
Abstract
Polycomb Repressive Complex 2 (PRC2) is a major repressive chromatin complex formed by the Polycomb Group (PcG) proteins. PRC2 mediates trimethylation of histone H3 lysine 27 (H3K27me3), a hallmark of gene silencing. PRC2 is a key regulator of development, impacting many fundamental biological processes, like stem cell differentiation in mammals and vernalization in plants. Misregulation of PRC2 function is linked to a variety of human cancers and developmental disorders. In correlation with its diverse roles in development, PRC2 displays a high degree of compositional complexity and plasticity. Structural biology research over the past decade has shed light on the molecular mechanisms of the assembly, catalysis, allosteric activation, autoinhibition, chemical inhibition, dimerization and chromatin targeting of various developmentally regulated PRC2 complexes. In addition to these aspects, structure-function analysis is also discussed in connection with disease data in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Polycomb repressive complex 2 (PRC2)
- Chromatin
- Epigenetics
- Histone methylation
- Gene repression
- Structural biology
Introduction
Polycomb (PC), the founding member of the family of Polycomb Group (PcG) genes, was originally identified in Drosophila melanogaster to control segmentation by repressing homeotic (HOX) genes (Lewis 1978), which are known to dictate the development of anatomical structures in different species, like insects, mammals and plants. Compared to their counterparts in Drosophila, PcG proteins are conserved, but more diverse in mammals and plants (Hennig and Derkacheva 2009; Schuettengruber et al. 2017). PcG proteins impact a variety of fundamental biological processes, ranging from stem cell differentiation and X-chromosome inactivation in mammals to vernalization in plants (Wutz 2011; Whittaker and Dean 2017; Aloia et al. 2013). On the molecular level, PcG proteins assemble into two major repressive chromatin complexes, Polycomb Repressive Complex 1 (PRC1) and PRC2 (Fig. 17.1). Both PRC1 and PRC2 are histone modifying enzyme complexes; whereas PRC1 is responsible for monoubiquitination of histone H2A lysine 119 (H2AK119ub) (Wang et al. 2004a), PRC2 mediates trimethylation of histone H3 lysine 27 (H3K27me3) (Cao et al. 2002; Czermin et al. 2002; Kuzmichev et al. 2002; Muller et al. 2002) (Fig. 17.1). Both H2AK119ub and H3K27me3 are repressive histone marks and in particular H3K27me3 is considered as a hallmark of silent chromatin. PcG proteins also form the Polycomb Repressive-Deubiquitinase complex (PR-DUB), which removes ubiquitin from H2AK119ub but, intriguingly, is also required for PcG-mediated gene silencing (Scheuermann et al. 2010).

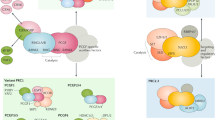
Compositions of distinct classes of PRC1 and PRC2. Compositions of six classes of PRC1 (PRC1.1–PRC1.6) and two classes of PRC2 (PRC2.1 and PRC2.2) are shown, with the core and accessory subunits clustered separately. Histone modifications mediated by PRC1 and PRC2 are also indicated. Proteins identified from proteomics are shown as the accessory subunits of PRC1. In contrast, only proteins and ncRNAs that are known to mediate direct physical interactions with the core subunits are defined as the accessory subunits of PRC2. This more restrictive definition makes PRC2 appear to be less complex than PRC1 in the figure
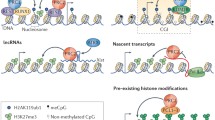
Polycomb Response Elements (PREs) containing clusters of DNA motifs direct specific chromatin targeting of PRC1 and PRC2 in Drosophila, via cognate transcription factors (Ringrose and Paro 2007). In contrast, mammalian counterparts of Drosophila PREs appear to be lacking (Ringrose and Paro 2007). Recruitment of PRC1 and PRC2 can be interdependent. For example, in a hierarchical recruitment model, PRC2 is targeted to chromatin to install H3K27me3, which in turn recruits PRC1 through an H3K27me3 reader protein (Wang et al. 2004b). Conversely, H2AK119ub can be essential for the recruitment of PRC2, which contains components recognizing H2AK119ub (Blackledge et al. 2014; Cooper et al. 2014; Kalb et al. 2014; Cooper et al. 2016; Kahn et al. 2016; Blackledge et al. 2020; Tamburri et al. 2020). Hypomethylated CpG islands (CGIs) are known sites of PRC2 enrichment on chromatin (Deaton and Bird 2011). In addition, de novo recruitment of PRC2 to CGIs can be induced by inhibition of transcription (Riising et al. 2014). Indeed, CGI sequences are sufficient to recruit PRC2 in vivo in the absence of DNA methylation and transcription factor binding (Mendenhall et al. 2010; Jermann et al. 2014; Wachter et al. 2014).
PRC2 enzymatic activity is subjected to cellular regulation in both normal and diseased cells. Particularly, PRC2 binds to and is allosterically activated by H3K27me3; this positive feedback mechanism partially accounts for epigenetic inheritance, for which PRC2 association with cis-acting DNA elements is also required (Hansen et al. 2008; Margueron et al. 2009; Coleman and Struhl 2017; Laprell et al. 2017). An oncogenic histone H3K27M harboring a lysine-to-methionine mutation is linked to diffuse midline gliomas, a type of high-grade lethal brain cancer found in both children and adults; PRC2 enzymatic activity is inhibited by H3K27M, leading to a global reduction of H3K27me3 in patient samples (Chan et al. 2013; Lewis et al. 2013). Intriguingly, both gain-of-function and loss-of-function mutations of PRC2 components have been associated with human cancers (Laugesen et al. 2016). Additionally, mutations in PRC2 components also cause overgrowth disorders manifesting a variable degree of intellectual disability, including Weaver syndrome and Cohen–Gibson syndrome (Cyrus et al. 2019).
This chapter aims to provide a structural perspective on the mechanism of gene repression by PRC2, with a focus on the regulation of the enzymatic activity and chromatin recruitment of mammalian PRC2. Interested readers are also referred to many other reviews that discuss PRC2 structure and function (Simon and Kingston 2009; Margueron and Reinberg 2011; Schwartz and Pirrotta 2013; Aranda et al. 2015; Vizan et al. 2015; Chittock et al. 2017; Schuettengruber et al. 2017; Laugesen et al. 2019; van Mierlo et al. 2019; Chammas et al. 2020). Understanding composition of functional PRC2 represents one of the very first steps towards structural characterization. Below, knowledge concerning discovery, sequence feature, domain structure, complex formation and cellular function of PRC2 components is discussed.
Composition of Functional PRC2
Subunit composition and interaction of macromolecular complexes are a determinant of biological function. Multifaceted roles of PRC2 in cell development are largely correlated with the tremendous complexity and plasticity of PRC2 composition. Core subunits of PRC2 remain relatively constant, and they assemble into diverse holo complexes together with a wide spectrum of developmentally regulated accessory subunits (Fig. 17.1). Historically, components of mammalian PRC2 holo complexes were mostly identified based on homology to Drosophila PcG proteins or by mass spectrometry following co-immunoprecipitation or affinity purification experiments. The accessory subunits were originally shown to be “substoichiometric”, sometimes suggesting weak or transient binding. However, biochemical and structural data based on fully reconstituted PRC2 holo complexes indicated otherwise: many of the accessory subunits are stable stoichiometric components of the holo complexes. Nuclear extracts of HeLa human cervical cancer cells, HEK293/HEK293T human embryonic kidney cells and mouse embryonic stem cells (mESCs) are among the most frequently used sources for PRC2 isolation in both focused and large-scale proteomics studies (Smits et al. 2013; Alekseyenko et al. 2014; Hein et al. 2015; Huttlin et al. 2015; Maier et al. 2015; Hauri et al. 2016; Huttlin et al. 2017). In addition, mESCs and NT1/D2 human embryonal carcinoma cells offer convenient model systems for a direct comparison of the dynamic interactome of PRC2 under undifferentiated and differentiated conditions (Kloet et al. 2016; Oliviero et al. 2016). Although mammalian PRC2 has been subjected to extensive studies, the current picture of its composition may still be incomplete, for at least two reasons: cell type-specific new components of PRC2 may exist in some of the formerly unexplored cell types; furthermore, it is not impossible that certain PRC2 interactions may entail a chromatin context—not captured in chromatin-free cell extracts—and may have been missed from most of the previous proteomics studies.
RNAs and in particular a diverse collection of non-coding RNAs (ncRNAs) can be considered as non-conventional components of PRC2 holo complexes (Fig. 17.1). RNAs were shown to mediate promiscuous binding to PRC2 in cells (Khalil et al. 2009; Zhao et al. 2010; Davidovich et al. 2013; Kaneko et al. 2013), influencing critical physiological processes, such as X-chromosome inactivation by XIST (Plath et al. 2003; Silva et al. 2003; Zhao et al. 2008), repression of HOX gene clusters by HOTAIR (Rinn et al. 2007; Tsai et al. 2010), and genomic imprinting by KCNQ1OT1 (Pandey et al. 2008; Zhao et al. 2010). PRC2 preferentially binds to guanine-rich RNA motifs that may form G-quadruplex structures (Wang et al. 2017a). Canonical RNA-binding domains are not found in PRC2; instead, RNA-binding surfaces contributed by multiple PRC2 subunits—both core and accessory subunits—appear to be dispersed across the entire complex (Kaneko et al. 2014a; Long et al. 2017; Wang et al. 2017b; Zhang et al. 2019). Lacking clear binding specificity on the sequence level, whether RNAs are able to mediate specific chromatin targeting of PRC2 is still under debate (Brockdorff 2013; Davidovich and Cech 2015; Ringrose 2017). PRC2 association with RNAs is antagonistic to chromatin binding (Beltran et al. 2016; Wang et al. 2017b), and this antagonism may also underlie RNA-mediated inhibition of the enzymatic activity of PRC2 towards nucleosomal substrates (Cifuentes-Rojas et al. 2014; Kaneko et al. 2014b; Wang et al. 2017b). Ideally, structural and mutagenesis data that reveal binding specificity in three-dimensional space will help clarify a defined role of RNA molecules in PRC2 recruitment. RNA components of PRC2 will not be covered in more detail in this chapter.
The Core Complex
The PRC2 core complex from mice and humans consists of four subunits: EZH1/EZH2 (aka ENX-2/ENX-1 or KMT6B/KMT6A), EED (aka WAIT-1), SUZ12 (aka CHET9 or JJAZ1), and RBBP4/RBBP7 (aka RBAP48/RBAP46) (Fig. 17.1). EZH1 and EZH2 are paralogs and both can function as the catalytic subunit of PRC2. EZH1 and EZH2 share overall 60% sequence identity, with the SET (SU(VAR)3-9, Enhancer-of-zeste and Trithorax) domain being one of the most homologous regions (Fig. 17.2 and Table 17.1) (Abel et al. 1996; Chen et al. 1996; Hobert et al. 1996; Laible et al. 1997). Less conserved regions confer functional divergence: for example, EZH1-containing PRC2 compacts chromatin to a greater extent, whereas EZH2-containing PRC2 is better stimulated by H3K27me3 (Margueron et al. 2008; Son et al. 2013; Lee et al. 2018). EZH2 expression is correlated with active cell proliferation and EZH2 acts as the principal H3K27 methyltransferase in mESCs (Bracken et al. 2003; Lee et al. 2018). EZH1 is ubiquitously expressed and partially complements EZH2 function during mESC differentiation (Margueron et al. 2008; Shen et al. 2008). H3K27 methylation is eliminated in mESCs lacking both EZH1 and EZH2 (Shen et al. 2008; Hojfeldt et al. 2018).

Domain structures of the core subunits of PRC2. Domain structures of EZH2, EED, SUZ12 and RBBP4 are shown and color-coded. Domains with unknown structures are colored in grey. The subunit definition of the catalytic module and the accessory subunit-binding module is indicated. The domain structure of EZH1 is not shown, but is predicted to be similar to that of EZH2. These domain structures are defined based on sequence or structural analysis. Full names corresponding to the acronyms of protein domains are provided in Table 17.1
Both EED and SUZ12 are necessary for the enzymatic activity of EZH2 (Cao and Zhang 2004; Nekrasov et al. 2005). EED was found to associate with EZH1 and EZH2 in yeast two-hybrid screens (Sewalt et al. 1998; van Lohuizen et al. 1998). A large portion of EED forms a seven-bladed β-propeller WD40 domain featuring short β-sheet structural repeats of 40 amino acids (aa) ending with a tryptophan–aspartate (WD) dipeptide (Fig. 17.2 and Table 17.1) (Han et al. 2007; Stirnimann et al. 2010). EED interacts with the H3K27me3 histone mark, leading to allosteric activation of PRC2 enzymatic activity (Hansen et al. 2008; Margueron et al. 2009). SUZ12 was identified in a biochemically isolated EZH2–EED complex (Cao et al. 2002; Kuzmichev et al. 2002). Sequence analysis indicates there are a Cys2His2-type Zinc finger domain and a VEFS (VRN2, EMF2, FIS2, and SU(Z)12) domain in SUZ12. Structural data reveal additional functional domains in both EZH2 and SUZ12, some of which undergo dramatic conformational change in distinct states of PRC2 (see below) (Fig. 17.2 and Table 17.1). Like EED, RBBP4 and RBBP7 are also WD40 domain proteins (Fig. 17.2 and Table 17.1). Although frequently co-purified with the EZH2–EED complex (Cao et al. 2002; Kuzmichev et al. 2002), RBBP4 and RBBP7 are not dedicated subunits of PRC2—the two paralogs are also components of other chromatin complexes, such as Mi-2/NuRD and Sin3A histone deacetylases (HDACs) and NURF chromatin remodeling complex (Tsukiyama and Wu 1995; Zhang et al. 1997, 1998; Barak et al. 2003). RBBP4 was shown to enhance the enzymatic activity of an EZH2–EED–SUZ12 ternary complex (Cao and Zhang 2004). RBBP4 and RBBP7 are almost 90% identical in protein sequence, and it is unclear to what extent they differ from each other in PRC2 structure and function.
Isoforms of some of the core subunits exist in cells. Knowledge on isoform-specific function is limited but nonetheless worth mentioning. EZH2isoform1 (746aa) and EZH2isoform2 (751aa, residues 297–298 of EZH2isoform1 replaced by another 7aa) are most frequently referred to in the literature, but functional distinction between these two isoforms has not been reported. In comparison, EZH2isoform3 (707aa, lacking residues 83–121 of EZH2isoform1) was found to display weaker interactions with EED and SUZ12 and to be correlated with the expression pattern of both redundant and unique gene targets in human pancreatic epithelial cells (Grzenda et al. 2013). In another study, a mouse EZH1 isoform that lacks the SET domain was shown to sequester EED in the cytosol and thereby impair the assembly of nuclear PRC2 and PRC2-dependent gene repression in differentiated C2C12 mouse myoblast cells (Bodega et al. 2017). In addition, EED isoforms with distinct N-termini that might be produced by alternative translation initiation were observed previously in HeLa nuclear extracts; however, unique PRC2 function conferred by these isoforms still remains to be elucidated (Kuzmichev et al. 2004, 2005; Martin et al. 2006).
