
Abstract
Since many decades, nonmelanoma skin cancer (NMSCs) is the most common malignancy worldwide. Basal cell carcinomas (BCC) and squamous cell carcinomas (SCC) are the major types of NMSCs, representing approximately 70% and 25% of these neoplasias, respectively. Because of their continuously rising incidence rates, NMSCs represent a constantly increasing global challenge for healthcare, although they are in most cases nonlethal and curable (e.g., by surgery). While at present, carcinogenesis of NMSC is still not fully understood, the relevance of genetic and molecular alterations in several pathways, including evolutionary highly conserved Notch signaling, has now been shown convincingly. The Notch pathway, which was first developed during evolution in metazoans and that was first discovered in fruit flies (Drosophila melanogaster), governs cell fate decisions and many other fundamental processes that are of high relevance not only for embryonic development, but also for initiation, promotion, and progression of cancer. Choosing NMSC as a model, we give in this review a brief overview on the interaction of Notch signaling with important oncogenic and tumor suppressor pathways and on its role for several hallmarks of carcinogenesis and cancer progression, including the regulation of cancer stem cells, tumor angiogenesis, and senescence.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Angiogenesis
- Cancer
- Cancer stem cells
- Cancer treatment
- Notch
- Nonmelanoma skin cancer
- Notch signaling
- Notch pathway
- Skin cancer
- Tumor angiogenesis
Ancient Friends, Revisited: A Short Introduction to the Relevance of Notch Signaling for Skin and Cancer
Although most types of nonmelanoma skin cancers (NMSCs) are not documented in the cancer registries of most countries, there is no doubt that they are since many decades the most common malignancies worldwide (Reichrath and Reichrath 2012a; Reichrath and Rass 2014). Basal cell carcinomas (BCCs) and cutaneous squamous cell carcinomas (SCCs), that are in general nonlethal and curable (e.g., by surgery), represent in many populations approximately 70% and 25% of NMSCs, respectively (Reichrath and Reichrath 2012a; Reichrath and Rass 2014). Because of their continuously rising incidence rates, NMSCs represent a constantly increasing challenge for global healthcare (Reichrath and Reichrath 2012a). BCCs and SCCs share many characteristics, including that both of them may be induced by solar or artificial ultraviolet radiation (UVR), but they are very different from etiology to progression (Reichrath and Reichrath 2012a; Reichrath and Rass 2014). Both UVA and UVB may cause DNA damage and immunosuppression, which play crucial roles in skin photocarcinogenesis (Reichrath and Reichrath 2012a; Reichrath and Rass 2014). UVB can be directly absorbed by DNA molecules, thereby resulting in characteristic UV-signature DNA damages (Reichrath and Reichrath 2012a; Reichrath and Rass 2014). UVA, on the other hand, may, to a lesser extent, also exert DNA damage, through inducing cellular reactive oxygen species (ROS) which then induces oxidative DNA damages (Reichrath and Reichrath 2012a; Reichrath and Rass 2014). Although the photocarcinogenesis of NMSC is still not fully understood, it has shown promise as a well-suited model to investigate the relevance of various signaling networks, including pathways that are also of relevance for embryonic development and for the multistep-carcinogenesis of solid tumors (Reichrath and Reichrath 2012a; Reichrath and Rass 2014). In NMSC, the relevance of genetic and molecular alterations in many pro- and anticarcinogenic pathways, including evolutionary highly conserved Notch signaling, has now been shown convincingly (Reichrath and Reichrath 2012a). The tale that earned the gene Notch, its name started over a century ago at Olivet College (Olivet, Michigan, USA), when the American Scientist John S. Dexter observed and described the characteristic notched-wing phenotype (a nick or notch in the wingtip) in his mutant fruit flies Drosophila melanogaster (Dexter 1914; Reichrath and Reichrath 2020a, b, c). Notably, the Notch pathway is simple in design but has a striking versatility in function (Andersson et al. 2011; Reichrath et al. 2010; Reichrath and Reichrath 2020a, b, c). Notch signaling first developed during evolution in metazoans and was first discovered in fruit flies (Drosophila melanogaster) (Gazave et al. 2009; Richards and Degnan 2009). It orchestrates and governs from sponges to humans during embryonic development and in adult tissues cell fate decisions and many other fundamental cellular processes (Andersson et al. 2011; Reichrath and Reichrath 2020a, b, c). Besides the ability to activate and orchestrate cell proliferation, thereby keeping precursor and stem cells in a nondifferentiated state as well as generating and maintaining stem cells, these flagship functions include regulation of important hallmarks during the multistep process of skin carcinogenesis, including initiation, promotion, and progression of cancer cells. In general, these functions involve canonical, ligand-dependent stimulation of Notch receptors (McIntyre et al. 2020). However, ligand-independent Notch activation has also been observed in several distinct cellular contexts (rev. in McIntyre et al. 2020; Reichrath and Reichrath 2020a, b, c). During the last decades, a huge mountain of new scientific information – ranging from the elucidation of the Notch pathway (Kidd et al. 1986; Kiernan et al. 2001, 2006, 2007; reviewed by Bray 2016; Kopan and Ilagan 2009; McIntyre et al. 2020; Reichrath and Reichrath 2020a, b, c), to the generation of knockouts in model organisms and the discovery of mutated Notch genes in humans (Gridley 2003) – has confirmed an essential role for Notch signaling for various types of cancers, including NMSC. As outlined above, environmental hazards, including solar and artificial UV-radiation, represent important risk factors for carcinogenesis of both melanoma and NMSC (Reichrath and Rass 2014). Notably, the skin (with the epidermis and its adjoining structures, including hair follicles (HF) and associated sebaceous glands; that together comprise the pilosebaceous unit) is not only the human body’s largest organ but also its first line of defense against UV-radiation and many other environmental hazards, providing protection from dehydration, injury, and infection (Shi et al. 2017). Hair follicles have been described as self-renewing structures that continuously generate new epithelial cells to guarantee skin integrity and to renew the skin and its epidermal appendages in response to injury or environmental hazards (Rishikaysh et al. 2014). It is well known that Notch signaling is of high importance for skin homeostasis and wound repair, that both depend on the presence of epithelial stem cells as the primary source for regenerative cells (Blanpain and Fuchs 2006; Shi et al. 2017). Multipotent stem cells that reside within the epidermis and in the bulge region of HFs can give rise to a variety of different cell types, including those forming HFs, interfollicular epidermis, and associated epithelial glands (Shi et al. 2017). Notch signaling has been shown to govern proliferation and differentiation of these cell types, two processes whose alterations have the potential to disrupt normal cell growth and skin homeostasis (Shi et al. 2017) and may finally result in malignant transformation of these cells.
Besides certain other disorders of the skin, such as chronic wounds, skin atrophy, skin fragility, hirsutism, and alopecia, NMSC show characteristic features that are well in line with a disorder of skin stem cells (Najafzadeh et al. 2015; Shi et al. 2017). In NMSC and many other malignancies, it has been hypothesized that tumor formation is caused by inappropriate stimulation/regulation of distinct cellular signaling pathways (including Notch signaling), thereby activating these stem cells or their immediate pluripotent progenitors (Burkert et al. 2006; Shi et al. 2017). In line with this concept is the observation that several types of NMSCs can obviously be derived from HFs, an assumption that is supported by characteristic histological findings and the detection of specific molecular markers both in HFs and in skin malignancies (Jahoda and Reynolds 2000; Shi et al. 2017). It has been concluded that understanding the molecular mechanisms by which proliferation and differentiation are regulated in skin appendages may provide important insights into the molecular basis of NMSC and other diseases, and may also identify promising targets for treatment intervention (Shi et al. 2017). In this review, we give a brief overview on the role of Notch signaling for the multistep process of photocarcinogenesis of NMSC (including the roles of the Notch pathway for the regulation of cancer stem cells (CSCs) and tumor angiogenesis), for progression and clinical management of NMSC (including Notch’s role as an emerging therapeutic target). Because understanding the fundamental role of Notch for embryonic development of skin, HFs, and other appendages is of critical importance for understanding the relevance of Notch signaling for skin carcinogenesis, we will also give a short introduction on this topic.
A Snapshot on the Role of Notch Signaling for Embryonic Development and Tissue Homeostasis of Skin and Hair Follicles
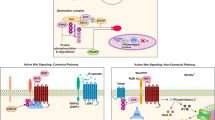
It has been convincingly shown that the evolutionary, highly conserved Notch pathway governs fundamental developmental processes that include binary decision, lateral inhibition, and boundary formation (Artavanis-Tsakonas et al. 1999; Reichrath and Reichrath 2020a, b, c). In general, Notch-mediated cell–cell communication is context and cell-type dependent and exerted by coordinated, differential expression of distinct Notch receptors and corresponding ligands on the surface of adjacent cells (reviewed in McIntyre et al. 2020). In mammals, four evolutionary, highly conserved transmembrane Notch receptors (Notch1–4) have been identified, that can be activated via five corresponding ligands of the Delta-like (Dll 1, 3, and 4) and Jagged (JAG1 and 2) families (Kiernan et al. 2001; reviewed in McIntyre et al. 2020), In general, neighboring cells stimulate each other to produce elevated levels of ligands, thereby inducing an increased activation of Notch receptors (Artavanis-Tsakonas et al. 1999; reviewed in McIntyre et al. 2020). In most cases, elevated expression of ligands with subsequent Notch activation results in cellular differentiation (and cell growth arrest), thereby regulating the cluster size of cell populations (Artavanis-Tsakonas et al. 1999; reviewed in McIntyre et al. 2020). At the molecular level, it has been shown that in the canonical Notch-signaling pathway, ligand-induced Notch receptor stimulation results in cleavage of the intracellular domain of the Notch receptor (NID) (reviewed in McIntyre et al. 2020). The NID then consecutively translocates to the nucleus where it forms a ternary complex with the transcriptional coactivator, Mastermind-like (MAML) protein, as well as the DNA-binding protein, CBF-1, Su (H), Lag-1-type transcription factor (CSL, also termed Recombinant recognition sequence binding protein at the Jκ site, RBP-J), which have been shown to direct specific binding to response elements in DNA regions of target genes and to regulate target gene expression (reviewed in McIntyre et al. 2020). Until today, only a limited number of Notch target genes have been identified and characterized, most importantly basic-helix–loop-helix proteins of the hairy and enhancer of split (Hes) and Hes-related transcription factor (Hrt) families, which function as transcriptional repressors (reviewed in McIntyre et al. 2020).
Notch Signaling in Skin: Simple in Design but Versatile in Function
Recent scientific findings indicate that Notch signaling orchestrates the process of epidermal differentiation and proliferation through the sequential activation of different Notch ligands, receptors, and downstream pathways. Notch receptors and corresponding ligands are present in the skin (Reichrath and Reichrath 2012a, b), although until today, most of their particular functions are still uncertain. It has been shown that Notch receptors and ligands are differentially expressed in the different cell layers of the viable epidermis (reviewed in Reichrath and Reichrath 2012a). In healthy skin Notch receptor 1 and its corresponding ligands, Dll1 and JAG1 are present in all cell layers of the viable epidermis, with pronounced expression of Dll1 and JAG 1 in the epidermal basal layer (Table 9.1). It has been observed in various cell types that Delta/Notch signaling is increased in cells that undergo a normal differentiation program, as in human keratinocytes of cell layers of the normal adult epidermis. In contrast, activity of Notch signaling has been described to be decreased in psoriasis vulgaris and other hyperproliferating skin diseases. In line with these investigations, it was reported that loss of Notch receptor1 in young mice induces hyperproliferation of the basal epidermal layer and deregulates expression of multiple differentiation markers, including reduced expression of p21 and elevated expression of Gli2. In epidermal keratinocytes, activation of Notch receptor 1 has been shown to induce p21 expression in a CBF-1, Su (H), Lag-1 (CSL)-type (also termed RBP-J) transcription factor–dependent manner, resulting in cell cycle withdrawal and terminal differentiation. In addition, stimulation of Notch receptor 1 directly promotes caspase 3 activity, that is required for terminal differentiation of embryonic keratinocytes.
