
Abstract
Multimodal sensorial disruption typically accompanies migraine attacks and patients may experience sensory stimuli such as light, sound, touch, and odors as unpleasant or even painful. Sensorimotor integration is an anatomical and functional process with motor orders and sensory feedback and also with cognitive events. Different sensory modalities need to be identified and translated into coding to plan an action where the high-order thalamus mediates synchronization of cortical oscillations. Preclinical research revealed the critical involvement of thalamic reticular nucleus and clinical studies in migraine patients disclosed reduced sensory gating, abnormal early high-frequency oscillations, impaired somatosensory temporal discrimination thresholds, and disrupted integrity of sensorial input with motor output in sensorimotor cortex indicating the dysfunctional inhibitor circuit. Current evidences suggest that central sensory processing and sensorimotor integration are affected in migraine patients probably reflecting an increased excitability state toward the headache attacks.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Migraine
- Sensory processing
- Sensorimotor integration
- Somatosensory temporal discrimination
- Thalamus
- Short afferent inhibition
- Sensorimotor cortex
- Cortical excitability
9.1 Introduction
Multimodal sensorial disruption is one of the characteristic features of migraine headache attacks. Up to 12 h before and during a migraine attack, patients may experience sensory stimuli (light, sound, and odors) as unpleasant or even painful. Sometimes, migraineurs report these as being triggers of their attacks as well [1, 2]. Photophobia is present in approximately 50–90% of migraineurs and often the intensity of the headache increases with light exposure or certain light patterns [3, 4]. About 25–45% experience osmophobia during attacks and half of them report that strong odors (e.g., perfume, deodorant, coffee, fried dishes, as well as cigarette smoke) may trigger attacks [5, 6].
When allodynia is present, pain may be induced by non-painful stimulation such as mild touch, shaving, dressing, and combing hair. In approximately 65%, this manifests primarily in the periorbital region and spreads to extra-cephalic regions [7, 8].
Photophobia, phonophobia, osmophobia, and allodynia are thought to be clinical correlates of central sensitization. An increased sensitivity to sensory stimuli during the premonitory phase (especially photophobia and phonophobia) is believed to occur independently from trigeminal inputs during an attack [9,10,11,12].
In addition, the ability to distinguish two identical somaesthetic stimuli applied to the same or different cutaneous region in short intervals is often impaired during migraine [13,14,15,16].
Sensorimotor integration is an anatomical and functional process with motor orders and sensory feedback. Different sensory modalities need to be identified and translated into coding to plan a movement. Sensorimotor integration is mostly associated with the somatosensory cortex, thalamus, posterior parietal cortex, and motor cortex. The sensorimotor cortex is highly dynamic and associated not only with motor learning but also with cognitive events.
Overall, there is evidence that sensory processing may be affected in migraine patients. Much research has been undertaken to elucidate the underlying mechanisms and to study the implications for sensorimotor integration. In this chapter, we will provide an overview of the involved anatomical structures and physiological processes and discuss the current knowledge.
9.2 The Anatomical and Physiological Basis of Sensory Processing and Sensorimotor Integration
Processing of sensory information occurs in the thalamus, the primary sensory cortex, as well as the posterior parietal cortex.
9.2.1 Thalamus
Neurons in the sensory thalamus receive information from the external environment through the medial lemniscus and transfer them to the cerebral cortex. Thalamic neurons and cortico-thalamic feedback neurons ensure a constant communication between the thalamus and cortex in sensory processing [17].
The thalamic nuclei are subdivided into first-order and higher-order relay nuclei. First-order thalamic nuclei transmit sensory inputs to first-order cortical areas such as the primary sensory cortex (S1) [18, 19]. Higher-order thalamic nuclei, on the other hand, primarily receive input from the fifth layer of the cerebral cortex and forward information to higher-order cortical areas [18]. Thereby, the thalamus mediates synchronization of cortico-cortical oscillations [18, 20].
Cortico-thalamic feedback neurons are located in the cortical layer 6 and their axons project on both the thalamus and the cerebral cortex. In addition to sending monosynaptic input to thalamic relay neurons, axons of cortico-thalamic neurons also provide polysynaptic inhibition relayed by local inhibitory neurons and neurons in the thalamic reticular nucleus (TRN). Therefore, the cortico-thalamic activity has both excitatory and inhibitory effects on thalamic function [18].
Furthermore, cortico-thalamic neurons strengthen the sensory signals transmitted from the periphery to the cortex and sharpen the receptive field [21, 22].
Cortico-thalamic feedback affects gain and responsiveness of thalamic neurons at both local and global levels. At the local level, cortico-thalamic feedback selectively increases the sensory response of individual thalamic neurons in somatosensory, auditory, and visual systems [23, 24]. At the global level, cortico-thalamic projections adjust the responsiveness of thalamic neurons during sleep and wakefulness. In addition, cortico-thalamic feedback modulates the sensory responses in the thalamus [25, 26]. More recently, cortico-thalamic feedback was found to increase thalamic response to painful stimulation and to increase the flow of information between the thalamus and somatosensory cortex [27, 28].
The thalamic reticular nucleus (TRN) is a layer of GABAergic neurons surrounding the sensory thalamus. Its neurons project to the thalamus and receive input from both thalamo-cortical neurons and the cortex. Neurons of the TRN are non-reciprocally linked to thalamo-cortical neurons [29, 30]. Multiple brain regions, such as the prefrontal cortex, the amygdala, other thalamic regions, and the basal forebrain region, indirectly affect cortical activation by projections to the TRN [31, 32]. Open-loop connections may be a potential substrate for long-range modulation of cortical activity, and, paradoxically, they are involved in increased thalamo-cortical signal current and signal propagation in the thalamus [33, 34]. Based upon these findings, it is thought that the TRN modulates transmission of information to the cerebral cortex [29, 35, 36]. The TRN controls the response mode of thalamo-cortical neurons, and the flow of sensory information to the cortex, mediates lateral inhibition and maintains the sleep [29, 36, 37].
The thalamo-cortical neurons have two spiking response modes—bursting and tonic activation [37]. Thalamic burst firing plays an important role in sensory gating. It occurs spontaneously in cases of neuropathic pain and noxious stimulation. Inhibition of thalamic burst firing can lead to changes in nociceptive responses [38]. Burst firing occurs during slow-wave sleep as well as sleep spindles [29] and may be associated with relief of migraine pain or migraine attacks during sleep.
