the continuation of the ED stay, since the AG may result from an insensitive screen for elevated lactate in critically ill patientsFootnote 5 [22]. Moreover it has long been recognized that the infusion of large volumes of “normal” (0.9% NaCl) saline can cause acidosis [23]. This has been traditionally explained as a “dilutional’‘acidosis in which the serum bicarbonate is diluted by the fluid, resulting in lower serum bicarbonate and a metabolic acidosis. However, the Stuart approach offers an alternative explanation [24]. Saline causes metabolic acidosis not by ‘diluting’ HCO3− but rather by its Cl−content. The reason why this occurs with saline administration is that, although saline contains equal amounts of both Na+ and Cl−, plasma does not. When large amounts of salt are added, the Cl− concentration increases much more than the sodium concentration [25]. Since normal saline has a strong ion difference (SID) of 0, its administration would tend to lower the serum SID resulting in a metabolic acidosisFootnote 6 [26]. A shift of free water from the intracellular volume to the extracellular volume in order to reach an osmolar equilibrium will result in additional dilution of the extracellular, and therefore an additional acidifying effect [27, 28]. That is why, in our case, neither hyponatraemia nor hyposmolarity were corrected with saline infusion; actually a marked mixed respiratory and metabolic acidosis on 48 h after hospital admission was highlighted (Table 33.1).Footnote 7
2.3 Ventilatory Therapy
The appearance of respiratory acidosis could be ascribed to hypoventilation as a sign of pump failure, either owing to an increased load of the respiratory system, or strictly related to decreased compliance due to interstitial/alveolar flooding [29]. CPAP—also known as spontaneous PEEP—is a mode of ventilation in which the patient is in spontaneous breathing (SB) and is exposed to a continuous pressure from the ventilator (equal to 5 cmH2O as in this case). This is the pressure in force at the end of expiration, that is named positive end-expiratory pressure (PEEP). The patient creates a negative pressure in inspiration and a positive pressure in expiration, thereby the circuit pressure varies throughout the cycle; but at the end of the expiration the pressure will return to the set PEEP value (5 cmH2O) (Fig. 33.3). Thus, in preventing alveolar collapse at end-expiration and reducing intrapulmonary shunting of blood, CPAP redistributes excess lung water to sites where it interferes less with gas exchange, hence improving oxygenation (arterial PO2 and lung compliance) [30]. According to Demling et al. (1975), PEEP cannot decrease EVLW, but increases the difference between pulmonary microvascular pressure and plasma colloid osmotic pressure (net intravascular filtration pressure), therefore impairing the venous return. In patients with volume overload, decreasing venous return will directly decrease the amount of pulmonary edema being generated, by decreasing right cardiac output [31].
It is noteworthy, in this regard, that the hypoxemia occurring in alveolar flooding is caused by right to-left shunting of blood that is reduced by PEEP (through a reduction in cardiac output) [32].
On the contrary, in interstitial pulmonary edema (as in ARDS), the hypoxemia is due to Va/Q mismatch—by the presence of shunt or units of very low Va/Q ratio; in these pts a fall of the cardiac output caused by PEEP leads to a decrease in the perfusion of unventilated lung and an increase in the ventilation of unperfused alveoli [32].
Moreover, when used in combination with furosemide, given intravenously, PEEP improves lung fluid resorption by increasing the plasma colloid osmotic pressure, as shown in experimental hydrostatic pulmonary edema [33].
“In patients with hydrostatic pulmonary edema, mechanical ventilation could have either beneficial or detrimental effects on alveolar fluid clearance. Beneficial effects of positive pressure ventilation that might indirectly increase alveolar fluid clearance include decreased preload and afterload and decreased myocardial oxygen consumption due to decreased work of breathing. These factors could reduce the formation of pulmonary edema and thus promote net alveolar fluid clearance” [34].
However, it is well established in medical literature that mechanical ventilation at high tidal volumes can promote lung injury [35]. Furthermore, high levels of PEEP may raise CVP, inhibiting lung lymphatic drainage from the interstitium and thereby limiting alveolar fluid clearance [34].