The importance of Notch signaling for skin embryogenesis is underlined by characteristic cutaneous findings in several inherited syndromes, including Alagille syndrome (Kamath et al. 2004, 2012, 2013; McCright et al. 2001, 2002, 2006) and Adams–Oliver syndrome. Adams–Oliver syndrome is a rare genetic disorder that has been linked to mutations in several different genes, including DLL4 (OMIM 605185; cytogenetic location: 15q15.1) and NOTCH1 (OMIM 190198; cytogenetic location: 9q34.3), as well as in RBP-J (OMIM 147183; cytogenetic location: 4q15.2), EOGT (OMIM 614789; cytogenetic location: 3p14.1), ARHGAP31 (OMIM 610911; cytogenetic location: 3q13.2–3q13.33), and DOCK6 (OMIM 614194; cytogenetic location: 19p13.2) (reviewed in Mašek and Andersson 2017; Meester et al. 2019; Reichrath and Reichrath 2020a). Adams–Oliver syndrome is diagnosed based on the presence of aplasia cutis congenita and several other clinical hallmarks, namely terminal transverse limb malformations and a partial absence of skull bones (reviewed in Mašek and Andersson 2017; Meester et al. 2019; Reichrath and Reichrath 2020a; Zanotti and Canalis 2016). Typically, aplasia cutis congenita is found in the skull region, however other body parts, including the abdomen, may also be affected (reviewed in Meester et al. 2019; Zanotti and Canalis 2016). The severity and symptoms of aplasia cutis congenita may greatly vary (reviewed in Meester et al. 2019; Zanotti and Canalis 2016). At birth, the affected skin region typically presents as healed but scarred skin, and skin histology shows characteristic findings that may include absent epidermis, dermal atrophy, and a lack of elastic fibers and other skin structures (reviewed in Meester et al. 2019; Reichrath and Reichrath 2020a; Zanotti and Canalis 2016). However, symptoms may range from a localized region with complete absence of skin to patches of skin that lack hair (reviewed in Meester et al. 2019; Zanotti and Canalis 2016).
A large body of convincing evidence from clinical and laboratory investigations has shown the importance of Notch signaling for the embryonic development of all anatomical structures of the skin, including the epidermal compartment, HFs, and other appendages. It was demonstrated that in response to external cues, embryonic skin cells have to make a cell fate decision whether or not to differentiate and generate stratified epidermis, or to invaginate and initiate morphogenesis of HF (Fuchs 2007). It has been demonstrated that the cell fate decisions of epidermal keratinocytes whether or not to transit from basal to suprabasal epidermal cell layers begin around embryonic day 13.5 (E13.5). At this time, the activation of Notch receptors by their corresponding ligands is associated with the stratification of the epidermis (Blanpain et al. 2006). On the molecular level, this process is mediated by enzymatic cleavage of the NID and its translocation to the nucleus, where it associates in keratinocytes of suprabasal cell layers with DNA-binding protein RBP-J to regulate downstream target genes (Kopan and Ilagan 2009; Lowell et al. 2000; Moriyama et al. 2008; Okuyama et al. 2004; Wang et al. 2008).
In skin, the epidermis is maintained throughout life through the proliferation of stem cells and differentiation of their progeny. The innermost (basal) layer of the epidermis consists of proliferative progenitor cells which give rise to multiple differentiating layers, a stratified epithel providing a barrier that keeps the inside of the body moist and protects the body from environmental hazards by physical, chemical, and biological factors, including ultraviolet (UV)-radiation (Massi and Panelos 2012). Investigations using transgenic mice have demonstrated that in contrast to embryonic development of the HF that can be achieved without Notch, its postnatal development requires an intact Notch signaling in two important compartments of the hair, the bulb, and the outer root sheath (reviewed in Aubin-Houzelstein 2012, reviewed in Massi and Panelos 2012). In the hair bulb, Notch governs cell differentiation, ensuring the proper development of every layer of both the hair shaft and the inner root sheath (reviewed in Aubin-Houzelstein 2012, reviewed in Massi and Panelos 2012). Among the many roles played by Notch in the skin and HF, it has to be highlighted that in the bulge, Notch controls a cell fate switch in HF stem cells or their progenitors, preventing them from adopting an epidermal fate (reviewed in Aubin-Houzelstein 2012). Notch function in the skin and HF is both cell autonomous and cell nonautonomous and involves intercellular communication between adjacent cell layers (reviewed in Aubin-Houzelstein 2012, reviewed in Massi and Panelos 2012).
The tightly regulated Notch function depends on a large network of contributing pathways that have also been shown to be of importance for skin carcinogenesis, including Wnt-mediated signals from adjacent epidermal cells and suppressing bone morphogenic protein (BMP)–mediated signals from underlying mesenchymal condensates, which converge to activate Sonic hedgehog (Shh) in the developing hair bud. Loss of Shh signaling widely disturbs this highly regulated epithelial–mesenchymal cross-talk, impairing HF down-growth and maturation in the embryo and distorting homeostasis throughout postnatal skin epithelium (Chiang et al. 1999; Gritli-Linde et al. 2007; Oro and Higgins 2003). Notably, it was shown that epidermal morphogenesis not only precedes but also may be observed independently of Hh signaling (Oro and Higgins 2003).
Notch Signaling and Nonmelanoma Skin Cancer (NMSC)
The physiological/pathophysiological function and the regulation of the Notch pathway in the pathogenesis of human NMSCs (Table 9.1) are at present not completely understood. Previous studies indicate an important role of Notch signaling both for pathogenesis and progression of SCCs and BCCs (reviewed in Reichrath and Reichrath 2012a).
It was demonstrated that in accordance with its function in inducing differentiation of keratinocytes, mice with an experimentally induced epidermal deletion of the Notch1 gene develop extensive epidermal hyperplasia and spontaneously develop BCCs (reviewed in Reichrath and Reichrath 2012a). Consequently, this finding has resulted in the hypothesis that Notch1 may act in the skin as a tumor suppressor (Table 9.1). Moreover, in mice with epidermal inactivation of Notch1, chemical injury promoted the formation of cutaneous lesions representing both BCCs and SCCs, in addition to inducing numerous papillomas. It has been shown that mice expressing a dominant negative MAML1 (DNMAML1) protein to inhibit RBP-J dependent Notch signaling in the epidermis exhibit multiple skin defects including diffuse alopecia, epidermal hyperplasia, and hyperkeratinization (reviewed in Reichrath and Reichrath 2012a). These mice develop spontaneous lesions resembling human SCC and actinic keratoses, but do not develop BCC. In contrast to normal epidermis, keratinocytes and lesional cells from DNMAML1 mutant mice express nuclear ß-catenin and cyclin D1 in a pattern similar to that observed in human cutaneous SCC, suggesting a conserved role for these molecules in SCC (reviewed in Reichrath and Reichrath 2012a). Taken together, these data strongly suggest that functional interactions between Notch signaling, ß-catenin, and cyclin D1 play critical roles in the pathogenesis of cutaneous SCC.
Epidemiology and Clinical Findings of NMSC
BCCs and SCCs, the two major types of NMSC, vary considerably in their clinical presentation, growth patterns, and metastatic capability. In general, most cases of both BCCs and SCCs have a good prognosis, especially when detected at their early stages (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). BCC cells resemble many characteristic features of epidermal basal cells (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). There is evidence that BCCs, at least in part, originate from the basal layer of the outer root sheath of the hair follicle, which closely resembles the interfollicular basal layer of the epidermis with respect to protein expression patterns, including members of the Notch signaling pathway (Table 9.1). BCCs have been described as the least aggressive type of NMSC. They very rarely metastasize and show a low degree of malignancy, despite of the capability of local invasion, tissue destruction, and recurrence (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). It was reported that the prevalence of BCCs and SCCs has increased by 35% and 133%, respectively, over 2 decades. BCCs contribute minimally to the NMSC mortality rate (MR). An incidence rate of 1 case per 14,000,000 for metastatic BCC, and 2 patients per 14,000,000 who die from locally advanced BCC have been reported. In consequence, a MR of 0.02 per 10,000 is to be expected (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). Individual risk factors for BCC include gender, age, immunosuppression, genetic diseases (e.g., Gorlin–Goltz syndrome), and Fitzpatrick skin types I and II (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). Ultraviolet (UV) radiation represents the most important environmental risk factor for BCC pathogenesis, although the precise relationship (chronic or intermittent (sunburn) UV exposure) between UV radiation and BCC development remains controversial (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). BCCs develop predominantly in elderly patients on sun-exposed skin areas. BCCs rarely develop on palmoplantar surfaces and are never found on the mucosa (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). Individuals who develop BCC have an elevated risk of developing new foci of BCC, as well as other types of skin cancer, including melanoma and SCC. Their incidence has increased strongly over time, also reflecting our aging population.
SCCs are characterized by atypical, invasive proliferation of squamous cells, which have the potential to metastasize. SCCs show a considerable potential for recurrence, which depends on many factors, including tumor size, degree of histological differentiation, depth of the lesion, perineural invasion, patient’s immune system, and anatomic localization (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). Several risk factors have been reported in SCC patients, including Fitzpatrick skin types I and II, outdoor occupation, human papillomavirus (HPV) types 16, 18, and 31, and several cutaneous genetically inherited skin diseases (including albinism, xeroderma pigmentosum, and epidermodysplasia verruciformis) (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). However, the most important environmental risk factor is UV radiation (artificial and solar) (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). A direct correlation between psoralen and UVA (PUVA) exposure and the incidence of SCC has been reported (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). In most cases, SCCs arise on sun-exposed areas, with about 55% and 18% of all SCCs presenting on the head and neck area and on the extensor surfaces of the hands and forearms, respectively (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). Nevertheless, up to 13% of SCC cases arise on the legs (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). Actinic keratoses (AKs) represent in situ SCCs, the earliest manifestation of SCCs. Although their prevalence varies according to geographical location and age, AKs are extremely common, showing a prevalence greater than 40% in many adult populations. AKs occur usually on chronically UV-exposed skin. AKs share several pathological features with SCC, and they represent a continuum in a multistep process over the years on chronically sun exposed fair skin. Normal-appearing skin that surrounds AKs may develop AKs, because of the UV exposure and expression of molecular alteration, including p53 mutations. This whole area is today known as “field cancerization.” SCCs show a variable metastatic rate of 0.1–9.9% and they account for about 75% of deaths due to NMSC (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014). Although the first-choice therapy is still surgical excision, plenty of alternative approaches have been reported to manage NMSC, including photodynamic therapy (PDT), cryotherapy, and topical imiquimod 5% (Apalla et al. 2017; Didona et al. 2018; Leiter et al. 2014).
