
Abstract
The concept that sleep is regulated by humoral factors dates back to the very beginning of experimental sleep research. Attempts to find a single key “hypnotoxin” which by itself would be responsible for eliciting sleep were unsuccessful, but the search led to the identification of several hormones and immune modulators with sleep-promoting properties. Modern theories about humoral sleep-promoting factors posit that these molecules constitute a complex signaling network, with significant overlaps and redundancies. Major sources of these signals are metabolic and immune organs, and the main function of this complex regulatory system is to help align sleep-wake activity with the metabolic and immune status of the organism. In the present chapter, we review the best characterized humoral sleep signals and the emerging role of metabolic tissues and the intestinal microbiota in sleep regulation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Interleukin
- Tumor necrosis factor
- Cholecystokinin
- Ghrelin
- Leptin
- Prolactin
- Prostaglandins
- Short-chain fatty acids
- Butyrate
- Brown adipose tissue
- White adipose tissue
- Liver
- Metabolism
- Inflammation
- Macrophages
- Feeding
- Fasting
- Endotoxin
- Vagus
- Microbiota
- Gut-brain signaling
The idea that circulating “humors” may be the ultimate cause of sleep dates back to ancient scientists and philosophers, but only at the beginning of the last century was the concept first addressed experimentally. With the advance of experimental techniques and the discovery of the first hormone, secretin, the search for humoral sleep factor(s) appeared to be technically possible and theoretically justified. Kuniomi Ishimori in Japan and Henri Pieron in France independently demonstrated that injection of brain extracts or cerebrospinal fluid (CSF) from sleep-deprived dogs to non-sleep-deprived recipients causes robust increases in behavioral sleep [1, 2]. Pieron named the putative sleep-inducing substance “hypnotoxin. ” Schnedorf and Ivy reproduced Pieron’s findings which gave further support to the hypnotoxin theory and inspired further search for the elusive sleep factor [3]. Kornmüller in 1961 and Monnier in 1963 used parabiotic cats and rabbits and demonstrated that thalamic stimulation of the donor elicits increased cortical slow-wave activity and sleep in the recipient animals [4, 5]. These experiments led to the isolation of delta sleep-inducing peptide (DSIP) [6]. A Japanese group led by Shojiro Inoue extracted a sleep-promoting substance from the brainstem of sleep-deprived rats in the early 1970s, and later they identified one component as uridine and another component as the oxidized form of glutathione (reviewed in [7]).
The above attempts led to the identification of substances with variable and generally modest effects on sleep, and the field all but gave up on pursuing their further investigation. One series of studies, however, left a lasting effect on sleep research. In the late 1960s, Pappenheimer and his coworkers undertook a series of studies on extracting sleep-promoting substance, which was initially found in the CSF of sleep-deprived goats [8], subsequently in bovine and rabbit brains and CSF, as well as in human urine (reviewed in [9]). The somnogenic component of the sleep-promoting material was identified as muramyl peptides [10]. Since muramyl peptides are bacterial cell wall fragments, not produced by eukaryotes, there was a general skepticism about their physiological role as endogenous sleep-promoting substances. Their identification as somnogenic substances, however, led to further investigation of their actions and to the important discovery that proinflammatory cytokines , endogenous peptides produced mainly by macrophages exposed to bacterial cell wall components, possess strong sleep-promoting activities. Furthermore, the recent recognition of the microbiota as a source of brain signaling and the recognition that products of the intestinal bacteria translocate to the internal environment of the host put the possible relevance of bacterial products in sleep regulation in a new perspective.
Proinflammatory Cytokines
It has been known since the 1950s that systemic infections and administration of components of bacteria, such as endotoxin, elicit fever through the stimulation of the production of a circulating pyrogen, which was called “endogenous pyrogen” at that time. Two lines of evidence suggested that the endogenous pyrogen could also be a sleep-inducing factor. One, in addition to fever, acute systemic infections are also characterized by increased sleepiness. Two, cell wall components of bacteria that elicit sleep also stimulate the production of endogenous pyrogen by immune cells.
In the 1980s, three laboratories characterized the sleep effects of purified endogenous pyrogen obtained from the supernatants of macrophages or astrocytes activated by heat-killed bacteria. Krueger and coworkers demonstrated that intravenous and intracerebroventricular (icv) administration of purified endogenous pyrogens induce prolonged increases in non-rapid eye movement sleep (NREMS) and fever in rabbits [11]. The effects were attributed to interleukin-1 (IL-1), which was the only known endogenous pyrogen at that time, but the actual structure of the active component of the injected sample was unknown. Tobler and coworkers demonstrated that icv administration of astrocyte-derived endogenous pyrogen elicits fever and enhanced slow-wave activity of the electroencephalogram (EEG), a measure of sleep intensity [12]. In cats, icv administration of purified human IL-1 caused dose-dependent changes in sleep, the lower doses being somnogenic, while higher doses wake-promoting [13].
Subsequent studies using recombinant IL-1 confirmed that central or systemic administration of IL-1 induces NREMS in rabbits [14], rats [15], and mice [16]. Circulating IL-1β acts on a peripheral target to induce sleep as evidenced by the effectiveness of vagotomy to block the somnogenic actions of moderate doses of systemically administered IL-1β [17, 18]. However, when high doses of IL-1β are administered peripherally or directly into the cerebral ventricles or brain tissue, it also has the potential to act on central sleep-promoting mechanisms. The site of the central actions is unclear, but the locus ceruleus [19], dorsal raphe [20], prostaglandin D2 (PGD2)-sensitive basal forebrain region [21], and median preoptic area [22] have been suggested, along with growth hormone-releasing hormone-receptive [23] and serotonergic mechanisms [24, 25]. The role of prostaglandins in IL-1β-induced fever has been well established, but their contribution to its somnogenic actions is controversial [26, 27]. Extensive evidence indicates, however, that sleep increases in response to IL-1β are not a direct consequence of its pyrogenic actions (reviewed in [28]).
In the second half of the 1980s, it became apparent that IL-1 is not the only endogenous pyrogen. White blood cells that are activated by components of the bacterial cell wall secrete other bioactive peptides into the circulation that induce fever and have complex effects on immune functions (reviewed in [29]). Screening of these peptides revealed that interferon α2 (IFNα2) [30] and tumor necrosis factor-α (TNFα) [14, 31] also have NREMS-promoting properties.
TNFα, similarly to IL-1β, is a proinflammatory cytokine with multiple target sites and complex effects on immune functions and metabolism. Its primary source is the immune cells, such as macrophages, dendritic cells, and T lymphocytes, but it is also expressed in the brain by microglia, astrocytes, and neurons [32,33,34]. After the identification of TNFα as another endogenous pyrogen produced by activated white blood cells [35], it was soon established that systemic or central administration of TNFα induces inflammatory responses, including sickness response characterized by increased sleep, fever, and loss of appetite (reviewed in [36]). The NREMS-promoting effects of TNFα are mediated by the TNF receptor 1 [16] and have been described in rabbits, sheep, mice, and rats [14, 16, 26, 31, 37]. TNFα production also correlates with sleep-wake activity. In rats, hypothalamic levels of TNFα mRNA and TNFα protein are higher during the rest phase [38, 39], and sleep loss elevates TNFα mRNA expression in the cerebral cortex and basal forebrain [40].
