
Abstract
Biophysical properties of membranes are dependent on their glycerophospholipid compositions. Lysophospholipid acyltransferases (LPLATs) selectively incorporate fatty chains into lysophospholipids to affect the fatty acid composition of membrane glycerophospholipids. Lysophosphatidic acid acyltransferases (LPAATs) of the 1-acylglycerol-3-phosphate O-acyltransferase (AGPAT) family incorporate fatty chains into phosphatidic acid during the de novo glycerophospholipid synthesis in the Kennedy pathway. Other LPLATs of both the AGPAT and the membrane bound O-acyltransferase (MBOAT) families further modify the fatty chain compositions of membrane glycerophospholipids in the remodeling pathway known as the Lands’ cycle. The LPLATs functioning in these pathways possess unique characteristics in terms of their biochemical activities, regulation of expressions, and functions in various biological contexts. Essential physiological functions for LPLATs have been revealed in studies using gene-deficient mice, and important roles for several enzymes are also indicated in human diseases where their mutation or dysregulation causes or contributes to the pathological condition. Now several LPLATs are emerging as attractive therapeutic targets, and further understanding of the mechanisms underlying their physiological and pathological roles will aid in the development of novel therapies to treat several diseases that involve altered glycerophospholipid metabolism.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

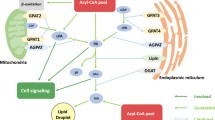
Keywords
2.1 Function and Diversity of Cellular Membranes
Cellular membranes are comprised mainly of protein and lipids; and glycerophospholipids, along with sphingolipids and cholesterol, are the major lipid components. The glycerophospholipids share a common glycerol-based structure with a phosphate-containing headgroup at the sn-3 position of the glycerol backbone, and two hydrophobic fatty chains attached to the sn-1 and sn-2 positions (Fig. 2.1). This basic structural unit supports formation of lipid bilayers, which serve various cellular functions: to separate the interior of cells from their physical environments; to form compartments within cells that define organelles and structures; to form domains of molecular interactions; and as storage of lipid mediator precursors. Some glycerophospholipids also form monolayers such as outer monolayers of HDL (high-density lipoprotein) particles, LDL (low-density lipoprotein) particles, and lipid droplets; as well as specialized surfactant monolayers such as pulmonary surfactant. A great diversity of glycerophospholipid species exists due to the many possible combinations of headgroups, fatty chains, and linkages of the fatty chains to the glycerol backbone. This diversity varies among cell types and tissues and imparts membranes with unique biophysical and biological properties.

Glycerophospholipid structure
Basic structures of diacyl-, alkyl-acyl-, and alkenyl-acyl (plasmalogen) -glycerophospholipids are shown. X represents -OH, choline, ethanolamine, serine, inositol, or glycerol for PA, PC, PE, PS, PI, and PG, respectively. Stereospecific numbering (sn) positions of the glycerol backbone are indicated by red arrows. R1 and R2 represent the fatty chains at sn-1 and sn-2, respectively. Acyl, alkyl, and alkenyl linkages at sn-1 are highlighted in blue. GPL, glycerophospholipid
2.1.1 Glycerophospholipid Classes
Glycerophospholipids possess a phosphate-containing headgroup esterified to sn-3 of their glycerol backbones. In the simplest glycerophospholipid, phosphatidic acid (PA), the phosphate is the headgroup and no further addition is made. PA is not very abundant in membranes but is a key intermediate in biosynthesis of all other classes of glycerophospholipids. The other classes are formed by esterification of the phosphate group to any of several alcohols (choline, ethanolamine, serine, inositol, or glycerol) forming the glycerophospholipid classes phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), and phosphatidylglycerol (PG). Cardiolipin (CL) is structurally unique in having a modified dimeric PG structure, with a glycerol backbone esterified to two PA moieties.
The relative amounts of the different glycerophospholipid classes in cellular membranes varies. PC followed by PE are the most abundant glycerophospholipids in membranes, and together typically account for over half of the glycerophospholipids. Other classes of glycerophospholipids are typically less abundant, PI and PS classes each typically account for ~5–10% of all glycerophospholipids, and lesser amounts of CL, PG, and PA (<5% each) are generally present [1]. However, the less abundant classes of glycerophospholipids may have specialized functions and important roles in various biological contexts, such as roles for PS in cell fusion, phagocytosis, and apoptosis [2] and roles for PI in vesicle trafficking [3]. These less abundant classes may also be concentrated within cells in certain structures, domains, or organelles; for example, CL is a major component of the inner mitochondrial membrane, where it has important roles in mitochondrial structure and function [4].
2.1.2 Fatty Chain Diversity
Fatty chains found in glycerophospholipids vary in terms of carbon chain length and double-bond number. The fatty chain compositions of membrane glycerophospholipids varies among organelles, cell types, tissues, and physiological states; and these compositions can strongly affect biophysical properties of the membranes in terms of fluidity, flexibility, fusion, fission, and curvature [5, 6]. Carbon chain lengths affect membrane thickness and microdomain formation, which can strongly affect functions of transmembrane proteins and membrane-associated proteins. Double-bond number affects membrane thickness, viscosity, flexibility, and strength. Compared to unsaturated and monounsaturated fatty chains, polyunsaturated fatty chains in membrane phospholipids impart flexibility and curvature to the membranes. Polyunsaturated fatty acids in membrane glycerophospholipids also represent important sources of precursor molecules that can be de-esterified by phospholipase A (PLA) 1/2 s and converted to lipid mediators [7, 8].
2.1.3 Glycerophospholipid Linkages
In addition to chain length and saturation, the linkages of the fatty chains to the glycerol backbone further increase the diversity of glycerophospholipid species. Acyl linkages are the most common at both sn-1 and sn-2. Alkyl and alkenyl linkages may also occur at sn-1, forming what are known as plasmanyl- or plasmenyl-phospholipids, respectively (Fig. 2.1). Plasmanyl- and plasmenyl-phospholipids have a different biosynthetic route than diacyl glycerophospholipids. Rather than being synthesized from a PA precursor, as are diacyl species, their synthesis begins in peroxisomes with acylation of dihydroxyacetone phosphate (DHAP) by the enzyme DHAP-acyltransferase (DHAPAT). This reaction produces 1-acyl-DHAP, which is enzymatically converted to 1-alkyl-DHAP and then 1-alkylglycerol-3-phosphate. 1-alkylglycerol-3-phosphate is acylated at sn-2 and further metabolized in the endoplasmic reticulum to form the plasmanyl- and plasmenyl-phospholipids [9].
The plasmanyl-phospholipids, with alkyl linkages at sn-1, usually are of the PC class of glycerophospholipids. A notable example is platelet-activating factor (PAF; 1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine). PAF is a potent inflammatory phospholipid mediator, and produced by lyso-PAF acetyltransferase activities of lysophosphatidylcholine acyltransferase (LPCAT)1 or LPCAT2 from lyso-PAF and acetyl-CoA [10, 11].
