
Abstract
Coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is highly pathogenic with relatively high mortality and morbidity. In addition to pneumonia, acute respiratory distress syndrome, and microembolic disorder, a high proportion of patients with SARS-CoV-2 develop lymphopenia and cytokine storm disorder. This review explores the underlying mechanisms behind the pathogenesis of SARS-CoV-2, especially the immune mechanisms, which could be potentially used as therapeutic targets for the management of COVID-19.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Infection of SARS-CoV-2 in the lungs results in stimulation of macrophages and monocytes, the release of cytokines, and adaptive immune responses [1, 2]. In certain instances, this immune response can overcome viral infection and the patient recovers. However, in some instances, a dysfunctional immune response will result in pneumonia and multiorgan failure [3]. Dysfunctional immune response in some patients activates a cytokine storm which results in widespread inflammation of the lungs. There is some evidence to show that lymphopenia and the cytokine storm result in a worse prognosis. Managing the inflammatory response due to dysfunctional immune response is found to be as crucial as controlling the infection. Medications that prevent viral infection as well as those which regulate defective immune defenses can potentially prevent development of multiorgan failure [4].
2 Virus Entry
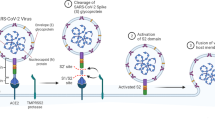
SARS-CoV-2 is a solitary strand RNA virus with four main basic proteins: spike (S), envelope (E), nucleocapsid (N), and membrane (M) proteins which infect human respiratory tract cells. The virus enters cells through binding of the S protein to the angiotensin-converting enzyme 2 (ACE2) receptor following S protein priming by the host transmembrane protease, serine 2 (TMPRSS2). Another receptor, CD147, is also associated with SARS-CoV-2 entry into the host cells [5]. After these processes, the virus enters the cells by endocytosis, and the viral RNA is discharged into the cytosol. The virus utilizes the cell hardware for multiplication and then erupts from the cell through exocytosis. Patients with relatively high viral burdens tend to develop more severe COVID-19 disease. Furthermore, downregulation and shedding of ACE2 by the viral S protein may disrupt the renin-angiotensin framework and increase vascular penetrability resulting in more severe lung injury [6].
3 Immune Responses in COVID-19 Disease
The immune responses induced by SARS-CoV-2 have two principal stages. The initial stage is the protective response, and the second stage is the inflammatory response. COVID-19 causes an imbalance of the immune system and hyperactivation of the immune response. The adaptive immune response is needed during the asymptomatic phase to get rid of infection [7]. Thus, strategies related to improving the immune system are essential at this point. Patients must be in good health and have a favorable genetic background (e.g., HLA) that could contribute to the first line of defense against the virus [8, 9]. However, if this response is not adequate, the virus will spread mainly to tissues with high ACE2 expression, such as the intestines and kidneys. The infected cells can cause latent pulmonary inflammation which is principally mediated by pro-inflammatory macrophages and granulocytes. At this stage, strategies to reduce inflammation could be potentially helpful. Effective intervention at this stage will bring down the virus load and prevent hyperinflammation. In this regard, type I interferon (IFN) is crucial for early viral clearance to minimize viral replication, T-cell exhaustion, and cytokine storms (Fig. 10.1) [10].

The invading SARS-CoV-2 virus induces nonserious symptoms during the incubation period, which elicits defensive immune responses. Successful clearance of viruses relies on the state of safety. If the affected individual’s general health condition does not remove the infection, then the patient reaches the severe stage of an intense and harmful inflammatory reaction, particularly in the lungs
3.1 Innate Immune Responses in COVID-19: Role of Cytokines and Chemokines
SARS-CoV-2 stimulates expression of numerous IFN-stimulated genes (ISGs) [11, 12]. These have an immunopathogenic capacity through overexpression of the inflammatory genes. Type I IFN is essential for protection against viral diseases as it facilitates intracellular destruction of RNA and recovery from viral infections, induces tissue repair, and activates a continuous adaptive immune response [13]. Type I IFN is delivered by plasmacytoid dendritic cells (pDCs) because they are less susceptible to active viral infection and virus-mediated cytotoxicity. They also release inflammatory cytokines, including tumor necrosis factor (TNF)-α and interleukin (IL)-6 to control the T-cell response. PDCs disperse immune cells that serve as guardians and are activated after physical contact within infected cells as part of a process called the interferogenic synapse. This results in transition of pathogen-associated molecular pattern molecules (PAMPs) to toll-like receptor 7 (TLR7) receptors in pDCs [14]. This synapse facilitates development of type I IFN at the infected area, therefore restricting viral replication and the potentially harmful systemic response. A reduced IFN type I response is associated with higher COVID-19 severity. Hypercytokinemia in COVID-19 patients is related to the severity of COVID-19 disease [15]. The most crucial cytokines in this regard are chemokines, such as neutrophil-recruiting chemokines, and monocyte attractants [16].
