
Abstract
Chronic pain is partially sustained by central sensitization, a synaptic plasticity phenomenon, and enhanced neuronal responsiveness following painful insults in the central pain pathways. Accumulating studies indicates that neuroinflammation in the peripheral and central nervous system also drives central sensitization. The activation of glial cells such as microglia and astrocytes in the spinal cord and brain is a distinctive feature of neuroinflammation, leading to the discharge of pro-inflammatory cytokines and chemokines. Recent studies suggest that modulating neuroinflammation could be used as therapeutic target in chronic pelvic pain. One of the promising formula is represented by a co-micronization of palmitoylethanolamide and polidatin, two strong natural compounds with a powerful anti-inflammatory and antioxidant activities. In this chapter we focused our attention on the role of neuroinflammation in chronic pelvic pain and its newest possible treatment.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keyword
1 Chronic Pelvic Pain
Chronic pelvic pain (CPP) is well defined by the European Association of Urology (EAU) as “chronic or persistent pain perceived in structures associated to the pelvis in both men and women. It is frequently correlated with negative behavioral, cognitive, emotional, and sexual effects as well as with suggestive signs of lower urinary tract, bowel, pelvic floor, or gynecological dysfunction. For documented nociceptive pain that becomes chronic/persistent over time, pain must have been permanent or recurrent for at least 6 months. If the sensitization mechanisms of pain are well documented, the pain may be considered chronic, regardless of the time period” [1, 2]. CPP in the female or male genital zone may be localized to the vulva, vagina, or perineum, or may involve intra-abdominal organs, including uterus, ovaries, and fallopian tubes (females), or can involve the prostate, epididymis, scrotum, penis, or testicles (males) [3] (see Table 3.1).
These conditions lead to a substantial burden on limited health care resources. For example, an estimated £158 million are spent every year for the treatment of this disorder in the UK National Health Service [4, 5].
Additionally, in Europe, a study undertaken in 2004 by Breivik and colleagues [6] found that chronic pain of moderate maximum severe intensity occurs in 19% of adult Europeans, extremely disturbing the quality of their lives. There are some changes between states but not much spread is seen.
Considering the complexity of CPP, it is very difficult to treat and these lead to frustration for both patients and their physicians. Treatment should include multifactorial approaches involving counseling, psychosocial support, medication management, physical therapy, and interventional procedures [1].
2 Neuroinflammation
Neuroinflammation is described as an inflammatory reaction inside the spinal cord or brain [7]. Inflammatory events in the peripheral nervous system (PNS) or in the central nervous system (CNS) happen at diverse levels from those of other tissues and involved different types of cells [8, 9]. In particular, the primary distinction relies in the lack of resident dendritic cells in the CNS parenchyma and perivascular macrophages and vascular pericytes take over the functions of mature dendritic cells in the CNS [10]. Secondary, the stimulation of the innate immune cells of CNS parenchyma, such as astrocytes, microglia, and, in some regions, mast cells, may be amplified in clinical conditions such as trauma, stroke, neurodegenerative disease, or growth of a tumor [11,12,13]. Furthermore, the extravasation of immune cells and molecules towards the inflamed region, indispensable to stimulate complement cascades and maintain the immunity reaction, is crucial for the inflammatory response of the total organism.
However, in the CNS, the blood–CNS barrier limited the permeability of microvessels, making thus the entire inflammatory response incredibly different and difficult. Just stimulated T cells may infiltrate the barrier, but they do not elicit an effective response to inflammation equivalent with that observed in peripheral tissues, where dendritic cells are responsible for the adaptive immune reaction [14]. Due to these features, it is curious to point out that CNS replies to inflammatory events when these exert a direct effect on CNS, for example, in the case of pathogens and tissue injury, and when the inflammatory events are so austere that penetrating T cells are involved. With these clarifications it is crucial to understand the “neuroinflammation” terms that differentiates inflammatory response in the CNS from inflammation reaction in different tissues.
In this view the neuroinflammation terms are a reply of the CNS to altered homeostasis. Principally, one maybe two cell systems are competent to intermediate this response: glia of the CNS, lymphocytes, macrophages of the hematopoietic system, and monocytes [15]. The actions encouraged by the neuroinflammations are classified as:
-
Homeostatic: when it involves different events such as vasodilation or the release of cytokines and neurotrophic factors
-
Maladaptive or neurotoxic: when it is characterized by the release of pro-inflammatory factors or the breakdown of blood–CNS barrier
-
Anti-inflammatory: when, contrary to what was said above, it involves the release of pro-inflammatory cytokines, neurotrophic factors, neurotransmitters, and cell adhesion molecules
After injury neuroinflammation is dynamically coordinated by a complex network of regulatory mechanisms, which confine the hypothetically damaging effects of persistent inflammation.
In particular, chronic, uncontrolled inflammation is characterized by overexpression of reactive oxygen species (ROS), cytokines, such as TNF-α and IL-1β, and other inflammatory mediators, such as inducible nitric oxide synthase (iNOS).
All these inflammatory molecules are detected following trauma to the CNS, and are involved by employment and trafficking of neutrophils and peripheral macrophages to the injury place. Anyhow, when the inflammatory event is protracted, and the hyperactivation of macrophages is continued, it overpowers the bounds of physiological control and leads to a series of deleterious effects that involve the activation of pro-inflammatory signaling pathways, increase oxidative stress, and death of nearby neurons that provide to the pathogenesis of chronic pain, such as neuropathic pain or neurodegeneration [16, 17].
Last but not least is the role played by neuroinflammation in animal pain models of neuropathic, incisional, inflammatory, and central pain and it is also closely associated with a number of comorbidities of chronic pain such as diabetes, sleep and anxiety disorders, obesity, and depression [18] and for these reasons targeting excessive neuroinflammation can offer new therapeutic approaches for the management of chronic pain and related neurological and psychiatric disorders.
3 Microglia and Astrocytes in Chronic Pain
The involvement of microglia and astrocytes in pain processing has been progressively recognized by many laboratories using varied procedures and animal models of temporary or persistent pain. These activations play a crucial role during neuronal recovery after central or peripheral injury [19]. Microglia are macrophage-like cells in the CNS that originate from bone marrow-derived monocytes and that migrate during perinatal development. They are heterogeneously disseminated throughout the CNS. Under physiological situations, microglia are not inactive as many researchers initially assumed, but it has been shown that microglia dynamically sense their environment with their ramified processes [20,21,22]. In particular, microglia energetically cooperate with synapses to regulate their organizations and functions in healthy brain [23]. During growth, microglial processes can engulf synapses, and synaptic pruning by microglia, which includes the activation of the complement system, is necessary for normal brain development [24, 25].
During activation, microglia exhibit morphological changes, such as a changing into the amoeboid form, from ramified, to and upregulation of microglial markers such as CCR3/CD11b, major histocompatibility complex II [MHC II], or ionized calcium-binding adaptor molecule-1 [IBA1] [20, 26,27,28].
Various studies have shown that microglia plays a critical role in neuropathic pain development as well as acute inflammatory pain [29,30,31,32,33]. For instance, it has been shown that minocycline, a nonselective microglia inhibitor, reduces inflammatory or postoperative or neuropathic pain. However, its function in decreasing neuropathic pain in the late phase is restricted [32, 34,35,36].
Astrocytes are the most abundant cells in the CNS and play several active functions in acute and chronic neurological diseases such as stroke or ischemia [37]. In contrast to microglia and oligodendrocytes, astrocytes formed physically coupled networks intermediated by gap junctions, which, among other roles, simplify intercellular transmission of Ca2+ signaling, exchange of cytosolic contents, and display oscillations in ion permeability across astrocytic networks. Gap junction communication is mediated by homo- and heteromeric associations of hemichannels, such as connexin-43 (Cx43), the most prevalent connexin expressed in astrocytes [38]. Although astrocytes are naturally immune labeled by glial fibrillary acidic protein (GFAP).
It is important to note that, every astrocyte forms a nonoverlapping territory or domain, which all together resemble a lattice framework, looking crystalline in nature. On the other hand, the implications of this organization are not fully understood; it becomes lost when astrocytes transition to reactive states [37, 39, 40]. Moreover, astrocytes have wide-ranging interactions with both cerebral blood vessels and synapses, and through these connections they control the increase in blood flow induced by synaptic activity. The astrocyte-mediated blood flow increased is fundamental to the blood-oxygen-level-dependent (BOLD) signal detected by functional magnetic resonance imaging (fMRI) [39]. It is assessed that, in rodents, a single astrocyte can enwrap 140,000 synapses and 4–6 neuronal somata, and can interact to 300–600 neuronal dendrites [40,41,42]. During synaptic transmission, close contact with neurons and synapses allows astrocytes not only to help and nourish neurons but also to control the external chemical environment. The increasing appreciation for active roles of astrocytes has led to the proposal of a “tripartite synapse” theory, founded on the facts that glia respond to neuronal activity with an increase of their internal Ca2+ concentration and cause the release of chemical transmitters from glia themselves, and glial transmitters through a feedback regulation of neuronal activity and synaptic strength [43, 44]. According to this, astrocytic processes are active components of synapses, in addition to pre- and postsynaptic components [45]. On the other hand, active contribution to synaptic activity remains just a possibility because several recent studies have challenged this theory, by demonstrating that alterations in astrocytic Ca2+ do not modulate synaptic transmission [46,47,48].
