
Abstract
Polyarteritis nodosa (PAN) is a rare systemic necrotizing vasculitis predominantly affecting the medium-sized arteries with widely variable presenting features, disease course, and outcomes. Recent updates regarding the nomenclature of PAN have resulted in the description of several PAN sub-phenotypes. Herein discussed are idiopathic PAN, Hepatitis B-associated PAN and monogenic disorders such as adenosine deaminase-2 deficiency. The current understanding of the pathogenesis, histopathological features, and treatment of these conditions are reviewed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
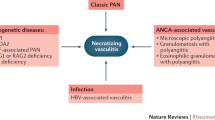
The terminology associated with polyarteritis nodosa (PAN) has undergone significant changes over the last century, particularly in the last four decades. Therefore, a review of the evolving nomenclature is integral to understanding this condition and its associated subgroups. The term “periarteritis nodosa” was first used by Kussmaul and Maier in 1866 during their description of a 27-year-old male with multiple nodules along the length of medium and small arteries in the thorax and abdomen [1]. Further histologic evaluation by Ferrari in 1903 revealed that the inflammatory process was not relegated to the adventitia, but rather was transmural, so use of “polyarteritis nodosa” was proposed [2]. Historically, the presence of necrotizing vasculitis on any biopsy was attributed to PAN. Little further distinction was made during the first half of the twentieth century until granulomatosis with polyangiitis (GPA, formerly Wegener’s granulomatosis) was initially described [3] and then subsequently considered as a separate vasculitic entity [4].
Discovery of anti-neutrophil cytoplasmic antibodies (ANCA) allowed for further distinguishing polyarteritis nodosa from the ANCA-associated vasculitides [GPA, microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA, formerly Churg–Strauss)] [5, 6]. A definitive distinction between PAN and MPA was outlined through the 1994 [7] and the revised 2012 International Chapel Hill Consensus Conference (CHCC) nomenclature of vasculitides [8] where PAN was defined as a necrotizing inflammation of medium-sized or small arteries without glomerulonephritis or vasculitis of the small vessels (i.e., arterioles, capillaries, and venules) and not associated with ANCA. MPA was defined as a necrotizing vasculitis with few or no immune deposits, predominantly affecting the small vessels and associated with myeloperoxidase (MPO) ANCA or proteinase 3 (PR3) ANCA.
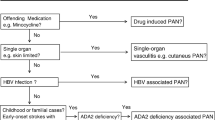
Further distinction between PAN and PAN-like conditions has reduced the number of patients classified as having systemic idiopathic PAN. These subgroups now include vasculitis associated with probable etiology (i.e., drug, viral) and monogenic disease-related PAN-like conditions. Given the presentation of hepatitis B virus (HBV)-associated PAN and systemic idiopathic PAN have the same clinical features, the chapter herein will focus on both systemic idiopathic PAN and HBV-associated PAN but will additionally note distinguishing characteristics of the presentation and treatment of other less common PAN-like variants.
2 Epidemiology
PAN can affect any age but more commonly affects adults between their fourth and sixth decades of life. There is a slight male to female predominance (1.5:1), but ethnic predilection has not been observed [9]. Annual incidence of systemic idiopathic PAN has been estimated to be 1–5 per 1,000,000 with a prevalence of approximately 30 per 1,000,000 [9,10,11]. Incidence of HBV-associated PAN has been reported as high as 77 per 1,000,000 in endemic areas [12]. Following the institution of safer transfusion practices, hospital hygiene, and HBV vaccination, the rates of HBV-associated PAN have dramatically reduced from 35% to less than 5% of PAN cases [10].
3 Etiopathogenesis
According to more recent understanding, necrotizing vasculitis likely represents a range of diseases with varying etiopathogenesis [13]. In systemic idiopathic PAN, the underlying mechanisms are not well understood, but the predominance of dendritic cells and CD4+ lymphocytes in vascular lesions suggest the possibility of an antigen-specific T-cell-mediated response [14]. Several infections have been identified as potential triggers. Hepatitis B virus is the most well-documented but hepatitis C [15], human immunodeficiency virus [16], parvovirus B19 [17], Epstein–Barr virus [18], and cytomegalovirus [19] have also been observed. In contrast to systemic idiopathic PAN, viral replication [20] and circulating immune-complex deposition [21, 22] have been noted to result in direct vascular inflammation among patients with HBV-associated PAN. Drug-induced causes are uncommon; however, systemic PAN-like disease in the context of chronic use of minocycline for treatment of acne vulgaris has been reported [23, 24].