The plasmenyl glycerophospholipids, commonly known as plasmalogens, have alkenyl linkages at sn-1 and are enriched with polyunsaturated fatty acids at sn-2. This unique structure may impart them with special properties, however determining the biological functions of plasmalogens has often been challenging. Overall, plasmalogens account for 15–20% of all phospholipids in cell membranes and are highly enriched in certain tissues. Plasmalogens of the PE class are generally the most abundant, as well as PC plasmalogens in certain tissues. For instance, in adult humans over half of the PE glycerophospholipids in heart and brain may be plasmalogen; and, while PC in brain is low in plasmalogen, ~25% of PC in heart may be plasmalogen. Other tissues rich in plasmalogens include kidney, lung, skeletal muscle, and ocular lens; and plasmalogens may also have unique roles during development and in immune cells [9].
2.1.4 De novo Glycerophospholipid Biosynthesis (Kennedy Pathway)
De novo glycerophospholipid production proceeds through pathways that involve production of PA as a precursor in production of all mammalian diacyl glycerophospholipids. These enzymatic reactions were first described by Kennedy and Weiss in 1956 and are now termed the Kennedy pathway [12]. First, glycerol-3-phosphate acyltransferases (GPATs) esterify an acyl chain to glycerol-3-phosphate (G3P) at the sn-1 position to produce lyso-PA (LPA; 1-acylglycerol-3-phosphate). Next, LPA-acyltransferases (LPAATs) esterify a second acyl chain to the sn-2 position to produce PA. Newly synthesized PA may be further converted by either of two metabolic routes active in glycerophospholipid biosynthesis. The PA may be dephosphorylated by PA phosphatases (PAPs; also known as lipins) to produce diacylglycerol (DAG), which may be further metabolized to produce PC, PE, PS, and triglyceride (TG). Alternatively, PA may be acted upon by cytidine diphosphate (CDP) -DAG synthase to produce CDP-DAG, which may be further metabolized to produce CL, PI and PG [13] (Fig. 2.2).

Glycerophospholipid de novo synthesis and remodeling pathways
Basic mammalian pathways to produce diacyl-glycerophospholipids is shown (see text for details). Acyl chains at sn-2 are specified during production of PA from LPA in Kennedy pathway (green circle), and may be extensively remodeled during Lands’ cycle reactions (orange circles). Abbreviations: CDP-DAG cytidine diphosphate-diacylglycerol, CDP-DGS cytidine diphosphate-diacylglycerol synthase, DAG diacylglycerol, DGAT diacylglycerol acyltransferase, G3P glycerol-3-phosphate, GPAT glycerol-3-phosphate acyltransferase, LPC lyso-PC, LPE lyso-PE, LPG lyso-PG, LPI lyso-PI, LPS lyso-PS, MAG monoacylglycerol, PAP phosphatidic acid phosphatase, PEMT PE methyltransferase, PSD PS decarboxylase, PSS1 PS synthase 1, PSS2 PS synthase 2, TG triglyceride
2.1.5 PC and PE Are Produced from Diacylglycerol
PC and PE are the most abundant glycerophospholipids, and in the Kennedy pathway two parallel pathways convert DAG to either PC or PE. The Kennedy pathway to produce PC proceeds in three sequential reactions: (a) choline kinase (CK) phosphorylates choline to form phosphocholine; (b) CTP:phosphocholine cytidyltransferase (PCYT1) forms CDP-choline from phosphocholine and cytidine triphosphate (CTP); and (c) CDP choline:1,2-DAG cholinephosphotransferase (CPT) utilizes CDP-choline and DAG as substrates to produce PC. In the analogous Kennedy pathway to produce PE, (a) ethanolamine kinase (EK) phosphorylates ethanolamine to form phosphoethanolamine; (b) CTP:phosphoethanolamine cytidylyltransferase (PCYT2) forms CDP-ethanolamine from CTP and phosphoethanolamine; and (c) CDP-ethanolamine:1,2-diacylglycerol ethanolaminephosphotransferase (EPT) utilizes CDP-ethanolamine and DAG to produce PE [1].
2.1.6 Additional Reactions to Generate Glycerophospholipid Classes
Other enzymatic reactions occur that mediate conversion of glycerophospholipids between classes. An alternative pathway to produce PC from PE exists that is called the PE methyltransferase (PEMT) pathway. In this pathway, PC production occurs via three sequential methylations of PE, all catalyzed by the enzyme PEMT. This pathway to PC production is only active in liver, where it accounts for ~30% of PC production [14].
PS is produced from PC and PE via base exchange reactions catalyzed by either of two enzymes, PS synthase (PSS) 1 or PSS2. Both enzymes produce PS in regions of the endoplasmic reticulum; PSS1 converts PC to PS, and PSS2 converts PE to PS. PS that has been transported to the mitochondria may be converted back to PE by the actions of another enzyme, PS decarboxylase (PSD) [1].
These pathways for production of PC, PE, and PS all proceed from DAG as a common intermediary. The pathways to produce the other classes of glycerophospholipids PI, CL, and PG all involve a series of enzymatic reactions that utilize CDP-DAG, produced from PA, as a precursor. Thus, PA is a common precursor molecule in de novo synthesis of all diacyl glycerophospholipids. PA is usually maintained at relatively low levels in cells, and newly synthesized PA may be rapidly converted to other glycerophospholipid classes. Therefore, the enzymes that regulate fatty chain compositions of PA also regulate the fatty chain compositions of all glycerophospholipids produced in Kennedy pathway [12].
2.2 Regulation of Acyl Chain Compositions
During Kennedy pathway of glycerophospholipid synthesis, acyl chains are determined first when GPAT incorporates an acyl chains into sn-1 position of G3P to produce LPA, and again when LPAAT incorporates an acyl chain into sn-2 to produce PA. GPAT enzymes preferentially utilize saturated or mono-unsaturated acyl-CoA substrates for incorporation into sn-1, and different LPAAT enzymes vary in their acyl-CoA selectivities to utilize saturated, mono-unsaturated, or poly-unsaturated acyl-CoA substrates for incorporation into sn-2. Thus, GPAT and LPAAT enzyme selectivities are major determinants of fatty chain compositions at sn-1 and -2 of glycerophospholipids formed during the de novo synthesis. However, these selectivities do not fully account for the fatty chain diversity found in cellular membranes. The fatty chain compositions at sn-2 are further modified as part of a glycerophospholipid acyl chain remodeling pathway known as the Lands’ cycle.
Rapid turnover of acyl chains in glycerophospholipids was originally described by William Lands in 1958 [15]. In radioactive tracer experiments of PC synthesis in lung tissue, Lands noticed the ratio of fatty acid incorporation to glycerol was several-fold higher in phospholipids compared to TGs. This suggested that in addition to the biosynthetic route leading to production of both TG and PC from a common DAG precursor, another mechanism to allow exchange of fatty acids on PC in absence of de novo synthesis may exist. Lands postulated existence of an enzyme system which catalyzes exchange of fatty acids via formation of a lysophospholipid intermediate followed by reacylation. This enzyme system is now known to catalyze the rapid turnover at sn-2 via cycles of PLA2-mediated deacylations and lysophospholipid acyltransferase (LPLAT) -mediated reacylations in a pathway distinct from de novo synthesis. PA production as part of the Kennedy pathway and glycerophospholipid remodeling during Lands’ cycle are two distinct mechanisms by which acyl chains of glycerophospholipids are specified. Different sets of LPLATs function in these two pathways, and together these pathways account for the acyl chain diversity that exists in membrane glycerophospholipids.
