
Abstract
Lung cysts are defined as low-attenuating, gas- or liquid-containing circumscribed spaces within the lung parenchyma that are enclosed by an epithelial or fibrous wall that forms a well-defined interface with the normal lung tissue. A limited number of infectious agents produce cystic lesions in the lung that can mimic the more common diffuse cystic lung diseases (DCLDs) seen in pulmonary clinics. The clinical history and acuity of illness often distinguish these disorders from the more indolent chronic DCLDs. This review describes infectious etiologies of cystic lung disease as well as the mechanisms of cyst development, the clinical and radiographic presentations, and the clinical management, with a special focus on the prototypical DCLD-associated infection, Pneumocystis jirovecii.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Infectious etiologies more typically produce single or multiple, focal or widely scattered cystic lesions than the extensive profusion of cysts that is typical for diffuse thin-walled cystic lung diseases [1, 2]. Mechanistically, three major pathways are postulated to result in infectious lung cyst formation [3], including (1) cystic dilatation of lung structures, (2) parenchymal necrosis, and (3) cystic expansion and displacement of lung structures. This classification will be used to outline the clinical and radiographic presentations of infectious cyst formation in the lung (Table 10.1).
Radiologic Definitions
As defined by the third Fleischner Society criteria [4, 5], a cyst is any low-attenuating circumscribed space, containing gas or liquid, enclosed by an epithelial or fibrous wall and with a well-defined interface with the normal lung tissue. A cavity, in contrast, is a gas-filled space within a pulmonary consolidation or mass that is characterized by thicker walls (> 4 mm) than a cyst (≤ 4 mm). Pneumatoceles are thin-walled gas-filled spaces associated with acute infection or trauma that tend to resolve over time. Other mimics of cystic lung disease include honeycombing and bronchiectasis. Viewed in certain planes, bronchiectatic air spaces may have a similar appearance to cysts; however, the presence of adjacent bronchovascular bundles can help distinguish bronchiectasis from true cysts.
Cystic Dilatation of Lung Structures
Pneumocystis jirovecii-Associated Cystic Lung Disease
Pneumocystis jirovecii , a human-specific fungal pathogen, is an important opportunistic pathogen in immunocompromised patients, particularly those with cell-mediated immunodeficiency [6]. Pneumocystis was first recognized as the pathogen that caused interstitial plasma cell pneumonia in premature, malnourished infants in European orphanages following World War II. Pneumocystis has since emerged as a leading cause of pneumonia in immunocompromised patients of diverse etiologies, such as those infected with the human immunodeficiency virus (HIV) [7, 8].
Pneumocystis pneumonia (PCP) is characterized by the triad of dyspnea, nonproductive cough, and fever [6]. Although a productive cough and chest tightness may occur, purulent sputum should raise suspicion of bacterial infection. HIV-infected patients frequently have prolonged prodromal periods with subtle clinical manifestations, in contrast to other immunocompromised hosts who typically have more acute symptoms of 1- or 2-week duration at presentation. Physical examination reveals varying degrees of respiratory distress. Lung auscultation is typically nonrevealing, although basilar rales may occasionally be present.
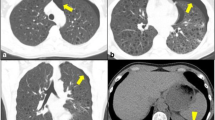
The radiographic findings seen in HIV- and non-HIV-infected individuals with PCP also differ [9]. On the chest radiograph, usual findings include diffuse infiltrates with symmetric reticular or granular opacities emanating from the perihilar regions. Patients with early disease may have a normal chest radiograph, but patchy ground-glass opacification may be detected on high-resolution computerized tomography [10]. The classical findings of extensive ground-glass opacifications are seen in both HIV-infected and non-HIV-infected individuals [11], but lung cysts occur primarily in HIV-infected patients (Fig. 10.1), perhaps related to the long-standing, low-grade inflammation that often exists before a clinical diagnosis of PCP is made in this subset of patients. This radiographic presentation has become less common due to earlier intervention with antiretroviral therapy and PCP prophylaxis. Other unusual manifestations include focal infiltrates, lobar consolidation, nodules, cavities, effusions, and lymphadenopathy. An increased frequency of pneumothorax and apical infiltrates has been noted with the administration of aerosolized pentamidine.

Axial chest CT of an HIV-infected patient with Pneumocystis jirovecii pneumonia showing diffuse cystic changes
Risk factors for PCP include deficiency in cell-mediated immunity; prematurity and malnutrition; primary immunodeficiency disorders, particularly severe combined immunodeficiency disease; infection with HIV; and cytotoxic or immunosuppressive drugs for the treatment of cancer, transplantation, or collagen vascular disorders [8]. Studies of HIV/AIDS patients have documented a significant risk of PCP when CD4 lymphocyte counts fall below 200 cells/μL; other factors associated with a higher risk for PCP included CD4+ cell percentage < 14%, previous episodes of PCP, oral thrush, recurrent bacterial pneumonia, unintentional weight loss, and higher plasma HIV RNA [12, 13]. Corticosteroids are by far the most commonly used immunosuppressive drugs that predispose to PCP in the non-HIV-infected patients. Symptoms often begin after the steroid dose has been tapered.
The principal laboratory abnormality is arterial hypoxemia with an increased alveolar-arterial (PAO2-PaO2) gradient; this is often accompanied by respiratory alkalosis. Blood oxygenation may be normal early in the course of PCP, but desaturation with exercise is common at this stage. Pulmonary function tests may reveal reversible airway obstruction, airway hyperreactivity, or a restrictive abnormality, and the diffusing capacity for carbon monoxide (DLCO) is sensitive in detecting alveolar-capillary block. Serum lactic dehydrogenase (LDH) levels, which reflect the degree of lung injury, increase with disease progression. 1,3-ß-D glucan has excellent sensitivity for PCP; however, it can be falsely elevated by a variety of infectious and noninfectious etiologies.
