
Abstract
Acquired autoantibodies targeting coagulation factors are fortunately rare. However, unfamiliarity with them can lead to misinterpretation of clinical presentations and screening laboratory tests, erroneous or delayed diagnoses, and inappropriate management decisions which may result in severe morbidity or mortality. Using acquired neutralizing antibodies to factor VIII (acquired hemophilia A) as a model for clinical presentation, laboratory diagnosis, management of bleeding, and immunosuppression treatment, this chapter reviews our current knowledge of acquired inhibitors of the coagulation system.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Presentation
Patients with clinically important coagulation factor inhibitors usually present with excessive hemorrhage and bruising exceeding the patient’s typical spontaneous or traumatic bleeding patterns. The symptoms may be subtle and localized at first, gradually exacerbating to life-threatening hemorrhage from multiple sites, or the first bleeding complication may compel the patient to seek medical attention. Healthcare providers may search for, identify, and treat an anatomic cause for bleeding, often via invasive procedures, before considering the possibility of a systemic bleeding condition or reviewing screening coagulation test results. For example, gross hematuria or rectal bleeding may lead to detection of a bleeding tumor. However, to biopsy or resect the tumor in the patient with an acquired factor deficiency can result in uncontrolled bleeding. While at least one screening coagulation test will be abnormal in almost all patients with a clinically important inhibitor, there are some exceptions. For example, an acquired factor XIII (FXIII, hereafter all factors are abbreviated to F) deficiency will not prolong aPTT or PT, but would make a patient vulnerable to spontaneous and post-invasive procedure bleeding. Finally, some patients with neutralizing coagulation factor autoantibodies can be asymptomatic, and the first clue is an unexplained prolonged aPTT, PT, or both.
Laboratory Detection and Confirmation of Coagulation Factor Autoantibodies
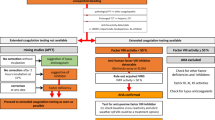
Bleeding complications are unlikely unless the reduction of the target factor activity is low enough to prolong screening clotting tests, aPTT, PT, or both, depending upon the inhibited factor. Figure 10.1 shows the coagulation cascade and the factors included in PT and aPTT screening tests. Figure 10.2 provides an algorithmic approach to evaluate a patient suspected of having an acquired inhibitor. Indirect evidence of a neutralizing autoantibody is incomplete correction of a 50:50 mixing study performed by repeating the prolonged screening clotting test using a mixture of equal volumes of patient plasma and normal pooled plasma (NPP). Acquired FVIII inhibitors are notorious for delayed inhibition of FVIII molecules in NPP [1]. An aPTT performed immediately after mixing plasma containing a FVIII inhibiting antibody and NPP may show considerable, or even complete, correction which could be interpreted as a simple factor deficiency. Therefore, it is a standard laboratory practice when evaluating an unexplained prolonged aPTT (and in some laboratories, PT as well) to perform both an immediate 50:50 aPTT and a second aPTT after incubating the 50:50 mixture of patient plasma and NPP for at least 60 min at 37 °C in order to observe the maximal inhibitory effect and prolongation of the aPTT. However, a delayed in vitro inhibition pattern in an aPTT 50:50 mixing study is not specific for a FVIII inhibitor or an acquired inhibitor of one of the other coagulation factors in the intrinsic pathway since a lupus anticoagulant autoantibody may occasionally mimic this pattern [2]. When the 50:50 mix results show incomplete correction, the next step is to perform additional coagulation testing (Fig. 10.2).