Similarly to IL-1β, vagotomy blocks the sleep-promoting effects of systemically administered TNFα pointing to a peripheral site of action [41, 42]. Direct delivery of TNFα into the brain also induces NREMS, possibly acting in the locus ceruleus [19], the preoptic region [43], and the PGD2-sensitive, sleep-promoting zone of the basal forebrain [21]. Nitric oxide and prostaglandins have an established role in mediating the sleep effects of TNFα [26, 44, 45]. While the role of increased secretion of proinflammatory cytokines in sleep responses to systemic infections and inflammatory processes is widely accepted, their contribution to the regulation of sleep-wake activity under healthy conditions appears to be less clear. The role of endogenous TNFα in sleep regulation was first studied by using anti-TNF antibodies, TNF-binding protein, and fragments of TNF receptor 1. The general outcome of those experiments was that molecules that bind to and neutralize endogenous TNFα decrease the time spent in NREMS, which pointed to the possibility that TNFα may contribute to the maintenance of spontaneous sleep (reviewed in [46]). Subsequent advances in TNFα biology revealed further complexities in TNFα biochemistry that may necessitate the reinterpretation of the initial findings. TNFα is a transmembrane protein which can function in its membrane-bound form, or it is proteolytically cleaved with the subsequent release of the trimeric soluble TNFα, which is the form of TNF present in the extracellular environment, including the plasma (reviewed in [34]). Antibodies and receptor fragments which were used in sleep studies (e.g., [47, 48]) not only bind and neutralize soluble TNFα molecules but also are ligands for membrane-bound TNF and may, in fact, trigger increased TNFα secretion (reviewed in [49]).
The role of endogenous TNFα in sleep regulation has also been addressed by using mouse strains with deficient TNF signaling. Studies with TNF receptor-deficient mice led to disparate findings on their sleep behavior. Both reduced and unchanged NREMS were reported in TNF receptor 1 knockout (TNF R1 KO) mice [16, 50] and TNF R1 and R2 double-KO mice [51, 52]. TNF R2 mice had decreased rapid eye movement sleep (REMS) but normal amounts of NREMS [50]. Deficiency of the receptor ligand, as demonstrated in TNFα KO [53] and TNFα and lymphotoxin-α double-KO mice [50], does not result in reduced NREMS. Regarding REMS, somewhat consistent results have been obtained in experiments using exogenous TNFα administration and in studies on spontaneous sleep in KO models. Icv or systemic injection of TNFα suppresses REMS in mice, rats, and rabbits [14, 41, 54], while REMS is increased in both TNF R1 and R2 double-KO mice [51] and in TNFα KO mice [53]. This suggests that endogenous TNFα may have a tonic REMS-suppressive activity.
Acute and chronic sleep loss or sleep fragmentation results in a proinflammatory state manifested as increased expression of IL-1β and TNFα and activation of NF-κB signaling in the brain and elevated circulating proinflammatory cytokine levels (e.g., [55,56,57,58,59,60,61]). Sleep rebound after sleep deprivation is attenuated by a TNF receptor fragment and anti-IL-1β antibodies [62, 63] and in TNF R1 and IL-1 R1 double-KO mice [64] suggesting a possible role for proinflammatory cytokines in response. Studies using TNF receptor and ligand KO models, however, did not confirm that TNF signaling is necessary for these rebound responses [50, 53].
In summary, TNFα and IL-1β have the potency to increase NREMS acting on both peripheral and central targets. A few cytokine-sensitive brain sites have been identified, but additional studies will be required to construct a comprehensive map of the central targets. Attempts to acutely neutralize soluble TNF and IL-1 and the constitutive KO models all have their limitations. To gain a clear picture of the role of endogenous cytokines in maintaining spontaneous sleep will require the use of inducible KO models or pharmacological approaches that do not have the potential to stimulate the release of proinflammatory cytokines. Three of the endogenous pyrogens, IFNα, TNFα, and IL-1β, show striking similarities in their effects on body temperature and NREMS. Endogenous prostaglandins provide a unifying mechanism for the pyrogenic actions of these molecules [29]. The identification of similar unifying mechanism for their somnogenic actions would provide a valuable insight into fundamental mechanisms of sleep regulation.
Adipokines
In addition to its role in energy storage, the white adipose tissue is also recognized as an endocrine organ. Hormones secreted by the adipose tissue are collectively referred to as adipokines. Adipokines, such as leptin, adiponectin, and resistin, have diverse effects on metabolism by acting on multiple tissues, including the brain (reviewed in [65]). Leptin is a 167-amino acid peptide produced by white adipose tissue in proportion to the amount of body fat, thereby reflecting the status of long-term energy stores. The most widely recognized action of leptin is its potency to reduce body weight by decreasing food intake and increasing energy expenditure, but leptin may also provide a link between metabolism and the regulation of sleep. Systemic or icv administration of leptin increases NREMS in rats, suggesting that leptin released from the adipose tissue may be a sleep-promoting molecule [66, 67]. Obesity, however, leads to increased sleep in rats and mice independently of leptin as shown by increased sleep amounts in obese rodents with deficient leptin signaling [68,69,70,71]. A major source of circulating TNFα is the white adipose tissue; thus TNFα is often regarded as an adipokine. Within adipose tissue, macrophages account for nearly all TNFα production [72]. Circulating TNFα concentration rises with increasing obesity and correlates with insulin resistance [73]. Increased sleep observed in obese rodents may be linked to enhanced production of TNFα [68,69,70, 74, 75].
In addition to adipokines, lipolysis itself may result in the production of sleep-promoting signals. In nocturnal rodents, the feeding period (dark phase) is characterized by lipogenesis, while in the rest phase (light), when feeding is minimal, lipolysis provides the main energy source. By using sequential infusions of lipolytic and lipogenic hormones, the lipogenic and lipolytic phases can be reversed in these nocturnal animals, which leads to the complete reversal of the sleep-wake cycles to a diurnal pattern [76]. Further, restricting feeding to the light period also causes the reversal of the lipolytic and lipogenic phases with the concomitant reversal of the sleep-wake pattern [77,78,79,80]. In addition, strong lipolytic signals, such as IL-1β, TNFα, epinephrine, β3-adrenergic receptor (β3-AR) activation, and 2-deoxy glucose, all enhance NREMS [14, 37, 53, 81,82,83].
Gastrointestinal Hormones
The relationship between sleep and energy balance has long been recognized. In general, feeding and positive energy states facilitate sleep, while fasting and negative energy balance induce wakefulness (e.g., [74, 84,85,86]). This relationship pointed to the possible role key metabolic organs as well as satiety and orexigenic hormones in sleep signaling. The best characterized satiety hormone is cholecystokinin (CCK), a product of the “I” enteroendocrine cells of the small intestines, which is produced postprandially in response to dietary fat and protein, and its systemic administration suppresses feeding [87]. The actions of ghrelin, a product of the X/A-like cells of the gastric mucosa, are the opposite of those of CCK. Ghrelin secretion increases under conditions of negative energy balance, such as starvation and cachexia, and declines under conditions of positive energy balance, such as feeding and obesity [88]. Ghrelin is the only known peripheral hormone that stimulates feeding [89]. In addition to their gastrointestinal sources, ghrelin and CCK are also produced by neurons in the brain, and their receptors are widely expressed both in the periphery and the central nervous system [87, 90]. Ghrelin and CCK have antagonistic actions not only on feeding but also on sleep. CCK was first identified as a sleep-inducing hormone in cats; two pioneering studies demonstrated that feeding or intraduodenal administration of fat elicits sleep, and the sleep-inducing effects were reproduced by the iv administration of CCK [91, 92]. Subsequently, NREMS-promoting effects of systemically injected CCK were also described in rats [93, 94], rabbits [95], and mice [96]. CCK elicits the complete behavioral sequence of satiety, and its effects are indistinguishable from the signs of naturally occurring postprandial state [97, 98]. REMS amounts are not affected by CCK in intact animals, but in para-chlorophenylalanine-induced insomniac cats, CCK restores REMS [99], and in normal rats, CCK increases REMS frequency [100] and decreases REMS latency [98].