The Holo Complexes: PRC2.1 and PRC2.2
Compositional complexity of mammalian PRC2 holo complexes is reflected by the increasing number of identified accessory subunits. These accessory subunits directly associate with the PRC2 core complex, and many of them facilitate PRC2 core complex binding to chromatin, via interactions with linker DNA, core nucleosome, or histone marks (Ballare et al. 2012; Musselman et al. 2012; Cai et al. 2013; Kalb et al. 2014; Cooper et al. 2016; Choi et al. 2017; Li et al. 2017; Wang et al. 2017b; Chen et al. 2018, 2020; Perino et al. 2018). Based on the mutually exclusive binding of the accessory subunits to the core complex, PRC2 holo complexes have so far been categorized into two classes, PRC2.1 and PRC2.2 (Fig. 17.1) (Grijzenhout et al. 2016; Hauri et al. 2016). EPOP (Elongin BC and Polycomb Repressive Complex 2-associated Protein; aka C17ORF96 or esPRC2p48), PALI1/PALI2 (PRC2 Associated LCOR Isoform 1 and 2; aka C10ORF12 as a part of PALI1) and PCL (Polycomb-Like) proteins, including PHF1 (aka PCL1), MTF2 (aka PCL2) and PHF19 (aka PCL3), are the known accessory subunits of PRC2.1 (Fig. 17.1). Those belonging to PRC2.2 include JARID2 (Jumonji/AT-Rich Interaction Domain containing 2) and AEBP2 (Adipocyte Enhancer-Binding Protein 2) (Fig. 17.1). Mutual exclusivity in PRC2 core complex binding was observed between PCL proteins and AEBP2 (Grijzenhout et al. 2016; Hauri et al. 2016; Chen et al. 2018), between PALI1 and AEBP2 (Hauri et al. 2016; Conway et al. 2018), and among EPOP, PALI1 and JARID2 (Alekseyenko et al. 2014; Beringer et al. 2016; Hauri et al. 2016; Liefke et al. 2016; Chen et al. 2018; Conway et al. 2018). PRC2.1 and PRC2.2 are complementary in maintenance of the H3K27me3 histone mark at specific chromatin loci in mESCs; combined ablation of the two classes of the accessory subunits abolishes normal H3K27me3 pattern on chromatin, accompanied by diffuse H3K27 trimethylation likely due to mislocalization of the PRC2 core complex (Healy et al. 2019; Hojfeldt et al. 2019). In line with these observations, PCL proteins and AEBP2 were indicated to reduce mobility and prolong residence time of the respective PRC2 holo complexes on chromatin (Choi et al. 2017; Youmans et al. 2018).
PRC2.1: PHF1, MTF2 and PHF19
PHF1 and PHF19 were identified based on homology to Drosophila PCL protein (Coulson et al. 1998; Wang et al. 2004c). MTF2 was originally cloned as a DNA-binding protein recognizing the metal response elements (MREs) on the promoters of metallothionein (MT) genes (Inouye et al. 1994). A series of additional studies indicated that all three PCL proteins are components of PRC2 holo complexes, colocalizing with EZH2 and SUZ12 on chromatin; in many cases, knockdown of PCL proteins was shown to reduce H3K27me3 deposition on specific targets, in parallel with change of gene expression (O’Connell et al. 2001; Cao et al. 2008; Sarma et al. 2008; Walker et al. 2010; Boulay et al. 2011; Casanova et al. 2011; Li et al. 2011; Ballare et al. 2012; Brien et al. 2012; Hunkapiller et al. 2012; Cai et al. 2013). Expression of PCL proteins is developmentally regulated. For example, MTF2 is the dominant form in mESCs; in comparison, MTF2 level is downregulated during mESC differentiation to neural progenitor cells (NPCs), whereas PHF1 and PHF19 become relatively enriched (Kloet et al. 2016).
PHF1, MTF2 and PHF19 share similar domain structures: the N-terminal half of these proteins contains one Tudor domain, two PHD fingers (PHD1 and PHD2), and one EH/WH (Extended Homology/Winged-Helix) domain; located at the C-terminus is one RC/CL (Reversed Chromo/Chromo-Like) domain (Fig. 17.3 and Table 17.2) (Ballare et al. 2012; Choi et al. 2017; Li et al. 2017). The Tudor domain of all three PCL proteins binds to H3K36me3, a histone mark associated with gene body of active genes; this interaction facilitates chromatin targeting of PHF19-containing PRC2.1 for de novo silencing of active genes, a process to which H3K36me3 demethylases like NO66 and KDM2b also contribute (Abed and Jones 2012; Ballare et al. 2012; Brien et al. 2012; Musselman et al. 2012; Cai et al. 2013). The EH/WH domain of PCL proteins interacts with double-stranded linker DNA, enhancing chromatin association of PRC2.1 (Choi et al. 2017; Li et al. 2017). Furthermore, DNA helical shape was proposed to dictate specific DNA binding by MTF2 (Perino et al. 2018). The RC/CL domain is responsible for the stable binding of PCL proteins to the PRC2 core complex (Ballare et al. 2012; Choi et al. 2017; Chen et al. 2018). In the case of MTF2 and PHF19, the RC domain was also shown to stabilize a dimeric state of PRC2.1 to promote chromatin binding, possibly through an avidity effect (Chen et al. 2020).

Domain structures of the accessory subunits of PRC2. Domain structures of the accessory subunits of PRC2.1 (PHF1, MTF2, PHF19, EPOP, PALI1) and PRC2.2 (AEBP2 and JARID2) are shown and color-coded. Domains with unknown structures are colored in grey. PALI2 is less characterized and its domain structure is not included. These domain structures are defined based on sequence or structural analysis. Full names corresponding to the acronyms of protein domains are provided in Table 17.2
PRC2.1: EPOP and PALI1/PALI2
EPOP and PALI1 (the C10ORF12 part) were originally noted in a pool of proteins that associate with certain core subunits of PRC2 (Zhang et al. 2011; Smits et al. 2013; Alekseyenko et al. 2014; Maier et al. 2015; Hauri et al. 2016; Kloet et al. 2016; Oliviero et al. 2016). Proteomics on EPOP or PALI1 pulldowns indicate they are exclusive members of PRC2.1 and are incompatible between themselves in PRC2 binding (Alekseyenko et al. 2014; Beringer et al. 2016; Hauri et al. 2016; Liefke et al. 2016; Conway et al. 2018). EPOP interacts with PRC2 via the CT (C-Terminal) domain, which also competes with JARID2 for PRC2 binding (Fig. 17.3 and Table 17.2) (Liefke and Shi 2015; Chen et al. 2018). A BC box located in the N-terminal portion of EPOP binds to the Elongin BC heterodimer, which together with Elongin A forms the three-subunit Elongin complex, a positive regulator of RNA polymerase II transcription elongation (Fig. 17.3 and Table 17.2) (Beringer et al. 2016; Liefke et al. 2016). EPOP colocalizes with EZH2 and SUZ12 on chromatin and moderately impedes SUZ12 binding to chromatin in mESCs (Beringer et al. 2016; Liefke et al. 2016). Loss of EPOP impairs low-level permissive transcription of PRC2 target genes in mESCs and reduces proliferation of certain human cancer cell lines (Beringer et al. 2016; Liefke et al. 2016).
PALI1 and PALI2 are vertebrate-specific proteins and they are encoded by isoforms of the LCOR (Ligand-dependent nuclear receptor Corepressor) and LCORL (Ligand-dependent nuclear receptor Corepressor-Like) gene loci, respectively (Conway et al. 2018). PALI1 bears one NRB (Nuclear Receptor Binding) box, two CB (CTBP Binding) motifs, one PIG (PALI Interaction with G9A) domain and one PIP (PALI Interaction with PRC2) domain (Fig. 17.3 and Table 17.2) (Conway et al. 2018). Intriguingly, the H3K9 methyltransferases G9A and GLP were previously shown to directly interact with the PRC2 core complex and control both PRC2 targeting and H3K27 trimethylation (Mozzetta et al. 2014). PALI1 promotes the methyltransferase activity of PRC2 towards oligonucleosomes in vitro (Conway et al. 2018). The antagonistic interplay between PALI1 and AEBP2 regulates the balance of PRC2.1 and PRC2.2 activities on chromatin, which influences PRC2 target gene expression during mESC differentiation (Conway et al. 2018).
PRC2.2: AEBP2
AEBP2 was originally identified as a transcriptional repressor harboring three Gli-Krüppel Cys2His2-type zinc fingers in mice (He et al. 1999). AEBP2 was found in PRC2 isolated from HeLa nuclear extracts and was shown to enhance PRC2 enzymatic activity towards oligonucleosomal substrates (Cao et al. 2002; Cao and Zhang 2004). There are both short and long isoforms of AEBP2, corresponding to embryonic and somatic isoforms in mice, respectively (Kim et al. 2009). AEBP2 contains multiple functional domains (Fig. 17.3 and Table 17.2), including a Zn (Zinc finger) domain that is thought to bind DNA and confer binding specificity towards methylated CpG DNAs (Kim et al. 2009; Wang et al. 2017b), a KR (K (lysine) and R (arginine)-rich) motif that enhances nucleosome binding to PRC2 (Lee et al. 2018), and a CT (C-Terminal) domain that mediates PRC2 binding and is involved in nucleosome binding as well (Cao and Zhang 2004; Chen et al. 2018). In support of PRC1-mediated PRC2 targeting (Blackledge et al. 2014; Cooper et al. 2014; Kahn et al. 2016; Blackledge et al. 2020; Tamburri et al. 2020), AEBP2 and JARID2 were shown to recognize the H2AK119ub histone mark on chromatin (Kalb et al. 2014; Cooper et al. 2016). The long somatic isoform of AEBP2 contains a unique, intrinsically disordered NT (N-Terminal) domain of over 200 amino acids marked by several glutamate/aspartate-rich, serine-rich, and glycine-rich patches, for which function remains unknown (Fig. 17.3 and Table 17.2) (Kim et al. 2009; Chen et al. 2020).
PRC2.2: JARID2
JARID2 was identified as a cDNA clone involved in development of the nervous system in mice and humans (Takeuchi et al. 1995; Berge-Lefranc et al. 1996). Although it belongs to the JmjC (Jumonji C) domain-containing histone lysine demethylase superfamily that depends on α-ketoglutarate and iron as cofactors for catalysis, JARID2 lacks the demethylation activity due to the absence of some critical catalytic residues mediating cofactor binding (Cloos et al. 2008; Landeira and Fisher 2011). JARID2 directly associates with the PRC2 core complex, helps recruit PRC2 to chromatin, and is required for proper differentiation of mESCs (Peng et al. 2009; Shen et al. 2009; Landeira et al. 2010; Li et al. 2010; Pasini et al. 2010). The pleiotropic roles of JARID2 in embryo development are largely connected to diverse functional domains within this protein (Fig. 17.3 and Table 17.2). From the N- to C-terminus, JARID2 harbors a UI (Ubiquitin-Interacting) motif that recognizes H2AK119ub (Cooper et al. 2016), a TR (Transrepression) domain that mediates PRC2 binding and is necessary for gene repression in vivo (Kim et al. 2003; Pasini et al. 2010; Chen et al. 2018), an R/NB (RNA/Nucleosome-Binding) domain that regulates chromatin association of PRC2 (Son et al. 2013; Kaneko et al. 2014a), a JmjN (Jumonji N) domain, an ARID (AT-Rich Interaction Domain) that plays a crucial role in chromatin targeting of JARID2 likely via direct DNA binding (Kim et al. 2003; Patsialou et al. 2005; Pasini et al. 2010), a JmjC domain, and a Zn (Zinc finger) domain that may also facilitate DNA binding (Li et al. 2010). It has not been established how the characteristic JmjN and JmjC domains of JARID2 may contribute to PRC2 function in gene repression. Notably, residue K116 of JARID2 is methylated by PRC2, and trimethylated JARID2K116 (JARID2K116me3) allosterically activates PRC2 enzymatic activity on chromatin where H3K27me3 is absent, by associating with EED at a site identical to that for H3K27me3 binding (Fig. 17.3 and Table 17.2) (Sanulli et al. 2015).
Structure-Function Analysis of PRC2
Overall Structure of PRC2.2
Electron microscopy (EM) studies have provided important insights on the overall structure of a class of PRC2 holo complexes, PRC2.2. The negative-stain EM envelope at 21 Å resolution provided an initial glimpse of the structure architecture of a PRC2.2 holo complex, PRC2–AEBP2 (Ciferri et al. 2012). With support from the chemical crosslinking and internal GFP labeling data, this structure outlines an overall bipartite organization of PRC2–AEBP2 (Ciferri et al. 2012, 2015), consisting of the catalytic module and the accessory subunit-binding module (Fig. 17.4). The catalytic module contains EZH2, EED and the C-terminal VEFS domain of SUZ12 [SUZ12(VEFS)] (Figs. 17.2 and 17.4). The VEFS domain is known to mediate EZH2 binding in Drosophila and mammalian PRC2 (Ketel et al. 2005; Yamamoto et al. 2004). EED and the VEFS domain of SUZ12 are minimally required for the enzymatic activity of EZH2 in Drosophila PRC2 (Birve et al. 2001; Ketel et al. 2005). RBBP4 and the N-terminal two-thirds of SUZ12 [SUZ12(N)] together form the accessory subunit-binding module, which is defined here based on the recent finding that it is sufficient to mediate the mutually exclusive binding of most accessory subunits of PRC2.1 and PRC2.2 (Figs. 17.2, 17.3 and 17.4) (Chen et al. 2018, 2020). The accessory subunit-binding module was also formerly referred to as the nucleosome-binding module given its involvement in nucleosome binding (Nekrasov et al. 2005; Chen et al. 2018). The version of AEBP2 used in the negative stain EM structure roughly corresponds to the shorter isoform of AEBP2; from the N to C-terminus, it spans from the catalytic module to the accessory subunit-binding module (Ciferri et al. 2012).

Overall structural model of PRC2.2 from cryo-EM and crystal structures. Negative-stain EM envelope (EMD-2236) and cryo-EM map (EMD-7334) of PRC2.2 holo complexes are shown. The catalytic module and the accessory subunit-binding module are defined based on a natural demarcation of the overall structure architecture of PRC2.2. The two modules are connected through SUZ12 [e.g. SUZ12(N)–SUZ12(VEFS)] (PDB 6C23, holo complex cartoon on the left). Protein domains responsible for the execution and regulation of diverse PRC2 functions are resolved to atomic resolution in the crystal structures of the catalytic module and the accessory subunit-binding module (subcomplex cartoons on the right). Throughout this book chapter, PyMOL (The PyMOL Molecular Graphics System, Version 2.3.5 Schrödinger, LLC.) and Chimera (http://www.rbvi.ucsf.edu/chimera) are used for displaying structures and rendering images
Cryogenic-EM (Cryo-EM) maps of higher resolutions were recently generated for a modified PRC2–AEBP2 complex additionally bound to a fragment of JARID2 (residues 106–450) (Fig. 17.4) (Kasinath et al. 2018). Flexible N-terminal domains of both EED and SUZ12 as well as residues C-terminal to the VEFS domain of SUZ12 were removed to improve the resolution (Kasinath et al. 2018). An extended active state and a compact active state were resolved to an overall resolution of 3.5 and 3.9 Å, respectively, for this PRC2–AEBP2–JARID2 complex (Kasinath et al. 2018). A C-terminal portion of AEBP2 and a short JARID2 fragment are visible in these structures (Kasinath et al. 2018). Available crystal structures were docked into corresponding regions of the cryo-EM maps to facilitate model building and interpretation of unassigned electron densities (Kasinath et al. 2018). Importantly, combination of the cryo-EM maps and X-ray crystal structures provides the most accurate and complete structural information of a PRC2.2 holo complex to date (Fig. 17.4). For example, it now becomes clear how SUZ12 orchestrates the assembly of PRC2.2: the VEFS domain of SUZ12 contacts multiple domains including the SET domain of EZH2, forming the catalytic module together with EED; the part of SUZ12 N-terminal to the VEFS domain associates with RBBP4 to form the accessory subunit-binding module, bridging AEBP2 and JARID2 to the core complex (Figs. 17.2, 17.3 and 17.4) (Justin et al. 2016; Chen et al. 2018; Kasinath et al. 2018). Structural mechanisms of PRC2 as a sum of functional domains of PRC2 components will be discussed below, when crystal structures of the catalytic module and the accessory subunit-binding module are examined in atomic details, with reference to the cryo-EM results wherever needed. In stark contrast, knowledge on the structure of PRC2.1 holo complexes has been limited, except that some latest results indicate MTF2 or PHF19-containing PRC2.1 displays a structural organization vastly different from that of PRC2.2, which may underlie a distinct mode of chromatin binding by PRC2.1 (see below) (Chen et al. 2020).