Tepe and colleagues showed for the first time that spreading depression waves in the cerebral cortex could invade and activate the TRN in awake rats [39]. While the TRN has seven anatomically distinct sectors of motor, limbic, and sensory processing, cortical spreading depression (CSD) selectively activates the visual sector of TRN. Administration of valproic acid as well as calcitonin gene-related peptide receptor antagonist inhibited CSD-induced activation of the TRN [39, 40]. Dysfunction of the GABAergic neurons in TRN by CSD results in enhanced transmission of sensory information to the cerebral cortex and hyper-responsiveness to sensorial stimulus, as seen in migraine. Given that TRN plays a key role in sleep, selective attention, lateral inhibition, and discrimination of sensory stimuli, studying the complex role of the thalamus in is crucial to understand clinical features and sensory impairment in migraine [41].
Thalamo-cortical somatosensory projections from the ventral posterolateral nucleus (VPL), the ventral posteromedial nucleus (VPM), and the lateral posterior nucleus (LP) reach primary and secondary somatosensory areas. Efferents of the ventro-posterior nucleus (VPN) transmit the sense of touch and pain. Recent paper evaluated the potential participation of thalamocortical network interruption in development of sensory dysfunction accompanying migraine headaches [41].
9.2.2 Sensorimotor Cortex
The posterior medial nucleus of the thalamus transmits somatosensory information to layer 1 of the primary somatosensory cortex (S1), which consists of Brodmann’s areas 3a, 3b, 1, and 2 [42,43,44]. Sensory neurons in S1 project to the primary motor cortex (M1) both directly and via thalamo-cortical connections [45, 46]. Precentral and postcentral gyri together are termed sensorimotor cortex.
Electrophysiological studies have shown that layer 4 of M1 receives excitatory inputs from the thalamus and sends unidirectional excitatory outputs to layers 2 and 3 [47]. While nearly half of the synapses in M1 are excitatory, synapses in S1 are both excitatory and inhibitory [48]. Feedforward inhibition occurs in layer 1 instead of layer 4 of M1 through thalamo-cortical projections [49].
Direct projections from S1 to M1 and projections from the thalamus to M1 are important for somatosensory motor integration that will be discussed below [50]. In addition, the somatosensory cortex is crucial for motor learning [51,52,53].
9.2.3 The Posterior Parietal Cortex
Different sensory modalities such as visual, somatosensory, prefrontal, and auditory are integrated in the posterior parietal cortex (PPC). It is connected to somatosensory and motor areas [54, 55], to prefrontal motor areas both directly and through networks [56], and to the M1 via monosynaptic projections [57]. PPC neurons play a role in planning, controlling, and correcting movements. These neurons encode dynamic information about motion [58, 59].
9.3 Somatosensory Processing
9.3.1 Habituation, Sensitization, and Allodynia
Somatosensory evoked potentials (SSEPs) have been used to study processing of somatosensory signals. To that end, the median nerve is stimulated using non-noxious stimuli and an evoked potential—the negative N20-peak—is recorded from the contralateral somatosensory cortex using EEG or magnetoencephalography [60].
The responsiveness of the brain to external stimuli varies. A decrement of responses to repetitive stimuli is referred to as habituation [61]. An increase in responsiveness on the other hand is termed sensitization. The presence of two independent systems (i.e., habituation and sensitization) influencing the reaction of a biological system has firstly been hypothesized by Groves and Thompson in their dual process theory of response habituation [61, 62]. When confronted with a new stimulus, sensitization may occur first; habituation follows later, if sensory stimulation persists.
These phenomena have been studied in migraine patients recording SSEPs. In one study, sensitization was assumed when the average amplitude of the first 100 stimulations was higher than in healthy controls. This was the case in patients suffering from migraine without aura examined during an attack [63]. When two further blocks, each with 100 stimulations were added, a decrement of the average amplitude per block compared to the amplitude of the first block was observed in healthy controls as well as in patients suffering from an acute migraine attack. These findings suggest that sensitization is present during a migraine attack [63]. In one study, allodynia was experimentally induced in healthy subjects and led to painful sensations and an increase in amplitude of potentials evoked by stimulation of Aβ fibers [64]. It is thus likely that central sensitization during migraine attacks is caused by painful afferents and manifests as allodynia.
While habituation can be observed in healthy subjects, in migraine patients it is present only during an attack [63]. An interictal lack of habituation has been reported for various sensory stimuli [65, 66]. The reason for this is less clear. According to the “ceiling theory,” signal intensity must reach a threshold (“ceiling”) in order to trigger habituation [67].
In order to investigate further the mechanism of the abnormalities of the SSEPs, some studies focused on high-frequency oscillations (HFO) [68]. These are superimposed on the N20-peak and may be subdivided in early and late bursts. It is believed that generators of the former are thalamo-cortical afferents and of the latter are inhibitory interneurons in the primary sensory cortex (area 3b) [69, 70]. The amplitude of early HFOs was significantly lower in migraineurs in the interictal period and normalized during an attack. No difference in late HFOs was found. A low amplitude of early HFOs had also been found in previous studies when SSEPs were recorded during non-REM sleep.
Given that the amplitude of early HFOs increases after administration of rivastigmine [71] and given that cholinergic systems are relevant for the sleep-wake cycle, it has been hypothesized that these early HFOs were generated by cholinergic reticulo-thalamic pathways [68]. Consequently, an interictal hypofunction of thalamo-cortical excitatory cholinergic afferents was assumed in migraine patients [68]. This finding again might indicate that the pre-activation of sensory cortices was too small to induce habituation.
In patients with chronic migraine, both sensitization and habituation are found in the interictal period, suggesting a permanent ictal-like state [72]. In addition, a correlation between the degree of sensitization and the number of headache days was found. This finding suggests that an increasing sensitization might indeed lead to habituation as predicted by the ceiling theory.
In patients with medication overuse headache, central sensitization is present, while habituation is absent. It has been suggested these patients are in locked a persistent pre-ictal state [63]. These findings are unexpected given the hypothesis that the absence of habituation in migraine patients is due to the low amplitude of the potential. Since that medication overuse is associated with sensitization, an insufficient signal amplitude seems unlikely [63]. Therefore, it has been suggested that the lack of habituation may be the consequence of a cortical hyper-reactivity [73]. The reduced amplitude of evoked potentials and the reduced activity of thalamo-cortical afferents might represent compensatory mechanism mediated by feedback loops [73]. The lack of habituation might not be due to too little input but to a cortical hyper-excitability. Thus, the ceiling theory may be invalid.
Overall, it is likely that the primary sensory cortex is hyper-excitable and the activity of thalamo-cortical afferents is reduced in migraine patients in the interictal period. During an attack, cortical excitability normalizes (as indicated by the appearance of habituation) and the activity of the thalamo-cortical afferents increases.
9.3.2 Energy Metabolism
Despite their name, evoked potentials probably do not represent stimulus-evoked brain events. Rather they document a phase resetting of different cortical rhythms – in particular alpha, mu, and frontal midline theta rhythms [74]. In migraine patients, SSEPs recorded during attacks have a higher amplitude, compared to healthy controls [63], indicating that sensory stimuli lead to a higher degree of phase resetting.