Studies using CCK2 agonists and CCK1 receptor antagonist indicate that CCK2 receptor activation is not sufficient, but CCK1 receptor activation is necessary for the somnogenic effects of CCK [101,102,103]. Spontaneous sleep of CCK1 receptor-deficient rats is not altered, suggesting that the activation of the receptor is not required for maintaining baseline sleep [104], but pharmacological blockade of CCK1 receptors abolished feeding-induced sleep, indicating a role for CCK in postprandial sleep increases [105].
Central administration of ghrelin triggers the dark onset syndrome, the behavioral sequence characteristic of the first hours of the active period in nocturnal rodents, including long bouts of wakefulness and increased feeding activity [106, 107]. In microinjection studies, the lateral hypothalamus, medial preoptic area, and hypothalamic paraventricular nucleus were identified as potential target sites for the wake-inducing actions of ghrelin [106]. It has been proposed that a hypothalamic circuit, containing ghrelin – orexin – neuropeptide-Y neurons, forms a shared mechanism for both the orexigenic and wake-promoting effects of orexigenic signals [108].
The extent to which peripheral ghrelin-sensitive targets are also involved in the wake-promoting actions of the hormone is unclear. In rats and mice, systemic administration of ghrelin induced both wakefulness [109] and sleep [110] or had no effect on vigilance [107]. In humans, intravenous administration of ghrelin also led to inconclusive findings. The most potent known synthetic agonist of the ghrelin receptors, hexarelin, suppressed deep sleep [111], whereas ghrelin itself was either wake- or sleep-promoting or had no effect depending on the gender of the subjects and the treatment paradigm [112,113,114,115]. Ghrelin receptor KO mice or mice with the deletion of the GHRL gene that encodes the peptide preproghrelin, a precursor to ghrelin and obestatin, have more fragmented sleep, but the amount of sleep and the rebound sleep response after sleep loss do not show any alteration suggesting that ghrelin signaling is not key to maintaining baseline sleep or to homeostatic sleep responses [116, 117]. Ghrelin signaling, however, plays a role in arousal responses to several physiological stimuli. Exposure to novel environment or fasting suppresses sleep in mice, a response that is absent in ghrelin receptor KO animals [117]. Furthermore, ghrelin plays a role in integrating sleep and thermoregulatory responses to metabolic challenges. In response to fasting at low ambient temperatures, wild-type mice enter short torpor bouts, but preproghrelin knockout mice develop protracted hypothermia associated with reduced sleep, which culminates in a marked drop in body temperature to near-ambient levels [118]. The sleep promoting effects of proinflammatory stimuli, such as lipopolysaccharide, are accentuated in preproghrelin KO mice suggesting that ghrelin-associated arousal circuits may have a role to balance sleep-wake activity in inflammatory conditions [119].
Prolactin
Circulating levels of most pituitary hormones show 24-h rhythms and thus correlate with sleep-wake cycles, and exogenous administration of some of these hormones affects sleep under certain conditions. These findings, while suggestive, do not necessarily indicate a role for a hormone in the regulation of sleep. The most complete set of evidence that a pituitary hormone may be a physiological modulator of sleep, mainly REMS, and therefore could be considered a humoral sleep factor has accumulated for prolactin. Plasma prolactin levels increase after sleep onset, independent of the time of the day when sleep occurs, suggesting that prolactin secretion is regulated by sleep-wake activity rather than circadian factors (reviewed in [120]).
Exogenous administration of prolactin induces selective REMS increases in rats and rabbits [121,122,123]. Stimulation of prolactin secretion by vasoactive intestinal peptide or by prolactin-releasing peptide and the induction of hyperprolactinemia by transplanted pituitary grafts led to increases in REMS [123,124,125]. Anti-prolactin antibodies suppress REMS in rats [126, 127]. In mutant hypoprolactinemic rats, REMS is suppressed during the light period [128]. Prolactin-deficient transgenic mice have reduced amounts of spontaneous REMS and diminished REMS rebound after sleep deprivation [129].
Increased circulating prolactin levels correlate with enhanced REMS amount in pregnancy/pseudopregnancy and during recovery after exposure to certain stressors, thus suggesting that prolactin may play a role in REMS responses in these conditions [130,131,132,133]. In fact, ether stress-induced REMS is diminished after anti-prolactin antibody treatment or in prolactin KO animals [129, 134].
Sleep Signaling by Metabolic Organs
The findings that phase shifts in metabolic processes greatly influence the diurnal distribution of sleep and wakefulness and that positive and negative energy balances affect sleep the opposite way suggested that signals from key metabolic organs may play a role in sleep regulation. Adipose tissue plays a central role in the interplay between nutrition, energy balance, and sleep. Two types of adipose tissue with fundamentally different functions exist. White adipose tissue is responsible for storing excess energy in the form of fat, whereas brown adipose tissue (BAT) dissipates energy as heat. The tightly regulated balance between the activities of the two fat tissues is critical in maintaining metabolic homeostasis [135]. BAT controls energy balance via regulated (adaptive) heat production. The thermogenic property of BAT is conferred by the tissue-specific presence of uncoupling protein 1 (UCP-1). UCP-1 is a H+ transporter on the inner mitochondrial membrane of brown adipocytes [136]. The thermogenic activity of the BAT is regulated by the sympathetic nervous system through β3-ARs expressed by brown adipocytes. Stimulation of BAT by β3-AR agonists elicits robust sleep increases [53, 81, 137], which is absent or attenuated in UCP-1 KO mice [81] indicating that thermogenic activation of BAT promotes sleep. The somnogenic actions of BAT activation are independent of changes in core body temperature and are mediated by capsaicin-sensitive afferents arising from the organ [81]. Systemic inflammation or warm ambient temperature-induced sleep and rebound sleep increases after sleep loss are abolished or attenuated in UCP-1 KO mice indicating that BAT-derived somnogenic signaling plays a role in sleep responses to systemic inflammation and thermoregulatory challenges, and it is required for the full activity of homeostatic sleep regulation [54, 138].
In the immune system, macrophages provide a significant source of sleep-inducing signals. After demonstrating the sleep-inducing effects of endogenous pyrogen(s), several studies focused on the role of macrophages in sleep responses during clinically manifest infections. It was determined that macrophages play a role in the production and metabolism of somnogenic substances, inflammatory cytokines, after microbial challenge [139, 140]. Subsequently, it was demonstrated that macrophages also play a role in sleep signaling under physiological conditions. Depletion of the peripheral macrophage pool by clodronate-containing liposomes suppresses rebound sleep responses after sleep loss in mice and macrophage-depleted animals unable to maintain normal sleep amounts when exposed to moderately cold temperatures. These findings indicate that in the absence of an inflammatory challenge, under normal physiological conditions, macrophage function/signaling is required for maintaining normal sleep [141]. The lack of alternatively activated (M2) macrophage subpopulation leads to similar deficiencies in sleep as the complete macrophage deficiency, which suggests a central role for M2 cells in sleep signaling [142].