3.2 Monocytes and Macrophages in COVID-19
Bronchoalveolar fluid (BALFs) from individuals with severe COVID-19 showed an increased expression of CCL2 and CCL7, two of the most essential chemokines for recruitment of CCR2+ monocytes. BALF analysis by single-cell RNA sequencing of moderate COVID-19 patients reported increased concentrations of mononuclear phagocytes [11]. In COVID-19 patients, there is an increased concentration of the group of macrophages that are enriched in tissue-repaired genes and promote generation of fibrosis, as found in liver cirrhosis. This suggests that the pathogenicity of invading macrophages may go farther than acute inflammation to fibrosis in ventilated patients [11, 17]. Park et al. have referenced macrophages as a Trojan horse in COVID-19. ACE2-expressive CD68+CD169+ macrophages were found in the splenic marginal zone and marginal sinuses of the lymph node which expresses nucleoprotein antigen SARS-CoV-2 and produces a significant rise in IL-6 concentrations. This suggests CD169+ macrophages can facilitate viral spread during SARS-CoV-2 disease, heighten inflammation, and activation-induced lymphocytic cell death [18].
3.3 Role of Complement in COVID-19
Complement is one of the essential factors helpful in shielding from pathogens. However, the excessive and deregulated response of complement can trigger injury to the tissue. Complement is both an integral part of the innate immune system of the pathogens and a pro-inflammatory reaction orchestrator. C3-lacking mice infected with SARS-CoV show reduced pulmonary injury, lower neutrophil and monocyte infiltration, and diminished cytokine and chemokine levels in both the lungs and the sera [19]. This suggests that the inactivation of C3 in the inflammatory lung may likewise reduce the severity of SARS-CoV-2 injury in tissues. The reduction in lung-invading neutrophils and the reduced intrapulmonary and plasma IL-6 levels observed in C3-deficient mice infected with SARS-CoV suggests the opportunities for utilizing C3 blockers with anti-IL-6 agents [19]. C3 inhibition can simultaneously block the development of C3a and C5a, as well as intrapulmonary activation of C3 and the release of IL-6 from alveolar macrophages or other cells expressing C3a (C3aR) and C5a (C5aR) receptors, thereby limiting lung injury. Ex vivo experiments of whole blood infection with the AMY-101 C3 inhibitor have demonstrated that it will reduce IL-6 levels. The lung biopsy specimens from individuals with extreme COVID-19 revealed extensive activation of complement, characterized by the production of C3a and deposition of C3 fragment. There was also a rise in the serum C5a levels [20]. Patients with an anti-C5a antibody showed better lung oxygenation and diminished inflammatory responses [21, 22].
3.4 B-Cell Immunity
In patients with COVID-19, B-cell reactions emerge around the same time as T follicular helper cell responses, beginning about 1 week after the inception of symptoms. A B-cell response mainly occurs in patients with SARS-CoV disease for the most part against the viral N protein [23]. Antibody responses to the S protein were seen within 4–8 days of the beginning of symptoms. Neutralizing responses of antibodies against the S antigen begin to increase by week 2, and by week 3, most patients develop neutralizing antibodies. However, this does not appear to result in durable SARS-CoV-2 antibodies [23]. The neutralization of the virus is viewed as a fundamental mode of action for antibodies although the specific titer of antibodies remains unresolved [10, 23].
