
Abstract
A significant application of microarray gene expression data is the classification and prediction of biological models. An essential component of data analysis is dimension reduction. This study presents a comparison study on a reduced data using Analysis of Variance (ANOVA) and Recursive Feature Elimination (RFE) feature selection dimension reduction techniques, and evaluates the relative performance evaluation of classification procedures of Support Vector Machine (SVM) classification technique. In this study, an accuracy and computational performance metrics of the processes were carried out on a microarray colon cancer dataset for classification, SVM-RFE achieved 93% compared to ANOVA with 87% accuracy in the classification output result.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
In biological learning, Next-generation sequencing (NGS) has been expansively utilized. General NGS information is the Ribonucleic Acid sequencing (RNA-seq); it is utilized to test the anomalies of mRNA expression in ailments. In difference with microarray advancements, microarray talks about significant data that presents explicit inventiveness of narrative protein isoforms with various compound scopes of uncovered qualities.
Microarray has become an expansively utilized genome-wide expression profile for figuring substance cells, because of their capacity of determining potential heterogeneities in cell populaces [1]. Since the advancement of RNA tasks as a notable intermediary among genome and proteome, finding and estimating gene expression have been the unmistakable conduct in biological science [2]. There is no foremost prospective or good channel for the assorted variety of claims and analysis state in which microarray can be utilized. Researches and adoption of systematic methodologies on living being and their objectives have advanced [3].
A flourishing microarray study must have a major prerequisite of creating information with the possibilities of responding to biological inquiries of concern. This is practiced by characterizing an investigational aim, series intensity and replicating reasonable biological plans under examination and by advancement of sequencing research, ensuring that information achievement does not end up being tainted with redundant views. One critical part of the microarray information is the expulsion of the scourge of high-dimension, for example, noises, commotions, repetition, redundancy, immaterial as well as irrelevant data, among others [4]. Because of high-measurement of biological information challenges, dimension reduction techniques are vital. Microarray information has turned out to be a potential high-throughput procedure to simultaneously profile transcriptomes of substantial information [5]. Microarray has key advantages, for example, the capacity to spot narrative transcripts, precision, and dynamic range [6]. Thousands of quality genes are simultaneously communicated and expressed in microarray, expression levels of genes are usually difficult, finding an effective low-dimensional representation of microarray information is important. A few dimension reduction methods utilized for gene expression data analysis and information investigation to expel noises related to explicit information exist [7]. Although many dimensionality reduction methods have been proposed and developed in this field, yet this study proposes efficient feature selection methods, by ranking the feature genes and selecting key to tackle principal drawbacks of high dimensional data. Overcoming this limitation, this study introduces an efficient implementation of SVM classification combined with the selected informative genes. This study proposes a simple method for preprocessing datasets, for informative dispersal of samples with a more credible classification result.
This study proposes a computational dimensionality reduction technique using ANOVA and RFE, to deal with the issue of curse of high dimensionality in gene expression space and analyzes SVM kernel classification methods. This study exhibits the robustness of this technique regarding to noises and sampling on RNA-Seq Anopheles Gambiae dataset.
2 Materials and Methods
2.1 Dataset Used for Analysis
Colon cancer dataset was used for this experiment, it contains an expression of 2000 genes with highest minimal intensity across 62 tissues, derived from 40 tumor and 22 normal colon tissue samples. The gene expression was analyzed with an Affymetrix oligonucleotide array complementary to more than 6,500 human genes. The gene intensity has been derived from about 20 feature pairs that correspond to the gene on the DNA microarray chip by using a filtering process. Details for data collection methods and procedures are described [8], and the data set is available from the website http://microarray.princeton.edu/oncology/.
MATLAB (Matrix Laboratory) is utilized to perform the experiment, due to its ease and beneficial programming environment for engineers, architects, scientists, researchers, among others. MATLAB is a multi-worldview numerical processing environment and exclusive programming language created by MathWorks. It permits framework controls, plotting of functions and information, execution of algorithms, production of User Interfaces, and interfacing with projects written in different languages, such as; C, C++, C#, Java, Fortran and Python [9]. The principle point of this study is the prediction of the RNA-Seq technology utilizing the MATLAB tool by utilizing the colon database. Table-1 demonstrates a concise description of the dataset.
