
Abstract
The sequencing of the genome of new virus, such as the coronavirus type 2 of the acute severe respiratory syndrome (SARS-CoV-2), is essential and of great importance to mitigate new zoonotic outbreaks, which are caused by mutations present in structural and non-structural proteins that make up the viruses. Sequencing allows tracking the behavior of the virus locally and globally, knowing the route of transmission and spread of the virus, and determine the virulence rate. Current studies have been carried out, using first, second or third generation sequencing techniques, which have allowed reading and analyzing the nucleotides that make up the virus genome. Thus, the benefits of effective technologies to know its genetic composition in the shortest possible time become evident. New technologies are able to monitor an epidemic in real time, monitor the evolution and efficacy of a drug, the development of a vaccine as well as epidemiological advances. This work addresses the Oxford Nanopore sequencing, which is considered the most efficient and applied method for sequencing viruses that cause epidemics. Some of the advantages of using this sequencing are highlighted in this work, such as the ability to perform long readings and be able to obtain sample responses in short time. It’s also able to discover as much information as possible about the pathogen, being an important feature to deal with public health emergencies, such is the case of the COVID-19.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others
Keywords
1 Introduction
The COVID-19 is a contagious infectious disease caused by the new SARS-CoV-2 virus (Severe Acute Respiratory Syndrome Coronavirus 2). The virus is thus known due to its taxonomic and genomic relationships with SARS-CoV-1, identified in 2002 as the etiological agent of an epidemic of the Severe Acute Respiratory Syndrome (SARS). The term COVID, designated in 2020 by the World Health Organization (WHO), means Corona Virus Disease (Coronavirus Disease), while “19” refers to the year 2019 when the first cases were reported in the city of Wuhan, China [1].
The new Coronavirus belongs to the order Nidoviral of the family Coronaviridae and to the ß subgroup genus [2, 3]. According to phylogenetic analyzes, its relationship with the ß genus of the Coronavirus family was evidenced specifically with SARS-CoV-1 [4]. On January 10, 2020, the first genome sequencing of this virus was published in Wuhan, and, until the month of August 2020, more than 78,000 genomic sequences of SARS-CoV-2 obtained worldwide have already been deposited in the GISAID database [5].
2 SARS-CoV-2
SARS-CoV-2 has a positive, linear, single-stranded RNA genome of approximately 30 kb, enveloped and with a diameter of 60–140 nm, with 14 6-11 open reading frames (ORFs) encoding 29 proteins. The ORF1ab encodes proteins for RNA replication and genes for non-structural proteins (nsp) and structural proteins [3]. Sixteen non-structural proteins (nsp 1-16) (Table 1) have been reported to be of great importance for their specific functions in Coronavirus replication. However, the functions of some of the nsps are still unknown or not well understood [6]. The virus also has accessory proteins that interfere with the host’s innate immune response [7].
SARS-CoV-2 still has four structural proteins: the main one, which is the protein S (spike), allows the virus to bind to the cell membrane, connecting the host’s ACE2 receptor, being an important determinant of the reach and pathogenicity of the host. This protein is functionally subdivided into the S1 domain, responsible for binding to the receptor and the S2 domain responsible for cell membrane fusion. Protein E (envelope) is associated with the formation of the coronavirus envelope and contains a hydrophobic domain. Finally, protein M (membrane) and protein N (nucleocapsid) interfere in cellular, viral replication and packaging processes [3].
3 Mutations
Genetic mutations are modifications that change the nucleotide sequence of an organism’s DNA (or RNA). In the SARS-CoV-2 genome, the most reported mutations are found, mainly, in spike (S) protein, RNA polymerase, primase RNA and nucleoprotein, being related to changes in the transmissibility and virulence of the virus. For example, the most significant mutations are found in the gene that encodes S protein, such as mentioned in [3]. It is also showed that primer-independent RNA primase (nsp8) contains more mutations than any other protein [8].
