
Abstract
ls1 mardyn is a molecular dynamics (MD) simulation framework that enables investigations of multicomponent and multiphase processes relevant to engineering applications, such as droplet coalescence or bubble formation. These scenarios require the simulation of ensembles containing a large number of molecules. We present recent advances in ls1 mardyn both from the software design and high-performance computing perspective. From the former we describe the recently introduced plugin framework, from the latter we will look at some recent load balancing improvements to ls1 mardyn. We further present preliminary results of the integration of AutoPas, a C++ node-level library employing auto-tuning to achieve optimal node-level performance for particle simulations, into ls1 mardyn.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


1 Introduction
Molecular dynamics (MD) simulations have become a valuable tool for engineering applications. They rest on molecular models that describe the molecular interactions and encode the macroscopic behavior of matter. Equilibrium MD simulations thus enable sampling of thermodynamic properties in a consistent manner. Such data can be used to develop either fully predictive equations of state (EOS) or hybrid EOS, where simulation data are combined with experimental data [11]. An important advantage is that simulations can straightforwardly be carried out under extreme conditions, i.e. high temperatures and pressures, that are hardly accessible with experiments. Beside classical equilibrium scenarios, MD simulations can also be employed to investigate systems that are not in global equilibrium so that imposed gradients drive processes like droplet coalescence [17], bubble formation [12] or interfacial flows [13]. For many phenomena concerning multi-phase systems, the interface between the phases plays a key role. The spatial extent of the interface region is often only a few molecular diameters and can therefore only be resolved on the atomistic level. Employing molecular simulation, there are no additional modeling approaches, the physical processes evolve naturally and hence can be investigated unbiasedly. For many fluids that are relevant for engineering applications, comparatively simple molecular force field models have been developed, consisting of a few interaction sites, e.g.. Lennard Jones (LJ) sites considering the dispersive interaction and point charges, dipoles or quadrupoles to model the electrostatic interaction. A typical example is the mixture of acetone (four LJ sites, one dipole and one quadrupole) and nitrogen (two LJ sites and one quadrupole) which is frequently used to model fuel injection-like scenarios in thermodynamic laboratories. However, the present simulations were conducted with a simpler molecular model, consisting of a single LJ site. This model can be parametrized such that it mimics the thermodynamic behavior of noble gases like argon, krypton or xenon as well as methane [22]. This model is well suited for investigations focusing on the basic understanding of processes like the droplet coalescence so that it was considered in the present work.
In a long-term interdisciplinary effort of computer scientists and mechanical engineers, the MD framework ls1 mardyn has evolved over the last decade to investigate such large systems of small molecules [14]. ls1 mardyn has been used in various studies [21] and has been continuously extended to optimally exploit current HPC architectures [6, 18, 20]. In the following, we detail recent developments within the framework to achieve optimal performance at node and multi-node level. After introducing the actual problem setting of short-range molecular dynamics, related work and the original implementation of ls1 mardyn in Sect. 2, we introduce the newly developed plugin framework of ls1 mardyn in Sect. 3. Improvements to the MPI load balancing are shown in Sect. 4. We report preliminary results on the integration of AutoPas in ls1 mardyn in Sect. 5, which have been published in [8]. We close with a summary and an outlook to future work in Sect. 6.
2 Short-Range Molecular Dynamics
2.1 Theory
In short-range MD, Newton’s equations of motion are solved numerically [16]. In the following, considerations are restricted to small molecules. Due to their negligible conformational changes, molecules undergo translational or rotational motion; both are included in the equations of motion and are solved simultaneously in ls1 mardyn using a leapfrog time integrator, without the need for iterative procedures (such as the SHAKE algorithm) to handle geometric constraints [16].
Molecules interact via force fields. In short-range MD, arising forces are only explicitly accounted for if the distance between two considered molecules is below a specified cut-off radius \(r_c\). There are basically two variants to efficiently implement the cut-off condition: linked cells and Verlet lists [16]. Both methods turn the actual molecule-molecule interaction complexity from \(O(N^2)\) to O(N). In the Verlet list approach, a list of all molecules within a surrounding \(r_c+h\) is stored per molecule and updated regularly. Computing interactions thus reduces to traversing the list. The choice of h dictates the frequency of necessary list rebuilds on the one hand and the overall size of interaction search volume on the other hand. ls1 mardyn makes use of the linked cell approach: a Cartesian grid with cell sizes \(\ge r_c\) is introduced and covers the computational domain. The molecules are sorted into these cells. Molecular interactions only need to be considered for molecules that reside within the same cell or in neighboring cells.
