
Abstract
The intracranial compartment syndrome, which is the clinical consequence of intracranial hypertension, is a condition characterized by high morbidity and mortality. The pathogenesis of the syndrome is very complex and is based on the Monro-Kellie doctrine. In a rigid, non-compliant structure (as is the skull), even slight increases in the contained volumes (brain, blood, cerebrospinal fluid) translate into wide increases of internal pressure. Many pathologic conditions (intra- and/or extracranial) can cause an intracranial compartment syndrome and, although some compensatory mechanisms exist, those are usually ineffective when the rise in intracranial pressure is acute or hyperacute. Consequently, any therapeutic action should be based on a deep knowledge of the pathophysiology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Definition
A compartment syndrome is, by definition, a clinical syndrome characterized by a severe increase of pressure in an enclosed body district (compartment) and subsequent hypoperfusion and tissue damage due to compression of vital structures such as vessels and nerves [1]. The prerequisite for a compartment syndrome to develop is that the compartment must be non-extensible above a certain limit, i.e., its compliance must be close to zero after a critical level of stretching of its structures has been reached. Some compartments in the body, due to their physical and physiological characteristics, are more prone than others to develop a compartment syndrome.
The intracranial compartment, completely surrounded by a rigid, noncompliant bony case, can be intuitively considered the ideal environment for a compartment syndrome to occur since slight increases in volume of the enclosed structures translate into wide increases in compartment pressure. A quick rise in intracranial pressure (ICP) can displace the brainstem, which can suffer from possibly irreversible ischemia, leading to rapid death. Slower (though acute) rises in ICP can result in brain tissue compression, hypoperfusion, and diffuse ischemic damage, leading to severe and irreversible neurologic damage [2].
The intracranial compartment syndrome (ICS) has been known and studied for the last two centuries, but it has usually been referred to as intracranial hypertension (IH). Depending on different classifications, some forms of IH can develop slowly (e.g., growing cerebral tumors), others can be chronic or even benign. In this chapter, we will consider ICS a synonym of IH caused by an acute brain injury (ABI).
2.1.1 Clinical Definition
A comprehensive dissertation about the clinical aspects of IH is outside the scope of this chapter. However, in order to define IH and ICS operatively, we need to introduce the concept of ICP monitoring, which is often applied in patients with ABI. Whichever the technology adopted (e.g., intraparenchymal probe, intraventricular catheter), the measure of ICP allows the clinician to react adequately if the patient reaches a critical threshold level, which defines IH [3]. Historically, the most frequently adopted ICP cutoff to define IH has been 20 mmHg, which is derived from the seminal Lundberg study [4]. More recent research tend to confirm this cutoff, showing that an ICP above 20 mmHg correlates with mortality and poor outcomes [5, 6]. However, the latest Guidelines for the Management of Severe Traumatic Brain Injury recommend treating ICP values above 22 mmHg [7], thus implying that lower values should not be considered as IH and suggesting that below that level the patient should not be at risk of ICS. This new threshold fired the debate since many experts do not agree with such strict limits and recommend that the diagnosis of IH (and of ICS) be based not only on a mere number but on a complete clinical picture of the patient [3, 8, 9]. IH should be considered in terms of insult severity and duration possibly with the aid of multimodal neuromonitoring [3, 8, 9].
2.2 Pathomechanism of the Intracranial Compartment Syndrome
2.2.1 The Monro-Kellie Doctrine
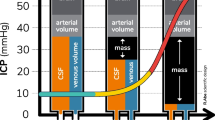
As stated above, IH leads to ICS, but what leads to IH in the first place? The basic concept of ICP is described by the Monro-Kellie Doctrine, which is based on separate studies by the Scottish physician Alexander Monroe secundus (1733–1817) and the surgeon George Kellie (1770–1829). Essentially, the doctrine states that total intracranial volume is constant and fixed although the relationships between its components may vary. Total intracranial volume is determined by the sum of the volumes of cerebrospinal fluid (CSF), blood, and brain tissue. We can roughly estimate that CSF and blood occupy each 10% of the volume, leaving to the brain tissue the remaining 80% (near 1900 mL in adults) [2].