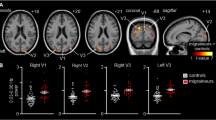
Higher amplitudes of evoked potentials have been linked to a higher energy consumption of sensory processing ([75], Fig. 9.1). This notion is supported by several studies in which the power consumption in the visual cortex was studied using MR spectroscopy before, during, and after stimulation [76,77,78,79,80].

The figure below shows that amplitude differences of visual evoked potentials at 8 Hz stimulation between migraine patients and control subjects are mainly explained by single-sweep amplitude, rather than phase modulation of EEG activity. Amplitude variation is thought to be more energy demanding than phase shift
In migraine without aura, lactate levels were normal at all times [77]. However, two studies reported low phosphocreatine, low adenosine triphosphate, and an increased adenosine diphosphate suggesting an altered energy metabolism [78, 79]. In migraine with aura, lactate and adenosine diphosphate levels were high and phosphocreatine was low even in the absence of sensory stimulation [76, 80]. In addition, muscles biopsies revealed an abnormal energy metabolism as well [80].
The precise pathophysiology of the energy metabolism in migraine is yet to be understood. Nevertheless, these findings led to the idea of migraine being due to a defect of the oxidative energy generation [81]. Therefore, it was hypothesized that strengthening the mitochondrial function may help to prevent attacks. Indeed, riboflavin and co-enzyme Q10 reduce the attack frequency in many patients [82,83,84,85].
9.3.3 Sensory Gating
Large amounts of sensory information are collected by the nervous system at every instant. While some of these data may be relevant, others are not. The process of selecting important input is referred to as sensory gating. It is studied by measuring the capacity of the brain to filter repetitive stimuli. To this end, two stimuli are commonly applied in close temporal relationship and cortical evoked potentials are registered using electro-encephalography or magnetoencephalography. The so-called gating ratio reflects the amount of attenuation of the second signal. It is calculated by dividing the amplitude of the second signal by the amplitude of the first. Cortical potentials may be evoked using different sensory input, like acoustic, visual, or sensory signals [86].
Two studies examined sensory gating in headache patients [87, 88]. Non-painful electric stimuli were applied to the left index finger and magnetoencephalography was used to record responses from the contralateral primary sensory cortex. The main finding was that the gating ratio was higher in patients with migraine than in controls and higher in chronic migraine than in episodic implying a reduced attenuation of the second stimulus in these groups. There was a positive correlation between the number of headache days and the gating ratio [87, 88].
The mechanisms underlying these findings are unclear. While a true gating deficit may be present (i.e., an incomplete suppression of repetitive stimuli because of a cortical hyper-reactivity), it should also be considered that the small amplitude of the first signal might have been insufficient to induce gating [87].
9.3.4 Somatosensory Temporal Discrimination
The ability to distinguish two identical somaesthetic stimuli applied to the same spot or different cutaneous regions in short intervals is referred to as somatosensory temporal discrimination (STD) [13]. It is crucial for somatosensory functions such as kinesthesia, graphesthesia, and stereognosis [89, 90]. The somatosensory temporal discrimination threshold (STDT) is defined as the shortest delay between two stimuli that still allows discrimination and differs between body areas [13].
Different studies evaluated STD in migraine patients [14,15,16]. Boran and colleagues examined the STDTs of the upper extremity (dermatome C7) and face (mandibular nerve) in patients with episodic migraine and healthy volunteers. While no difference could be found interictally, an approximately two- to fourfold prolongation was observed in all regions during an attack [14]. In the contralateral upper extremity and the ipsilateral face, STDTs were significantly longer compared to other regions. These prolongations were thought to have resulted from alterations of central pain perception. However, it has been suggested that a significant prolongation of the contralateral upper extremity and ipsilateral face cannot be explained by the impairment of sensory networks alone [14].
In patients with chronic migraine (CM), STDTs are approximately three times longer than in healthy volunteers—even on days without headache. This finding stands in contrast to episodic migraine where normal interictal STDTs were found. This suggests that the impairment of sensorial processing is sustained in chronic migraine [15]. Again, patients with chronic migraine seem to be locked in an ictal state. In patients with tension-type headache (TTH), on the other hand, STDTs are unaltered [16]. Therefore, STD may be used as a biomarker for chronic migraine as well as to differentiate migraine from TTH [15, 16].
Abnormal STD does not only occur in migraine; it has been linked to cerebral damage in different locations. Lesions in the primary somatosensory cortex, the internal capsule, and the thalamus may cause impairment of both sensory perception and STD. Lesions in the posterior parietal cortex, head of the caudate nucleus, putamen, medial thalamus, and lenticular nucleus do not affect sensory perception but lead to abnormal STD. Finally, a bilateral lesion of the supplementary motor area may be associated with impaired STD as well [91]. In healthy subjects, fMRI imaging revealed the inferior parietal lobule, the middle and inferior frontal gyrus, the anterior part of the right insula, the right anterior cingulate gyrus, as well as the cerebellum to be relevant for STD. The pre-SMA and the anterior cingulate gyrus are thought to be specific for the task [92].
Higher STDTs also occur in patients suffering from movement disorders such as Parkinson’s disease, dystonia, and multisystem atrophy [93, 94]. Consequently, basal ganglia are likely to play an important role in temporal discrimination. The affection of STD in Parkinson’s disease is thought to be due to an impairment of sensorimotor integration, timing, and projections to the supplementary motor area [95]. Finally, STDTs are higher in patients with cerebellar atrophy [96].
Some of the structures involved in pain perception during migraine attacks such as the insula, cingulate gyrus, cerebellum, and basal ganglia are known to play a role in STD impairment [97]. Thus, changes of STDTs in migraine patients could arise from a transient impairment in these areas [14].
9.3.5 Non-dermatomal Sensory Deficits
About 20–40% of patients suffering from chronic pain complain about sensory deficits ipsilateral to their pain, so-called non-dermatomal sensory deficits (NDSD), and migraine patients are not spared from these constraints [98, 99].
The absence of anatomical lesions in the peripheral and central nervous system often led to speculation about “hysteria” or a conversion disorder [100]. However, careful sensory testing revealed significantly higher thresholds for mechanical and painful stimuli ipsilateral to the pain suggesting functional changes of sensory processing [98].
Imaging studies were undertaken to help understanding these findings. Riederer and co-workers investigated gray matter changes in patients with NDSD [101]. They discovered an increase in gray matter in the right primary sensory cortex, in the thalamus, and bilaterally in lateral temporal regions and the hippocampus. Patients with chronic pain but without NDSD had been included in the study as controls and had changes in similar areas but to a lesser extent. An association with psychiatric disorders was not found. Egloff et al. studied NDSD using FDG-PET imaging and found a significant hypometabolism in the contralateral post-central gyrus, posterior insula, putamen, medial temporal gyrus, cuneus, superior and inferior temporal gyrus, as well as ipsilateral putamen and precuneus [102].