All simulations reported in this contribution rest on the truncated and shifted form of the LJ potential [22]
with species-dependent parameters for size \(\sigma \) and energy \(\epsilon \) and the distance \(r_{ij}\) between molecules i and j, as well as the cutoff radius \(r_c\). Due to the truncation of the potential, no long range corrections have to be considered. This simplifies the treatment of multi-phase systems, where the properties of the interface can be strongly dependent on the cut-off radius [24]. The force calculation is typically by far the most expensive part of MD simulations that often contributes \(\ge 90\%\) to the overall compute time and hence is the preferential target for code optimizations.
2.2 Related Work
HPC and Related MD Implementations
Various packages efficiently and flexibly implement (short-range) molecular dynamics algorithms, with the most popular ones given by Gromacs,Footnote 1 LAMMPSFootnote 2 and NAMD.Footnote 3 Gromacs leverages particularly GPUs but also supports OpenMP and large-scale MPI parallelism, and it also exploits SIMD instructions via a new particle cluster-based Verlet list method [1, 15]. A LAMMPS-based short-range MD implementation for host-accelerator systems is reported in [2] with speedups for LJ scenarios of 3–4. A pre-search process to improve neighbor list performance at SIMD level and an OpenMP slicing scheme are presented in [10, 23]. The arising domain slices, however, need to be thick enough, to actually boost performance at shared-memory level. This restricts the applicability of the method to rather large (sub-)domains per process.
ls1 mardyn
An approach to efficient vectorization built on top of the linked cell data structure within ls1 mardyn is presented for single [5] and multi-siteFootnote 4 molecules [4]. This method, combined with a memory-efficient storage, compression and data management scheme [7], allowed for a four-trillion atom simulation in 2013 on the supercomputer SuperMUC, phase 1 [6]. A multi-dimensional, OpenMP-based coloring approach that operates on the linked cells is provided in [20]. The method has been evaluated on both Intel Xeon and Intel Xeon Phi architectures and exhibits good scalability up to the hyperthreading regime. ls1 mardyn further supports load balancing. It uses k-d trees for this purpose. Recently, this approach has been employed to balance computational load on heterogeneous architectures [18]. A detailed overview of the original release of ls1 mardyn is provided in [14]. Various applications from process and energy engineering, including several case studies that exploit ls1 mardyn, are discussed in [21]. Recently, ls1 mardyn was used to simulate twenty trillion atoms at up to 1.33 PFLOPS performance [19].
3 Plugin Framework
ls1 mardyn has many users with different backgrounds (process engineering, computer science) which have very differing levels of C++ knowledge. To implement new features, developers had to first understand considerable parts of the program before being able to contribute to the further development of ls1 mardyn. Additionally, most changes were done on a local copy or a private branch within the main simulation loop of the program or within some deeply coupled classes. Consequently, integrating the new code into the main source tree became a major difficulty and therefore often was rejected. If such changes were integrated anyways, they cluttered the source code and made it harder to understand. Moreover, new features often were not easily configurable and could only be disabled or enabled at compile-time.

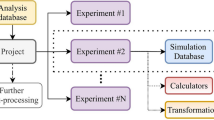
Extension points of ls1 mardyn through plugins. The extension points are marked by dashed arrows, normal steps of the simulation are shown with solid lines
To prevent the mentioned drawbacks, we have performed major code refactoring steps within ls1 mardyn to allow for both easier maintainability and extendability by introducing a plugin framework. Most user code can now be expressed as plugins that can be easily implemented, maintained, extended, integrated into ls1 mardyn and enabled upon startup of the simulation. Additionally, the user code is now mostly removed from the main simulation loop and main classes from ls1 mardyn, making maintainability more affordable and the code more readable.
ls1 mardyn provides a total of five different extension points that each prove their own purpose (c.f. Fig. 1):
- beforeEventNewTimeStep:
-
This extension point (EP) is used as legacy support for some older code parts. Mostly endStep can be used instead.