The natural history of untreated PCP in HIV-infected and other immunocompromised patients is progressive respiratory impairment ending in death. Since recovery from PCP does not confer immunity, patients are at risk of recurrence as long as the predisposing conditions exist; HIV-infected patients are at highest risk for this complication. The cystic lesions typically seen in HIV-infected individuals with late presentation of PCP may regress or disappear following treatment of the P. jirovecii infection and the underlying HIV [14]. Chronic airway disease can develop following PCP, which may be further complicated by bacterial infections.
Histopathologically, PCP is characterized by the presence of a foamy, vacuolated exudate filling the alveoli in lung sections stained with hematoxylin and eosin [15]. The use of stains such as methenamine silver reveals masses of Pneumocystis cystic forms. With severe disease, there may be interstitial fibrosis, edema, and the development of hyaline membranes. Hypertrophy of type II alveolar cells, which suggests tissue repair, is often present; however, other aspects of the host inflammatory response in HIV-infected and other immunocompromised patients are mild and nonspecific. In contrast, premature or malnourished infants display an interstitial plasma cell infiltrate, which led to the historical classification of PCP as an interstitial plasma cell pneumonia. Extrapulmonary lesions of PCP display the typical foamy material found in the lungs [16].
Serological studies have revealed that Pneumocystis has a worldwide distribution and that exposure to the organism occurs early in life [17]. The HIV pandemic changed PCP from a sporadic disease to a problem of major medical and public health importance. Although the incidence of PCP has fallen with widespread chemoprophylaxis and antiretroviral therapy, the organism remains the leading cause of opportunistic infection in HIV patients in industrialized countries. Despite difficulties in obtaining accurate incidence rates in developing countries because of lack of access to medical care and the higher frequency of more virulent infections such as tuberculosis, Pneumocystis is now recognized with increasing frequency in tropical and developing countries, and cystic manifestations may be more prevalent in those populations given the late presentation and the prolonged duration of subclinical disease that occur in under-resourced clinical settings [18].
Trimethoprim-sulfamethoxazole (TMP-SMX) , which acts by inhibiting folic acid synthesis, is considered the drug of choice for all forms of PCP [19]. Therapy is continued for 14 days in non-HIV-infected patients and for 21 days in persons infected with HIV. TMP-SMX is well tolerated by non-HIV-infected patients, whereas more than half of HIV-infected patients experience serious adverse reactions [20]. Several alternative regimens are available for the treatment of mild to moderate cases of PCP (PaO2 > 70 mmHg or a PAO2 – PaO2 < 35 mmHg on breathing room air). TMP plus dapsone and clindamycin plus primaquine are as effective as TMP-SMX [21]. Alternative regimens that are recommended for the treatment of moderate to severe PCP (PaO2 ≤ 70 mmHg or a PAO2 – PaO2 ≥ 35 mmHg) are parenteral pentamidine, parenteral clindamycin plus primaquine, or trimetrexate plus leucovorin [21]. Parenteral clindamycin plus primaquine may be more efficacious than pentamidine [22].
Molecular evidence of resistance to sulfonamides and to atovaquone has emerged among human Pneumocystis isolates [23]. Although prior sulfonamide exposure is a risk factor, this resistance has also occurred in HIV-infected patients who have never received sulfonamides. The outcome of therapy appears to be linked more strongly to traditional measures – e.g., high Acute Physiology, Age, and Chronic Health Evaluation III (APACHE III) scores, need for positive-pressure ventilation, delayed intubation, and development of pneumothorax – than to the presence of molecular markers of sulfonamide resistance [24].
Early institution of antiretroviral therapy in HIV patients presenting with PCP has been associated with improved survival, but careful attention should be devoted to the possible development of the immune reconstitution inflammatory syndrome [25]. HIV-infected patients frequently experience deterioration of respiratory function shortly after receiving anti-Pneumocystis drugs. The adjunctive administration of tapering doses of glucocorticoids to HIV-infected patients with moderate to severe PCP can prevent this problem and improve the rate of survival [26]. For maximal benefit, this adjunctive therapy should be started early in the course of the illness. The use of steroids as adjunctive therapy in HIV-infected patients with mild PCP or in non-HIV-infected patients is currently being evaluated.
Prophylaxis is indicated for HIV-infected patients with CD4+ T cell counts of <200/μL or a history of oropharyngeal candidiasis and for both HIV-infected and non-HIV-infected patients who have recovered from PCP [19]. Prophylaxis may be discontinued in HIV-infected patients once CD4+ T cell counts have risen to >200/uL and remained at that level for ≥3 months. Primary prophylaxis guidelines for immunocompromised hosts who are not infected with HIV are less clear. TMP-SMX is the drug of choice for primary and secondary prophylaxis [27]. This agent also provides protection against toxoplasmosis and some bacterial infections. Alternative regimens are available for individuals intolerant of TMP-SMX, including dapsone, atovaquone, or pentamidine regimens.
Cystic Suppurative/Necrotic Bronchiectatic Air Spaces
Bronchiectasis is defined as irreversible localized or diffuse bronchial dilatation usually resulting from chronic infection, airway obstruction, or congenital bronchial abnormalities. Repeated infection cycles result in failure of normal mucosal and muscular layer healing, and transmural inflammation causes destruction of supportive mural tissues such as smooth muscle and cartilage resulting in bronchomalacia and scarring. Airway dilatation can extend to the lung periphery. Cystic or saccular forms of bronchiectasis result in dilated airways that can be difficult to distinguish from true pulmonary cysts. Infections such as Bordetella pertussis and Mycobacterium tuberculosis are frequently implicated in the generation of bronchiectatic airways; secondary colonization and infection with agents such as Pseudomonas aeruginosa, Staphylococcus aureus, and Aspergillus fumigatus may perpetuate the infection/inflammation cycle resulting in ongoing airway damage.