The Role of Notch Signaling for Carcinogenesis of Basal Cell Carcinomas
Several studies, including investigations from Thélu and coworkers, compared the expression of Notch receptor family members and their corresponding ligands in BCCs and unaffected and healthy skin (Table 9.1). Expression of Notch1 and its ligands varies in the different layers of the epidermis (Fig. 9.1d–f). In unaffected, healthy skin Notch receptor 1, Dll1 and JAG1 are detectable in the whole epidermis, with pronounced expression of the latter two in the basal layer. Thélu and coworkers demonstrated that the protein expression of Notch receptor 1 and its corresponding ligands Dll1 and JAG1 was markedly reduced in BCC tumor regions as compared with unaffected normal skin (Thelu et al. 2002). Moreover, in that study, all three proteins were undetectable in BCC regions comprised of palisading cells penetrating the dermis. The authors concluded that because in normal human skin Notch receptor 1 and its corresponding ligands Dll1 and JAG1 are detectable in all cell layers of the viable epidermis (with pronounced expression of the latter two in the basal layer), in absence of Notch1, Dll1 and JAG1, missing or reduced Notch signaling activity may cause disordered epidermal terminal differentiation and proliferation (Thelu et al. 2002). It has been speculated that during malignant transformation of BCCs, keratinocytes may enter a pathological status when they neither transcribe Notch receptor family members nor corresponding ligands, thereby resulting in abolishing a fundamental signal for terminal differentiation (Thelu et al. 2002).

Immunohistochemical detection in cutaneous squamous (a–c) and basal cell (d–f) carcinomas of Notch receptors 1 (b, f;  ), 2 (c;
), 2 (c;  ) and corresponding ligand JAG1 (a, d, e;
) and corresponding ligand JAG1 (a, d, e;  ). Note weak-to-moderate cytoplasmic and/or nuclear immunoreactivity (representative tumor areas marked as examples with arrows)
). Note weak-to-moderate cytoplasmic and/or nuclear immunoreactivity (representative tumor areas marked as examples with arrows)
In another immunohistochemical study, immunoreactivity for Notch receptor 1 was absent in viable normal epidermis and in BCCs, while Notch receptor 2 and its downstream target gene Hes1 could be detected in both cytoplasm and nuclei of normal skin epithelia (8/8) and, with reduced detection rates, of BCCs (Liu et al. 2008). The authors of this study (Liu et al. 2008) concluded that Notch signaling in normal epidermis may be mediated mainly by Notch receptor 2, and that Notch signaling might have twofold bioactivities: It may benefit normal maintenance/renewal of epidermal cells, while its attenuation may favor epidermal tumor growth due to its tumor inhibitory effects (Bolós et al. 2007; Liu et al. 2008). In contrast to other organs, Notch receptor 1 appears to act as a tumor suppressor in the skin (Table 9.1). It was shown that ablation of Notch receptor 1 from epidermal cells in mice leads to an uncontrolled proliferation of the basal epidermal layer and finally results in BCC-like tumors. The immunohistochemical detection of Notch receptor 1 and JAG1 in BCCs is shown in Fig. 9.1d–f, respectively.
Previous findings indicate that at least a significant subset of BCCs is directly HF derived (Table 9.1) (Shi et al. 2017; Crowson 2006; Grachtchouk et al. 2011; López-Takegami et al. 2016; Peterson et al. 2015). It has been reported that the stem cells of the HF bulge region and adjacent cells represent a potential primary source of BCCs derived from HFs (Shi et al. 2017; Grachtchouk et al. 2011; Peterson et al. 2015). In some regard, HFs and BCCs represent “ordered” and “disordered” variants of skin appendage growths, respectively (Shi et al. 2017). An important property both of BCCs and HFs is the ability of their cells to indefinitely and repeatedly proliferate, a key mechanism that is responsible for maintaining a tumor mass or regular hair fiber production (Shi et al. 2017). It has been speculated that all BCCs, HF-derived or not, may express similar fundamental growth mechanisms to those that regulate HF growth and cycling (Shi et al. 2017). Several specific molecular mechanisms involved in this process of self-renewal, including the Notch and other pathways, including sonic hedgehog (Shh) and Wingless-related integration site (Wnt) signaling, have been found to be active in normal HFs and in BCCs (Thélu et al. 1998; McMahon et al. 2003; Jayaraman et al. 2014; Wuest et al. 2007; Rishikaysh et al. 2014; Shi et al. 2017). Notably, the roles of these signaling networks for BCC growth, particularly the Notch pathway, and their crosstalk, remain until today only poorly understood. Mutations in genes encoding for components of the sonic hedgehog (Shh) and patched (PTCH) signaling pathways are the hallmarks of BCC pathogenesis (Hutchin et al. 2005; Shi et al. 2017). The primary mechanism, by which most BCCs develop, a constitutive activation of the Hedgehog pathway, is a fundamental regulatory mechanism in HF development (Hutchin et al. 2005; Shi et al. 2017). Shh signaling is required for the proliferation and normal cycling of HF epithelium. Modifications of Shh signaling can result in tumor development in tissues of different origins (McMahon et al. 2003). Hyperactivation of the Shh signaling pathway is found in several HF-derived tumors and in BCCs (Dahmane et al. 1997; Oro et al. 1997; Shi et al. 2017). Overexpression of GLI1 and GLI2 gene products have been reported in BCCs, indicating increased Shh signaling (Dahmane et al. 1997; Grachtchouk et al. 2000; Shi et al. 2017). In line with previous reports (Dahmane et al. 1997; Grachtchouk et al. 2000; Shi et al. 2017), enhanced expression of glioma-associated oncogene homolog 1 (GLI1) and GLI2 was demonstrated in BCCs and in HF root sheaths, compared with normal skin. In these investigations, HF root sheaths exhibited significantly elevated expression levels of GLI1 and GLI2 compared with BCCs. Interestingly, several reports (Jayaraman et al. 2014; Shi et al. 2017) demonstrated the significance of mutations in NOTCH1 and NOTCH2 in BCCs.
It has been speculated that defining gene expression patterns and pathways in BCCs that are distinct from HF growth and cycling may lead to a better understanding of the abnormal proliferation that these cells undergo in the development of skin cancer. A recent study identified specific molecular mechanisms that are involved in the process of cell self-renewal in HFs and BCCs, including Notch and Hedgehog signaling pathways. Interestingly several key Notch signaling factors showed in that study significant differential expression in BCCs compared with HFs. In that study, a number of genes were uniquely expressed in HFs or BCCs only, indicating that selected Notch pathway genes were differentially activated and inhibited in BCCs, which may be due to positive feedback, and reciprocal negative feedback, from differences in Delta and Notch cell surface expression, or the irregular activation of downstream Notch signaling pathway genes.
Moreover, examination of downstream components of the Notch pathway revealed that the transcription factor RBP-J and downstream target genes of the Hes and Deltex families exhibited a high expression in hair shafts compared with BCCs and normal skin. By contrast, two genes that affect the co-repression of RBP-J, CTBP1, and CREBBP were observed to have a significantly lower expression in HFs compared with BCCs. Deletion of RBP-J from follicular stem cells results in an aberrant cell fate switch that leads to the establishment of epidermal progenitors and basal cells (Yamamoto et al. 2003; Shi et al. 2017). This result, therefore, demonstrated that the Notch/RBP-J signaling pathway is strongly activated in HFs. Since the Notch signaling pathway promotes a stem cell phenotype in skin (Lowell et al. 2000; Shi et al. 2017), the degree of Notch signaling pathway activation may be important for HF stem cell proliferation and differentiation. It has been speculated that the high level of Notch/RBP-J signaling pathway activation may be required for the formation and maintenance of HF.
Importantly, results of this study suggest a reduced expression of downstream genes of the Notch/RBP-J signaling pathway in BCCs. This may allow basal cells to escape from the normal regulation of proliferation that is normally found in the absence of Notch signaling activity, as observed in mammary epithelium cell lineages (Buono et al. 2006; Shi et al. 2017). Loss of RBP-J action in BCCs may promote cells toward a more stem, or progenitor, cell-like status, enabling basal cell tumor growth. Notch signaling via NID translocation into the nucleus, and subsequent binding to the transcription factor RBP-J, may be an important stage of BCC development. As such, RBP-J signaling may be a focus for the introduction of promising, new BCC therapies, as has been suggested for other types of cancer (Garber 2007; Purow 2012; Shi et al. 2017).
Notably, Notch receptor family members 1 and 2 have been reported to exert equivalent or reverse biological effects in cell type-dependent fashions (Weng and Aster 2004). JAG1 plays an important role in the differentiation of keratinocytes, as the activation of Notch pathway triggers terminal keratinocytes differentiation. It was demonstrated that stimulating Notch signaling with JAG1 induced apoptosis of BCC cells by increasing Fas ligand expression and downstream caspase-8 activation. In that study, activation of the Notch signaling pathway by adding exogenous JAG1 into BCC cell culture resulted in increased Fas ligand mRNA and protein expression. This further activated downstream caspase-8 to initiate BCC cell apoptosis. Fas ligand is a type II transmembrane protein that can induce apoptosis upon binding to Fas (Wang et al. 2012; Shi et al. 2017). However, some tumors can decrease Fas expression to resist Fas ligand–mediated T-cell cytotoxicity, and simultaneously upregulate the expression of Fas ligand to induce apoptosis in Fas-expressing T-cells (Satchell et al. 2004; Shi et al. 2017). BCCs strongly express Fas ligand, which may help prevent attack from surrounding immune effector cells, while also lacking Fas, potentially to make the tumor cells resistant to apoptosis (Erb et al. 2008; Shi et al. 2017). Further investigation is required to characterize the exact role of elevated Fas ligand expression induced by Notch signaling activation by in vivo experiments.
This study adds to the body of evidence that Notch signaling pathway activity is, in contrast to HFs, where it is very strong, suppressed in BCCs. As outlined above, animal models have shown that epidermal skin tumors that spontaneously develop in mice lacking Notch1 display basal cell carcinoma (BCC)–like phenotype. In line with these findings, it has been shown that in BCCs, the protein expression of Notch receptors and corresponding ligands, Dll1 and JAG1 is markedly lowered in tumor regions as compared to healthy epidermis. Interestingly, Thélu and coworkers also reported that they were unable to detect these proteins in the regions with palisading cells penetrating the dermis. In summary, an increasing body of evidence indicates that in the absence of Notch1, Dll1, and JAG1, missing or decreased Notch signaling leads to disorder in epidermal differentiation and proliferation, and promotes formation of BCCs. Impaired Notch signaling is also reported to promote the development of cutaneous squamous cell carcinoma (SCC), as outlined above, and malignant melanoma (MM). In summary, it can be assumed that in contrast to other tissues, Notch seems to function in the skin as a tumor suppressor, as shown by Nicolas et al. (2003).
It has been speculated that pharmacologic modulation of Notch signaling could be a new promising target for the treatment of skin cancer, including BCC, and potentially for hair follicle engineering (Shi et al. 2017). Different therapeutic options are available to treat BCC pharmacologically, including topical immunotherapy (Wuest et al. 2007) and oral treatment with the hedgehog-inhibitor vismodegib. Imiquimod is a small synthetic compound that has been approved for the topical treatment of superficial BCC (sBCC), representing a strong immune response modifier via stimulation of Toll-like-receptor 7 (TLR-7), which is present in plasmacytoid dendritic cells, macrophages, and monocytes (Wuest et al. 2007). This activation of toll-like receptor 7 (TLR7) results in an activation of NF-κB, increased synthesis of proinflammatory cytokines (including interferon-α), and a potent stimulation of antitumor Th1 immunity that finally results in tumor destruction. In a clinical and laboratory investigation, six patients with BCC were evaluated for expression of Notch receptor 1 and corresponding ligands JAG1 and Dll1 before and along with topical treatment with imiquimod using real-time PCR and immunohistochemistry. Interestingly, selective transcriptional upregulation of Notch pathway members (Notch1, JAG1 and Delta1) was detected in that study post-treatment in tumor cells of the BCCs (Wuest et al. 2007). Furthermore, a minor increase of Notch1 protein expression on infiltrating cells as well as strong increase in Jagged1 protein expression was detected in regressing sBCCs post-treatment. Interestingly, Jagged1 is implicated to represent a downstream target of NF-κB activation providing a link between these two signaling pathways. It was speculated that via these mechanisms imiquimod may act as a stimulator of the Notch pathway in sBCC tumor cells by upregulating protein expression of the Notch ligand, JAG1 (Wuest et al. 2007). Moreover, imiquimod may exert tumor suppressor function via induction of Notch signaling, which together with its proinflammatory properties may result in tumor regression.