Model of the classic coagulation cascade and screening coagulation tests. aPTT reagent activates FXII to initiate the intrinsic pathway. PT reagent activates FVII to initiate the extrinsic pathway. Factor deficiencies or inhibitors of the intrinsic pathway prolong the aPTT and of the extrinsic pathway prolong the PT. Factor deficiencies or inhibitors of the common pathway prolong both the aPTT and PT. The exception is FXIII deficiency or inhibition, which will not prolong the aPTT or PT. From Lefkowitz JB. Coagulation pathway and physiology (From Kottke-Marchant [41]. Reproduced with permission)

A diagnostic algorithm for laboratory evaluation of a suspected acquired bleeding disorder
A specific neutralizing antibody will result in a moderate-to-severe in vitro deficiency of its target factor. However, neighboring factor assays may show inhibition due to partial interference of the specific inhibitor in these assays (Table 10.1). Having confirmed the specificity of the inhibitory antibody, its potency is measured by performing either a Bethesda or Nijmegen inhibiter titer assay [1]. Serial dilutions of the patient’s plasma are mixed with NPP (50:50 mix) and incubated for 2 h at 37 °C followed by measurement of the residual activity of the inhibited factor. Results are expressed as the reciprocal of the patient’s plasma dilution which neutralized 50% of the target factor activity in the diluted NPP. This method was originally developed to measure FVIII inhibitors in hemophilia A patients who developed alloantibodies to infused FVIII. The in vitro behavior of FVIII alloantibodies typically shows simple, irreversible inhibition of FVIII, and interpretation of the inhibitor assay results is straightforward, while acquired FVIII autoantibodies may show a more complex pattern of inhibition [1]. The Bethesda or Nijmegen assay can be adapted to measure the potency of other coagulation factor inhibitors by measuring the recovery of the factor of interest.
Acquired Hemophilia A
The most common coagulation factor target for neutralizing autoantibodies is FVIII with an estimated incidence of 1.4/million [3]. The results of several large prospective registries or population-based cohorts show similar findings for clinical presentations [3,4,5,6] (Table 10.2). Affected patients are typically elderly, slightly biased toward men. About half of acquired FVIII inhibitor patients have an underlying autoimmune disorder, malignancy, or other condition (suspected medication [7], infection, dermatologic disease), which may be causally related to inhibitor development, while the other 50% of patients’ inhibitors are idiopathic. One unique FVIII inhibitor population is post-partum women representing 2–3% of all cases. The median onset of bleeding is 2.5 months post-partum. They have excellent responses to hemostatic and immunosuppression treatment with >85% complete remissions and much lower mortality rates compared to older patients [8].
Symptoms at Presentation
Patients typically present with new onset of spontaneous bruising and bleeding at one or more sites, while intracranial and joint hemorrhages are uncommon. Bleeding can be spontaneous, post-traumatic, or post-invasive procedures and is usually severe. However, up to a third of patients are asymptomatic when an inhibitor is discovered based on an incidentally prolonged aPTT [3, 4].
Laboratory Findings
FVIII activities range from <1 to ~50% and most inhibitor results are >10 units (Table 10.2). Other coagulation factor activities based on aPTT testing may show varying degrees of inhibition due to the partial neutralization of the factor VIII during the 3–5 min incubation stage of an aPTT-based FXII, FXI, or FIX (Table 10.1). The inhibitor behavior is diminished when the factor assay is performed on serial dilutions of the patients’ plasma. Patients with FVIII inhibitors may also have positive lupus anticoagulant (LA) results using an aPTT-based method [9]. While it is possible for a patient to have both a non-specific LA and a specific FVIII inhibitor, most often the positive LA is an artifact due to interference from the factor VIII neutralizing antibody rather than evidence for an additional autoantibody since the DRVVT-based LA test, which activates FX and FV of the common pathway (Fig. 10.1), is negative in most patients with FVIII inhibitors.