Structure of the Catalytic Module
Assembly
PRC2 components are not conserved in the yeast model organisms Saccharomyces cerevisiae (S. cerevisiae, baker’s yeast) and Schizosaccharomyces pombe (S. pombe, fission yeast). However, PRC2 is known to mediate H3K27 methylation in other fungi, like Neurospora crassa (N. crassa, red bread mold) (Jamieson et al. 2013), Fusarium graminearum (F. graminearum, cereal pathogen) (Connolly et al. 2013), and Cryptococcus neoformans (C. neoformans, human pathogen) (Dumesic et al. 2015). The high-resolution crystal structures of an active PRC2 were first determined for the EZH2–EED–SUZ12(VEFS) catalytic module from a thermophilic fungus Chaetomium thermophilum, a recently sequenced model organism for structural biology (Fig. 17.5a and b) (Amlacher et al. 2011; Jiao and Liu 2015). The overall conserved Chaetomium thermophilum PRC2 (ctPRC2) is active in H3K27 trimethylation and recapitulates H3K27me3-mediated stimulation of human PRC2 enzymatic activity in a reconstituted system (Jiao and Liu 2015). Crystal structures capture ctPRC2 in both basal and H3K27me3-stimulated states at 2.7 and 2.3 Å resolution, respectively (Fig. 17.5a and b) (Jiao and Liu 2015). The crystal structure of the corresponding catalytic module of human PRC2 at 2.9 Å resolution was determined in the H3K27M and JARID2K116me3-bound state (Fig. 17.5c), sharing a remarkable resemblance to that of its Chaetomium thermophilum counterpart (Justin et al. 2016).

Crystal structures of the catalytic module of ctPRC2 and human PRC2. Structural comparison of a crystal structure of ctPRC2 in the basal state (PDB 5KJI), b crystal structure of ctPRC2 in the H3K27me3-stimulated state (PDB 5KJH), and c crystal structure of human PRC2 in the JARID2K116me3-stimulated, H3K27M-inhibited state (PDB 5HYN). Corresponding domains in ctPRC2 and human PRC2 are colored the same, according to the color code used for the domain structures shown in Fig. 17.2. In the ctPRC2 structures, an internal linker sequence is trapped in the active site, mimicking the histone substrate
These ctPRC2 and human PRC2 structures reveal that EZH2 contains ten structurally distinct domains or motifs, which are dispersed across the entire catalytic module (Figs. 17.2 and 17.5 and Table 17.1) (Jiao and Liu 2015; Justin et al. 2016). The SBD (SANT1-Binding Domain) at the N-terminus forms an intramolecular complex with the SANT1 (SWI3, ADA2, N-CoR, and TFIIIB 1) domain (Jiao and Liu 2015; Justin et al. 2016). The SBD, EBD (EED-Binding Domain), BAM (β-Addition Motif), SAL (SET-Activation Loop), SRM (Stimulation-Responsive Motif) and SANT1 together adopt a belt-like structure that entraps EED (Jiao and Liu 2015; Justin et al. 2016). The SANT1 domain and SANT2 (SWI3, ADA2, N-CoR, and TFIIIB 2) domain are linked by the MCSS (Motif Connecting SANT1 and SANT2), which is followed by the CXC domain and the SET domain (Jiao and Liu 2015; Justin et al. 2016). The VEFS domain of SUZ12 primarily associates with the SANT2, CXC and SET domain (Figs. 17.2 and 17.5 and Table 17.1) (Jiao and Liu 2015; Justin et al. 2016). In addition to the two three-atom zinc clusters, Zn3Cys8His and Zn3Cys9, in the CXC domain, the MCSS–SANT2 region also contains a ZnCys3His and a ZnCys4 zinc-binding site (Figs. 17.2 and 17.5 and Table 17.1) (Jiao and Liu 2015; Justin et al. 2016). The SAL extends to the back of the SET domain to stabilize the active conformation (Figs. 17.2 and 17.5 and Table 17.1) (Jiao and Liu 2015; Justin et al. 2016). In the presence of H3K27me3 or JARID2K116me3, the SRM helix is juxtaposed with the SET domain to bridge the stimulating signal to the active site (Figs. 17.17.2, 17.5b and c and Table 17.1) (Jiao and Liu 2015; Justin et al. 2016).
Catalytic Mechanism, Cancer Mutations and H3K27M-Mediated Inhibition of Enzymatic Activity
Two crystal structures of an isolated inactive CXC–SET region of human EZH2 at 2.0 Å resolution were available before structure determination of the active catalytic module (Antonysamy et al. 2013; Wu et al. 2013). The inactive conformation of the SET domain features an inaccessible histone substrate-binding groove blocked by the SET-I region and an incomplete SAM-binding pocket due to the largely disordered post-SET region (Fig. 17.6a) (Antonysamy et al. 2013; Wu et al. 2013). These structures provided valuable initial insights on many recurrent disease mutations, for example, mutation of residue Y641 to a phenylalanine, serine, asparagine, histidine, or cysteine residue in follicular and diffuse large B-cell lymphomas (Antonysamy et al. 2013; Wu et al. 2013; Morin et al. 2010). In addition, structure of the Zn3Cys8His and Zn3Cys9 zinc cluster-containing CXC domain was revealed for the first time (Fig. 17.6a) (Antonysamy et al. 2013; Wu et al. 2013). Similar zinc cluster structures also exist in some other lysine methyltransferases, like N. crassa DIM-5 and S. pombe CLR4 (Min et al. 2002; Zhang et al. 2002; Dillon et al. 2005). Missense mutation of the zinc-coordinating residues in the CXC domain compromises histone methyltransferase activity and is associated with myelodysplastic syndromes (MDS) (Ketel et al. 2005; Ernst et al. 2010).

Structural basis of the catalytic mechanism of PRC2. a Crystal structure of an inactive catalytic domain of EZH2 consisting of the CXC and SET domain (PDB 4MI5). Zinc clusters are labeled. The SET-I region of the SET domain is indicated. The post-SET region of the SET domain is disordered in the inactive conformation. b Structure of an active catalytic domain of EZH2 (PDB 5HYN). The post-SET region becomes ordered. The SAL is shown in transparent surface representation. SAH and H3K27M mimicking SAM and the histone substrate, respectively, are indicated. c Structure alignment of the inactive and active catalytic domain. Rotation of the SET-I region makes space for histone substrate binding and also completes the SAM-binding pocket together with the post-SET region. d Close-up view of the active site. The current view is rotated from the view in (b) according to the provided matrix. The substrate lysine is modeled in silico from the H3K27M structure. Aromatic residues forming the lysine-binding channel are shown in sticks representation. The dotted oval circle indicates the pore structure connecting the lysine-binding channel and SAM-binding pocket. Carbonyl oxygens and hydroxyl groups surrounding the pore structure are not shown. Sites of gain-of-function cancer mutations, Y641 and A677, are highlighted by red labels
Structural rearrangement of the SET domain was predicted to occur upon formation of an active PRC2 (Antonysamy et al. 2013; Wu et al. 2013). The crystal structures of the minimally active ctPRC2 and human PRC2 confirmed this prediction (Fig. 17.6b and c) (Jiao and Liu 2015; Justin et al. 2016). In the active conformation, the conserved NHSXXPN and EELXXDY motifs meet at the characteristic pseudoknot fold, directly contacting both SAM and the substrate lysine (Fig. 17.6b) (Dillon et al. 2005; Jiao and Liu 2015; Justin et al. 2016). Notably, the active PRC2 contains a split catalytic domain: the SAL region located in N-terminal portion of EZH2 interacts with the SET domain at the C-terminus of EZH2, and this structural configuration is likely required to maintain the active conformation of the SET domain (Fig. 17.6b) (Jiao and Liu 2015; Justin et al. 2016). Compared to the active conformation, a counterclockwise rotation of the SET-I region in the inactive conformation opens up the otherwise blocked histone substrate-binding groove (Fig. 17.6c) (Jiao and Liu 2015; Justin et al. 2016). In addition, the SAM-binding pocket becomes complete with the rotation of the SET-I and the placement of the post-SET (Fig. 17.6c) (Jiao and Liu 2015; Justin et al. 2016).
Information about substrate specificity can be partially derived from the crystal structure of JARID2K116me3 and H3K27M-bound human PRC2 (Justin et al. 2016). In addition to the specificity conferred by steric exclusion, residue R26 of histone H3 (H3R26) next to the substrate lysine is specifically recognized by residues Q648 and D652 of the SET domain (Fig. 17.6d) (Justin et al. 2016). Broader target sequences and in particular non-histone ones were recently characterized by peptide array data, suggesting a preferred substrate sequence of (A/C/V/P)−3−(A/V/L)−2−(R/K)−1−(K)0−(F/Y/H)+1−(A/V/C/T/S)+2, which is also readily rationalized based on the structure (Ardehali et al. 2017).
Spatial arrangement of active site residues provides insights on the mechanism of the methyl transfer reaction, which appears to be similar to that of other lysine methyltransferases (Dillon et al. 2005; Justin et al. 2016). The model-built substrate lysine side chain is inserted through the lysine-binding channel formed mostly by aromatic residues, including Y641, F667, F724, Y726 and Y728; the nitrogen atom receiving the methyl group is positioned towards the SAM binding pocket, through a pore structure surrounded by several main chain carbonyl oxygens and side chain hydroxyl groups of conserved tyrosine residues, which are thought to facilitate the methyl transfer process (Fig. 17.6d) (Dillon et al. 2005; Justin et al. 2016).
Mutation of some active site residues in other SET domains is known to change methylation multiplicity, by altering size and conformation of the active site (Xiao et al. 2003; Zhang et al. 2003; Collins et al. 2005). Notably, coordinated actions of the wild-type and Y641F/N/S/H/C mutant EZH2 drive H3K27 hypertrimethylation in subsets of heterozygous human B-cell lymphoma: whereas efficient in making H3K27me1 from H3K27me0, the wild-type EZH2 displays limited activity in converting H3K27me2 into H3K27me3 due to a spatially restricted active site; conversely, the Y641F/N/S/H/C mutation results in a slightly expanded active site, which disfavors H3K27 monomethylation but enhances H3K27 trimethylation (Fig. 17.6d) (Sneeringer et al. 2010). An A677G mutation proximal to residue Y641 in lymphoma cells also causes H3K27 hypertrimethylation: this mutation may promote alternative conformation of residue Y641 and enlarge the active site indirectly (Fig. 17.6d) (McCabe et al. 2012a).
The oncogenic histone mutant H3K27M was proposed to inhibit PRC2 activity, by contacting the active site directly (Chan et al. 2013; Lewis et al. 2013). Another study on PRC2 inhibition by H3K27 peptides bearing various substitutions at the K27 position suggests that the enzyme active site binds strongly to linear, hydrophobic side chains (Brown et al. 2014). Congruently, the crystal structure reveals that side chain of the methionine residue is accommodated in the lysine-binding channel by mimicking the aliphatic portion of the substrate lysine (Fig. 17.6b and d) (Justin et al. 2016). Compared to its wild-type H3K27 counterpart, an H3K27M peptide binds PRC2 with an over 10-fold higher affinity in the presence of SAM in vitro (Justin et al. 2016). In addition, H3K27me3-bound PRC2 is particularly sensitive to the inhibition by H3K27M, which also depends on SAM concentration (Stafford et al. 2018; Diehl et al. 2019). Intriguingly, a non-histone protein EZHIP (EZH1/2 Inhibitory Protein) was found to associate with PRC2 and inhibit PRC2 enzymatic activity with an H3K27M-like protein sequence in Posterior Fossa A (PFA) ependymomas (Pajtler et al. 2018; Hubner et al. 2019; Jain et al. 2019; Piunti et al. 2019).
Allosteric Activation by H3K27me3 and Weaver Syndrome Mutations
EED is a major mediator of H3K27me3-based allosteric activation of PRC2. The top surface of EED WD40 domain provides an aromatic cage, formed by residues F97, Y148, W364 and Y365, to recognize trimethylated lysine, preferentially in the A–R−K–S sequence context found for H3K27me3, H3K9me3 and H1K26me3 (Fig. 17.7a) (Margueron et al. 2009; Xu et al. 2010). The binding affinity between EED and histone peptides bearing these repressive histone marks lies in the tens of micromolar range, and aromatic cage mutations greatly diminish the binding in vitro (Margueron et al. 2009; Xu et al. 2010). The essential role of the aromatic cage in PRC2 allosteric activation by H3K27me3 in vivo is supported by the observation that mutation of the corresponding residues in Drosophila ESC, an equivalent of human EED, causes massive reduction of H3K27me3 level in extracts of mutant larvae, accompanied by severe developmental defects (Margueron et al. 2009). In addition, an intact aromatic cage on EED is required for formation of H3K27me3 chromatin domains in mESCs (Oksuz et al. 2018). At a locus lacking H3K27me3, JARID2K116me3 can also stimulate PRC2 enzymatic activity for proper H3K27me3 deposition, by binding to the aromatic cage of EED with a 10-fold greater binding affinity compared with that for H3K27me3 (Fig. 17.7b and c) (Sanulli et al. 2015).

Structural basis of the allosteric activation of PRC2. a Crystal structure of EED bound to an H3K27me3 peptide (PDB 3IIW). Aromatic cage residues are shown as sticks representation. b The SRM of EZH2. Proteins and domains from the H3K27M and JARID2K116me3-bound catalytic module of human PRC2 (PDB 5HYN) that are involved in the allosteric activation are shown. Rotation matrix that relates the current view to the view in (a) is indicated. c Close-up view of the interactions between the SRM and JARID2K116me3. Mutation of residues P132, Y133, M134 and Y153 is found in Weaver syndrome. d Cryo-EM Structural model of a PRC2–AEBP2–JARID2 holo complex in compact active conformation (PDB 6C23). Only the catalytic module is shown. e The same as (d) except that extended active conformation (PDB 6C24) is shown. f Crystal structure of the GSK126-bound catalytic module of human PRC2 (PDB 5WG6). The dotted circle indicates the disordered SBD–SANT1 region
The SRM of EZH2 functions to transmit the stimulating signal from H3K27me3 or JARID2K116me3 to the SET domain (Jiao and Liu 2015; Justin et al. 2016). In the ctPRC2 structure, the SRM is positioned to simultaneously interact with H3K27me3 and an SET-I helix (Fig. 17.5b); the SRM becomes completely disordered in the absence of H3K27me3 (Fig. 17.5a) (Jiao and Liu 2015). Likewise, in the human PRC2 structure, the JARID2K116me3 peptide promotes the ordered conformation of the SRM, which also makes contacts with the corresponding SET-I helix (Fig. 17.7b and c) (Justin et al. 2016). The interaction between the SRM and the SET-I helix triggered by H3K27me3 or JARID2K116me3 binding may stabilize the overall conformation of the SET domain and cause dynamic changes of the active site conformation, such that mono-, di- and trimethylation of H3K27 are all enhanced (Fig. 17.7b and c) (Jiao and Liu 2015; Justin et al. 2016). Importantly, a few de novo germline mutations of human EZH2 found in Weaver syndrome are clustered in the SRM region and are predicted to impair allosteric activation of PRC2 (Fig. 17.7c); some Weaver syndrome mutations are also mapped to the SET domain, potentially affecting the basal enzymatic activity of PRC2 (Tatton-Brown et al. 2011; Gibson et al. 2012; Al-Salem et al. 2013; Cohen et al. 2016). It remains to be studied whether these two types of Weaver syndrome mutations are associated with different disease mechanisms.