As we pointed out earlier, CPAP can reduce inspiratory work of breathing both by decreasing pulmonary resistance, related to an increase in functional residual capacity, and by increasing lung volume to a more favourable position on the pressure-volume curve [36]. Reducing the work from respiratory muscles also reduces the generation of CO2 and lactate from these muscles, helping improve acidosis. At the same time, the decreased return may improve over-distension in the left ventricle, placing it at a more advantageous point in the Frank-Starling curve and possibly improving cardiac output [37]. Furthermore, inspiratory muscle unloaded reduce intrathoracic and left ventricular transmural pressures (and afterload), as seen in patients with congestive heart failure, thereby improving oxygenation [37]. Although CPAP decreases ventilation/min and respiratory rate, PaCO2 value generally does not increase, thanks to the decrease in dead space ventilation [36]. As reported in Aliberti et al. (2010), in case of a mixed acidosis (as in ours), pH increased very fast during the first hours of CPAP treatment, possibly owing to beneficial effects on the heart and hemodynamics, as well as to tissue perfusion [29]. However, in our case, the slower decrease in PaCO2 levels related to the lung decreased compliance (“stiff lung”) forced to apply a low PEEP value (5 cmH2O), given the risk of barotrauma. A light increase in PaCO2 value at the 12th hour of CPAP can be explained by the fact that, unlike pressure support ventilation (PSV), CPAP does not provide inspiratory assistance to the rest of the muscles of respiration [38]. It is now established that the rise in intrathoracic pressure and not the modality of ventilation (CPAP vs PSV) seems to be important in acute cardiogenic pulmonary edema patients [39, 40].
2.4 Outcome
Thanks to medical therapy, the increasing in urine output dramatically reduced edema in few hours (Fig. 33.4) and CPAP reduced PaCO2 within 7 h (Table 33.1).

CRX at RHDCU discharge
A “post hoc” analysis according to the “risk scale score” named HACOR, at 1 h of NIV, on a value obtained by analysing five variables—i.e. heart rate (H), acidosis (pH), consciousness (GCs), oxygenation (P/F), and respiratory rate (R)—showed a low (5) prediction (NIV) failure [41].
3 Limits of the Present Contribution
A faster and more accurate diagnosis would have been certainly made, were hemodynamic factors (including pulmonary arterial wedge pressure and left ventricular ejection fraction) routinely measured, along with monitoring EVLW by single transpulmonary thermal dilution. Likewise, the availability of main electrolytes (Na+, K+, Cl−, Latt−) and serum osmolarity, together with ABG values in the same blood sample, would have allowed a more rapid assessment of the acid-base status. Most notably, a quasi-continuous monitoring of urinary pH and principal urinary electrolytes available by the applications of the K.IN.G® urinary analyzer would have helped “to disentangle the great mosaic concerning acid-base chemistry” [42]. It is also important to highlight that, hemodynamic factors being routinely monitored, some therapeutic errors, i.e. the administration of diuretics and of other infusion drugs, such as NaCl 9% saline (instead of an early conservative fluid management strategy) which caused overload and the onset of “iatrogenic” metabolic acidosis, could have been successfully avoided. The availability of NIV-ventilators which can provide air-leaks compensation could have proved equally useful. Despite there are no significant differences in clinical outcomes when comparing CPAP vs BiPAP, would a Bi-level mode have been applied at the first occurrence of hypercapnia, the overall time of mechanical ventilation could have been perhaps reduced.
4 Conclusions
It has been shown that central active substances may cause pulmonary edema by increased permeability of the alveolar-capillary membrane [43,44,45].
The evolution of the chest radiographic findings in the present case are compatible with an initial acute increase of vascular permeability due to the intoxication. The increase in density in the peripheral regions of the lung is an indication of alveolar involvement. There are no findings compatible with an interstitial phase preceding the alveolar flooding as it happens in conditions of severe cardiogenic pulmonary edema. According to this interpretation, despite the presence of alveolar edema, there are no findings of septal lines, peribronchovascular cuffing, perihylar haze and pleural effusion that usually are hallmarks of cardiogenic pulmonary edema. From the pathophysiologic point of view this type of edema occurring after drug overdose can be considered as a normal pressure injury edema that could be reversible after the cessation of the pharmacologic injury [10]. The evolution of this case is conditioned by the presence of over-hydration with marked enlargement of the vascular pedicle and further involvement of the central regions of the lungs. Over-hydration could cause an increased hydrostatic pressure within the pulmonary capillaries favoring further development of edema. The quite rapid resolution of the radiographic findings could be related on the one hand to the removal of the toxic condition with consequent reduction of the injury component of pulmonary edema and on the other hand to the effect of CPAP facilitating the re-absorption of edema of hydrostatic origin. In summary, the radiographic aspect of this case can be speculatively defined as toxic injury edema (ARDS) with superimposed over-hydration hydrostatic edema [10].