While these findings support the hypothesis of a dysfunction of the central nervous system having caused the symptoms, the precise pathophysiology remains elusive. It has been suggested that NDSD may be the consequence of the central nervous system trying to reduce pain by suppressing sensory afferents [103]. Another hypothesis is based upon the increase in gray matter in and the hypometabolism of the lateral temporal gyrus. Given that dysfunction of this region may be associated with a neglect, a perception disorder might explain the sensory complaints [101]. In the past, the finding of neglect-like symptoms in patients suffering from complex regional pain syndrome had led to the question whether we are “neglecting neglect” [104, 105]. Based upon these findings, one may wonder whether the possibility of an acquired neglect in NDSD should receive greater attention.
9.4 Sensorimotor Integration
Relevant sensory data need to be identified and different sensory qualities be translated into a common coding to plan a movement. This process is referred to as sensorimotor integration.
Much information on executed movements such as muscle contraction and body kinematics converge on the sensorimotor cortex [106,107,108]. Irrelevant information having been filtered by sensory gating, the remaining data are integrated to plan, monitor, and optimize current and future movements [109]. This complex process is influenced by different factors such as training, motivation, purpose, and neuronal excitability [109].
Some studies tried to evaluate whether altered sensory information may have repercussions in motion planning in migraine patients. To this end, the motor cortex was investigated using transcranial magnetic stimulation (TMS).
Increased excitability was found and suggested that neurophysiological findings have a role in the mechanism of migraine [110, 111]. Increased motor threshold and increased cortical excitability or decreased inhibition in migraine patients was shown [110]. Contradictory results were found in other studies. It is thought that there is dysregulation of cortical excitability in migraine patients whether cortical excitability is decreased or increased. This change in cortical excitability may play a role in explaining the different symptoms in migraine patients.
The amplitude of motor evoked potentials (MEPs) can be measured in peripheral muscles after cortical stimulation is applied using TMS. Low MEP threshold and high MEP amplitude suggest higher cortical excitability [112]. MEP thresholds were found to be normal, increased, or decreased in migraineurs [113].
The resting motor threshold (RMT) reflects the excitability of cortico-motor projections. RMT was assessed in the interictal period and directly after an attack-day in one study. RMT was negatively correlated with the number of days after the migraine attacks in migraine patients [113].
An inhibitory effect is expected in low-frequency repetitive TMS (rTMS) (≤1 Hz), while an excitatory effect is expected in high-frequency rTMS (≥5 Hz) in healthy people [114]. The intracortical facilitation circuit of the motor cortex of migraine patients with aura was significantly activated with 1 Hz rTMS at 90% of the RMT even though it should have been inhibited [115]. Excitatory systems are easily activated in migraine patients with aura at 5 Hz rTMS at 110% and 120% of RMT compared to migraine patients without aura and healthy volunteers. On the other hand, inhibition was observed in migraine patients with 130% of RMT at 5 Hz rTMS, while MEP facilitation was observed in healthy individuals [116]. These paradoxical responses were interpreted as being due to cortical homeostatic metaplasticity. In a hyperactivated cortex, the excitability of the cortex should be maintained in the physiological range when stimulating with low-frequency or high-frequency rTMS [117, 118].
Paired pulse TMS activates inhibiting or facilitating intracortical interneurons, which project to the corticospinal tract. To inhibit the cerebral cortex, short interstimulus interval (ISI) of 1–5 ms (short-interval intracortical inhibition, SICI) and long ISI of 50–400 ms (long-interval intracortical inhibition, LICI) are used. Whereas ISI of 6–30 ms is used to facilitate the cortex (intracortical facilitation, ICF) [119, 120]. Some investigators found increased ICF [121] or decreased SICI [115, 122] supporting the increased hyper-excitability hypothesis in migraine, while others did not. Changing the severity of the test stimulus has a significant effect on cortical inhibition and facilitation in healthy individuals in this paradigm [123, 124]. Test stimulation at 110%, 130%, and 150% of the RMT and 10 ms of ISI for ICF, 2 ms of ISI for SICI, and 100 ms of ISI for LICI was used in another study. Significantly, facilitation was observed by using test stimulation at 110% of the RMT compared to controls in ICF paradigm [125]. ICF is mediated by glutamatergic functions while SICI is mediated by GABAA receptors and LICI is mediated by GABAB receptors. The decrease of postsynaptic activity decreases the threshold for long-term potentiation (LTP) and increases the threshold for long-term depression (LTD). Because of the homeostatic plasticity, cortical neurons adjust the postsynaptic activity level through presynaptic stimulation response, thereby changing cortical excitability [126, 127].
It was shown that cortical excitability is reduced after using anodal transcranial direct current stimulation (tDCS), while cortical excitability is increased using cathodal tDCS [128]. MEP decrease was observed after cathodal tDCS was used in migraine patients with visual aura similar to healthy volunteers, MEP amplitudes returned to baseline at 5th and 15th min by following 5 Hz rTMS. Both groups showed significant facilitation when anodal TDCS was applied. After the 5 Hz rTMS, inhibition was observed at fifth and 15th min in healthy volunteers, while facilitation continued in migraine patients [128]. Inhibitor dysfunction was found to be more prominent in migraine patients with aura than in migraine patients without aura and healthy subjects [129]. Taken together, when evaluating cortical excitability in migraine patients using external modulation, an inhomogeneous group was reported.
Applying stimuli to the periphery and the cortex at different times, inhibition is observed with 10 ms of ISI and facilitation is observed with 25 ms of ISI in the sensorimotor cortex in healthy subjects. This lasts for at least 30–60 min [130, 131] and arises from LTP and LTD.
In one study, long-term synaptic plasticity was investigated. Inhibition of MEP was found to be significantly associated with paired associative stimulation (PAS) 10. While inhibiting, facilitation was observed with PAS 25 in healthy volunteers. However, PAS 10 increased MEP rather than inhibit; PAS25 increased MEP non-significantly in migraine patients without aura.
Short-latency afferent inhibition (SAI) is a modulation of motor response by a sensory stimulus and known to be associated with sensorimotor integration and cognitive functions. SAI is probably related to thalamo-cortical output from cholinergic paramedian thalamic nuclei to M1 or by the direct output from S1 to inhibitory M1 interneurons [132, 133]. A preceding electrical stimulation of a peripheral nerve (conditioning afferent stimulus) transiently suppresses transcranial magnetic stimulation (TMS)-induced motor output. Inhibition of the motor response occurs if the interstimulus interval between the electrical stimulation and TMS is 19 and 50 ms. Thereby, Alaydin and colleagues evaluated the sensorimotor cortex integrity by using SAI paradigm in migraine for the first time [134].