3.5 T-Cell Immunity
CD8+ T cells are expected to attack and destroy virus-infected cells specifically, while CD4+ T cells are essential in the activation of both CD8+ T cells and B cells. CD4+ T cells likewise produce cytokines to activate immune cells [10]. It seems that SARS-CoV-2 can cause a protective immune response mediated by the T lymphocyte, in comparison to other CoVs. Patients with COVID-19 have increased monocytes and T cells in the lungs and a significant reduction in the amounts of CD4+ and CD8+ T cells in the peripheral blood due to insufficient activation as seen by elevated HLA-DR and CD38 double-positive fractions [9]. Such outcomes demonstrate that T cells are attracted to monitor virus infection away from the blood and toward the affected region. Likewise, the intense stage response in patients with SARS-CoV is associated with a significant reduction in CD4+ T and CD8+ T cells [8]. Albeit additional precautionary measures ought to be taken in patients determined to have SARS-CoV-2 who are hospitalized with lymphopenia, cellular immune reactions also appear to be reduced. A cellular immune response efficiently destroys SARS-CoV-2 in the safest-case scenario without any (or mild) clinical signs of infection. However, this is not always the case as the virus also induces immunosuppression that reduces and sometimes overcomes the host’s defenses [8].
4 Pro-inflammatory Th17 Lymphocytes and Disease Progression
Xu et al. observed that patients with severe COVID-19 infection had high concentrations of CCR4+ CCR6+ TH17 cells in the peripheral blood, thereby indicating a TH17-type cytokine storm. This research demonstrated a crucial role of Th17 inflammatory response in the pathogenesis of COVID-19 pneumonia [24]. This involves releasing essential cytokines such as IL-17 and other factors to intensify viral immunopathogenesis by downregulating Treg cells, facilitating neutrophil relocation, and simultaneously inciting Th2 reactions. IL-17 can induce severe eosinophilic reactions, allergic disease, and, to some degree, extravasation into the lungs [24]. Most recent outcomes show that the N protein is a potential inducer of IL-6 reactions that could intervene in coronavirus lung pathology [24, 25].
5 Lymphopenia and COVID-19
Lymphopenia is one of the most noteworthy markers of COVID-19. All lymphocyte subsets, which incorporate CD4+ and CD8+ cytotoxic T cells, natural killer (NK) cells, memory and Treg cells along with B cells, have been shown to be diminished in COVID-19 disease. This is critical as lower levels of lymphocytes are strongly related to the seriousness of disease [26]. T-cell numbers are inversely related with serum levels of IL-6, IL-10, and TNF-α and elevated levels of programmed cell death protein 1(PD-1) or T-cell immunoglobulin (Ig) and mucin domain-containing molecule-3 (TIM-3) [27].
The evidence suggests that SARS-CoV-2 targets T cells by receptor-dependent, S-protein-mediated membrane fusion. T cells have a low level of ACE2 expression, suggesting both an alternate receptor and a strong S-sensitivity to the protein. Invasion of T cells is abortive, showing that replication of SARS-CoV-2 inside T cells is not possible yet causes cell death instead [28]. Second, impaired lung macrophages or epithelial cells (in the first stage of hypercytokinemia) build up a variety of inhibitory cytokines, particularly TNF-α causing T-cell apoptosis, IL-10-restraining T-cell expansion, and type I IFN in the regulation of lymphocyte distribution. Thirdly, lymphopenia has been accepted to be the result of immune cell redistribution, with lymphocytes proliferating in the lungs or lymphoid glands [28]. Immunohistochemical staining of spleen and lymph nodes has shown decreased levels of CD4+ and CD8+ T cells. Finally, metabolic molecules generated by metabolic disorders such as lactic acid can block lymphocytes. Severe cases of COVID-19 patients have been found to have high blood lactic acid levels which can limit lymphocyte expansion [29].