2.2 Experimental Methodology
This study summarizes the proposed framework in Fig. 1 below. The fundamental idea is to predict machine learning task on high dimensional microarray data, for cells and genes into lower dimensional dataset. The plan is adjusted to fetch out important data in a given dataset by utilizing ANOVA and RFE feature selection methods and evaluate the performance of colon cancer microarray dataset on SVM classification algorithm.

Proposed framework.
Microarray data is the next generation sequencing technology to think about in transcriptome. It is utilized as an option to microarrays for gene expression analysis, without the need to earlier realize the RNA/DNA succession. RNA-seq offers progressively precise information and applications including identification of gene fusion, variations, alternative joining, post-transcriptional changes as well as analysis of small RNAs, such as; tRNA or miRNA profiles [10,11,12]. A total image of the RNA/DNA substance can be gotten from low quantity biological samples. A few expository advances are basic for an effective portrayal and evaluation of the transcriptome. Bioinformatics tools are proposed for quality control, information handling, annotation, quantification and representation for translation and biological science investigation for understanding gene information.
2.3 Analysis of Variance (ANOVA)
ANOVA algorithm simplifies the value of intensity as a sum of components. ANOVA algorithm helps in normalization and gene-specific model. The normalization eliminates properties due to total differences in intensity among diverse arrays. ANOVA normalization is trivial and basically deducts the mean of the log-transformed intensity from each array and refer to the distorted and normalized intensity values as Y [13]. ANOVA test is used to compare the ‘multiple means’ values of the dataset, and visualize whether there exists any significant difference between mean values of multiple groups (classes). The statistic for ANOVA is called the F-statistic, which can be calculated using following steps [14, 35, 36]:
The input to the algorithm is a matrix of the form N × M, where N is the total number of feature sets and M is the number of samples in the dataset.
2.4 Recursive Feature Elimination (RFE)
Guyon introduce RFE [15], RFE makes feature selection by iteratively training a set of data with the current set of features and eliminating the least significant feature indicated. In the linear case, the separating hyperplane (decision function)is \( D (\vec{x}) = (\vec{w} \cdot \vec{x}) + b \). The feature with the smallest weight w2 contributes the least to the resulting hyperplane and can be discarded. Due to the heavy computational cost of RFE, several variants have been introduced to speed up the algorithm. Instead of removing only one least important feature at every iteration, removing a big chunk of features in each iteration will speed up the process. The goal is to remove more features during each iteration, but not to eliminate the important features. [16].
2.5 Classification
A few classification algorithms exist, for example, Logistic Regression, SVM, K-Nearest Neighbor, among others [17]. After reducing the dimensional complexity of data, the subsequent stage is the classification procedure. Classification is the fundamental goal; the analyzed data is classified. Two SVM kernels techniques were utilized: Polynomial Kernel and Gaussian Kernel. The results of the algorithms are analyzed and compared based on computational time, training time and performance metrics such as accuracy.
2.6 Support Vector Machine (SVM)
SVM is a learning machine algorithm presented by Vapnik in 1992 [18]. The algorithm works with the point of finding the best hyperplane that isolates between classes in the input space. SVM is a linear classifier; it is created to work with nonlinear problems by joining the kernel ideas in high-dimensional workspaces. In non-linear issues, SVM utilizes a kernel in training the data with the goal of spreading the dimension widely. When the dimensions are tweaked, SVM will look for the optimal hyperplane that can separate a class from different classes [19] (Chang, and Lin 2011). As indicated by the adoption of Aydadenta and Adiwijaya (2018) [18], the procedure to locate the best hyperplane utilizing SVM is as follows:
-
i.
Let \( y_{i} \in \left\{ {y_{1, } y_{2} , \ldots ,y_{n} } \right\}, where\,y_{i} \,is\,the\,p - attributes\,and\,target\,class\,z_{i} \in \left\{ { + 1, - 1} \right\} \)
-
ii.
Assuming the classes +1 and −1 can be separated completely by hyperplane, as defined in Eq. 2 below
From Eq. (2), Eqs. (3) and (4) are gotten:
Where, y is the input data, v is the ordinary plane and c is the positive relative to the center field coordinates.
SVM intends to discover hyperplanes that maximizes margins between two classes. Expanding margins is a quadratic programming issue that is solved by finding the minimal point. The advantage of SVM is its capacity to manage wide assortment of classification problems in high dimensional data [20].