- beforeForces:
-
At this point the positions have been updated. Using this EP you can change positions of particles, for example to realign a droplet at the center of the domain.
- siteWiseForces:
-
This EP can be used to apply forces on specific sites of the molecules. One existing plugin uses it to implement a site-wise potential that prevents Lennard-Jones sites from moving through a wall.
- afterForces:
-
At this point additional forces to entire molecules can be added.
- endStep:
-
This step is mostly used for output. Most plugins only use this extension point.
Even though less than a year has passed since these changes were implemented (as of March 2019), we have already seen a lot of user code to actually find its way into the main source tree. Additionally, the user-base has provided very positive feedback on these changes, as their life got easier as well.
4 Load Balancing
In the previous report we presented preliminary results on the coalescence of two droplets with a diameter of \(d=50\,\)nm containing a number of \(N=10^6\) particles, cf. Fig. 2. These simulations were, however, only run on a fairly small amount of processes. When we tried scaling the simulation to more processes we discovered that the k-d tree-based load balancing implementation (kdd, see [3, 14, 18], Fig. 3) in ls1 mardyn at that point did not provide the performance we expected, as the load-unaware Cartesian domain decomposition (sdd, Fig. 4) outperformed the load-balancing kdd starting at around 32 nodes (see old, sdd in Fig. 5).

Snapshot of two argon droplets with a diameter of \(d=50\,\)nm containing a number of \(N=10^6\) particles in equilibrium with their vapor at a temperature of \(T=110\,\)K, rendered by the cross-platform visualization framework MegaMol [9]. It shows the time instance where a liquid bridge starts to grow, spanning over the initial gap of \(1\,\)nm between the droplets’ interfaces. The colors red and green were selected to be able to distinguish between particles that initially constituted either the left or right droplet. To provide a clear view through the vapor, particles were rendered with a diameter of \(\sigma /3\)

Space-partitioning using kdd. The different colors represent the different levels of the splitting hyperplanes

Space-partitioning using a 2-d Cartesian grid (sdd). Shown is a splitting into 12 subdomains
The kdd distributes the domain by splitting the overall domain into a grid of cells. A load \(c_\text {cell}\) is assigned to each cell. The grid is then split into N disjunct subdomains, such that each subdomain j contains roughly the same load
where \(C_\text {total}\) is the total combined cost for the entire domain
To get the loads per cell a load estimation model was used, that takes the number of particles in the current cell \(n_\text {cell}\) and its neighbors \(n_\text {neighbor cell}\) into account and uses a quadratic model:
The investigation of the observed performance drops showed that the distribution of the loads \(C_\text {subdomain}\) was appropriate, but the actual time spent on the calculations of a specific subdomain did not properly match the loads, indicating a poor estimation of the loads \(c_\text {cell}\). We henceforth introduced three additional load estimators:
- vecTuner:
-
This load estimator evaluates the time needed for each cell by doing a reference simulation at the beginning of the simulation. Therefore, for each particle count \(n_\text {cell}\) the time needed to calculate the interactions within a cell and the interactions across cells is measured.
- measureLoadV1:
-
This load estimator uses dynamic runtime measurements within the actual simulation. Therefore, the time needed to calculate all interactions within each process is measured. This time is the sum of the times needed for each cell, similar to Eq. (3):
$$\begin{aligned} T_\text {subdomain} = \sum _\text {cells in subdomain}t_\text {cell} \end{aligned}$$(5)The time for each cell \(t_\text {cell}\) cannot be easily measured, because these times are very small and exhibit a high level of noise and inaccuracy. Instead of determining the values \(t_\text {cell}\) we decided to introduce cell types to get better statistical properties. One typical cell type would be characterized by the number of particles per cell, but other characterizations are possible. Using the cell types, Eq. (5) becomes
$$\begin{aligned} T_\text {subdomain} = \sum _\text {cell types}n_\text {cell type}\cdot t_\text {cell type}. \end{aligned}$$(6)Assuming that the processes need the same amount of time for each cell of the same type, we can derive the matrix equation
$$\begin{aligned} \forall i:\quad T_i = \sum _j n_{i,j} \cdot t_j, \end{aligned}$$(7)where \(T_i\) is the time needed by process i, \(n_{i,j}\) is the amount of cells of type j within rank i and \(t_j\) is the time needed to calculate the interactions of cell type j. Hereby only \(t_j\) is an unknown and can thus be estimated by solving the matrix equation of the typically overdetermined system through a least squares fit. We are always using the characterization of cell type by particle number, i.e. cell type j resembles all cells with j particles.