6 Exhausted T Lymphocytes Associated with COVID-19 Disease Severity
Immune system homeostasis represents a vital role in preventing COVID-19 pneumonia [28]. Yong-Tang Zheng provides substantial differences in the levels of exhaustion modules between the three target groups (healthy, mild, severe), in particular PD-1, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and T-cell immunoglobulin and ITIM domain (TIGIT), and functional modules, such as IFN-γ, TNF-α, and IL-2. In the severe group, the amount of multifunctional CD4+ T cells declined significantly relative to the levels in healthy controls and a mild group, whereas the number of nonfunctional subgroups (IFN − TNF-α − IL-2) increased. The disturbance of CD4+ T cells could also have predisposed COVID-19 patients to severe diseases [28]. Prior research from Guang Chen et al. demonstrated that the Treg (CD4+ CD25+ CD127low+) and CD45RA+ Treg rates were lower in practically every severe and moderate case, with CD45RA+ Treg cells falling more significantly in severe cases than in moderate ones. It should be remembered that in certain patients with extreme and moderate COVID-19, CD4+T, CD8+T, and NK cells, the levels of IFN-γ secretion are reduced [30]. An early IFN response is fundamental for an effective T-cell reaction as a delayed IFN response may reduce T-cell expansion or T-cell departure from lymphoid organs. It might also bring about T-cell depletion and cell death. In patients with extreme COVID-19, lung damage was found to be correlated with cytokine release syndrome, suggesting an expected failure to trigger opportune immunosuppressive systems. Even so, Treg cell counts have been suggested to be associated inversely with the seriousness of the disease in COVID-19 patients [15]. IFNs are viewed as significant controllers for Treg cell development. Zheng et al. showed that the total amount of NK and CD8+ T cells diminished significantly in patients with SARS-CoV-2 disease. With the increased production of NKG2A in patients with COVID-19, the activity of the NK and CD8+ T cells was found to be reduced. Interestingly, the amount of NK and CD8+ T cells with decreased NKG2A expression has been shown to be increased in patients convalescing following treatment [31]. Consequently, infection with SARS-CoV-2 can destroy antiviral immunity rapidly. Hence, expression of SARS-CoV-2 induced NKG2A in COVID-19 patients with serious pulmonary inflammation associated with an initial phase of functional exhaustion of cytotoxic lymphocytes, which may lead to progression of the disease. Persistent infection and cancer have been found to have immune-inhibitory “checkpoint” receptors that lead to dysfunction of NK and T cells. It is important to take note that checkpoint inhibitors, such as anti-PD-1 and anti-TIGIT, help in the case of chronic infection and cancer and revitalize depleted T- or NK-cell responses [32]. NKG2A is believed to be another inhibitory molecule in the immune-checkpoint blockade. Such results show the importance of improving the immune response of NK cells and CTLs underlying SARS-CoV-2 infection and maintaining strategies for avoiding cytotoxic lymphocyte exhaustion [33].
7 Cytokine Storm, a Lethal Phase
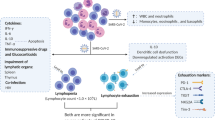
Cytokine release syndrome (CRS) tends to involve severely affected individuals with COVID-19 disease. Considering that lymphocytopenia is frequently seen in severe COVID-19 patients, leukocytes other than T cells will mediate the CRS induced by the SARS-CoV-2 virus. Cytokines are highly significant for COVID-19 pathophysiology [4]. Although some are protective (type I interferon, IL-7), others seem hazardous (IL-1β, IL-6, and TNF-α) and result in cytokine storms. This cytokine storm appears to be more likely to occur by a combination of the defective or delayed first line of protection, accompanied by chronic hypercytokinemia and an abnormal T-cell response [4]. This tends to result in incomplete removal of apoptotic cells or affected macrophages, an increase in viral proliferation and expansion, accompanied by an IL-18-/IFN-γ-activating macrophages, and which leads to massive secretion of cytokines, hemophagocytosis, coagulopathy, and acute respiratory distress syndrome (ARDS). By examining the immunopathology of SARS-CoV-2-related ARDS more closely, two mechanisms of immune failure have been identified as COVID-19 worsens: (1) an IL-1β-driven macrophage activation syndrome and (2) an immune dysregulation process guided by IL-6 [34]. The latter has been depicted by a combination of hypercytokinemia, immunoparalysis (CD14 monocytes with reduced HLA-DR molecules), and generalized lymphopenia (such as CD4+ and NK cells). Nonetheless, the blockade of IL-6 tocilizumab preserved HLA-DR expression on CD14 monocytes and increased the number of circulating lymphocytes. Cytokine storms could cause ARDS, severe cardiac attack, and secondary infection, culminating in systemic sepsis, and multisystem failure that may result in death (Fig. 10.2) [17, 35].