Authors detected a marked decrease in SAI during pre-ictal and ictal periods in migraine without aura patients, which points toward a prominent facilitation to a conditioned stimulus instead of inhibition taking place in the sensorimotor cortex (Fig. 9.2). SAI results in the interictal period in migraine patients were comparable to that of healthy controls. An impairment of the sensorimotor integrity and increased excitability state begins several hours prior to the headache phase in migraine without aura patients. Authors suggested that decreased sensorimotor integration occurs at cortical level and cortical inhibitory volley from S1 to M1 may play an important role in SAI impairment in migraine [134]. This phenomenon could be related to the cortical hyper-responsivity to sensory stimuli and cognitive disturbances accompanying migraine attacks because SAI is modulated by cholinergic activity. In support of the latter, a positive effect of a cholinergic drug on SAI was reported [135]. Transient cholinergic dysfunction may play a role in both abnormal sensory processing and cortical excitability in migraine patients. Cholinergic activity of the cortex is also associated with cognitive functions. It is thought that SAI impairment may be related to prodromal and ictal cognitive symptoms [134].

Figure shows typical traces of SAI. MEP amplitudes were reduced with SAI paradigm (5) compared to MEP amplitudes without SAI paradigm (2) in a healthy volunteer (A) and in a migraine patient during interictal period (B). MEP amplitude facilitation was detected instead of inhibition in the sensorimotor cortex during a headache attack in a migraine patient (C). (1) Stimulus artifact of TMS without SAI paradigm. (2) Average of MEP amplitudes before SAI paradigm. (3) Peripheral stimulus artifact in SAI paradigm. (4) Stimulus artifact of TMS in SAI paradigm. (5) Average of MEP amplitudes in SAI paradigm. MEP motor evoked potential, SAI short-latency afferent inhibition, TMS transcranial magnetic stimulation
9.5 Conclusions
Sensory processing and sensorimotor integration are affected in migraine patients. Clinically, both allodynia and the prolonged somatosensory temporal discrimination during migraine attacks suggest an impairment of sensory processing. Electrophysiological studies pointed toward a hyper-reactivity of the sensory cortex in migraine. The primary sensory cortex of patients with chronic migraine is sensitized and therefore seems to be fixed in an “ictal state.” In addition, the integration of sensory input is impaired as well, because, both before and during a migraine attack, sensory input does not lead to an inhibition of the motor response, but to a facilitation.
Overall, changes of sensory processing and sensorimotor integration in migraine patients probably reflect a cortical hyper-responsivity. The precise pathophysiology, however, remains to be elucidated.
Abbreviations
- CM:
-
Chronic migraine
- CSD:
-
Cortical spreading depression
- GABA:
-
Gamma amino butyric acid
- HFO:
-
High-frequency oscillations
- ICF:
-
Intracortical facilitation
- ISI:
-
Interstimulus interval
- LICI:
-
Long-interval intracortical inhibition
- LP:
-
Lateral posterior nucleus
- LTD:
-
Long-term depression
- LTP:
-
Long-term potentiation
- M1:
-
Primary motor cortex
- MEP:
-
Motor evoked potential
- NDSD:
-
Non-dermatomal sensory deficits
- PAS:
-
Paired associative stimulation
- PPC:
-
Posterior parietal cortex
- REM:
-
Rapid eye movement
- RMT:
-
Resting motor threshold
- rTMS:
-
Repetitive TMS
- S1:
-
Primary somatosensory cortex
- SAI:
-
Short-latency afferent inhibition
- SICI:
-
Short-interval intracortical inhibition
- SSEP:
-
Somatosensory evoked potential
- STD:
-
Somatosensory temporal discrimination
- STDT:
-
Somatosensory temporal discrimination threshold
- tDCS:
-
Transcranial direct current stimulation
- TMS:
-
Transcranial magnetic stimulation
- TRN:
-
Thalamic reticular nucleus
- TTH:
-
Tension-type headache
- VPL:
-
Ventral posterolateral nucleus
- VPM:
-
Ventral posteromedial nucleus
- VPN:
-
Ventro-posterior nucleus
References
Schoonman GG, et al. The prevalence of premonitory symptoms in migraine: a questionnaire study in 461 patients. Cephalalgia. 2006;26(10):1209–13.
Quintela E, et al. Premonitory and resolution symptoms in migraine: a prospective study in 100 unselected patients. Cephalalgia. 2006;26(9):1051–60.
Russell MB, et al. Migraine without aura and migraine with aura are distinct clinical entities: a study of four hundred and eighty-four male and female migraineurs from the general population. Cephalalgia. 1996;16(4):239–45.
Wober-Bingol C, et al. Clinical features of migraine: a cross-sectional study in patients aged three to sixty-nine. Cephalalgia. 2004;24(1):12–7.
De Carlo D, et al. Osmophobia in migraine classification: a multicentre study in juvenile patients. Cephalalgia. 2010;30(12):1486–94.
Demarquay G, et al. Rating of olfactory judgements in migraine patients. Cephalalgia. 2006;26(9):1123–30.
Burstein R, Cutrer MF, Yarnitsky D. The development of cutaneous allodynia during a migraine attack clinical evidence for the sequential recruitment of spinal and supraspinal nociceptive neurons in migraine. Brain. 2000;123(Pt 8):1703–9.
Lipton RB, et al. Cutaneous allodynia in the migraine population. Ann Neurol. 2008;63(2):148–58.
Aurora SK, et al. The occipital cortex is hyperexcitable in migraine: experimental evidence. Headache. 1999;39(7):469–76.
Aurora SK, Welch KM, Al-Sayed F. The threshold for phosphenes is lower in migraine. Cephalalgia. 2003;23(4):258–63.
Schoenen J. Deficient habituation of evoked cortical potentials in migraine: a link between brain biology, behavior and trigeminovascular activation? Biomed Pharmacother. 1996;50(2):71–8.
Afra J, et al. Interictal cortical excitability in migraine: a study using transcranial magnetic stimulation of motor and visual cortices. Ann Neurol. 1998;44(2):209–15.
Hoshiyama M, Kakigi R, Tamura Y. Temporal discrimination threshold on various parts of the body. Muscle Nerve. 2004;29(2):243–7.
Boran HE, Cengiz B, Bolay H. Somatosensory temporal discrimination is prolonged during migraine attacks. Headache. 2016;56(1):104–12.
Vuralli D, et al. Chronic migraine is associated with sustained elevation of somatosensory temporal discrimination thresholds. Headache. 2016;56(9):1439–47.