- measureLoadV2:
-
This load estimator is based on measureLoadV1, but additionally assumes a quadratic dependency of \(t_j\) on the particle count j.
$$\begin{aligned} t_j = a_0 + a_1 \cdot j + a_2\cdot j^2 \end{aligned}$$(8)The resulting matrix equation
$$\begin{aligned} \forall i&:\quad T_i = \sum _j n_{i,j} \cdot \sum _{k=0}^2 j^k \cdot a_k \end{aligned}$$(9)$$\begin{aligned} \forall i&:\quad T_i = \sum _{k=0}^2 (\sum _j n_{i,j} \cdot j^k) \cdot a_k \end{aligned}$$(10)is then solved using a non-negative least squares algorithm to obtain \(a_k\).

The different load estimation techniques for a droplet coalescence scenario with 8 million particles using 8 OpenMP threads per rank and the full shell method

Comparison of the different scenarios for the vecTuner load estimator. The eight-shell method has been used for the kdd, marked by es in the legend

Time evolution of the droplet contour of the scenario with 25 million particles
A comparison of the results using the different load estimation techniques is shown in Fig. 5 for a droplet coalescence scenario with 3 million particles, showing a clear improvement of all new load estimators compared to the old one. While for 64 nodes a speedup of roughly 4x over the old load estimation techniques and an improvement of 2x over the standard domain decomposition (sdd) is visible, the sdd still performs best for large process counts. This is due to better communication schemes and sub-optimal load balancing even when using the new estimators with the kdd.
Scaling results using vecTuner for scenarios with 25 million and 200 million particles are shown in Fig. 6. For these scenarios the kdd always outperforms the standard domain decomposition if the new load estimators are used.
Simulations over a longer time-scale have been calculated for all three scenarios. For the scenario with 25 million particles, the evolution of the droplets is show in Fig. 7. In contrast to the previous simulations, the larger simulation was able to visualize the wiggling within the droplet formation nicely.
5 Preliminary Results: AutoPas Integration
Our work further concentrated on the integration of the C++ library AutoPas [8] into ls1 mardyn. The library employs auto-tuning to provide close to optimal node-level performance for particle simulations, which is expected to complement the distributed-memory load balancing approach. Early studies have shown successful automatic adaptions of the employed algorithms to both varying inputs as well as dynamically changing scenarios.

Spinodal decomposition scenario with 4 million particles calculated with ls1 mardyn and AutoPas. The images on the top show the end configuration of the system from the side (top left) and a slice of it (top right). The bottom figure shows the time needed for each iteration for two different shared-memory parallelization strategies. AutoPas is able to automatically choose between these two strategies
Figure 8 shows how AutoPas can already be used to calculate a spinodal decomposition scenario using ls1 mardyn. The simulation starts with a supercritical temperature and a density close to the critical one. Then, the temperature is controlled immediately to a temperature far below the critical one by the velocity scaling thermostat, so that the state of the fluid suddenly becomes physically unstable, resulting in the decomposition of the fluid into stable vapor and liquid phases (see Fig. 8, top). During the simulation the system thus changes from a homogeneous state to a very heterogeneous one. Looking at shared-memory parallelization strategies, in the beginning, while the system is still homogeneous, a load-unaware strategy can be used that simply splits the subdomain into even parts to be calculated by each thread of a cpu. Later on, when the system becomes increasingly heterogeneous a load-balancing strategy is needed. In the shown figure AutoPas is allowed to choose between two shared-memory parallelization strategies, here called traversals [19, 20]:
- c08:
-
This traversal uses coloring to split the domain into multiple groups of cells (colors), where calculation on all cells in one group can be done in parallel without any data races. The cells of each color are then distributed to the threads using OpenMP’s dynamic scheduling. After one color is finished, the next color is started.