The immune responses mediated by SARS-CoV-2 are two main phases: the protective immune step and the second damage processing step done by inflammation and the cytokine storm
8 Coronavirus Treatments: Which Therapies Could Be Effective for COVID-19?
8.1 Apoptosis Inhibitors
Currently, there are no definite treatment protocols developed for COVID-19. Traditional therapies are mostly directed at the symptom level. Evidence shows that lymphopenia is generally identical in SARS-COV-2, respiratory syncytial virus infection, measles, and sepsis. The main triggers of sepsis and measles is apoptosis, which is expected to promote lymphopenia. For example, apoptosis inhibitors ameliorate inflammation and reduce mortality in sepsis models. These results have given us valuable insights concerning SARS-CoV-2 patients [36]. The proliferation of lymphocytes or targeting drugs for apoptosis (PD1/PD-L1 inhibitors) may forestall lymphopenia or recuperate lymphocyte in severe COVID-19 patients [37]. Restoration of the leukomonocyte populations of COVID-19-hospitalized patients appears to be associated with viral clearance. By comparing the numbers of leukomonocytes in COVID-19 patients at different periods of the sicknesses, research showed that CD3+, CD4+, and CD8+ T cells and B cells appear to play significant roles in viral clearance. It has been proposed that stabilization of leukomonocyte levels may be used as a guide for releasing and discharging patients in the COVID-19 diagnostic guidelines [37].
8.2 Convalescent Plasma and COVID-19
Immune or convalescent plasma is plasma obtained from patients after recovering from infection and antibody production. As can be shown, there are also many issues concerning convalescent plasma or immunoglobulins regardless of their wide approval. They had been used to increase the recovery rate of patients with SARS, human immunodeficiency virus (HIV), and severe H1N1 influenza whose conditions worsened despite the treatment with pulsed methylprednisolone [38]. One possible rationale behind the viability of convalescent plasma therapy might be that the antibodies could forestall viremia and provide passive immunity [39]. An in vivo examination likewise found that these neutralizer activities were not only restrictive for virus clearance and prevented new invasion but also increased infected cell clearance. Convalescent plasma, acquired from recovered COVID-19 patients with humoral immunity to the virus, has a large amount of neutralizing antibodies which could neutralize SARS-CoV-2 and eliminate the pathogen from the blood circulation and pulmonary tissues [40]. The outcome might be particularly advantageous for individuals with severe or life-threatening COVID-19 disease and, by using this medication, reduce the length or extent of the illness. The neutralization antibody titers correlate with the numbers of virus-specific T cells. Given the unavailability of data of SARS-CoV-2 fundamental biology, particularly virus heterogeneity and mutation, locally acquired plasma that better represents the circulating virus in the community may be a viable treatment choice. However, for this to work, we need appropriate donor selection with significantly higher serum titers of antibody that are neutralizing [40].
8.3 Intravenous Immunoglobulin (IVIG)
Individuals with debilitated immune systems, in general , have a greater danger of the related complications of COVID-19. Coupled with antiviral medications, IVIG-utilizing immunotherapy can be utilized to control or eliminate COVID-19 and improve the immune response to this virus. IVIG antibodies have two fundamental parts. These are the F (ab′) 2 piece, which is essential for the recognition of antigens, and the crystallizable fragment (Fc), which is essential for activating the immune response by communicating with B cells as well as other innate human immune cells with Fcγ receptors [41]. The Fc section additionally plays a pivotal role in enacting complement and evacuating the microorganisms. Elective treatment may, for the most part, be given with safe IVIG as an adjunctive prescription combined with antivirals. Tolerant IVIG antibodies acquired from healing patients would be successful against COVID-19 by reinforcing the immune response reaction in recently infected patients. However, no COVID-19 immunization is authoritatively accessible, and the mix of insusceptible IVIG antibodies with antiviral medications only gives short- and medium-term protection against COVID-19 [41, 42].
8.4 Cytokine-Based Interventions
8.4.1 Type I Interferon
Patient immune status will establish the efficacy of the COVID-19 treatments. Emerging data indicates that SARS-CoV-2 can activate a range of immune processes, allowing immunosuppressive agents in clinical trials to be beneficial in certain patients but dangerous to others [43]. IFN-α and IFN-β have recognized as potentially beneficial anti-SARS-CoV-2 medications. Type I IFN should be given as early as possible following diagnosis (ideally before the initiation of manifestations), however not in the late stage because of likely disturbance to tissues. Until peak viral replication, early IFN therapy was found to save mice from fatal SARS-CoV or MERS-CoV challenge, while late IFN therapy disrupted viral clearance and exacerbated immunopathology [14].