Limited information is known regarding genetic abnormalities and risk of PAN. However, a monogenic polyarteritis with similar clinical and histological characteristics to systemic idiopathic PAN has been recently observed. This predominantly childhood-onset PAN-like variant called deficiency of adenosine deaminase 2 (DADA2) is caused by an autosomal recessive mutation in the Adenosine Deaminase 2 (ADA2) gene [formerly known as the Cat Eye Syndrome Chromosome Region 1 (CECR1) gene] [25], which encodes for the adenosine deaminase 2 enzyme (ADA2). ADA2 has been hypothesized to be a key growth factor for endothelial cells and a regulator in the differentiation of monocytes. Deficiency of ADA2 results in endothelial damages and skewing of monocytes to a pro-inflammatory macrophage subset [26]. Preliminary findings from evaluation of 117 adult-onset, HBV-negative systemic idiopathic PAN patients have also shown the presence of heterozygous or biallelic missense variants in ADA2 among 8 (7%) patients resulting in reduced ADA2 activity. These findings suggest a potential genetic basis among a subset of systemic idiopathic PAN patients [27].
4 Clinical Manifestations and Laboratory Markers
4.1 Clinical Manifestations
Because medium- and small-sized arteries are involved in PAN, a wide spectrum of clinical manifestations has been reported (Table 14.1). Constitutional symptoms of fever, weight loss, myalgia, and arthralgia are common and are present in 30–70% [28,29,30]. Cutaneous findings are lower extremity predominant and occur in up to 50–60% of patients as demonstrated by purpura, livedo reticularis/racemosa, nodules, and ulcers [28]. Digital infarction resulting in gangrene can also occur (6%) but limb ischemia is rare [29, 31] (Fig. 14.1). Peripheral nerve involvement results from arteriolar occlusion of the vasa nervosum [32]. Mononeuritis multiplex, typically presenting as wrist drop or foot drop, is the most common neurologic feature. Patients will often note pain or change in sensation (hypo/hyper/dysesthesia) prior to the onset of a motor deficit, but palsy can develop suddenly without sensory prodrome. Symmetric sensorimotor peripheral neuropathy and pure sensory neuropathy are also seen; among which, the sciatic, peroneal, tibial, ulnar, median, and radial nerves appear to have a higher likelihood of involvement [33]. Cranial nerve palsies have been reported but are infrequent and affect less than 2% of patients [33]. Stroke can occur in systemic idiopathic PAN but is rare. If present, particularly in a child or young adult, DADA2 variant should be considered.

Dry gangrene of the distal second and fourth phalanx on a background of livedo reticularis and acrocyanosis
Abdominal pain is the most frequent gastrointestinal feature but is nonspecific. If associated with or exacerbated by meals, this raises concern for intestinal angina secondary to mesenteric arteritis. Ischemia appears to be more common in the small intestine compared to the colon and can result in nausea, vomiting, diarrhea, melena, or hematochezia. Vasculitic involvement of visceral organs such as the gallbladder and appendix can be seen and mimic cholecystitis and appendicitis, respectively. Upon surgical removal, histologic evaluation confirms arteritic involvement if present. Both ischemic bowel perforation and rupture of a visceral artery aneurysm can manifest as a surgical abdomen; a presentation which carries high morbidity and mortality [34].
Renal abnormalities in PAN differ from ANCA-associated vasculitis as the former does not cause glomerulonephritis [8]. Renal infarcts (Fig. 14.2), resulting from either occlusion of intrarenal arteries or rupture of microaneurysms (Fig. 14.3), can produce micro- or macroscopic hematuria, but dysmorphia is generally absent. Proteinuria, if detected, is typically sub-nephrotic [28]. Renal insufficiency is uncommon but may develop as a consequence of significant renal parenchymal loss due to infarction or as a result of severe renovascular hypertension from renal artery stenosis. Urologic involvement has been noted in 17% of cases and is rarely the initial manifestation; nevertheless, non-infectious orchitis secondary to testicular arteritis is a characteristic feature of PAN [28, 29].

Renal infarct, right inferior pole (CT, coronal view)

Selective conventional right renal angiogram demonstrating multiple segmental microaneurysms
Cardiac involvement has been noted since the index description of PAN in 1866 but is an underreported finding as many patients may be asymptomatic. Indeed, only 2–10% of patients have clinically diagnosed cardiac findings [28, 29], whereas histologic evidence has been reported in up to 78% of patients in autopsy studies [35]. Left ventricular heart failure is the most frequently observed abnormality, and the etiology is likely multifactorial with coronary arteritis, myocardial infarction, and renovascular hypertension as potential contributors [36]. Coronary arteritis has been described in 50% of autopsy cases [35] but clinically symptomatic coronary angina (2–18%) and myocardial infarctions (1–12%) are less often reported [36]. Giant coronary aneurysms (Fig. 14.4) have been observed but are considered rare [37, 38]. These are likely sequelae of untreated disease and angiographically may be challenging to distinguish from patients with history of Kawasaki’s Disease.

Conventional coronary angiogram with alternating stenotic and aneurysmal segments of the left anterior descending (top) and giant aneurysm of the left circumflex (arrow)
Myalgias occur in 60–70% [28, 29] of patients but inflammatory myopathy is rare [39]. Creatine kinase levels may be elevated but are generally less than 2000 IU/L. Pain can be present due to arterial insufficiency of the medium-sized muscular vessels. Thigh and calf muscle involvement is typical. Magnetic resonance imaging of the musculature can demonstrate diffuse or patchy hyperintensity of the affected muscle on T2-weighted imaging and contrast-enhanced images may demonstrate small fluffy enhancing lesions centered on the vessels (“cotton wool appearance”) [40].