2.2.1 Distinct Sets of LPLATs Function During de novo and Remodeling Pathways
The first identified mammalian LPAATs, LPAAT1 and LPAAT2, were cloned by several independent groups in 1997 based on motifs conserved in homologous sequences from plants, bacteria, yeast, nematodes and viruses [16,17,18,19,20]. These motifs included regions of high homology, now known as 1-acylglycerol-3-phosphate O-acyltransferase (AGPAT) motifs I-IV, that are important for substrate binding and enzymatic activity [19, 21, 22]. Originally, eleven mammalian AGPAT family members were identified [23]; however, biochemical characterization of the recombinant enzymes indicated many of these AGPAT family members primarily produce glycerolipids other than PA. Now five AGPAT family members are thought to function primarily as LPAATs by acylating LPA at sn-2 to produce PA [18, 24,25,26,27]. The other AGPAT family members have been reclassified according to their biochemical activities as GPATs, DHAPAT, or Lands’ cycle LPLATs (such as LPCATs, LPEATs, LPIATs, LCLATs, and LPGATs) [21, 28]. Subsequently, non-AGPAT family LPLATs were also identified that are membrane bound O-acyltransferase (MBOAT) family members [29]. MBOAT family enzymes have activation motifs that are different from AGPAT motifs [30]. Currently four MBOAT family members are known to function as LPLATs in the Lands’ cycle. The LPLATs of AGPAT and MBOAT families which function in the Kennedy pathway and Lands’ cycle are summarized in Fig. 2.3.

LPLATs of the AGPAT and MBOAT families
LPLATs of the AGPAT family function in the Kennedy pathway (green box), and LPLATs of the AGPAT and MBOAT families function during Lands’ cycle remodeling (orange box). Both LPCAT4/MBOAT2 and LPEAT2/LPCAT4 (in red letters) are referred to as “LPCAT4” and sometimes induce confusion in the literature
2.3 LPAAT Enzymes Determine Acyl Chains During de novo Synthesis
In de novo synthesis of PA, following esterification of a fatty chain to G3P by GPATs, the next step involving esterification of a second fatty chain, to the sn-2 position, is catalyzed by LPAAT enzymes of the AGPAT family (Fig. 2.2). The first identified mammalian isoforms, LPAAT1 and LPAAT2, were cloned nearly simultaneously by several groups [16,17,18,19]. Eberhardt et al. identified motifs in human LPAAT2 that were conserved in homologous sequences from plants, bacteria, yeast, nematodes and viruses [19], and Aguado et al. reported similarly conserved sequences in human LPAAT1 the following year [20]. These regions included the regions now referred to as AGPAT motifs that are conserved in all AGPAT family members and important in substrate binding and enzymatic activity [21]. Mammalian LPAAT1 and LPAAT2 are the most evolutionarily conserved isoforms and suggested to have evolutionarily arisen as part of the earliest TG synthesis pathway, indicating the importance of these isoforms in TG as well as glycerophospholipid production [31].
2.3.1 LPAAT1 Has Essential Functions in Multiple Tissues
LPAAT1 and LPAAT2 share high homology and have similar but not identical biochemical activities to utilize a variety of fatty chain-CoAs in producing PA [32]. LPAAT1 is broadly expressed, while LPAAT2 shows more restricted expression. Both LPAAT1 and LPAAT2 are expressed in adipocytes [32], however genetic loss of LPAAT2 activity causes congenital generalized lipodystrophy (CGL) [33], indicating that LPAAT2 has essential functions in adipocytes that are not compensated for by LPAAT1 [34].
To understand the biological functions of LPAAT1, LPAAT1 knockout (KO) mice were generated and phenotypically analyzed [35]. LPAAT1 KO mice had widespread disturbances in metabolism and glycerophospholipid homeostasis, causing pathological effects in multiple organ system. The LPAAT1 KO pups were born at less than Mendelian frequency, and those born had reduced body weights that almost did not increase after day 12. The LPAAT1 KO pups had decreased leptin and decreased plasma glucose, and about half died by 4 weeks of age. KO mice had markedly reduced epididymal fat pads, and cervical brown adipose tissue was also reduced but maintained in proportion to the reduced body weights. The LPAAT1 KO mice that survived to adult had reproductive abnormalities including impaired sperm development in males, and ovulation defects in females especially in late follicular maturation. LPAAT1 KO mice had features of seizures/epilepsy, and in hippocampus, where LPAAT1 is normally expressed, LPAAT1 KO mice tended to have reduced CA-region thickness, reduced neuron number, and impaired neurological function. Overall these results indicate LPAAT1 has essential functions in brain and testes, and is required for normal lipid homeostasis [35].
2.3.2 LPAAT2 and Lipodystrophy
LPAAT2 is expressed in several tissues, showing highest expression in adipose tissue, pancreas and liver, and has broad specificity to utilize a variety of fatty acid-CoAs as substrates; including C14:0, C16:0, C18:1, and C18:2 -CoAs to produce PA [32]. LPAAT2 mutations are associated with congenital generalized lipodystrophy (CGL), also known as Berardinelli-Seip lipodystrophy [33]. Four subtypes of the disease are recognized based upon the causative gene mutation (LPAAT2, BSCL2, CAV1, or CAVIN1/PTRF), and LPAAT2 was identified as the causative gene of type 1 CGL through positional cloning [33, 36]. Disease-causing homozygous or compound heterozygous LPAAT2 mutations have been identified that include deletions, nonsense, missense, splice-site and those in the 3′-UTR mutations [34, 37].
CGL is a rare autosomal recessive disorder and the most striking feature is a total lack of subcutaneous body fat from birth. Children with this disorder have increased appetites, undergo accelerated growth, and develop metabolic complications including severe insulin resistance, hypertriglyceridemia, hepatic steatosis and early onset of diabetes. LPAAT2 KO mice have been used to elucidate the pathological mechanisms involved [38]. LPAAT2 KO mice had almost a complete lack of both white and brown adipose tissue, and just 2% body fat compared to 24–29% in wild-type mice. The LPAAT2 KO mice developed extreme insulin resistance, diabetes, and hepatic steatosis. Lipogenic gene expression was increased and fatty acid biosynthesis was accelerated in LPAAT2 KO mouse livers, accompanied by increased monoacylglycerol (MAG) acyltransferase isoform 1, suggesting the MAG pathway for TG production may be hyper-activated under conditions of LPAAT2 deficiency, possibly causing or contributing to the hepatic steatosis. Both LPAAT1 and LPAAT2 are normally expressed in adipose tissue, however LPAAT1 cannot compensate for the observed phenotypic abnormalities in LPAAT2 KO mice, underscoring the essential function of LPAAT2 in this tissue [38].