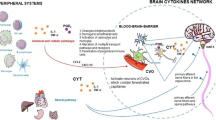
The liver has a central role in the regulation of metabolism and immune defenses. It harbors about 80% of the body’s macrophage population and is responsible for removing circulating microbial molecules, such as endotoxin and other products of the microbiota or intruding pathogens. The first direct evidence about the role of liver in sleep signaling came for studies demonstrating that local, passive warming of the organ results in increased NREMS [143]. Depletion of liver macrophages diminishes feeding-induced sleep [144] and recovery sleep responses after sleep loss [141]. More recently it was demonstrated that the hepatoportal region contains a sleep-promoting viscerosensory mechanism which is sensitive to butyrate, a short-chain fatty acid, a product of the intestinal microbiota [145]. This finding suggests that products of the intestinal microbiota, after translocating from the gut lumen to the portal circulation, may enhance sleep by acting in the hepatoportal region (Fig. 11.1). Consistent with this notion are the findings that the depletion of the intestinal microbiota results in decreased sleep in rats and mice [146, 147].

Hepatoportal viscerosensory sleep signaling. The effects of commensal bacteria on extra-intestinal organs, including the brain, are explained by the exchange of microbial molecules to virtually all tissues of the body. Bacterial translocation is defined as the migration of viable bacteria or bacterial products from the intestinal lumen to the portal circulation. During bacterial growth, division, and death, components of bacterial cell wall are released and translocated through the intestinal microcirculation to the portal blood in biologically significant quantities. Fragments of the bacterial cell wall, such as lipopolysaccharide, and bacterial metabolites, such as short-chain fatty acids, are detected in the portal and systemic circulation even under physiological conditions [155,156,157]. Low doses of intraportally, but not systemically, administered short-chain fatty acids and lipopolysaccharide induce robust sleep increases, indicating that the hepatoportal region is a privileged site for the somnogenic actions of microbial products [145, 158]. Portally circulating microbial molecules reach the liver, which may act directly on hepatic afferents or may activate liver macrophages and other hepatic cells to secrete bioactive molecules, such as prostaglandins and tumor necrosis factor-α. These secretory products may reach brain sleep circuits through the systemic circulation, or they may also act on local sensory nerves
It has been proposed that somnogenic signals for the liver reach core sleep circuits of the brain via sensory vagus innervation. The role of vagus in peripheral sleep signaling has been known for long. For example, stimulation of the vagus elicits EEG synchronization and complete sleep cycles [148,149,150]. Vagotomy attenuates or abolishes the sleep-promoting actions of systemically administered lipopolysaccharide, IL-1β, and TNFα [17, 18, 41, 42, 151].
Conclusions
For long, it has been thought that sleep is of the brain, by the brain, and for the brain [152]. In light of the recent advances in the field and after careful reinterpretation of old findings, this notion needs revision. Sleep is a complex behavior, in which the entire organism, including the brain, participates. Core circuits of sleep regulation, just like core circuits of all behavioral manifestations, are located in the brain. These circuits receive extensive ascending somatic and visceral inputs from metabolic organs and the immune and endocrine systems through humoral and neural pathways (Fig. 11.2). The significance of these inputs is to help align the timing and intensity of sleep behavior with the actual metabolic status of the body. Sleep loss and misalignment between sleep and the circadian system have a negative impact not only on the brain but on the function of metabolic organs and immune system [153, 154].

Integration of peripheral sleep-promoting signals with core sleep circuits in the brain. Metabolic tissues, such as the brown adipose tissue, and the liver as well as cells of the immune system and intestinal bacteria generate sleep-promoting signals. Somnogenic signals may be carried to core sleep circuits of the brain by the circulation or by the activation of neuronal afferent pathways. The hypothalamus, where the best characterized sleep circuits are located, and the brainstem, where visceral afferent systems project, are likely targets of peripherally produced sleep-inducing signals
References
Kubota K. Kuniomi Ishimori and the first discovery of sleep-inducing substances in the brain. Neurosci Res. 1989;6(6):497–518.
Pieron H, Legendre R. Recherches sur le besoin de sommeil consecutif a une veille prolongée. Z Allg Physiol. 1913;14:235–62.
Schnedorf JG, Ivy AC. An examination of the hypnotoxin theory of sleep. Am J Physiol. 1939;25(4):491–505.
Kornmüller AE, Lux HB, Winkel K, Klee M. Neurohumoral ausgelöste Schlafzustände an Tieren mit gekreuztem Kreislauf unter der Kontrolle von EEG-Ableitungen. Naturwissenschaften. 1961;48:503–5.
Monnier M, Koller TH, Graber S. Humoral influences of induced sleep and arousal upon electrical brain activity of animals with crossed circulation. Exp Neurol. 1963;8:264–77.
Schoenenberger GA, Maier PF, Tobler JH, Monnier M. A naturally occurring delta-EEG enhancing nonapeptide in rabbits. X. Final isolation, characterization and activity test. Pflugers Arch. 1977;369(2):99–109.
Inoué S, Honda K, Komoda Y. Sleep as neuronal detoxification and restitution. Behav Brain Res. 1995;69(1–2):91–6.
Pappenheimer JR, Miller TB, Goodrich CA. Sleep-promoting effects of cerebrospinal fluid from sleep- deprived goats. Proc Natl Acad Sci U S A. 1967;58(2):513–7.
Krueger JM, Karnovsky ML. Sleep as a neuroimmune phenomenon: a brief historical perspective. Adv Neuroimmunol. 1995;5(1):5–12.
Krueger JM, Karnovsky ML, Martin SA, Pappenheimer JR, Walter J, Biemann K. Peptidoglycans as promoters of slow-wave sleep. II. Somnogenic and pyrogenic activities of some naturally occurring muramyl peptides; correlations with mass spectrometric structure determination. J Biol Chem. 1984;259(20):12659–62.
Krueger JM, Walter J, Dinarello CA, Wolff SM, Chedid L. Sleep-promoting effects of endogenous pyrogen (interleukin-1). Am J Physiol. 1984;246(6 Pt 2):R994–9.
Tobler I, Borbély AA, Schwyzer M, Fontana A. Interleukin-1 derived from astrocytes enhances slow wave activity in sleep EEG of the rat. Eur J Pharmacol. 1984;104(1–2):191–2.
Susic V, Totić S. “Recovery” function of sleep: effects of purified human interleukin-1 on the sleep and febrile response of cats. Metab Brain Dis. 1989;4(1):73–80.
Shoham S, Davenne D, Cady AB, Dinarello CA, Krueger JM. Recombinant tumor necrosis factor and interleukin 1 enhance slow-wave sleep. Am J Physiol. 1987;253(1 Pt 2):R142–9.
Opp MR, Obál F Jr, Krueger JM. Interleukin 1 alters rat sleep: temporal and dose-related effects. Am J Physiol. 1991;260(1 Pt 2):R52–8.
Fang J, Wang Y, Krueger JM. Mice lacking the TNF 55 kDa receptor fail to sleep more after TNFα treatment. J Neurosci. 1997;17(15):5949–55.