The Role of Notch Signaling for Carcinogenesis and Progression of Cutaneous Squamous Cell Carcinomas
An Introduction to the Molecular Biology of Cutaneous Squamous Cell Carcinomas: Crosstalk Between Notch and p53 Signaling
Interestingly, cross-regulation among the p53 family members and the Notch signaling pathway has been shown (Missero and Antonini 2014; Roemer 2012). The individual functions of the tumor suppressor p53 (TP53) and its family members, p63 (TP63) and p73 (TP73), and their interactions in the photocarcinogenesis of cutaneous SCCs have been extensively investigated (Kouwenhoven et al. 2010; Lang et al. 2004; Lokshin et al. 2007; Missero and Antonini 2014; Yang et al. 2010; Roemer 2012). Many different functions have been assigned to p53, which is often referred to as the “guardian of the genome” because of its ability to prevent mutations (that may be induced upon DNA-damaging stress, including UV-radiation or by other environmental hazards) in the genome by promoting cell cycle exit, senescence or apoptosis (Erb et al. 2008; Hoare and Narita 2018; Lane and Levine 2010; Missero and Antonini 2014; Roemer 2012). The architecture of the p53 protein contains the following domains: transactivation domain (TA), DNA-binding domain (DBD), and oligomerization domain (OD) (Missero and Antonini 2014; Roemer 2012). Due to its crucial role in maintaining genomic stability, inactivation of p53 is the most common event in human cancers, being mutated in over half of human cancers, and often indirectly inactivated via its regulators in other half (Edlund et al. 2012; Lane and Levine 2010; Missero and Antonini 2014; Muller and Vousden 2013; Roemer 2012). Notably, the percentage of C → T transition in the most common mutated amino acids is higher in cSCC as compared to all cancers (consistent with an UV-B radiation signature. No C are found in the codon for R249 (−); data obtained from the UMD TP53 mutation database, release: June 2012_R1; http://p53.fr) (Edlund et al. 2012). Most p53 mutations observed in human tumors fall into the DNA-binding domain (DBD) and inhibit p53 binding to its consensus DNA sequence (Missero and Antonini 2014; Muller and Vousden 2013). As p53 binds DNA as a tetramer, mutant p53 proteins can act with a dominant-negative mechanism on the wild-type protein by heterotetramer formation (Missero and Antonini 2014; Roemer 2012). In addition, some p53 mutants have been shown experimentally to gain novel activities in the absence of a wild-type p53 (Lang et al. 2004; Missero and Antonini 2014; Muller and Vousden 2013; Olive et al. 2004; Roemer 2012).
Each p53 family member encodes several protein isoforms, generated by the presence of alternative promoters, translation initiation sites, and splicing sites. The canonical transactivation domain (TA) present at the N-terminus of the longer protein isoforms is required for transcription of a number of canonical target genes with antiproliferative, pro-senescence or pro-apoptotic, and DNA repair functions. A second internal promoter drives expression of ∆N proteins that lack the TA domain, can exert dominant-negative functions toward the TA proteins and can transactivate a number of specific target genes. This is the case of ∆Np63 and ∆Np73, whereas ∆Np53 (∆133p53) lacks a small portion of the DNA-binding domain and thus is a selective modulators of some TAp53 functions without binding to canonical p53-binding sites (Missero and Antonini 2014; Roemer 2012). As p53 family members can regulate each other at the transcriptional level (Antonini et al. 2006; Chen et al. 2001; Harmes et al. 2003; Kartasheva et al. 2002; Marcel et al. 2012; Missero and Antonini 2014; Wang and El-Deiry 2006; Roemer 2012), this may in part explain the unbalance expression of their transcripts in cancer.
The structural similarity among the p53 family members and their property to function as tetramers allow heterotetramerization between p63 and p73 isoforms (Davison et al. 1999; Della Gatta et al. 2008; Missero and Antonini 2014; Rocco et al. 2006) and between mutant p53 and p63 or p73 (Di Como et al. 1999; Gaiddon et al. 2001; Missero and Antonini 2014; Strano et al. 2000, 2002; Roemer 2012). The interaction with mutant p53 can lead to p63/p73 inactivation (Di Como et al. 1999; Adorno et al. 2009; Missero and Antonini 2014). Competition for a virtually identical DNA binding site is another crucial level of regulation among p53 family members (Kouwenhoven et al. 2010; Lokshin et al. 2007; Missero and Antonini 2014; Yang et al. 2010). In p53-mutant keratinocytes, p63 and mutant p53 bind to partially overlapping elements, some of which are different from the canonical p63 binding elements found in normal keratinocytes (Martynova et al. 2012; Missero and Antonini 2014). In addition, even when wild-type p53 and p63 bind to the same genomic sites, they regulate largely nonoverlapping gene sets as shown in a lung SCC cell line (Gallant-Behm et al. 2012; Missero and Antonini 2014).
Interestingly, cross-regulation among the p53 family members and the Notch signaling pathway has been shown (Missero and Antonini 2014; Roemer 2012). In keratinocytes, p53 exerts its tumor suppressive function by inducing expression of the pro-differentiation Notch1 and the cell cycle inhibitor p21/CDKN1A, among other target genes (Missero and Antonini 2014; Roemer 2012). A more complex crosstalk exists between p63 and the Notch signaling pathway. p63 directly induces JAG1 and Notch expression, as well as the Notch target IRF6, favoring the initial steps of terminal differentiation (Missero and Antonini 2014). At the same time, p63 suppresses expression of the Notch downstream target genes, p21/CDKN1A and Hes1, sustaining cell cycle progression and repressing late stages of differentiation (Missero and Antonini 2014). Importantly, Notch and IRF6 counteract p63 activity in a negative feedback loop. Relatively little is known about p73, although in other cell types it has been reported to positively regulate JAG1 and JAG2 (Missero and Antonini 2014; Sasaki et al. 2002; Roemer 2012). The p53 family member can potentially regulate each other as indicated and described above.
Notch, p53, and Senescence
Recent findings indicate that Notch signaling is intimately involved in the development of cellular senescence (Hoare and Narita 2018). In contrast to earlier assumptions that thought of cellular senescence as an autonomous tumor suppressor mechanism, cellular senescence has recently been emerging as a phenotype and effector present throughout the life of an organism from embryogenesis to senile decline (Hoare and Narita 2018). Senescent cells exert powerful nonautonomous effects upon multiple players within their microenvironment that they orchestrate mainly through their secretory phenotype (Hoare and Narita 2018). How senescent cells coordinate numerous, sometimes functionally contrasting, outputs through their secretome and/or other mechanisms is still not completely understood, but recent findings indicate a key role of the complex physical and functional interplay between Notch and p53 for the regulation of cellular senescence in both non-malignant and in cancer cells (Hoare and Narita 2018). It has been suggested that a better understanding of the interplay between Notch, p53, and senescence, and how it acts to coordinate the composition and functional effects of the senescence secretome could allow us to develop promising new therapeutics to improve cancer treatment (Hoare and Narita 2018).
The Effects of Notch1 Deletion on Multistage Events in Skin Carcinogenesis
As outlined above, it has been shown convincingly that Notch1 deletion in epidermal keratinocytes causes skin carcinogenesis, while in contrast Notch1 acts in most other tissues as a proto-oncogene (Koch and Radtke 2007). Figure 9.1 shows the immunohistochemical detection of Notch1 in human SCC (b) and BCC (f). The mechanisms underlying the carcinogenesis-promoting characteristics of Notch1-deficient skin have been analyzed in mice with a global or chimeric deletion pattern in their epidermis (Demehri et al. 2009). Results of this study (Demehri et al. 2009) obtained by deleting Notch1 either before or after DMBA treatment in the K14CreERT system indicate that loss of Notch1 is not involved in the initiating event of multistage skin carcinogenesis (Demehri et al. 2009; Zoumpourlis et al. 2003). However, it was shown that Notch1 loss acts as a skin cancer-promoting event. In this study, delaying Notch1 deletion in K14CreERT mice until after the tumor-promotion stage of carcinogenesis demonstrated that late deletion of Notch1 contributed to malignant progression of benign papillomas (Demehri et al. 2009), a phenotype that is observed upon loss of p53 but not loss of p21WAF1/Cip1, a specific Notch1 target in the skin. In summary, the authors concluded that the main effect of Notch1 loss in skin carcinogenesis is to provide the initiated cells with a proliferative signal to promote tumor growth and proceed to invasive skin cancer. It has been speculated that this proliferative signal is located downstream of Notch1 loss and could be originated from within the initiated cells, supporting Notch1’s role as a classical tumor suppressor in epidermal keratinocytes. As an alternative pathway, it has been hypothesized that this signal could be delivered by the skin microenvironment reacting to Notch1 loss in the epidermis. The experimental system used by Demehri et al. allowed to distinguish between these two possibilities (Demehri et al. 2009). In their study, the chimeric pattern of Notch1 deletion by Msx2-Cre created neighboring territories of Notch1-expressing and Notch1-deficient keratinocytes coexisting in the same microenvironment. Examining a large number of tumors isolated from DMBA/TPA-treated Msx2-N1CKO mice clearly demonstrated that tumors comprised mostly (>99%) of Notch1-expressing cells were as likely to form as tumors comprised predominantly of Notch1-deleted cells in the same environment. The authors concluded that Notch1 loss in the epidermis generates a non-cell autonomous signal, promoting tumorigenesis from any initiated cell exposed to the microenvironment conditioned by Notch1-deficient keratinocytes. These results underline the relevance of the microenvironment as an active contributor to tumor development by demonstrating that it can be the primary source of proliferative signals to initiated cells (Demehri et al. 2009).