Cryo-EM maps indicate that JARID2K116me3 is bound to EED in both compact active and extended active states of a PRC2–AEBP2–JARID2 holo complex (Kasinath et al. 2018). The two states primarily differ in the position of the SBD–SANT1 intramolecular complex of EZH2 relative to EED (Fig. 17.7d and e). Compared to the extended active state, the SBD–EBD long helix is bent at the junction between the SBD and EBD in the compact active state (Fig. 17.7d and e) (Kasinath et al. 2018). This difference was also previously noted between the crystal structures of the H3K27M and JARID2K116me3-bound human PRC2 (compact active) and an inhibitor-bound human/chameleon hybrid PRC2 at 2.6 Å resolution (extended active) (Brooun et al. 2016; Justin et al. 2016). In another crystal structure of an inhibitor-bound human PRC2 at 3.9 Å resolution with a different crystal lattice, the SBD–SANT1 region becomes disordered (Fig. 17.7f), highlighting a dynamic nature of this structural unit (Bratkowski et al. 2018).
Autoinhibition and Automethylation
In addition to the basal and H3K27me3-stimuated states, the catalytic module of ctPRC2 also exists in an autoinhibited state in the absence of the histone substrate and SAM, which represents another distinct stable conformation (Fig. 17.8a) (Bratkowski et al. 2017). Compared to the basal and H3K27me3-stimulated states (Fig. 17.8b), the post-SET region of the SET domain becomes stably associated with the active site in the autoinhibited state, occupying the substrate-binding groove and blocking histone substrate binding (Fig. 17.8a) (Bratkowski et al. 2017). SAM binding is largely impeded in the autoinhibited state as well, due to the lack of stabilization by the post-SET region and the steric clash to the SET-I region (Fig. 17.8a and b) (Bratkowski et al. 2017). SAM binding induces local conformational change of the SET-I region and partially alleviates the autoinhibition (Bratkowski et al. 2017). A series of intramolecular interactions of EZH2 maintain the autoinhibited conformation: whereas residue F922 from the post-SET region is inserted into the lysine-binding channel, residues E840 and K852 from the SET-I region mediate hydrogen bonding interactions with the post-SET (Fig. 17.8a). Disruption of these interactions by mutagenesis results in an overactivated ctPRC2 (Bratkowski et al. 2017). The autoinhibition may provide a mechanism for ctPRC2 to sense the effective SAM or histone substrate concentration in cells (Bratkowski et al. 2017). However, critical residues mediating the autoinhibition are not conserved in human EZH2, suggesting autoinhibition either may not exist or may involve a different mechanism for human PRC2 (Bratkowski et al. 2017).

Structural basis of PRC2 autoinhibition and automethylation. a Crystal structure of ctPRC2 in an autoinhibited conformation (PDB 5BJS). Close-up view is shown on the right, with residues that maintain the autoinhibited conformation highlighted in sticks representation. The post-SET region is colored in violet. Rotation matrix that relates the close-up view to the overall view is indicated. b Active conformation of the SET domain to be compared with the autoinhibited conformation. The SET domain of an active ctPRC2 (PDB 5KJH) is shown. The post-SET region is colored in violet. c Automethylation of EZH2 (based on PDB 5HYN). Three lysine residues located on a disordered loop that are subjected to automethylation are represented by filled yellow circles. The dotted arrow indicates the lysine residues are predicted to occupy the active site prior to automethylation, likely causing autoinhibition. d Crystal structure of the CLR4 SET domain in an autoinhibited conformation (PDB 6BOX). The view is aligned to that in (c). Residue K455 on a loop preceding the post-SET region occupies the active site
The enzymatic activity of human PRC2 is subjected to the regulation by automethylation (Lee et al. 2019; Wang et al. 2019), reminiscent of the regulation of kinases by autophosphorylation and of histone acetyltransferases by autoacetylation (Beenstock et al. 2016; Thompson et al. 2004). PRC2 automethylation occurs intramolecularly on three lysine residues, K505, K509 and K510, on the unstructured loop connecting the SANT2 and CXC domain, (Fig. 17.8c) (Lee et al. 2019; Wang et al. 2019). Prior to the automethylation, these lysine residues may occupy the active site of EZH2, hinder histone substrate binding, and thus confer autoinhibition; the automethylation process promotes PRC2 enzymatic activity towards the histone substrate both in vitro and in cells, conceivably by releasing the automethylation loop from the active site (Fig. 17.8c) (Lee et al. 2019; Wang et al. 2019). The proposed conformational switch during PRC2 activation by the automethylation is in line with the crystal structures of the autoinhibited and automethylated conformations of the fission yeast histone H3K9 methyltransferase CLR4 (aka SUV39H), although PRC2 enzymatic activity responds differently to alanine and arginine mutations of these automethylated lysine residues, compared to the case of CLR4 (Fig. 17.8d) (Iglesias et al. 2018; Lee et al. 2019; Wang et al. 2019).
Chemical and Stapled Peptide Inhibitors of PRC2
A family of SAM-competitive pyridone inhibitors, like GSK126, EPZ-6438 (Tazemetostat) and CPI-1205, exhibits potent (e.g. Ki or IC50 at low nanomolar range) and selective (e.g. > 1000-fold selective for EZH2 against many other methyltransferase families) inhibition of PRC2 enzymatic activity (Fig. 17.9a). Many of them are being tested in clinical trials for cancer treatment (McCabe et al. 2012b; Knutson et al. 2013; Vaswani et al. 2016). In particular, Tazemetostat (brand name Tazverik) has already been approved as a medication for metastatic or locally advanced epithelioid sarcoma and for relapsed or refractory follicular lymphoma. The unique structure of the split catalytic domain of EZH2 accounts for the remarkable selectivity of these drugs: they primarily bind to an extended pocket located on the interface between the SET and SAL domain of EZH2 that also borders EED; only a small portion of these drugs protrudes into the SAM-binding pocket and competes with SAM (Fig. 17.9b and c) (Brooun et al. 2016; Vaswani et al. 2016; Bratkowski et al. 2018). Mutation of the ‘gating residues’ of the drug-binding pocket, including residues I109, Y111 and Y661, results in acquired drug resistance in lymphoma cell line models (Fig. 17.9c) (Baker et al. 2015; Brooun et al. 2016; Gibaja et al. 2016).

Structural basis of chemical inhibition of PRC2. a Chemical structures of selected PRC2 inhibitors. b Aligned crystal structures of drug-bound PRC2 (PDBs 5LS6 and 5GSA). The shown structures include CPI-1205 bound to PRC2 catalytic module and EED226 bound to EED. c Close-up view of CPI-1205 binding pocket. Residues critical for CPI-1205 binding are shown in sticks representation and labeled. Rotation matrix that relates the current view to the view in (b) is indicated. d Close-up view of EED226 binding pocket. Aromatic cage residues mediating EED226 binding are shown. Aromatic cage residue W364 involved in H3K27me3 or JARID2K116me3 binding moves away from the EED226-binding site (compare to Fig. 17.7a)
A new class of chemical inhibitors of PRC2 became available recently. A-395 and EED226 target the aromatic cage of EED, which antagonizes H3K27me3 binding and thereby prevents the allosteric activation of PRC2 enzymatic activity (Fig. 17.9b and d) (He et al. 2017; Qi et al. 2017). These inhibitors cause similar phenotypes as the SAM-competitive inhibitors of EZH2 do in lymphoma cells; notably, they display robust PRC2 inhibition activity and strong antitumor efficacy even in the presence of the acquired mutations that confer resistance to the SAM-competitive inhibitors (He et al. 2017; Qi et al. 2017).
Stapled peptide inhibitors inhibit PRC2 enzymatic activity by disrupting complex assembly. Stapling of α-helical peptides is a strategy in drug development that allows modulation of protein-protein interactions, with the added advantage of being cell penetrating and resistant to proteolytic degradation (Verdine and Hilinski 2012). A helix of 30 amino acids of mouse EZH2 was previously mapped to be the minimal EED-binding domain (corresponding to the EBD in the catalytic module), occupying the bottom face of the EED WD40 fold in the crystal structure of a minimal EZH2–EED binary complex (Han et al. 2007). This early structural study laid the foundation for the rational design of hydrocarbon-stapled EZH2 (EBD) peptides for targeted disruption of PRC2, which impairs the growth of EZH2-dependent MLL-AF9 leukemia cells and Karpas-422 B cell lymphoma cells (Kim et al. 2013).
Structure of the Accessory Subunit-Binding Module
An Apo State
The accessory subunit-binding module of the PRC2 core complex consists of the N-terminal two thirds of SUZ12 [SUZ12(N)] and RBBP4. The Drosophila homologs of mammalian SUZ12 and RBBP4 are SU(Z)12 and NURF55, respectively. In the 2.3 Å resolution crystal structure of NURF55 bound to a minimal NURF55-binidng epitope of SU(Z)12, NURF55 adopts a WD40 fold and the SU(Z)12 fragment is engaged in hydrophobic interactions with a binding cleft formed by the N-Terminal (NT) helix and the PP loop of NURF55 (Fig. 17.10a) (Schmitges et al. 2011). An overlapping surface in Drosophila NURF55 and human RBBP7 from the Chromatin Assembly Factor 1 (CAF-1) complex is occupied by Helix 1 of the histone fold of histone H4 based on the corresponding crystal structures of 2.4–3.2 Å resolution (Fig. 17.10a); this interaction is presumed to facilitate nucleosome assembly during DNA replication and DNA repair (Murzina et al. 2008; Song et al. 2008). A more complete structure of a human SUZ12(N)–RBBP4 binary complex at 3.3 Å resolution was solved recently: four functional domains of SUZ12 are engaged in self and RBBP4 interactions, including the ZnB (Zinc finger-Binding) helix, WDB1 (WD40-Binding domain 1), WDB2 (WD40-Binding domain 2), and Cys2His2-type Zn (Zinc finger) domain (Figs. 17.2 and 17.10b and Table 17.1) (Chen et al. 2018). The WDB1 harbors an equivalent of the minimal NURF55-binidng epitope of SU(Z)12 mentioned above (Fig. 17.10a and b) (Chen et al. 2018). The ZnB and Zn domains form an intramolecular complex, which also contacts RBBP4 and the WDB1 (Fig. 17.10b) (Chen et al. 2018). Notably, a large fragment of over 250 amino acids between the WDB1 and Zn domains is mostly disordered in the current structure (Figs. 17.2 and 17.10b) (Chen et al. 2018).

Crystal structure of the apo accessory subunit-binding module. a Aligned crystal structures of Drosophila NURF55 bound to fragments of SU(Z)12 and histone H4 (PDBs 2YB8 and 3C9C). b Crystal structure of human RBBP4 bound to SUZ12(N) (PDB 5WAK). Ordered protein domains of SUZ12(N) are color coded the same as the domain structure shown in Fig. 17.2
An AEBP2 and JARID2-Bound Monomeric State
The crystal structure of a human SUZ12(N)–RBBP4–AEBP2–JARID2 complex was determined to 3.0 Å resolution (Fig. 17.11a and b), which fits well into the cryo-EM maps of a PRC2–AEBP2–JARID2 holo complex (Fig. 17.4) (Chen et al. 2018; Kasinath et al. 2018). The crystallographic and cryo-EM data support a monomeric structure architecture of the AEBP2 and JARID2-bound accessory subunit-binding module or a corresponding PRC2.2 holo complex (Chen et al. 2018; Kasinath et al. 2018). Notably, in association with AEBP2, the large disordered region between the WDB1 and Zn domains of SUZ12 becomes largely ordered (Fig. 17.11a, and compare to Fig. 17.10b), and is folded into a noncanonical type II C2 domain featuring an eight-strand β-sandwich structure, as exemplified in PLCδ, PTEN, and PI3Kα (Fig. 17.11c and d) (Chen et al. 2018). The C2 domain is known to mediate phospholipid binding and protein-protein interactions (Nalefski and Falke 1996). There are cases in which phospholipids associate with phospholipid-binding domains in chromatin complexes to impact enzymatic activity or chromatin binding activity of these complexes (Kutateladze 2012; Watson et al. 2012; Hamann and Blind 2018). However, whether the C2 domain of SUZ12 is merely a protein-protein interaction domain or it also binds phospholipids to regulate PRC2 function remains to be further studied.

Crystal structure of the accessory subunit-binding module of AEBP2 and JARID2-containing PRC2.2. a Overall view of the crystal structure of the SUZ12(N)–RBBP4–AEBP2–JARID2 heterotetrameric complex (PDB 5WAI). Protein domains are color coded as in Figs. 17.2 and 17.3. Compared to the structure of the apo complex shown in Fig. 17.10b, the C2 domain of SUZ12 becomes ordered in the current structure. b A different view of the heterotetrameric complex. Rotation matrix relates (b) to (a). c Close-up view of the C2 domain of SUZ12. The indicated rotation matrix relates (c) to (b). d Topology diagram of the C2 domain. e RBBP4 binding to the H3K4D domain of AEBP2. Electrostatic potential surface of RBBP4 is shown. Crystal structure of histone H3K4-bound Drosophila NURF55 (PDB 2YBA) is aligned to the current structure and the H3K4 peptide is colored in cyan. AEBP2 residues competing with H3K4 for RBBP4 binding are highlighted in the close-up view. f Binding surface for JARID2. Rotation matrix from (b) is provided. The breakpoint of SUZ12 found in the JAZF1-SUZ12 fusion oncoprotein is indicated by a red arrow
Around 100 amino acids at the C-terminus of AEBP2 (residues 407-503) are present in this heterotetrameric crystal structure, containing the C2B (C2-Binding) domain and the H3K4D (Histone H3K4 Displacement) domain (Figs. 17.3 and 17.11b and Table 17.2) (Chen et al. 2018). The C2B domain of AEBP2 forms a stable complex with the C2 domain and contacts the ZnB helix of SUZ12 as well (Fig. 17.11b) (Chen et al. 2018). The H3K4D domain sits on the top surface of RBBP4; particularly, residues K502 and R503 within this domain occupy an acidic central cavity in RBBP4 (Fig. 17.11e) (Chen et al. 2018). A comparable structural configuration of these two AEBP2 residues is also present in the cryo-EM structural model (Kasinath et al. 2018). Residues R2 and K4 on the histone H3K4 tail were previously shown to bind the corresponding acidic central cavity in Drosophila NURF55 in an H3K4 trimethylation-sensitive manner (Fig. 17.11e) (Schmitges et al. 2011). Structural comparison and binding assays in solution indicate AEBP2 is not compatible with histone H3K4 in RBBP4 binding (Fig. 17.11e) (Chen et al. 2018). The reason why AEBP2-containing PRC2 evolves to block H3K4 binding to RBBP4 is unclear; it is hypothesized that this structural mechanism can potentially ensure sequential recruitment of NuRD histone deacetylase and PRC2: during de novo repression of active loci, NuRD as the major binder of the unmethylated histone H3K4 tail is initially recruited to remove the acetylated histone H3K27 (H3K27ac) active histone mark, before PRC2 can be targeted to mediate histone methylation at the same site (Reynolds et al. 2012; Chen et al. 2018).
In addition to AEBP2, the crystal structure of the heterotetrameric complex also captures a JARID2 peptide (residues 147–165), corresponding to the minimal transrepression (TR) domain of JARID2 (Figs. 17.3 and 17.11f and Table 17.2) (Kim et al. 2003; Pasini et al. 2010; Chen et al. 2018). The ZnB–Zn intramolecular complex of SUZ12 is found in both the crystal and cryo-EM structural models (Chen et al. 2018; Kasinath et al. 2018). It provides a composite surface for the binding of the TR domain, which is largely folded into an α-helix structure (Fig. 17.11f) (Chen et al. 2018). The TR domain of JARID2 and the C-Terminal (CT) domain of EPOP compete for the binding of the SUZ12(N)–RBBP4 binary complex, in line with the mutually exclusive binding of JARID2 and EPOP in holo complex formation (Chen et al. 2018). In endometrial stromal tumors, a JAZF1–SUZ12 fusion oncoprotein is generated by a recurrent chromosomal translocation with a breakpoint located in the middle of the ZnB helix of SUZ12 (Fig. 17.11f) (Koontz et al. 2001; Chen et al. 2018). Based on the structural and biochemical data, JAZF1–SUZ12 is predicted to disrupt JARID2 association with PRC2, which may compromise chromatin targeting of PRC2 and contribute to the global reduction of H3K27me3 in patient samples (Ma et al. 2017; Chen et al. 2018).