Due to important modifications in the expression of membrane proteins as well as neural circuits during growth, it is feasible that the notion of receptor-mediated Ca2+ signaling will be extended to include other intracellular signaling pathways as a main element defining astrocytic involvement in greater neural function. For example, in the young or adult rodent brain, glutamate-dependent neuroglial Ca2+ signaling is different [49,50,51]. Freshly, it has been demonstrated that receptor-mediated increases in astrocytic Ca2+ can control neural network activity by active uptake of extracellular K+ [52]. Considering that the extracellular concentration of K+ is an important determinant of the resting membrane potential and thereby of neuronal activity, active uptake of K+ represents a simple yet powerful tool for rapid variation of neural networks.
Studies using astroglial toxins or astroglial aconitase inhibitor or inhibitors of the astroglial enzyme glutamine synthetase in adult animals suggest that astrocytes play a key role both for the stimulation and preservation of inflammatory and for neuropathic pain [41, 53,54,55,56,57,58,59,60].
Models of neuropathic pain such as rhizotomy and spinal nerve ligation have shown proliferation of spinal cord astrocytes [61, 62]. Conversely, inhibiting astrocyte proliferation in the spinal cord was shown to reduce neuropathic pain [61].
4 Molecular Mediators in Chronic Pain
A main problem with regard to glial pain control is understanding how glial mediators are generated and released. In particular, glia produce large molecules such as chemokines, cytokines, proteases, and growth factors, as well as small molecules like glutamate, prostaglandin E2 (PGE2), ATP, and D-serine. These glial mediators can control neuronal and synaptic activity and, most important, pain sensitivity. Among the most well-studied glial mediators are pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IL-1β.
These cytokines are upregulated in spinal cord glia after nerve injury, inflammation, and others, and they are involved in the development and maintenance of inflammatory, neuropathic, and cancer pain and morphine tolerance [63,64,65,66].
In relation to its well-documented role in modulating peripheral sensitization, TNF-α plays a main role in generating central sensitization and persistent pain [67,68,69,70,71,72,73]. IL-1β is induced in astrocytes and microglia after bone cancer, inflammation, and nerve injury [55, 67, 74,75,76,77,78]. It was clearly demonstrated that the inhibition of spinal and brain IL-1β signaling reduces inflammatory, neuropathic, and cancer pain and enhances morphine analgesia [55, 66, 74, 76, 79,80,81]. Also, chemokines are produced by glial cells, particularly in astrocytes and in neurons [82, 83]. In primary cultures of astrocytes, TNF-α induced rapid expression of (C-C motif) Ligand 2 (CCL2), C-X-C motif chemokine 10 (CXCL10), and C-X-C motif chemokine 1(CXCL1) [84]. Spinal injection of TNF-α-activated astrocytes leads to constant mechanical allodynia through CCL2 release [85]. Additionally, CCL2 expression is further increased in astrocytes of the medullary dorsal horn and contributes to trigeminal neuropathic pain and in spinal nerve ligation induces CCL2 release in spinal astrocytes, and it was observed that intrathecal administration of an MCP-1 neutralizing antibody diminished neuropathic pain [84, 86]. In fact, mice with CCL2 overexpression in astrocytes display pain hypersensitivity [87].
Moreover, growth factors are well known to be induced in spinal glia by nerve injury. In particular, brain-derived neurotrophic factor (BDNF) was upregulated during nerve ligation in spinal microglia, via activation of P2X4 and p38 [88, 89]. Additionally, spinal injection of ATP-activated microglia is sufficient to stimulate mechanical allodynia via releasing BDNF, and, equally, neuropathic pain is repressed by spinal blockade of the BDNF receptor TrkB [30]. Furthermore, treatment of microglial cultures with morphine increases BDNF release, which does not require l-opioid receptor and TLR [90]. BDNF is also induced in dorsal root ganglion (DRG) neurons after nerve injury and can be produced from primary afferents in the spinal cord [91, 92]. Unlike BDNF, basic fibroblast growth factor (bFGF or FGF-2) is produced in activate astrocytes of the spinal cord in the late phase (3 weeks) of nerve injury [56].
Intrathecal infusion of bFGF produces persistent activation of spinal astrocytes through the upregulation of P-JNK and GFAP and sustained mechanical allodynia and chronic pain [56]. On the other hand, intrathecal administration of a bFGF-neutralizing antibody reduces established neuropathic pain [93].
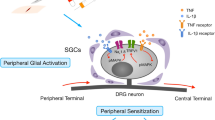
After nerve injury, also proteases are upregulated in spinal glia. It is well known that, spinal nerve ligation induces matrix metalloprotease-2 (MMP-2) in spinal cord astrocytes and DRG and satellite glial cells (SGCs) in the late phase of neuropathic pain to maintain neuropathic state, via activation of IL-1β and ERK [94].
Nerve injury additionally stimulates the production of cathepsin S in spinal microglia [95] and tissue-type plasminogen activator (tPA) in spinal astrocytes to enhance neuropathic pain [96]. A recent study indicated that nerve injury also increased the production of thrombospondin-4 (TSP4), an extracellular matrix glycoprotein, in spinal cord astrocytes correlated for the development of neuropathic pain and for synaptogenesis [97, 98].
To increase and to maintain the pain state, astrocytes produce small molecule mediators such as D-serine, ATP, and glutamate [41].
On the other hand, the anti-inflammatory and antinociceptive mediators IL-4, IL-10, and TGF-β were produced by glial cells for the recovery and resolution of pain [20, 99,100,101,102]. Improvement of endogenous production of IL-10 via gene therapy has been demonstrated to produce long-term relief in neuropathic pain. Of interest, a possible off-target effect of high doses of siRNAs is to induce IFN-α in spinal astrocytes for eliciting antinociceptive effects [103, 104].
5 Targeting Excessive Inflammation as a Therapy for Neuropathic Pain
There is currently strong suggestion from preclinical studies, and more restricted evidence in clinical studies, that damage to the nervous system can lead to a maladaptive inflammatory reaction contributing to the generation of persistent pain. There remain various obstacles to making an interpretation of this information into patient benefit. For this reason, there are several challenges in the design of appropriate clinical trials.
Using pain animal models are most successful at the time of injury, while delayed treatment is a more likely clinical scenario. On the other hand, only a subset of patients develop neuropathic pain after a lesion and we do not yet have effective predictive models. Increasingly evidences suggest that there are multiple pathophysiological mechanisms leading to persistent pain after nerve injury. It would be of great benefit to use either clinical or molecular biomarkers to individualize treatment, for example, targeting excessive inflammation only in those patients where there is evidence of an ongoing inflammatory response [105]. Some agents that modulate inflammation are already being used in selected groups of patients with neuropathic pain, although there is often a lack of evidence from the trial. It is well known that corticosteroids suppress pro-inflammatory cytokine expression and cell-mediated immunity. They are administered by several routes for the treatment of several neuropathic pain conditions, such as post-herpetic neuralgia or radicular back pain; however, definitive evidence for their efficacy is absent because of the scarcity of placebo-controlled studies and, in some cases, trials have shown side effects [106,107,108,109].
Another approach, recently studied, involved the use of select cytokines inhibitors [110, 111]. One probable trouble is the significant redundancy in the action of cytokines. Furthermore, as with corticosteroid suppression of the immune system, if these agents are given systemically they may be associated with an appreciably amplified risk of infection. The use of pro-resolution agents such as resolvins would be one strategy that could use a wide anti-inflammatory intervention [112].
The inhibition of microglial function is another novel option. Minocycline clinical trials for the prevention of postoperative intercostal pain, an optimal situation for testing this agent, are ongoing (NCT0131 4482).
Propentofylline decreases the production of free radicals and microglia activation. A randomized controlled trial of this agent did not find efficacy in the treatment of post-herpetic neuralgia [62]. Further approaches would be to target key ligand-gated ion channels expressed in microglia such as P2X4 and P2X7 or downstream signaling pathways that drive microglia towards an effector state such as p38 MAP kinase.
In a small double-blind crossover trial, the p38 mitogen-activated protein kinase inhibitor SB-681323 significantly decreased the daily pain score in patients with neuropathic pain [8, 113].
6 Clinical Significance and Future Perspectives
The delivery of anti-inflammatory drugs to the CNS is critical, given the significant role of key neuroinflammation in keeping chronic pain. Neuroinflammation consequential from neuroglial and neuro-immune interactions not only assists as a driving force for chronic pain but is also involved in other neurological and psychiatric diseases such as Alzheimer’s and Parkinson’s disease, multiple sclerosis (SM), autism, and others, as well as in cognitive deficits after major surgeries [114, 115]. Chronic pain is, in fact, commonly linked with depression, anxiety, sleep disorders, and cognitive decline, which are clinical sequelae of particular concern to the growing aging population which has increasingly high prevalence of chronic pain. Neuroinflammation and astrocyte reactivity is also connected with chronic pain in postmortem human spinal cord samples [116]. The development of effective new treatments for the prevention and resolution of neuroinflammation and postoperative pain is mandatory. Actually to counteract neuroinflammatory processes a new therapeutic approach is represented by the use of natural compound. In this chapter we focused our attention on some recent evidences that involved the use of aliamides, alone, or in association with antioxidant molecules.