ICP is normally below 10 mmHg, and it is relatively constant through all the brain regions. The introduction of an additional volume in one of the three components must be compensated by changes in the other two components; however, a volume increase of more than 10% leads inevitably to the upper limit of the system’s compliance and so to an increase in ICP. Since the skull is rigid, from this point on, minimal increases in intracranial volume translate into wide increases in ICP (exponential relationship, see Fig. 2.1).

Cerebral pressure–volume curve
2.2.2 Compensatory Mechanisms
As stated before, the limit of compliance of the intracranial compartment is an increase in volume of about 10%. This means that the brain is not normally able to swell more than 10% of its volume and the same is true for the CSF and the blood component. According to the Monro-Kellie Doctrine, these changes modify the total volume and must be counterbalanced by a correspondent reduction of the other components. Namely, both the CSF and the blood components are able to shift outside the intracranial compartment as explained below. Displacement of CSF and blood explains the horizontal part of the pressure–volume curve represented in Fig. 2.1. Of course, individual patients may show better compensation than others, for example, due to cerebral atrophy, which warrants higher volume reserve by shifting the pressure–volume curve to the right.
2.2.2.1 Shifts of CSF
The CSF is able to shift from the ventricular or subarachnoid space to the spinal compartment. However, the spinal compartment has limited distensibility and may be insufficient to compensate for pathologic changes in intracranial volume.
Moreover, in case of increasing intracranial volume, CSF outflow and resorption may be forced through the low-resistance arachnoid villi.
2.2.2.2 Shifts of Cerebral Blood Volume (CBV)
Most of the blood (about two thirds) in the intracranial compartment is contained in the dural sinuses and venules, while the remaining one-third is in the arteriolar system. Cerebral blood volume (CBV) in the intracranial compartment is regulated both on the arteriolar side (inflow) and on the venous side (outflow). The blood-based compensation mechanism is based on the reduction in volume of the venules and dural sinuses (up to their collapse) and on the vasoconstriction/vasodilation of the arterioles and other vessels. Although it may seem a rather ineffective mechanism, especially when compared to fast shifts of CSF between the intracranial and the intraspinal compartments, shifts in intracranial blood content are in fact ample enough to compensate for wide variations in intracranial volume: intracranial blood volume can oscillate between 15 and 70 mL, thus making it a really important compensation mechanism [2, 3].
Cerebral blood flow (CBF) in the healthy brain is strictly regulated according to the autoregulation mechanism, that is able to maintain a physiologic blood flow (~750 mL/min in a normal adult) in a wide range of mean arterial pressures (~50–150 mmHg). The mechanism is based on the continuous fine-tuning of cerebrovascular resistances, mediated by myogenic reflexes in the endothelium and by vasodilating agents released by the tissues [2].
On the contrary, in the acutely injured brain, autoregulation is often impaired (in a variable manner, according to the type of lesion and its site). Brain injury can disrupt cerebral autoregulation (the degree of the disruption is generally directly proportional to the degree of brain injury), and this translates in a pathologically linear relationship between CBF and cerebral perfusion pressure (CPP).
Other factors influencing CBF are the arterial partial pressures of oxygen (PaO2) and carbon dioxide (PaCO2). In particular, hypercapnia (and, to a lesser extent, hypoxemia) increases CBF through vasodilation and may lower the upper limit of autoregulation. Reactivity to CO2 is continuous up to partial pressures of about 80 mmHg, for which cerebral vasodilation is about 100% of the baseline value; above 80 mmHg, no further vasodilation occurs. Reactivity to hypoxemia is much less pronounced and is significant only below 50 mmHg of PaO2 [2].