Overall, the clinical features of patients with classical PAN and HBV-associated PAN are similar. However, a few noted differences have been observed with HBV-associated PAN demonstrating a higher frequency of myalgia, neurologic manifestations, abdominal pain, and vasculitis-related cardiomyopathy but a lower frequency of livedo and nodular skin lesions [28].
4.2 Laboratory Markers
There are no specific laboratory markers for PAN. An inflammatory state with normocytic anemia, thrombocytosis, and elevated erythrocyte sedimentation rate and/or C-reactive protein is common. Leukocytosis may be seen. If peripheral eosinophilia is noted, particularly if >10%, then the possibility of eosinophilic granulomatosis with polyangiitis should be assessed. Renal insufficiency can be present, but is not typically severe. Urinalysis may show sub-nephrotic proteinuria and non-dysmorphic microscopic hematuria. ANCA serologies (cANCA/PR3, pANCA/MPO) should be negative. Cryoglobulins, complements (C3, C4), and rheumatoid factor should be evaluated to assess for possibility of cryoglobulinemia. The presence of HIV, hepatitis B, and hepatitis C should be investigated. Lactate levels may be of assistance in patients presenting with severe abdominal pain or surgical abdomen to assess for tissue ischemia.
5 Histopathology
Cutaneous findings observed in classical systemic PAN are indistinguishable from the isolated cutaneous variant (see histopathology 13.4). The vascular abnormalities in PAN demonstrate a segmental pattern with a predilection for arterial branch points of muscular arteries [41]. The cause for predisposition of branch points is unknown but an increase in expression of adhesion molecules and intimal macrophages at these locations has been proposed [42]. The vascular infiltrates observed vary depending on the stage of the inflammatory process. In the early, active phase a transmural inflammatory infiltrate composed of an admixture of lymphocytes, macrophages, neutrophils and eosinophils are seen along with findings of fibrinoid necrosis of the vessel wall [41]. Subsequently, vascular remodeling occurs with dense vessel wall fibrosis as well as intimal hyperplasia. Thrombosis can lead to vascular occlusion whereas disruption of the elastic lamina results in aneurysmal dilation.
6 Diagnosis and Differential Diagnosis
There are no validated or approved diagnostic criteria for PAN. The American College of Rheumatology has developed classification criteria for PAN (Table 14.2) [43]. Unfortunately, these criteria are of limited utility for two reasons. First, these criteria are intended for research purposes to distinguish what subtype of vasculitis a patient has once they have a confirmed vasculitis diagnosis. As such they should not be used in the clinical setting to determine if a patient does or does not have vasculitis. Second, these criteria are not useful in differentiating patients with PAN from microscopic polyangiitis, given the latter was not considered a separate entity at the time of drafting. Because of these noted limitations, an ongoing international collaborative effort is currently underway to develop diagnostic and classification criteria for PAN [44].
Consequently, the diagnosis of PAN requires the combination of characteristic clinical manifestations, laboratory parameters, angiographic features, and histopathology in a suspected individual. Biopsy of an affected organ confirming the presence of focal, segmental, transmural, necrotizing inflammation of the medium- or small-sized arteries is considered the gold standard for diagnosis. If affected, the highest yield is typically observed in skin, nerve, muscle, and testicle [45]. Combined nerve and muscle biopsy appears to be superior to muscle biopsy alone for diagnosis. In a large series of patients with suspected PAN, vasculitis confirmation from dual nerve/muscle biopsy was obtained in 83% (90/108) of patients with peripheral neuropathy and 81% (17/21) of patients without peripheral neuropathy; compared with 68% (41/60) positive biopsy specimens in patients with peripheral neuropathy and 60% (24/40) without peripheral neuropathy among those with isolated muscle biopsy performed [28]. While this study highlights the potential utility of blind nerve and/or muscle biopsy even in asymptomatic patients, it is suggested that evaluation with electromyogram and/or muscle MRI be performed to identify if pathologic findings are present in order to guide biopsy location. Although confirmatory findings of PAN can be observed on kidney and liver biopsy specimens, these locations carry a high risk of post-procedure hemorrhage and therefore should not be considered as first-line targets.
Angiography provides additional diagnostic utility in patients with suspected PAN, particularly among those with abdominal or renal symptoms for which biopsy was not able to be obtained or was non-diagnostic. The typical angiographic features of PAN include saccular or fusiform microaneurysms (1–5 mm diameter) usually coinciding with stenotic lesions [46] (Fig. 14.5). Larger aneurysm may also be present within which dissections may occur (Figs. 14.6, 14.7, and 14.8). The most frequent arterial territories affected include the celiac, hepatic, renal, and mesenteric branches. Visceral organ infarcts, bowel wall thickening, and perinephric hematoma are commonly seen but are less specific for PAN and must be differentiated from diseases causing in situ thrombosis or thromboembolism [47]. With the advancements in noninvasive imaging, computed tomography angiography (CTA) is a reasonable initial screening modality given it has spatial resolution detail that is sufficient to evaluate for the majority of findings suggestive of PAN including stenosis/occlusion, infarcts, and aneurysms >2 mm diameter. However, if CTA is negative or equivocal and suspicion remains, then conventional angiography is required. If characteristic angiographic findings are identified by an experienced radiologist the diagnosis may be confirmed, even without biopsy, provided there is appropriate clinical context and mimicking conditions (Table 14.3) have been ruled out.