A PHF19-Bound Dimeric State
In contrast to the monomeric structure of PRC2.2, data from a number of studies over the years suggest the existence of PRC2 dimers in Drosophila, mice and humans (O’Connell et al. 2001; Tie et al. 2003; Margueron et al. 2008; Casanova et al. 2011; Ballare et al. 2012; Son et al. 2013; Davidovich et al. 2014; Grijzenhout et al. 2016). Finally, a recent structural study provides a molecular basis for dimerization of MTF2 (based on sequence homology) or PHF19-containing PRC2.1 (Chen et al. 2020). The crystal structure of a human SUZ12(N)–RBBP4–PHF19–JARID2 complex was determined to 2.9 Å resolution, revealing the SUZ12(N)–RBBP4 accessory subunit-binding module adopts a dimeric structure architecture when bound to the RC/CL domain of PHF19 (Figs. 17.3, 17.12a and b and Table 17.2) (Chen et al. 2020). Although the TR domain of JARID2 is also present in the crystal structure, it was only used to improve the crystal quality, without interfering with PHF19 binding (Fig. 17.12a and b) (Chen et al. 2020). Intriguingly, formation of an atypical hybrid PRC2 holo complex containing both MTF2 and JARID2 was previously reported in mESCs in the absence of AEBP2 (Grijzenhout et al. 2016). More specifically, the SUZ12(N)–RBBP4 part forms a dimer by swapping the C2 domain of SUZ12 between each symmetry-related protomer (Fig. 17.12a and b) (Chen et al. 2020). On the major dimer interface, three positively charged residues, K195, R196 and K197, from the C2 domain of SUZ12 engage with the electronegative central cavity of RBBP4 through a series of hydrophobic, cation-π, charge-charge and hydrogen-bonding interactions (Fig. 17.12c) (Chen et al. 2020).

Crystal structure of the accessory subunit-binding module of PHF19-containing PRC2.1. (a) Overall view of the crystal structure of the SUZ12(N)–RBBP4–PHF19–JARID2 heterotetrameric complex (PDB 6NQ3). Protein domains are color coded as in Figs. 17.2 and 17.3. Corresponding protein domains of symmetry-related protomers in the dimer are distinguished by a prime symbol in the name. b A different view of the heterotetrameric complex. Rotation matrix relates (b) to (a). c Interactions on the dimer interface. Important residues are labeled in the close-up view on the right. Rotation matrix relates (c) to (b). d Interactions mediating PHF19 binding to SUZ12(N)–RBBP4. The view is the same as that in (a). The ‘R-W-L’ triad is highlighted in the close-up view on the left
PHF19 does not participate in dimerization per se, but instead stabilizes the intrinsic dimer of SUZ12(N)–RBBP4, by associating with the C2 domain of SUZ12 from one protomer and with the ZnB and WDB2 domains of SUZ12 and the NT helix of RBBP4 from the other protomer (Fig. 17.12d) (Chen et al. 2020). The RC/CL domain of PHF19 can be further divided into four functional regions: a DS (Dimer Stabilization) helix, an SC (Short Connecting) helix, a C2B (C2-Binding) domain and a CT (C-Terminal) tail (Figs. 17.3 and 17.12d and Table 17.2). The C2B domain of PHF19 is ‘locked’ to the C2 domain of SUZ12 from one protomer via an ‘R-W-L’ triad involving residues R561 and L571 of the C2B and residue W334 of the C2; the DS and CT regions of PHF19 associate with the other protomer: the DS helix packs against the ZnB helix of SUZ12 and the NT helix of RBBP4, whereas the CT tail fits into a surface cleft formed by the WDB2 domain of SUZ12 (Fig. 17.12d) (Chen et al. 2020). Intriguingly, both MTF2 and Drosophila PCL contain the conserved sequence of the DS helix found in PHF19; the corresponding sequence is not conserved in PHF1, suggesting it may not stabilize the intrinsic PRC2 dimer.
Results from in vitro dimerization assays in solution are congruent with the structural observations. Mutation of residues at the dimer interface, including the K195D/R196D/K197D triple mutation and R196A single mutation of the C2 domain of SUZ12, blocks formation of the intrinsic PRC2 dimer (Chen et al. 2020). Deletion of the DS helix prevents PHF19-mediated stabilization of the intrinsic PRC2 dimer; alanine mutation of residue W334 of SUZ12 causes a similar phenotype, due to the loss of RC/CL domain binding to SUZ12(N)–RBBP4 (Chen et al. 2020). The dimer-disrupting mutations of SUZ12 also impair the binding of MTF2 or PHF19-containing PRC2 to a piece of natural CGI DNA in vitro (Chen et al. 2020). In addition, replacing the endogenous SUZ12 with the dimer-disrupting K195D/R196D/K197D triple mutant in mESCs compromises H3K27me3 enrichment on PRC2 targets, suggesting a potentially critical role of PRC2 dimerization in the chromatin recruitment of PRC2 (Chen et al. 2020).
Distinct Structure Architectures and Modes of Chromatin Binding
DNA or nucleosome-bound structures of PRC2 subcomplexes or protein domains have recently started to emerge, providing important insights on the mechanism of chromatin binding by PRC2 holo complexes. The cryo-EM structural model of an AEBP2-containing PRC2.2 holo complex engaging an H3K27me3–H3K27 bifunctional dinucleosome at an overall resolution of 6.2 Å reveals how histone H3K27 methylation and H3K27me3-mediated allosteric activation of the enzyme may be coordinated in a chromatin context (Fig. 17.13a) (Poepsel et al. 2018). H3K27me3 is mimicked by a chemically installed methylation analog in the study (Poepsel et al. 2018). Whereas the DNA backbone of the H3K27me3 nucleosome directly contacts the SBD of EZH2, the DNA backbone of the H3K27 substrate nucleosome interacts with the CXC domain of EZH2, guiding the histone H3K27 tail to the enzyme active site (Fig. 17.13a) (Poepsel et al. 2018). Only the catalytic module but not the accessory subunit-binding module is refined to the final resolution, together with the bound dinucleosome (Poepsel et al. 2018). 2D class averages of negative-stain EM particles indicate the AEBP2-bound accessory subunit-binding module loosely associates with a nucleosome (Poepsel et al. 2018), in line with the mononucleosome binding EMSA (Electrophoresis Mobility Shift Assay) data reported elsewhere (Chen et al. 2018). The Zn finger domain of AEBP2 is not visible in the structure, suggesting the dinucleosome used may not be optimal for the binding of this domain (Kim et al. 2009; Wang et al. 2017b). Intriguingly, it was recently proposed that the AEBP2 Zn finger might have functions other than DNA binding, based on the sequence feature and NMR solution structure (Sun et al. 2018).

Modes of chromatin binding of PRC2.1 and PRC2.2. a Cryo-EM map and structural model of a PRC2.2 holo complex, PRC2-AEBP2, bound to an H3K27me3−H3K27 bifunctional dinucleosome (EMD-7306). Only the catalytic module is resolved in the map. The SBD and CXC domains of EZH2 that contact DNA directly are indicated. b Crystal structure of MTF2 bound to DNA (PDB 5XFR). The Tudor, PHD1, PHD2 and EH/WH domains of MTF2 are colored according to the domain structure shown in Fig. 17.3. Crystal structure of the PHD2–EH/WH cassette of Drosophila PCL is aligned to the MTF2 structure and colored in gray. Interactions between the W1 loop and DNA major groove are highlighted in the close-up view. c Relocation of the C2 domain from the PHF19-bound to AEBP2-bound state. The two structures are aligned based on RBBP4. For clarity, only one copy of the C2 domain in the PHF19-bound dimeric structure is colored and labeled (C2′ in this case). AEBP2 binding structurally relocates the C2′ domain from the dimer interface and results in dimer disruption, as indicated by the dotted red arrow. Also refer to Fig. 17.12b. d Schematic of chromatin binding by the dimeric PRC2.1 and monomeric PRC2.2 holo complexes. H3K36me3 binding by PCL proteins and H2AK119ub binding by AEBP2 and JARID2 are not depicted.
The recent 2–2.5 Å resolution crystal structure of a CpG-containing duplex DNA bound to PHF1 or MTF2 harboring the Tudor–PHD1–PHD2–EH/WH cassette reveals how PCL proteins may directly facilitate chromatin targeting of PRC2.1 (Fig. 17.13b) (Li et al. 2017). Residues K323 and K324 from the W1 loop of the EH/WH domain in PHF1 are responsible for recognition of the unmethylated CpG sequence content (Li et al. 2017). The corresponding residues, K338 and K339, in MTF2 contact DNA in the same manner (Fig. 17.13b) (Li et al. 2017). Compared with the 2.4 Å resolution crystal structure of the PHD2–EH/WH cassette of Drosophila PCL in the absence of DNA, the partially conserved W1 loop of PHF1 and MTF2 is reshaped to insert into the major groove of the DNA, which is distorted to accommodate the W1 loop (Fig. 17.13b) (Choi et al. 2017; Li et al. 2017). DNA binding by the EH/WH domain appears to occur independently of H3K36me3 binding by the Tudor domain of PHF1 or MTF2, although both mechanisms are thought to contribute to chromatin targeting of PRC2.1 (Li et al. 2017).
Structural comparison highlights the vastly different structure architectures of the accessory subunit-binding module from PRC2.1 and PRC2.2: AEBP2 disrupts the intrinsic dimer of the accessory subunit-binding module, whereas MTF2 and PHF19 stabilizes it; in parallel, the C2 domain of SUZ12 is placed in completely different locations in the complexes to accommodate the transition between the monomeric and dimeric state (Fig. 17.13c) (Chen et al. 2020). Together with the respective accessory subunits, the accessory subunit-binding module may also control the oligomerization state of PRC2.1 and PRC2.2 holo complexes and thereby dictate the mode of chromatin binding by PRC2.1 and PRC2.2 (Fig. 17.13d) (Chen et al. 2020). As shown by the cryo-EM study, an AEBP2-containing PRC2.2 holo complex exists as a monomer (Kasinath et al. 2018). In contrast, the actual structure of PRC2.1 holo complexes is not yet available, although MTF2 or PHF19-containing ones are predicted to be a dimer based on the existing biochemical data and partial structures (Chen et al. 2020). It is proposed that PRC2 dimerization may at least enhance MTF2 or PHF19 binding to CGI DNA in pairs, possibly through an avidity effect (Fig. 17.13d) (Chen et al. 2020). Likewise, H3K27me3 or substrate nucleosome binding by the catalytic module may also be promoted in this dimeric structural framework of PRC2.1 holo complexes.
Concluding Remarks and Future Perspectives
The past decade has witnessed a growing interest and rapid progress in structural biology towards understanding the molecular basis of the assembly, catalysis and targeting of PRC2, a key mediator of gene repression during development. A series of structural studies have generated important mechanistic insights, which verify hypothetical models from functional studies, deepen the understanding of disease mechanisms, and also lay a foundation for new lines of research.
Structural data need to be interpreted critically. Whereas low-resolution structures can be valuable in offering information about the overall shape and assembly of the target, a medium to high-resolution structure is ideal to provide accurate side chain positions, for example, to allow hypotheses to be tested by specific mutations. In the 3–4 Å medium resolution range, de novo model building of large chromatin complexes like PRC2 can be found challenging and sometimes intimidating in realms of both X-ray crystallography and cryo-EM. Whenever necessary, the register of structural models should be validated and other biochemical or functional data should be taken into consideration to support model building.
Many pressing questions remain to be answered regarding structure and function of PRC2. For example, structures of nucleosome-bound PRC2.1 holo complexes are still lacking. How do functional nucleosomes communicate with each other and with the active site of PRC2.1 to influence de novo installation and spreading of the H3K27me3 repressive histone mark? Can different modes of chromatin binding by PRC2.1 and PRC2.2 be translated into distinct mechanisms of gene repression? How may ncRNAs fit into the picture of chromatin-bound PRC2.1 or PRC2.2? Another important topic is concerned with epigenetic inheritance. Does PRC2 dimerization play a role in the faithful transmission of the H3K27me3 histone mark during mitosis? Are there functional crosstalk and physical contact between PRC2 and DNA replication machinery? In answering these questions and many others, a whole new world of structural biology research on PRC2 is anticipated to unfold.