Drugs may also significantly alter CBF. Most notably, barbiturates act as potent cerebral vasoconstrictors, but their effect on CBF is only partly explained by their effect on the cerebral circulation. In fact, they also reduce CBF by reducing neuronal metabolism. Inhalational anesthetics can instead induce cerebral vasodilation and increase CBF.
Other acute conditions, such as fever and seizures, are associated with CBF augmentation, which often translates in IH in patients with ABI and impaired autoregulation.
Chronic arterial hypertension significantly modifies cerebral autoregulation, resulting in a rightward shift of the pressure–CBF curve (meaning that constant CBF is maintained at higher arterial pressures).
2.2.3 Intracranial Causes of IH
Intracranial causes of IH are summarized in Table 2.1.
2.2.3.1 Brain-Related Causes of IH
The most important brain-related causes of IH are expansive processes inside the brain or between the brain and the meninges. Excluding neoplasms, which normally cause a slow and chronic increase in ICP, hemorrhages are the most frequent expansive processes. They are usually classified according to their location into the following: epidural, subdural, subarachnoid, and intraparenchymal. Regardless of the site of the hemorrhage, their effect on the ICP is mediated by the mass effect they produce on the brain and the other intracranial structures.
The other common cause of acute increase in brain volume is brain edema, which can be classified into two major pathologic entities: vasogenic and cytotoxic [2].
The origin of vasogenic edema lies in a disruption of the blood–brain barrier (BBB). This causes swelling and subsequent breakdown of the myelin. CBF and cellular functions tend to remain mostly unaltered since edema is mostly extracellular. This type of edema is usually caused by inflammatory/infective processes.
On the other hand, cytotoxic edema is intracellular and mostly localized in the astrocytes. Thus, it involves the gray matter more than the white matter and is typically observed in ischemic/anoxic injury and in serum electrolytes imbalances (see below).
2.2.3.2 CSF-Related Causes of IH
Increases in CSF volume may be due to increased CSF production, decreased CSF absorption, and/or obstructed outflow of CSF toward the spinal subarachnoid space (see above). In ABI, and especially in subarachnoid hemorrhage (SAH), obstructive hydrocephalus is a common condition, due to blood accumulation into the ventricular system; in this situation, a decreased CSF absorption can also be observed due to an involvement of the arachnoid granulations [10, 11]. Another typical mechanism of acute obstructive hydrocephalus is the fourth ventricle compression, for example, due to a posterior fossa hematoma.
2.2.3.3 Blood-Related Causes of IH
Increased in CBV has been previously described. The main mechanism is arteriolar vasodilation, most commonly caused by hypercapnia and/or hypoxemia. Fever and seizures can cause wide increases in CBF and CBV especially when they are associated to an impaired CBF autoregulation.
On the other hand, an increase in CBV can be determined by an obstructed venous outflow; this is often iatrogenic and due to jugular veins compression (erroneous head positioning, cervical compression) or to elevated intrathoracic/intra-abdominal pressures (see below).
2.2.4 Extracranial Causes of IH
In Fig. 2.2, we summarize the complex network of extracranial organs that can possibly influence and increase the ICP.

Extracranial organs and ICP. ICP intracranial pressure, IAP intra-abdominal pressure, ITP intrathoracic pressure, VR venous return, CO cardiac output, CPP cerebral perfusion pressure, JVP jugular venous pressure, VVS vertebral venous system, CBV cerebral blood volume
2.2.4.1 Intrathoracic Pressure (ITP)
The interaction between ITP and ICP is fundamentally based on one mechanism: venous outflow obstruction. Indeed, an increase in ITP directly translates to an increase in central venous pressure (CVP) and so in jugular and intracerebral venous pressure. This in turn expands CBV and impairs CSF absorption (see above). Moreover, the increased ITP can also lower the CPP by impairing venous return and lowering cardiac output (CO).