Computed tomography angiography (axial view) demonstrating superior mesenteric artery branch with alternating stenotic and aneurysmal segments (thick arrow), mesenteric artery branches with arterial thickening (thin arrow), and mid-pole left renal infarct (dashed arrow)

Computed tomography angiography highlighting proximal celiac artery aneurysm (arrow) [Axial view, left pane; 3D formatted, right pane]

Multiple aneurysms in the superior mesenteric artery, bilateral common iliac arteries, and common femoral arteries [computed tomography 3D formatted, right pane] with complex dissection of the left femoral artery (arrow) [axial view, left pane]

Selective superior mesenteric artery conventional angiogram with long segment proximal dissection (arrow)
7 Prognosis
If left untreated, systemic PAN carries a high mortality with a 5-year survival of 13% [48]. Conversely, those receiving treatment have a notably improved outcome with 5-year survival nearing 80–90% [28, 49]. The overall outcome is largely dependent on the severity of disease at time of diagnosis. A prognostic scoring system called the Five-Factor Score (FFS) was devised by the French Vasculitis Study Group from evaluation of a prospective study of 342 patients with polyarteritis nodosa, microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis (Table 14.4) [50]. For patients with an FFS = 0, 5-year mortality was 12%, whereas the mortality rate for FFS = 1 was 26% and FFS ≥ 2 was 46% [50]. The same group re-evaluated this scoring system evaluating 1108 total patients with systemic necrotizing vasculitis, this time including granulomatosis with polyangiitis [51]. The updated 2009 FFS (Table 14.4) added age > 65 year at diagnosis as a poor prognostic factor but no longer includes CNS involvement among these parameters. Given patients with ANCA-vasculitis were included, ENT symptoms were evaluated and the absence of such findings were considered to carry a poorer prognosis; however, this is not applicable to patients with PAN. The updated rates are similar to the original FFS prediction model, demonstrating reliability of this prognostic tool [51]. Death in the first year is more commonly due to poorly controlled vasculitis, whereas subsequent mortality is more often attributable to complications resulting from sequelae of vasculitis-associated organ damage, cardiovascular disease, or consequences of immunosuppressive treatments, particularly infection [52, 53].
PAN has been noted to have a more frequent monophasic pattern when compared to other systemic necrotizing vasculitides; nevertheless, a proportion of patients will undergo a relapsing course. Among a cohort of 348 patients with PAN, 76 (22%) relapsed within 5 years of follow-up [28]. Patients with HBV-associated PAN have been observed to have less frequent relapse than those with non-HBV-associated disease with 5-year relapse-free survival rates being seen in 59.4% of patients with non-HBV-associated PAN, compared to 67% in those with HBV-associated disease [28]. Childhood-onset PAN has been reported to have a more benign course when compared to adults with less renal and neurologic involvement noted and shorter duration of induction treatment required [54].
Patients with PAN require long-term follow-up with specialists that are familiar with the disease process and its multi-system clinical manifestations, as well as clinicians that are comfortable with the immunosuppressive therapies required for induction and maintenance. During the active phase, patients should be closely observed with visits every 2–4 weeks for the first 3–6 months. Once stabilized, visits can occur at less frequent intervals of every 2–6 months for the 2 years following diagnosis. Because of possible late-stage relapse as well as potential development of comorbidities due to sequelae from vascular damage or immunosuppressive therapy, patients should be followed life-long at intervals of every 6–12 months, even during remission. At each visit, patients should (at a minimum) have a blood pressure evaluation, comprehensive multi-system physical examination, creatinine and urinalysis with microscopy. Additional labs may be needed for immunosuppressive drug monitoring. In asymptomatic patients, routine repeat angiography is not requisite but should be considered if there are new or progressive symptoms of abdominal or cardiac pain or if there are known arterial dilatations/aneurysms that require routine monitoring.