Despite an occurrence of mild hypercapnia in the continuation of the treatment, CPAP appeared to be an effective treatment. Furthermore, the existence of a metabolic acidosis associated to the acute respiratory acidosis, also due to the high degree of hypoxemia, can affect its outcome. Careful interpretation of AB status is recommended.
Key Teaching Points
-
In patients with acute pulmonary edema an initial acute increase of vascular permeability due to drug intoxication can lead to radiological finding of ARDS.
-
The superimposed over-hydration can lead to hydrostatic pulmonary edema.
-
The diagnosis of hydrostatic pulmonary edema usually relies on clinical information and a rapid improvement of oxygenation and of the radiograph findings after increased diuresis.
-
In patients with hydrostatic pulmonary edema CPAP can reduce edema owing to beneficial effects on the heart and hemodynamics as well as on work of breathing and tissue perfusion which lead to a reduction in acidosis.
-
High tidal volume ventilation and high levels of PEEP must be avoid to prevent the consequent lung injury and reduction of alveolar fluid clearance.
-
Similarly, since inducing metabolic acidosis both I.V. rapidly infused saline (NaCl 0.9%) and loop diuretics (thiazides) must be avoided, or they should be administered very carefully.
Questions and Answers
-
1.
During continuous positive airway pressure (CPAP) ventilation:
-
(a)
The pressure is delivered by the ventilator in the expiratory phase only and represents the positive end-expiratory pressure (PEEP)
-
(b)
The airways pressure has no different value both in inspiration and in expiration
-
(c)
The pressure delivered by the ventilator is the airways pressure at the end of expiration
-
(d)
None of them above
Answer: (c) The pressure delivered by the ventilator is the airways pressure at the end of expiration
-
(a)
-
2.
In Acute Hydrostatic Pulmonary Edema the occurrence of a Respiratory Alkalosis is due to:
-
(a)
Fluid overload involved in leading to hypocapnia
-
(b)
The rise in serum lactate produced by a marked reduction of splachnic and muscle blood flow, caused by hypoxemia
-
(c)
Can be managed with a conservative fluid strategy and oxygen therapy
-
(d)
All of them above
Answer: (d) All of them above
-
(a)
-
3.
In Acute Hydrostatic Pulmonary Edema the occurrence of a metabolic acidosis is due to:
-
(a)
Administration of diuretics (especially thiazides)
-
(b)
Infusion of a large volumes of fluids (NaCl 9% saline)
-
(c)
High degree of hypoxemia
-
(d)
All of them above
Answer: (d) All of them above
-
(a)
-
4.
In Acute Hydrostatic Pulmonary Edema the occurrence of a hyponatremia:
-
(a)
Can be transient or “idiopathic” (secondary to diuretics, especially thiazides)
-
(b)
Is described in situations where ADH secretion may be increased (as a postoperative ADH release due to barbiturates)
-
(c)
When equal to or below 120 mEq/L causes drowsiness owing to overhydration of the central nervous system
-
(d)
All of them above
Answer: (d) All of them above
-
(a)
-
5.
In Acute Hydrostatic Pulmonary Edema beneficial effects of CPAP that might indirectly increase alveolar fluid clearance are:
-
(a)
Decreased preload and afterload
-
(b)
Decreased work of breathing
-
(c)
Decreased myocardial oxygen consumption
-
(d)
All of them above
Answer: (d) All of them above
-
(a)
Notes
- 1.