Parenchymal Necrosis
Infectious cavities may occur during or following a necrotizing pneumonic process due to aerobic and anaerobic infections. Infectious cavities vary in size and number and are characterized by an area of surrounding pulmonary consolidation on chest imaging [28].
Suppurative
Lung Abscess
Lung abscesses are formed by necrosis of pulmonary parenchyma followed by expulsion or resorption of the caseous material. They are classically round or ovoid in shape with air fluid levels and thick walls and may be classified as primary or secondary based on the presence of underlying conditions such as airway obstruction, bronchiectasis, or hematogenous spread and as acute or chronic based on the duration of symptoms prior to presentation. Pneumonia complicated by the formation of multiple small (<2 cm) abscesses is referred to as necrotizing pneumonia [28, 29]. Cavitation and abscess formation are often associated with aspiration, which results in polymicrobial infections of mixed gram-positive and gram-negative anaerobic bacteria and aerobic flora derived from the gingival crevices. Inocula that reach the lower airways may be inadequately cleared because of impaired consciousness or large, recurring bacterial challenges that can occur in the setting of swallowing disorders [28]. Cavitation may also be seen with aggressive gram-positive pathogens such as Streptococcus pneumoniae, Streptococcus anginosus, or Staphylococcus aureus or enteric gram-negative pathogens such as Escherichia coli, Enterobacter cloacae, Klebsiella pneumoniae, and Pseudomonas aeruginosa [30, 31]. Cavities are common features of septic embolization especially with aggressive suppurative organisms such as Staphylococcus aureus following tricuspid valve endocarditis or following embolization from jugular vein thrombophlebitis associated with Fusobacterium necrophorum in Lemierre’s syndrome.
Most patients with lung abscess , especially those with anaerobic polymicrobial infections, present with indolent symptoms evolving over weeks or months. Symptoms include fever, cough, and sputum production often with evidence of chronic systemic disease such as night sweats and weight loss. Physical findings often include evidence of poor dentition, associated conditions that reduce consciousness, digital clubbing, and lung sounds reflecting parenchymal disease and/or pleural effusion.
Microbiological sampling is important in guiding therapy with the caveat that anaerobes may be difficult to isolate and the antimicrobial therapy should usually include anaerobic coverage unless a clear monobacterial process is confirmed [30]. Many empiric combinations have been used, including clindamycin, beta-lactam/beta-lactamase combinations, beta-lactams with metronidazole, or carbapenems. Targeted therapy directed at a single organism without anaerobic coverage should only be considered when no oral flora is isolated. Recent influenza infection increases the risk for a monomicrobial Staphylococcus aureus abscess. Duration of therapy should be at least 3 to 4 weeks, but many will continue until radiographic stability or clearance. Therapy may consist of initial intravenous therapy with conversion to an oral regimen as the infection responds. Surgery is rarely required for uncomplicated lung abscess management but may be required in patients who fail to respond to medical management, suspected malignancy, hemorrhage, extremely large abscesses, or abscesses involving highly resistant organisms [31, 32].
Postinfectious Pneumatocele
These thin-walled , air-filled cavities arise from an inflammatory process that causes central necrosis with subsequent retraction by surrounding pulmonary parenchyma [33]. The process is thought to result from parenchymal necrosis and check valve or ball valve obstruction resulting in air trapping. The most common cause is infection in patients with localized bacterial pneumonia although similar findings may be seen as a complication of blunt trauma or hydrocarbon aspiration [34]. Infectious pneumatoceles are most commonly seen in staphylococcal and streptococcal infections but are also reported with pneumococcal pneumonia and in association with gram-negative infections (Fig. 10.2). Management consists of appropriate treatment of the underlying infection. These transient air-filled cavities typically resolve over time and persistence should raise concern for an alternative diagnosis.

Axial CT of a 57-year-old female with no underlying health issues following recovery from Staphylococcus aureus pneumonia demonstrating bilateral thin-walled pneumatoceles
Chronic Fungal Infections
Chronic fungal infections such as pulmonary aspergillosis and pulmonary mucormycosis can result in dense focal parenchymal consolidation with cavitation [35,36,37,38]. Although the radiographic appearance of fungal cystic lesions can be similar to that of bacterial necrotizing pneumonias, fungal infections classically present with a more insidious onset and are more commonly seen in immunocompromised individuals or patients with underlying chronic lung disease. The foci of parenchymal breakdown within the area of consolidation are often small and multiple in number. Regional hilar adenopathy is not a prominent feature associated with pulmonary aspergillosis or mucormycosis. Pulmonary aspergillus infection may present with a variety of clinical syndromes that vary based on host inflammatory response and immune status. These may range from allergic bronchopulmonary aspergillosis with reactive airways, increased IgE levels, and central bronchiectasis to invasive pulmonary aspergillosis (IPA) in severely immunocompromised individuals. In invasive and semi-invasive aspergillosis, a halo of low attenuation may be seen with early invasive lesions, and this may progress to cavitary lesions with a characteristic air crescent sign due to cavitary necrosis on CT imaging [39]. These features are characteristic of IPA, but similar findings may be seen with other angioinvasive organisms including Mucorales, Fusarium, and Scedosporium [40]. Chronic necrotizing aspergillosis associated with cavitary infiltrates may also be seen in individuals with chronic lung disease and lesser degrees of immunosuppression such as alcoholism or diabetes [37].