7 PEA
N-Acylethanolamines are classified as naturally occurring lipidic mediator molecules composed of a fatty acid and ethanolamine, collectively namely “fatty acid ethanolamines” (FAEs). They are endogenous molecules involved in endogenous protective mechanisms, activated in the body as a result of different types of tissue damage or stimulation of inflammatory responses and nociceptive fibers. The members of FAE family are the endocannabinoid N-arachidonoylethanolamine (anandamide, or 5Z,8Z,11Z,14Z)-N-(2-hydroxyethyl)icosa-5,8,11,14-tetraenamide) and its congeners N-stearoylethanolamine (N-(2-hydroxyethyl)-stearamide), N-oleoylethanolamine (N-2-hydroxyethyl- 9(Z)-octadecenamide), and N-palmitoylethanolamine (PEA, or palmitoylethanolamide) (N-(2-hydroxyethyl)- hexadecanamide).
PEA and its congeners are formed from N-acylated phosphatidylethanolamine (NAPE) by several enzymatic pathways [117], the principal one involving a membrane-associated NAPE-phospholipase D which generates the respective NAE and phosphatidic acid. This enzyme is able to convert N-palmitoyl-phosphatidyl-ethanolamine into PEA. In the mammalian brain, NAEs are hydrolyzed by: (1) fatty acid amide hydrolase in the endoplasmic reticulum, which breaks down NAEs into the corresponding fatty acid and ethanolamine; (2) lysosomal NAE-hydrolyzing acid amidase (NAAA) [118]. NAAA is found mainly in macrophages, where it hydrolyzes NAEs with less than 18 carbon atoms, i.e., PEA, but not N-oleoylethanolamine and N-stearoylethanolamine. In contrast, fatty acid amide hydrolase hydrolyzes all three NAEs. PEA is abundant in mammals; there are evidences for the presence of PEA as well as other FAEs in marine species and sea urchin ovaries [119]. Biologically, PEA is produced and hydrolyzed by microglia [120], inhibits mast cell activation [121], and increases in glutamate-treated neocortical neurons ex vivo and in cortex after CNS injury, as well as in muscle dialysate from women with chronic neck/shoulder pain [122]. PEA levels are also increased in a mouse model of experimental allergic encephalomyelitis [123].
Mechanistically PEA may be a ligand for peroxisome proliferator activated receptor α (PPARα), one of a group of nuclear receptor proteins that function as transcription factors regulating the expression of genes. In particular, the α- and γ-isoforms of PPAR are associated with pro-inflammatory effects. Moreover, in PPARα null mice or blocked by PPARα antagonists the anti-inflammatory, antinociceptive/anti-neuropathic, and neuroprotective effect of PEA were not detected [124]. PEA is produced through an “on-demand” synthesis within the lipid bilayer where N -phosphatidylethanolamine-specific phospholipase D (NAPE-PLD) releases it from its membrane precursor, N-palmitoyl phosphatidylethanolamine.
An “entourage effect” has also been hypothesized to clarify the pharmacological actions of PEA, whereby PEA enhances the anti-inflammatory and antinociceptive activity of other endogenous compounds by potentiating their affinity for a receptor or by inhibiting their metabolic degradation.
Anandamide and its congeners like PEA have in common the transient receptor potential vanilloid type 1 (TRPV1) receptor that is activated by noxious heat, low pH, and capsaicin. Anandamide itself is a TRPV1 receptor agonist, and PEA enhances anandamide stimulation of the human TRPV1 receptor in a cannabinoid CB2 receptor antagonist-sensitive fashion—which could be interpreted as PEA acting indirectly by potentiating anandamide actions. Mast cells and microglia reportedly express TRPV1 receptors [125].
8 Polydatin
Polydatin (PO), also called piceid, is a traditional Chinese medicine, detected in many daily diets food that has wide-ranging pharmacological activities [126, 127]. There are four main derivatives of PO in nature, including trans-polydatin, trans-resveratrol, cis-polydatin, and cis-resveratrol [128].
PO has a range of biological effects, such as the ability to protect lung, brain, heart, and intestine against ischemia-reperfusion (I/R) injury, anti-platelet aggregation, as well as anti-inflammatory, anti-shock, and anti-oxidation effects [129,130,131,132,133,134,135]. Additionally, two studies done in the last year demonstrated that PO protects against acetaminophen-induced hepatotoxicity in mice and suppresses nucleus pulposus cell senescence, promoting matrix homeostasis and attenuating intervertebral disc degeneration in rats [136, 137].
9 PEA and Polydatin as Future Treatment of Chronic Pelvic Pain
Preclinical studies about the management of chronic pain with the association of PEA and PO showed a significant reduction in the inflammatory process and pain associated with an experimental rat model of surgically induced endometriosis or carrageenan-induced acute inflammation as well as possess the ability to decrease prostate weight, DHT production, inflammation and oxidative stress process and apoptosis dysregulation in an experimental model of testosterone induced benign prostatic hyperplasia [138,139,140].
Clinical trials in which PEA/PO was first used was published in 2010 [141, 142], suggesting that a combination of micronized PEA/PO was efficient in endometriosis-related chronic pelvic pain. Indraccolo et al. [142] reported only 4 cases of endometriosis treatment with oral micronized PEA/PO (400 mg/40 mg) twice a day for 3 months, while Cobellis et al. [143] treated 18 patients in one arm of a randomized trial with micronized PEA/PO (200 mg/20 mg) orally, three times a day for 3 months. Both studies showed an improvement in mean pain visual analog scale (VAS) scores for chronic pelvic pain and other endometriotic pains (with improvement in the micronized PEA/PO arm versus placebo arm in the randomized trial [143]). The above observations were substantiated by results of VAS score improvement in a study on 610 patients [144] treated with micronized PEA (600 mg twice daily) for chronic pain due to several causes, leading us to speculate that micronized PEA is effective also on chronic pelvic pain, even in the presence of endometriosis.
Additionally, another study provides preliminary evidence on the efficacy and safety of um-PEA/PO as an add-on treatment to conventional pharmacological regimens in patients suffering from IC/BPS, showing a significantly decreased pain in 75% of patients [145].
In another set of experiment, Tartaglia and colleagues considered the effectiveness of an oral combination of PEA and trans-polydatin in the treatment of primary dysmenorrhea in healthy adolescents and young women and found a reduction in symptoms, exerting a neuroprotective and antinociceptive effect during primary dysmenorrhea [146].
Interestingly, all mechanistic studies showing a benefit of active treatment in the management of several pathologies failed to exactly clarify the exact mechanism of action of the active compound, confirming the complexity of these type of studies [147,148,149]. Whether the PEA/PO effect is centrally related, secondary to mast cell stabilization or to modulation of the endocannabinoid system remains to be further investigated [150, 151].
In fact, confirmation of these initial findings will require randomized, double-blind, placebo-controlled clinical trials of sufficient power to assess rates of respondents in subgroups of patients, in order to fully appreciate the efficacy of micronized PEA/PO combination as a therapy for endometriosis, together with cohort studies to assess long-term effects of such therapy [142].
References
Engeler DS, Baranowski AP, Dinis-Oliveira P, Elneil S, Hughes J, Messelink EJ, et al. The 2013 EAU guidelines on chronic pelvic pain: is management of chronic pelvic pain a habit, a philosophy, or a science? 10 years of development. Eur Urol. 2013;64(3):431–9.
Passavanti MB, Pota V, Sansone P, Aurilio C, De Nardis L, Pace MC. Chronic pelvic pain: assessment, evaluation, and Objectivation. Pain Res Treat. 2017;2017:9472925.
Roy H, Offiah I, Dua A. Neuromodulation for pelvic and urogenital pain. Brain Sci. 2018;8(10):180.
Mathias SD, Kuppermann M, Liberman RF, Lipschutz RC, Steege JF. Chronic pelvic pain: prevalence, health-related quality of life, and economic correlates. Obstet Gynecol. 1996;87(3):321–7.
Latthe P, Latthe M, Say L, Gulmezoglu M, Khan KS. WHO systematic review of prevalence of chronic pelvic pain: a neglected reproductive health morbidity. BMC Public Health. 2006;6:177.
Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain. 2006;10(4):287–333.
DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139(Suppl 2):136–53.
Ellis A, Bennett DL. Neuroinflammation and the generation of neuropathic pain. Br J Anaesth. 2013;111(1):26–37.
Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov. 2014;13(7):533–48.
Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239(4837):290–2.
Skaper SD, Giusti P, Facci L. Microglia and mast cells: two tracks on the road to neuroinflammation. FASEB J. 2012;26(8):3103–17.
Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50(4):427–34.
Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1(1):14.
Melchior B, Puntambekar SS, Carson MJ. Microglia and the control of autoreactive T cell responses. Neurochem Int. 2006;49(2):145–53.
Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58(3):233–47.
Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354(6312):572–7.
Chen G, Zhang YQ, Qadri YJ, Serhan CN, Ji RR. Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron. 2018;100(6):1292–311.
Sandu RE, Buga AM, Uzoni A, Petcu EB, Popa-Wagner A. Neuroinflammation and comorbidities are frequently ignored factors in CNS pathology. Neural Regen Res. 2015;10(9):1349–55.
DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10(1):40–52.
Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–94.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8.
Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28(11):571–3.
Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31(45):16064–9.
Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for Normal brain development. Science. 2011;333(6048):1456–8.