Immediate Management: Stop Severe Bleeding
Neither the severity of FVIII deficiency nor the potency of the inhibitor predicts bleeding severity [3]. Most symptomatic acquired FVIII inhibitor patients require infusions of hemostatic products to stop acute hemorrhaging (Table 10.3). Rarely, weak inhibitors (inhibitor titer <5 unit) may be overwhelmed with larger doses of FVIII concentrate or stimulation of endogenous FVIII release by vasopressin analog DDAVP (Desmopressin™). An effective alternate strategy to generate sufficient fibrin clot to stop bleeding is to bypass the inhibited function of FVIII by infusing recombinant factor VIIa (rFVIIa) or an anti-inhibitor plasma concentrate containing FVIIa and mainly nonactivated FII, FIX, and FX: FVIII inhibitor bypass activity (FEIBA) [10]. While no study has directly compared these two approaches to stop bleeding, findings from case series and registries show they are very similar in efficacy and safety [5, 10]. For both bypass therapies, effectiveness is based on clinical monitoring of bleeding and not based on correction of in vitro coagulation tests. Most FVIII autoantibodies do not avidly inhibit porcine FVIII, at least upon initial exposure, and porcine FVIII is a suitable substitute for human FVIII in patients with inhibitors [11]. However, concern for transmission of porcine parvovirus leads to its discontinuation around 2000. In 2015, the US FDA approved recombinant porcine FVIII (rPFVIII) for the treatment of life-threatening bleeding in patients with acquired FVIII inhibitors [12]. Unlike rFVIIa and FEIBA, rPFVIII can be monitored by FVIII activity testing to permit dose adjustments to obtain desired peak and trough levels in addition to clinical monitoring of bleeding sites. These three hemostatic products are expensive and could be associated with thrombotic events in elderly patients. Therefore, the goal of hemostatic treatment is to stop life-threatening bleeding. Persistent minor bleeding and bruising is not an indication for continued bypass therapy. FVIII acquired inhibitor patients experiencing mucosal bleeding may also benefit from antifibrinolytic treatment with tranexamic acid or epsilon aminocaproic acid. In the setting of life-threatening bleeding not responding to hemostatic therapies, extracorporeal immunoabsorption [13], which in not available in the United States, and plasma exchange [14] may be effective interventions to reduce inhibitor titers. In 2019, the US FDA approved emicizumab, a bispecific antibody which binds FIXa and FX, for subcutaneous injection to prevent bleeding in hemophilia A patients with or without inhibitors [15]. One can anticipate clinical studies will be performed to evaluate emicizumab treatment for bleeding complications in non-hemophiliacs with acquired FVIII inhibitors.
Concurrent Management: Immunosuppressant Therapy to Eliminate Factor VIII Inhibitor
While acquired FVIII inhibitors occasionally spontaneously remit, immunosuppression therapy should be started once laboratory testing confirms the diagnosis to reduce the risk of future bleeding complications. Clinical remission (CR) is defined as FVIII activity >50–70% and no inhibitor activity detected in plasma following withdrawal of immunosuppression treatment. Experts agree all patients should initially receive prednisone (typically 1 mg/kg/day). But there is no consensus on whether cytotoxic immunosuppression with oral cyclophosphamide 1–2 mg/day should be started with prednisone or reserved for patients who do not respond to several weeks of prednisone monotherapy [4, 5, 16].
Due to the rarity of acquired FVIII inhibitors, an adequately powered prospective randomized trial comparing first-line prednisone and cyclophosphamide to prednisone is not feasible. Comparing outcomes for different treatment regimens from registry databases may be skewed by confounding variables and clinician biases as well. However, there are several consistent observations: (1) Initial prednisone + cyclophosphamide produces inhibitor remission faster than prednisone alone [16], but does not affect long-term outcomes of clinical remission (CR) and survival [3, 16]. (2) Intravenous immunoglobulin (IVIG) is not effective as a monotherapy or in combination with prednisone and cyclophosphamide [3, 16]. (3) Rituximab is not an effective first-line monotherapy [16]. (4) Patients with initial severe FVIII deficiency and high inhibitor titers have lower CR rates and poorer survival [5, 16, 17]. (5) Relapses do occur, ranging from 15% to 24% during the first year of follow-up [3, 16]. Therefore, initial immunosuppression treatment decisions require assessing the likely risks and benefits of the alternative approaches specific to each patient. If prednisone monotherapy is chosen, median time to CR is about 5 weeks [17].
For the minority of patients who do not respond to prednisone or prednisone + cyclophosphamide, there are alternative immunosuppression therapies to consider including cyclosporine, azathioprine, and mycophenolate [18]. Rituximab has grown in popularity as a second-line treatment [19].