LPAAT2 functions in TG as well as glycerophospholipid synthesis, and it is possible that LPAAT2 mutations cause CGL primarily by inhibiting TG biosynthesis and storage in adipocytes. The lack of functional adipose tissue in CGL results in TG accumulations in other tissues like skeletal muscle and liver and contributes to the disrupted metabolic homeostasis [39]. It has been suggested that the lipodystrophy due to mutations in LPAAT2 and other CGL-causing genes might be mechanistically caused by defective lipid droplet formation in adipocytes. However, LPAAT2-generated phospholipids may also impact adipocyte function and TG storage, and further studies are required to understand the relative contribution of the LPAAT2-generated TGs and glycerophospholipids to metabolic homeostasis that is disrupted in CGL [34].
2.3.3 LPAAT3 Incorporates DHA into Membranes
Consumption of fish oil, which is rich in very long-chain omega-3 fatty acids such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), is well-known to promote cardiovascular health and have beneficial effects in other tissues [40]. Incorporation of very long-chain, highly unsaturated chains like DHA and EPA into membrane glycerophospholipids may impart fluidity and flexibility to the membranes, as well as impact functions of membrane proteins [41]. DHA and EPA also have other biological functions as ligands for lipid sensing molecules such as PPARs [42] and as precursor molecules for specialized pro-resolving mediators [43]. Thus, incorporation of DHA and EPA into membrane glycerophospholipids may both alter membrane properties as well regulate their availability as ligands or precursors of bioactive molecules. We have focused on the functions of DHA in biological membranes, because DHA-CoA is selectively utilized as a substrate by LPAAT3 during glycerophospholipid synthesis [44, 45].
LPAAT3 was originally identified as an enzyme broadly expressed in tissues, regulated by PPAR-alpha in mouse heart [24], and involved in golgi dynamics [46]. Substrate specificity of LPAAT3 was shown to incorporate very long-chain polyunsaturated fatty acids into LPA [25, 44, 45] and to selectively incorporate DHA to increase production of DHA-containing glycerophospholipids [45, 47,48,49]. LPAAT3 KO mice were generated, and they show marked and specific reductions in DHA-containing glycerophospholipids in several tissues, demonstrating a central role for LPAAT3 in generation of DHA-containing glycerophospholipids [47, 48].
DHA-containing glycerophospholipids are abundant in several tissues including retina, testes, brain, heart, and skeletal muscle where they may have important physiological functions. DHA deficiency is associated with several human diseases including hyperlipidemia, cardiovascular disease, cognitive dysfunction, retinal degeneration, and male infertility [50]. We have investigated regulation and biological functions of LPAAT3-generated DHA-containing glycerophospholipids in sperm, retina, and skeletal muscle cells [47,48,49].
2.3.4 LPAAT3 Incorporates DHA into Glycerophospholipids During Sperm Cell Development
Male infertility frequently occurs due to declines in sperm function. Mammalian sperm contains a high amount of very long-chain polyunsaturated fatty acids including DHA, and defective human sperm populations show declines in DHA in both esterified and unesterified fatty acid fractions [51]. Thus, high DHA content in sperm may be required to impart sperm cell plasma membranes with necessary properties for specialized functions involved in sperm cell maturation, morphology, motility, or fusion with the egg [52].
LPAAT3 was investigated as a candidate enzyme to incorporate DHA into sperm cell membranes. LPAAT3 was highly expressed in mouse testicles, and expression increased during development, coinciding with increased enzymatic activities to incorporate DHA into PC and PE [25, 45]. Immunohistochemical analyses indicated LPAAT3 was expressed both in Leydig cells as well as in spermatids, where LPAAT3 levels increased during spermatogenesis. DHA levels and metabolism in the testes were investigated in LPAAT3 KO mice. Microsome fractions from testes of LPAAT3 KO mice had moderately decreased LPAAT activities to utilize arachidonic acid-CoA but greatly decreased activities to utilize DHA-CoA. Moreover, DHA-containing PC, PE, and PS were highly and selectively decreased in testes, establishing the physiological role of LPAAT3 to incorporate DHA into membrane glycerophospholipids [47].
LPAAT3 KO mice showed male infertility due to defective spermiogenesis. LPAAT3 KO sperm had morphological defects, including sperm heads that bent backwards. Scanning electron microscopy revealed abnormal membranous structures wrapped around the sperm heads which should have been removed when the sperm were released into the seminiferous tubules. This suggests DHA-containing glycerophospholipids generated by LPAAT3 impart flexibility to sperm membranes that allows rapid endocytosis required for normal spermiation [47] and may partly explain why omega-3 supplementation promotes male fertility [53].
2.3.5 LPAAT3 and DHA in Visual Function
Retinal tissue is abundant in DHA-containing glycerophospholipids, and DHA may have protective roles to prevent or delay the progression of several retinal disorders [54, 55]. The role of LPAAT3 in visual function was examined in wild-type and LPAAT3 KO mice [48]. In wild-type mice, LPAAT3 was highly expressed in retinas, and protein expression increased during development from age 11 days through 8 weeks. Compared to wild-type tissues, retinal tissues from LPAAT3 KO mice had decreased levels of DHA in several glycerophospholipids (PA, PC, PE, and PS) accompanied by corresponding increases in several arachidonic acid-containing species. Imaging mass spectrometry showed DHA-containing PC was present in outer segments of photoreceptors in wild-type mice but nearly absent in KO mice. Histologically, LPAAT3 KO retinas appeared normal until 2 weeks of age, but by 3–8 weeks of age they showed abnormal retinal layer morphologies including incomplete elongation of the outer segment, decreased thickness of the outer nuclear layers, and disordered disc morphology in photoreceptor cells. LPAAT3 KO mice showed attenuation of visual function as assessed by electroretinography, with 50–80% decreases in a- and b-wave amplitudes at 8 weeks of age. These results established that LPAAT3 has an essential role in visual function by producing DHA-containing glycerophospholipids, which are required to form, organize, and maintain proper disc shape in photoreceptor cells [48].
2.3.6 LPAAT3 and DHA in Skeletal Muscle
Skeletal muscle is adaptive tissue, and adaptation of skeletal muscle to endurance exercise training is associated with metabolic benefits including reduced obesity and improved glucose handling. Although the biological function and mechanism is unknown, several studies indicate that DHA-containing glycerophospholipids are also increased in endurance-trained muscle and correlates with oxidative status of the muscles [56,57,58,59,60]. In mice, exercise training increased phospholipid-DHA in glycolytic extensor digitorum longus muscle, while phospholipid-DHA was constitutively high in oxidative soleus muscle even without training [56]. In rats, PE-DHA content was enriched in the oxidative compared to glycolytic vastus lateralis muscles [60]. In human volunteers, a 4 weeks regime of one-leg exercise training resulted in increased DHA-containing GPLs that was accompanied by increased citrate synthase activity compared to the untrained leg [58].