Compared to other classification methods, SVM is outstanding, with its exceptional classification adequacy [21]. SVM is grouped into linear and non-linear separable. SVM’s has kernel functions that change data into a higher dimensional space to make it conceivable to perform seperations. Kernel functions are a class of algorithms for pattern analysis or recognition. Training vectors xi is mapped into higher dimensional space by the capacity Ф. SVM finds a linear seperating hyperplane with the maximal in this higher dimension space. C > 0 is the penalty parameter of the error term.
There are several SVM kernels that exist such as; the polynomial kernel, Radial basis function (RBF), linear kernel, Sigmoid, Gaussian kernel, String Kernels, among others. The decision of a Kernel relies upon the current issue at hand, since it relies upon what models are to be analyzed, a couple of kernel functions have been found to function admirably in for a wide assortment of applications [22]. The prescribed kernel function for this study is the SVM-Polynomial Kernel and Gaussian Kernel.
SVM-Gaussian Kernel
Gaussian kernel [23] compare to a general smoothness supposition in all k-th order subordinates. Kernels coordinating a certain prior recurrence substance of the data can be developed to reflect earlier issues in learning. Each input vector x is mapped to an interminable dimensional vector including all degree polynomial extensions of x’s components. For instance, a polynomial kernel model features conjunction up to the order of the polynomial. Radial basis functions permit circles in contrast with the linear kernel, which permits just selecting lines (or hyperplanes).
For instance, polynomial kernel is the least complex kernel function. It is given by the inner product (a, b) in addition to a discretionary constant K.
In SVM kernel functions, γ, a, and b are kernel parameters, RBF is the fundamental kernel function due to the nonlinearly maps tests in higher dimensional space unlike the linear kernel, it has less hyper parameters than the polynomial portion.
3 Related Works
Dimensionality reduction approaches have established important consideration recently, with evolving new algorithms and variant combinations. SVM has engrossed researchers’ interests due to its viable performance in classification and intrinsic capability.
A dimensionality reduction model was proposed [4] for zero inflated single cell gene expression analysis, they built a dimensionality reduced technique, zero inflated factor analysis (ZIFA), which expressly models the dropout attributes, and demonstrate that it improves modelling precision on biological and simulated datasets. They modified the PPCA and FA framework to represent dropout and deliver a safe technique for the dimensionality reduction of single-cell gene expression data that gives robustness against such vulnerabilities.
Without dropouts, the method is basically equal to PPCA or FA. Hence, users could utilize ZIFA as an immediate substitute with the advantage that it will consequently represent dropouts while remedial endeavors might be required with standard PCA. There procedure varies from methodologies, for example, the numerous variations of strong PCA, which mean to show corrupted perceptions. ZIFA regards dropouts as genuine perceptions, not exceptions, whose event properties have been described utilizing an observationally educated factual model.
A novel hybrid dimension reduction method was proposed [24], for small high dimensional gene expression datasets with information intricacy principle for cancer classification. Their study addressed the restrictions inside the setting of Probabilistic PCA (PPCA) by presenting and building up new and novel methodology utilizing most extreme entropy covariance matrix and its hybridized smoothed covariance estimators. To diminish the dimensionality of the data and pick the quantity of probabilistic PCs (PPCs) to be held, they further presented and created observed Akaike’s information criterion (AIC), consistent Akaike’s information criterion (CAIC), and the information theoretic measure of complexity (ICOMP) rule of Bozdogan. Six openly accessible undersized benchmark informational collections were breaking down to demonstrate the utility, adaptability, and flexibility of their methodology with hybridized smoothed covariance matrix estimators, which does not decline to play out the PPCA to diminish the measurement and to do regulated characterization of malignancy bunches in high measurements. Their proposed technique can be utilized to take care of new issues and difficulties present in the investigation of NGS information in bioinformatics and other biomedical applications.