8 Treatment
The level of clinical trial evidence guiding the therapeutic decisions in the management of PAN is low [55]. In addition, trials evaluating this condition must be interpreted carefully as they commonly include an admixture of other systemic necrotizing vasculitides such as EGPA and MPA [49, 56,57,58,59]. In general, treatment for systemic PAN is determined based on the severity of disease at time of presentation as well as the presence or absence of HBV. Patients with systemic idiopathic PAN with mild disease (FFS = 0) may be treated with glucocorticoid monotherapy with initial doses of 1 mg/kg/day (up to 60 mg) with subsequent tapering over 6–8 months [28, 49]. For patients with glucocorticoid-resistant disease and in those that develop major relapse despite the use of adequate glucocorticoid doses, the addition of an adjunct disease-modifying agent may be required. Among patients with mild PAN, cyclophosphamide is generally avoided and medications such as azathioprine (up to 2 mg/kg/day) have shown similar efficacy to pulse dose cyclophosphamide but with lower risk of side effects [49]. While often used, the overall long-term benefit of azathioprine is debated. In a recent study evaluating 95 systemic necrotizing vasculitis patients (51, EGPA, 25 MPA, 19 PAN) with FFS = 0, the addition of azathioprine to a glucocorticoid remission-induction regimen did not significantly improve the rates of remission and failed to reduce the risk of relapse or overall steroid exposure [57]. Methotrexate (up to 25 mg/week) and mycophenolate (up to 1500 mg twice daily) have also been used in the management of glucocorticoid-resistant disease, but supportive data for these agents is limited to observational studies [29, 60] and largely extrapolated from their use in the treatment of other systemic necrotizing vasculitidies such as ANCA-associated vasculitis.
The treatment of patients with poor prognostic factors (FFS ≥1) requires more aggressive management. In such circumstances, cyclophosphamide is advocated in addition to high-dose glucocorticoids. Both oral (target 2 mg/kg/day) and intravenous pulse (600 mg/m2 monthly) regimens have shown efficacy, but the latter has demonstrated a more tolerable side effect profile [59]. The duration of cyclophosphamide treatment is less well understood and has only been evaluated in the context of a single clinical trial evaluating 47 patients with MPA and 18 patients with PAN [58]. The results of this study suggest that 6 months of cyclophosphamide was less effective than 12 months of therapy; however, remission maintenance therapies were not utilized. Conventionally, patients with severe PAN are treated with cyclophosphamide for a minimum of 6 months, after which if they are in remission are transitioned to a lower level immunosuppression agent such as azathioprine, methotrexate, or mycophenolate for ongoing remission maintenance. The therapeutics options for patients with severe systemic idiopathic PAN failing to respond to cyclophosphamide are limited. Rituximab [61, 62] and tocilizumab [63] have been used with reported success, but results are limited to case reports and small case series and are considered currently experimental. Inhibitors of tumor necrosis factor alpha (infliximab, adalimumab, etanercept) have also shown preliminary benefit in systemic PAN [64,65,66,67] and appear to have a greater observed role in managing patients with the PAN-like DADA2 variant [68].
In patients with a potential precipitant for the development of PAN or PAN-like illness control or removal of the offending agent is imperative. For example, in patients with minocycline-induced PAN-like illness, cessation of minocycline may be sufficient to result in disease remission. However, in severe cases additional immunosuppressive therapy may be required [24].
Management of patients with HBV-associated PAN is focused on initial control of severe life-threatening manifestations (if present) followed by removal of immune complexes and subsequent clearance of viremia. Although prolonged use of glucocorticoids is contraindicated due to an increased risk of viral replication, short-term use of glucocorticoids (1 mg/kg/day for 1 week then tapered off over 1 additional week) has been safely utilized [69, 70]. Plasma exchange has not demonstrated improvement in outcomes among patients with systemic idiopathic PAN [71] but is considered integral in the treatment of HBV-associated PAN because clearance of immune complexes attenuates vessel inflammation [69, 70]. Suggestions for plasma exchange frequency are 3/week for 3 weeks, 2/week for 2 weeks, then weekly until hepatitis B e antigen to hepatitis B e antibody seroconversion is observed, or until 2–3 months of sustained clinical recovery has been obtained [69]. Antiviral therapy should be initiated at the time of diagnosis. Selection of the antiviral agent and determination of duration should be guided through coordination with hepatology. Interferon alpha2b and lamivudine have shown efficacy in prospective open-label trials [69, 72]. While newer nucleos(t)ide analogs (entecavir and tenofovir) have not been formally evaluated in patients with HBV-associated PAN their efficacy in patients with chronic hepatitis B viral infections is well established [73] and may be considered for assistance with viral clearance. Prolonged vasculitis control occurs in 90–100% of patients for which viral replication has ceased and seroconversion has occurred [69, 70].
References
Kussmaul A, Maier K. Ueber eine nicht bisher beschriebene eigenhümliche Arterienerkrankung (Periarteritis Nodosa), die mit Morbus Brightii und rapid fortschreitender allgemeiner Muskellähmung einhergeht. Dtsch Arch Klin Med. 1866;1:484–518.
Ferrari E. Uber polyarteritis acuta nodosa (sogenannte periarteritis nodosa) und ihre Beziehungen zur Polymyositis und Polyneuritis acuta. Zieglers Beitr. 1930;34:350–86.
Wegener F. Über eine eigenartige rhinogene Granulomatose mit besonderer Beteilgung des Arteriensystems und der Nieren. Beitr Path Anat. 1939;102:36–8.
Godman GC, Churg J. Wegener’s granulomatosis: pathology and review of the literature. AMA Arch Pathol. 1954;58(6):533–53.