Therefore, it is important to sequence and analyze, phylogenetically, the origin of the new coronavirus to understand and prevent new outbreaks of zoonoses caused by mutations that can occur constantly. Thus, we trace in this work relationships with other coronaviruses of wild origin. In addition, this work also shows that knowing the mutation rate, defined as the probability that a change in genetic information will pass to the next generation, helps to predict the infectious capacity of the virus and the damage to human health [9, 10].
4 Sequencing
Sequencing a genome literally means writing a sequence of letters A, C, G and T (or G, C, U and A in the case of RNA), where each letter refers to one of the four types of base, or nucleotides present in DNA, i.e., the substances:
-
A: Adenine
-
C: Cytosine
-
G: Guanine
-
T: Thymine (or U: Uracil, in the case of RNA).
The genetic material of SARS-CoV-2 is more specifically formed by RNA [11].
Nowadays, there are the first, second and third generation sequencing, being the last two called next-generation sequencing, which allow the reading and analysis of these nucleotides for molecular studies. Originally, first-generation sequencing was developed by Sanger in 1975, being a method that generates small sequencing fragments of different sizes, starting from the same point. It was the basic method used for the sequencing of the human genome, however, it has limitations related to the high cost for long size sequences, in addition to the high processing time, since it processes little genetic material per unit of time [12].
Second-generation sequencing (SGS), which emerged in the world in 2005, is able to supply the high cost and low performance of first-generation sequencing, and has a higher yield, with the capacity to sequence a large number of molecules in parallel. In fact, SGS techniques have a low cost for the amount of data it generates, however, they also need polymerase chain reaction (PCR) amplification, which can cause errors, in addition to increasing the complexity and time required for sample preparation [13].
The Sanger sequencing and SGS deliver only short-read DNA fragments within the range of 50–1000 bases [14]. So, due to the need for technologies that are capable of performing long and high-speed readings, third generation sequencing (TGS) is the more suitable. Unlike the other ones, TGS technologies have a direct target on genetic molecules, allowing sequencing in real-time, and making readings available for analysis as soon as they pass through the sequencer [13].
TGS has a low sequencing cost and easy sample preparation without the need of PCR amplification and an execution time significantly faster than SGS technologies. In addition, TGS is able to produce long reads (exceeding several kilobases) for the resolution of the assembly problem and repetitive regions of complex genomes, however [15].
The three commercially available TGS technologies are Pacific Biosciences (PacBio) real-time sequencing, Illumina’s Illuminate Tru-seq Synthetic Long-Read technology, and Oxford Nanopore Technologies’ sequencing platform (available on devices MiniON, GridION and PromethION [16]).
Oxford Nanopore Technologies (ONT), like the others TGS, is cheaper and faster than SGS methods. This has potential advantages over current widely used sequencing technologies (Ion Torrent and Illumina), which depend on sequencing groups of amplified molecules [17]. MinION has the capacity to produce >1,000,000 sequences per day, with average reading lengths of around 20,000 bases and maximum reading lengths close to 1,000,000 bases [18], however, the speed error is greater than SGS methods [17].
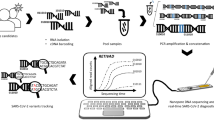
ONT sequencing has been increasingly applied in clinical virology, due to its ability to perform long readings. It is also able to get, in the shortest time possible, sample responses to discover as much information about the pathogen candidate, which is an important feature to deal with public health emergencies. Li et al. [19] noticed that Nanopore is efficient in terms of time, both for detection (of nucleotides) and for data analysis, and has been used in different organisms, from the simplest to the more complex (like humans) because there is no maximum limit on the size of the sequence, as occurs in other techniques [20].
Nanopore sequencing has other advantages, such as tracking biomarkers or genes, requiring a low volume of samples with low concentration, and the fact that it does not require complex steps such as amplifications and conversions of genetic material. Thus, Nanopore proves to be a cheaper and more efficient technique than techniques that use PCR [21]. It can be also used for real-time detection of pathogens in complex clinical samples [22]. In addition, one of the great advantages demonstrated by this technique is the direct RNA sequencing. This method is attractive because it eliminates the need for reverse transcription and, therefore, can reduce initialization errors and non-random copies introduced by reverse transcriptase [23].