Tumor Angiogenesis and Cancer Stem Cells: Emerging Therapeutic Targets in NMSC
Tumor Angiogenesis: An Introduction
Malignant tumors, including BCCs and SCCs, consist of a population of constantly and rapidly dividing cancer cells that have lost their ability to control cell division and that progressively accumulate mutations. However, in order to grow and to expand beyond a certain size, malignant tumors need sufficient vascularization (McDougall et al. 2006; Spill et al. 2015). It was reported that malignant solid tumors are unable to grow any more than 2–3 mm in diameter without a sufficient blood supply which corresponds to about 50–100 cells (Nishida et al. 2006). While some scientists believe that the major task of these capillaries is to supply cancer cells with the oxygen and with the essential nutrients that they require, other researchers are convinced that angiogenesis really represents a waste pathway, taking away the biological end products secreted by rapidly dividing cancer cells. To accomplish these needs of supply and/or waste disposal, SCCs and BCCs, like other malignant solid tumors, induce blood vessel growth (angiogenesis), by secreting various growth factors and proteins, that may exert endocrine and paracrine effects (Djokovic et al. 2015; Folkman and Klagsbrun 1987; Folkman 1996). These pro-angiogenic stimulators are then transported to endothelial cells of already existing, nearby located blood vessels, where they cause, via receptor activation, the release of proteolytic enzymes from the vasculature. These enzymes target a particular point on the blood vessel and induce the formation of a characteristic pore that represents the starting point where the new blood vessel will grow from (Nishida et al. 2006). Unlike normal blood vessels, tumor blood vessels are in general dilated with an irregular shape (Gonzalez-Perez and Rueda 2013). It can be emphasized that in either case angiogenesis is an obligate requirement both for transition from small harmless clusters of cells to large life-threatening tumors, and for the metastatic spread of these malignant tumors. Tumor angiogenesis may provide the transport vehicle that enables single cancer cells after these cells have separated from a localized solid tumor have then migrated to and entered these newly build blood vessels, to travel via the bloodstream to distant sites, where they can implant and start the growth of metastases. Evidence from some investigations indicates that the blood vessels in a malignant solid tumor may, in fact, may represent mosaic vessels that are composed not only of endothelial cells but also of tumor cells (Allard et al. 2004). This mosaicity may enable substantial shedding of tumor cells into the vasculature, possibly promoting the distribution of circulating tumor cells in the peripheral blood of cancer patients (Allard et al. 2004). The subsequent growth of the resulting metastases will also need both the supply of nutrients and oxygen and a waste disposal pathway as obligate requirements (Allard et al. 2004).
The angiogenesis-modulating growth factors produced by tumor cells include fibroblast growth factor (FGF – governs via binding to corresponding cell surface FGF receptors in the presence of heparin proteoglycans a variety of cellular functions such as proliferation and differentiation of all cell types required for building arterial vessels, including endothelial cells and smooth muscle cells), vascular endothelial growth factor (VEGF – stimulates primarily the formation of new capillaries; induces endothelial cells via binding to VEGF receptor-2 a tyrosine kinase signaling cascade that promotes the production of factors that variously stimulate vessel permeability [eNOS, producing NO], proliferation/survival [bFGF], migration [ICAMs/VCAMs/MMPs], and finally differentiation into mature blood vessels), platelet-derived growth factor (PDGF), angiopoietins (Ang – required for the formation of mature blood vessels), matrix metalloproteinases (MMPs – required for the formation of new capillaries; promote the degradation of proteins that keep the walls of blood vessels solid, enabling by this proteolysis endothelial cells to migrate into the interstitial matrix, for example, in sprouting angiogenesis), class 3 semaphorins (SEMA3s – regulate angiogenesis by modulating endothelial cell adhesion, survival, proliferation, migration, and the recruitment of pericytes), and the transmembrane ligand Dll4 of the Notch receptor family. Interestingly, potent negative regulatory effects of Dll4 on angiogenesis have been reported (Hellström et al. 2007; Lee et al. 2016; Lobov et al. 2007; Segarra et al. 2008). One study investigated the effects of DII4 both on tumor vascularity and growth. It was shown that the combined inhibition of VEGF and DII4 in endothelial cells blocks proliferation and sprouting of these cells, thereby blocking angiogenesis throughout the tumor and tumor progression. With this inhibition, cancer growth is stopped very effectively. However, after lifting the blockade, cancer cells will start again to proliferate (Hellström et al. 2007; Lee et al. 2016; Lobov et al. 2007; Segarra et al. 2008). It has been demonstrated that, in contrast to normal blood vessels, tumor blood vessels are dilated with an irregular shape.
As outlined above, pro-angiogenic growth factors, such as FGF and VEGF, can induce capillary growth into malignant tumors. Anti-angiogenic therapies are being employed to fight cancer and malignancies (Folkman and Klagsbrun 1987; Folkman 1996), which require an abundance of oxygen and nutrients to proliferate. Several anti-angiogenic therapeutic approaches can be discriminated regarding their mechanism of action including gene therapy (targeting genes of interest for amplification or inhibition) and protein replacement therapy (which primarily targets pro-angiogenic growth factors like FGF-1 or VEGF). A large number of preclinical studies have investigated efficacy and safety of protein-, gene-, and cell-based therapies in animal models of angiogenesis, including models of cardiac ischemia and of peripheral artery disease. Reproducible, promising, and credible successes in these early in vitro and animal studies led to high enthusiasm that this new therapeutic approach could be rapidly translated to a clinical benefit for millions of patients in the Western world suffering from these disorders. However, several decades of clinical testing both gene- and protein-based treatment modalities designed to stimulate angiogenesis in underperfused tissues and organs, has resulted from one disappointment to another.
Notably, there are still serious, fundamental questions and unsolved problems related to gene therapy that are in part due to the complexity of the genetic and molecular basis of angiogenesis. These obstacles include the potentially increased risk for oncogenesis that may be caused by the viral vectors used for successful and effective integration of the therapeutic genes into the genome of target cells, and for other undesired adverse events (AEs), such as inflammatory autoimmune responses and potential toxicity. In contrast, anti-angiogenic protein therapies are in general based on well-defined, precisely structured proteins, with previously defined optimal therapeutic doses and with well-known biological effects of the individual protein that may vary for different disease states. On the other hand, protein therapy is also associated with difficulties that include the optimal mode of delivery. Oral, intravenous, intra-arterial, or intramuscular routes of protein administration are not always comparable in their safety and efficacy, because the therapeutically applied protein may be cleared or metabolized before it can enter the target tissue. For many years, there was the assumption that endothelial cells are genetically more stable than cancer cells. This difference in genomic stability may represent an advantage to targeting endothelial cells using anti-angiogenic therapy, as compared to targeting cancer cells, which rapidly mutate and thereby may acquire so called “drug resistance” to therapy with conventional chemotherapy. For this reason, it has been concluded that endothelial cells represent an optimal therapeutic target for gene therapy.
Angiogenesis in BCCs: Lessons Learned from Embryology
Angiogenesis is a characteristic feature of BCCs that are clinically characterized by the presence of telangiectasias (Table 9.1). It has convincingly been shown that in embryology, components of the Notch signaling pathway govern various important aspects of vascular development, from vascular growth and endothelial tip and stalk cell selection to vascular smooth muscle cell (vSMC) development (Table 9.1).
Notably, Dll4/Notch signaling is absolutely required for normal arterial specification during embryonic development and is a key regulator of embryonic, postnatal, developmental, regenerative, and tumor-sprouting angiogenesis (Djokovic et al. 2015; Lobov et al. 2007). It mediates communication between adjacent endothelial cells (ECs) that lead the sprout formation and adjacent ECs that under Dll4/Notch control remain in the quiescent state in preexisting vasculature or rather proliferate and then migrate, thereby forming the trunk of the new vessel (Djokovic et al. 2015). Mechanistically, Dll4/Notch enables the selective EC departure from preexisting activated endothelium and organized sprout outgrowth by decreasing the VEGFR2/VEGFR1 ratio and therefore reducing the sensitivity of signal-receiving ECs to VEGF (Djokovic et al. 2015). Balanced sprouting is achieved by Dll4-induced “high” Notch signaling and inhibition of sprouting, via suppression of VEGFR signaling in tip cells, which is antagonized in stalk endothelial cells exhibiting JAG1-mediated “low” Notch signaling activity (Djokovic et al. 2015). Although Dll4/Notch blockade potentiates the tumor-driven angiogenic response, it inhibits tumor growth due to the formation of immature and poorly functional vessels that result in reduced tumor perfusion (Djokovic et al. 2015).
Because of resulting defects in angiogenesis of the embryonic and yolk sac vasculature, the systemic knockout of Jag1 is embryonic lethal in mice at ∼E11.5 (Kiernan et al. 2007, reviewed in Mašek and Andersson 2017; reviewed in Reichrath and Reichrath 2020b). A similar picture is found in homozygous Notch2 knockout mice that are characterized by widespread apoptosis and die at ∼E10.5 (Hamada et al. 1999; McCright et al. 2006; reviewed in Mašek and Andersson 2017; reviewed in Reichrath and Reichrath 2020b). The endothelial-specific ablation (via Tie1- or Tie2-Cre) of JAG1 phenocopies systemic Jag1 deletion, demonstrating that a lack of JAG1 signaling from the vascular endothelium likely results in the differentiation defects, loss of vSMCs, and severe disruption of angiogenesis that can be found in JAG1 mutants (Benedito et al. 2009; High et al. 2008; reviewed in Reichrath and Reichrath 2020b). A similar loss of vSMCs has been demonstrated in embryos with homozygous hypomorphic Notch2 (McCright et al. 2001; Wang et al. 2012; reviewed in Reichrath and Reichrath 2020b). Additionally, it has been speculated that the perivascular coverage of newly formed vessels by vSMCs and pericytes is mediated by JAG1-induced expression of integrin αvβ3, which facilitates binding to a basement membrane-specific von Willebrand factor protein (reviewed in Reichrath and Reichrath 2020b; Scheppke et al. 2012). In adults, JAG1 instead functions downstream of Dll4/Notch1 signaling to stimulate maturation of vSMCs after injury through P27kip1-mediated inhibition of proliferation (Boucher et al. 2013; Pedrosa et al. 2015; reviewed in Mašek and Andersson 2017; reviewed in Reichrath and Reichrath 2020b).
JAG1 also governs angiogenesis-associated sprouting; both gain- and loss-of-function investigations in endothelial cells demonstrate that JAG1 stimulates the sprouting of new tip cells during retinal angiogenesis (High et al. 2008; reviewed in Benedito and Hellström 2013; reviewed in Mašek and Andersson 2017; reviewed in Reichrath and Reichrath 2020b). Notably, balanced sprouting is achieved by Dll4-induced “high” Notch signaling and inhibition of sprouting, via suppression of VEGFR signaling in tip cells, which is antagonized in stalk endothelial cells exhibiting JAG1-mediated “low” Notch signaling activity (Benedito et al. 2009; Pedrosa et al. 2015; reviewed in Mašek and Andersson 2017; reviewed in Reichrath and Reichrath 2020b). Although these different aspects of JAG1 and Notch2 signaling have not yet been connected to Alagille or Hajdu–Cheney syndromes (reviewed in Reichrath and Reichrath 2020a, b), they may be of relevance for the severity of these conditions, and the risk for vascular accidents, such as ruptured aneurysms and bleeding (Kamath et al. 2004, 2013; reviewed in Mašek and Andersson 2017; reviewed in Reichrath and Reichrath 2020a, b).