- sli:
-
The sliced traversal (sli) slices the domain into multiple equally sized subdomains. Each subdomain is then calculated by one thread. Locks are employed to prevent data races.
Henceforth, the c08 traversal is better suited for heterogeneous scenarios, as it provides dynamic scheduling, while the sli traversal is better suited for homogeneous scenarios, as it uses less overhead. As expected, AutoPas switches the shared-memory parallelization strategy for the mentioned scenario at time step \(\sim \)9 000 from sli to c08.
6 Summary and Outlook
We have outlined recent progress in usability (plugin concept), load balancing (kdd-based decomposition and load estimation approaches) and auto-tuning (library AutoPas) to improve the molecular dynamics software ls1 mardyn. Load balancing improvements enabled unprecedented large-scale droplet coalescence simulations leveraging the supercomputer Hazel Hen. Yet, more work and effort is required to improve scalability of the scheme beyond O(200) nodes. The auto-tuning approach we follow by the integration of AutoPas appears promising in terms of both scenario as well as hardware-aware HPC algorithm adoption. More work in this regard is in progress, focusing amongst others on the incorporation of Verlet list options and different OpenMP parallelization schemes.
Notes
- 1.
- 2.
- 3.
- 4.
Molecules that consist of several interaction sites, e.g.. two LJ sites.
References
M. Abraham, T. Murtola, R. Schulz, S. Páll, J. Smith, B. Hess, E. Lindahl, GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015)
W. Brown, P. Wang, S. Plimpton, A. Tharrington, Implementing molecular dynamics on hybrid high performance computers—short range forces. Comput. Phys. Commun. 182(4), 898–911 (2011)
M. Buchholz, Framework zur Parallelisierung von Molekulardynamiksimulationen in verfahrenstechnischen Anwendungen. Dissertation, Institut für Informatik, Technische Universität München, 2010
W. Eckhardt, Efficient HPC implementations for large-scale molecular simulation in process engineering. Dissertation, Dr. Hut, Munich, 2014
W. Eckhar, A. Heinecke, An efficient Vectorization of Linked-Cell Particle Simulations, in ACM International Conference on Computing Frontiers (ACM, New York, NY, USA, 2012) pp. 241–243
W. Eckhardt, A. Heineck, R. Bader, M. Brehm, N. Hammer, H. Huber, H.G. Kleinhenz, J. Vrabec, H. Hasse, M. Horsch, M. Bernreuther, C. Glass, C. Niethammer, A. Bode, H.J. Bungartz, 91 TFLOPS Multi-trillion Particles Simulation on SuperMUC (Springer, Berlin, Heidelberg, 2013), pp. 1–12
W. Eckhardt, T. Neckel, Memory-efficient implementation of a rigid-body molecular dynamics simulation, in Proceedings of the 11th International Symposium on Parallel and Distributed Computing (ISPDC 2012) (IEEE, Munich 2012) , pp. 103–110
F.A. Gratl, S. Seckler, N. Tchipev, H.J. Bungartz, P. Neumann, Autopas: auto-tuning for particle simulations, in 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS) (2019)
S. Grottel, M. Krone, C. Müller, G. Reina, T. Ertl, MegaMol—a prototyping framework for particle-based visualization. IEEE Trans. Visual Comput. Graph. 21(2), 201–214 (2015)
C. Hu, X. Wang, J. Li, X. He, S. Li, Y. Feng, S. Yang, H. Bai, Kernel optimization for short-range molecular dynamics. Comput. Phys. Commun. 211, 31–40 (2017)
A. Köster, T. Jiang, G. Rutkai, C. Glass, J. Vrabec, Automatized determination of fundamental equations of state based on molecular simulations in the cloud. Fluid Phase Equilib. 425, 84–92 (2016)
K. Langenbach, M. Heilig, M. Horsch, H. Hasse, Study of homogeneous bubble nucleation in liquid carbon dioxide by a hybrid approach combining molecular dynamics simulation and density gradient theory. J. Chem. Phys. 148, 124702 (2018)