A feature selection for cancer classification for disease utilizing microarray data expression was proposed [25]. This paper used information on microarray gene expression level to decide marker genes that are pertinent to a sort of malignancy. They researched a separation-based element choice strategy for two-gather grouping issue. So as to choose marker genes, the Bhattacharyya separation is actualized to quantify the uniqueness in gene expression levels. They used SVM for classification with utilization of the selected marker genes. The execution of marker gene selection and classification are represented in both recreation studies and two genuine information analyses by proposing a new gene selection method for classification based on SVMs. In the proposed method, they firstly ranked every gene according to the importance of their Bhattacharyya distances between the two indicated classes. The optimal gene subset is chosen to accomplish the least misclassification rate in the developed SVMs following a forward selection algorithm. 10-fold cross-validation is connected to locate the optimal parameters for SVM with the final optimal gene subset. Subsequently, the classification model is trained and built. The classification model is evaluated by its prediction performance for testing set. The execution of the proposed B/SVM technique with that of SVM-RFE and SWKC/SVM gives normal misclassification rate (1.1%) and high normal recovery rate (95.7%).
An Alzheimer’s infection determination by utilizing dimensionality reduction was proposed [26], based on KNN classification algorithm for analyzing and classifying the Alzheimer malady and mild cognitive mutilation are available in the datasets. Their study gave more precision rate, accuracy rate and sensitivity rate to give a better output. This paper proposed a narrative dimensionality reduction based KNN classification Algorithm dissected the Alzheimer’s illness present in the datasets. With the algorithm, the dataset was separated into 3 classes; first class having the Alzheimer’s disease (AD), second class was having the normal outcome, third class having the mild cognitive impairment. The information’s were taken from the researcher’s data dictionary - Uniform Data Set (RDD-UDS).
The relative investigations between the current PNN classification procedures with the proposed KNN classification demonstrated that high measure of normal accuracy, sensitivity, specificity precision, recall, jaccard and dice coefficients furthermore diminish the information dimensionality and computational multifaceted nature. Their future work, stated that the feature extraction and classification algorithm will improve the classification performance.
PCA and Factor Analysis for dimensionality reduction of bio-informatics data was proposed [27], they utilized the dimensionality reduction model of bioinformatics information. These systems were applied on Leukemia dataset and the number of attributes was decreased. An investigation was exhibited on reducing the number of attributes using PCA and Factor Analysis. Leukemia data was used for the analyses. PCA was carried out on the dataset and 9 components were chosen out of the 500 components. The Factor Analysis was used to extract the critical features.
A simulation study for the RNA-Seq data classification was proposed [28], they contrasted a few classifiers including PLDA renovation, NBLDA, single SVM, bagging SVM, CART, and random forest (RF). They analyzed the impact of a few parameters, for example, over-dispersion, sample size, number of genes and classes, differential expression rate, and the transform technique on model performances. A broad modeled study was conducted and the outcomes were contrasted using the consequences of two miRNA and two mRNA exploratory datasets. The outcomes uncovered that expanding the sample size, differential expression rate and transformation method on model presentation. RNA-Seq data classification requires cautious consideration when taking care of data over-scattering. They ended up that count-based classifier, the power changed PLDA and as classifiers, vst or rlog changed RF and SVM classifiers might be a decent decision for classification.
A neural network algorithm to reduce the dimensions of single cell RNA-Seq data was proposed [9], containing a few new computational complexities. These incorporate inquiries concerning the best strategies for clustering scRNA-Seq data, how to recognize unique cells, and deciding the state or capacity of explicit cells dependent on their expression profile. To address these issues, they created and tested a technique based on neural network (NN) for the analysis and recovery of single cell RNA-Seq data. They showed different NN structures, some of which fuse prior biological learning, and utilized these to acquire a reduced dimension representation of the single cell expression data. They demonstrate that the NN technique enhances earlier strategies in the capacity to accurately group cells in analyses not utilized in the training and the capacity to effectively derive cell type or state by questioning a database of a huge number of single cell profiles. Such database queries (which can be performed utilizing a web server) will empower researchers to better characterize cells while investigating heterogeneous scRNASeq tests.
A review of recent ongoing advancements in PCA as a strategy for diminishing the dimensionality of RNA-Seq datasets was proposed [29], for expanding interpretability and yet limiting data misfortune by making new uncorrelated factors that progressively maximize variance. This study presented the essential thoughts of PCA, talking about what it can, can’t do and after that depict a few variations of PCA and their application.