Endurance training activates transcriptional responses that mediate adaptive responses in skeletal muscle. PPAR-delta and AMPK are activated by exercise and promote increased oxidative metabolic capacity and transitioning of glycolytic myofibers to more oxidative fiber types [61, 62]. PGC1-alpha is activated downstream of AMPK and is a coactivator of PPAR-delta. In one study, mice overexpressing PGC1-alpha in skeletal muscle had enhanced levels of several DHA-containing glycerophospholipid species that were also increased by endurance training, indicating exercise-induced transcriptional responses may also enhance DHA incorporation into glycerophospholipids of muscle [56].
The role of LPAAT3 to produce DHA-containing glycerophospholipids in skeletal muscle was investigated using primary skeletal muscle myoblasts. LPAAT3 expression was transiently upregulated during differentiation into myotubes and functioned to increase DHA-containing PC and PE [49]. Treatment of differentiated myofibers with PPAR-delta and AMPK agonists also enhanced LPAAT3 expression and increased levels of DHA-containing PC and PE. These results showed LPAAT3 expression is regulated by two mechanisms in myoblasts, transient upregulation as part of the myogenic transcriptional program during myogenesis, and in differentiated myotubes in response to PPAR-delta and AMPK agonists. The PPAR-delta agonist GW1516 and AMPK agonist AICAR are exercise mimicking drugs, and when administered to mice promote endurance and transcriptionally upregulate genes that promote oxidative metabolism [63, 64]. LPAAT3 expression and DHA incorporation was upregulated by these same compounds in cultured myotubes, suggesting LPAAT3 may also be upregulated by exercise-induced pathways to increase DHA in endurance-trained muscle [49].
2.4 Acyl Chain Remodeling by Lands’ Cycle Enzymes
In glycerolipids, an asymmetric distribution of fatty acids at sn-1 and sn-2 positions imparts diversity to cellular membranes that cannot be fully explained enzymatic reactions of de novo glycerophospholipid synthesis, and further diversity at sn-2 especially is generated by cycles of deacylation and reacylation reactions known as Lands’ cycle (remodeling pathway). PC, the most abundant glycerophospholipid in membranes, may undergo extensive Lands’ cycle remodeling mediated by the actions of PLA2s and LPCAT enzymes. Several LPCAT enzymes of both the AGPAT and MBOAT families have been identified, and a growing list of functional roles pertinent to physiological and/or pathological conditions is being assigned to these enzymes, garnering much interest in their biological activities and therapeutic potentials. Similar remodeling of other classes of glycerophospholipids is mediated by other LPLATs classified as LPEATs, LCLATs, LPIATs, or LPGATs, and as their biological activities are being increasingly uncovered, interest in their potential as therapeutic targets is also emerging (Table 2.1).
2.4.1 LPCAT1 Produces Lung Surfactant Essential for Respiratory Function
Pulmonary is surfactant essential for breathing and comprised mainly of lipids (~90%) and protein (~10%), and nearly half of the lipid is PC(16:0/16:0), dipalmitoyl-PC (DPPC) [65]. DPPC along with surfactant proteins form a specialized monolayer in alveolar spaces which reduces surface tension, facilitates gas exchange, and prevents alveolar collapse [66, 67]. Pulmonary surfactant is produced by alveolar type II cells, and deficiency of pulmonary surfactant is associated with several pulmonary diseases. Respiratory distress syndrome, the leading cause of death in pre-term infants, is caused by lack of surfactant in premature lung structures [68, 69].
Identification of the biosynthetic routes of DPPC production in lung had been long sought, and both the Kennedy pathway and Lands’ cycle were thought to have significant roles. Involvement of Lands’ cycle enzymes was indicated by biochemical activities present in alveolar type II cells and rat lung tissue to incorporate labelled palmitate (C16:0) preferentially into the sn-2 position of DPPC rather than TGs, presumably through acylation of LPC [70, 71]. LPCAT activities had been known to exist in several tissues, and in 2006 the first mammalian LPCAT enzyme was independently identified by two groups [72, 73]. Both studies identified LPCAT1 as the candidate enzyme present in alveolar type II cells that produces DPPC.
LPCAT1 has AGPAT motifs, and Nakanishi et al. cloned mouse LPCAT1 based on homology to other AGPAT family members [72]. Mouse LPCAT1 was highly expressed in lung and had activity to produce PC using saturated acyl-CoA as donors. Rat LPCAT1 was also highly expressed in lung and enriched in alveolar type II cells rather than macrophages. Later, they also characterized the human LPCAT1 homolog, which was suggested to also produce DPPC in lung [74]. Chen et al. also identified LPCAT1 as an enzyme that was upregulated in mouse and rat lungs shortly before birth. They cloned both the mouse and rat enzymes, which were 99% identical at amino acid level. The enzymes both possessed a H(X)4D activation domain sequence that was known to be present in other AGPAT family members [75]. Rat LPCAT1 was overexpressed in mammalian cells and produced DPPC [73].
These studies indicated that LPCAT1 may be responsible for production of DPPC in pulmonary surfactant [72,73,74]. Additional studies utilizing LPCAT1-deficient mice generated by two different groups have confirmed that LPCAT1 is a major producer of DPPC in pulmonary surfactant [76, 77]. The two LPCAT1-deficient mouse strains showed varying degrees of respiratory dysfunction; this variation may be due to the genetic backgrounds or different gene deletion strategies. Bridges et al. generated mice bearing a hypomorphic allele of LPCAT1. These mice had difficulty to transition to air breathing and showed varying degrees of perinatal lethality due to respiratory distress. Perinatal lethality correlated with decreased LPCAT1 mRNA, decreased disaturated PC in lungs, and functionally poor surfactant [76]. Harayama et al. generated LPCAT1 KO mice lacking exon 3 which contains the H(X)4D AGPAT activation domain. These mice had dramatically reduced LPCAT activity to utilize C16:0-CoA. The KO mice had ~40% reduced DPPC in lung but did not show major signs of neonatal lung distress. However, the mice had enhanced susceptibility to a ventilator-induced lung injury model, which was partially rescued by intratracheal administration of DPPC [77]. These studies establish a critical function for LPCAT1 in producing sufficient levels of DPPC in pulmonary surfactant to prevent respiratory distress or failure under different physiological challenges.
2.4.2 LPCAT1 and Cancer
Besides producing DPPC in pulmonary surfactant, LPCAT1 has additional important roles in physiological and disease states. LPCAT1 is important for visual function, and a natural mutation in LPCAT1 in rd11 mice causes rod and cone cell loss, followed by retinal degeneration [78]. LPCAT1 also has a role to produce PAF under non-inflammatory conditions [22]. Another reported function of LPCAT1 is to protect against polyunsaturated fatty acid-mediated cytotoxicity [79].