Notch and Cancer Stem Cells: Challenge and Promise to Cure Cancer
An increasing body of evidence indicates an important role of cancer stem cells (CSC) for pathogenesis and progression of many malignancies, including NMSC (Chatterjee and Sil 2019; Espinoza et al. 2013; Quan et al. 2018; Venkatesh et al. 2018). The CSC hypothesis postulates that malignant tumors are characterized by a hierarchical structure that consists of different cell subpopulations, including so called cancer stem–like cells or tumor-initiating cells (TIC), that have the capacity for self-renewal and to develop heterogeneous lineages of cancer cells that comprise the tumor (Quan et al. 2018). Moreover, these cells have in general a slow growth rate, reveal colony- and tumor-forming capacities, as well as altered differentiation, migration, and treatment sensitivity (Quan et al. 2018). They are often resistant to chemotherapy and radiotherapy, thereby resulting in the failure of these conventional therapies (Chatterjee and Sil 2019; Venkatesh et al. 2018). Preventing cancer recurrence and progression by targeting the CSCs is at present a promising perspective to reach the ultimate goal to cure cancer. Although the development of new systemic pharmaceuticals for the treatment of advanced NMSC does not belong to the most urgently needed advances in oncology, the analysis of biology and function of CSC in NMSC has proven to be of particular importance for this rapidly growing field, opening new avenues for the development of new treatment options for a broad variety of different types of cancers (Chatterjee and Sil 2019; Venkatesh et al. 2018). Like any other stem cells, CSCs activate distinct signal transduction pathways, including the Notch signaling pathway, that are of importance for embryonic development and for tissue homeostasis (Chatterjee and Sil 2019; Reichrath and Reichrath 2020a, b, c; Venkatesh et al. 2018). At present, new therapeutic options that target Notch signaling and other pathways that govern stem-cell replication, survival, growth, and differentiation are being developed (Amin et al. 2015; Espinoza et al. 2013; Quan et al. 2018). Notch inhibitors have been introduced to fight cancer and its recurrence either single or in combination with chemotherapy drugs. Targeting Notch and other relevant signaling pathways in CSCs represents a promising future direction for the ultimate therapeutic goal to cure cancer (Espinoza et al. 2013). Great progress in defining the existence and the biological function of distinct CSCs and TICs was achieved in different types of human solid tumors through identification of specific subpopulations characterized by expression of distinct surface determinants and other molecular markers, including CD133, CD44, CXCR4, and ALDH1 (Quan et al. 2018). Recent studies have revealed in cSCC (and also in HNSCC and lung SCC tumors) significant genetic alterations, involving components of Notch, and inactivation or activation of several other common and distinct canonical pathways important for cellular growth, death or survival, senescence migration, and epithelial/mesenchymal differentiation/transition, including WNT, HEDGEHOG, NF-κB, growth factor receptors, RAS-mitogen–activated protein kinase, PI3K–Akt–mTOR, and TP53 (Chatterjee and Sil 2019; Quan et al. 2018; Venkatesh et al. 2018), but their expression and role in CSC and TIC versus other populations in cSCC have until today not been clearly elucidated. Interestingly, it was demonstrated in primary human cSCC tumors and cell line models that the small and distinct CD133+ subpopulation (live CD133+ cells that form spheroid colonies in vitro and tumors in vivo) differentially expresses stem-like and cancer gene signatures linked to Notch1-mediated NF-κB modulation, NF-κB, and WNT pathways (Quan et al. 2018). Furthermore, characterization of the landscape of gene signatures in these CD133+ stem cells revealed activation of a highly orchestrated, complex network of multiple pathways, which were linked to Notch and NF-κB signaling and demonstrated sensitivity to genetic and pharmacologic inhibitors of Notch and NF-κB. The authors of this investigation concluded that their functional, genetic, and pharmacologic studies uncovered a linkage between Notch1, IKKα, and NF-κB pathway activation in maintaining the CD133+ population and its self-renewal ability in established primary cSCC and cell lines (Quan et al. 2018). IKK and NF-κB signaling has been implicated in promoting tumor cell survival, inflammatory, and angiogenesis responses. However, how the molecular components of these signaling pathways are orchestrated to comprise the functionally versatile network that governs the induction and regulation of the different phenotypes that are associated with the distinct CSC/TIC subpopulations in cSCC tumors are not well understood (Quan et al. 2018). It has been suggested that identification of significant molecular/genetic alterations in key pathways that govern the maintenance of the CD133+ CSC phenotype could potentially help identify promising new targets for pharmacological cancer prevention and therapy (Quan et al. 2018). Wnt pathway is an evolutionarily conserved signaling pathway determining patterning of animal embryos, cell fate, cell polarity, and a substantial role in the origin and maintenance of stem cells. Wnt signaling has been found to crosstalk with Notch, and another major developmental pathway, Hedgehog, in many embryological development cascades and in maintaining stemness of stem cells (Chatterjee and Sil 2019). Research has shown that every single one of these three pathways is potent in inducing tumorigenesis, driving tumor progression, and aiding epithelial to mesenchymal transition in malignant cells, apart from maintaining cancer stem cells population inside the tumor tissue (Espinoza et al. 2013). The role of the crosstalks between Wnt, Hedgehog, and Notch signaling in cancer is under intensive research. Inhibition of all the three pathways individually have resulted in tumor regression, but not optimally, as treatment failure and cancer relapse have been found to occur (Chatterjee and Sil 2019). Hence, instead of targeting a single pathway, targeting the crosstalk network could be a better alternative to conventional cancer treatment. Also, elimination of both tumor cells as well as cancer stem cells implies a reduced risk of relapse. Drugs developed to target these crosstalking networks, when used in combinatorial therapy, can hopefully increase the efficacy of the therapy to a very large extent (Chatterjee and Sil 2019).
Conclusion
Notch signaling controls tissue development during embryonal organogenesis, while in adult tissues it contributes to maintenance of cellular differentiation, proliferation, and apoptosis. Moreover, it governs tumor angiogenesis, maintenance of cancer stem cells, and senescence, thereby playing an important role for pathogenesis and progression of skin cancer. Recent findings demonstrate that Notch signaling has in cancer a dual action (either as an oncogene or as a tumor suppressor), depending on the tumor type and the synchronous modulation of other intracellular signaling pathways. Further understanding of the pleiotropic roles of the complex network of Notch and other signaling pathways (including hedgehog, Wnt) in BCC and SCC will hopefully finally result in the development of successful new therapies for NMSC.
Abbreviations
- AE:
-
Adverse event
- AK:
-
Actinic keratosis
- Ang:
-
Angiopoietin
- BCC:
-
Basal cell carcinoma
- BMP:
-
Bone morphogenic protein
- cKO:
-
Conditional knockout
- CSC:
-
Cancer stem cell
- CSL:
-
CBF-1, Su (H), Lag-1-type transcription factor (also termed RBP-J)
- DBD:
-
DNA-binding domain
- Dll:
-
Delta-like
- E:
-
Embryonic day
- FGF:
-
Fibroblast growth factor
- GLI1:
-
Glioma-associated oncogene homolog 1
- Hes:
-
Hairy and enhancer of split
- Hf:
-
Hair follicle
- HPV:
-
Human papilloma virus
- Hrt:
-
Hes-related transcription factor
- IP:
-
Intermediate progenitor
- IPC:
-
Intermediate progenitor cell
- JAG:
-
Jagged
- KO:
-
Knockout
- MAML:
-
Mastermind-like
- MET:
-
Mesenchymal-to-epithelial transition
- MMP:
-
Matrix metalloproteinase
- MR:
-
Mortality rate
- NID:
-
Notch intracellular domain
- NMSC:
-
Nonmelanoma skin cancer
- NO:
-
Nitric oxide
- OD:
-
Oligomerization domain
- PDGF:
-
Platelet-derived growth factor
- PDT:
-
Photodynamic therapy
- PTCH:
-
Patched
- RBP-J:
-
Recombinant recognition sequence-binding protein at the Jκ site (also termed CSL)
- SCC:
-
Squamous cell carcinoma
- Sema:
-
Semaporin
- Shh:
-
Sonic hedgehog
- SMC:
-
Smooth muscle cell
- TA:
-
Transactivation domain
- TLR:
-
Toll-like-receptor
- UVR:
-
Ultraviolet radiation
- VEGF:
-
Vascular endothelial growth factor
- Wnt:
-
Wingless-related integration site
References
Adorno M, Cordenonsi M, Montagner M et al (2009) A Mutant-p53/Smad complex opposes p63 to empower TGF beta-induced metastasis. Cell 137(1):87–98. https://doi.org/10.1016/j.cell.2009.01.039
Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW, Terstappen LW (2004) Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res 10(20):6897–6904. https://doi.org/10.1158/1078-0432.CCR-04-0378. PMID 15501967
Amin ARMR, Karpowicz PA, Carey TE et al (2015) Evasion of anti-growth signaling: a key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin Cancer Biol 35(Suppl):S55–S77. https://doi.org/10.1016/j.semcancer.2015.02.005
Andersson ER, Sandberg R, Lendahl U (2011) Notch signaling: simplicity in design, versatility in function. Development 138:3593–3612. https://doi.org/10.1242/dev.063610
Antonini D, Rossi B, Han R, et al. (2006) An autoregulatory loop directs the tissue-specific expression of p63 through a long-range evolutionarily conserved enhancer. Mol Cell Biol 26(8):3308–3318. https://doi.org/10.1128/MCB.26.8.3308-3318.2006
Apalla Z, Nashan D, Weller RB, Castellsagué X (2017) Skin cancer: epidemiology, disease burden, pathophysiology, diagnosis, and therapeutic approaches. Dermatol Ther 7:5–19. https://doi.org/10.1007/s13555-016-0165-y
Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284:770–776
Aubin-Houzelstein G (2012) Notch signaling and the developing hair follicle. Adv Exp Med Biol 727:142–160. https://doi.org/10.1007/978-1-4614-0899-4_11
Benedito R, Hellström M (2013) Notch as a hub for signaling in angiogenesis. Exp Cell Res 319:1281–1288. https://doi.org/10.1016/j.yexcr.2013.01.010
Benedito R, Roca C, Sörensen I, et al (2009) The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137(6):1124–1135. https://doi.org/10.1016/j.cell.2009.03.025
Blanpain C, Fuchs E (2006) Epidermal stem cells of the skin. Annu Rev Cell Dev Biol 22:339–373. https://doi.org/10.1146/annurev.cellbio.22.010305.104357
Blanpain C, Lowry WE, Pasolli HA, Fuchs E (2006) Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev 20(21):3022–3035. https://doi.org/10.1101/gad.1477606. PMCID: PMC1620020
Bolós V, Grego-Bessa J, de la Pompa JL (2007) Notch signaling in development and cancer. Endocr Rev 28:339–363