8.4.2 Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF)
GM-CSF performs a primary controlling function in cytokine production and myeloid cell-induced hyperinflammation. In addition, the late phases of COVID-19 disease will most likely be controlled not by forceful viral replication and cell lysis but by immunopathology, especially myeloid cell immunopathology [44]. In this manner, the putative pathogenic function of GM-CSF in immune hyperactivation opens up the possibility for starting continuous clinical investigations using GM-CSF focusing on monoclonal antibodies (mAbs) for treating COVID-19 patients. The reasoning and the risks for both therapeutic administration and inhibition of GM-CSF in COVID-19 have been established. The use of GM-CSF in COVID-19 patients may improve lung capacity by supporting the alveolar wall and enhancing viral removal and thereby offer a positive advantage in early COVID-19 phases. Conversely, GM-CSF or GM-CSFR inhibition can be a protective treatment for the cytokine storm and myeloid cell tissue invasion. As it may influence the release of several pro-inflammatory cytokines and chemokines by myeloid cells, the GM-CSF approach may have noteworthy immunomodulatory implications [44].
8.5 Anti-Cytokine Interventions
8.5.1 IL-6 Inhibition
Inflammation caused by SARS-CoV-2 results in a dose-dependent release of IL-6 from bronchial epithelial cells [45]. In patients with SARS-CoV-2 infections, alterations in IL-6 levels could potentially be a crucial mediator when severe systemic inflammatory reactions occur. IL-6 is involved in two particular pathways in SARS-CoV-2 pathology. The first of these is thought to be anti-inflammatory (induced by the membrane-bound variant of the IL-6 receptor, mIL6R) and the second is the trans-signaling pathway (induced by the soluble IL6R form) which is presumed to have a pro-inflammatory role [25]. Specific blockers of these pathways should be investigated to determine which is the most effective as a treatment for COVID-19 disease.
8.5.2 TNF Inhibitors
As mentioned above, SARS-CoV-2 infection is related to downregulation of ACE2 expression combined with activation of the renin-angiotensin system liable for the lung injury [6]. In addition, the viral spike protein will cause a TNF-α-converting enzyme (TACE)-dependent shedding of the ectodomain ACE2, which is fundamental for the viral entry into the cell. For these reasons, it has been speculated that the use of TNF inhibitors may be efficient in lowering both SARS-CoV-2 infection and the resulting organ damage. Subsequently, the Chinese Clinical Trial Registry (ChiCTR2000030089) announced an investigation of adalimumab in patients with COVID-19 infection [46].
8.5.3 Targeting Chemokine Receptors
In patients with COVID-19, a significant rise of CCL2 and CCL3 expression in macrophages has been seen alongside diminished expression of CCR1, the receptor for both chemokines. Since binding of CCL2 or CCL3 to CCR1, CCR2, or CCR5 causes monocyte recruitment into the lung parenchyma with eventual differentiation into inflammatory macrophages and resulting recruitment and activation of multiple immune cells, and epithelial injury, CCR1, CCR2, and CCR5 could be potent anti-inflammatory targets in COVID-19 [11]. HIV and other viral diseases target the CCR2/CCL2 axis. However, the evidence has not verified the production of CCR2 in the respiratory tract of COVID-19 patients (possibly due to its accelerated inhibition in monocytes as they leave the circulation and reach tissues). This leaves CCR1 and CCR5 as potential targets, which should be investigated further [11, 16].
8.6 Nontargeted Therapies
8.6.1 Corticosteroids
Corticosteroids are effective cytokine inhibitors that act by various pathways but essentially through inhibition of the transcription factor NF-κB. These drugs are the foundation of therapies for autoimmune and autoinflammatory disorders with cytokine storms. Dexamethasone is a medication that has been utilized in a variety of treatments since the 1960s to minimize inflammation involving autoimmune diseases and certain cancers [47]. According to early findings in a clinical trial conducted in the United Kingdom, dexamethasone was found to reduce mortality by about one-third for patients on ventilators, and mortality was decreased by around one-fifth for patients needing only oxygen [47].
8.6.2 Remdesivir
Remdesivir is a nucleotide-analog prodrug that prevents polymerases of viral RNA, which has been shown to have efficacy against SARS-CoV-2 in vitro [48]. Remdesivir is intracellularly metabolized to an analog of adenosine triphosphate that suppresses viral RNA polymerases. Remdesivir has broad actions on a variety of viral agents, including filoviruses (Ebola) and coronaviruses (SARS-CoV and MERS-CoV). The US Food and Drug Administration has provided an urgent usage permit for the investigational antiviral medication remdesivir for the care of suspected or laboratory-affirmed COVID-19 in hospitalized adults and children with serious illness. One study showed that remdesivir reduced the period of rehabilitation in some instances of COVID-19 [49].