Eriksson NP, Persson JKE, Svensson M, Arvidsson J, Molander C, Aldskogius H. A quantitative-analysis of the microglial cell reaction in central primary sensory projection territories following peripheral-nerve injury in the adult-rat. Exp Brain Res. 1993;96(1):19–27.
Suter MR, Wen YR, Decosterd I, Ji RR. Do glial cells control pain? Neuron Glia Biol. 2007;3:255–68.
Suter MR, Wen YR, Decosterd I, Ji RR. Do glial cells control pain?. (vol 3, pg 255, 2007). Neuron Glia Biol. 2007;3:389.
Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, et al. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86(6):1534–44.
Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017–21.
Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33.
Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306(2):624–30.
Zhou Z, Peng X, Hao S, Fink D, Mata M. HSV-mediated transfer of interleukin-10 reduces inflammatory pain through modulation of membrane tumor necrosis factor alpha in spinal cord microglia. Gene Ther. 2008;15(3):183–90.
Beggs S, Currie G, Salter MW, Fitzgerald M, Walker SM. Priming of adult pain responses by neonatal pain experience: maintenance by central neuroimmune activity. Brain. 2012;135:404–17.
Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26(16):4308–17.
Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci. 2005;22(10):2431–40.
Kimelberg HK, Nedergaard M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics. 2010;7(4):338–53.
Chen MJ, Kress B, Han X, Moll K, Peng W, Ji RR, et al. Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia. 2012;60(11):1660–70.
Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10(11):1369–76.
Oberheim NA, Takano T, Han X, He W, Lin JHC, Wang F, et al. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009;29(10):3276–87.
Gao YJ, Ji RR. Targeting astrocyte Signaling for chronic pain. Neurotherapeutics. 2010;7(4):482–93.
Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22(1):183–92.
Abbadie C, Bhangoo S, De Koninck Y, Malcangio M, Melik-Parsadaniantz S, White FA. Chemokines and pain mechanisms. Brain Res Rev. 2009;60(1):125–34.
Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100(13):7947–52.
Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22(5):208–15.
Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;327(5970):1250–4.
Nedergaard M, Verkhratsky A. Artifact versus reality-how astrocytes contribute to synaptic events. Glia. 2012;60(7):1013–23.
Petravicz J, Fiacco TA, McCarthy KD. Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J Neurosci. 2008;28(19):4967–73.
Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, et al. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339(6116):197–200.
Fitzgerald M. The development of nociceptive circuits. Nat Rev Neurosci. 2005;6(7):507–20.
Benn SC, Costigan M, Tate S, Fitzgerald M, Woolf CJ. Developmental expression of the TTX-resistant voltage-gated sodium channels Nav1.8 (SNS) and Navl.9 (SNS2) in primary sensory neurons. J Neurosci. 2001;21(16):6077–85.
Wang F, Smith NA, Xu Q, Fujita T, Baba A, Matsuda T, et al. Astrocytes modulate neural network activity by Ca 2+ −dependent uptake of extracellular K +. Sci Signal. 2012;5(218):ra26.
Chiang CY, Sessle BJ, Dostrovsky JO. Role of astrocytes in pain. Neurochem Res. 2012;37(11):2419–31.
Chiang CY, Wang J, Xie YF, Zhang S, Hu JW, Dostrovsky JO, et al. Astroglial glutamate-glutamine shuttle is involved in central sensitization of nociceptive neurons in rat medullary dorsal horn. J Neurosci. 2007;27(34):9068–76.
Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27(22):6006–18.
Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006;2(4):259–69.
Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33(11):1471–8.
Okada-Ogawa A, Suzuki I, Sessle BJ, Chiang CY, Salter MW, Dostrovsky JO, et al. Astroglia in medullary dorsal horn (trigeminal spinal subnucleus caudalis) are involved in trigeminal neuropathic pain mechanisms. J Neurosci. 2009;29(36):11161–71.
Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010;16(11):1267–76.
Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Armijo LM, et al. Intrathecal cannabilactone CB <inf>2</inf>R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain. 2012;153(5):1091–106.
Tsuda M, Kohro Y, Yano T, Tsujikawa T, Kitano J, Tozaki-Saitoh H, et al. JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain. 2011;134(4):1127–39.
Landry RP, Jacobs VL, Romero-Sandoval EA, DeLeo JA. Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Exp Neurol. 2012;234(2):340–50.
DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90(1–2):1–6.
Sommer C, Schäfers M, Marziniak M, Toyka KV. Etanercept reduces hyperalgesia in experimental painful neuropathy. J Peripher Nerv Syst. 2001;6(2):67–72.
Svensson CI, Schäfers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005;379(3):209–13.
Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24(8):450–5.
Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–55.
Xu JT, Xin WJ, Zang Y, Wu CY, Liu XG. The role of tumor necrosis factor-alpha in the neuropathic pain induced by lumbar 5 ventral root transection in rat. Pain. 2006;123(3):306–21.
Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain. 2011;152(2):419–27.
Zheng W, Ouyang H, Zheng X, Liu S, Mata M, Fink DJ, et al. Glial TNFα in the spinal cord regulates neuropathic pain induced by HIV gp120 application in rats. Mol Pain. 2011;7:40.
Jin X, Gereau Iv RW. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J Neurosci. 2006;26(1):246–55.
Schäfers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-α after spinal nerve ligation. J Neurosci. 2003;23(7):3028–38.
Sorkin LS, Xiao WH, Wagner R, Myers RR. Tumour necrosis factor-α induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81(1):255–62.
Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci. 2008;28(42):10482–95.
Weyerbacher AR, Xu Q, Tamasdan C, Shin SJ, Inturrisi CE. N-methyl-d-aspartate receptor (NMDAR) independent maintenance of inflammatory pain. Pain. 2010;148(2):237–46.
Zhang RX, Liu B, Wang L, Ren K, Qiao JT, Berman BM, et al. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain. 2005;118(1–2):125–36.
Clark AK, Staniland AA, Marchand F, Kaan TKY, McMahon SB, Malcangio M. P2X7-dependent release of interleukin-1β and nociception in the spinal cord following lipopolysaccharide. J Neurosci. 2010;30(2):573–82.
Deleo JA, Colburn RW, Rickman AJ. Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Res. 1997;759(1):50–7.
Milligan ED, Twining C, Chacur M, Biedenkapp J, O'Connor K, Poole S, et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23(3):1026–40.
Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001;103(2):529–39.
Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, et al. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24(33):7353–65.
Guo W, Wang H, Zou S, Dubner R, Ren K. Chemokine signaling involving chemokine (C-C motif) ligand 2 plays arole in descending pain facilitation. Neurosci Bull. 2012;28(2):193–207.
Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126(1):56–68.
Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29(13):4096–108.
Gao YJ, Zhang L, Ji RR. Spinal injection of TNF-α-activated astrocytes produces persistent pain symptom mechanical allodynia by releasing monocyte chemoattractant protein-1. Glia. 2010;58(15):1871–80.
Zhang ZJ, Dong YL, Lu Y, Cao S, Zhao ZQ, Gao YJ. Chemokine CCL2 and its receptor CCR2 in the medullary dorsal horn are involved in trigeminal neuropathic pain. J Neuroinflammation. 2012;9:136.
Menetski J, Mistry S, Lu M, Mudgett JS, Ransohoff RM, DeMartino JA, et al. Mice overexpressing chemokine ligand 2 (CCL2) in astrocytes display enhanced nociceptive responses. Neuroscience. 2007;149(3):706–14.
Trang T, Beggs S, Salter MW. ATP receptors gate microglia signaling in neuropathic pain. Exp Neurol. 2012;234(2):354–61.
Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, et al. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28(44):11263–8.
Ferrini F, Trang T, Mattioli TAM, Laffray S, Del'Guidice T, Lorenzo LE, et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl-homeostasis. Nat Neurosci. 2013;16(2):183–92.
Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21(13):4891–900.
Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, et al. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001;21(12):4469–77.
Madiai F, Goettl VM, Hussain SR, Clairmont AR, Stephens RL Jr, Hackshaw KV. Anti-fibroblast growth factor-2 antibodies attenuate mechanical allodynia in a rat model of neuropathic pain. J Mol Neurosci. 2005;27(3):315–24.
Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14(3):331–6.
Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104(25):10655–60.
Kozai T, Yamanaka H, Dai Y, Obata K, Kobayashi K, Mashimo T, et al. Tissue type plasminogen activator induced in rat dorsal horn astrocytes contributes to mechanical hypersensitivity following dorsal root injury. Glia. 2007;55(6):595–603.
Kim DS, Li KW, Boroujerdi A, Yu YP, Zhou CY, Deng P, et al. Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. J Neurosci. 2012;32(26):8977–87.
Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Özkan E, et al. Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139(2):380–92.
Correa F, Hernangómez-Herrero M, Mestre L, Loría F, Docagne F, Guaza C. The endocannabinoid anandamide downregulates IL-23 and IL-12 subunits in a viral model of multiple sclerosis: evidence for a cross-talk between IL-12p70/IL-23 axis and IL-10 in microglial cells. Brain Behav Immun. 2011;25(4):736–49.
Hao S, Mata M, Glorioso JC, Fink DJ. HSV-mediated expression of interleukin-4 in dorsal root ganglion neurons reduces neuropathic pain. Mol Pain. 2006;2:6.
Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26(12):696–705.
Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10(1):23–36.
Sloane EM, Soderquist RG, Maier SF, Mahoney MJ, Watkins LR, Milligan ED. Long-term control of neuropathic pain in a non-viral gene therapy paradigm. Gene Ther. 2009;16(4):470–5.
Tan PH, Gao YJ, Berta T, Xu ZZ, Ji RR. Short small-interfering RNAs produce interferon-α-mediated analgesia. Br J Anaesth. 2012;108(4):662–9.
von Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron. 2012;73(4):638–52.
Han Y, Zhang JJ, Chen N, He L, Zhou MK, Zhu CR. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2013;3:CD005582.
Kotani N, Kushikata T, Hashimoto H, Kimura F, Muraoka M, Yodono M, et al. Intrathecal methylprednisolone for intractable postherpetic neuralgia. N Engl J Med. 2000;343(21):1514–9.
Kikuchi A, Kotani N, Sato T, Takamura K, Sakai I, Matsuki A. Comparative therapeutic evaluation of intrathecal versus epidural methylprednisolone for long-term analgesia in patients with intractable postherpetic neuralgia. Reg Anesth Pain Med. 1999;24(4):287–93.
Tran DQH, Duong S, Bertini P, Finlayson RJ. Treatment of complex regional pain syndrome: a review of the evidence. Can J Anaesth. 2010;57(2):149–66.
Okoro T, Tafazal SI, Longworth S, Sell PJ. Tumor necrosis alpha-blocking agent (Etanercept) a triple blind randomized controlled trial of its use in treatment of sciatica. J Spinal Disord Tech. 2010;23(1):74–7.
Korhonen T, Karppinen J, Paimela L, Malmivaara A, Lindgren KA, Jarvinen S, et al. The treatment of disc herniation-induced sciatica with infliximab—results of a randomized, controlled, 3-month follow-up study. Spine. 2005;30(24):2724–8.
Ji RR, Xu ZZ, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 2011;34(11):599–609.
Anand P, Shenoy R, Palmer JE, Baines AJ, Lai RY, Robertson J, et al. Clinical trial of the p38 MAP kinase inhibitor dilmapimod in neuropathic pain following nerve injury. Eur J Pain. 2011;15(10):1040–8.
Terrando N, Eriksson LI, Ryu JK, Yang T, Monaco C, Feldmann M, et al. Resolving postoperative neuroinflammation and cognitive decline. Ann Neurol. 2011;70(6):986–95.
Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology. 2018;129(2):343–66.
Shi Y, Gelman BB, Lisinicchia JG, Tang SJ. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci. 2012;32(32):10833–40.
Ueda N, Tsuboi K, Uyama T. Metabolism of endocannabinoids and related N-acylethanolamines: canonical and alternative pathways. FEBS J. 2013;280(9):1874–94.
Tsuboi K, Takezaki N, Ueda N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA). Chem Biodivers. 2007;4(8):1914–25.
Bisogno T, Ventriglia M, Milone A, Mosca M, Cimino G, Di Marzo V. Occurrence and metabolism of anandamide and related acyl-ethanolamides in ovaries of the sea urchin Paracentrotus lividus. Biochim Biophys Acta. 1997;1345(3):338–48.
Muccioli GG, Stella N. Microglia produce and hydrolyze palmitoylethanolamide. Neuropharmacology. 2008;54(1):16–22.
Facci L, Dal Toso R, Romanello S, Buriani A, Skaper SD, Leon A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc Natl Acad Sci U S A. 1995;92(8):3376–80.
Hansen HS, Lauritzen L, Strand AM, Vinggaard AM, Frandsen A, Schousboe A. Characterization of glutamate-induced formation of N-acylphosphatidylethanolamine and N-acylethanolamine in cultured neocortical neurons. J Neurochem. 1997;69(2):753–61.
Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Makriyannis A, et al. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001;15(2):300–2.
de Novellis V, Luongo L, Guida F, Cristino L, Palazzo E, Russo R, et al. Effects of intra-ventrolateral periaqueductal grey palmitoylethanolamide on thermoceptive threshold and rostral ventromedial medulla cell activity. Eur J Pharmacol. 2012;676(1–3):41–50.
Kim SR, Kim SU, Oh U, Jin BK. Transient receptor potential vanilloid subtype 1 mediates microglial cell death in vivo and in vitro via Ca2+−mediated mitochondrial damage and cytochrome c release. J Immunol. 2006;177(7):4322–9.
Gao Y, Chen T, Lei X, Li Y, Dai X, Cao Y, et al. Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury. Mol Med Rep. 2016;14(6):5481–8.
Du QH, Peng C, Zhang H. Polydatin: a review of pharmacology and pharmacokinetics. Pharm Biol. 2013;51(11):1347–54.
Mikulski D, Molski M. Quantitative structure-antioxidant activity relationship of trans-resveratrol oligomers, trans-4,4 '-dihydroxystilbene dimer, trans-resveratrol-3-O-glucuronide, glucosides: trans-piceid, cis-piceid, trans-astringin and trans-resveratrol-4 '-O-beta-D-glucopyranoside. Eur J Med Chem. 2010;45(6):2366–80.
Cheng Y, Zhang HT, Sun L, Guo S, Ouyang S, Zhang Y, et al. Involvement of cell adhesion molecules in polydatin protection of brain tissues from ischemia-reperfusion injury. Brain Res. 2006;1110(1):193–200.
Wang FY, Xu ZJ, Zhang XL, Wang WT, Ha ML, Wang Y. Protective effects of polydatin against lung ischemia/reperfusion injury and the initial exploration for its mechanism. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2008;24(1):62–5.
Wang X, Song R, Bian HN, Brunk UT, Zhao M, Zhao KS. Polydatin, a natural polyphenol, protects arterial smooth muscle cells against mitochondrial dysfunction and lysosomal destabilization following hemorrhagic shock. Am J Physiol Regul Integr Comp Physiol. 2012;302(7):R805–14.
Lanzilli G, Cottarelli A, Nicotera G, Guida S, Ravagnan G, Fuggetta MP. Anti-inflammatory effect of resveratrol and polydatin by in vitro IL-17 modulation. Inflammation. 2012;35(1):240–8.
Kerem Z, Bilkis I, Flaishman MA, Sivan L. Antioxidant activity and inhibition of alpha-glucosidase by trans-resveratrol, piceid, and a novel trans-stilbene from the roots of Israeli Rumex bucephalophorus L. J Agric Food Chem. 2006;54(4):1243–7.
Sheng C, Yu YH, Zhao KS, Qin W, Wang CH. Hypotensive resuscitation combined with polydatin improve microcirculation and survival in a rabbit model of uncontrolled hemorrhagic shock in pregnancy. J Surg Res. 2011;168(1):103–10.
Zhang PW, Yu CL, Wang YZ, Luo SF, Sun LS, Li RS. Influence of 3,4′,5-trihydroxystibene-3-beta-mono-D-glucoside on vascular endothelial epoprostenol and platelet aggregation. Zhongguo Yao Li Xue Bao. 1995;16(3):265–8.
Wang J, Huang C, Lin Z, Pan X, Chen J, Zheng G, et al. Polydatin suppresses nucleus pulposus cell senescence, promotes matrix homeostasis and attenuates intervertebral disc degeneration in rats. J Cell Mol Med. 2018;22(11):5720–31.
Liu YH, Huang QH, Wu X, Wu JZ, Liang JL, Lin GS, et al. Polydatin protects against acetaminophen-induced hepatotoxicity in mice via anti-oxidative and anti-apoptotic activities. Food Funct. 2018;9(11):5891–902.
Di Paola R, Fusco R, Gugliandolo E, Crupi R, Evangelista M, Granese R, et al. Co-micronized Palmitoylethanolamide/Polydatin treatment causes endometriotic lesion regression in a rodent model of surgically induced endometriosis. Front Pharmacol. 2016;7:382.
Cordaro M, Impellizzeri D, Siracusa R, Gugliandolo E, Fusco R, Inferrera A, et al. Effects of a co-micronized composite containing palmitoylethanolamide and polydatin in an experimental model of benign prostatic hyperplasia. Toxicol Appl Pharmacol. 2017;329:231–40.
Esposito E, Impellizzeri D, Bruschetta G, Cordaro M, Siracusa R, Gugliandolo E, et al. A new co-micronized composite containing palmitoylethanolamide and polydatin shows superior oral efficacy compared to their association in a rat paw model of carrageenan-induced inflammation. Eur J Pharmacol. 2016;782:107–18.
Indraccolo U, Barbieri F. Effect of palmitoylethanolamide-polydatin combination on chronic pelvic pain associated with endometriosis: preliminary observations. Eur J Obstet Gynecol Reprod Biol. 2010;150(1):76–9.
Indraccolo U, Indraccolo SR, Mignini F. Micronized palmitoylethanolamide/trans-polydatin treatment of endometriosis-related pain: a meta-analysis. Annali dell'Istituto superiore di sanita. 2017;53(2):125–34.
Cobellis L, Castaldi MA, Nocerino A, Boccia O, Pisani I, Salzillo ME, et al. N-Palmitoiletanolamide micronizzata e transpolidatina nel trattamento del dolore pelvico cronico associato all’endometriosi. Giornale italiano di ostetricia e ginecologia. 2010;32(3):160–5.