When pt was in spontaneous breathing (SB) all the ABG samples were measured in the air room (FiO2: 21%) for at least 15 min after discontinuation of O2T. PaO2 Standard: 1.66 × PaCO2 + PaO2 – 66.4 = 41.93 mmHg (rounded up: 42 mmHg).
- 2.
Da-aO2 = PAO2 − PaO2; PAO2 = (PB − PH2O) × FiO2 − (1.25 × PaCO2).
- 3.
The vascular pedicle width (VPW) is the mediastinal silhouette of the great vessels. It is the distance between parallel lines drawn from the point at which the superior vena cava intersects the right main bronchus and a line drawn at the takeoff of the left subclavian artery from the aorta. It can provide a clue in the assessment of patients’ intravascular volume status. Its value varies depending on the position; at the supine position the VPW can increase up to nearly 20%, compared to the upright position. Thus when the patient is supine, the “normal” VPW spans between 58 and 62 mm. A VPW value of 72 or higher is likely due to volume overload [10, 11].
- 4.
This is a primary disorder: in (acute) respiratory alkalosis, the decrease in expected HCO3− is equal to 2 mmol/L for a decrease in PaCO2 equal to 10 mmHg; thus for a decrease in PaCO2 equal to [40 – 29.6] = 10.4 mmHg, the expected HCO3− is ≈23; OR (it is easier): for a decrease in PaCO2 equal to 1 mmHg, the pH increase is equal to 0.01 U: thus, for a decrease in PaCO2 equal to ≈10 (40 – 29.6) mmHg, the increase in pH is 0.1: that is 7.50, as in our case.
- 5.
Anion Gap (AG) = [(Na+ + K +) − (Cl− + HCO3−)] = [(129 + 3.1) − (98 + 23.3)] = 10.8 mEq/L.
- 6.
The difference between the sum of all strong cations [Na+, K+, Ca2+, Mg2+] and all strong anions (mainly Cl−, Lactate and Albumine) is known as the strong ion difference (SID).
- 7.
This corresponds to a mixed disorder. In fact the expected HCO3− in acute respiratory acidosis: [(PCO2 − 40)/10] + 24 should be 25.64 mmol/L instead of 22.9 mmol/L (actual value); thus there is an associated metabolic acidosis.
Abbreviations
- ABG:
-
Arterial blood gas
- ADH:
-
Anti-diuretic hormone
- AFC:
-
Alveolar fluid clearance
- AG:
-
Anion gap
- APACHE II:
-
Acute physiology and chronic health evaluation
- APE:
-
Acute pulmonary edema
- ARDS:
-
Acute respiratory distress syndrome
- CPAP:
-
Continuous positive airway pressure
- CRX:
-
Chest radiograph
- CVP:
-
Central venous pressure
- ED:
-
Emergency department
- EVLW:
-
Extra-vascular lung water
- FiO2:
-
Inspiratory oxygen fraction
- GCs:
-
Glasgow Coma score
- HR:
-
Heart rate
- ICU:
-
Intensive care unit
- MV:
-
Mechanical ventilation
- NIV:
-
Noninvasive ventilation
- O2T:
-
Oxygen therapy
- P/F:
-
PaO2/FiO2 ratio
- PaCO2:
-
Carbon dioxide arterial pressure
- PaO2:
-
Oxygen arterial pressure
- PAWP:
-
Pulmonary arterial wedge pressure
- PEEP:
-
Positive end-expiratory pressure
- PSV:
-
Pressure support ventilation
- pts:
-
Patient/s
- RHDCU:
-
Respiratory high-dependency care unit
- RR:
-
Respiratory rate
- SB:
-
Spontaneous breathing
- SID:
-
Strong ion difference
- SpO2:
-
Trans-cutaneous pulse oximetry saturation
- Va/Q:
-
Ratio of ventilation to perfusion
- VPW:
-
Vascular pedicle width
References
Bone RC, Balk RA. Non-invasive respiratory care unit: a cost-effective solution for the future. Chest. 1988;93:390–4.
Knaus WA, Draper EA, Wagner DP, et al. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–29.
Erdmann AJ 3rd, Vaughan TL Jr, Brigham KL, et al. Effect of increased vascular pressure on lung fluid balance in unanesthetized sheep. Circ Res. 1975;37:271–84.