9 Conclusions
The occurrence and development of SARS-CoV-2 depend on the interaction between virus infection and the immune system. Dysregulation of the immune system in COVID-19 patients can contribute to serious illness. Dysregulation of the immune system such as lymphopenia and cytokine storm could be a crucial factor related to the severity of COVID-19. Decreased T lymphocytes and elevated cytokines could potentially serve as COVID-19 prognostic markers. Antiviral or immunomodulatory therapies have not been shown to be useful for treatment of COVID-19 to date. In clinical trials, interventions could be timed based on immune response. For example, antivirals and immune boosters should be started soon after the start of symptoms, whereas immunosuppressants should be delivered at the beginning of the cytokine storm.
References
Shoenfeld Y (2020) Corona (COVID-19) time musings: our involvement in COVID-19 pathogenesis, diagnosis, treatment and vaccine planning. Autoimmun Rev 19(6):102538. https://doi.org/10.1016/j.autrev.2020.102538
Fauci AS, Lane HC, Redfield RR (2020) Covid-19 – navigating the uncharted. N Engl J 382(13):1268–1269
Li X, Geng M, Peng Y, Meng L, Lu S (2020) Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal 10(2):102–108
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ et al (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395(10229):1033–1034
Ulrich H, Pillat MM (2020) CD147 as a target for COVID-19 treatment: suggested effects of azithromycin and stem cell engagement. Stem Cell Rev Rep 16(3):434–440
Acosta FASAD (2020) Entry of SARS-CoV2 through the basal surface of alveolar endothelial cells–A proposed mechanism mediated by CD147 in COVID-19. Preprints 2020050359. https://doi.org/10.20944/preprints202005.0359.v1
Tufan A, GÜLER AA, Matucci-Cerinic M (2020) COVID-19, immune system response, hyperinflammation and repurposing antirheumatic drugs. Turk J Med Sci 50(SI-1):620–632
Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X et al (2020) COVID-19 infection: the perspectives on immune responses. Cell Death Differ 27(5):1451–1454
Nguyen A, David JK, Maden SK, Wood MA, Weeder BR, Nellore A et al (2020) Human leukocyte antigen susceptibility map for SARS-CoV-2. J Virol 94(13):e00510–e00520. https://doi.org/10.1128/JVI.00510-20
Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N et al (2020) Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 27(6):992–1000.e3
Merad M, Martin JC (2020) Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 20(6):355–362
Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z et al (2020) Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27(6):883–890.e2
Trouillet-Assant S, Viel S, Gaymard A, Pons S, Richard JC, Perret M et al (2020) Type I IFN immunoprofiling in COVID-19 patients. J Allergy Clin Immunol 146(1):206–208.e2
Acharya D, Liu G, Gack MU (2020) Dysregulation of type I interferon responses in COVID-19. Nat Rev Immunol 20(7):397–398
Park A, Iwasaki A (2020) Type I and type III interferons–induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 27(6):870–878
Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M (2020) The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev 53:25–32
McGonagle D, Sharif K, O’Regan A, Bridgewood C (2020) Interleukin-6 use in COVID-19 pneumonia related macrophage activation syndrome. Autoimmun Rev 19(6):102537. https://doi.org/10.1016/j.autrev.2020.102537
Park MD (2020) Macrophages: a Trojan horse in COVID-19? Nat Rev Immunol Apr 17:1. https://doi.org/10.1038/s41577-020-0317-2. [Epub ahead of print]
Campbell CM, Kahwash R (2020) Will complement inhibition be the new target in treating COVID-19–related systemic thrombosis? Circulation 141(22):1739–1741
Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC et al (2020) The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol 215:108450. doi: https://doi.org/10.1016/j.clim.2020.108450
Risitano AM, Mastellos DC, Huber-Lang M, Yancopoulou D, Garlanda C, Ciceri F et al (2020) Complement as a target in COVID-19? Nat Rev Immunol 20(6):343–344
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J et al (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res 220:1–13
Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LF (2020) The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol 20:1–12
Wu D, Yang XO (2020) TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect 53(3):368–370
Zhang C, Wu Z, Li JW, Zhao H, Wang GQ (2020) The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int J Antimicrob Agents 55(5):105954. https://doi.org/10.1016/j.ijantimicag.2020.105954
Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang YQ et al (2020) Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct Target Ther 5(1):1–3
Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L et al (2020) Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol 11:827. https://doi.org/10.3389/fimmu.2020.00827
Zheng HY, Zhang M, Yang CX, Zhang N, Wang XC, Yang XP et al (2020) Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol 17(5):541–543
Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL et al (2020) A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 26(7):1070–1076
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H et al (2020) Clinical and immunologic features in severe and moderate forms of coronavirus disease 2019. J Clin Invest 130(5):2620–2629
Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D et al (2020) Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol 17(5):533–535
Pickles OJ, Lee LY, Starkey T, Freeman-Mills L, Olsson-Brown A, Cheng V et al (2020) Immune checkpoint blockade: releasing the breaks or a protective barrier to COVID-19 severe acute respiratory syndrome? Br J Cancer Jun 16;1–3. https://doi.org/10.1038/s41416-020-0930-7. Online ahead of print
Bersanelli M (2020) Controversies about COVID-19 and anticancer treatment with immune checkpoint inhibitors. Immunotherapy 12(5):269–273
Ye Q, Wang B, Mao J (2020) The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J Inf 80(6):607–613
Henderson LA, Canna SW, Schulert GS, Volpi S, Lee PY, Kernan KF et al (2020) On the alert for cytokine storm: immunopathology in COVID-19. Arthritis Rheum 72:1059–1063
Cao X (2020) COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol 20(5):269–270
Bermejo-Martin JF, Almansa R, Menéndez R, Mendez R, Kelvin DJ, Torres A (2020) Lymphopenic community acquired pneumonia as signature of severe COVID-19 infection. J Inf Secur 80(5):e23–e24
Chen L, Xiong J, Bao L, Shi Y (2020) Convalescent plasma as a potential therapy for COVID-19. Lancet Infect Dis 20(4):398–400
Duan K, Liu B, Li C, Zhang H, Yu T, Qu J et al (2020) Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci U S A 117(17):9490–9496
Rojas M, Rodríguez Y, Monsalve DM, Acosta-Ampudia Y, Camacho B, Gallo JE et al (2020) Convalescent plasma in Covid-19: possible mechanisms of action. Autoimmun Rev 19(7):102554. https://doi.org/10.1016/j.autrev.2020.102554
Jawhara S (2020) Could intravenous immunoglobulin collected from recovered coronavirus patients protect against COVID-19 and strengthen the immune system of new patients? Int J Mol Sci 21(7):2272. https://doi.org/10.3390/ijms21072272
Xie Y, Cao S, Dong H, Li Q, Chen E, Zhang W et al (2020) Effect of regular intravenous immunoglobulin therapy on prognosis of severe pneumonia in patients with COVID-19. J Infect Apr 10;S0163-4453(20):30172–30179. https://doi.org/10.1016/j.jinf.2020.03.044. Online ahead of print
Ritchie AI, Singanayagam A (2020) Immunosuppression for hyperinflammation in COVID-19: a double-edged sword? Lancet 395(10230):1111. https://doi.org/10.1016/S0140-6736(20)30691-7
Lang FM, Lee KMC, Teijaro JR, Becher B, Hamilton JA (2020) GM-CSF-based treatments in COVID-19: reconciling opposing therapeutic approaches. Nat Rev Immunol Jun 23;1–8. https://doi.org/10.1038/s41577-020-0357-7. Online ahead of print
Liu B, Li M, Zhou Z, Guan X, Xiang Y (2020) Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun 111:102452. https://doi.org/10.1016/j.jaut.2020.102452
Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, Rowland M et al (2020) Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 395(10234):1407–1409
Theoharides T, Conti P (2020) Dexamethasone for COVID-19? Not so fast. J Biol Regul Homeost Agents Jun 4;34(3). https://doi.org/10.23812/20-EDITORIAL_1-5. Online ahead of print
Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A et al (2020) Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med 382(24):2327–2336
Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y et al (2020) Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 395(10236):1569–1578
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tavasolian, F. et al. (2021). The Immune Response and Effectiveness of COVID-19 Therapies. In: Guest, P.C. (eds) Clinical, Biological and Molecular Aspects of COVID-19. Advances in Experimental Medicine and Biology(), vol 1321. Springer, Cham. https://doi.org/10.1007/978-3-030-59261-5_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-59261-5_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-59260-8
Online ISBN: 978-3-030-59261-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)