Acquired Inhibitors of Other Coagulation Factors
FVIII is by far the most immunogenic coagulation factor and yet the incidence of FVIII inhibitors is <2.0/million/year. Information on the presentation, management, and outcomes for acquired inhibitors to other coagulation factors is limited to sporadic case reports or small case series, which makes it difficult to provide definitive descriptions of their presentations and prognoses, or recommendations for management [20, 21]. In addition, reporting bias may affect the accuracy of the information. However, experience gained from diagnosis and treatment of acquired hemophilia A patients can generally be applied to these much rarer inhibitors. Since screening coagulation test results are a crucial first step toward diagnosis of an acquired inhibitor, it is appropriate to organize a review around the different patterns which can occur (Fig. 10.1).
Acquired Inhibitors Other Than Factor VIII Which Prolong the aPTT: Factor IX, Factor XI, and Factor XII
Information about inhibitory autoantibodies to FIX (acquired hemophilia B) consists of a handful of case reports. Bleeding sites, association with autoimmune disorders or malignancies, response to rFVIIa or FEIBA to control bleeding, and immunosuppressive therapy to eliminate the inhibitor are similar to findings from acquired FVIII case series [22]. The aPTT is prolonged and only partially corrects with immediate 50:50 mixing, and there is no progressive inhibition with prolonged incubation as is typically seen with FVIII inhibitors. The FIX inhibitor potency can be measured with the Bethesda or Nijmegen method technique by measuring residual FIX activity with shorter incubation times [1].
To date, most, but not all, reports of acquired FXI inhibitory antibodies have occurred in patient with systemic lupus erythematosus [20]. In a review of 14 cases 9 presented with spontaneous or trauma-induced bleeding and 4 with thrombotic events, and 1 was asymptomatic. Immunosuppression therapy resolved all inhibitors [23].
Reports of acquired inhibitors to FXII usually are in the context of antiphospholipid syndrome or liver disease [20]. Since congenital FXII deficiency does not cause excessive bleeding, it is not surprising that case reports of acquired FXII deficiency have not been associated with bleeding either.
Acquired Inhibitors Which Prolong the PT: Factor VII
While inherited FVII deficiency is estimated to have a prevalence of 1 per 500,000, acquired, isolated FVII deficiencies are extremely rare and associated with underlying malignancies, infections, and hematopoietic stem cell transplants; and inhibitory antibodies are identified in a minority of cases [24, 25]. Bleeding complications range from mild to severe [26]. One patient’s plasma demonstrated a time-dependent in vitro inhibition of FVII in NPP, similar to acquired FVIII inhibitors [27]. Given the rarity of acquired FVII inhibitors, management of immune suppression strategy would be empiric, borrowing from the experience with acquired FVIII inhibitors.
Bleeding complications have been managed with fFVIIa, fresh frozen plasma, and prothrombin complex concentrates [25]. However, there is insufficient evidence to determine if there is superior treatment.
Acquired Inhibitors Which Prolong Both aPTT and PT: Factor V, Factor X, Factor II, and Fibrinogen
Historically, factor V (FV) was a relatively frequent target of acquired inhibitory autoantibodies, due to iatrogenic exposure to bovine FV. An effective surgical hemostasis technique was to apply an aerosol mixture of bovine thrombin and fibrinogen to produce a fibrin sealant on a diffusely oozing surface. However, bovine thrombin contains bovine FV, and some patients would produce neutralizing antibodies which would cross-react with human FV. The presentation could be delayed, post-operative prolongation of PT and aPTT with or without bleeding complications [28]. Some patients suffered major complications or death. Fortunately, the incidence of acquired FV inhibitors has dramatically declined following replacement of bovine thrombin with either human plasma-derived or recombinant human thrombin. Acquired FV inhibitors are associated with additional triggering factors including surgery without exposure to thrombin glue; a wide range of antibiotics, including beta lactams; malignancies; autoimmune disorders; and infections [29]. Laboratory findings include a prolonged aPTT and PT, which do not completely correct with 50:50 mixing and do not demonstrate time-dependent neutralization; selected deficiency of FV; and an inhibitor based on a Bethesda or Nijmegen methods [30]. In addition to the interventions used to stop bleeding in acquired hemophilia A patients, platelet transfusions are probably effective in patients with FV inhibitors by delivering FV released from activated platelet alpha granules at the site of vascular injury. Frequently, FV inhibitors spontaneously remit, especially when a suspected antibiotic is discontinued, and not all patients required immunosuppression [29].