Gatti A, Lazzari M, Gianfelice V, Di Paolo A, Sabato E, Sabato AF. Palmitoylethanolamide in the treatment of chronic pain caused by different etiopathogenesis. Pain Med. 2012;13(9):1121–30.
Gubbiotti M, Illiano E, Costantini E, Giannantoni A. Palmitoylethanolamide/polydatin as add-on therapy in pain resistant patients with interstitial cystitis/bladder painful syndrome. Eur Urol Suppl. 2019;18(1):e1970.
Tartaglia E, Armentano M, Giugliano B, Sena T, Giuliano P, Loffredo C, et al. Effectiveness of the association N-palmitoylethanolamine and transpolydatin in the treatment of primary dysmenorrhea. J Pediatr Adolesc Gynecol. 2015;28(6):447–50.
Wouters MM, Balemans D, Van Wanrooy S, Dooley J, Cibert-Goton V, Alpizar YA, et al. Histamine receptor H1-mediated sensitization of TRPV1 mediates visceral hypersensitivity and symptoms in patients with irritable bowel syndrome. Gastroenterology. 2016;150(4):875–87. e9
Lam C, Tan W, Leighton M, Hastings M, Lingaya M, Falcone Y, et al. A mechanistic multicentre, parallel group, randomised placebo-controlled trial of mesalazine for the treatment of IBS with diarrhoea (IBS-D). Gut. 2016;65(1):91–9.
Klooker TK, Braak B, Koopman KE, Welting O, Wouters MM, van der Heide S, et al. The mast cell stabiliser ketotifen decreases visceral hypersensitivity and improves intestinal symptoms in patients with irritable bowel syndrome. Gut. 2010;59(9):1213–21.
Iuvone T, Affaitati G, De Filippis D, Lopopolo M, Grassia G, Lapenna D, et al. Ultramicronized palmitoylethanolamide reduces viscerovisceral hyperalgesia in a rat model of endometriosis plus ureteral calculosis: role of mast cells. Pain. 2016;157(1):80–91.
Esposito G, Capoccia E, Turco F, Palumbo I, Lu J, Steardo A, et al. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-alpha activation. Gut. 2014;63(8):1300–12.
Cheah PY, Liong ML, Yuen KH, Teh CL, Khor T, Yang JR, et al. Terazosin therapy for chronic prostatitis/chronic pelvic pain syndrome: a randomized, placebo controlled trial. J Urol. 2003;169(2):592–6.
Gul O, Eroglu M, Ozok U. Use of terazosine in patients with chronic pelvic pain syndrome and evaluation by prostatitis symptom score index. Int Urol Nephrol. 2001;32(3):433–6.
Mehik A, Alas P, Nickel JC, Sarpola A, Helstrom PJ. Alfuzosin treatment for chronic prostatitis/chronic pelvic pain syndrome: a prospective, randomized, double-blind, placebo-controlled, pilot study. Urology. 2003;62(3):425–9.
Tugcu V, Tasci AI, Fazlioglu A, Gurbuz G, Ozbek E, Sahin S, et al. A placebo-controlled comparison of the efficiency of triple- and monotherapy in category III B chronic pelvic pain syndrome (CPPS). Eur Urol. 2007;51(4):1113–7. discussion 8
Evliyaoglu Y, Burgut R. Lower urinary tract symptoms, pain and quality of life assessment in chronic non-bacterial prostatitis patients treated with alpha-blocking agent doxazosin; versus placebo. Int Urol Nephrol. 2002;34(3):351–6.
Chen Y, Wu X, Liu J, Tang W, Zhao T, Zhang J. Effects of a 6-month course of tamsulosin for chronic prostatitis/chronic pelvic pain syndrome: a multicenter, randomized trial. World J Urol. 2011;29(3):381–5.
Nickel JC, Downey J, Pontari MA, Shoskes DA, Zeitlin SI. A randomized placebo-controlled multicentre study to evaluate the safety and efficacy of finasteride for male chronic pelvic pain syndrome (category IIIA chronic nonbacterial prostatitis). BJU Int. 2004;93(7):991–5.
Nickel JC, O'Leary MP, Lepor H, Caramelli KE, Thomas H, Hill LA, et al. Silodosin for men with chronic prostatitis/chronic pelvic pain syndrome: results of a phase II multicenter, double-blind, placebo controlled study. J Urol. 2011;186(1):125–31.
Lee JC, Muller CH, Rothman I, Agnew KJ, Eschenbach D, Ciol MA, et al. Prostate biopsy culture findings of men with chronic pelvic pain syndrome do not differ from those of healthy controls. J Urol. 2003;169(2):584–7. discussion 7-8
Nickel JC, Downey J, Clark J, Casey RW, Pommerville PJ, Barkin J, et al. Levofloxacin for chronic prostatitis/chronic pelvic pain syndrome in men: a randomized placebo-controlled multicenter trial. Urology. 2003;62(4):614–7.
Zhou Z, Hong L, Shen X, Rao X, Jin X, Lu G, et al. Detection of nanobacteria infection in type III prostatitis. Urology. 2008;71(6):1091–5.
Thakkinstian A, Attia J, Anothaisintawee T, Nickel JC. Alpha-blockers, antibiotics and anti-inflammatories have a role in the management of chronic prostatitis/chronic pelvic pain syndrome. BJU Int. 2012;110(7):1014–22.
Zhao WP, Zhang ZG, Li XD, Yu D, Rui XF, Li GH, et al. Celecoxib reduces symptoms in men with difficult chronic pelvic pain syndrome (category IIIA). Braz J Med Biol Res. 2009;42(10):963–7.
Goldmeier D, Madden P, McKenna M, Tamm N. Treatment of category III a prostatitis with zafirlukast: a randomized controlled feasibility study. Int J STD AIDS. 2005;16(3):196–200.
Bates SM, Hill VA, Anderson JB, Chapple CR, Spence R, Ryan C, et al. A prospective, randomized, double-blind trial to evaluate the role of a short reducing course of oral corticosteroid therapy in the treatment of chronic prostatitis/chronic pelvic pain syndrome. BJU Int. 2007;99(2):355–9.
Nickel JC, Atkinson G, Krieger JN, Mills IW, Pontari M, Shoskes DA, et al. Preliminary assessment of safety and efficacy in proof-of-concept, randomized clinical trial of tanezumab for chronic prostatitis/chronic pelvic pain syndrome. Urology. 2012;80(5):1105–10.
Nickel JC. Opioids for chronic prostatitis and interstitial cystitis: lessons learned from the 11th World Congress on Pain. Urology. 2006;68(4):697–701.
Kaplan SA, Volpe MA, Te AE. A prospective, 1-year trial using saw palmetto versus finasteride in the treatment of category III prostatitis/chronic pelvic pain syndrome. J Urol. 2004;171(1):284–8.
Nickel JC, Downey J, Ardern D, Clark J, Nickel K. Failure of a monotherapy strategy for difficult chronic prostatitis/chronic pelvic pain syndrome. J Urol. 2004;172(2):551–4.
Nickel JC, Roehrborn C, Montorsi F, Wilson TH, Rittmaster RS. Dutasteride reduces prostatitis symptoms compared with placebo in men enrolled in the REDUCE study. J Urol. 2011;186(4):1313–8.
Persson BE, Ronquist G, Ekblom M. Ameliorative effect of allopurinol on nonbacterial prostatitis: a parallel double-blind controlled study. J Urol. 1996;155(3):961–4.
McNaughton CO, Wilt T. Allopurinol for chronic prostatitis. Cochrane Database Syst Rev. 2002;(4):CD001041.
Ziaee AM, Akhavizadegan H, Karbakhsh M. Effect of allopurinol in chronic nonbacterial prostatitis: a double blind randomized clinical trial. Int Braz J Urol. 2006;32(2):181–6.
Elist J. Effects of pollen extract preparation Prostat/Poltit on lower urinary tract symptoms in patients with chronic nonbacterial prostatitis/chronic pelvic pain syndrome: a randomized, double-blind, placebo-controlled study. Urology. 2006;67(1):60–3.
Wagenlehner FM, Schneider H, Ludwig M, Schnitker J, Brahler E, Weidner W. A pollen extract (Cernilton) in patients with inflammatory chronic prostatitis-chronic pelvic pain syndrome: a multicentre, randomised, prospective, double-blind, placebo-controlled phase 3 study. Eur Urol. 2009;56(3):544–51.
Shoskes DA, Zeitlin SI, Shahed A, Rajfer J. Quercetin in men with category III chronic prostatitis: a preliminary prospective, double-blind, placebo-controlled trial. Urology. 1999;54(6):960–3.
Nickel JC, Forrest JB, Tomera K, Hernandez-Graulau J, Moon TD, Schaeffer AJ, et al. Pentosan polysulfate sodium therapy for men with chronic pelvic pain syndrome: a multicenter, randomized, placebo controlled study. J Urol. 2005;173(4):1252–5.
Aboumarzouk OM, Nelson RL. Pregabalin for chronic prostatitis. Cochrane Database Syst Rev. 2012;(8):CD009063.
Pontari MA, Krieger JN, Litwin MS, White PC, Anderson RU, McNaughton-Collins M, et al. Pregabalin for the treatment of men with chronic prostatitis/chronic pelvic pain syndrome: a randomized controlled trial. Arch Intern Med. 2010;170(17):1586–93.