Its functioning is based on the polarization of the membranes of the Nanopore, allowing to perceive changes in the signal of the electric current of the RNA or DNA molecules when they pass through the pores [24]. Basically, the ionic current depends on the nitrogenous base that is passing at the time of reading, making it possible to know the sequence of nucleotides, as changes in ionic currents are happening [21].
In Brazil, scientists from the Adolfo Lutz Institute, in collaboration with University of São Paulo (USP) and the University of Oxford, used the Nanopore methodology to carry out the first genetic sequencing of the new coronavirus (SARS-CoV-2) in Latin America, carried out in just 48 h after the first confirmed case [25].
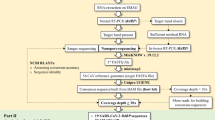
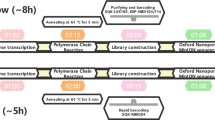
The estimated time to sequence SARS-CoV-2 using another technique is longer, so it is evident that the ONT has been used on several occasions. For example, the entire MinION workflow, from sample preparation to DNA extraction, sequencing, bioinformatics and interpretation, was carried out in approximately 2.5 h [26].
There is no doubt about the importance of the MinION sequencer to determine the SARS-CoV-2 sequence, however, its error rate must be considered, which ranges from 5 to 20% [26]. That is why currently there are different protocols to reduce the error rate, such as nanoCORR—Error-correction tool for nanopore sequence data [27], NanoOK—Software for nanopore data, quality and error profiles [28], and Nanocorrect—Error-correction tool for nanopore sequence data [29, 30]. Also, in ARTIC network—Real-time molecular epidemiology for outbreak response—the protocol starting a MinION sequencing run using MinKNOW [31]. Otherwise, without these protocols it is difficult to carry out an analysis and/or trace the phylogeny of this virus. Thus, with different protocols and sequencing methods in tandem repetition (concatemer), reduction of error rates from 1 to 3% is obtained [26].
This NGS used in Brazil to sequence the new coronavirus shows its benefits, allowing the monitoring of the epidemic in real-time and tracing the behavior of the virus locally and globally. Also, through the analysis of genetic variations, it is possible to predict the route of transmission and dispersion of the virus (allowing, in the future, the development of vaccines), verifying the evolution of the effectiveness of drugs on it, and conducting epidemiological research. Thus, as previously mentioned, sequencing SARS-CoV-2 using an efficient and cost-effective technology, such as is the case of portable sequencing devices as the Oxford Nanopore MinION, is of great importance for the humanity today [23].
5 Conclusions
After analyzing the various studies available in the literature so far, it is concluded that sequencing and analyzing the new coronavirus, in order to detect its mutations, is extremely necessary to prevent new outbreaks (of zoonoses). From this kind of analysis, it is possible to trace a phylogenetic relationship between the virus that causes COVID-19 and other coronaviruses present in the wild to predict its possible damage to human health and its capacity for infection, as well as to know and analyze its mutation rate, as it is transmitted from human to human, and predict future therapeutic targets [9].