A confirmed diagnosis requires tissue biopsy showing hyphal invasion and a positive culture for the appropriate organisms , although invasive procedures to achieve this level of certainty may be contraindicated in severely ill immunosuppressed patients due to potential risks. Cultures of Aspergillus spp. or other invasive fungi from expectorated sputum of high-risk individuals with compatible radiographic findings are supportive of the diagnosis, although Aspergillus is often cultured from patients in whom no clinical illness is apparent so positive cultures with low risk for invasive fungal infection should be interpreted with caution. Antibody detection plays little role in the diagnosis of IPA as immunocompromised individuals at greatest risk may mount poor serological responses. Detection of circulating galactomannan has contributed substantially to the diagnosis of invasive aspergillosis. Studies report sensitivity of 40%–89% depending on the extent of disease, prior antifungal therapy, number of samples per patient, and other variables. Specificity is also high although false positives have been reported associated with dietary intake, antibiotics such as piperacillin/tazobactam, and bacterial infection with Bifidobacterium . Other potential markers such as B-glucan, approved for clinical diagnosis of invasive fungal infections, may be of value although species-specific sensitivity varies [41].
Historically, the efficacy of antifungal therapy has been poor, likely related in part to the often compromised immune status of the host and the advanced stage of infection at the time of diagnosis. Triazoles such as voriconazole, posaconazole, and isavuconazole remain primary therapy for IPA, while amphotericin B preparations remain the first line for Mucorales infections. Triazoles may also be used for these infections although experience is more limited and variable. Adjunctive measures such as reduction in immunosuppressive agents and improvement in the underlying host defenses are crucial.
Chronic Bacterial Infections
Chronic bacterial infections such as actinomycosis may present with persistent consolidation or a mass which may contain areas of cavitation [42]. Pulmonary actinomycosis is caused by microaerophilic or anaerobic actinomycetes and may cross tissue planes resulting in pleural disease or invade the chest wall resulting in pleuropulmonary fistulas and sinuses draining purulent fluid containing characteristic sulfur granules. Similar subacute presentations of pulmonary infection with cavitation may be seen with organisms such as the aerobic actinomycetes, Nocardia, and Rhodococcus. As with other suppurative cavitary infections, management requires an accurate microbiological diagnosis and prolonged antimicrobial therapy.
Granulomatous Parenchymal Necrosis
Infections resulting in granulomatous inflammatory response may manifest as nodules which cavitate during primary infections or cavitary lesions associated with postprimary infection and secondary reactivation disease. Tuberculous cavitary lesions are usually seen in postprimary disease, while cavitary nodules in nontuberculous mycobacterial infection are predominantly seen as a manifestation of progressive primary disease. Cavitation of fungal nodules may be seen during both primary infection and in reactivation disease.
Tuberculous Cavitary Lung Disease
Cavitary pulmonary tuberculous lesions are seen primarily in the apical-posterior portion of the upper lobes and less frequently in the apex of the lower lobes [43]. The pathologic features of tuberculosis are the result of the balance between the degree of tissue hypersensitivity and bacterial antigen load. When the population of activated lymphocytes and macrophages with enhanced microbicidal activity is associated with a low bacterial antigen load, highly organized collections of lymphocytes, macrophages, Langerhans giant cells, and fibroblasts form granulomas resulting in containment of infection. When both antigen load and tissue hypersensitivity are high, epithelioid and giant cells are sparse, and exudative reactions with poorly organized granulomas with central caseous necrosis form. The caseous necrosis is unstable and may liquify and discharge through the bronchial tree resulting in tuberculous cavitary disease with extremely high bacterial burdens. Apical-posterior distribution is attributed to the higher oxygen tensions and lower lymphatic flow in the apices, resulting in enhanced bacterial proliferation and bacterial antigen retention, respectively. As tissue hypersensitivity develops during postprimary infection or reactivation, the environment is conducive to tissue necrosis and cavitation. Fibrous encapsulation and inelasticity of the surrounding lung tissue prevent collapse of the cavity. The cavitary lesions may progress due to the tendency of apical foci to liquify, the high organism burden within pulmonary cavities, and the spread of caseous material through the bronchial tree. Progression from limited infiltrates to advanced cavitary disease may occur rapidly over a period of months.
Radiographic findings of patchy or nodular infiltrates in the subapical-posterior areas of the upper lobe are suggestive of tuberculosis in at-risk populations especially if bilateral and associated with cavitation. Cavities may be better visualized by CT. Air-fluid levels are uncommon in tuberculous cavitation. Active disease is often characterized by a mixture of small sharply defined granulomatous lesions, softer exudative lesions with indistinct borders, and fibrous scars with sharp borders.
The diagnosis of tuberculosis is made by the demonstration of M. tuberculosis by acid-fast stain or auramine-rhodamine fluorescent staining in respiratory secretions and confirmed by growth and speciation [44]. Induced sputum or bronchoscopically derived samples may increase the yield if the diagnosis is not obtained from expectorated samples. Nucleic acid amplification tests offer an additional technique for the direct detection of M. tuberculosis in clinical specimens and distinction between tuberculous and nontuberculous mycobacterium cultured from clinical samples. Treatment typically consists of four drug therapy, followed by two drug-directed therapy based on sensitivities; however, the regimen may vary based on resistance patterns within the community.