Acquired factor X (FX) deficiency is most commonly due to absorption of the coagulation protein to amyloid deposits in patients with amyloidosis [31]. Management of bleeding due to acquired FX deficiency associated with amyloidosis includes infusions of prothrombin complex concentrates [32] and plasma-derived FX concentrate [33], which is approved for the treatment of congenital FX deficiency (Coagadex™).
Neutralizing FX autoantibodies are extremely rare [34]. A literature review identified 34 case reports, but only 26% provided convincing laboratory evidence of a FX inhibitor [35]. Cases were associated with malignancies and infectious and inflammatory diseases or considered idiopathic. Interestingly, 38% of cases were preceded by a non-specific respiratory viral illness. Bleeding complications varied from none to life-threatening with multiple interventions to stop bleeding. All patients survived and their inhibitors resolved including 16% with spontaneous remissions.
Prothrombin (FII) autoantibodies are a common laboratory finding in patients with lupus anticoagulants, and they have been associated with increased thrombosis risk. They are typically non-neutralizing and do not affect aPTT or PT, but if antibody-mediated clearance of prothrombin is greatly enhanced, patients may experience bleeding complications with laboratory findings of prolonged aPTT and PT and very low factor II activity (see Chapter 23 Lupus anticoagulant hypoprothrombinemia syndrome).
Acquired inhibitors of thrombin are very rare and, when described, are usually associated with exposure to bovine thrombin glue and less often with underlying autoimmune disorders or monoclonal gammopathies [20]. In addition to prolonging aPTT and PT, thrombin inhibitors prolong the thrombin time (TT), which is performed by adding purified thrombin to patient plasma and monitoring the time to fibrin clot formation. A TT mixing study would show incomplete correction. Management of bleeding complications due to a thrombin inhibitor is empiric, but plasma exchange may be an important therapeutic intervention to reduce antibody potency acutely since bypass coagulation products may not be as effective as they are for “upstream” factor inhibitors.
Hypofibrinogenemia due to dilution or consumption is common, but acquired inhibitors of fibrinogen are very rare. They may interfere with fibrin monomer formation or fibrin polymerization. Bleeding symptoms may be as severe as seen in patients with congenital afibrinogenemia. Typical laboratory findings include prolonged aPTT, PT, and TT, which do not correct with 50:50 mixing. A reptilase time would distinguish a fibrinogen inhibitor from a thrombin inhibitory autoantibody. Reptilase time uses the batroxobin snake venom to activate fibrinogen instead of thrombin, and it would be prolonged in the presence of a fibrinogen inhibitor but not a thrombin inhibitor [36].
Acquired Inhibitors Which Do Not Prolong aPTT or PT: Factor XIII
Both inherited and acquired severe deficiencies of FXIII are extremely rare. Presenting symptoms include spontaneous bleeding, including intracranial hemorrhage, delayed bleeding after trauma, and impaired wound healing. FXIII circulates as a pair of alpha and beta chain heterodimers. Thrombin activates the alpha chain, which stabilizes fibrin monomers via formation of multiple covalent cross-linking bonds (Fig. 10.1). Clot-based coagulation methods such as PT and aPTT detect initial polymerization of fibrin and are insensitive to FXIII deficiency. Therefore, testing for acquired FXIII inhibitor is appropriate in a patient who presents with an abrupt onset of severe unprovoked hemorrhage, normal platelet number and function, and normal PT and aPTT, fibrinogen, and thrombin time. Many laboratories screen for FXIII deficiency with the urea clot solubility test. A fibrin clot is produced from a patient’s plasma by adding thrombin. Then the visible clot is placed in a 5 M solution of urea. If fibrin is not cross-linked by FXIIIa, it will dissolve in the urea. However qualitative assays are insensitive and may yield false negative results for a clinically important FXIII deficiency. The preferred initial screening test is a sensitive, qualitative assay for FXIIIa transglutaminase activity [37, 38]. Confirmatory studies to demonstrate inhibitor activity and to measure FXIII antigen concentration are provided by a few reference laboratories. Notable findings from a systematic literature review of 28 cases of FXIII inhibitors included as follows: median age 65.5; 70% associated with a medication, specifically isoniazid in 30%; and 30% were idiopathic [39]. There were five fatal intracranial bleeds and overall mortality rate was 29%. Various strategies were employed to stop bleeding (FXIII concentrate, cryoprecipitate, FFP, plasma exchange) and suppress inhibitors (steroids, cyclophosphamide, rituximab, IVIG). A majority of patients obtained a remission, of which 25% were spontaneous after withdrawing a suspected medication.