Hansen MK, Krueger JM. Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1β. Am J Physiol. 1997;273(4):R1246–53.
Opp MR, Toth LA. Somnogenic and pyrogenic effects of interleukin-1β and lipopolysaccharide in intact and vagotomized rats. Life Sci. 1998;62(10):923–36.
De Sarro G, Gareri P, Sinopoli VA, David E, Rotiroti D. Comparative, behavioural and electrocortical effects of tumor necrosis factor-α and interleukin-1 microinjected into the locus coeruleus of rat. Life Sci. 1997;60(8):555–64.
Manfridi A, Brambilla D, Bianchi S, Mariotti M, Opp MR, Imeri L. Interleukin-1β enhances non-rapid eye movement sleep when microinjected into the dorsal raphe nucleus and inhibits serotonergic neurons in vitro. Eur J Neurosci. 2003;18(5):1041–9.
Terao A, Matsumura H, Saito M. Interleukin-1 induces slow-wave sleep at the prostaglandin D2-sensitive sleep-promoting zone in the rat brain. J Neurosci. 1998;18(16):6599–607.
Baker FC, Shah S, Stewart D, Angara C, Gong H, Szymusiak R, et al. Interleukin 1β enhances non-rapid eye movement sleep and increases c-Fos protein expression in the median preoptic nucleus of the hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2005;288(4):R998–R1005.
Obál F Jr, Fang J, Payne LC, Krueger JM. Growth-hormone-releasing hormone mediates the sleep- promoting activity of interleukin-1 in rats. Neuroendocrinology. 1995;61(5):559–65.
Imeri L, Bianchi S, Mancia M. Muramyl dipeptide and IL-1 effects on sleep and brain temperature after inhibition of serotonin synthesis. Am J Physiol. 1997;273(5):R1663–8.
Imeri L, Mancia M, Opp MR. Blockade of 5-hydroxytryptamine (serotonin)-2 receptors alters interleukin- 1-induced changes in rat sleep. Neuroscience. 1999;92(2):745–9.
Terao A, Matsumura H, Yoneda H, Saito M. Enhancement of slow-wave sleep by tumor necrosis factor-α is mediated by cyclooxygenase-2 in rats. Neuroreport. 1998;9(17):3791–6.
Zhang BJ, Shao SR, Aritake K, Takeuchi A, Urade Y, Huang ZL, et al. Interleukin-1β induces sleep independent of prostaglandin D2 in rats and mice. Neuroscience. 2017;340:258–67.
Krueger JM, Majde JA. Microbial products and cytokines in sleep and fever regulation. Crit Rev Immunol. 2017;37(2–6):291–315.
Dinarello CA. Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J Endotoxin Res. 2004;10(4):201–22.
Krueger JM, Dinarello CA, Shoham S, Davenne D, Walter J, Kubillus S. Interferon alpha-2 enhances slow- wave sleep in rabbits. Int J Immunopharmacol. 1987;9(1):23–30.
Dickstein JB, Moldofsky H, Lue FA, Hay JB. Intracerebroventricular injection of TNF-α promotes sleep and is recovered in cervical lymph. Am J Physiol. 1999;276(4):R1018–22.
Breder CD, Tsujimoto M, Terano Y, Scott DW, Saper CB. Distribution and characterization of tumor necrosis factor-α-like immunoreactivity in the murine central nervous system. J Comp Neurol. 1993;337(4):543–67.
Cearley C, Churchill L, Krueger JM. Time of day differences in IL1β and TNFα mRNA levels in specific regions of the rat brain. Neurosci Lett. 2003;352(1):61–3.
Waters JP, Pober JS, Bradley JR. Tumour necrosis factor in infectious disease. J Pathol. 2013;230(2):132–47.
Dinarello CA, Cannon JG, Wolff SM, Bernheim HA, Beutler B, Cerami A, et al. Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J Exp Med. 1986;163(6):1433–50.
Tizard I. Sickness behavior, its mechanisms and significance. Anim Health Res Rev. 2008;9(1):87–99.
Kapás L, Hong L, Cady AB, Opp MR, Postlethwaite AE, Seyer JM, et al. Somnogenic, pyrogenic, and anorectic activities of tumor necrosis factor-α and TNF-α fragments. Am J Physiol. 1992;263(3 Pt 2):R708–15.
Bredow S, Guha-Thakurta N, Taishi P, Obál F Jr, Krueger JM. Diurnal variations of tumor necrosis factor alpha mRNA and alpha-tubulin mRNA in rat brain. Neuroimmunomodulation. 1997;4(2):84–90.
Floyd RA, Krueger JM. Diurnal variation of TNFα in the rat brain. Neuroreport. 1997;8(4):915–8.
Zielinski MR, Kim Y, Karpova SA, McCarley RW, Strecker RE, Gerashchenko D. Chronic sleep restriction elevates brain interleukin-1 beta and tumor necrosis factor-alpha and attenuates brain-derived neurotrophic factor expression. Neurosci Lett. 2014;580:27–31.
Kubota T, Fang J, Guan Z, Brown RA, Krueger JM. Vagotomy attenuates tumor necrosis factor-α-induced sleep and EEG δ-activity in rats. Am J Physiol Regul Integr Comp Physiol. 2001;280(4):R1213–20.
Zielinski MR, Dunbrasky DL, Taishi P, Souza G, Krueger JM. Vagotomy attenuates brain cytokines and sleep induced by peripherally administered tumor necrosis factor-α and lipopolysaccharide in mice. Sleep. 2013;36(8):1227–38, 1238A.
Kubota T, Li N, Guan Z, Brown RA, Krueger JM. Intrapreoptic microinjection of TNF-α enhances non- REM sleep in rats. Brain Res. 2002;932(1–2):37–44.
Chen L, Taishi P, Majde JA, Peterfi Z, Obal F Jr, Krueger JM. The role of nitric oxide synthases in the sleep responses to tumor necrosis factor-α. Brain Behav Immun. 2004;18(4):390–8.
Yoshida H, Kubota T, Krueger JM. A cyclooxygenase-2 inhibitor attenuates spontaneous and TNF-α- induced non-rapid eye movement sleep in rabbits. Am J Physiol Regul Integr Comp Physiol. 2003;285(1):R99–109.
Rockstrom MD, Chen L, Taishi P, Nguyen JT, Gibbons CM, Veasey SC, et al. Tumor necrosis factor alpha in sleep regulation. Sleep Med Rev. 2018;40:69–78.
Takahashi S, Tooley DD, Kapás L, Fang J, Seyer JM, Krueger JM. Inhibition of tumor necrosis factor in the brain suppresses rabbit sleep. Pflugers Arch. 1995;431(2):155–60.
Takahashi S, Kapás L, Fang J, Krueger JM. An anti-tumor necrosis factor antibody suppresses sleep in rats and rabbits. Brain Res. 1995;690(2):241–4.
Szondy Z, Pallai A. Transmembrane TNF-alpha reverse signaling leading to TGF-beta production is selectively activated by TNF targeting molecules: therapeutic implications. Pharmacol Res. 2017;115:124–32.
Deboer T, Fontana A, Tobler I. Tumor necrosis factor (TNF) ligand and TNF receptor deficiency affects sleep and the sleep EEG. J Neurophysiol. 2002;88(2):839–46.
Kapás L, Bohnet SG, Traynor TR, Majde JA, Szentirmai E, Magrath P, et al. Spontaneous and influenza virus-induced sleep are altered in TNF-α double-receptor deficient mice. J Appl Physiol (1985). 2008;105(4):1187–98.