Nontuberculous Mycobacterial Infections
Nontuberculous mycobacterial (NTM) infections may present with a variety of radiographic findings including bronchiectasis and fibronodular infiltrates often involving the right middle lobe and left lingular segments [43]. Cavitation of nodules is not uncommonly observed. Large thin-walled cysts may also be seen [45]. These may occur predominantly in the upper lobes in individuals with chronic underlying lung disease, chronic smokers, and alcoholics. Multiple nontuberculous mycobacterial species have been associated with chronic pulmonary infection including rapidly growing mycobacteria, such as M. fortuitum, M. abscessus, and M. chelonae, and slow-growing mycobacteria, such as Mycobacterium avium complex and M. kansasii. Predisposing factors for pulmonary NTM disease include underlying lung disease, alcoholism, and cystic fibrosis. Mycobacterium avium infection is also found in a subset of middle-aged and elderly women with no predisposing conditions (Lady Windermere syndrome, named after the leading lady in an Oscar Wilde play). The diagnosis is made based on the isolation of NTMs from multiple samples of respiratory secretions in the presence of compatible radiographic and clinical findings [46, 47]. Treatment varies based on the mycobacterium species. As environmental organisms, NTMs are not uncommonly isolated as incidental findings in individuals without compatible radiographic findings or clinical symptoms, and this asymptomatic colonization or transient infection does not warrant treatment.
Endemic Fungal Infections
Endemic pulmonary fungal infections can arise through the inhalation of fungal spores of regionally prevalent organisms such as Histoplasma capsulatum in the Midwest, Coccidioides spp. in the Southwestern United States, and Blastomyces dermatitidis in the Southeastern United States and upper Midwest [48,49,50,51]. Most acute pulmonary infections due to endemic fungi resolve spontaneously, but nodules and cavities may be seen as residual findings [52, 53]. Solitary peripheral nodules are seen in about 4% of individuals with coccidioidomycosis [54, 55]. These may liquify and drain resulting in a solitary thin-walled peripheral cavity which may be asymptomatic or present with cough or localized pain. About half close spontaneously over a period of years. Underlying lung disease such as COPD, chronic smoking, diabetes, and alcohol abuse predispose to the development of chronic pulmonary infection, such as chronic fibrocavitary coccidioidal pneumonia or chronic cavitary or non-cavitary histoplasmosis. Blastomycosis more commonly presents as a chronic pneumonia or mass lesion and may mimic bronchogenic carcinoma [48]. Cavitation may be found with the primary lobar pneumonia, as solitary cavitary nodules or as fibronodular infiltrates with cavitation (Fig. 10.3). Similar findings may be seen in paracoccidioidosis and sporotrichosis. The diagnosis is dependent on the isolation and identification of the specific fungal pathogen, histological diagnosis, or specific serological assays [56, 57].

Axial CT of a 58-year-old male with history of pulmonary blastomycosis with right-sided cystic lesion
Cystic Expansion with Lung Displacement
Hydatid Cysts
Hydatid cysts caused by infection with the metacestode stage of Echinococcus tapeworms are most commonly described in sheep-raising areas and are endemic around the Mediterranean basin, Australia, and South America. Approximately 50% of detected cases occur in asymptomatic individuals, and many more cases likely remain undiagnosed [58,59,60,61]. Children and young adolescents are more likely to remain asymptomatic for reasons that are not fully clear. When symptomatic, presenting symptoms may include cough, chest pain, and hemoptysis. Hydatid cysts in the lung are characteristically smooth-walled ranging from 2 mm to 1 cm in thickness. Internal scolexes may be visible on CT imaging. Pulmonary cysts are more commonly multiple than solitary. The diagnosis is based on appropriate epidemiological exposure and supported by serological assays. Unfortunately, sensitivity of available serological assays is generally lower for pulmonary disease than hepatic disease, ranging from 51% to 84% [62]. There is no correlation between serological results and the size or number of cysts, and negative serology does not rule out the diagnosis of echinococcosis. The principal complication of pulmonary Echinococcus infection is cyst rupture with spilling of cyst contents into the bronchial tree or pleural cavity. Cyst rupture may be associated with fever and acute hypersensitivity reactions or a more insidious increase in cough or chest pain with bronchial rupture and pleural effusion, empyema, or pneumothorax with pleural rupture. Management usually consists of surgical removal of the cyst in conjunction with antiparasitic therapy with albendazole or mebendazole.
Paragonimiasis
Paragonimiasis is an infection caused by the lung flukes of the Paragonimus genus transmitted via consumption of raw or undercooked crab or crayfish. All forms of Paragonimus result in similar disease and clinical presentations including Paragonimus westermani from Asia, P. africanus from West Africa, and P. mexicanus from Central and South America. Most infections are subclinical, but symptoms can occur with heavy organism burdens [63,64,65] and include fever, malaise, diarrhea, and abdominal pain, especially early in infection. Symptoms typically resolve with no therapy but may be followed by the development of chest pain and dyspnea associated with exudative eosinophilic pleural effusions. Late phases of pulmonary infection may last for many years without outward evidence of chronic illness, although recurrent hemoptysis can occur. Fever and peripheral eosinophilia are generally absent. Chest radiograph or CT may include ring shadow lesions due to the relative lucency of cystic cavities. The cystic lesions are often located peripherally and are more common in the mid and lower lung zones helping distinguish them from tuberculous cavitation. The presence of pleural effusions (reported in 20–60% of cases) in conjunction with multiple cysts is suggestive of the diagnosis. Perilesional nodules and worm migration tracts are also reported.