G. Nagayama, P. Cheng, Effects of interface wettability on microscale flow by molecular dynamics simulation. Int. J. Heat Mass Transf. 47, 501–513 (2004)
C. Niethammer, S. Becker, M. Bernreuther, M. Buchholz, W. Eckhardt, A. Heinecke, S. Werth, H.J. Bungartz, C. Glass, H. Hasse, J. Vrabec, M. Horsch, ls1 mardyn: the massively parallel molecular dynamics code for large systems. J. Chem. Theory Comput. 10(10), 4455–4464 (2014)
S. Páll, B. Hess, A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 184(12), 2641–2650 (2013)
D. Rapaport, The Art of Molecular Dynamics Simulation (Cambridge University Press, Cambridge, 2004)
L. Rekvig, D. Frenkel, Molecular simulations of droplet coalescence in oil/water/surfactant systems. J. Chem. Phys. 127, 134701 (2007)
S. Seckler, N. Tchipev, H.J. Bungartz, P. Neumann, Load balancing for molecular dynamics simulations on heterogeneous architectures, in 2016 IEEE 23rd International Conference on High Performance Computing (HiPC) (2016), pp. 101–110
N. Tchipev, S. Seckler, M. Heinen, J. Vrabec, F. Gratl, M. Horsch, M. Bernreuther, C.W. Glass, C. Niethammer, N. Hammer, B. Krischok, M. Resch, D. Kranzlmüller, H. Hasse, H.J. Bungartz, P. Neumann, Twetris: twenty trillion-atom simulation. Int. J. High Perform. Comput. Appl. 1094342018819,741 (0). DOI https://doi.org/10.1177/1094342018819741
N. Tchipev, A. Wafai, C. Glass, W. Eckhardt, A. Heinecke, H.J. Bungartz, P. Neumann, Optimized force calculation in molecular dynamics simulations for the Intel Xeon Phi (Springer International Publishing, Cham, 2015), pp. 774–785
J. Vrabec, M. Bernreuther, H.J. Bungartz, W.L. Chen, W. Cordes, R. Fingerhut, C. Glass, J. Gmehling, R. Hamburger, M. Heilig, M. Heinen, M. Horsch, C.M. Hsieh, M. Hülsmann, P. Jäger, P. Klein, S. Knauer, T. Köddermann, A. Köster, K. Langenbach, S.T. Lin, P. Neumann, J. Rarey, D. Reith, G. Rutkai, M. Schappals, M. Schenk, A. Schedemann, M. Schönherr, S. Seckler, S. Stephan, K. Stöbener, N. Tchipev, A. Wafai, S. Werth, H. Hasse, Skasim—scalable hpc software for molecular simulation in the chemical industry. Chem. Ing. Tech. 90(3), 295–306 (2018)
J. Vrabec, G.K. Kedia, G. Fuchs, H. Hasse, Comprehensive study of the vapour-liquid coexistence of the truncated and shifted lennard-jones fluid including planar and spherical interface properties. Mol. Phys. 104(9), 1509–1527 (2006)
X. Wang, J. Li, J. Wang, X. He, N. Nie, Kernel optimization on short-range potentials computations in molecular dynamics simulations. (Springer, Singapore, 2016) , pp. 269–281
S. Werth, G. Rutkai, J. Vrabec, M. Horsch, H. Hasse, Long-range correction for multi-site lennard-jones models and planar interfaces. Mol. Phys. 112(17), 2227–2234 (2014)
Acknowledgements
The presented work was carried out in the scope of the Large-Scale Project Extreme-Scale Molecular Dynamics Simulation of Droplet Coalescence, acronym GCS-MDDC, of the Gauss Centre for Supercomputing. Financial support by the Federal Ministry of Education and Research, project Task-based load balancing and auto-tuning in particle simulations (TaLPas), grant numbers 01IH16008A/B/E, is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this paper

Cite this paper
Seckler, S. et al. (2021). Load Balancing and Auto-Tuning for Heterogeneous Particle Systems Using Ls1 Mardyn. In: Nagel, W.E., Kröner, D.H., Resch, M.M. (eds) High Performance Computing in Science and Engineering '19. Springer, Cham. https://doi.org/10.1007/978-3-030-66792-4_35
Download citation
DOI: https://doi.org/10.1007/978-3-030-66792-4_35
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-66791-7
Online ISBN: 978-3-030-66792-4
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)