Gottsch HP, Yang CC, Berger RE. A pilot study of botulinum toxin a for male chronic pelvic pain syndrome. Scand J Urol Nephrol. 2011;45(1):72–6.
Zermann D, Ishigooka M, Schubert J, Schmidt RA. Perisphincteric injection of botulinum toxin type A. A treatment option for patients with chronic prostatic pain? Eur Urol. 2000;38(4):393–9.
Rowe E, Smith C, Laverick L, Elkabir J, Witherow RO, Patel A. A prospective, randomized, placebo controlled, double-blind study of pelvic electromagnetic therapy for the treatment of chronic pelvic pain syndrome with 1 year of followup. J Urol. 2005;173(6):2044–7.
Kastner C, Hochreiter W, Huidobro C, Cabezas J, Miller P. Cooled transurethral microwave thermotherapy for intractable chronic prostatitis--results of a pilot study after 1 year. Urology. 2004;64(6):1149–54.
Montorsi F, Guazzoni G, Bergamaschi F, Galli L, Consonni P, Matozzo V, et al. Is there a role for transrectal microwave hyperthermia of the prostate in the treatment of abacterial prostatitis and prostatodynia? Prostate. 1993;22(2):139–46.
Zimmermann R, Cumpanas A, Miclea F, Janetschek G. Extracorporeal shock wave therapy for the treatment of chronic pelvic pain syndrome in males: a randomised, double-blind, placebo-controlled study. Eur Urol. 2009;56(3):418–24.
Lee SH, Lee BC. Electroacupuncture relieves pain in men with chronic prostatitis/chronic pelvic pain syndrome: three-arm randomized trial. Urology. 2009;73(5):1036–41.
Kabay S, Kabay SC, Yucel M, Ozden H. Efficiency of posterior tibial nerve stimulation in category IIIB chronic prostatitis/chronic pelvic pain: a sham-controlled comparative study. Urol Int. 2009;83(1):33–8.
Fitzgerald MP, Anderson RU, Potts J, Payne CK, Peters KM, Clemens JQ, et al. Randomized multicenter feasibility trial of myofascial physical therapy for the treatment of urological chronic pelvic pain syndromes. J Urol. 2013;189(1 Suppl):S75–85.
Leskinen MJ, Kilponen A, Lukkarinen O, Tammela TL. Transurethral needle ablation for the treatment of chronic pelvic pain syndrome (category III prostatitis): a randomized, sham-controlled study. Urology. 2002;60(2):300–4.
Aaltomaa S, Ala-Opas M. The effect of transurethral needle ablation on symptoms of chronic pelvic pain syndrome--a pilot study. Scand J Urol Nephrol. 2001;35(2):127–31.
Nickel JC, Alexander RB, Anderson R, Berger R, Comiter CV, Datta NS, et al. Category III chronic prostatitis/chronic pelvic pain syndrome: insights from the National Institutes of Health Chronic Prostatitis Collaborative Research Network studies. Curr Urol Rep. 2008;9(4):320–7.
Tripp DA, Nickel JC, Katz L. A feasibility trial of a cognitive-behavioural symptom management program for chronic pelvic pain for men with refractory chronic prostatitis/chronic pelvic pain syndrome. Can Urol Assoc J. 2011;5(5):328–32.
Badenoch AW. Chronic interstitial cystitis. Br J Urol. 1971;43(6):718–21.
Pool TL. Interstitial cystitis: clinical considerations and treatment. Clin Obstet Gynecol. 1967;10(1):185–91.
Soucy F, Gregoire M. Efficacy of prednisone for severe refractory ulcerative interstitial cystitis. J Urol. 2005;173(3):841–3. discussion 3
Theoharides TC. Hydroxyzine in the treatment of interstitial cystitis. Urol Clin North Am. 1994;21(1):113–9.
Seshadri P, Emerson L, Morales A. Cimetidine in the treatment of interstitial cystitis. Urology. 1994;44(4):614–6.
Theoharides TC. Hydroxyzine for interstitial cystitis. J Allergy Clin Immunol. 1993;91(2):686–7.
Warren JW, Horne LM, Hebel JR, Marvel RP, Keay SK, Chai TC. Pilot study of sequential oral antibiotics for the treatment of interstitial cystitis. J Urol. 2000;163(6):1685–8.
Forsell T, Ruutu M, Isoniemi H, Ahonen J, Alfthan O. Cyclosporine in severe interstitial cystitis. J Urol. 1996;155(5):1591–3.
Moran PA, Dwyer PL, Carey MP, Maher CF, Radford NJ. Oral methotrexate in the management of refractory interstitial cystitis. Aust N Z J Obstet Gynaecol. 1999;39(4):468–71.
Sairanen J, Forsell T, Ruutu M. Long-term outcome of patients with interstitial cystitis treated with low dose cyclosporine a. J Urol. 2004;171(6 Pt 1):2138–41.
Sairanen J, Tammela TL, Leppilahti M, Multanen M, Paananen I, Lehtoranta K, et al. Cyclosporine A and pentosan polysulfate sodium for the treatment of interstitial cystitis: a randomized comparative study. J Urol. 2005;174(6):2235–8.
Hansen HC. Interstitial cystitis and the potential role of gabapentin. South Med J. 2000;93(2):238–42.
Sasaki K, Smith CP, Chuang YC, Lee JY, Kim JC, Chancellor MB. Oral gabapentin (neurontin) treatment of refractory genitourinary tract pain. Tech Urol. 2001;7(1):47–9.
Sonnett TE, Setter SM, Campbell RK. Pregabalin for the treatment of painful neuropathy. Expert Rev Neurother. 2006;6(11):1629–35.
Ueda T, Tamaki M, Ogawa O, Yamauchi T, Yoshimura N. Improvement of interstitial cystitis symptoms and problems that developed during treatment with oral IPD-1151T. J Urol. 2000;164(6):1917–20.
Katske F, Shoskes DA, Sender M, Poliakin R, Gagliano K, Rajfer J. Treatment of interstitial cystitis with a quercetin supplement. Tech Urol. 2001;7(1):44–6.
Theoharides TC, Sant GR. A pilot open label study of Cystoprotek in interstitial cystitis. Int J Immunopathol Pharmacol. 2005;18(1):183–8.
Evans RJ, Moldwin RM, Cossons N, Darekar A, Mills IW, Scholfield D. Proof of concept trial of tanezumab for the treatment of symptoms associated with interstitial cystitis. J Urol. 2011;185(5):1716–21.
Cornel EB, van Haarst EP, Schaarsberg RW, Geels J. The effect of biofeedback physical therapy in men with chronic pelvic pain syndrome type III. Eur Urol. 2005;47(5):607–11.
Hetrick DC, Glazer H, Liu YW, Turner JA, Frest M, Berger RE. Pelvic floor electromyography in men with chronic pelvic pain syndrome: a case-control study. Neurourol Urodyn. 2006;25(1):46–9.
Anderson RU, Wise D, Sawyer T, Chan C. Integration of myofascial trigger point release and paradoxical relaxation training treatment of chronic pelvic pain in men. J Urol. 2005;174(1):155–60.
Srinivasan AK, Kaye JD, Moldwin R. Myofascial dysfunction associated with chronic pelvic floor pain: management strategies. Curr Pain Headache Rep. 2007;11(5):359–64.
Strebel RT, Leippold T, Luginbuehl T, Muentener M, Praz V, Hauri D. Chronic scrotal pain syndrome: management among urologists in Switzerland. Eur Urol. 2005;47(6):812–6.
Strom KH, Levine LA. Microsurgical denervation of the spermatic cord for chronic orchialgia: long-term results from a single center. J Urol. 2008;180(3):949–53.
Heidenreich A, Olbert P, Engelmann UH. Management of chronic testalgia by microsurgical testicular denervation. Eur Urol. 2002;41(4):392–7.
Sweeney CA, Oades GM, Fraser M, Palmer M. Does surgery have a role in management of chronic intrascrotal pain? Urology. 2008;71(6):1099–102.
Granitsiotis P, Kirk D. Chronic testicular pain: an overview. Eur Urol. 2004;45(4):430–6.
Calleary JG, Masood J, Hill JT. Chronic epididymitis: is epididymectomy a valid surgical treatment? Int J Androl. 2009;32(5):468–72.
Sweeney P, Tan J, Butler MR, McDermott TE, Grainger R, Thornhill JA. Epididymectomy in the management of intrascrotal disease: a critical reappraisal. Br J Urol. 1998;81(5):753–5.
Leslie TA, Illing RO, Cranston DW, Guillebaud J. The incidence of chronic scrotal pain after vasectomy: a prospective audit. BJU Int. 2007;100(6):1330–3.
Padmore DE, Norman RW, Millard OH. Analyses of indications for and outcomes of epididymectomy. J Urol. 1996;156(1):95–6.
Fall M, Baranowski AP, Elneil S, Engeler D, Hughes J, Messelink EJ, et al. EAU guidelines on chronic pelvic pain. Eur Urol. 2010;57(1):35–48.
Nangia AK, Myles JL, Thomas AJ. Vasectomy reversal for the post-vasectomy pain syndrome: a clinical and histological evaluation. J Urol. 2000;164(6):1939–42.
Myers SA, Mershon CE, Fuchs EF. Vasectomy reversal for treatment of the post-vasectomy pain syndrome. J Urol. 1997;157(2):518–20.