The diagnosis is made based on the combination of appropriate environmental exposure, positive serologies, microscopy, and imaging. Early disease may manifest with peripheral eosinophilia though this is rarely seen in later disease. Microscopic examination of expectorated sputum or bronchoalveolar lavage will usually reveal the characteristic eggs with a thick yellowish-brown shell and prominent operculum. A 24-hour sputum collection enhances the sensitivity of detection of eggs. ELISA assays are highly sensitive and specific (92% and > 90%, respectively). These assays are not useful for monitoring response to therapy as antibody levels may not decrease for many years after treatment. Therapy with praziquantel (25 mg/kg three times a day for 3 days) is the treatment of choice for all species of paragonimiasis. Treatment is indicated for symptomatic as well as asymptomatic infections, given the potential for later complications. Cure rates of pulmonary infection approach 100%. Infection can be prevented by avoiding eating raw or undercook crab and crayfish, and strict hygiene measures to prevent fecal contamination of water sources in order to limit transmission.
Conclusion
A number of infectious diseases can produce cystic changes in the lung parenchyma by a variety of pathophysiological mechanisms. While some of these disorders can mimic the common DCLDs, typically, the tempo of disease progression and other associated constitutional symptoms can provide a clue to the underlying etiology. A predominance of cavitary lesions and focal rather than diffuse cystic change can also help ascertain the underlying cause. It is important for clinicians to have a high index of suspicion for infectious etiologies, as these may persist in the chronic phase and may benefit from definite antimicrobial treatments.
Key Learning Points
-
A variety of pulmonary infections can produce cystic radiologic changes that may mimic other diffuse cystic lung diseases.
-
Clues that might lead clinicians to think about underlying infections may include acuity of symptoms, associated constitutional symptoms such as fever, other symptoms such as purulent sputum, underlying immunosuppressed states such as HIV, coexistence of cavitary and/or nodular lesions, and focal rather than diffuse disease.
-
Pneumocystis jirovecii is the typical example of infectious cystic lung disease. Cystic lung disease from pneumocystis is seen more commonly in patients with HIV as opposed to other immunocompromised states and is more of a late manifestation.
-
Bacterial infections such as Staphylococcus aureus, fungal infections such as coccidioidomycosis, and parasitic infections such as paragonimiasis are other common infectious etiologies of cystic lung disease.
-
The treatment involves antimicrobials directed at the causative organisms and often leads to resolution of the radiologic findings.
-
It is important for clinicians to consider infectious causes when evaluating patients with cystic lung disease as cysts may persist in the chronic phase and definitive antimicrobial treatment may be beneficial.
Abbreviations
- COPD:
-
Chronic obstructive pulmonary disease
- CT:
-
Computerized tomography
- HIV:
-
Human immunodeficiency virus
- IPA:
-
Invasive pulmonary aspergillosis
- MRI:
-
Magnetic resonance imaging
- NTM:
-
Nontuberculous mycobacteria
- PCP:
-
Pneumocystis pneumonia
- TMP-SMX:
-
Trimethoprim-sulfamethoxazole
References
Ryu JH, Tian X, Baqir M, Xu K. Diffuse cystic lung diseases. Front Med. 2013;7(3):316–27.
Seaman DM, Meyer CA, Gilman MD, McCormack FX. Diffuse cystic lung disease at high-resolution CT. AJR Am J Roentgenol. 2011;196(6):1305–11.
Boddu P, Parimi V, Taddonio M, Kane JR, Yeldandi A. Pathologic and radiologic correlation of adult cystic lung disease: a comprehensive review. Pathol Res Int. 2017;2017:3502438.
Tuddenham WJ. Glossary of terms for thoracic radiology: recommendations of the nomenclature Committee of the Fleischner Society. AJR Am J Roentgenol. 1984;143(3):509–17.
Hansell DM, Bankier AA, MacMahon H, McLoud TC, Muller NL, Remy J. Fleischner society: glossary of terms for thoracic imaging. Radiology. 2008;246(3):697–722.
Thomas CF Jr, Limper AH. Current insights into the biology and pathogenesis of Pneumocystis pneumonia. Nat Rev Microbiol. 2007;5(4):298–308.
Cisse OH, Pagni M, Hauser PM. De novo assembly of the Pneumocystis jirovecii genome from a single bronchoalveolar lavage fluid specimen from a patient. MBio. 2012;4(1):e00428–12.
Morris A, Norris KA. Colonization by Pneumocystis jirovecii and its role in disease. Clin Microbiol Rev. 2012;25(2):297–317.
Hardak E, Brook O, Yigla M. Radiological features of Pneumocystis jirovecii pneumonia in immunocompromised patients with and without AIDS. Lung. 2010;188(2):159–63.
Huang L, Stansell J, Osmond D, Turner J, Shafer KP, Fulkerson W, et al. Performance of an algorithm to detect Pneumocystis carinii pneumonia in symptomatic HIV-infected persons. Pulmonary complications of HIV infection study group. Chest. 1999;115(4):1025–32.
Kanne JP, Yandow DR, Meyer CA. Pneumocystis jiroveci pneumonia: high-resolution CT findings in patients with and without HIV infection. AJR Am J Roentgenol. 2012;198(6):W555–61.
Kaplan JE, Hanson DL, Navin TR, Jones JL. Risk factors for primary Pneumocystis carinii pneumonia in human immunodeficiency virus-infected adolescents and adults in the United States: reassessment of indications for chemoprophylaxis. J Infect Dis. 1998;178(4):1126–32.
Kaplan JE, Hanson DL, Jones JL, Dworkin MS. Viral load as an independent risk factor for opportunistic infections in HIV-infected adults and adolescents. AIDS. 2001;15(14):1831–6.