In addition, LPCAT1 has a role in cancer. LPCAT1 expression was reported to be high or involved in progression of a variety of cancers including colorectal adenocarcinomas, prostate cancer, lung adenocarcinoma, hepatocellular carcinoma, oral squamous cell carcinoma, breast cancer, esophageal cancer, gastric cancer, clear cell renal cell carcinoma, and glioblastoma [80,81,82]. Recently Bi et al. reported LPCAT1 is highly upregulated in a wide array of cancers, and LPCAT1-produced disaturated PCs may be required for proliferative signaling via oncogenic growth factor signaling [81]. Initially they examined the impact of EGFRvIII, an activating mutation of EGFR and common driver in glioblastoma, on membrane compositions in glioblastoma cells. In EGFRvIII-transduced glioblastoma cells, disaturated PC species and LPCAT1 mRNA were increased, and shRNA-knockdown of LPCAT1 reduced disaturated PC species, particularly PC(28:0), PC(30:0), and PC(32:0). Knockdown of LPCAT1 in glioblastoma cancer cells harboring EGFRvIII mutations inhibited EGFR phosphorylation and signaling, and this could be reversed by addition of either DPPC liposomes or saturated C14:0/C16:0 fatty acids. Knockdown of LPCAT1 also inhibited glioblastoma cell viability and colony-forming ability, and this was reversed by supplementation of DPPC or a constitutively-active AKT allele. Their results suggest LPCAT1 may promote glioblastoma growth by maintaining high saturated PC levels that support EGFR oncogenic signaling.
The authors went on to show that LPCAT1 amplification is a widespread occurrence across all human cancers. They examined LPCAT1 expression in publicly available databases, and LPCAT1 was found to have increased copy number in 30% of all cancer patients, and inverse correlations between LPCAT1 expression and overall survival was detected in multiple cancer types. LPCAT1 knockdown reduced cancer cell viability in vitro, and inducible genetic depletion of LPCAT1 inhibited tumor growth in mouse xenograft models. Their study suggests LPCAT1 inhibition may be a promising therapeutic strategy for a variety of oncogenic growth factor driven cancers [81].
2.4.3 LPCAT2 Produces PAF
LPCAT2 was identified in 2007 as a PAF-producing enzyme in the AGPAT family [83]. In the following year, LPCAT1, which had already been identified as the first LPCAT enzyme [72], was also reported to also have PAF-producing activity [22, 77]. The enzymes having lyso-PAF acetyltransferase activity had been long-sought following the first identification of PAF in 1972 as the substance responsible for platelet aggregation [84]. LPCAT2 can synthesize not only PAF but also polyunsaturated fatty acid-containing PC [77, 83]. Unlike most other LPLATs, the activity of LPCAT2 is calcium ion-dependent. LPCAT2 is expressed in inflammatory cells, such as macrophages and neutrophils, and the enhancement of its PAF biosynthesis activity by lipopolysaccharide (LPS) stimulation is another characteristic of LPCAT2 [83].
Recent studies demonstrated three distinct mechanisms that regulate LPCAT2 activity and expression level in macrophages (Fig. 2.4). First, LPCAT2 is rapidly activated (within 30 s) after either PAF or adenosine triphosphate (ATP) stimulation via intracellular calcium-stimulated activation of PLCβ-PKCα signaling. These pathways induce phosphorylation of LPCAT2 at Ser-34, which enhances LPCAT2’s PAF biosynthesis activity [85]. Another mechanism of phosphorylation is via toll-like receptor 4 (TLR4) activation. Thirty minutes of stimulation by the TLR4 ligand LPS activates the downstream pathway, MyD88-p38 MAPK-MK2, leading to phosphorylation of LPCAT2 at the same site Ser-34 as Ca2+-PLCβ-PKCα signaling [86]. Finally, LPS stimulation for 16 h increases mRNA expression of LPCAT2, which is attenuated by co-treatment with the steroidal anti-inflammatory drug dexamethasone [83]. These studies indicate LPCAT2 is upregulated and shows enhanced PAF-producing activity as part of inflammatory responses, but this is not the case for LPCAT1.

Regulatory mechanisms of LPCAT2-produced PAF levels and the PAF-Pain Loop hypothesis
(i) PAFR- and/or ATP receptors-mediated second-scale phosphorylation of LPCAT2, (ii) TLR4 and its downstream pathways-mediated minute-scale phosphorylation, and (iii) hour-scale LPCAT2 mRNA upregulation via TLR4. Nerve injury may activate these regulatory systems and increase PAF levels. The increased PAF further enhances PAFR-mediated PAF production, and may result in exacerbation of neuropathic pain (PAF-Pain Loop)
Although it has been clearly demonstrated that LPCAT1 produces PAF in in vitro assays, LPCAT1’s activity was not enhanced by inflammatory stimuli [85, 86]. It has been suggested that LPCAT1 may have a role to limit PAF production during the onset of diabetic retinopathy by catalyzing synthesis of alkyl-PC from lyso-PAF and acyl-CoA, thereby limiting lyso-PAF availability for LPCAT2-mediated PAF production [87]. However, further investigations are needed to clarify the roles of LPCAT1 to produce or regulate production of PAF in vivo.
2.4.4 LPCAT2 and PAF Production in Neuropathic Pain
Several reports have demonstrated LPCAT2 is involved in various pathological conditions such as allergic asthma [88], colorectal cancer resistance [89], nonalcoholic fatty liver disease [90], and multiple sclerosis [91]. It was recently reported that LPCAT2 is involved in neuropathic pain, a highly debilitating chronic pain syndrome [92]. Neuropathic pain occurs from damage to the nervous system; such as by trauma, cancer, diabetes, infection or chemotherapy; which does not resolve even after normal tissue healing. Unfortunately, although clinical neuropathic pain affects 7–10% of the human population [93], the cardinal symptom allodynia, pain caused by innocuous stimuli, is refractory to currently available treatments [94, 95].
In the 1980s, the involvement of PAF in pain processes was indicated by studies that observed enhancement of pain sensitivity following local injection of PAF into skin of animals [96, 97] and humans [98]. In addition, intrathecal PAF injection into the spinal cord also produced allodynia, but injection of its precursor lyso-PAF did not [99, 100]. The biological activities of PAF are elicited via its cognate G protein-coupled receptor, PAF receptor (PAFR) [101]. In rodent models of nerve injury-induced neuropathic pain, it has been reported that treatment with PAFR antagonists, such as WEB2086 or CV-3988, alleviate allodynia symptoms [100, 102]. In accord with this, neuropathic allodynia was attenuated in PAFR KO mice [100]. Both PAFR and LPCAT2 are highly expressed in macrophages (in the periphery) and microglia (in the spinal cord). Peripheral nerve injury increases the number of macrophages in the dorsal root ganglia, where cell bodies of primary sensory neurons are present, and also increases microglia in the spinal dorsal horn. Along with these increases, the protein expressions of PAFR and LPCAT2 also rise [92, 100]. Recently, LPCAT2 KO mice were established, and while the abundance of other PCs were similar to wild-type mice, PAF levels in the spinal cord of this mouse were decreased to below detectable levels under normal physiological conditions. Moreover, nerve injury-induced neuropathic allodynia was attenuated in LPCAT2 KO mice, similar to the PAFR KO mice [92]. These results indicate the importance of LPCAT2-PAFR axis in the pathology of neuropathic pain.