4 Results
The colon cancer dataset extracted were classified, the classification results obtained show the features capability for classifying the colon’s status. The average classification accuracy, which is using features with ANOVA and RFE are recorded in tabular form below. The proposed methodology was applied to the publicly available colon cancer database, the classification algorithm applies SVM kernel by utilizing MATLAB tools to implement the model.
Using ANOVA as a dimensionality reduction method, 416 features where fetched from the 2001 attributes of colon cancer dataset obtained from Alon, 2001 [8]. Using ANOVA, the output of the analysis is a statistically significant difference between group means. The significance value is 0.05 which is the mean length of time to complete the spreadsheet problem between the different courses taken.
ANOVA is appropriate when the model holds, have a single “treatment” with, say, k levels. “Treatment” may be interpreted in the loosest possible sense as any categorical explanatory variable. There is a population of interest for which there is a true quantitative outcome for each of the k levels of treatment. The selected features a processed for classification.
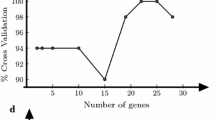
A supervised SVM kernel classifier methods, is among the most well-established and popular machine learning approaches in bioinformatics and genomics, 10-folds cross validation was used to evaluate the execution of the performance of the classification models, using 0.05 parameter holdout of data for training and 5% for testing to check the accuracy of the classifiers.
To each of the classifiers, a basic supervised learning assessment protocol is carried out. In particular, the training and testing stages are assessed as a 10-fold cross validation to eliminate the sampling bias. This protocol is implemented using MATLAB. The reported result of assessment is based on the computational time and performance metrics (Accuracy, Sensitivity, Specificity, F-score, Precision and Recall) [30,31,32,33,34].
Figure 2 and Fig. 3 shows the confusion matrix table comprising of the TP, TN, FP and FN which are used extensively for the evaluation of the performance metrics.

Confusion matrix and performance metrics for SVM-ANOVA

Confusion matrix and performance metrics for SVM-RFE classification
RFE-SVM algorithm was used to fetch out relevant data in the colon cancer dataset, 868 features were selected. SVM-RFE improve the computational performance of recursive feature elimination by eliminating chunks of features at a time with as little effect on the quality of the reduced feature set as possible. The RFE algorithm is implemented using an SVM to assist in identifying the least useful gene(s) to eliminate. Using SVM-RFE, the selected data was classified and accomplish 93.3% Accuracy.
In this study, data analysis of a well-known dataset colon cancer dataset by Alon [8], consisting of expression levels of 2000 genes describing 62 samples (40 tumor and 22 normal colon tissues, was analyzed using MATLAB tool. The dataset was used to compare the performance of the One-Way-ANOVA and SVM-RFE. The dataset was trained and tested. A different number of genes were selected by each of the algorithms, 416 and 868 respectively. The SVM was trained on the training data that was trimmed to the selected genes from each algorithm respectively. The SVM model produced was evaluated by its performance to predict the class labels (since cross validation results on the training data tend to be optimistic). Comparisons of the two algorithms in terms of prediction rate and time required are made. A comparison between ANOVA and SVM-RFE is also performed. The performance of ANOVA was comparable to the SVM-RFE algorithms in terms of prediction accuracy rate (each achieving around 87% and 93% accuracy on the test data). Experiment on the Alon colon cancer data sets also show that ANOVA has similar performance when compared with SVM-RFE with respect to accuracy, when comparing computational time, ANOVA is much faster than the SVM-RFE.
In general, SVM-RFE allows an enormous increase in the efficiency of the algorithm without a decrease of classification accuracy. Thus, the gene selection process is made much more practical in domains with a large number of features, such as gene expression data. This improvement is especially important as the number of samples available increases. The Table 1 below shows the comparative analysis of ANOVA and SVM-RFE feature selection algorithm using SVM classification algorithm to improve the performance of Colon Cancer data in microarray technology.
The performance analysis of classification using Support Vector Machine on colon cancer dataset shows that, SVM-RFE feature selection technique method achieves necessary higher value in the datasets on performance parameters such as the accuracy, timing, sensitivity, specificity, and prediction when compared to the ANOVA feature selection method. When the dataset is of high dimensional, by application of dimensionality reduction, some valuable data are considered and the accuracy of an algorithm increases by removing unnecessary data. The feature selection algorithm using ANOVA for high dimensional datasets plays an important role, it improves the performance of feature extraction methods, and SVM-RFE also enhances the classification algorithm “SVM” performance, in terms of accuracy, sensitivity, specificity and precision.