Ferre C, Baguena F, Podzamczer D, Sanchez C, Viladrich PF, Garau J, et al. Lung cavitation associated with Pneumocystis carinii infection in the acquired immunodeficiency syndrome: a report of six cases and review of the literature. Eur Respir J. 1994;7(1):134–9.
Saldana MJ, Mones JM. Pulmonary pathology in AIDS: atypical Pneumocystis carinii infection and lymphoid interstitial pneumonia. Thorax. 1994;49(Suppl):S46–55.
Karam MB, Mosadegh L. Extra-pulmonary Pneumocystis jiroveci infection: a case report. Braz J Infect Dis. 2014;18(6):681–5.
Peglow SL, Smulian AG, Linke MJ, Pogue CL, Nurre S, Crisler J, et al. Serologic responses to Pneumocystis carinii antigens in health and disease. J Infect Dis. 1990;161(2):296–306.
Lowe DM, Rangaka MX, Gordon F, James CD, Miller RF. Pneumocystis jirovecii pneumonia in tropical and low and middle income countries: a systematic review and meta-regression. PLoS One. 2013;8(8):e69969.
White PL, Backx M, Barnes RA. Diagnosis and management of Pneumocystis jirovecii infection. Expert Rev Anti-Infect Ther. 2017;15(5):435–47.
Safrin S, Finkelstein DM, Feinberg J, Frame P, Simpson G, Wu A, et al. Comparison of three regimens for treatment of mild to moderate Pneumocystis carinii pneumonia in patients with AIDS. A double-blind, randomized, trial of oral trimethoprim-sulfamethoxazole, dapsone-trimethoprim, and clindamycin-primaquine. ACTG 108 study group. Ann Intern Med. 1996;124(9):792–802.
Toma E, Fournier S, Dumont M, Bolduc P, Deschamps H. Clindamycin/primaquine versus trimethoprim-sulfamethoxazole as primary therapy for Pneumocystis carinii pneumonia in AIDS: a randomized, double-blind pilot trial. Clin Infect Dis. 1993;17(2):178–84.
Smego RA Jr, Nagar S, Maloba B, Popara M. A meta-analysis of salvage therapy for Pneumocystis carinii pneumonia. Arch Intern Med. 2001;161(12):1529–33.
Kazanjian PH, Fisk D, Armstrong W, Shulin Q, Liwei H, Ke Z, et al. Increase in prevalence of Pneumocystis carinii mutations in patients with AIDS and P. carinii pneumonia, in the United States and China. J Infect Dis. 2004;189(9):1684–7.
Weng L, Huang X, Chen L, Feng LQ, Jiang W, Hu XY, et al. Prognostic factors for severe Pneumocystis jiroveci pneumonia of non-HIV patients in intensive care unit: a bicentric retrospective study. BMC Infect Dis. 2016;16(1):528.
Fei MW, Kim EJ, Sant CA, Jarlsberg LG, Davis JL, Swartzman A, et al. Predicting mortality from HIV-associated Pneumocystis pneumonia at illness presentation: an observational cohort study. Thorax. 2009;64(12):1070–6.
National Institutes of Health-University of California Expert Panel for Corticosteroids as Adjunctive Therapy for Pneumocystis P. Consensus statement on the use of corticosteroids as adjunctive therapy for pneumocystis pneumonia in the acquired immunodeficiency syndrome. N Engl J Med. 1990;323(21):1500–4.
Ahuja J, Kanne JP. Thoracic infections in immunocompromised patients. Radiol Clin N Am. 2014;52(1):121–36.
Pennza PT. Aspiration pneumonia, necrotizing pneumonia, and lung abscess. Emerg Med Clin North Am. 1989;7(2):279–307.
Landay MJ, Christensen EE, Bynum LJ, Goodman C. Anaerobic pleural and pulmonary infections. AJR Am J Roentgenol. 1980;134(2):233–40.
Bartlett JG. The role of anaerobic bacteria in lung abscess. Clin Infect Dis. 2005;40(7):923–5.
Duncan C, Nadolski GJ, Gade T, Hunt S. Understanding the lung abscess microbiome: outcomes of percutaneous lung parenchymal abscess drainage with microbiologic correlation. Cardiovasc Intervent Radiol. 2017;40(6):902–6.
Hirshberg B, Sklair-Levi M, Nir-Paz R, Ben-Sira L, Krivoruk V, Kramer MR. Factors predicting mortality of patients with lung abscess. Chest. 1999;115(3):746–50.
Al-Saleh S, Grasemann H, Cox P. Necrotizing pneumonia complicated by early and late pneumatoceles. Can Respir J. 2008;15(3):129–32.
Yang TC, Huang CH, Yu JW, Hsieh FC, Huang YF. Traumatic pneumatocele. Pediatr Neonatol. 2010;51(2):135–8.
Danion F, Aguilar C, Catherinot E, Alanio A, DeWolf S, Lortholary O, et al. Mucormycosis: new developments into a persistently devastating infection. Semin Respir Crit Care Med. 2015;36(5):692–705.
Gefter WB, Weingrad TR, Epstein DM, Ochs RH, Miller WT. “semi-invasive” pulmonary aspergillosis: a new look at the spectrum of aspergillus infections of the lung. Radiology. 1981;140(2):313–21.
Parra I, Remacha A, Rezusta A, Suarez D, Suarez J, Herreras JA, et al. Chronic necrotizing pulmonary aspergillosis. Med Mycol. 2004;42(4):369–71.
Soeiro Ade M, Hovnanian AL, Parra ER, Canzian M, Capelozzi VL. Post-mortem histological pulmonary analysis in patients with HIV/AIDS. Clinics (Sao Paulo). 2008;63(4):497–502.