Several studies on the pathogenesis of neuropathic pain have reported that peripheral nerve injury significantly increases the expression levels of several ATP receptors (i.e. P2X4R, P2X7R, P2Y12R) and induces ATP release, via vesicular nucleotide transporter, in the spinal dorsal horn [103,104,105,106]. Furthermore, increased TLR4 expression and phosphorylation of p38 MAPK in microglia were also observed in response to nerve injury [107, 108]. Nerve injury also induces LPCAT2 PAF-producing activity by activating ATP receptors and TLR4 signaling, and PAF stimulation induces activation of LPCAT2 via PAFR to further enhance PAF production. Therefore, a positive feedback “PAF-pain loop” is suggested, as PAF-induced increases in PAF production resulted in exacerbation and chronicity of neuropathic pain [92] (Fig. 2.4).
Many pharmaceutical companies have already developed PAFR antagonists as possible drug candidates to treat inflammatory conditions such as asthma, but in most cases these drugs have so far shown limited efficacy [10, 109]. However, in light of LPCAT2’s role in the “PAF-pain loop”, it may be warranted to re-evaluate the possible clinical utility of these PAFR antagonists as analgesics. Moreover, LPCAT2 may also be a novel therapeutic target for analgesic drugs, suggesting that PAF loop blockers such as PAFR antagonists or LPCAT2 inhibitors might represent a new class of analgesic drugs different from the current classes of nonsteroidal anti-inflammatory drugs (NSAIDs) and opioids. Also, because PAF feedback loop may operate not only in neuropathic pain but also in various other pathological conditions with PAF involvement, a recently identified selective LPCAT2 inhibitor, TSI-01 [110], and its derivatives also have potential to contribute to the development of novel drugs for PAF-related diseases. Importantly, the biological significance of the activities of LPCAT2 to produce PCs other than PAF requires further investigation, which may expand our understanding of LPCAT2 as a therapeutic target [92].
2.4.5 LPCAT3 Functions in Small Intestine, Liver and Several Cells
LPCAT3 is a member of the MBOAT family that catalyzes the transfer reaction of acyl group to proteins and lipids [30]. LPCAT3 synthesizes linoleic acid (C18:2) and arachidonic acid (C20:4) containing PC, PE and PS from LPC, LPE and LPS [29, 111,112,113]. Recently, several reports suggested that LPCAT3 has several important roles in lipid homeostasis [82, 114]. LPCAT3 global KO mice showed neonatal lethality and died between postnatal day 2 and 3 weeks [115,116,117]. It was found to be caused by nutritional failure in small intestine because the pups were partially rescued by injections of 10% glucose solution or oral gavage with PCs/olive oil [115, 117]. However, the different studies reported different effects of LPCAT3 deficiency on TG accumulation in small intestine. We reported LPCAT3 deletion caused dramatic accumulation of lipid droplets in enterocytes of global LPCAT3 KO pups [116], and Rong et al. observed a similar accumulation in intestine-specific KO pups [115]. In contrast, Li et al. reported no TG accumulation in intestines of LPCAT3 global KO pups [117], nor following intestine-specific LPCAT3 depletion in adult mice [118]. It is unclear why intestinal lipid accumulation was observed in some studies but not others; the differences might reflect the different gene deletion strategies, gene-targeted-regions, or other factors.
The expression of proteins related to lipid absorption in enterocytes, such as NPC1L1, CD36 and FATP4, were decreased in LPCAT3 global KO mice [117] as well as following inducible intestine-specific depletion [118]. Body weight was decreased following intestine-specific LPCAT3 depletion, and total TG and cholesterol levels were also decreased in plasma and serum [115, 119]. In addition, small intestines were longer and intestinal stem cells showed hyperproliferation in the intestine-specific LPCAT3-KO mice [117, 119]. These results suggest that arachidonic acid rich cell membranes synthesized by LPCAT3 have an essential role in intestinal absorption of dietary lipid and may also have a function to limit proliferation of intestinal stem cells. The role in lipid absorption suggests that intestine-specific inhibition of LPCAT3 might be a useful therapeutic strategy to treat hyperlipidemia.
LPCAT3 was also highly expressed in liver [29, 111]. Its expression was regulated by LXR and PPAR-delta in hepatocytes [120,121,122,123]. LXR activates lipogenic genes such as SREBP-1c, SCD-1 and FAS [124, 125]; and plasma TG and phospholipids were changed in LXR agonist-treated mouse as well as LXR-alpha/beta double-KO mice [125]. These results suggest that LPCAT3 could contribute to fatty acid homeostasis in liver. In fact, plasma VLDL secretion was increased by hepatic LPCAT3 knockdown in mice [126]; and glucose tolerance was improved and plasma TG levels were decreased by transient overexpression of LPCAT3 in liver [127].
Compared to the changes observed following transient knockdown and overexpression, liver TG was increased and plasma TG, VLDL secretion levels, and VLDL particle size were decreased in LPCAT3 KO mice [115, 116]. These differences between transient LPCAT3 knockdown and LPCAT3 KO mice may reflect residual LPCAT3-generated arachidonic acid- and linoleic acid-containing phospholipids and lysophospholipids that are present under knockdown conditions, due to limited duration or incomplete efficiency of knockdown by the LPCAT3-targeting shRNAs. Both the effective transport of TG by MTP (microsomal triglyceride transfer protein) as well as SREBP-1c maturation in the endoplasmic reticulum by protein cleavage can be promoted by arachidonic acid-containing phospholipids synthesized by LPCAT3 [116, 128], further supporting that LPCAT3 inhibition might improve dyslipidemia.
LPCAT3 has also been shown to function or have roles in other pathological conditions. LPCAT3 expression levels inversely correlated with the disease stage of atherosclerosis, and atherosclerosis was facilitated by LPCAT3 deletion in macrophages [123, 129, 130]. LPCAT3 may also function in cancer because intestinal tumorigenesis was promoted by LPCAT3 deletion [119]. LPCAT3 also has important roles in adipocyte differentiation and macrophage polarization [131, 132], indicating LPCAT3 has diverse physiological roles and may be a promising therapeutic target in several diseases.
2.4.6 LPIAT1 Functions in Brain Development and Modifies Liver Disease Risk
LPIAT1 is a MBOAT family member and incorporates arachidonic acid-CoA into LPI to produce arachidonic acid-containing PI [133]. Two independent groups generated global LPIAT1 KO mice and reported the KO mice are small, born at low frequency, and have poor survival. Both groups also reported abnormal brain development and decreased arachidonic acid-containing PI and PI phosphates in brain [134, 135]. Lee et al. measured LPLAT activities in the KO tissues, and activity to incorporate arachidonic acid-CoA into LPI was almost abolished in several tissues including brain, whereas the activity to incorporate arachidonic acid-CoA into other lysophospholipids in brain was almost unchanged, establishing that LPIAT1 is a predominant LPIAT involved in production of arachidonic acid-containing PI in mice [134]. Lee et al. also performed detailed analyses of the defective brain development and reported delayed neuronal migration, decreased sizes of cerebral cortex and hippocampus, and disordered cortical lamination and neuronal processes [134]. Anderson et al. reported abnormal brain development as well, and arachidonic acid-containing PI and PI phosphates were decreased in both brain and liver. They also reported that in brain, but not in liver, arachidonic acid-containing PC(38:4) and PE(38:4) were both decreased by about half, which might also contribute to the defective brain development [135]. In humans, homozygous inactivating mutations of LPIAT1 are associated with intellectual disability, autism-like symptoms, and epilepsy; indicating a critical role for LPIAT1-generated arachidonic acid-containing PI in brain development in humans as well as mice [136].