In the setting of ABI, the most common cause of increased ITP is mechanical ventilation, and in particular the application of positive end-expiratory pressure (PEEP). Mechanical ventilation is also responsible for other transient, but significantly high surges in ITP, during lung recruitment maneuvers or during coughing in patients who are poorly adapted to mechanical ventilation. The third most important cause of increase of ITB is the increase in intra-abdominal pressure (IAP), which will be discussed in the next paragraph.
The use of PEEP in ABI patients has been debated for a long time [12]. Adequate levels of PEEP keep lung units open during expiration and avoid the so-called atelectrauma (lung injury due to cyclic opening and closing of lung units), thus improving oxygenation and, according to some authors, helping to protect the lung from further damage (“open lung approach”). Accordingly, many patients with ABI may theoretically benefit from the use of PEEP, given that lung damage is often present (due to direct trauma, aspiration, infection, etc.), but some concerns still exist as for the safety of PEEP in patients with IH. Many studies show that PEEP has minimal effects on ICP, especially if it is titrated to be lower than ICP itself [13,14,15]. A recent retrospective study (reflecting the current trends in ventilatory settings) on 341 patients and 28,644 paired PEEP and ICP measurements confirms no significant relation between PEEP and ICP or CPP, except in the subgroup of severely lung-injured patients. However, even in those patients with highly impaired lung compliance (in whom airway pressure increases are more easily transmitted to the vessels and so to the brain), the direct relation between PEEP and ICP appears to be statistically significant but not very relevant from a clinical point of view. Indeed, this retrospective study shows that on average a 5 cmH2O increase in PEEP translates in modest 1.6 mmHg increase in ICP and 4.3 mmHg decrease in CPP, and the authors conclude that PEEP can be safely applied in most of the ABI patients [16].
To date, only a very small number of studies have investigated the relationship between lung recruitment maneuvers and ICP increase. In 11 patients with ABI and acute lung injury, Bein and colleagues showed that aggressive recruitment maneuvers (sustained pressures up to 60 cmH2O for 30 s) have detrimental effects on cerebral hemodynamics and metabolism [17]. Another study on 16 patients with SAH and acute respiratory distress syndrome (ARDS) compared recruitment maneuvers in pressure control mode from PEEP 15 cmH2O up to an inspiratory pressure of 50 cmH2O for 120 s with recruitment maneuvers at a constant positive airway pressure of 35 cmH2O for 40 s. The results of this study show that recruitment in pressure control mode has a much smaller impact on ICP and CPP and much higher impact on oxygenation [18]. Nevertheless, larger studies are certainly needed to better define the “pros and cons” of lung recruitment maneuvers in patients with ABI.
2.2.4.2 Intra-abdominal Pressure (IAP)
The anatomical foundation of the complex interplay between IAP and ICP lies in the vertebral venous system (VVS). This system was firstly described almost two centuries ago, but it has continued to be thoroughly studied even in the last decade although under different names [19, 20]. The VVS is constituted by valveless veins that allow free flow of blood between the head and the distal end of the spinal canal [19]. It has been described as a large and valveless “venous lake,” where blood can move freely, and its flow direction is strongly influenced by the pressures in the different body compartments [21]. Indeed, the VVS is widely anastomosed with the inferior and superior caval system (through the azygos and lumbar veins) and to other cranial, cervical, thoracic, abdominal, and sacral venous plexuses [22]. Consequently, two main mechanisms explain the interplay between IAP and ICP: according to the first, an increase in IAP produces a blood shift into the VVS, which directly translates into a blood shift into the intracranial compartment. The second mechanism involves a transfer of IAP to the ITP through the diaphragm muscle, thus producing a back pressure on the jugular system, as already described in the previous paragraph [22].
Whichever the mechanism involved (normally both of them), the interaction between IAP and ICP has been studied in animal models and in human patients, and it has shown that increases in IAP invariably translate into raises in ICP by increasing the volume of intracranial venous blood [23].