Westcott J, Davis SD, Fleishon H, Gefter WB, Henschke CI, McLoud TC, et al. Acute respiratory illness in HIV-positive patients. American College of Radiology. ACR Appropriateness Criteria. Radiology. 2000;215(Suppl):649–53.
Rahman FU, Irfan M, Fasih N, Jabeen K, Sharif H. Pulmonary scedosporiosis mimicking aspergilloma in an immunocompetent host: a case report and review of the literature. Infection. 2016;44(1):127–32.
Smith JA, Kauffman CA. Pulmonary fungal infections. Respirology. 2012;17(6):913–26.
Smego RA Jr, Foglia G. Actinomycosis. Clin Infect Dis. 1998;26(6):1255–61. quiz 62–3
Goo JM, Im JG. CT of tuberculosis and nontuberculous mycobacterial infections. Radiol Clin N Am. 2002;40(1):73–87. viii
Nahid P, Dorman SE, Alipanah N, Barry PM, Brozek JL, Cattamanchi A, et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America clinical practice guidelines: treatment of drug-susceptible tuberculosis. Clin Infect Dis. 2016;63(7):e147–e95.
Larsson LO, Polverino E, Hoefsloot W, Codecasa LR, Diel R, Jenkins SG, et al. Pulmonary disease by non-tuberculous mycobacteria – clinical management, unmet needs and future perspectives. Expert Rev Respir Med. 2017;11(12):977–89.
Philley JV, DeGroote MA, Honda JR, Chan MM, Kasperbauer S, Walter ND, et al. Treatment of non-tuberculous mycobacterial lung disease. Curr Treat Options Infect Dis. 2016;8(4):275–96.
Haworth CS, Banks J, Capstick T, Fisher AJ, Gorsuch T, Laurenson IF, et al. British Thoracic Society guidelines for the management of non-tuberculous mycobacterial pulmonary disease (NTM-PD). Thorax. 2017;72(Suppl 2):ii1–ii64.
Bradsher RW Jr. Pulmonary blastomycosis. Semin Respir Crit Care Med. 2008;29(2):174–81.
Bradsher RW. Clinical features of blastomycosis. Semin Respir Infect. 1997;12(3):229–34.
Galgiani JN, Ampel NM, Blair JE, Catanzaro A, Johnson RH, Stevens DA, et al. Coccidioidomycosis. Clin Infect Dis. 2005;41(9):1217–23.
Kauffman CA. Endemic mycoses: blastomycosis, histoplasmosis, and sporotrichosis. Infect Dis Clin N Am. 2006;20(3):645–62. vii
Valdivia L, Nix D, Wright M, Lindberg E, Fagan T, Lieberman D, et al. Coccidioidomycosis as a common cause of community-acquired pneumonia. Emerg Infect Dis. 2006;12(6):958–62.
Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev. 2007;20(1):115–32.
Kim MM, Blair JE, Carey EJ, Wu Q, Smilack JD. Coccidioidal pneumonia, Phoenix, Arizona, USA, 2000–2004. Emerg Infect Dis. 2009;15(3):397–401.
Jude CM, Nayak NB, Patel MK, Deshmukh M, Batra P. Pulmonary coccidioidomycosis: pictorial review of chest radiographic and CT findings. Radiographics. 2014;34(4):912–25.
Galgiani JN, Ampel NM, Blair JE, Catanzaro A, Geertsma F, Hoover SE, et al. 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of Coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112–46.
Chapman SW, Dismukes WE, Proia LA, Bradsher RW, Pappas PG, Threlkeld MG, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46(12):1801–12.
Arinc S, Kosif A, Ertugrul M, Arpag H, Alpay L, Unal O, et al. Evaluation of pulmonary hydatid cyst cases. Int J Surg. (London, UK. 2009;7(3):192–5.
Morar R, Feldman C. Pulmonary echinococcosis. Eur Respir J. 2003;21(6):1069–77.
Kervancioglu R, Bayram M, Elbeyli L. CT findings in pulmonary hydatid disease. Acta Radiol. 1999;40(5):510–4.
Santivanez S, Garcia HH. Pulmonary cystic echinococcosis. Curr Opin Pulm Med. 2010;16(3):257–61.
Ortona E, Rigano R, Buttari B, Delunardo F, Ioppolo S, Margutti P, et al. An update on immunodiagnosis of cystic echinococcosis. Acta Trop. 2003;85(2):165–71.
Lane MA, Marcos LA, Onen NF, Demertzis LM, Hayes EV, Davila SZ, et al. Paragonimus kellicotti flukes in Missouri, USA. Emerg Infect Dis. 2012;18(8):1263–7.
Kanpittaya J, Sawanyawisuth K, Vannavong A, Intapan PM, Maleewong W, Zhang W, et al. Different chest radiographic findings of pulmonary paragonimiasis in two endemic countries. Am J Trop Med Hyg. 2010;83(4):924–6.
Kim TS, Han J, Shim SS, Jeon K, Koh WJ, Lee I, et al. Pleuropulmonary paragonimiasis: CT findings in 31 patients. AJR Am J Roentgenol. 2005;185(3):616–21.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Miller-Handley, H., Smulian, A.G. (2021). Infectious Etiologies of Diffuse Cystic Lung Diseases. In: Gupta, N., Wikenheiser-Brokamp, K.A., McCormack, F.X. (eds) Diffuse Cystic Lung Diseases. Respiratory Medicine. Humana, Cham. https://doi.org/10.1007/978-3-030-63365-3_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-63365-3_10
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-63364-6
Online ISBN: 978-3-030-63365-3
eBook Packages: MedicineMedicine (R0)