References
Wang C, Liu Z, Chen Z et al (2020) The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. https://doi.org/10.1002/jmv.25762
Fehr AR, Perlman S (2015) Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 1282:1–23
Yin C (2020) Genotyping coronavirus SARS-CoV-2. Genomics. https://doi.org/10.1016/j.ygeno.2020.04.016
Ji W, Wang W, Zhao X, Zai J, Li X (2020) Cross-species transmission of the newly identified coronavirus 2019-nCoV. J Med Virol 92:433–440
GISAID at https://www.gisaid.org/
Chen Y et al (2020) Emerging coronaviruses: genome structure, replication and pathogenesis. J Med Virol 92:418–423
Dawood AA (2020) Mutated CoVID-19 may foretell a great risk for making the future. New Microbes New Infect 35:1000673
Phan T (2020) Genetic diversity and evolution of SARS-CoV-2. Infect Genet Evol 81:104260
Zhamg J et al (2020) The continuous evolutions and disseminations of 2019 novel human coronavirus. J Infect 80:671–693
Sanjuán R, Domingo-Calap P (2016) Mechanisms of viral mutation. Cell Mol Life Sci 73:4433–4448
Rye C et al (2017) Biology. OpenStax, Houston
Schadt EE, Turner S, Kasarskis A (2010) A window into third-generation sequencing. Hum Mol Genet 19:R227–R240
Lu H et al (2016) Oxford nanopore MinION sequencing and genome assembly. Genomics Proteomicis Bioinform 14:265–279
Kraft F, Kurth I (2019) Long-read sequencing in human genetics. medizinische genetik 31:198–204
Kchouk M, Jean-François Gibrat Elloumi M (2017) Generations of sequencing technologies: from first to next generation. Biol Med 9:395
Pillai S et al (2017) Review of sequencing platforms and their applications in phaeochromocytoma and paragangliomas. Crit Rev Oncol/Hematol 116:58–67
Laver T, Harrison J, O’Neill PA, Moore K, Farbos A, Paszkiewicz K et al (2015) Assessing the performance of the Oxford nanopore technologies MinION. Biomol Detect Quantif 1(3):1–8
Huang J, Liang X, Xuan Y, Geng C, Li Y, Lu H et al (2017) Real-time DNA barcoding in a rainforest using nanopore sequencing: opportunities for rapid biodiversity assessments and local capacity building. Gigascience 6(5):1 [cited 2020 Jul 30]
Li Y, He X, Li M et al (2020) Comparison of third-generation sequencing approaches to identify viral pathogens under public health emergency conditions. Virus Genes 56(3):228–297
Roach NP et al (2020) The full-length transcriptome of C. elegans using direct RNA sequencing. Genome Res 30(2):299–312
Venkatesan B (2011) Nanopore sensors for nucleic acid analysis. Nat Nanotechnol 6:615–624
Greninger AL, Naccache SN, Federman S et al (2015) Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med 7:99
Quick J, Grubaugh N, Pullan S et al (2017) Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 112:1261–1276
Li Y, Han R, Bi C, Li M, Wang S, Gao X (2018) DeepSimulator: a deep simulator for nanopore sequencing. Bioinformatics 34(17):2899–2908
Jesus J (2020) Importation and early local transmission of COVID-19 on Brazil, 2020. J São Paulo Inst Trop Med 62:e30
Loit K, Adamson K, Bahram M, Puusepp R, Anslan S, Kiiker R et al (2019) Relative performance of MinION (Oxford Nanopore Technologies) versus Sequel (Pacific Biosciences) thirdgeneration sequencing instruments in identification of agricultural and forest fungal pathogens. Appl Environ Microbiol 85(21)
NANOCORR at https://github.com/jgurtowski/nanocorr
NANOOK at https://documentation.tgac.ac.uk/display/NANOOK/NanoOK
NANOCORRECT at https://github.com/jts/nanocorrect/
Lu H, Giordano F, Ning Z (2016) Oxford nanopore MinION sequencing and genome assembly. Genomics Proteomics Bioinf 14:265–279
MINKNOW at https://www.protocols.io/view/starting-a-minion-sequencing-run-using-minknow-7q6hmze
Acknowledgements
The authors thank CAPES and CNPq for their scholarships.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this paper

Cite this paper
Corredor-Vargas, A.M., Torezani, R., Paneto, G., Bastos-Filho, T.F. (2022). Importance of Sequencing the SARS-CoV-2 Genome Using the Nanopore Technique to Understand Its Origin, Evolution and Development of Possible Cures. In: Bastos-Filho, T.F., de Oliveira Caldeira, E.M., Frizera-Neto, A. (eds) XXVII Brazilian Congress on Biomedical Engineering. CBEB 2020. IFMBE Proceedings, vol 83. Springer, Cham. https://doi.org/10.1007/978-3-030-70601-2_199
Download citation
DOI: https://doi.org/10.1007/978-3-030-70601-2_199
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-70600-5
Online ISBN: 978-3-030-70601-2
eBook Packages: EngineeringEngineering (R0)