Vuralli D, et al. Somatosensory temporal discrimination remains intact in tension-type headache whereas it is disrupted in migraine attacks. Cephalalgia. 2017;37(13):1241–7.
Briggs F, Usrey WM. Emerging views of corticothalamic function. Curr Opin Neurobiol. 2008;18(4):403–7.
Hwang K, et al. The human thalamus is an integrative hub for functional brain networks. J Neurosci. 2017;37(23):5594–607.
Sherman SM. Thalamus plays a central role in ongoing cortical functioning. Nat Neurosci. 2016;19(4):533–41.
Destexhe A, Contreras D, Steriade M. Cortically-induced coherence of a thalamic-generated oscillation. Neuroscience. 1999;92(2):427–43.
Sherman SM, Guillery RW. Exploring the thalamus and its role in cortical function. 2nd ed. Cambridge, MA: Massachusetts Institute of Technology; 2006.
Jones EG. The thalamus. 2nd ed. Cambridge: Cambridge University Press; 2007. p. 1708.
Suga N, Ma X. Multiparametric corticofugal modulation and plasticity in the auditory system. Nat Rev Neurosci. 2003;4(10):783–94.
Temereanca S, Simons DJ. Functional topography of corticothalamic feedback enhances thalamic spatial response tuning in the somatosensory whisker/barrel system. Neuron. 2004;41(4):639–51.
O’Connor DH, et al. Attention modulates responses in the human lateral geniculate nucleus. Nat Neurosci. 2002;5(11):1203–9.
McAlonan K, Cavanaugh J, Wurtz RH. Attentional modulation of thalamic reticular neurons. J Neurosci. 2006;26(16):4444–50.
Monconduit L, et al. Corticofugal output from the primary somatosensory cortex selectively modulates innocuous and noxious inputs in the rat spinothalamic system. J Neurosci. 2006;26(33):8441–50.
Wang JY, et al. Corticofugal influences on thalamic neurons during nociceptive transmission in awake rats. Synapse. 2007;61(5):335–42.
Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262(5134):679–85.
Warren NM, et al. Muscarinic receptors in the thalamus in progressive supranuclear palsy and other neurodegenerative disorders. J Neuropathol Exp Neurol. 2007;66(5):399–404.
Crabtree JW, Collingridge GL, Isaac JT. A new intrathalamic pathway linking modality-related nuclei in the dorsal thalamus. Nat Neurosci. 1998;1(5):389–94.
Lee SC, Cruikshank SJ, Connors BW. Electrical and chemical synapses between relay neurons in developing thalamus. J Physiol. 2010;588(Pt 13):2403–15.
Brown JW, et al. A computational model of intrathalamic signaling via open-loop thalamo-reticular-thalamic architectures. 2019; https://doi.org/10.1101/574178.
Willis AM, et al. Open-loop organization of thalamic reticular nucleus and dorsal thalamus: a computational model. J Neurophysiol. 2015;114(4):2353–67.
Williams RW, Rakic P. Elimination of neurons from the rhesus monkey’s lateral geniculate nucleus during development. J Comp Neurol. 1988;272(3):424–36.
Wimmer RD, et al. Thalamic control of sensory selection in divided attention. Nature. 2015;526(7575):705–9.
Sherman SM. Tonic and burst firing: dual modes of thalamocortical relay. Trends Neurosci. 2001;24(2):122–6.
Kim D, et al. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science. 2003;302(5642):117–9.
Tepe N, et al. The thalamic reticular nucleus is activated by cortical spreading depression in freely moving rats: prevention by acute valproate administration. Eur J Neurosci. 2015;41(1):120–8.
Filiz A, et al. CGRP receptor antagonist MK-8825 attenuates cortical spreading depression induced pain behavior. Cephalalgia. 2019;39(3):354–65.
Bolay H. Thalamocortical Network Interruption: A Fresh View for Migraine Symptoms [published online ahead of print, 2020 May 18]. Turk J Med Sci. 2020;10.3906/sag-2005-21. https://doi.org/10.3906/sag-2005-21.
Kaneko T, Caria MA, Asanuma H. Information processing within the motor cortex. I. Responses of morphologically identified motor cortical cells to stimulation of the somatosensory cortex. J Comp Neurol. 1994;345(2):161–71.
Boivie J. Anatomical observations on the dorsal column nuclei, their thalamic projection and the cytoarchitecture of some somatosensory thalamic nuclei in the monkey. J Comp Neurol. 1978;178(1):17–48.
Boivie J. An anatomical reinvestigation of the termination of the spinothalamic tract in the monkey. J Comp Neurol. 1979;186(3):343–69.
Chakrabarti S, Zhang M, Alloway KD. MI neuronal responses to peripheral whisker stimulation: relationship to neuronal activity in si barrels and septa. J Neurophysiol. 2008;100(1):50–63.
Rocco-Donovan M, et al. Characteristics of synaptic connections between rodent primary somatosensory and motor cortices. Somatosens Mot Res. 2011;28(3–4):63–72.
Yamawaki N, et al. A genuine layer 4 in motor cortex with prototypical synaptic circuit connectivity. elife. 2014;3:e05422.
Bopp R, et al. An ultrastructural study of the thalamic input to layer 4 of primary motor and primary somatosensory cortex in the mouse. J Neurosci. 2017;37(9):2435–48.
Kuramoto E, et al. Two types of thalamocortical projections from the motor thalamic nuclei of the rat: a single neuron-tracing study using viral vectors. Cereb Cortex. 2009;19(9):2065–77.
Cash RF, et al. The influence of sensory afferent input on local motor cortical excitatory circuitry in humans. J Physiol. 2015;593(7):1667–84.
Matsuzaka Y, Picard N, Strick PL. Skill representation in the primary motor cortex after long-term practice. J Neurophysiol. 2007;97(2):1819–32.
Chouinard PA, Goodale MA. Functional reorganization in the adult brain. Neuron. 2007;54(3):352–3.
Carpenter AF, Georgopoulos AP, Pellizzer G. Motor cortical encoding of serial order in a context-recall task. Science. 1999;283(5408):1752–7.
Whitlock JR. Posterior parietal cortex. Curr Biol. 2017;27(14):R691–5.
Stepniewska I, et al. Organization of the posterior parietal cortex in galagos: II. Ipsilateral cortical connections of physiologically identified zones within anterior sensorimotor region. J Comp Neurol. 2009;517(6):783–807.
Gharbawie OA, Stepniewska I, Kaas JH. Cortical connections of functional zones in posterior parietal cortex and frontal cortex motor regions in new world monkeys. Cereb Cortex. 2011;21(9):1981–2002.
Chao CC, et al. Induction of motor associative plasticity in the posterior parietal cortex-primary motor network. Cereb Cortex. 2015;25(2):365–73.