Summary
Coagulation factor inhibitory antibodies are fortunately uncommon. However, when patients present with abnormal bleeding and bruising due to acquired inhibitors, their rarity can lead to incorrect or delayed diagnoses, greater morbidity, and fatalities. The first step is to rapidly perform laboratory investigations similar to the algorithm outlined in Fig. 10.2. Once an inhibitor is identified, interventions to restore hemostasis and initiate immunosuppression are indicated for most patients (Table 10.3). The majority of inhibitor management experience is derived from patients with acquired hemophilia A, which may be empirically applied to most other acquired inhibitors. A search for an underlying cause should be done concurrently with hemostatic treatment, but about 50% of acquired coagulation inhibitors are idiopathic. If diagnosed promptly and treated effectively, most patients will achieve a clinical remission. However, since many patients with acquired inhibitors are elderly, deaths from bleeding, treatment-related sepsis, and other comorbidities are fairly common [40].
Abbreviations
- aPTT:
-
Activated partial thromboplastin time
- FEIBA:
-
Factor eight inhibitor bypass activity
- INR:
-
International normalized ratio
- LA:
-
Lupus anticoagulant
- NPP:
-
Normal pooled plasma
- PT:
-
Prothrombin time
- rFVIIa:
-
Recombinant factor VIIa
- TT:
-
Thrombin time
References
Adcock DM, Favaloro EJ. Pearls and pitfalls in factor inhibitor assays. Int J Lab Hematol. 2015;37(Suppl 1):52–60.
Clyne LP, White PF. Time dependency of lupuslike anticoagulants. Arch Intern Med. 1988;148(5):1060–3.
Collins PW, et al. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007;109(5):1870–7.
Knoebl P, et al. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012;10(4):622–31.
Tiede A, et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): results from the GTH-AH 01/2010 study. Blood. 2015;125(7):1091–7.
Borg JY, et al. Outcome of acquired haemophilia in France: the prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hemophilie Acquise) registry. Haemophilia. 2013;19(4):564–70.
Franchini M, et al. Drug-induced anti-factor VIII antibodies: a systematic review. Med Sci Monit. 2007;13(4):RA55–61.
Tengborn L, et al. Pregnancy-associated acquired haemophilia A: results from the European Acquired Haemophilia (EACH2) registry. BJOG. 2012;119(12):1529–37.
Ames PR, et al. Prolonged activated partial thromboplastin time: difficulties in discriminating coexistent factor VIII inhibitor and lupus anticoagulant. Clin Appl Thromb Hemost. 2015;21(2):149–54.
Baudo F, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry. Blood. 2012;120(1):39–46.
Morrison AE, Ludlam CA, Kessler C. Use of porcine factor VIII in the treatment of patients with acquired hemophilia. Blood. 1993;81(6):1513–20.
Kruse-Jarres R, et al. Efficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Haemophilia. 2015;21(2):162–70.
Jansen M, et al. Treatment of coagulation inhibitors with extracorporeal immunoadsorption (Ig-Therasorb). Br J Haematol. 2001;112(1):91–7.
Losos M, et al. The tipping point: the critical role of therapeutic apheresis in a case of refractory acquired hemophilia. J Clin Apher. 2017;32(6):564–6.
Mahlangu J, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. 2018;379(9):811–22.
Collins P, et al. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood. 2012;120(1):47–55.
Collins PW. Therapeutic challenges in acquired factor VIII deficiency. Hematology Am Soc of Hematol Educ Program. 2012;2012:369–74.