Huppert LA, Matthay MA. Alveolar fluid clearance in pathologically relevant conditions: in vitro and in vivo models of acute respiratory distress syndrome. Front Immunol. 2017;8:371.
Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600.
Canessa CM, Schild L, Buell G, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367(6462):463–7.
Vivona ML, Matthay M, Chabaud MB, et al. Hypoxia reduces alveolar epithelial sodium and fluid transport in rats: reversal by β-adrenergic agonist treatment. Am J Respir Cell Mol Biol. 2001;25:554–61.
Solymosi EA, Kaestle-Gembardt SM, Vadász I, et al. Chloride transport-driven alveolar fluid secretion is a major contributor to cardiogenic lung edema. Proc Natl Acad Sci U S A. 2013;18(110):E2308–16.
Bernard GR, Brigham KL. Pulmonary edema. Pathophysiologic mechanisms and new approaches to therapy. Chest. 1986;89:594–600.
Milne ENC, Pistolesi M. Reading the chest radiograph. A physiologic approach. St. Louis: Mosby-Year Book Inc.; 1993.
Milne EN, Pistolesi M, Miniati M, et al. The radiologic distinction of cardiogenic and noncardiogenic edema. Am J Roentgenol. 1985;144:879–94.
Groeneveld AB, Polderman KH. Acute lung injury, overhydration or both? Crit Care. 2005;9:136–7.
Thomason JW, Ely EW, Chiles C, et al. Appraising pulmonary edema using supine chest roentgenograms in ventilated patients. Am J Respir Crit Care Med. 1998;57:1600–8.
Murray JF. Pulmonary edema: pathophysiology and diagnosis. Int J Tuberc Lung Dis. 2011;15:155–60.
ARDS Definition Task Force, Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome. The Berlin definition. JAMA. 2012;307:2526–33.
Weitzman RE, Kleeman CR. The clinical physiology of water metabolism-part III: the water depletion (hyperosmolar) and water excess (hyposmolar) syndromes (medical Progress). West J Med. 1980;132:16–38.
Widdicombe J. Reflexes from the lungs and airways: historical perspective. J Appl Physiol. 2006;101:628–34.
Agostoni A. Acid-base disturbances in pulmonary edema. Arch Intern Med. 1967;120:307–10.
Aberman A, Fulop M. The metabolic and respiratory acidosis of acute pulmonary edema. Ann Intern Med. 1972;76:173–84.
Asch MJ, Dell RB, Williams GS, et al. Time course for development of respiratory compensation in metabolic acidosis. J Lab Clin Med. 1969;73:610–5.
Arbus GS, Herbert LA, Levesque PR, et al. Characterization and clinical application of the "significance band" for acute respiratory alkalosis. N Engl J Med. 1969;280:117–23.
Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol. 2007;2:162–74.
Shires GT, Tolman J. Dilutional acidosis. Ann Intern Med. 1948;28:557–9.
Stewart PA. Modern quantitative acid–base chemistry. Can J Physiol Pharmacol. 1983;61:1444–61.
Kellum JA. Determinants of blood pH in health and disease. Crit Care. 2000;4:6–14.
Gauthier PM, Szerlip HM. Metabolic acidosis in the intensive care unit. Crit Care Clin. 2002;18:289–308.
Yunos NM, Kim IB, Bellomo R, et al. The biochemical effects of restricting chloride-rich fluids in intensive care. Crit Care Med. 2011;39:2419–24.
Langer T, Ferrari M, Zazzeron L, et al. Effects of intravenous solutions on acid-base equilibrium: from crystalloids to colloids and blood components. Anaesthesiol Intensive Ther. 2014;46:350–36.
Aliberti S, Piffer F, Brambilla AM, et al. Acidemia does not affect outcomes of patients with acute cardiogenic pulmonary edema treated with continuous positive airway pressure. Crit Care. 2010;14:R196.
Rizk NW, Murray JF. PEEP and pulmonary edema. Am J Med. 1982;72:381–3.
Demling RH, Staub NC, Edmunds LH Jr. Effect of end-expiratory airway pressure on accumulation of extravascular lung water. J Appl Physiol. 1975;38:907–12.