Some of the most important clinical studies on the interaction between IAP and ICP have been conducted in traumatic brain injury (TBI) patients. In an elegant physiologic study in 2001, Citerio and colleagues externally increased IAP (by positioning a 15-L water bag on the abdomen) in 15 TBI patients after stabilization and resolution of IH. This acute rise in IAP translated almost immediately in concomitant and rapid increases of CVP, internal jugular pressure, and finally ICP, which rose significantly from an average of 12 mmHg to an average of 15.5 mmHg [24]. In a retrospective study on 102 patients with severe TBI who underwent decompressive craniotomy (DC) or decompressive laparotomy or both, Scalea and colleagues found statistically significant decreases in ICP after DC and laparotomy, regardless of whether laparotomy was done before or after craniotomy. In addition, they found that decompressive laparotomy was successful in reducing ITP regardless of whether it was done before or after DC [25].
2.2.4.3 Fluids and Electrolytes
Plasma osmolarity is a key determinant of ICP. The BBB acts as a real barrier to the entry of molecules and solutes into the brain. An intact BBB is completely impermeable to sodium [26] and so any water flow across the barrier is sodium-free [27]. The concept of osmolarity and its relationship to ICP is important in the normal brain but has the utmost importance in the setting of ABI, when the BBB loses, at least partially, its function. When the function of the BBB is disrupted, hydraulic permeability and conductivity to solutes increase, raising the flow of water (accompanied by proteins) across the capillary membranes. This increase in water is the vasogenic edema, which was already described in a previous paragraph [28, 29]. This is especially important for fluid management in ABI patients and is the foundation of osmotherapy for increased ICP.
2.3 Conclusions
The ICS has a complex pathogenesis, which is often influenced by the presence of other coexisting compartment syndromes. The understanding of brain physiology and of the compensatory mechanisms to IH may help the physician in making the right choices for protecting the brain from secondary injuries and avoiding iatrogenic damage.
References
Merriam-Webster. Compartment syndrome. Merriam-Webster’s Med. Dict. 2016.
Wijdicks EFM. The practice of emergency and critical care neurology. 2nd ed. New York: Oxford University Press; 2016.
Chesnut RM. Intracranial pressure. In: Le Roux PD, Levine JM, Kofke WA, editors. Monitoring in neurocritical care. Philadelphia: Elsevier Saunders; 2013. p. 338–47.
Lundberg N. Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psychiatr Scand Suppl. 1960;36:1–193.
Marmarou A, Anderson RL, Ward JD, Choi SC, Young HF, Eisenberg HM, Foulkes MA, Marshall LF, Jane JA. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. J Neurosurg. 1991;75:S59–66.
Schreiber MA, Aoki N, Scott BG, Beck JR. Determinants of mortality in patients with severe blunt head injury. Arch Surg. 2002;137:285–90.
Carney N, Totten AM, OʼReilly C, et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery. 2016;81:1.
Meyfroidt G, Citerio G. Letter: guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery. 2017;81:E1.
Picetti E, Iaccarino C, Servadei F. Letter: guidelines for the management of severe traumatic brain injury fourth edition. Neurosurgery. 2017;81:E2.
van Gijn J, Hijdra A, Wijdicks EF, Vermeulen M, van Crevel H. Acute hydrocephalus after aneurysmal subarachnoid hemorrhage. J Neurosurg. 1985;63:355–62.
Graff-Radford NR, Torner J, Adams HP, Kassell NF. Factors associated with hydrocephalus after subarachnoid hemorrhage. A report of the Cooperative Aneurysm Study. Arch Neurol. 1989;46:744–52.
Lowe GJ, Ferguson ND. Lung-protective ventilation in neurosurgical patients. Curr Opin Crit Care. 2006;12:3–7.
Frost EAM. Effects of positive end-expiratory pressure on intracranial pressure and compliance in brain-injured patients. J Neurosurg. 1977;47:195–200.