Kaushal N, Ramesh V, Gozal D. TNF-α and temporal changes in sleep architecture in mice exposed to sleep fragmentation. PLoS One. 2012;7(9):e45610.
Szentirmai É, Kapás L. Sleep and body temperature in TNFα knockout mice: the effects of sleep deprivation, β3-AR stimulation and exogenous TNFα. Brain Behav Immun. 2019;81:260–71.
Szentirmai É, Kapás L. Brown adipose tissue plays a central role in systemic inflammation-induced sleep responses. PLoS One. 2018;13(5):e0197409.
Mackiewicz M, Sollars PJ, Ogilvie MD, Pack AI. Modulation of IL-1β gene expression in the rat CNS during sleep deprivation. Neuroreport. 1996;7(2):529–33.
Taishi P, Chen Z, Obál F Jr, Hansen MK, Zhang J, Fang J, et al. Sleep-associated changes in interleukin-1β mRNA in the brain. J Interferon Cytokine Res. 1998;18(9):793–8.
Basheer R, Rainnie DG, Porkka-Heiskanen T, Ramesh V, McCarley RW. Adenosine, prolonged wakefulness, and A1-activated NF-κB DNA binding in the basal forebrain of the rat. Neuroscience. 2001;104(3):731–9.
Manchanda S, Singh H, Kaur T, Kaur G. Low-grade neuroinflammation due to chronic sleep deprivation results in anxiety and learning and memory impairments. Mol Cell Biochem. 2018;449(1–2):63–72.
Moldofsky H, Lue FA, Davidson JR, Gorczynski R. Effects of sleep deprivation on human immune functions. FASEB J. 1989;3(8):1972–7.
Hu J, Chen Z, Gorczynski CP, Gorczynski LY, Kai Y, Lee L, et al. Sleep-deprived mice show altered cytokine production manifest by perturbations in serum IL-1ra, TNFa, and IL-6 levels. Brain Behav Immun. 2003;17(6):498–504.
Gao T, Wang Z, Dong Y, Cao J, Lin R, Wang X, et al. Role of melatonin in sleep deprivation-induced intestinal barrier dysfunction in mice. J Pineal Res. 2019;67(1):e12574.
Takahashi S, Kapás L, Seyer JM, Wang Y, Krueger JM. Inhibition of tumor necrosis factor attenuates physiological sleep in rabbits. Neuroreport. 1996;7(2):642–6.
Opp MR, Krueger JM. Interleukin-1 is involved in responses to sleep deprivation in the rabbit. Brain Res. 1994;639(1):57–65.
Baracchi F, Opp MR. Sleep-wake behavior and responses to sleep deprivation of mice lacking both interleukin-1 β receptor 1 and tumor necrosis factor-α receptor 1. Brain Behav Immun. 2008;22(6):982–93.
Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156(1–2):20–44.
Chang HY, Kapás L. The effects of leptin on sleep and temperature in rats. Soc Neurosci Abstr. 1997;23(2):1846.
Sinton CM, Fitch TE, Gershenfeld HK. The effects of leptin on REM sleep and slow wave delta in rats are reversed by food deprivation. J Sleep Res. 1999;8(3):197–203.
Danguir J. Sleep patterns in the genetically obese Zucker rat: effect of acarbose treatment. Am J Physiol. 1989;256(1 Pt 2):R281–3.
Laposky AD, Shelton J, Bass J, Dugovic C, Perrino N, Turek FW. Altered sleep regulation in leptin- deficient mice. Am J Physiol Regul Integr Comp Physiol. 2006;290(4):R894–903.
Laposky AD, Bradley MA, Williams DL, Bass J, Turek FW. Sleep-wake regulation is altered in leptin- resistant (db/db) genetically obese and diabetic mice. Am J Physiol Regul Integr Comp Physiol. 2008;295(6):R2059–66.
Wang Y, He J, Kastin AJ, Hsuchou H, Pan W. Hypersomnolence and reduced activity in pan-leptin receptor knockout mice. J Mol Neurosci. 2013;51(3):1038–45.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.
Tsigos C, Kyrou I, Chala E, Tsapogas P, Stavridis JC, Raptis SA, et al. Circulating tumor necrosis factor alpha concentrations are higher in abdominal versus peripheral obesity. Metabolism. 1999;48(10):1332–5.
Jenkins JB, Omori T, Guan Z, Vgontzas AN, Bixler EO, Fang J. Sleep is increased in mice with obesity induced by high-fat food. Physiol Behav. 2006;87(2):255–62.
Guan Z, Vgontzas AN, Bixler EO, Fang J. Sleep is increased by weight gain and decreased by weight loss in mice. Sleep. 2008;31(5):627–33.
Danguir J, Nicolaidis S. Circadian sleep and feeding patterns in the rat: possible dependence on lipogenesis and lipolysis. Am J Physiol. 1980;238(3):E223–30.
Mouret JR, Bobillier P. Diurnal rhythms of sleep in the rat: augmentation of paradoxical sleep following alterations of the feeding schedule. Int J Neurosci. 1971;2(6):265–9.
Roky R, Kapás L, Taishi TP, Fang J, Krueger JM. Food restriction alters the diurnal distribution of sleep in rats. Physiol Behav. 1999;67(5):697–703.
Bodosi B, Gardi J, Hajdu I, Szentirmai E, Obál F Jr, Krueger JM. Rhythms of ghrelin, leptin, and sleep in rats: effects of the normal diurnal cycle, restricted feeding, and sleep deprivation. Am J Physiol Regul Integr Comp Physiol. 2004;287(5):R1071–9.
Szentirmai É, Kapás L, Sun Y, Smith RG, Krueger JM. Restricted feeding-induced sleep, activity, and body temperature changes in normal and preproghrelin-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2010;298(2):R467–77.
Szentirmai É, Kapás L. The role of the brown adipose tissue in β3-adrenergic receptor activation-induced sleep, metabolic and feeding responses. Sci Rep. 2017;7(1):958.
Key BJ, Marley DE. The effect of the sympathomimetic amines on behaviour and electrocortical activity of the chicken. Electroencephalogr Clin Neurophysiol. 1962;14:90–105.
Panksepp J, Jalowiec JE, Zolovick AJ, Stern WC, Morgane PJ. Inhibition of glycolytic metabolism and sleep-waking states in cats. Pharmacol Biochem Behav. 1973;1(1):117–9.
Danguir J. Cafeteria diet promotes sleep in rats. Appetite. 1987;8(1):49–53.
Jacobs BL, McGinty DJ. Effects of food deprivation on sleep and wakefulness in the rat. Exp Neurol. 1971;30(2):212–22.
Danguir J, Nicolaidis S. Dependence of sleep on nutrients’ availability. Physiol Behav. 1979;22(4):735–40.
Sayegh AI. The role of cholecystokinin receptors in the short-term control of food intake. Prog Mol Biol Transl Sci. 2013;114:277–316.
Stengel A, Taché Y. Ghrelin - a pleiotropic hormone secreted from endocrine x/a-like cells of the stomach. Front Neurosci. 2012;6:24.
Al-Massadi O, Heppner KM, Nogueiras R, Perez-Tilve D, Tschöp M, Ghrelin. In: Kastin AJ, editor. Handbook of biologically active peptides. 2nd ed. Amsterdam: Academic Press; 2013. p. 1104–10.