Boucher JM, Harrington A, Rostama B, Lindner V, Liaw L (2013) A receptor-specific function for Notch2 in mediating vascular smooth muscle cell growth arrest through cyclin-dependent kinase inhibitor 1B. Circ Res 113:975–985. https://doi.org/10.1161/CIRCRESAHA.113.301272
Bray SJ (2016) Notch signalling in context. Nat Rev Mol Cell Biol 17:722–735. https://doi.org/10.1038/nrm.2016.94
Buono KD, Robinson GW, Martin C, Shi S, Stanley P, Tanigaki K, Honjo T, Hennighausen L (2006) The canonical Notch/RBP-J signaling pathway controls the balance of cell lineages in mammary epithelium during pregnancy. Dev Biol 293:565–580
Burkert J, Wright NA, Alison MR (2006) Stem cells and cancer: an intimate relationship. J Pathol 209:287–297
Chatterjee S, Sil PC (2019) Targeting the crosstalks of Wnt pathway with Hedgehog and Notch for cancer therapy. Pharmacol Res 142:251–261. https://doi.org/10.1016/j.phrs.2019.02.027
Chen X, Zheng Y, Zhu J, Jiang J, Wang J (2001) p73 is transcriptionally regulated by DNA damage, p53 and p73. Oncogene 20:769–774
Chiang C, Swan RZ, Grachtchouk M, Bolinger M, Litingtung Y, Robertson EK, Cooper MK, Gaffield W, Westphal H, Beachy PA, Dlugosz AA (1999) Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol 205:1–9
Crowson AN (2006) Basal cell carcinoma: biology, morphology and clinical implications. Mod Pathol 19(Suppl 2):S127–S147. Review. PubMed PMID: 16446711
Dahmane N, Lee J, Robins P, Heller P, Ruiz i Altaba A (1997) Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 389:876–881
Davison TS, Vagner C, Kaghad M, Ayed A, Caput D, Arrowsmith CH (1999) p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem 274:18709–18714
Della Gatta G, Bansal M, Ambesi-Impiombato A, Antonini A, Missero C, di Bernardo D (2008) Direct targets of the TRP63 transcription factor revealed by a combination of gene expression profiling and reverse engineering. Genome Res 18:939–948
Demehri S, Turkoz A, Kopan R (2009) Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 16(1):55–66
Dexter JS (1914) The analysis of a case of continuous variation in Drosophila by a study of its linkage relations. Am Nat 48:712–758. https://doi.org/10.1086/279446
Di Como CJ, Gaiddon C, Prives C (1999) p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol 19:1438–1449
Didona D, Paolino G, Bottoni U, Cantisani C (2018) Non-melanoma skin cancer pathogenesis overview. Biomedicines 6(1):pii: E6. https://doi.org/10.3390/biomedicines6010006. Review. PubMed PMID: 29301290; PubMed Central PMCID: PMC5874663
Djokovic D, Trindade A, Gigante J, Pinho M, Harris AL, Duarte A (2015) Incomplete Dll4/Notch signaling inhibition promotes functional angiogenesis supporting the growth of skin papillomas. BMC Cancer 15:608. https://doi.org/10.1186/s12885-015-1605-2
Edlund K, Larsson O, Ameur A, Bunikis I, Gyllensten U, Leroy B, Sundström M, Micke P, Botling J, Soussi T (2012) Data-driven unbiased curation of the TP53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proc Natl Acad Sci U S A 109(24):9551–9556. https://doi.org/10.1073/pnas.1200019109. Epub 2012 May 24. PubMed PMID: 22628563; PubMed Central PMCID: PMC3386058
Erb P, Ji J, Kump E, Mielgo A, Wernli M (2008) Apoptosis and pathogenesis of melanoma and nonmelanoma skin cancer. Adv Exp Med Biol 624:283–295. https://doi.org/10.1007/978-0-387-77574-6_22. Review. PubMed PMID: 18348464
Espinoza I, Pochampally R, Xing F, Watabe K, Miele L (2013) Notch signaling: targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco Targets Ther 6:1249–1259. https://doi.org/10.2147/OTT.S36162
Folkman J (1996) Fighting cancer by attacking its blood supply. Sci Am 275(3):150–154. https://doi.org/10.1038/scientificamerican0996-150. PMID 8701285
Folkman J, Klagsbrun M (1987) Angiogenic factors. Science 235(4787):442–447. https://doi.org/10.1126/science.2432664. PMID 2432664
Fuchs E (2007) Scratching the surface of skin development. Nature 445(7130):834–842. https://doi.org/10.1038/nature05659
Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C (2001) A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol 21:1874–1887
Gallant-Behm CL, Ramsey MR, Bensard CL et al (2012) ΔNp63α represses anti-proliferative genes via H2A.Z deposition. Genes Dev 26(20):2325–2336. https://doi.org/10.1101/gad.198069.112
Garber K (2007) Notch emerges as new cancer drug target. J Natl Cancer Inst 99:1284–1285
Gazave E, Lapébie P, Richards GS, Brunet F, Ereskovsky AV, Degnan BM, Borchiellini C, Vervoort M, Renard E (2009) Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC Evol Biol 9:249. https://doi.org/10.1186/1471-2148-9-249
Gonzalez-Perez RR, Rueda BR (2013) Tumor angiogenesis regulators, 1st edn. Taylor & Francis, Boca Raton, p 347. ISBN 978-1-4665-8097-8
Grachtchouk M, Mo R, Yu S, Zhang X, Sasaki H, Hui CC, Dlugosz AA (2000) Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet 24:216–217
Grachtchouk M, Pero J, Yang SH et al (2011) Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. J Clin Invest 121(5):1768–1781. https://doi.org/10.1172/JCI46307
Gridley T (2003) Notch signaling and inherited disease syndromes. Hum Mol Genet 12:R9–R13. https://doi.org/10.1093/hmg/ddg052
Gritli-Linde A, Hallberg K, Harfe BD, Reyahi A, Kannius-Janson M, Nilsson J, Cobourne MT, Sharpe PT, McMahon AP, Linde A (2007) Abnormal hair development and apparent follicular transformation to mammary gland in the absence of hedgehog signaling. Dev Cell 12:99–112
Hamada Y, Kadokawa Y, Okabe M, Ikawa M, Coleman JR, Tsujimoto Y (1999) Mutation in ankyrin repeats of the mouse Notch2 gene induces early embryonic lethality. Development 126:3415–3424
Harmes DC, Bresnick E, Lubin EA, Watson JK, Heim KE, Curtin JC, Suskind AM, Lamb J, DiRenzo J (2003) Positive and negative regulation of ΔN-p63 promoter activity by p53 and ΔN-p63-α contributes to differential regulation of p53 target genes. Oncogene 22:7607–7616
Hellström M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalén M, Gerhardt H, Betsholtz C (2007) Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445(7129):776–780. https://doi.org/10.1038/nature05571. PMID 17259973
High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA (2008) Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A 105:1955–1959. https://doi.org/10.1073/pnas.0709663105
Hoare M, Narita M (2018) Notch and senescence. Adv Exp Med Biol 1066:299–318. https://doi.org/10.1007/978-3-319-89512-3_15
Hutchin ME, Kariapper MS, Grachtchouk M, Wang A, Wei L, Cummings D, Liu J, Michael LE, Glick A, Dlugosz AA (2005) Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev 19(2):214–223. Epub 2004 Dec 29. PubMed PMID: 15625189; PubMed Central PMCID: PMC545881
Jahoda C, Reynolds A (2000) Skin stem cells-a hairy issue. Nat Med 6:1095–1097
Jayaraman SS, Rayhan DJ, Hazany S, Kolodney MS (2014) Mutational landscape of basal cell carcinomas by whole-exome sequencing. J Invest Dermatol 134(1):213–220. https://doi.org/10.1038/jid.2013.276. Epub 2013 Jun 17. PubMed PMID: 23774526
Kamath BM, Spinner NB, Emerick KM, Chudley AE, Booth C, Piccoli DA, Krantz ID (2004) Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation 109:1354–1358. https://doi.org/10.1161/01.CIR.0000121361.01862.A4
Kamath BM, Piccoli DA, Magee JC, Sokol RJ (2012) Pancreatic insufficiency is not a prevalent problem in Alagille syndrome. J Pediatr Gastroenterol Nutr 55:612–614. https://doi.org/10.1097/MPG.0b013e31825eff61
Kamath BM, Spinner NB, Rosenblum ND (2013) Renal involvement and the role of Notch signalling in Alagille syndrome. Nat Rev Nephrol 9:409–418. https://doi.org/10.1038/nrneph.2013.102
Kartasheva N, Contente A, Lenz-Stoppler C, Roth J, Dobbelstein M (2002) p53 induces the expression of its antagonist p73ΔN, establishing an autoregulatory feedback loop. Oncogene 21:4715–4727
Kidd S, Kelley MR, Young MW (1986) Sequence of the notch locus of Drosophila melanogaster: relationship of the encoded protein to mammalian clotting and growth factors. Mol Cell Biol 6(9):3094–3108
Kiernan AE, Ahituv N, Fuchs H, Balling R, Avraham KB, Steel KP, Hrabé de Angelis M (2001) The Notch ligand Jagged1 is required for inner ear sensory development. Proc Natl Acad Sci U S A 98:3873–3878. https://doi.org/10.1073/pnas.071496998
Kiernan AE, Xu J, Gridley T (2006) The Notch ligand JAG1 is required for sensory progenitor development in the mammalian inner ear. PLoS Genet 2:e4. https://doi.org/10.1371/journal.pgen.0020004
Kiernan AE, Li R, Hawes NL, Churchill GA, Gridley T (2007) Genetic background modifies inner ear and eye phenotypes of Jag1 heterozygous mice. Genetics 177(1):307–311
Koch U, Radtke F (2007) Notch and cancer: a double-edged sword. Cell Mol Life Sci 64:2746–2762
Kopan R, Ilagan MXG (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137:216–233. https://doi.org/10.1016/j.cell.2009.03.045
Kouwenhoven EN, van Heeringen SJ, Tena J, Oti M, Dutilh BE, Alonso ME, de la Calle-Mustienes E, Smeenk L, Rinne T, Parsaulian L, Bolat E, Jurgelenaite R, Huynen MA, Hoischen A, Veltman JA, Brunner HG, Roscioli T, Oates E, Wilson M, Manzanares M, Gomez-Skameta JL, Stunnenberg HG, Lohrum M, van Bokhoven H, Zhou H (2010) Genome-wide profiling of p63 DNA–binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet 6(8):e1001065
Lane D, Levine A (2010) p53 research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol 2:a000893
Lang GA, Iwakuma T, Suh Y-A, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El-Naggar AK, Lozano G (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119:861–872
Lee D, Kim D, Bin Choi Y, Kang K, Sung ES, Ahn JH, Goo J, Yeom DH, Sook Jang H, Duk Moon K, Hoon Lee S, You WK (2016) Simultaneous blockade of VEGF and Dll4 by HD105, a bispecific antibody, inhibits tumor progression and angiogenesis. mAbs 8:892–904
Leiter U, Eigentler T, Garbe C (2014) Epidemiology of skin cancer. Adv Exp Med Biol 810:120–140
Liu ZL, Li Y, Kong QY, Zhan C, Wang Q, Chen XY, Sun Y, Wen S, Tu CX, Liu J, Li H (2008) Immunohistochemical profiling of Wnt, NF-kappaB, Stat3 and Notch signaling in human epidermal tumors. J Dermatol Sci 52(2):133–136. https://doi.org/10.1016/j.jdermsci.2008.06.011. Epub 2008 Aug 13. PubMed PMID: 18703315
Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ (2007) Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci U S A 104(9):3219–3224. https://doi.org/10.1073/pnas.0611206104. PMC 1805530. PMID 17296940
Lokshin M, Li Y, Gaiddon C, Prives C (2007) p53 and p73 display common and distinct requirements for sequence specific binding to DNA. Nucleic Acids Res 35:340–352
López-Takegami JC, Wolter M, Löser C, Maiweg C, Jones M, Metze D, Böer-Auer A (2016) Classification of cysts with follicular germinative differentiation. J Cutan Pathol 43(3):191–199. https://doi.org/10.1111/cup.12619. Epub 2015 Oct 12. PubMed PMID: 26347259
Lowell S, Jones P, Le Roux I, Dunne J, Watt FM (2000) Stimulation of human epidermal differentiation by delta-notch signalling at the boundaries of stem-cell clusters. Curr Biol 10:491–500
Marcel V, Petit I, Murray-Zmijewski F, Goullet de Rugy T, Fernandes K, Meuray V, Diot A, Lane DP, Aberdam D, Bourdon J-C (2012) Diverse p63 and p73 isoforms regulate Δ133p53 expression through modulation of the internal TP53 promoter activity. Cell Death Differ 19:816–826