References
Abed JA, Jones RS (2012) H3K36me3 key to Polycomb-mediated gene silencing in lineage specification. Nat Struct Mol Biol 19(12):1214–1215. https://doi.org/10.1038/nsmb.2458
Abel KJ, Brody LC, Valdes JM, Erdos MR, McKinley DR, Castilla LH, Merajver SD, Couch FJ, Friedman LS, Ostermeyer EA, Lynch ED, King MC, Welcsh PL, Osborne-Lawrence S, Spillman M, Bowcock AM, Collins FS, Weber BL (1996) Characterization of EZH1, a human homolog of Drosophila Enhancer of zeste near BRCA1. Genomics 37(2):161–171. https://doi.org/10.1006/geno.1996.0537
Alekseyenko AA, Gorchakov AA, Kharchenko PV, Kuroda MI (2014) Reciprocal interactions of human C10orf12 and C17orf96 with PRC2 revealed by BioTAP-XL cross-linking and affinity purification. Proc Natl Acad Sci USA 111(7):2488–2493. https://doi.org/10.1073/pnas.1400648111
Al-Salem A, Alshammari MJ, Hassan H, Alazami AM, Alkuraya FS (2013) Weaver syndrome and defective cortical development: a rare association. Am J Med Genet A 161A(1):225–227. https://doi.org/10.1002/ajmg.a.35660
Aloia L, Di Stefano B, Di Croce L (2013) Polycomb complexes in stem cells and embryonic development. Development 140(12):2525–2534. https://doi.org/10.1242/dev.091553
Amlacher S, Sarges P, Flemming D, van Noort V, Kunze R, Devos DP, Arumugam M, Bork P, Hurt E (2011) Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 146(2):277–289. https://doi.org/10.1016/j.cell.2011.06.039
Antonysamy S, Condon B, Druzina Z, Bonanno JB, Gheyi T, Zhang F, MacEwan I, Zhang A, Ashok S, Rodgers L, Russell M, Gately Luz J (2013) Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PLoS ONE 8(12):e84147. https://doi.org/10.1371/journal.pone.0084147
Aranda S, Mas G, Di Croce L (2015) Regulation of gene transcription by Polycomb proteins. Science advances 1(11):e1500737. https://doi.org/10.1126/sciadv.1500737
Ardehali MB, Anselmo A, Cochrane JC, Kundu S, Sadreyev RI, Kingston RE (2017) Polycomb repressive complex 2 Methylates Elongin A to regulate transcription. Mol cell 68(5):872–884 e876. https://doi.org/10.1016/j.molcel.2017.10.025
Baker T, Nerle S, Pritchard J, Zhao B, Rivera VM, Garner A, Gonzalvez F (2015) Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget 6(32):32646–32655. https://doi.org/10.18632/oncotarget.5066
Ballare C, Lange M, Lapinaite A, Martin GM, Morey L, Pascual G, Liefke R, Simon B, Shi Y, Gozani O, Carlomagno T, Benitah SA, Di Croce L (2012) Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat Struct Mol Biol 19(12):1257–1265. https://doi.org/10.1038/nsmb.2434
Barak O, Lazzaro MA, Lane WS, Speicher DW, Picketts DJ, Shiekhattar R (2003) Isolation of human NURF: a regulator of Engrailed gene expression. The EMBO journal 22(22):6089–6100. https://doi.org/10.1093/emboj/cdg582
Beenstock J, Mooshayef N, Engelberg D (2016) How do protein kinases take a selfie (Autophosphorylate)? Trends Biochem Sci 41(11):938–953. https://doi.org/10.1016/j.tibs.2016.08.006
Beltran M, Yates CM, Skalska L, Dawson M, Reis FP, Viiri K, Fisher CL, Sibley CR, Foster BM, Bartke T, Ule J, Jenner RG (2016) The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res 26(7):896–907. https://doi.org/10.1101/gr.197632.115
Berge-Lefranc JL, Jay P, Massacrier A, Cau P, Mattei MG, Bauer S, Marsollier C, Berta P, Fontes M (1996) Characterization of the human jumonji gene. Hum Mol Genet 5(10):1637–1641. https://doi.org/10.1093/hmg/5.10.1637
Beringer M, Pisano P, Di Carlo V, Blanco E, Chammas P, Vizan P, Gutierrez A, Aranda S, Payer B, Wierer M, Di Croce L (2016) EPOP functionally links Elongin and Polycomb in pluripotent stem cells. Mol Cell 64(4):645–658. https://doi.org/10.1016/j.molcel.2016.10.018
Birve A, Sengupta AK, Beuchle D, Larsson J, Kennison JA, Rasmuson-Lestander A, Muller J (2001) Su(z)12, a novel Drosophila Polycomb group gene that is conserved in vertebrates and plants. Development 128(17):3371–3379
Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LL, Ito S, Cooper S, Kondo K, Koseki Y, Ishikura T, Long HK, Sheahan TW, Brockdorff N, Kessler BM, Koseki H, Klose RJ (2014) Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157(6):1445–1459. https://doi.org/10.1016/j.cell.2014.05.004
Blackledge NP, Fursova NA, Kelley JR, Huseyin MK, Feldmann A, Klose RJ (2020) PRC1 catalytic activity is central to Polycomb system function. Mol cell 77(4):857–874 e859. https://doi.org/10.1016/j.molcel.2019.12.001
Bodega B, Marasca F, Ranzani V, Cherubini A, Della Valle F, Neguembor MV, Wassef M, Zippo A, Lanzuolo C, Pagani M, Orlando V (2017) A cytosolic Ezh1 isoform modulates a PRC2-Ezh1 epigenetic adaptive response in postmitotic cells. Nat Struct Mol Biol 24(5):444–452. https://doi.org/10.1038/nsmb.3392
Boulay G, Rosnoblet C, Guerardel C, Angrand PO, Leprince D (2011) Functional characterization of human Polycomb-like 3 isoforms identifies them as components of distinct EZH2 protein complexes. Biochem J 434(2):333–342. https://doi.org/10.1042/BJ20100944
Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K (2003) EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22(20):5323–5335. https://doi.org/10.1093/emboj/cdg542
Bratkowski M, Yang X, Liu X (2017) Polycomb repressive complex 2 in an autoinhibited state. J Biol Chem 292(32):13323–13332. https://doi.org/10.1074/jbc.M117.787572
Bratkowski M, Yang X, Liu X (2018) An evolutionarily conserved structural platform for PRC2 inhibition by a class of Ezh2 inhibitors. Sci Rep 8(1):9092. https://doi.org/10.1038/s41598-018-27175-w
Brien GL, Gambero G, O’Connell DJ, Jerman E, Turner SA, Egan CM, Dunne EJ, Jurgens MC, Wynne K, Piao L, Lohan AJ, Ferguson N, Shi X, Sinha KM, Loftus BJ, Cagney G, Bracken AP (2012) Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat Struct Mol Biol 19(12):1273–1281. https://doi.org/10.1038/nsmb.2449
Brockdorff N (2013) Noncoding RNA and Polycomb recruitment. RNA 19(4):429–442. https://doi.org/10.1261/rna.037598.112
Brooun A, Gajiwala KS, Deng YL, Liu W, Bolanos B, Bingham P, He YA, Diehl W, Grable N, Kung PP, Sutton S, Maegley KA, Yu X, Stewart AE (2016) Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat Commun 7:11384. https://doi.org/10.1038/ncomms11384
Brown ZZ, Muller MM, Jain SU, Allis CD, Lewis PW, Muir TW (2014) Strategy for “detoxification” of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J Am Chem Soc 136(39):13498–13501. https://doi.org/10.1021/ja5060934
Cai L, Rothbart SB, Lu R, Xu B, Chen WY, Tripathy A, Rockowitz S, Zheng D, Patel DJ, Allis CD, Strahl BD, Song J, Wang GG (2013) An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol Cell 49(3):571–582. https://doi.org/10.1016/j.molcel.2012.11.026
Cao R, Zhang Y (2004) SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 15(1):57–67. https://doi.org/10.1016/j.molcel.2004.06.020
Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298(5595):1039–1043. https://doi.org/10.1126/science.1076997
Cao R, Wang H, He J, Erdjument-Bromage H, Tempst P, Zhang Y (2008) Role of hPHF1 in H3K27 methylation and Hox gene silencing. Mol Cell Biol 28(5):1862–1872. https://doi.org/10.1128/MCB.01589-07
Casanova M, Preissner T, Cerase A, Poot R, Yamada D, Li X, Appanah R, Bezstarosti K, Demmers J, Koseki H, Brockdorff N (2011) Polycomblike 2 facilitates the recruitment of PRC2 Polycomb group complexes to the inactive X chromosome and to target loci in embryonic stem cells. Development 138(8):1471–1482. https://doi.org/10.1242/dev.053652
Chammas P, Mocavini I, Di Croce L (2020) Engaging chromatin: PRC2 structure meets function. Br J Cancer 122(3):315–328. https://doi.org/10.1038/s41416-019-0615-2
Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, Sarkaria J, Zhang Z (2013) The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes & Dev 27(9):985–990. https://doi.org/10.1101/gad.217778.113
Chen H, Rossier C, Antonarakis SE (1996) Cloning of a human homolog of the Drosophila enhancer of zeste gene (EZH2) that maps to chromosome 21q22.2. Genomics 38(1):30–37. https://doi.org/10.1006/geno.1996.0588
Chen S, Jiao L, Shubbar M, Yang X, Liu X (2018) Unique structural platforms of Suz12 dictate distinct classes of PRC2 for chromatin binding. Mol Cell 69(5):840–852 e845. https://doi.org/10.1016/j.molcel.2018.01.039
Chen S, Jiao L, Liu X, Yang X, Liu X (2020) A dimeric structural scaffold for PRC2-PCL targeting to CpG Island chromatin. Mol Cell 77(6):1265–1278 e1267. https://doi.org/10.1016/j.molcel.2019.12.019
Chittock EC, Latwiel S, Miller TC, Muller CW (2017) Molecular architecture of polycomb repressive complexes. Biochem Soc Trans 45(1):193–205. https://doi.org/10.1042/BST20160173
Choi J, Bachmann AL, Tauscher K, Benda C, Fierz B, Muller J (2017) DNA binding by PHF1 prolongs PRC2 residence time on chromatin and thereby promotes H3K27 methylation. Nat Struct Mol Biol. https://doi.org/10.1038/nsmb.3488
Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E (2012) Molecular architecture of human polycomb repressive complex 2. eLife 1:e00005. https://doi.org/10.7554/elife.00005
Ciferri C, Lander GC, Nogales E (2015) Protein domain mapping by internal labeling and single particle electron microscopy. J Struct Biol 192(2):159–162. https://doi.org/10.1016/j.jsb.2015.09.016
Cifuentes-Rojas C, Hernandez AJ, Sarma K, Lee JT (2014) Regulatory interactions between RNA and polycomb repressive complex 2. Mol Cell 55(2):171–185. https://doi.org/10.1016/j.molcel.2014.05.009
Cloos PA, Christensen J, Agger K, Helin K (2008) Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev 22(9):1115–1140. https://doi.org/10.1101/gad.1652908
Cohen AS, Yap DB, Lewis ME, Chijiwa C, Ramos-Arroyo MA, Tkachenko N, Milano V, Fradin M, McKinnon ML, Townsend KN, Xu J, Van Allen MI, Ross CJ, Dobyns WB, Weaver DD, Gibson WT (2016) Weaver syndrome-associated EZH2 protein variants show impaired histone methyltransferase function in vitro. Hum Mutat 37(3):301–307. https://doi.org/10.1002/humu.22946
Coleman RT, Struhl G (2017) Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science 356(6333). https://doi.org/10.1126/science.aai8236
Collins RE, Tachibana M, Tamaru H, Smith KM, Jia D, Zhang X, Selker EU, Shinkai Y, Cheng X (2005) In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J Biol Chem 280(7):5563–5570. https://doi.org/10.1074/jbc.M410483200
Connolly LR, Smith KM, Freitag M (2013) The Fusarium graminearum histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genet 9(10):e1003916. https://doi.org/10.1371/journal.pgen.1003916
Conway E, Jerman E, Healy E, Ito S, Holoch D, Oliviero G, Deevy O, Glancy E, Fitzpatrick DJ, Mucha M, Watson A, Rice AM, Chammas P, Huang C, Pratt-Kelly I, Koseki Y, Nakayama M, Ishikura T, Streubel G, Wynne K, Hokamp K, McLysaght A, Ciferri C, Di Croce L, Cagney G, Margueron R, Koseki H, Bracken AP (2018) A family of vertebrate-specific polycombs encoded by the LCOR/LCORL genes balance PRC2 subtype activities. Mol Cell 70(3):408–421 e408. https://doi.org/10.1016/j.molcel.2018.03.005
Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP, De Marco V, Elderkin S, Koseki H, Klose R, Heger A, Brockdorff N (2014) Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep 7(5):1456–1470. https://doi.org/10.1016/j.celrep.2014.04.012
Cooper S, Grijzenhout A, Underwood E, Ancelin K, Zhang T, Nesterova TB, Anil-Kirmizitas B, Bassett A, Kooistra SM, Agger K, Helin K, Heard E, Brockdorff N (2016) Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat Commun 7:13661. https://doi.org/10.1038/ncomms13661
Coulson M, Robert S, Eyre HJ, Saint R (1998) The identification and localization of a human gene with sequence similarity to Polycomblike of Drosophila melanogaster. Genomics 48(3):381–383. https://doi.org/10.1006/geno.1997.5201
Cyrus S, Burkardt D, Weaver DD, Gibson WT (2019) PRC2-complex related dysfunction in overgrowth syndromes: a review of EZH2, EED, and SUZ12 and their syndromic phenotypes. Am J Med Genet Part C, Semin Med Genet 181(4):519–531. https://doi.org/10.1002/ajmg.c.31754
Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111(2):185–196
Davidovich C, Cech TR (2015) The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA 21(12):2007–2022. https://doi.org/10.1261/rna.053918.115
Davidovich C, Zheng L, Goodrich KJ, Cech TR (2013) Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol 20(11):1250–1257. https://doi.org/10.1038/nsmb.2679
Davidovich C, Goodrich KJ, Gooding AR, Cech TR (2014) A dimeric state for PRC2. Nucleic Acids Res 42(14):9236–9248. https://doi.org/10.1093/nar/gku540
Deaton AM, Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25(10):1010–1022. https://doi.org/10.1101/gad.2037511
Diehl KL, Ge EJ, Weinberg DN, Jani KS, Allis CD, Muir TW (2019) PRC2 engages a bivalent H3K27M-H3K27me3 dinucleosome inhibitor. Proc Natl Acad Sci USA 116(44):22152–22157. https://doi.org/10.1073/pnas.1911775116
Dillon SC, Zhang X, Trievel RC, Cheng X (2005) The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol 6(8):227. https://doi.org/10.1186/gb-2005-6-8-227
Dumesic PA, Homer CM, Moresco JJ, Pack LR, Shanle EK, Coyle SM, Strahl BD, Fujimori DG, Yates JR 3rd, Madhani HD (2015) Product binding enforces the genomic specificity of a yeast polycomb repressive complex. Cell 160(1–2):204–218. https://doi.org/10.1016/j.cell.2014.11.039
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, Grand FH, Cross NC (2010) Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42(8):722–726. https://doi.org/10.1038/ng.621
Gibaja V, Shen F, Harari J, Korn J, Ruddy D, Saenz-Vash V, Zhai H, Rejtar T, Paris CG, Yu Z, Lira M, King D, Qi W, Keen N, Hassan AQ, Chan HM (2016) Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 35(5):558–566. https://doi.org/10.1038/onc.2015.114
Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, Marra MA, Consortium FC, Chitayat D, Boycott KM, Weaver DD, Jones SJ (2012) Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet 90(1):110–118. https://doi.org/10.1016/j.ajhg.2011.11.018
Grijzenhout A, Godwin J, Koseki H, Gdula MR, Szumska D, McGouran JF, Bhattacharya S, Kessler BM, Brockdorff N, Cooper S (2016) Functional analysis of AEBP2, a PRC2 Polycomb protein, reveals a Trithorax phenotype in embryonic development and in ESCs. Development 143(15):2716–2723. https://doi.org/10.1242/dev.123935
Grzenda A, Lomberk G, Svingen P, Mathison A, Calvo E, Iovanna J, Xiong Y, Faubion W, Urrutia R (2013) Functional characterization of EZH2beta reveals the increased complexity of EZH2 isoforms involved in the regulation of mammalian gene expression. Epigenetics Chromatin 6(1):3. https://doi.org/10.1186/1756-8935-6-3
Hamann BL, Blind RD (2018) Nuclear phosphoinositide regulation of chromatin. J Cell Physiol 233(1):107–123. https://doi.org/10.1002/jcp.25886
Han Z, Xing X, Hu M, Zhang Y, Liu P, Chai J (2007) Structural basis of EZH2 recognition by EED. Structure 15(10):1306–1315. https://doi.org/10.1016/j.str.2007.08.007
Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 10(11):1291–1300. https://doi.org/10.1038/ncb1787
Hauri S, Comoglio F, Seimiya M, Gerstung M, Glatter T, Hansen K, Aebersold R, Paro R, Gstaiger M, Beisel C (2016) A high-density map for navigating the human polycomb complexome. Cell Rep 17(2):583–595. https://doi.org/10.1016/j.celrep.2016.08.096
He GP, Kim S, Ro HS (1999) Cloning and characterization of a novel zinc finger transcriptional repressor. A direct role of the zinc finger motif in repression. J Biol Chem 274(21):14678–14684. https://doi.org/10.1074/jbc.274.21.14678