Kageyama H, Takenoya F, Shiba K, Shioda S. Neuronal circuits involving ghrelin in the hypothalamus- mediated regulation of feeding. Neuropeptides. 2010;44(2):133–8.
Fara JW, Rubinstein EH, Sonnenschein RR. Visceral and behavioral responses to intraduodenal fat. Science. 1969;166(3901):110–1.
Rubinstein EH, Sonnenschein RR. Sleep cycles and feeding behavior in the cat; role of gastrointestinal hormones. Acta Dent Venez. 1971;22:125–8.
Kapás L, Obál F Jr, Alföldi P, Rubicsek G, Penke B, Obál F. Effects of nocturnal intraperitoneal administration of cholecystokinin in rats: simultaneous increase in sleep, increase in EEG slow-wave activity, reduction of motor activity, suppression of eating, and decrease in brain temperature. Brain Res. 1988;438(1–2):155–64.
de Saint Hilaire-Kafi Z, Depoortere H, Nicolaïdis S. Does cholecystokinin induce physiological satiety and sleep? Brain Res. 1989;488(1–2):304–10.
Kapás L, Obál F Jr, Opp MR, Johannsen L, Krueger JM. Intraperitoneal injection of cholecystokinin elicits sleep in rabbits. Physiol Behav. 1991;50(6):1241–4.
Szentirmai É, Kapás L, Sun Y, Smith R, Krueger JM. Sleep responses to ghrelin, leptin and cholecystokinin in ghrelin knockout mice. Sleep. 2007;30:A19–20.
Antin J, Gibbs J, Holt J, Young RC, Smith GP. Cholecystokinin elicits the complete behavioral sequence of satiety in rats. J Comp Physiol Psychol. 1975;89(7):784–90.
Mansbach RS, Lorenz DN. Cholecystokinin (CCK-8) elicits prandial sleep in rats. Physiol Behav. 1983;30(2):179–83.
Prospéro-García O, Ott T, Drucker-Colín R. Cerebroventricular infusion of cholecystokinin (CCK-8) restores REM sleep in parachlorophenylalanine (PCPA)-pretreated cats. Neurosci Lett. 1987;78(2):205–10.
DeMesquita S, Haney WH. Effect of chronic intracerebroventricular infusion of cholecystokinin on respiration and sleep. Brain Res. 1986;378(1):127–32.
De Saint Hilaire Z, Roques BP, Nicolaïdis S. Effect of a highly selective central CCK-B receptor agonist: BC-264 on rat sleep. Pharmacol Biochem Behav. 1991;38(3):545–8.
Chang HY, Kapás L. Selective activation of CCK-B receptors does not induce sleep and does not affect EEG slow-wave activity and brain temperature in rats. Physiol Behav. 1997;62(1):175–9.
Chang HY, Kapás L. L-364,718, a cholecystokinin (CCK)-A receptor antagonist, inhibits the sleep-inducing effects of CCK. Sleep Res. 1997;26:138.
Sei M, Sei H, Shima K. Spontaneous activity, sleep, and body temperature in rats lacking the CCK-A receptor. Physiol Behav. 1999;68(1–2):25–9.
Shemyakin A, Kapás L. L-364,718, a cholecystokinin-A receptor antagonist, suppresses feeding-induced sleep in rats. Am J Physiol Regul Integr Comp Physiol. 2001;280(5):R1420–6.
Szentirmai É, Kapás L, Krueger JM. Ghrelin microinjection into forebrain sites induces wakefulness and feeding in rats. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R575–85.
Szentirmai É. Central but not systemic administration of ghrelin induces wakefulness in mice. PLoS One. 2012;7(7):e41172.
Szentirmai É, Kapás L. Interactive regulation of sleep and feeding. In: Kryger MH, editor. Atlas of clinical sleep medicine. Philadelphia: Saunders Elsevier; 2013. p. 56–8.
Tolle V, Bassant MH, Zizzari P, Poindessous-Jazat F, Tomasetto C, Epelbaum J, et al. Ultradian rhythmicity of ghrelin secretion in relation with GH, feeding behavior, and sleep-wake patterns in rats. Endocrinology. 2002;143(4):1353–61.
Obál F Jr, Alt J, Taishi P, Gardi J, Krueger JM. Sleep in mice with nonfunctional growth hormone-releasing hormone receptors. Am J Physiol Regul Integr Comp Physiol. 2003;284(1):R131–9.
Frieboes RM, Antonijevic IA, Held K, Murck H, Pollmächer T, Uhr M, et al. Hexarelin decreases slow- wave sleep and stimulates the secretion of GH, ACTH, cortisol and prolactin during sleep in healthy volunteers. Psychoneuroendocrinology. 2004;29(7):851–60.
Weikel JC, Wichniak A, Ising M, Brunner H, Friess E, Held K, et al. Ghrelin promotes slow-wave sleep in humans. Am J Physiol Endocrinol Metab. 2003;284(2):E407–15.
Kluge M, Schüssler P, Zuber V, Yassouridis A, Steiger A. Ghrelin administered in the early morning increases secretion of cortisol and growth hormone without affecting sleep. Psychoneuroendocrinology. 2007;32(3):287–92.
Kluge M, Schüssler P, Zuber V, Kleyer S, Yassouridis A, Dresler M, et al. Ghrelin enhances the nocturnal secretion of cortisol and growth hormone in young females without influencing sleep. Psychoneuroendocrinology. 2007;32(8–10):1079–85.
Kluge M, Gazea M, Schüssler P, Genzel L, Dresler M, Kleyer S, et al. Ghrelin increases slow wave sleep and stage 2 sleep and decreases stage 1 sleep and REM sleep in elderly men but does not affect sleep in elderly women. Psychoneuroendocrinology. 2010;35(2):297–304.
Szentirmai E, Kapás L, Sun Y, Smith RG, Krueger JM. Spontaneous sleep and homeostatic sleep regulation in ghrelin knockout mice. Am J Physiol Regul Integr Comp Physiol. 2007;293(1):R510–7.
Esposito M, Pellinen J, Kapás L, Szentirmai É. Impaired wake-promoting mechanisms in ghrelin receptor- deficient mice. Eur J Neurosci. 2012;35(2):233–43.
Szentirmai E, Kapás L, Sun Y, Smith RG, Krueger JM. The preproghrelin gene is required for the normal integration of thermoregulation and sleep in mice. Proc Natl Acad Sci U S A. 2009;106(33):14069–74.
Szentirmai É, Krueger JM. Sickness behaviour after lipopolysaccharide treatment in ghrelin deficient mice. Brain Behav Immun. 2014;36:200–6.
Morris CJ, Aeschbach D, Scheer FAJL. Circadian system, sleep and endocrinology. Mol Cell Endocrinol. 2012;349(1):91–104.
Obál F Jr, Opp M, Cady AB, Johannsen L, Krueger JM. Prolactin, vasoactive intestinal peptide, and peptide histidine methionine elicit selective increases in REM sleep in rabbits. Brain Res. 1989;490(2):292–300.
Roky R, Valatx JL, Jouvet M. Effect of prolactin on the sleep-wake cycle in the rat. Neurosci Lett. 1993;156(1–2):117–20.
Obál F Jr, Payne L, Kacsoh B, Opp M, Kapás L, Grosvenor CE, et al. Involvement of prolactin in the REM sleep-promoting activity of systemic vasoactive intestinal peptide (VIP). Brain Res. 1994;645(1–2):143–9.