Mulliken GH, Musallam S, Andersen RA. Forward estimation of movement state in posterior parietal cortex. Proc Natl Acad Sci U S A. 2008;105(24):8170–7.
Snyder LH, Batista AP, Andersen RA. Coding of intention in the posterior parietal cortex. Nature. 1997;386(6621):167–70.
Nuwer MR. Fundamentals of evoked potentials and common clinical applications today. Electroencephalogr Clin Neurophysiol. 1998;106(2):142–8.
Rankin CH, et al. Habituation revisited: an updated and revised description of the behavioral characteristics of habituation. Neurobiol Learn Mem. 2009;92(2):135–8.
Groves PM, Thompson RF. Habituation: a dual-process theory. Psychol Rev. 1970;77(5):419–50.
Coppola G, et al. Abnormal cortical responses to somatosensory stimulation in medication-overuse headache. BMC Neurol. 2010;10:126.
Baron R, et al. Activation of the somatosensory cortex during Aβ-fiber mediated hyperalgesia. Brain Res. 2000;871(1):75–82.
Coppola G, Pierelli F, Schoenen J. Is the cerebral cortex hyperexcitable or hyperresponsive in migraine? Cephalalgia. 2007;27(12):1427–39.
Ozkul Y, Uckardes A. Median nerve somatosensory evoked potentials in migraine. Eur J Neurol. 2002;9(3):227–32.
Knott JR, Irwin DA. Anxiety, stress, and the contingent negative variation. Arch Gen Psychiatry. 1973;29(4):538–41.
Coppola G, et al. Somatosensory evoked high-frequency oscillations reflecting thalamo-cortical activity are decreased in migraine patients between attacks. Brain. 2005;128(Pt 1):98–103.
Klostermann F, et al. Spatiotemporal characteristics of human intrathalamic high-frequency (>400 Hz) SEP components. Neuroreport. 1999;10(17):3627–31.
Hashimoto I, Mashiko T, Imada T. Somatic evoked high-frequency magnetic oscillations reflect activity of inhibitory interneurons in the human somatosensory cortex. Electroencephalogr Clin Neurophysiol. 1996;100(3):189–203.
Restuccia D, et al. Influence of cholinergic circuitries in generation of high-frequency somatosensory evoked potentials. Clin Neurophysiol. 2003;114(8):1538–48.
Coppola G, et al. Electrophysiological correlates of episodic migraine chronification: evidence for thalamic involvement. J Headache Pain. 2013;14:76.
Cosentino G, Fierro B, Brighina F. From different neurophysiological methods to conflicting pathophysiological views in migraine: a critical review of literature. Clin Neurophysiol. 2014;125(9):1721–30.
Makeig S, et al. Dynamic brain sources of visual evoked responses. Science. 2002;295(5555):690–4.
Gantenbein AR, et al. Sensory information processing may be neuroenergetically more demanding in migraine patients. Neuroreport. 2013;24(4):202–5.
Sandor PS, et al. MR-spectroscopic imaging during visual stimulation in subgroups of migraine with aura. Cephalalgia. 2005;25(7):507–18.
Reyngoudt H, et al. Does visual cortex lactate increase following photic stimulation in migraine without aura patients? A functional (1)H-MRS study. J Headache Pain. 2011;12(3):295–302.
Montagna P, et al. 31P-magnetic resonance spectroscopy in migraine without aura. Neurology. 1994;44(4):666–9.
Reyngoudt H, et al. 31P-MRS demonstrates a reduction in high-energy phosphates in the occipital lobe of migraine without aura patients. Cephalalgia. 2011;31(12):1243–53.
Barbiroli B, et al. Abnormal brain and muscle energy metabolism shown by 31P magnetic resonance spectroscopy in patients affected by migraine with aura. Neurology. 1992;42(6):1209–14.
Montagna P, et al. Migraine as a defect of brain oxidative metabolism: a hypothesis. J Neurol. 1989;236(2):124–5.
Boehnke C, et al. High-dose riboflavin treatment is efficacious in migraine prophylaxis: an open study in a tertiary care centre. Eur J Neurol. 2004;11(7):475–7.
Sandor PS, et al. Prophylactic treatment of migraine with beta-blockers and riboflavin: differential effects on the intensity dependence of auditory evoked cortical potentials. Headache. 2000;40(1):30–5.
Sandor PS, et al. Efficacy of coenzyme Q10 in migraine prophylaxis: a randomized controlled trial. Neurology. 2005;64(4):713–5.
Rozen TD, et al. Open label trial of coenzyme Q10 as a migraine preventive. Cephalalgia. 2002;22(2):137–41.
Cromwell HC, et al. Sensory gating: a translational effort from basic to clinical science. Clin EEG Neurosci. 2008;39(2):69–72.
Hsiao FJ, et al. Somatosensory gating is altered and associated with migraine chronification: a magnetoencephalographic study. Cephalalgia. 2018;38(4):744–53.
Chen WT, et al. Comparison of somatosensory cortex excitability between migraine and “strict-criteria” tension-type headache: a magnetoencephalographic study. Pain. 2018;159(4):793–803.
Lyoo CH, et al. Abnormal temporal discrimination threshold in patients with multiple system atrophy. Mov Disord. 2007;22(4):556–9.
Costa J, et al. Subcortical interactions between somatosensory stimuli of different modalities and their temporal profile. J Neurophysiol. 2008;100(3):1610–21.
Lacruz F, et al. The anatomical basis of somaesthetic temporal discrimination in humans. J Neurol Neurosurg Psychiatry. 1991;54(12):1077–81.
Pastor MA, et al. The functional neuroanatomy of temporal discrimination. J Neurosci. 2004;24(10):2585–91.
Abbruzzese G, Berardelli A. Sensorimotor integration in movement disorders. Mov Disord. 2003;18(3):231–40.
Artieda J, et al. Temporal discrimination is abnormal in Parkinson’s disease. Brain. 1992;115(Pt 1):199–210.
Scontrini A, et al. Somatosensory temporal discrimination in patients with primary focal dystonia. J Neurol Neurosurg Psychiatry. 2009;80(12):1315–9.
Manganelli F, et al. Somatosensory temporal discrimination threshold is increased in patients with cerebellar atrophy. Cerebellum. 2013;12(4):456–9.
Bolay H, et al. Anatomy of headache, in pathophysiology of headaches: from molecule to man. M. Ashina and P. Geppetti, Editors. 2015, Springer, pp. 1–29.
Landmann G, et al. Bilateral sensory changes and high burden of disease in patients with chronic pain and unilateral nondermatomal somatosensory deficits: a quantitative sensory testing and clinical study. Clin J Pain. 2017;33(8):746–55.