5 Conclusion
In the past few years, remarkable works have been done on the innovation of microarray, improvement as far as the execution measurements and productivity that are extraordinarily influenced by exploratory plan, activity and the data analysis forms are in trends to enhance the performance. cancer is a deadly insect comprising of various kinds. Small sample sizes of high dimensionality are main characteristics of microarray data and they are challenging data analysis criterion. The significance of classification of colon cancer into gatherings has prompted numerous researches. By examination, this study classifies a colon cancer data by using SVM on reduced dimensional data that employs RFE and ANOVA algorithms. The experiment accomplished a comparable result that shows that SVM-RFE outperforms ANOVA-SVM with 93%. Further studies should be conducted to improve performance of Machine Learning based methods by using more data and hybridized models.
References
Aaron, T.L., Davis, J.M, John, C.M.: A step-by-step workflow for low-level analysis of single-cell RNA-seq data. Research 1(5), 1–62. https://doi.org/10.12688/f1000research.9501.2
Ana, C., et al.: A survey of best practices for RNA-seq data analysis. Genome Biol. 17(13), 1–19 (2016). https://doi.org/10.1186/s13059-016-0881-8
Levin, J.Z., et al.: Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 7, 709–715 (2010)
Pierson, E., Yau, C.: ZIFA: dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 16, 241–257 (2015)
Dongfang, W., Jin, G.: VASC: dimension reduction and visualization of single-cell RNA-seq data by deep variation autoencoder. Genom. Proteom. Bioinform. (2018). https://doi.org/10.1016/j.gpb.2018.08.03
Junhyong, K.: Computational Analysis of RNA-Seq Data: From Quantification to High-Dimensional Analysis. University of Pennsylvania, pp. 35–43 (2012)
Bacher, R., and Kendziorski, C.: Design and computational analysis of single-cell RNA-seq experiments. Genome Biol. 17(63) (2016)
Alon, U., et al.: Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc. Natl. myAcad. Sci. USA 8; 96(12), 6745–6750 (1999)
Chieh, L., Siddhartha, J., Hannah, K., Ziv, B.: Using neural networks for reducing the dimensions of single-cell RNA-Seq data. Nucleic Acids Res. 45(17), 1–11 (2017). https://doi.org/10.1093/nar/gkx681
Mariangela, B., et al.: RNA-seq analyses of changes in the Anopheles gambiae transcriptome associated with resistance to pyrethroids in Kenya: identification of candidate-resistance genes and candidate-resistance SNPs. Paras. Vector 8(474), 1–13 (2015). https://doi.org/10.1186/s13071-015-1083-z
Bezanson, J., Karpinski, S., Shah, V., Edelman, A.: Julia: a fast-dynamic language for technical computing (2012). arXiv:1209.5145
Gary, A.C.: Using ANOVA to analyze microarray data. Biotechn. Future Sci. 37(2), 1–5 (2018)
Mukesh, K., Nitish, K.R., Amitav, S., Santanu, K.R.: Feature selection and classification of microarray data using MapReduce Based ANOVA and KNN. Procedia Comput. Sci. 54, 301–310 (2015)
Ding, Y., Dawn, W.: Improving the performance of SVM-RFE to select genes in microarray data. BMC Bioinform. 2(12), 1–11 (2015)
Shruti, M., Mishra, D.: SVM-BT-RFE: an improved gene selection framework using Bayesian T-test embedded in support vector machine (recursive feature elimination) algorithm. Karbala Int. J. Modern Sci. 1(2), 86–96 (2015)
Rimah, A., Dorra, B.A., Noureddine, E.: An empirical comparison of SVM and some supervised learning algorithms for vowel recognition. Int. J. Intell. Inf. Process. (IJIIP) 3(1), 1–5 (2012)
Aydadenta, H., Adiwijaya: On the classification techniques in data mining for microarray data classification. In: International Conference on Data and Information Science, Journal of Physics: Conf. Series vol. 971. pp. 1–10 (2018). https://doi.org/10.1088/1742-6596/971/1/012004
Chang, C., Lin, C.: LIBSVM: a library for support vector machines. ACM TIST. 2(3), 27