Davies DJ, Moran JE, Niall JF, Ryan GB. Segmental necrotising glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? Br Med J (Clin Res Ed). 1982;285(6342):606.
van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet. 1985;1(8426):425–9.
Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37(2):187–92.
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
Watts RA, Lane SE, Scott DG, Koldingsnes W, Nossent H, Gonzalez-Gay MA, et al. Epidemiology of vasculitis in Europe. Ann Rheum Dis. 2001;60(12):1156–7.
Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum. 2004;51(1):92–9.
Mohammad AJ, Jacobsson LT, Mahr AD, Sturfelt G, Segelmark M. Prevalence of Wegener’s granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology (Oxford). 2007;46(8):1329–37.
McMahon BJ, Bender TR, Templin DW, Maynard JE, Barrett DH, Berquist KR, et al. Vasculitis in Eskimos living in an area hyperendemic for hepatitis B. JAMA. 1980;244(19):2180–2.
Ozen S. The changing face of polyarteritis nodosa and necrotizing vasculitis. Nat Rev Rheumatol. 2017;13(6):381–6.
Cid MC, Grau JM, Casademont J, Campo E, Coll-Vinent B, Lopez-Soto A, et al. Immunohistochemical characterization of inflammatory cells and immunologic activation markers in muscle and nerve biopsy specimens from patients with systemic polyarteritis nodosa. Arthritis Rheum. 1994;37(7):1055–61.
Saadoun D, Terrier B, Semoun O, Sene D, Maisonobe T, Musset L, et al. Hepatitis C virus-associated polyarteritis nodosa. Arthritis Care Res (Hoboken). 2011;63(3):427–35.
Font C, Miro O, Pedrol E, Masanes F, Coll-Vinent B, Casademont J, et al. Polyarteritis nodosa in human immunodeficiency virus infection: report of four cases and review of the literature. Br J Rheumatol. 1996;35(8):796–9.
Corman LC, Dolson DJ. Polyarteritis nodosa and parvovirus B19 infection. Lancet. 1992;339(8791):491.
Caldeira T, Meireles C, Cunha F, Valbuena C, Aparicio J, Ribeiro A. Systemic polyarteritis nodosa associated with acute Epstein-Barr virus infection. Clin Rheumatol. 2007;26(10):1733–5.
Kouchi M, Sato S, Kamono M, Taoda A, Iijima K, Mizuma A, et al. A case of polyarteritis nodosa associated with cytomegalovirus infection. Case Rep Rheumatol. 2014;2014:604874.
Trepo C, Guillevin L. Polyarteritis nodosa and extrahepatic manifestations of HBV infection: the case against autoimmune intervention in pathogenesis. J Autoimmun. 2001;16(3):269–74.
Guillevin L, Ronco P, Verroust P. Circulating immune complexes in systemic necrotizing vasculitis of the polyarteritis nodosa group. Comparison of HBV-related polyarteritis nodosa and Churg Strauss Angiitis. J Autoimmun. 1990;3(6):789–92.
Michalak T. Immune complexes of hepatitis B surface antigen in the pathogenesis of periarteritis nodosa. A study of seven necropsy cases. Am J Pathol. 1978;90(3):619–32.
Katada Y, Harada Y, Azuma N, Matsumoto K, Terada H, Kudo E, et al. Minocycline-induced vasculitis fulfilling the criteria of polyarteritis nodosa. Mod Rheumatol. 2006;16(4):256–9.
Kermani TA, Ham EK, Camilleri MJ, Warrington KJ. Polyarteritis nodosa-like vasculitis in association with minocycline use: a single-center case series. Semin Arthritis Rheum. 2012;42(2):213–21.
Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370(10):911–20.
Fayand A, Sarrabay G, Belot A, Hentgen V, Kone-Paut I, Grateau G, et al. [Multiple facets of ADA2 deficiency: vasculitis, auto-inflammatory disease and immunodeficiency: a literature review of 135 cases from literature]. Rev Med Interne. 2018;39(4):297–306.
Schnappauf O, Stoffels M, Aksentijevich I, Kastner D, Grayson P, Cuthbertson D, et al. Screening of patients with adult-onset idiopathic polyarteritis nodosa for deficiency of adenosine deaminase 2. Arthritis Rheumatol. 2018;70.
Pagnoux C, Seror R, Henegar C, Mahr A, Cohen P, Le Guern V, et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa: a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis Rheum. 2010;62(2):616–26.
Alibaz-Oner F, Koster MJ, Crowson CS, Makol A, Ytterberg SR, Salvarani C, et al. Clinical spectrum of medium-sized vessel vasculitis. Arthritis Care Res (Hoboken). 2017;69(6):884–91.
Lhote F, Cohen P, Guillevin L. Polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome. Lupus. 1998;7(4):238–58.
Merlin E, Mouy R, Pereira B, Mouthon L, Bourmaud A, Piette JC, et al. Long-term outcome of children with pediatric-onset cutaneous and visceral polyarteritis nodosa. Joint Bone Spine. 2015;82(4):251–7.