Dantzker RD, Brook CJ, Dehart P, et al. Ventilation-perfusion distributions in the adult respiratory distress syndrome. Am Rev Respir Dis. 1979;120:1039–52.
Wickerts CJ, Blomqvist H, Berg B, et al. Furosemide, when used in combination with positive end-expiratory pressure, facilitates the resorption of extravascular lung water in experimental hydrostatic pulmonary oedema. Acta Anaesthesiol Scand. 1991;35:776–83.
Verghese GM, Ware LB, Matthay BA, et al. Alveolar epithelial fluid transport and the resolution of clinically severe hydrostatic pulmonary edema. J Appl Physiol. 1999;87:1301–12.
Tremblay L, Valenza F, Ribeiro SP, et al. Injurious ventilatory strategies increase cytokines and c-fos mRNA expression in an isolated rat lung model. J Clin Invest. 1997;99:944–52.
Katz JA, Marks JA. Inspiratory work with and without continuous positive airway pressure in patients with acute respiratory failure. Anaesthesiology. 1985;63:598–607.
Naughton MT, Rahman MA, Hara K, et al. Effect of continuous positive airway pressure on intrathoracic and left ventricular transmural pressures in patients with congestive heart failure. Circulation. 1995;91:1725–31.
Mehta S, Jay GD, Woolard RH, et al. Randomized, prospective trial of bilevel versus continuous positive airway pressure in acute pulmonary edema. Crit Care Med. 1997;25:620–8.
Pinsky MR. Cardiovascular issues in respiratory care. Chest. 2005;128:592S–7S.
Esquinas AM, Ferrari G, Bellone A. Noninvasive ventilation in patients with acute cardiogenic pulmonary edema with acute coronary syndrome: is the debate still? Circ J. 2013;77:1920.
Duan J, Han X, Bai L, et al. Assessment of heart rate, acidosis, consciousness, oxygenation, and respiratory rate to predict noninvasive ventilation failure in hypoxemic patients. Intensive Care Med. 2017;43:192–9.
Caironi P, Langer T, Taccone P, et al. Kidney instant monitoring (K.IN.G): a new analyzer to monitor kidney function. Minerva Anestesiol. 2010;76:316–24.
Gottlieb LS, Boylen T. Pulmonary complications of drug abuse. West J Med. 1974;120(1):8–1.
Kitson GE, Wauchob TD. Pulmonary oedema following carbamazepine overdose. Anaesthesia. 1988;43:967–9.
Pereda J, Gómez-Cambronero L, Alberola A, et al. Co-administration of pentoxifylline and thiopental causes death by acute pulmonary oedema in rats. Br J Pharmacol. 2006;149:450–5.
Ventrella F. Insufficienza respiratoria acuta cardiogena—ruolo della ventilazione non invasiva. In: Nardi R, editor. QUADERNI—Italian Journal of Medicine, vol. 3. Pavia: PAGE Press; 2015. p. e56, 427–441.
Acknowledgments
My gratitude goes to Andrea Rossi (MD, PhD) and Thomas Langer (MD, PhD), for their precious advice and suggestions, and to Fabrizio Bigotti (PhD), for revising the final draft of this paper. I would like also to thank Giuseppe Brunetti, MD, Mario Carlo Buscajoni, MD, Paola Marazzi (MD) and Maurizio Morandi (MD) who assisted me in the patients’ treatment. Finally, I wish to dedicate this paper to Vittorio Emanuele Antonini, Claudio Balderi, and Roberto Sabato, whose memory shall for ever live with me.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mollica, C., Pedicelli, G., Spadaro, S., Pistolesi, M. (2022). Unusual Case of Acute Pulmonary Edema Treated by Non Invasive Ventilation: A 30 Years Ago “Cold Case”!. In: Esquinas, A.M. (eds) Teaching Pearls in Noninvasive Mechanical Ventilation. Springer, Cham. https://doi.org/10.1007/978-3-030-71298-3_33
Download citation
DOI: https://doi.org/10.1007/978-3-030-71298-3_33
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-71297-6
Online ISBN: 978-3-030-71298-3
eBook Packages: MedicineMedicine (R0)