Obál F Jr, Kacsóh B, Bredow S, Guha-Thakurta N, Krueger JM. Sleep in rats rendered chronically hyperprolactinemic with anterior pituitary grafts. Brain Res. 1997;755(1):130–6.
Zhang SQ, Inoué S, Kimura M. Sleep-promoting activity of prolactin-releasing peptide (PrRP) in the rat. Neuroreport. 2001;12(15):3173–6.
Obál F Jr, Kacsóh B, Alföldi P, Payne L, Markovic O, Grosvenor C, et al. Antiserum to prolactin decreases rapid eye movement sleep (REM sleep) in the male rat. Physiol Behav. 1992;52(6):1063–8.
Roky R, Valatx JL, Paut-Pagano L, Jouvet M. Hypothalamic injection of prolactin or its antibody alters the rat sleep-wake cycle. Physiol Behav. 1994;55(6):1015–9.
Lobo LL, Claustrat B, Debilly G, Paut-Pagano L, Jouvet M, Valatx JL. Hypoprolactinemic rats under conditions of constant darkness or constant light. Effects on the sleep-wake cycle, cerebral temperature and sulfatoxymelatonin levels. Brain Res. 1999;835(2):282–9.
Obál F Jr, Garcia-Garcia F, Kacsóh B, Taishi P, Bohnet S, Horseman ND, et al. Rapid eye movement sleep is reduced in prolactin-deficient mice. J Neurosci. 2005;25(44):10282–9.
Zhang SQ, Kimura M, Inoué S. Sleep patterns in cyclic and pseudopregnant rats. Neurosci Lett. 1995;193(2):125–8.
Kimura M, Zhang SQ, Inoué S. Pregnancy-associated sleep changes in the rat. Am J Physiol. 1996;271(4 Pt 2):R1063–9.
Meerlo P, Easton A, Bergmann BM, Turek FW. Restraint increases prolactin and REM sleep in C57BL/6J mice but not in BALB/cJ mice. Am J Physiol Regul Integr Comp Physiol. 2001;281(3):R846–54.
Machado RB, Tufik S, Suchecki D. Chronic stress during paradoxical sleep deprivation increases paradoxical sleep rebound: association with prolactin plasma levels and brain serotonin content. Psychoneuroendocrinology. 2008;33(9):1211–24.
Bodosi B, Obál F Jr, Gardi J, Komlódi J, Fang J, Krueger JM. An ether stressor increases REM sleep in rats: possible role of prolactin. Am J Physiol Regul Integr Comp Physiol. 2000;279(5):R1590–8.
Cinti S. The adipose organ at a glance. Dis Model Mech. 2012;5(5):588–94.
Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell. 2012;151(2):400–13.
Dewasmes G, Loos N, Delanaud S, Dewasmes D, Géloën A. Activation of brown adipose tissue thermogenesis increases slow wave sleep in rat. Neurosci Lett. 2003;339(3):207–10.
Szentirmai É, Kapás L. Intact brown adipose tissue thermogenesis is required for restorative sleep responses after sleep loss. Eur J Neurosci. 2014;39(6):984–98.
Fincher EF 4th, Johannsen L, Kapás L, Takahashi S, Krueger JM. Microglia digest Staphylococcus aureus into low molecular weight biologically active compounds. Am J Physiol. 1996;271(1 Pt 2):R149–56.
Johannsen L, Wecke J, Obál F Jr, Krueger JM. Macrophages produce somnogenic and pyrogenic muramyl peptides during digestion of staphylococci. Am J Physiol. 1991;260(1 Pt 2):R126–33.
Ames C, Boland E, Szentirmai É. Effects of macrophage depletion on sleep in mice. PLoS One. 2016;11(7):e0159812.
Massie A, Boland E, Kapás L, Szentirmai É. Mice lacking alternatively activated (M2) macrophages show impairments in restorative sleep after sleep loss and in cold environment. Sci Rep. 2018;8(1):8625.
El Hajjaji FZ, Pelletier A, Delanaud S, Libert JP, Bach V, Loos N. Sleep structure and feeding pattern changes induced by the liver’s thermal status in the rat. J Sleep Res. 2012;21(2):204–11.
Hansen MK, Krueger JM. Gadolinium chloride pretreatment prevents cafeteria diet-induced sleep in rats. Sleep. 1999;22(6):707–15.
Szentirmai É, Millican NS, Massie AR, Kapás L. Butyrate, a metabolite of intestinal bacteria, enhances sleep. Sci Rep. 2019;9(1):7035.
Brown R, Price RJ, King MG, Husband AJ. Are antibiotic effects on sleep behavior in the rat due to modulation of gut bacteria? Physiol Behav. 1990;48(4):561–5.
Millican NS, Massie AR, Szentirmai É, Kapás L. The effects of antibiotic-induced gut-microbiome depletion on sleep in mice. 2018 Neuroscience meeting planner. San Diego, CA: Society for Neuroscience meeting; 2018.
Dell P. Afferences baroreceptives, phases de synchronisation corticale et sommeil. Arch Ital Biol. 1973;111:553–63.
Puizillout JJ, Foutz AS. Characteristics of the experimental reflex sleep induced by vago-aortic nerve stimulation. Electroencephalogr Clin Neurophysiol. 1977;42:552–63.
Chase MH, Nakamura Y, Clemente CD, Sterman MB. Afferent vagal stimulation: neurographic correlates of induced EEG synchronization and desynchronization. Brain Res. 1967;5:236–49.
Kapás L, Hansen MK, Chang HY, Krueger JM. Vagotomy attenuates but does not prevent the somnogenic and febrile effects of lipopolysaccharide in rats. Am J Physiol. 1998;274:R406–11.
Hobson JA. Sleep is of the brain, by the brain and for the brain. Nature. 2005;437(7063):1254–6.
Schmid SM, Hallschmid M, Schultes B. The metabolic burden of sleep loss. Lancet Diabetes Endocrinol. 2015;3(1):52–62.
Irwin MR, Opp MR. Sleep health: reciprocal regulation of sleep and innate immunity. Neuropsychopharmacology. 2017;42(1):129–55.
Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. 2007;86:1286–92.
Neal MD, Leaphart C, Levy R, Prince J, Billiar TR, Watkins S, Li J, Cetin S, Ford H, Schreiber A, Hackam DJ. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol. 2006;176:3070–9.
Stilling RM, van de Wouw M, Clarke G, Stanton C, Dinan TG, Cryan JF. The neuropharmacology of butyrate: the bread and butter of the microbiota-gut-brain axis? Neurochem Int. 2016;99:110–32.
Millican NS, Massie AR, Szentirmai É, Kapás L. Lipopolysaccharide-induced sleep in rats: the role of hepatoportal sensory mechanisms. Program No. 148.12. 2019 Neuroscience meeting planner. Chicago, IL: Society for Neuroscience; 2019. Online
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Szentirmai, É., Kapás, L. (2021). Humoral and Other Sleep-Promoting Factors. In: Gozal, D., Kheirandish-Gozal, L. (eds) Pediatric Sleep Medicine. Springer, Cham. https://doi.org/10.1007/978-3-030-65574-7_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-65574-7_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65573-0
Online ISBN: 978-3-030-65574-7
eBook Packages: MedicineMedicine (R0)