Minagar A, Fowler M, Harris MK, Jaffe SL. Neurologic presentations of systemic vasculitides. Neurol Clin. 2010;28(1):171–84.
de Boysson H, Guillevin L. Polyarteritis nodosa neurologic manifestations. Neurol Clin. 2019;37(2):345–57.
Levine SM, Hellmann DB, Stone JH. Gastrointestinal involvement in polyarteritis nodosa (1986-2000): presentation and outcomes in 24 patients. Am J Med. 2002;112(5):386–91.
Schrader ML, Hochman JS, Bulkley BH. The heart in polyarteritis nodosa: a clinicopathologic study. Am Heart J. 1985;109(6):1353–9.
Pagnoux C, Guillevin L. Cardiac involvement in small and medium-sized vessel vasculitides. Lupus. 2005;14(9):718–22.
Ebersberger U, Rieber J, Wellmann P, Goebel C, Gansera B. Polyarteritis nodosa causing a vast coronary artery aneurysm. J Am Coll Cardiol. 2015;65(5):e1–2.
Wi J, Choi HH, Lee CJ, Kim T, Shin S, Ko YG, et al. Acute myocardial infarction due to polyarteritis nodosa in a young female patient. Korean Circ J. 2010;40(4):197–200.
Calvo R, Negri M, Ortiz A, Roverano S, Paira S. Myositis as the initial presentation of panarteritis nodosa. Reumatol Clin. 2019;15(5):e24–6.
Kang Y, Hong SH, Yoo HJ, Choi JY, Park JK, Park J, et al. Muscle involvement in polyarteritis nodosa: report of eight cases with characteristic contrast enhancement pattern on MRI. AJR Am J Roentgenol. 2016;206(2):378–84.
Lie JT. Systemic and isolated vasculitis. A rational approach to classification and pathologic diagnosis. Pathol Annu. 1989;24(Pt 1):25–114.
Jennette JC. Implications for pathogenesis of patterns of injury in small- and medium-sized-vessel vasculitis. Cleve Clin J Med. 2002;69(Suppl 2):SII33–8.
Lightfoot RW Jr, Michel BA, Bloch DA, Hunder GG, Zvaifler NJ, McShane DJ, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum. 1990;33(8):1088–93.
Craven A, Robson J, Ponte C, Grayson PC, Suppiah R, Judge A, et al. ACR/EULAR-endorsed study to develop Diagnostic and Classification Criteria for Vasculitis (DCVAS). Clin Exp Nephrol. 2013;17(5):619–21.
Hernandez-Rodriguez J, Alba MA, Prieto-Gonzalez S, Cid MC. Diagnosis and classification of polyarteritis nodosa. J Autoimmun. 2014;48–49:84–9.
Jee KN, Ha HK, Lee IJ, Kim JK, Sung KB, Cho KS, et al. Radiologic findings of abdominal polyarteritis nodosa. AJR Am J Roentgenol. 2000;174(6):1675–9.
Singhal M, Gupta P, Sharma A, Lal A, Rathi M, Khandelwal N. Role of multidetector abdominal CT in the evaluation of abnormalities in polyarteritis nodosa. Clin Radiol. 2016;71(3):222–7.
Frohnert PP, Sheps SG. Long-term follow-up study of periarteritis nodosa. Am J Med. 1967;43(1):8–14.
Ribi C, Cohen P, Pagnoux C, Mahr A, Arene JP, Puechal X, et al. Treatment of polyarteritis nodosa and microscopic polyangiitis without poor-prognosis factors: a prospective randomized study of one hundred twenty-four patients. Arthritis Rheum. 2010;62(4):1186–97.
Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore). 1996;75(1):17–28.
Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P, et al. The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore). 2011;90(1):19–27.
Bourgarit A, Le Toumelin P, Pagnoux C, Cohen P, Mahr A, Le Guern V, et al. Deaths occurring during the first year after treatment onset for polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: a retrospective analysis of causes and factors predictive of mortality based on 595 patients. Medicine (Baltimore). 2005;84(5):323–30.
Jardel S, Puechal X, Le Quellec A, Pagnoux C, Hamidou M, Maurier F, et al. Mortality in systemic necrotizing vasculitides: a retrospective analysis of the French Vasculitis Study Group registry. Autoimmun Rev. 2018;17(7):653–9.
Erden A, Batu ED, Sonmez HE, Sari A, Armagan B, Arici ZS, et al. Comparing polyarteritis nodosa in children and adults: a single center study. Int J Rheum Dis. 2017;20(8):1016–22.
Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009;68(3):310–7.
Pagnoux C, Quemeneur T, Ninet J, Diot E, Kyndt X, de Wazieres B, et al. Treatment of systemic necrotizing vasculitides in patients aged sixty-five years or older: results of a multicenter, open-label, randomized controlled trial of corticosteroid and cyclophosphamide-based induction therapy. Arthritis Rheumatol. 2015;67(4):1117–27.