Burchiel KJ, Steege TD, Wyler AR. Intracranial pressure changes in brain-injured patients requiring positive end-expiratory pressure ventilation. Neurosurgery. 1981;8:443–9.
Cooper KR, Boswell PA, Choi SC. Safe use of PEEP in patients with severe head injury. J Neurosurg. 1985;63:552–5.
Boone MD, Jinadasa SP, Mueller A, Shaefi S, Kasper EM, Hanafy KA, O’Gara BP, Talmor DS. The effect of positive end-expiratory pressure on intracranial pressure and cerebral hemodynamics. Neurocrit Care. 2017;26:174–81.
Bein T, Kuhr LP, Bele S, Ploner F, Keyl C, Taeger K. Lung recruitment maneuver in patients with cerebral injury: effects on intracranial pressure and cerebral metabolism. Intensive Care Med. 2002;28:554–8.
Nemer SN, Caldeira JB, Azeredo LM, et al. Alveolar recruitment maneuver in patients with subarachnoid hemorrhage and acute respiratory distress syndrome: a comparison of 2 approaches. J Crit Care. 2011;26:22–7.
Parkinson D. Extradural neural axis compartment. J Neurosurg. 2000;92:585–8.
Tobinick E, Vega CP. The cerebrospinal venous system: anatomy, physiology, and clinical implications. MedGenMed. 2006;8:53.
Epstein HM, Linde HW, Crampton AR, Ciric IS, Eckenhoff JE. The vertebral venous plexus as a major cerebral venous outflow tract. Anesthesiology. 1970;32:332–7.
Depauw PRAM, Groen RJM, Van Loon J, Peul WC, Malbrain MLNG, De Waele JJ. The significance of intra-abdominal pressure in neurosurgery and neurological diseases: a narrative review and a conceptual proposal. Acta Neurochir. 2019;161:855. https://doi.org/10.1007/s00701-019-03868-7.
De Laet I, Citerio G, Malbrain MLNG. The influence of intra-abdominal hypertension on the central nervous system: current insights and clinical recommendations, is it all in the head? Acta Clin Belg. 2007;62(Suppl 1):89–97.
Citerio G, Vascotto E, Villa F, Celotti S, Pesenti A. Induced abdominal compartment syndrome increases intracranial pressure in neurotrauma patients: a prospective study. Crit Care Med. 2001;29:1466–71.
Scalea TM, Bochicchio GV, Habashi N, McCunn M, Shih D, McQuillan K, Aarabi B. Increased intra-abdominal, intrathoracic, and intracranial pressure after severe brain injury: multiple compartment syndrome. J Trauma. 2007;62:647–56; discussion 656.
Qureshi AI, Suarez JI. Use of hypertonic saline solutions in treatment of cerebral edema and intracranial hypertension. Crit Care Med. 2000;28:3301–13.
Rossi S, Picetti E, Zoerle T, Carbonara M, Zanier ER, Stocchetti N. Fluid management in acute brain injury. Curr Neurol Neurosci Rep. 2018;18:74.
Hladky SB, Barrand MA. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS. 2014;11:26.
Hladky SB, Barrand MA. Fluid and ion transfer across the blood-brain and blood-cerebrospinal fluid barriers; a comparative account of mechanisms and roles. Fluids Barriers CNS. 2016;13:19.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tonetti, T., Biondini, S., Minardi, F., Rossi, S., Picetti, E. (2021). Definition and Pathomechanism of the Intracranial Compartment Syndrome. In: Coccolini, F., Malbrain, M.L., Kirkpatrick, A.W., Gamberini, E. (eds) Compartment Syndrome. Hot Topics in Acute Care Surgery and Trauma. Springer, Cham. https://doi.org/10.1007/978-3-030-55378-4_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-55378-4_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-55377-7
Online ISBN: 978-3-030-55378-4
eBook Packages: MedicineMedicine (R0)