Martynova E, Pozzi S, Basile V, et al (2012) Gain-of-function p53 mutants have widespread genomic locations partially overlapping with p63. Oncotarget 3(2):132–143. https://doi.org/10.18632/oncotarget.447
Mašek J, Andersson ER (2017) The developmental biology of genetic Notch disorders. Development 144:1743–1763. https://doi.org/10.1242/dev.148007
Massi D, Panelos J (2012) Notch signaling and the developing skin epidermis. Adv Exp Med Biol 727:131–141. https://doi.org/10.1007/978-1-4614-0899-4_10. Review. PubMed PMID: 22399344
McCright B, Gao X, Shen L, Lozier J, Lan Y, Maguire M, Herzlinger D, Weinmaster G, Jiang R, Gridley T (2001) Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development 128:491–502
McCright B, Lozier J, Gridley T (2002) A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 129:1075–1082
McCright B, Lozier J, Gridley T (2006) Generation of new Notch2 mutant alleles. Genesis 44:29–33. https://doi.org/10.1002/gene.20181
McDougall SR, Anderson ARA, Chaplain MAJ (2006) Mathematical modeling of dynamic adaptive tumor-induced angiogenesis: clinical implications and therapeutic targeting strategies. J Theor Biol 241:564–589
McIntyre B, Asahara T, Alev C (2020) Overview of basic mechanisms of notch signaling in development and disease. Adv Exp Med Biol 1227:9–27
McMahon AP, Ingham PW, Tabin CJ (2003) Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol 53:1–114. Review. PubMed PMID: 12509125
Meester JAN, Verstraeten A, Alaerts M, Schepers D, Van Laer L, Loeys BL (2019) Overlapping but distinct roles for NOTCH receptors in human cardiovascular disease. Clin Genet 95(1):85–94. https://doi.org/10.1111/cge.13382. Epub 018 Jun 10. Review. PubMed PMID: 2976
Missero C, Antonini D (2014) Cross-talk among p53 family members in cutaneous squamous cell carcinoma. Exp Dermatol 23:143–146
Moriyama M, Durham AD, Moriyama H, Hasegawa K, Nishikawa S, Radtke F, Osawa M et al (2008) Multiple roles of Notch signaling in the regulation of epidermal development. Dev Cell 14(4):594–604
Muller PA, Vousden KH (2013) p53 mutations in cancer. Nat Cell Biol 15:2–8
Najafzadeh N, Esmaeilzade B, Dastan Imcheh M (2015) Hair follicle stem cells: in vitro and in vivo neural differentiation. World J Stem Cells 7:866–872
Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F (2003) Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 33:416–421
Nishida N, Yano H, Nishida T, Kamura T, Kojiro M (2006) Angiogenesis in cancer. Vasc Health Risk Manag 2(3):213–219. https://doi.org/10.2147/vhrm.2006.2.3.213. PMC 1993983. PMID 17326328
Okuyama R, Nguyen BC, Talora C, Ogawa E, Tommasi di Vignano A, Lioumi M, Chiorino G, Tagami H, Woo M, Dotto GP (2004) High commitment of embryonic keratinocytes to terminal differentiation through a Notch1-caspase 3 regulatory mechanism. Dev Cell 6:551–562
Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA (2004) Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119:847–860
Oro AE, Higgins K (2003) Hair cycle regulation of Hedgehog signal reception. Dev Biol 255:238–248
Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH Jr, Scott MP (1997) Basal cell carcinomas in mice overexpressing sonic hedgehog. Science 276:817–821
Pedrosa A-R, Trindade A, Fernandes A-C, Carvalho C, Gigante J, Tavares AT, Diéguez-Hurtado R, Yagita H, Adams RH, Duarte A (2015) Endothelial jagged1 antagonizes Dll4 regulation of endothelial branching and promotes vascular maturation downstream of Dll4/Notch1. Arterioscler Thromb Vasc Biol 35:1134–1146. https://doi.org/10.1161/ATVBAHA.114.304741
Peterson SC, Eberl M, Vagnozzi AN, Belkadi A, Veniaminova NA, Verhaegen ME, Bichakjian CK, Ward NL, Dlugosz AA, Wong SY (2015) Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 16(4):400–412. https://doi.org/10.1016/j.stem.2015.02.006. PubMed PMID: 25842978; PubMed Central PMCID: PMC4387376
Purow B (2012) Notch inhibition as a promising new approach to cancer therapy. Adv Exp Med Biol 727:305–319
Quan XX, Hawk NV, Chen W, Coupar J, Lee SK, Petersen DW, Meltzer PS, Montemarano A, Braun M, Chen Z, Van Waes C (2018) Targeting Notch1 and IKKα enhanced NF-κB activation in CD133(+) skin cancer stem cells. Mol Cancer Ther 17(9):2034–2048. https://doi.org/10.1158/1535-7163.MCT-17-0421. Epub 2018 Jun 29. PubMed PMID: 29959199; PubMed Central PMCID: PMC6461743
Reichrath J, Rass K (2014) Ultraviolet damage, DNA repair and vitamin D in nonmelanoma skin cancer and in malignant melanoma: an update. Adv Exp Med Biol 810:208–233. Review. PubMed PMID: 25207368
Reichrath J, Reichrath S (2012a) Notch-signaling and nonmelanoma skin cancer: an ancient friend, revisited. Adv Exp Med Biol 727:265–271. https://doi.org/10.1007/978-1-4614-0899-4_20
Reichrath S, Reichrath J (2012b) No evidence for induction of key components of the Notch signaling pathway (Notch-1, Jagged-1) by treatment with UV-B, 1,25(OH)(2)D(3), and/or epigenetic drugs (TSA, 5-Aza) in human keratinocytes in vitro. Dermatoendocrinol 4(1):44–52. https://doi.org/10.4161/derm.19027
Reichrath J, Reichrath S (2020a) Notch pathway and inherited diseases: challenge and promise. Adv Exp Med Biol 1218:159–187. https://doi.org/10.1007/978-3-030-34436-8_9
Reichrath J, Reichrath S (2020b) Notch signaling and embryonic development: an ancient friend, revisited. Adv Exp Med Biol 1218:9–37. https://doi.org/10.1007/978-3-030-34436-8_2
Reichrath J, Reichrath S (2020c) A snapshot of the molecular biology of Notch signaling: challenges and promises. Adv Exp Med Biol 1227:1–7. https://doi.org/10.1007/978-3-030-36422-9_1
Reichrath S, Müller CS, Gleissner B, Pfreundschuh M, Vogt T, Reichrath J (2010) Notch- and vitamin D signaling in 1,25(OH)2D3-resistant glioblastoma multiforme (GBM) cell lines. J Steroid Biochem Mol Biol 121(1–2):420–424. https://doi.org/10.1016/j.jsbmb.2010.02.028
Richards GS, Degnan BM (2009) The dawn of developmental signaling in the metazoa. Cold Spring Harb Symp Quant Biol 74:81–90. https://doi.org/10.1101/sqb.2009.74.028
Rishikaysh P, Dev K, Diaz D, Qureshi WM, Filip S, Mokry J (2014) Signaling involved in hair follicle morphogenesis and development. Int J Mol Sci 15(1):1647–1670. https://doi.org/10.3390/ijms15011647. Review. PubMed PMID: 24451143; PubMed Central PMCID: PMC3907891
Rocco JW, Leong CO, Kuperwasser N, De Young MP, Ellisen LW (2006) p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 9:45–56
Roemer K (2012) Notch and the p53 clan of transcription factors. Adv Exp Med Biol 727:223–240. https://doi.org/10.1007/978-1-4614-0899-4_17
Sasaki Y, Ishida S, Morimoto I et al (2002) The p53 family member genes are involved in the Notch signal pathway. J Biol Chem 277(1):719–724. https://doi.org/10.1074/jbc.M108080200
Satchell AC, Barnetson RS, Halliday GM (2004) Increased Fas ligand expression by T cells and tumour cells in the progression of actinic keratosis to squamous cell carcinoma. Br J Dermatol 151:42–49
Scheppke L, Murphy EA, Zarpellon A, Hofmann JJ, Merkulova A, Shields DJ, Weis SM, Byzova TV, Ruggeri ZM, Iruela-arispe ML et al (2012) Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood 119:2149–2158. https://doi.org/10.1182/blood-2011-04-348706
Segarra M, Williams CK, Sierra ML, Bernarndo M, McCormick PJ, Meric D, Regino C, Choyke P, Tosato G (2008) Dll4 activation of Notch signaling reduces tumore vascularity and inhibits tumor growth. Blood J 112(5):1904–1911
Shi FT, Yu M, Zloty D, Bell RH, Wang E, Akhoundsadegh N, Leung G, Haegert A, Carr N, Shapiro J, McElwee KJ (2017) Notch signaling is significantly suppressed in basal cell carcinomas and activation induces basal cell carcinoma cell apoptosis. Mol Med Rep 15(4):1441–1454. https://doi.org/10.3892/mmr.2017.6163
Spill F, Guerrero P, Alarcon T, Maini PK, Byrne HM (2015) Mesoscopic and continuum modelling of angiogenesis. J Math Biol 70(3):485–532. https://doi.org/10.1007/s00285-014-0771-1
Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y, Castagnoli L, Levine AJ, Sacchi A, Cesareni G, Oren M, Blandino G (2000) Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem 275:29503–29512
Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, Del Sar G, Levrero M, Sacchi A, Oren M, Blandino G (2002) Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J Biol Chem 277:18817–18826
Thélu J, Viallet JP, Dhouailly D (1998) Differential expression pattern of the three Fringe genes is associated with epidermal differentiation. J Invest Dermatol 111(5):903–906. https://doi.org/10.1046/j.1523-1747.1998.00372.x
Thelu J, Rossio P, Favier B (2002) Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma, psoriasis and wound healing. BMC Dermatol 2:7
Venkatesh V, Nataraj R, Thangaraj GS et al (2018) Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig 5:5. https://doi.org/10.21037/sci.2018.02.02
Wang S, El-Deiry WS (2006) p73 or p53 directly regulates human p53 transcription to maintain cell cycle checkpoints. Cancer Res 66:6982–6989
Wang X, Pasolli HA, Williams T, Fuchs E (2008) AP-2 factors act in concert with Notch to orchestrate terminal differentiation in skin epidermis. J Cell Biol 183:37–48
Wang Q, Zhao N, Kennard S, Lilly B (2012) Notch2 and Notch3 function together to regulate vascular smooth muscle development. PLoS One 7:e37365. https://doi.org/10.1371/journal.pone.0037365
Weng AP, Aster JC (2004) Multiple niches for Notch in cancer: context is everything. Curr Opin Genet Dev 14(1):48–54. Review. PubMed PMID: 15108805
Wuest M, Dummer R, Urosevic M (2007) Induction of the members of Notch pathway in superficial basal cell carcinomas treated with imiquimod. Arch Dermatol Res 299(10):493–498. Epub 2007 Oct 6. PubMed PMID: 17922128
Yamamoto N, Tanigaki K, Han H, Hiai H, Honjo T (2003) Notch/RBP-J signaling regulates epidermis/hair fate determination of hair follicular stem cells. Curr Biol 13(4):333–338. PubMed PMID: 12593800
Yang A, Zhu Z, Kettenbach A, Kapranov P, McKeon F, Gingeras TR, Struhl K (2010) Genome-wide mapping indicates that p73 and p63 co-occupy target sites and have similar DNA-binding profiles In Vivo. PLoS One 5:e11572
Zanotti S, Canalis E (2016) Notch signaling and the skeleton. Endocr Rev 37:223–253. https://doi.org/10.1210/er.2016-1002
Zoumpourlis V, Solakidi S, Papathoma A, Papaevangeliou D (2003) Alterations in signal transduction pathways implicated in tumour progression during multistage mouse skin carcinogenesis. Carcinogenesis 24:1159–1165
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Reichrath, J., Reichrath, S. (2021). The Impact of Notch Signaling for Carcinogenesis and Progression of Nonmelanoma Skin Cancer: Lessons Learned from Cancer Stem Cells, Tumor Angiogenesis, and Beyond. In: Reichrath, J., Reichrath, S. (eds) Notch Signaling in Embryology and Cancer. Advances in Experimental Medicine and Biology, vol 1287. Springer, Cham. https://doi.org/10.1007/978-3-030-55031-8_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-55031-8_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-55030-1
Online ISBN: 978-3-030-55031-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)