Puechal X, Pagnoux C, Baron G, Quemeneur T, Neel A, Agard C, et al. Adding azathioprine to remission-induction glucocorticoids for eosinophilic granulomatosis with polyangiitis (Churg-Strauss), microscopic polyangiitis, or polyarteritis nodosa without poor prognosis factors: a randomized, controlled trial. Arthritis Rheumatol. 2017;69(11):2175–86.
Guillevin L, Cohen P, Mahr A, Arene JP, Mouthon L, Puechal X, et al. Treatment of polyarteritis nodosa and microscopic polyangiitis with poor prognosis factors: a prospective trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in sixty-five patients. Arthritis Rheum. 2003;49(1):93–100.
Gayraud M, Guillevin L, Cohen P, Lhote F, Cacoub P, Deblois P, et al. Treatment of good-prognosis polyarteritis nodosa and Churg-Strauss syndrome: comparison of steroids and oral or pulse cyclophosphamide in 25 patients. French Cooperative Study Group for Vasculitides. Br J Rheumatol. 1997;36(12):1290–7.
Eleftheriou D, Dillon MJ, Tullus K, Marks SD, Pilkington CA, Roebuck DJ, et al. Systemic polyarteritis nodosa in the young: a single-center experience over thirty-two years. Arthritis Rheum. 2013;65(9):2476–85.
Ribeiro E, Cressend T, Duffau P, Grenouillet-Delacre M, Rouanet-Lariviere M, Vital A, et al. Rituximab efficacy during a refractory polyarteritis nodosa flare. Case Rep Med. 2009;2009:738293.
Neel A, Masseau A, Hervier B, Bossard C, Cacoub P, Pagnoux C, et al. Life-threatening hepatitis C virus-associated polyarteritis nodosa successfully treated by rituximab. J Clin Rheumatol. 2011;17(8):439–41.
Krusche M, Ruffer N, Kotter I. Tocilizumab treatment in refractory polyarteritis nodosa: a case report and review of the literature. Rheumatol Int. 2019;39(2):337–44.
Capuozzo M, Ottaiano A, Nava E, Cascone S, Fico R, Iaffaioli RV, et al. Etanercept induces remission of polyarteritis nodosa: a case report. Front Pharmacol. 2014;5:122.
Wang CR, Yang CC. Adalimumab therapy in hepatitis B virus-negative polyarteritis nodosa: a case report. Medicine (Baltimore). 2018;97(25):e11053.
Lerkvaleekul B, Treepongkaruna S, Ruangwattanapaisarn N, Treesit T, Vilaiyuk S. Recurrent ruptured abdominal aneurysms in polyarteritis nodosa successfully treated with infliximab. Biologics. 2019;13:111–6.
Ginsberg S, Rosner I, Slobodin G, Rozenbaum M, Kaly L, Jiries N, et al. Infliximab for the treatment of refractory polyarteritis nodosa. Clin Rheumatol. 2019;38:2825.
Ombrello AK, Qin J, Hoffmann PM, Kumar P, Stone D, Jones A, et al. Treatment strategies for deficiency of adenosine deaminase 2. N Engl J Med. 2019;380(16):1582–4.
Guillevin L, Mahr A, Cohen P, Larroche C, Queyrel V, Loustaud-Ratti V, et al. Short-term corticosteroids then lamivudine and plasma exchanges to treat hepatitis B virus-related polyarteritis nodosa. Arthritis Rheum. 2004;51(3):482–7.
Guillevin L, Lhote F, Leon A, Fauvelle F, Vivitski L, Trepo C. Treatment of polyarteritis nodosa related to hepatitis B virus with short term steroid therapy associated with antiviral agents and plasma exchanges. A prospective trial in 33 patients. J Rheumatol. 1993;20(2):289–98.
Guillevin L, Lhote F, Cohen P, Jarrousse B, Lortholary O, Genereau T, et al. Corticosteroids plus pulse cyclophosphamide and plasma exchanges versus corticosteroids plus pulse cyclophosphamide alone in the treatment of polyarteritis nodosa and Churg-Strauss syndrome patients with factors predicting poor prognosis. A prospective, randomized trial in sixty-two patients. Arthritis Rheum. 1995;38(11):1638–45.
Guillevin L, Lhote F, Sauvaget F, Deblois P, Rossi F, Levallois D, et al. Treatment of polyarteritis nodosa related to hepatitis B virus with interferon-alpha and plasma exchanges. Ann Rheum Dis. 1994;53(5):334–7.
Lok AS, McMahon BJ, Brown RS Jr, Wong JB, Ahmed AT, Farah W, et al. Antiviral therapy for chronic hepatitis B viral infection in adults: a systematic review and meta-analysis. Hepatology. 2016;63(1):284–306.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Koster, M.J. (2021). Systemic Polyarteritis Nodosa. In: Salvarani, C., Boiardi, L., Muratore, F. (eds) Large and Medium Size Vessel and Single Organ Vasculitis. Rare Diseases of the Immune System. Springer, Cham. https://doi.org/10.1007/978-3-030-67175-4_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-67175-4_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-67174-7
Online ISBN: 978-3-030-67175-4
eBook Packages: MedicineMedicine (R0)