Eisert S, et al. Successful use of mycophenolate mofetil and prednisone in a 14-year-old girl with acquired hemophilia A. Thromb Haemost. 2005;93(4):792–3.
Franchini M, et al. Acquired inhibitors of clotting factors: AICE recommendations for diagnosis and management. Blood Transfusion Trasfusione del sangue. 2015. p. 1–16.
Hay CR, editor. Hemostasis and thrombosis basic principles and clinical practice. In: Acquired disorders of coagulation: the immune coagulopathies. 6th ed. Philadelphia: Wolters Kluwer/Lippincott Wiliams & Wilkins: 2013. p. 723–37.
Franchini M, Lippi G, Favaloro EJ. Acquired inhibitors of coagulation factors: part II. Semin Thromb Hemost. 2012;38(5):447–53.
Krishnamurthy P, et al. A rare case of an acquired inhibitor to factor IX. Haemophilia. 2011;17(4):712–3.
Bortoli R, et al. Acquired factor XI inhibitor in systemic lupus erythematosus--case report and literature review. Semin Arthritis Rheum. 2009;39(1):61–5.
Girolami A, et al. Acquired isolated FVII deficiency: an underestimated and potentially important laboratory finding. Clin Appl Thromb Hemost. 2016;22(8):705–11.
Mulliez SM, Devreese KM. Isolated acquired factor VII deficiency: review of the literature. Acta Clin Belg. 2015.
Kamikubo Y, et al. Purification and characterization of factor VII inhibitor found in a patient with life threatening bleeding. Thromb Haemost. 2000;83(1):60–4.
Delmer A, et al. Life-threatening intracranial bleeding associated with the presence of an antifactor VII autoantibody. Blood. 1989;74(1):229–32.
Lippi G, et al. Inherited and acquired factor V deficiency. Blood Coagul Fibrinolysis. 2011;22(3):160–6.
Franchini M, Lippi G. Acquired factor V inhibitors: a systematic review. J Thromb Thrombolysis. 2011;31(4):449–57.
Siekanska-Cholewa A, et al. Acquired factor V inhibitor in a woman following aortic aneurysm surgery. Blood Coagul Fibrinolysis. 2014;25(5):515–7.
Mumford AD, et al. Bleeding symptoms and coagulation abnormalities in 337 patients with AL-amyloidosis. Br J Haematol. 2000;110(2):454–60.
Veneri D, et al. Use of prothrombin complex concentrate for prophylaxis of bleeding in acquired factor X deficiency associated with light-chain amyloidosis. Blood Transfus. 2016;14(6):585–6.
Mahmood S, et al. Utility of factor X concentrate for the treatment of acquired factor X deficiency in systemic light-chain amyloidosis. Blood. 2014;123(18):2899–900.
Broze GJ Jr. An acquired, calcium-dependent, factor X inhibitor. Blood Cells Mol Dis. 2014;52(2–3):116–20.
Lee G, Duan-Porter W, Metjian AD. Acquired, non-amyloid related factor X deficiency: review of the literature. Haemophilia. 2012;18(5):655–63.
Karapetian H. Reptilase time (RT). Methods Mol Biol. 2013;992:273–7.
Kohler HP, et al. Diagnosis and classification of factor XIII deficiencies. J Thromb Haemost. 2011;9(7):1404–6.
Yan MTS, et al. Acquired factor XIII deficiency: a review. Transfus Apher Sci. 2018;57(6):724–30.
Franchini M, et al. Acquired FXIII inhibitors: a systematic review. J Thromb Thrombolysis. 2013;36(1):109–14.
Godaert L, et al. Acquired hemophilia A in aged people: a systematic review of case reports and case series. Semin Hematol. 2018;55(4):197–201.
Kottke-Marchant K, editor. An algorithmic approach to hemostasis testing. Northfield, IL: College of American Pathologists; 2008.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Eby, C. (2021). Bleeding Associated with Coagulation Factor Inhibitors. In: Teruya, J. (eds) Management of Bleeding Patients. Springer, Cham. https://doi.org/10.1007/978-3-030-56338-7_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-56338-7_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-56337-0
Online ISBN: 978-3-030-56338-7
eBook Packages: MedicineMedicine (R0)