Soofi, A.A., Awan, A.: Classification techniques in. machine learning: applications and issues. J. Basic Appl. Sci. 13, 459–465 (2017)
Khan, A., Baharudin, B., Lee, L.H., Khan, K.: A review of machine learning algorithms for text-documents classification. J. Adv. Inf. Technol. 1(1), 1–17 (2010)
Bhavsar, H., Panchal, M.H.: A review on support vector machine for data classification. Int. J. Adv. Res. Comput. Eng. Technol. (IJARCET) 1(2), 185–189 (2012)
Devi, A.V., Devaraj, D.V.: Gene expression data classification using support vector machine and mutual information-based gene selection. Procedia Comput. Sci. 47, 13–21 (2015)
Esra, P., Hamparsum, B., Sinan, Ç.: A novel hybrid dimension reduction technique for undersized high dimensional gene expression data sets using information complexity criterion for cancer classification. Comput. Math. Methods Med. 1, 1–14 (2015). https://doi.org/10.1155/2015/370640
Wenyan, Z., Xuewen, L., Jingjing, W.: Feature selection for cancer classification using microarray gene expression data. Biostat. Biometr. J. 1(2), 1–7 (2017)
Balamurugan, M., Nancy, A., Vijaykumar, S.: Alzheimer’s disease diagnosis by using dimensionality reduction based on KNN classifier. Biomed. Pharmacol. J. 10(4), 1823–1830 (2017)
Usman, A., Shazad, A., Javed, F.: Using PCA and factor analysis for dimensionality reduction of bio-informatics data. (IJACSA) Int. J. Adv. Comput. Sci. Appl. 8(5), 515–426 (2017)
Gökmen, Z., et al.: A comprehensive simulation study on classification of RNASeq data. PLoS ONE J. 12(8), 1–24 (2017)
Ian, T.J., Jorge, C.: Principal component analysis: a review and recent developments. Philosoph. Trans. Math. Phys. Eng. Sci. 374, 1–21 (2017)
Nathan, T.J., Andi, D., Katelyn, J.H., Dmitry, K.: Biological classification with RNA-Seq data: Can alternative splicing enhance machine learning classifier? bioRxiv. doi:http://dx.doi.org/10.1101/146340 (2017)
Keerthi, K.V., Surendiran, B.: Dimensionality reduction using Principal Component Analysis for network intrusion detection. Perspect. Sci. 8, 510–512 (2016)
Sofie, V.: A comparative review of dimensionality reduction methods for high-throughput single-cell transcriptomics. Master’s dissertation submitted to Ghent University to obtain the degree of Master of Science in Biochemistry and Biotechnology. Major Bioinformatics and Systems Biology, pp. 1–88 (2017)
Elavarasan, Mani, K.: A survey on feature extraction techniques. Int. J. Innov. Res. Comput. Commun. Eng. 3(1), 1–4 (2015)
Divya, J., Vijendra, S.: Feature selection and classification systems for chronic disease prediction: a review. Egyptian Inform. J. (2018). https://doi.org/10.1016/j.eij.2018.03.002
Awotunde, J.B., Ogundokun, R.O., Ayo, Femi E., Ajamu, Gbemisola J., Adeniyi, E.A., Ogundokun, E.O.: Social media acceptance and use among university students for learning purpose using UTAUT model. In: Borzemski, L., Świątek, J., Wilimowska, Z. (eds.) ISAT 2019. AISC, vol. 1050, pp. 91–102. Springer, Cham (2020). https://doi.org/10.1007/978-3-030-30440-9_10
Ogundokun, R.O.: Evaluation of the scholastic performance of students in 12 programs from a private university in the south-west geopolitical zone in Nigeria. Research 8 (2019)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this paper

Cite this paper
Abdulsalam, S.O. et al. (2020). Performance Evaluation of ANOVA and RFE Algorithms for Classifying Microarray Dataset Using SVM. In: Themistocleous, M., Papadaki, M., Kamal, M.M. (eds) Information Systems. EMCIS 2020. Lecture Notes in Business Information Processing, vol 402. Springer, Cham. https://doi.org/10.1007/978-3-030-63396-7_32
Download citation
DOI: https://doi.org/10.1007/978-3-030-63396-7_32
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-63395-0
Online ISBN: 978-3-030-63396-7
eBook Packages: Computer ScienceComputer Science (R0)