Costantini E, Zucchi A, Del Zingaro M, Mearini L. Treatment of urethral syndrome: a prospective randomized study with Nd:YAG laser. Urol Int. 2006;76(2):134–8.
Baldoni F, Baldaro B, Trombini G. Psychotherapeutic perspectives in urethral syndrome. Stress Med. 1995;11(1):79–84.
Kaur H, Arunkalaivanan AS. Urethral pain syndrome and its management. Obstet Gynecol Surv. 2007;62(5):348–51. quiz 53-4
Brown J, Crawford TJ, Allen C, Hopewell S, Prentice A. Nonsteroidal anti-inflammatory drugs for pain in women with endometriosis. Cochrane Database Syst Rev. 2017;1:CD004753.
Allen C, Hopewell S, Prentice A, Gregory D. Nonsteroidal anti-inflammatory drugs for pain in women with endometriosis. Cochrane Database Syst Rev. 2009;2:CD004753.
Ness RB, Soper DE, Holley RL, Peipert J, Randall H, Sweet RL, et al. Effectiveness of inpatient and outpatient treatment strategies for women with pelvic inflammatory disease: results from the pelvic inflammatory disease evaluation and clinical health (PEACH) randomized trial. Am J Obstet Gynecol. 2002;186(5):929–37.
Corey L, Adams HG, Brown ZA, Holmes KK. Genital herpes simplex virus infections: clinical manifestations, course, and complications. Ann Intern Med. 1983;98(6):958–72.
Jarrell J, Brant R, Leung W, Taenzer P. Women’s pain experience predicts future surgery for pain associated with endometriosis. J Obstet Gynaecol Can. 2007;29(12):988–91.
Jarrell J, Mohindra R, Ross S, Taenzer P, Brant R. Laparoscopy and reported pain among patients with endometriosis. J Obstet Gynaecol Can. 2005;27(5):477–85.
Daniels J, Gray R, Hills RK, Latthe P, Buckley L, Gupta J, et al. Laparoscopic uterosacral nerve ablation for alleviating chronic pelvic pain: a randomized controlled trial. JAMA. 2009;302(9):955–61.
Margulies RU, Lewicky-Gaupp C, Fenner DE, McGuire EJ, Clemens JQ, Delancey JO. Complications requiring reoperation following vaginal mesh kit procedures for prolapse. Am J Obstet Gynecol. 2008;199(6):678. e1-4
Walid MS, Heaton RL. Laparoscopic apical mesh excision for deep dyspareunia caused by mesh banding in the vaginal apex. Arch Gynecol Obstet. 2009;280(3):347–50.
Damsted-Petersen C, Boyer SC, Pukall CF. Current perspectives in vulvodynia. Womens Health (Lond). 2009;5(4):423–36.
Lotery HE, McClure N, Galask RP. Vulvodynia. Lancet. 2004;363(9414):1058–60.
Masheb RM, Kerns RD, Lozano C, Minkin MJ, Richman S. A randomized clinical trial for women with vulvodynia: cognitive-behavioral therapy vs. supportive psychotherapy. Pain. 2009;141(1–2):31–40.
Brown SR, Watson A. Comments to 'Rubber band ligation versus excisional haemorrhoidectomy for haemorrhoids'. Tech Coloproctol. 2016;20(9):659–61.
Shanmugam V, Thaha MA, Rabindranath KS, Campbell KL, Steele RJ, Loudon MA. Rubber band ligation versus excisional haemorrhoidectomy for haemorrhoids. Cochrane Database Syst Rev. 2005;(3):CD005034.
Jayaraman S, Colquhoun PH, Malthaner RA. Stapled versus conventional surgery for hemorrhoids. Cochrane Database Syst Rev. 2006;(4):CD005393.
Nelson RL, Thomas K, Morgan J, Jones A. Non surgical therapy for anal fissure. Cochrane Database Syst Rev. 2012;(2):CD003431.
Samim M, Twigt B, Stoker L, Pronk A. Topical diltiazem cream versus botulinum toxin a for the treatment of chronic anal fissure: a double-blind randomized clinical trial. Ann Surg. 2012;255(1):18–22.
Valizadeh N, Jalaly NY, Hassanzadeh M, Kamani F, Dadvar Z, Azizi S, et al. Botulinum toxin injection versus lateral internal sphincterotomy for the treatment of chronic anal fissure: randomized prospective controlled trial. Langenbeck's Arch Surg. 2012;397(7):1093–8.
Halpert A, Dalton CB, Diamant NE, Toner BB, Hu Y, Morris CB, et al. Clinical response to tricyclic antidepressants in functional bowel disorders is not related to dosage. Am J Gastroenterol. 2005;100(3):664–71.
Ianiro G, Eusebi LH, Black CJ, Gasbarrini A, Cammarota G, Ford AC. Systematic review with meta-analysis: efficacy of faecal microbiota transplantation for the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2019;50:240.
Bohn L, Storsrud S, Tornblom H, Bengtsson U, Simren M. Self-reported food-related gastrointestinal symptoms in IBS are common and associated with more severe symptoms and reduced quality of life. Am J Gastroenterol. 2013;108(5):634–41.
Johannesson E, Simren M, Strid H, Bajor A, Sadik R. Physical activity improves symptoms in irritable bowel syndrome: a randomized controlled trial. Am J Gastroenterol. 2011;106(5):915–22.
Johannesson E, Ringstrom G, Abrahamsson H, Sadik R. Intervention to increase physical activity in irritable bowel syndrome shows long-term positive effects. World J Gastroenterol. 2015;21(2):600–8.
Shahabi L, Naliboff BD, Shapiro D. Self-regulation evaluation of therapeutic yoga and walking for patients with irritable bowel syndrome: a pilot study. Psychol Health Med. 2016;21(2):176–88.
Camilleri M. Management options for irritable bowel syndrome. Mayo Clin Proc. 2018;93(12):1858–72.
Ford AC, Talley NJ, Spiegel BM, Foxx-Orenstein AE, Schiller L, Quigley EM, et al. Effect of fibre, antispasmodics, and peppermint oil in the treatment of irritable bowel syndrome: systematic review and meta-analysis. BMJ. 2008;337:a2313.
Tack J, Fried M, Houghton LA, Spicak J, Fisher G. Systematic review: the efficacy of treatments for irritable bowel syndrome—a European perspective. Aliment Pharmacol Ther. 2006;24(2):183–205.
Khanna R, MacDonald JK, Levesque BG. Peppermint oil for the treatment of irritable bowel syndrome: a systematic review and meta-analysis. J Clin Gastroenterol. 2014;48(6):505–12.
Brennan BP, Fogarty KV, Roberts JL, Reynolds KA, Pope HG Jr, Hudson JI. Duloxetine in the treatment of irritable bowel syndrome: an open-label pilot study. Hum Psychopharmacol. 2009;24(5):423–8.
Kaplan A, Franzen MD, Nickell PV, Ransom D, Lebovitz PJ. An open-label trial of duloxetine in patients with irritable bowel syndrome and comorbid generalized anxiety disorder. Int J Psychiatry Clin Pract. 2014;18(1):11–5.
Lewis-Fernandez R, Lam P, Lucak S, Galfalvy H, Jackson E, Fried J, et al. An open-label pilot study of duloxetine in patients with irritable bowel syndrome and comorbid major depressive disorder. J Clin Psychopharmacol. 2016;36(6):710–5.
Camilleri M, Lembo A, Katzka DA. Opioids in gastroenterology: treating adverse effects and creating therapeutic benefits. Clin Gastroenterol Hepatol. 2017;15(9):1338–49.
Ragnarsson G, Bodemar G. Treatment of irritable bowel syndrome with loperamide oxide. An open study to determine optimal dosage. J Intern Med. 2000;248(2):165–6.
Amarenco G, Kerdraon J, Bouju P, Le Budet C, Cocquen AL, Bosc S, et al. Treatments of perineal neuralgia caused by involvement of the pudendal nerve. Rev Neurol (Paris). 1997;153(5):331–4.
Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol. 2006;108(4):915–23.
Hibner M, Desai N, Robertson LJ, Nour M. Pudendal neuralgia. J Minim Invasive Gynecol. 2010;17(2):148–53.
Choi SS, Lee PB, Kim YC, Kim HJ, Lee SC. C-arm-guided pudendal nerve block: a new technique. Int J Clin Pract. 2006;60(5):553–6.
Khoder W, Hale D. Pudendal neuralgia. Obstet Gynecol Clin N Am. 2014;41(3):443–52.
Beco J, Climov D, Bex M. Pudendal nerve decompression in perineology: a case series. BMC Surg. 2004;4:15.
Shafik A. Pudendal canal syndrome: a cause of chronic pelvic pain. Urology. 2002;60(1):199.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Crupi, R., Cordaro, M., Cuzzocrea, S. (2021). Neuroinflammation and Chronic Pelvic Pain Syndrome. In: Giammò, A., Biroli, A. (eds) Chronic Pelvic Pain and Pelvic Dysfunctions. Urodynamics, Neurourology and Pelvic Floor Dysfunctions. Springer, Cham. https://doi.org/10.1007/978-3-030-56387-5_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-56387-5_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-56386-8
Online ISBN: 978-3-030-56387-5
eBook Packages: MedicineMedicine (R0)