He Y, Selvaraju S, Curtin ML, Jakob CG, Zhu H, Comess KM, Shaw B, The J, Lima-Fernandes E, Szewczyk MM, Cheng D, Klinge KL, Li HQ, Pliushchev M, Algire MA, Maag D, Guo J, Dietrich J, Panchal SC, Petros AM, Sweis RF, Torrent M, Bigelow LJ, Senisterra G, Li F, Kennedy S, Wu Q, Osterling DJ, Lindley DJ, Gao W, Galasinski S, Barsyte-Lovejoy D, Vedadi M, Buchanan FG, Arrowsmith CH, Chiang GG, Sun C, Pappano WN (2017) The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat Chem Biol 13(4):389–395. https://doi.org/10.1038/nchembio.2306
Healy E, Mucha M, Glancy E, Fitzpatrick DJ, Conway E, Neikes HK, Monger C, Van Mierlo G, Baltissen MP, Koseki Y, Vermeulen M, Koseki H, Bracken AP (2019) PRC2.1 and PRC2.2 synergize to coordinate H3K27 trimethylation. Mol Cell 76(3):437–452 e436. https://doi.org/10.1016/j.molcel.2019.08.012
Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163(3):712–723. https://doi.org/10.1016/j.cell.2015.09.053
Hennig L, Derkacheva M (2009) Diversity of Polycomb group complexes in plants: same rules, different players? Trends Genet 25(9):414–423. https://doi.org/10.1016/j.tig.2009.07.002
Hobert O, Sures I, Ciossek T, Fuchs M, Ullrich A (1996) Isolation and developmental expression analysis of Enx-1, a novel mouse Polycomb group gene. Mech Dev 55(2):171–184
Hojfeldt JW, Hedehus L, Laugesen A, Tatar T, Wiehle L, Helin K (2019) Non-core subunits of the PRC2 complex are collectively required for its target-site specificity. Mol Cell 76(3):423–436 e423. https://doi.org/10.1016/j.molcel.2019.07.031
Hojfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A, Mohammad F, Jensen ON, Helin K (2018) Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat Struct Mol Biol 25(3):225–232. https://doi.org/10.1038/s41594-018-0036-6
Hubner JM, Muller T, Papageorgiou DN, Mauermann M, Krijgsveld J, Russell RB, Ellison DW, Pfister SM, Pajtler KW, Kool M (2019) EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro-oncology 21(7):878–889. https://doi.org/10.1093/neuonc/noz058
Hunkapiller J, Shen Y, Diaz A, Cagney G, McCleary D, Ramalho-Santos M, Krogan N, Ren B, Song JS, Reiter JF (2012) Polycomb-like 3 promotes polycomb repressive complex 2 binding to CpG islands and embryonic stem cell self-renewal. PLoS Genet 8(3):e1002576. https://doi.org/10.1371/journal.pgen.1002576
Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wuhr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP (2015) The BioPlex network: a systematic exploration of the human interactome. Cell 162(2):425–440. https://doi.org/10.1016/j.cell.2015.06.043
Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, Szpyt J, Tam S, Zarraga G, Pontano-Vaites L, Swarup S, White AE, Schweppe DK, Rad R, Erickson BK, Obar RA, Guruharsha KG, Li K, Artavanis-Tsakonas S, Gygi SP, Harper JW (2017) Architecture of the human interactome defines protein communities and disease networks. Nature 545(7655):505–509. https://doi.org/10.1038/nature22366
Iglesias N, Currie MA, Jih G, Paulo JA, Siuti N, Kalocsay M, Gygi SP, Moazed D (2018) Automethylation-induced conformational switch in Clr4 (Suv39h) maintains epigenetic stability. Nature 560(7719):504–508. https://doi.org/10.1038/s41586-018-0398-2
Inouye C, Remondelli P, Karin M, Elledge S (1994) Isolation of a cDNA encoding a metal response element binding protein using a novel expression cloning procedure: the one hybrid system. DNA Cell Biol 13(7):731–742. https://doi.org/10.1089/dna.1994.13.731
Jain SU, Do TJ, Lund PJ, Rashoff AQ, Diehl KL, Cieslik M, Bajic A, Juretic N, Deshmukh S, Venneti S, Muir TW, Garcia BA, Jabado N, Lewis PW (2019) PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10(1):2146. https://doi.org/10.1038/s41467-019-09981-6
Jamieson K, Rountree MR, Lewis ZA, Stajich JE, Selker EU (2013) Regional control of histone H3 lysine 27 methylation in neurospora. Proc Natl Acad Sci USA 110(15):6027–6032. https://doi.org/10.1073/pnas.1303750110
Jermann P, Hoerner L, Burger L, Schubeler D (2014) Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc Natl Acad Sci USA 111(33):E3415–E3421. https://doi.org/10.1073/pnas.1400672111
Jiao L, Liu X (2015) Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 350(6258):aac4383. https://doi.org/10.1126/science.aac4383
Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, Wilson JR, Gamblin SJ (2016) Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 7:11316. https://doi.org/10.1038/ncomms11316
Kahn TG, Dorafshan E, Schultheis D, Zare A, Stenberg P, Reim I, Pirrotta V, Schwartz YB (2016) Interdependence of PRC1 and PRC2 for recruitment to Polycomb response elements. Nucleic Acids Res 44(21):10132–10149. https://doi.org/10.1093/nar/gkw701
Kalb R, Latwiel S, Baymaz HI, Jansen PW, Muller CW, Vermeulen M, Muller J (2014) Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 21(6):569–571. https://doi.org/10.1038/nsmb.2833
Kaneko S, Son J, Shen SS, Reinberg D, Bonasio R (2013) PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat Struct Mol Biol 20(11):1258–1264. https://doi.org/10.1038/nsmb.2700
Kaneko S, Bonasio R, Saldana-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D (2014a) Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell 53(2):290–300. https://doi.org/10.1016/j.molcel.2013.11.012
Kaneko S, Son J, Bonasio R, Shen SS, Reinberg D (2014b) Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev. https://doi.org/10.1101/gad.247940.114
Kasinath V, Faini M, Poepsel S, Reif D, Feng XA, Stjepanovic G, Aebersold R, Nogales E (2018) Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science 359(6378):940–944. https://doi.org/10.1126/science.aar5700
Ketel CS, Andersen EF, Vargas ML, Suh J, Strome S, Simon JA (2005) Subunit contributions to histone methyltransferase activities of fly and worm polycomb group complexes. Mol Cell Biol 25(16):6857–6868. https://doi.org/10.1128/MCB.25.16.6857-6868.2005
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA 106(28):11667–11672. https://doi.org/10.1073/pnas.0904715106
Kim TG, Kraus JC, Chen J, Lee Y (2003) JUMONJI, a critical factor for cardiac development, functions as a transcriptional repressor. J Biol Chem 278(43):42247–42255. https://doi.org/10.1074/jbc.M307386200
Kim H, Kang K, Kim J (2009) AEBP2 as a potential targeting protein for Polycomb repression complex PRC2. Nucleic Acids Res 37(9):2940–2950. https://doi.org/10.1093/nar/gkp149
Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH (2013) Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol 9(10):643–650. https://doi.org/10.1038/nchembio.1331
Kloet SL, Makowski MM, Baymaz HI, van Voorthuijsen L, Karemaker ID, Santanach A, Jansen PW, Di Croce L, Vermeulen M (2016) The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat Struct Mol Biol 23(7):682–690. https://doi.org/10.1038/nsmb.3248
Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H (2013) Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA 110(19):7922–7927. https://doi.org/10.1073/pnas.1303800110
Koontz JI, Soreng AL, Nucci M, Kuo FC, Pauwels P, van Den Berghe H, Dal Cin P, Fletcher JA, Sklar J (2001) Frequent fusion of the JAZF1 and JJAZ1 genes in endometrial stromal tumors. Proc Natl Acad Sci USA 98(11):6348–6353. https://doi.org/10.1073/pnas.101132598
Kutateladze TG (2012) Histone deacetylation: IP4 is an epigenetic coregulator. Nat Chem Biol 8(3):230–231. https://doi.org/10.1038/nchembio.795
Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of Zeste protein. Genes Dev 16(22):2893–2905. https://doi.org/10.1101/gad.1035902
Kuzmichev A, Jenuwein T, Tempst P, Reinberg D (2004) Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell 14(2):183–193
Kuzmichev A, Margueron R, Vaquero A, Preissner TS, Scher M, Kirmizis A, Ouyang X, Brockdorff N, Abate-Shen C, Farnham P, Reinberg D (2005) Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc Natl Acad Sci USA 102(6):1859–1864. https://doi.org/10.1073/pnas.0409875102
Laible G, Wolf A, Dorn R, Reuter G, Nislow C, Lebersorger A, Popkin D, Pillus L, Jenuwein T (1997) Mammalian homologues of the Polycomb-group gene Enhancer of zeste mediate gene silencing in Drosophila heterochromatin and at S. cerevisiae telomeres. EMBO J 16(11):3219–3232. https://doi.org/10.1093/emboj/16.11.3219
Landeira D, Fisher AG (2011) Inactive yet indispensable: the tale of Jarid2. Trends Cell Biol 21(2):74–80. https://doi.org/10.1016/j.tcb.2010.10.004
Landeira D, Sauer S, Poot R, Dvorkina M, Mazzarella L, Jorgensen HF, Pereira CF, Leleu M, Piccolo FM, Spivakov M, Brookes E, Pombo A, Fisher C, Skarnes WC, Snoek T, Bezstarosti K, Demmers J, Klose RJ, Casanova M, Tavares L, Brockdorff N, Merkenschlager M, Fisher AG (2010) Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat Cell Biol 12(6):618–624. https://doi.org/10.1038/ncb2065
Laprell F, Finkl K, Muller J (2017) Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science 356(6333):85–88. https://doi.org/10.1126/science.aai8266
Laugesen A, Hojfeldt JW, Helin K (2016) Role of the Polycomb repressive complex 2 (PRC2) in transcriptional regulation and cancer. Cold Spring Harb Perspect Med 6(9). https://doi.org/10.1101/cshperspect.a026575
Laugesen A, Hojfeldt JW, Helin K (2019) Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol Cell 74(1):8–18. https://doi.org/10.1016/j.molcel.2019.03.011
Lee CH, Holder M, Grau D, Saldana-Meyer R, Yu JR, Ganai RA, Zhang J, Wang M, LeRoy G, Dobenecker MW, Reinberg D, Armache KJ (2018) Distinct stimulatory mechanisms regulate the catalytic activity of polycomb repressive complex 2. Mol Cell 70(3):435–448 e435. https://doi.org/10.1016/j.molcel.2018.03.019
Lee CH, Yu JR, Granat J, Saldana-Meyer R, Andrade J, LeRoy G, Jin Y, Lund P, Stafford JM, Garcia BA, Ueberheide B, Reinberg D (2019) Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev 33(19–20):1428–1440. https://doi.org/10.1101/gad.328773.119
Lewis EB (1978) A gene complex controlling segmentation in Drosophila. Nature 276(5688):565–570
Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340(6134):857–861. https://doi.org/10.1126/science.1232245
Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D (2010) Jarid2 and PRC2, partners in regulating gene expression. Genes Dev 24(4):368–380. https://doi.org/10.1101/gad.1886410
Li X, Isono K, Yamada D, Endo TA, Endoh M, Shinga J, Mizutani-Koseki Y, Otte AP, Casanova M, Kitamura H, Kamijo T, Sharif J, Ohara O, Toyada T, Bernstein BE, Brockdorff N, Koseki H (2011) Mammalian polycomb-like Pcl2/Mtf2 is a novel regulatory component of PRC2 that can differentially modulate polycomb activity both at the Hox gene cluster and at Cdkn2a genes. Mol Cell Biol 31(2):351–364. https://doi.org/10.1128/MCB.00259-10
Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P, Zhang W, He Q, Patel DJ, Bulyk ML, Shi Y, Wang Z (2017) Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549(7671):287–291. https://doi.org/10.1038/nature23881
Liefke R, Shi Y (2015) The PRC2-associated factor C17orf96 is a novel CpG island regulator in mouse ES cells. Cell Discov 1:15008. https://doi.org/10.1038/celldisc.2015.8
Liefke R, Karwacki-Neisius V, Shi Y (2016) EPOP Interacts with Elongin BC and USP7 to modulate the chromatin landscape. Mol Cell 64(4):659–672. https://doi.org/10.1016/j.molcel.2016.10.019
Long Y, Bolanos B, Gong L, Liu W, Goodrich KJ, Yang X, Chen S, Gooding AR, Maegley KA, Gajiwala KS, Brooun A, Cech TR, Liu X (2017) Conserved RNA-binding specificity of polycomb repressive complex 2 is achieved by dispersed amino acid patches in EZH2. eLife 6. https://doi.org/10.7554/elife.31558
Ma X, Wang J, Wang J, Ma CX, Gao X, Patriub V, Sklar JL (2017) The JAZF1-SUZ12 fusion protein disrupts PRC2 complexes and impairs chromatin repression during human endometrial stromal tumorogenesis. Oncotarget 8(3):4062–4078. https://doi.org/10.18632/oncotarget.13270
Maier VK, Feeney CM, Taylor JE, Creech AL, Qiao JW, Szanto A, Das PP, Chevrier N, Cifuentes-Rojas C, Orkin SH, Carr SA, Jaffe JD, Mertins P, Lee JT (2015) Functional proteomic analysis of repressive histone methyltransferase complexes reveals ZNF518B as a G9A regulator. Mol Cell Proteomics: MCP 14(6):1435–1446. https://doi.org/10.1074/mcp.M114.044586
Margueron R, Reinberg D (2011) The Polycomb complex PRC2 and its mark in life. Nature 469(7330):343–349. https://doi.org/10.1038/nature09784
Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D (2008) Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32(4):503–518. https://doi.org/10.1016/j.molcel.2008.11.004
Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ (2009) Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461(7265):762–767. https://doi.org/10.1038/nature08398
Martin C, Cao R, Zhang Y (2006) Substrate preferences of the EZH2 histone methyltransferase complex. J Biol Chem 281(13):8365–8370. https://doi.org/10.1074/jbc.M513425200
McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, Chen SB, Della Pietra A, 3rd, Dul E, Hughes AM, Gilbert SA, Thrall SH, Tummino PJ, Kruger RG, Brandt M, Schwartz B, Creasy CL (2012a) Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Nat Acad Sci USA 109(8):2989–2994. https://doi.org/10.1073/pnas.1116418109
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL (2012b) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492(7427):108–112. https://doi.org/10.1038/nature11606
Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, Bernstein BE (2010) GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet 6(12):e1001244. https://doi.org/10.1371/journal.pgen.1001244
Min J, Zhang X, Cheng X, Grewal SI, Xu RM (2002) Structure of the SET domain histone lysine methyltransferase Clr4. Nat Struct Biol 9(11):828–832. https://doi.org/10.1038/nsb860
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R, Tam A, Varhol R, Smailus D, Moksa M, Zhao Y, Delaney A, Qian H, Birol I, Schein J, Moore R, Holt R, Horsman DE, Connors JM, Jones S, Aparicio S, Hirst M, Gascoyne RD, Marra MA (2010) Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42(2):181–185. https://doi.org/10.1038/ng.518
Mozzetta C, Pontis J, Fritsch L, Robin P, Portoso M, Proux C, Margueron R, Ait-Si-Ali S (2014) The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol Cell 53(2):277–289. https://doi.org/10.1016/j.molcel.2013.12.005
Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111(2):197–208
Murzina NV, Pei XY, Zhang W, Sparkes M, Vicente-Garcia J, Pratap JV, McLaughlin SH, Ben-Shahar TR, Verreault A, Luisi BF, Laue ED (2008) Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure 16(7):1077–1085. https://doi.org/10.1016/j.str.2008.05.006
Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde ME, Hong Z, Allen C, Roy S, Nunez JK, Nickoloff J, Kulesza CA, Yasui A, Cote J, Kutateladze TG (2012) Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat Struct Mol Biol 19(12):1266–1272. https://doi.org/10.1038/nsmb.2435
Nalefski EA, Falke JJ (1996) The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci Publ Protein Soc 5(12):2375–2390. https://doi.org/10.1002/pro.5560051201
Nekrasov M, Wild B, Muller J (2005) Nucleosome binding and histone methyltransferase activity of Drosophila PRC2. EMBO Rep 6(4):348–353. https://doi.org/10.1038/sj.embor.7400376
O’Connell S, Wang L, Robert S, Jones CA, Saint R, Jones RS (2001) Polycomblike PHD fingers mediate conserved interaction with enhancer of zeste protein. J Biol Chem 276(46):43065–43073. https://doi.org/10.1074/jbc.M104294200
Oksuz O, Narendra V, Lee CH, Descostes N, LeRoy G, Raviram R, Blumenberg L, Karch K, Rocha PP, Garcia BA, Skok JA, Reinberg D (2018) Capturing the Onset of PRC2-mediated repressive domain formation. Mol Cell 70(6):1149–1162 e1145. https://doi.org/10.1016/j.molcel.2018.05.023
Oliviero G, Brien GL, Waston A, Streubel G, Jerman E, Andrews D, Doyle B, Munawar N, Wynne K, Crean J, Bracken AP, Cagney G (2016) Dynamic protein interactions of the polycomb repressive complex 2 during differentiation of pluripotent cells. Mol Cell Proteomics MCP 15(11):3450–3460. https://doi.org/10.1074/mcp.M116.062240
Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, Hubner JM, Ramaswamy V, Jia S, Dalton JD, Haupfear K, Rogers HA, Punchihewa C, Lee R, Easton J, Wu G, Ritzmann TA, Chapman R, Chavez L, Boop FA, Klimo P, Sabin ND, Ogg R, Mack SC, Freibaum BD, Kim HJ, Witt H, Jones DTW, Vo B, Gajjar A, Pounds S, Onar-Thomas A, Roussel MF, Zhang J, Taylor JP, Merchant TE, Grundy R, Tatevossian RG, Taylor MD, Pfister SM, Korshunov A, Kool M, Ellison DW (2018) Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol 136(2):211–226. https://doi.org/10.1007/s00401-018-1877-0
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32(2):232–246. https://doi.org/10.1016/j.molcel.2008.08.022
Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K (2010) JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 464(7286):306–310. https://doi.org/10.1038/nature08788
Patsialou A, Wilsker D, Moran E (2005) DNA-binding properties of ARID family proteins. Nucleic Acids Res 33(1):66–80. https://doi.org/10.1093/nar/gki145
Peng JC, Valouev A, Swigut T, Zhang J, Zhao Y, Sidow A, Wysocka J (2009) Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 139(7):1290–1302. https://doi.org/10.1016/j.cell.2009.12.002
Perino M, van Mierlo G, Karemaker ID, van Genesen S, Vermeulen M, Marks H, van Heeringen SJ, Veenstra GJC (2018) MTF2 recruits polycomb repressive complex 2 by helical-shape-selective DNA binding. Nat Genet 50(7):1002–1010. https://doi.org/10.1038/s41588-018-0134-8
Piunti A, Smith ER, Morgan MAJ, Ugarenko M, Khaltyan N, Helmin KA, Ryan CA, Murray DC, Rickels RA, Yilmaz BD, Rendleman EJ, Savas JN, Singer BD, Bulun SE, Shilatifard A (2019) CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci Adv 5(7):eaax2887. https://doi.org/10.1126/sciadv.aax2887
Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300(5616):131–135. https://doi.org/10.1126/science.1084274
Poepsel S, Kasinath V, Nogales E (2018) Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat Struct Mol Biol 25(2):154–162. https://doi.org/10.1038/s41594-018-0023-y
Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, Zhang M, Zhang J, Yu Z, Li L, Teng L, Chuai S, Zhang C, Zhao M, Chan H, Chen Z, Fang D, Fei Q, Feng L, Feng L, Gao Y, Ge H, Ge X, Li G, Lingel A, Lin Y, Liu Y, Luo F, Shi M, Wang L, Wang Z, Yu Y, Zeng J, Zeng C, Zhang L, Zhang Q, Zhou S, Oyang C, Atadja P, Li E (2017) An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol 13(4):381–388. https://doi.org/10.1038/nchembio.2304
Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, Behrens A, Bertone P, Hendrich B (2012) NuRD-mediated deacetylation of H3K27 facilitates recruitment of polycomb repressive complex 2 to direct gene repression. EMBO J 31(3):593–605. https://doi.org/10.1038/emboj.2011.431
Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K (2014) Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell 55(3):347–360. https://doi.org/10.1016/j.molcel.2014.06.005
Ringrose L (2017) Noncoding RNAs in polycomb and trithorax regulation: a quantitative perspective. Annu Rev Genet 51:385–411. https://doi.org/10.1146/annurev-genet-120116-023402
Ringrose L, Paro R (2007) Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 134(2):223–232. https://doi.org/10.1242/dev.02723
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129(7):1311–1323. https://doi.org/10.1016/j.cell.2007.05.022
Sanulli S, Justin N, Teissandier A, Ancelin K, Portoso M, Caron M, Michaud A, Lombard B, da Rocha ST, Offer J, Loew D, Servant N, Wassef M, Burlina F, Gamblin SJ, Heard E, Margueron R (2015) Jarid2 methylation via the PRC2 complex regulates H3K27me3 deposition during cell differentiation. Mol Cell 57(5):769–783. https://doi.org/10.1016/j.molcel.2014.12.020
Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D (2008) Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol 28(8):2718–2731. https://doi.org/10.1128/MCB.02017-07
Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Muller J (2010) Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465(7295):243–247. https://doi.org/10.1038/nature08966
Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, Bunker RD, Wirth U, Bouwmeester T, Bauer A, Ly-Hartig N, Zhao K, Chan H, Gu J, Gut H, Fischle W, Muller J, Thoma NH (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42(3):330–341. https://doi.org/10.1016/j.molcel.2011.03.025
Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G (2017) Genome regulation by polycomb and trithorax: 70 years and counting. Cell 171(1):34–57. https://doi.org/10.1016/j.cell.2017.08.002
Schwartz YB, Pirrotta V (2013) A new world of Polycombs: unexpected partnerships and emerging functions. Nat Rev Genet 14(12):853–864. https://doi.org/10.1038/nrg3603
Sewalt RG, van der Vlag J, Gunster MJ, Hamer KM, den Blaauwen JL, Satijn DP, Hendrix T, van Driel R, Otte AP (1998) Characterization of interactions between the mammalian polycomb-group proteins Enx1/EZH2 and EED suggests the existence of different mammalian polycomb-group protein complexes. Mol Cell Biol 18(6):3586–3595
Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH (2008) EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 32(4):491–502. https://doi.org/10.1016/j.molcel.2008.10.016
Shen X, Kim W, Fujiwara Y, Simon MD, Liu Y, Mysliwiec MR, Yuan GC, Lee Y, Orkin SH (2009) Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell 139(7):1303–1314. https://doi.org/10.1016/j.cell.2009.12.003
Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, Brockdorff N (2003) Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell 4(4):481–495
Simon JA, Kingston RE (2009) Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10(10):697–708. https://doi.org/10.1038/nrm2763
Smits AH, Jansen PW, Poser I, Hyman AA, Vermeulen M (2013) Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res 41(1):e28. https://doi.org/10.1093/nar/gks941
Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA (2010) Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci USA 107(49):20980–20985. https://doi.org/10.1073/pnas.1012525107
Son J, Shen SS, Margueron R, Reinberg D (2013) Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev 27(24):2663–2677. https://doi.org/10.1101/gad.225888.113
Song JJ, Garlick JD, Kingston RE (2008) Structural basis of histone H4 recognition by p55. Genes Dev 22(10):1313–1318. https://doi.org/10.1101/gad.1653308
Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-Meyer R, Yu JR, Leroy G, Oksuz O, Chapman JR, Suarez F, Modrek AS, Bayin NS, Placantonakis DG, Karajannis MA, Snuderl M, Ueberheide B, Reinberg D (2018) Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 4(10):eaau5935. https://doi.org/10.1126/sciadv.aau5935
Stirnimann CU, Petsalaki E, Russell RB, Muller CW (2010) WD40 proteins propel cellular networks. Trends Biochem Sci 35(10):565–574. https://doi.org/10.1016/j.tibs.2010.04.003
Sun A, Li F, Liu Z, Jiang Y, Zhang J, Wu J, Shi Y (2018) Structural and biochemical insights into human zinc finger protein AEBP2 reveals interactions with RBBP4. Protein Cell 9(8):738–742. https://doi.org/10.1007/s13238-017-0483-6
Takeuchi T, Yamazaki Y, Katoh-Fukui Y, Tsuchiya R, Kondo S, Motoyama J, Higashinakagawa T (1995) Gene trap capture of a novel mouse gene, jumonji, required for neural tube formation. Genes Dev 9(10):1211–1222. https://doi.org/10.1101/gad.9.10.1211
Tamburri S, Lavarone E, Fernandez-Perez D, Conway E, Zanotti M, Manganaro D, Pasini D (2020) Histone H2AK119 Mono-ubiquitination is essential for polycomb-mediated transcriptional repression. Mol Cell 77(4):840–856 e845. https://doi.org/10.1016/j.molcel.2019.11.021
Tatton-Brown K, Hanks S, Ruark E, Zachariou A, Duarte Sdel V, Ramsay E, Snape K, Murray A, Perdeaux ER, Seal S, Loveday C, Banka S, Clericuzio C, Flinter F, Magee A, McConnell V, Patton M, Raith W, Rankin J, Splitt M, Strenger V, Taylor C, Wheeler P, Temple KI, Cole T, Childhood Overgrowth C, Douglas J, Rahman N (2011) Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2(12):1127–1133. https://doi.org/10.18632/oncotarget.385
Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, Levrero M, Sartorelli V, Cotter RJ, Cole PA (2004) Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol 11(4):308–315. https://doi.org/10.1038/nsmb740
Tie F, Prasad-Sinha J, Birve A, Rasmuson-Lestander A, Harte PJ (2003) A 1-megadalton ESC/E(Z) complex from Drosophila that contains polycomblike and RPD3. Mol Cell Biol 23(9):3352–3362
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329(5992):689–693. https://doi.org/10.1126/science.1192002
Tsukiyama T, Wu C (1995) Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 83(6):1011–1020. https://doi.org/10.1016/0092-8674(95)90216-3
van Lohuizen M, Tijms M, Voncken JW, Schumacher A, Magnuson T, Wientjens E (1998) Interaction of mouse polycomb-group (Pc-G) proteins Enx1 and Enx2 with Eed: indication for separate Pc-G complexes. Mol Cell Biol 18(6):3572–3579
van Mierlo G, Veenstra GJC, Vermeulen M, Marks H (2019) The complexity of PRC2 subcomplexes. Trends Cell Biol 29(8):660–671. https://doi.org/10.1016/j.tcb.2019.05.004
Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M, Iyer P, Balasubramanian S, Zhao F, Good AC, Campbell R, Lee C, Cantone N, Cummings RT, Normant E, Bellon SF, Albrecht BK, Harmange JC, Trojer P, Audia JE, Zhang Y, Justin N, Chen S, Wilson JR, Gamblin SJ (2016) Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1 -(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-cell lymphomas. J Med Chem 59(21):9928–9941. https://doi.org/10.1021/acs.jmedchem.6b01315
Verdine GL, Hilinski GJ (2012) Stapled peptides for intracellular drug targets. Methods Enzymol 503:3–33. https://doi.org/10.1016/B978-0-12-396962-0.00001-X
Vizan P, Beringer M, Ballare C, Di Croce L (2015) Role of PRC2-associated factors in stem cells and disease. FEBS J 282(9):1723–1735. https://doi.org/10.1111/febs.13083
Wachter E, Quante T, Merusi C, Arczewska A, Stewart F, Webb S, Bird A (2014) Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. eLife 3:e03397. https://doi.org/10.7554/elife.03397
Walker E, Chang WY, Hunkapiller J, Cagney G, Garcha K, Torchia J, Krogan NJ, Reiter JF, Stanford WL (2010) Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell 6(2):153–166. https://doi.org/10.1016/j.stem.2009.12.014
Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y (2004a) Role of histone H2A ubiquitination in Polycomb silencing. Nature 431(7010):873–878. https://doi.org/10.1038/nature02985
Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS (2004b) Hierarchical recruitment of polycomb group silencing complexes. Mol Cell 14(5):637–646. https://doi.org/10.1016/j.molcel.2004.05.009
Wang S, Robertson GP, Zhu J (2004c) A novel human homologue of Drosophila polycomblike gene is up-regulated in multiple cancers. Gene 343(1):69–78. https://doi.org/10.1016/j.gene.2004.09.006
Wang X, Goodrich KJ, Gooding AR, Naeem H, Archer S, Paucek RD, Youmans DT, Cech TR, Davidovich C (2017a) Targeting of polycomb repressive complex 2 to RNA by short repeats of consecutive guanines. Mol Cell 65(6):1056–1067 e1055. https://doi.org/10.1016/j.molcel.2017.02.003
Wang X, Paucek RD, Gooding AR, Brown ZZ, Ge EJ, Muir TW, Cech TR (2017b) Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat Struct Mol Biol. https://doi.org/10.1038/nsmb.3487
Wang X, Long Y, Paucek RD, Gooding AR, Lee T, Burdorf RM, Cech TR (2019) Regulation of histone methylation by automethylation of PRC2. Genes Dev 33(19–20):1416–1427. https://doi.org/10.1101/gad.328849.119
Watson PJ, Fairall L, Santos GM, Schwabe JW (2012) Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 481(7381):335–340. https://doi.org/10.1038/nature10728
Whittaker C, Dean C (2017) The FLC locus: a platform for discoveries in epigenetics and adaptation. Annu Rev Cell Dev Biol 33:555–575. https://doi.org/10.1146/annurev-cellbio-100616-060546
Wu H, Zeng H, Dong A, Li F, He H, Senisterra G, Seitova A, Duan S, Brown PJ, Vedadi M, Arrowsmith CH, Schapira M (2013) Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS ONE 8(12):e83737. https://doi.org/10.1371/journal.pone.0083737
Wutz A (2011) Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet 12(8):542–553. https://doi.org/10.1038/nrg3035
Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ (2003) Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 421(6923):652–656. https://doi.org/10.1038/nature01378
Xu C, Bian C, Yang W, Galka M, Ouyang H, Chen C, Qiu W, Liu H, Jones AE, MacKenzie F, Pan P, Li SS, Wang H, Min J (2010) Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2). Proc Natl Acad Sci USA 107(45):19266–19271. https://doi.org/10.1073/pnas.1008937107
Yamamoto K, Sonoda M, Inokuchi J, Shirasawa S, Sasazuki T (2004) Polycomb group suppressor of zeste 12 links heterochromatin protein 1alpha and enhancer of zeste 2. J Biol Chem 279(1):401–406. https://doi.org/10.1074/jbc.M307344200
Youmans DT, Schmidt JC, Cech TR (2018) Live-cell imaging reveals the dynamics of PRC2 and recruitment to chromatin by SUZ12-associated subunits. Genes Dev 32(11–12):794–805. https://doi.org/10.1101/gad.311936.118
Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, Reinberg D (1997) Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell 89(3):357–364
Zhang Y, LeRoy G, Seelig HP, Lane WS, Reinberg D (1998) The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 95(2):279–289. https://doi.org/10.1016/s0092-8674(00)81758-4
Zhang X, Tamaru H, Khan SI, Horton JR, Keefe LJ, Selker EU, Cheng X (2002) Structure of the neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell 111(1):117–127. https://doi.org/10.1016/s0092-8674(02)00999-6
Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X (2003) Structural basis for the product specificity of histone lysine methyltransferases. Mol Cell 12(1):177–185. https://doi.org/10.1016/s1097-2765(03)00224-7
Zhang Z, Jones A, Sun CW, Li C, Chang CW, Joo HY, Dai Q, Mysliwiec MR, Wu LC, Guo Y, Yang W, Liu K, Pawlik KM, Erdjument-Bromage H, Tempst P, Lee Y, Min J, Townes TM, Wang H (2011) PRC2 complexes with JARID2, MTF2, and esPRC2p48 in ES cells to modulate ES cell pluripotency and somatic cell reprogramming. Stem Cells 29(2):229–240. https://doi.org/10.1002/stem.578
Zhang Q, McKenzie NJ, Warneford-Thomson R, Gail EH, Flanigan SF, Owen BM, Lauman R, Levina V, Garcia BA, Schittenhelm RB, Bonasio R, Davidovich C (2019) RNA exploits an exposed regulatory site to inhibit the enzymatic activity of PRC2. Nat Struct Mol Biol 26(3):237–247. https://doi.org/10.1038/s41594-019-0197-y
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322(5902):750–756. https://doi.org/10.1126/science.1163045
Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT (2010) Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell 40(6):939–953. https://doi.org/10.1016/j.molcel.2010.12.011
Acknowledgements
I thank members of the Liu laboratory for critical reading of this book chapter. This work was supported by Welch Foundation research grant I-1790 and NIH grants GM121662 and GM136308. X.L. is a W. W. Caruth, Jr. Scholar in Biomedical Research at UT Southwestern Medical Center.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Liu, X. (2021). A Structural Perspective on Gene Repression by Polycomb Repressive Complex 2. In: Harris, J.R., Marles-Wright, J. (eds) Macromolecular Protein Complexes III: Structure and Function. Subcellular Biochemistry, vol 96. Springer, Cham. https://doi.org/10.1007/978-3-030-58971-4_17
Download citation
DOI: https://doi.org/10.1007/978-3-030-58971-4_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-58970-7
Online ISBN: 978-3-030-58971-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)