Mailis A, et al. Unexplainable nondermatomal somatosensory deficits in patients with chronic nonmalignant pain in the context of litigation/compensation: a role for involvement of central factors? J Rheumatol. 2001;28(6):1385–93.
Egloff N, et al. Nondermatomal somatosensory deficits in chronic pain patients: are they really hysterical? Pain. 2012;153(9):1847–51.
Riederer F, et al. Nondermatomal somatosensory deficits in chronic pain are associated with cerebral grey matter changes. World J Biol Psychiatry. 2017;18(3):227–38.
Egloff N, et al. Nondermatomal somatosensory deficits in patients with chronic pain disorder: clinical findings and hypometabolic pattern in FDG-PET. Pain. 2009;145(1–2):252–8.
Mailis Gagnon A, Nicholson K. Nondermatomal somatosensory deficits (NDSDs): a neuropsychobiological phenomenon? Pain. 2009;145(1–2):12–3.
Frettlöh J, Huppe M, Maier C. Severity and specificity of neglect-like symptoms in patients with complex regional pain syndrome (CRPS) compared to chronic limb pain of other origins. Pain. 2006;124(1–2):184–9.
Schattschneider J. Complex regional pain syndrome—are we neglecting neglect? Nat Clin Pract Neurol. 2007;3(1):16–7.
Moran DW, Schwartz AB. Motor cortical representation of speed and direction during reaching. J Neurophysiol. 1999;82(5):2676–92.
Kalaska JF, et al. A comparison of movement direction-related versus load direction-related activity in primate motor cortex, using a two-dimensional reaching task. J Neurosci. 1989;9(6):2080–102.
Alexander GE, Crutcher MD. Preparation for movement: neural representations of intended direction in three motor areas of the monkey. J Neurophysiol. 1990;64(1):133–50.
Edwards LL, et al. Putting the “sensory” into sensorimotor control: the role of sensorimotor integration in goal-directed hand movements after stroke. Front Integr Neurosci. 2019;13:16.
de Noordhout AM, et al. Percutaneous magnetic stimulation of the motor cortex in migraine. Electroencephalogr Clin Neurophysiol. 1992;85(2):110–5.
Bettucci D, et al. Menstrual migraine without aura: cortical excitability to magnetic stimulation. Headache. 1992;32(7):345–7.
Rossini PM, et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord, roots and peripheral nerves: basic principles and procedures for routine clinical and research application. An updated report from an I.F.C.N. committee. Clin Neurophysiol. 2015;126(6):1071–107.
Cortese F, et al. Excitability of the motor cortex in patients with migraine changes with the time elapsed from the last attack. J Headache Pain. 2017;18(1):2.
Houdayer E, et al. The effects of low- and high-frequency repetitive TMS on the input/output properties of the human corticospinal pathway. Exp Brain Res. 2008;187(2):207–17.
Brighina F, et al. Facilitatory effects of 1 Hz rTMS in motor cortex of patients affected by migraine with aura. Exp Brain Res. 2005;161(1):34–8.
Brighina F, et al. Abnormal facilitatory mechanisms in motor cortex of migraine with aura. Eur J Pain. 2011;15(9):928–35.
Cosentino G, et al. Transcranial direct current stimulation preconditioning modulates the effect of high-frequency repetitive transcranial magnetic stimulation in the human motor cortex. Eur J Neurosci. 2012;35(1):119–24.
Lang N, et al. Preconditioning with transcranial direct current stimulation sensitizes the motor cortex to rapid-rate transcranial magnetic stimulation and controls the direction of after-effects. Biol Psychiatry. 2004;56(9):634–9.
Kujirai T, et al. The effect of transcranial magnetic stimulation on median nerve somatosensory evoked potentials. Electroencephalogr Clin Neurophysiol. 1993;89(4):227–34.
Valls-Solé J, et al. Human motor evoked responses to paired transcranial magnetic stimuli. Electroencephalogr Clin Neurophysiol. 1992;85(6):355–64.
Siniatchkin M, et al. Intracortical inhibition and facilitation in migraine–a transcranial magnetic stimulation study. Headache. 2007;47(3):364–70.
Brighina F, et al. High-frequency transcranial magnetic stimulation on motor cortex of patients affected by migraine with aura: a way to restore normal cortical excitability? Cephalalgia. 2010;30(1):46–52.
Ilic TV, et al. Short-interval paired-pulse inhibition and facilitation of human motor cortex: the dimension of stimulus intensity. J Physiol. 2002;545(1):153–67.
Sanger TD, Garg RR, Chen R. Interactions between two different inhibitory systems in the human motor cortex. J Physiol. 2001;530(Pt 2):307–17.
Cosentino G, et al. Intracortical facilitation within the migraine motor cortex depends on the stimulation intensity. A paired-pulse TMS study. J Headache Pain. 2018;19(1):65.
Huang YY, et al. The influence of prior synaptic activity on the induction of long-term potentiation. Science. 1992;255(5045):730–3.
Wang H, Wagner JJ. Priming-induced shift in synaptic plasticity in the rat hippocampus. J Neurophysiol. 1999;82(4):2024–8.
Antal A, et al. Homeostatic metaplasticity of the motor cortex is altered during headache-free intervals in migraine with aura. Cereb Cortex. 2008;18(11):2701–5.
Chadaide Z, et al. Transcranial direct current stimulation reveals inhibitory deficiency in migraine. Cephalalgia. 2007;27(7):833–9.
Stefan K, et al. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain. 2000;123(Pt 3):572–84.
Wolters A, et al. A temporally asymmetric Hebbian rule governing plasticity in the human motor cortex. J Neurophysiol. 2003;89(5):2339–45.
Turco CV, et al. Short- and long-latency afferent inhibition; uses, mechanisms and influencing factors. Brain Stimul. 2018;11(1):59–74.
Oliviero A, et al. Reduced sensorimotor inhibition in the ipsilesional motor cortex in a patient with chronic stroke of the paramedian thalamus. Clin Neurophysiol. 2005;116(11):2592–8.
Alaydin HC, et al. Reduced short-latency afferent inhibition indicates impaired sensorimotor integrity during migraine attacks. Headache. 2019;59(6):906–14.
Di Lazzaro V, et al. Neurophysiological predictors of long term response to AChE inhibitors in AD patients. J Neurol Neurosurg Psychiatry. 2005;76(8):1064–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Boran, H.E., Bolay, H., Gantenbein, H.A.R., Pohl, H. (2021). Sensory Processing and Sensorimotor Integration in Migraine. In: Coppola, G., Chen, WT. (eds) Neurophysiology of the Migraine Brain. Headache. Springer, Cham. https://doi.org/10.1007/978-3-030-56538-1_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-56538-1_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-56537-4
Online ISBN: 978-3-030-56538-1
eBook Packages: MedicineMedicine (R0)