LPIAT1 is also suggested to be a genetic modifier of liver disease risk. Liver cirrhosis is a major disease burden worldwide in terms of disability and death and may develop due to a number of different factors that cause chronic liver injury such as hepatitis C, hepatitis B, alcoholism, non-alcoholic steatohepatitis, and autoimmune hepatitis. High variabilities in disease progression exist due partly to genetic modifiers, and a common genetic variant of LPIAT1 (rs641738) has been reported to be associated with an array of alcohol and non-alcoholic fatty liver diseases. The role of LPIAT1 rs641738 variant in modifying fatty liver disease is not well understood but apparently involves altering LPIAT1 expression and the acyl chain compositions of membrane glycerophospholipids, which is proposed to affect inflammation-driven hepatic fibrosis and other symptoms in a variety of contexts including alcohol-related cirrhosis [137], non-alcoholic fatty liver disease [138], and hepatitis C [139]. However, results among different studies with different cohorts of patients have been inconsistent with some studies not supporting LPIAT1 rs641738 as a disease-modifying variant, suggesting that liver disease may have different genetic factors in different populations [140]. Additional mechanistic studies are required to understand the roles of LPIAT1 in liver disease and possible function as a genetic modifier.
2.5 Summary and Future Directions
It has become increasingly clear that the glycerophospholipid compositions of cell membranes, including their fatty chain compositions, affects a variety of physiological and disease states. Identification and biochemical characterization of the key enzymes involved, including LPLATs of the AGPAT and MBOAT families, has shed light on how the membrane compositions may be regulated in different biological contexts. Genetic deletion of these enzymes in mice has revealed unique physiological roles of many of these enzymes, and is expected to translate into new understanding of related human physiological and pathological conditions. Development of potent and selective inhibitors/activators of LPLATs will allow further discovery and elucidation of their biological functions, and pave the way to realize their promise as therapeutic targets.
References
Vance JE (2015) Phospholipid synthesis and transport in mammalian cells. Traffic 16(1):1–18. https://doi.org/10.1111/tra.12230
Schlegel RA, Williamson P (2001) Phosphatidylserine, a death knell. Cell Death Differ 8(6):551–563. https://doi.org/10.1038/sj.cdd.4400817
Ohashi M, Jan de Vries K, Frank R, Snoek G, Bankaitis V, Wirtz K, Huttner WB (1995) A role for phosphatidylinositol transfer protein in secretory vesicle formation. Nature 377(6549):544–547. https://doi.org/10.1038/377544a0
Schlame M, Greenberg ML (2017) Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim Biophys Acta Mol Cell Biol Lipids 1862(1):3–7. https://doi.org/10.1016/j.bbalip.2016.08.010
Barelli H, Antonny B (2016) Lipid unsaturation and organelle dynamics. Curr Opin Cell Biol 41:25–32. https://doi.org/10.1016/j.ceb.2016.03.012
Hishikawa D, Hashidate T, Shimizu T, Shindou H (2014) Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res 55(5):799–807. https://doi.org/10.1194/jlr.R046094
Antonny B, Vanni S, Shindou H, Ferreira T (2015) From zero to six double bonds: phospholipid unsaturation and organelle function. Trends Cell Biol 25(7):427–436. https://doi.org/10.1016/j.tcb.2015.03.004
Shimizu T (2009) Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu Rev Pharmacol Toxicol 49:123–150. https://doi.org/10.1146/annurev.pharmtox.011008.145616
Braverman NE, Moser AB (2012) Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta 1822(9):1442–1452. https://doi.org/10.1016/j.bbadis.2012.05.008
Ishii S, Shimizu T (2000) Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog Lipid Res 39(1):41–82
Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM (2000) Platelet-activating factor and related lipid mediators. Annu Rev Biochem 69:419–445. https://doi.org/10.1146/annurev.biochem.69.1.419
Kennedy EP, Weiss SB (1956) The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem 222(1):193–214
Reue K, Wang H (2019) Mammalian lipin phosphatidic acid phosphatases in lipid synthesis and beyond: metabolic and inflammatory disorders. J Lipid Res 60(4):728–733. https://doi.org/10.1194/jlr.S091769
Vance DE, Ridgway ND (1988) The methylation of phosphatidylethanolamine. Prog Lipid Res 27(1):61–79
Lands WE (1958) Metabolism of glycerolipides; a comparison of lecithin and triglyceride synthesis. J Biol Chem 231(2):883–888
Kume K, Shimizu T (1997) cDNA cloning and expression of murine 1-acyl-sn-glycerol-3-phosphate acyltransferase. Biochem Biophys Res Commun 237(3):663–666. https://doi.org/10.1006/bbrc.1997.7214
Stamps AC, Elmore MA, Hill ME, Kelly K, Makda AA, Finnen MJ (1997) A human cDNA sequence with homology to non-mammalian lysophosphatidic acid acyltransferases. Biochem J 326(Pt 2):455–461. https://doi.org/10.1042/bj3260455
West J, Tompkins CK, Balantac N, Nudelman E, Meengs B, White T, Bursten S, Coleman J, Kumar A, Singer JW, Leung DW (1997) Cloning and expression of two human lysophosphatidic acid acyltransferase cDNAs that enhance cytokine-induced signaling responses in cells. DNA Cell Biol 16(6):691–701. https://doi.org/10.1089/dna.1997.16.691
Eberhardt C, Gray PW, Tjoelker LW (1997) Human lysophosphatidic acid acyltransferase. cDNA cloning, expression, and localization to chromosome 9q34.3. J Biol Chem 272(32):20299–20305. https://doi.org/10.1074/jbc.272.32.20299
Aguado B, Campbell RD (1998) Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class III region of the human major histocompatibility complex. J Biol Chem 273(7):4096–4105. https://doi.org/10.1074/jbc.273.7.4096
Yamashita A, Nakanishi H, Suzuki H, Kamata R, Tanaka K, Waku K, Sugiura T (2007) Topology of acyltransferase motifs and substrate specificity and accessibility in 1-acyl-sn-glycero-3-phosphate acyltransferase 1. Biochim Biophys Acta 1771(9):1202–1215. https://doi.org/10.1016/j.bbalip.2007.07.002
Harayama T, Shindou H, Ogasawara R, Suwabe A, Shimizu T (2008) Identification of a novel noninflammatory biosynthetic pathway of platelet-activating factor. J Biol Chem 283(17):11097–11106. https://doi.org/10.1074/jbc.M708909200
Bradley RM, Duncan RE (2018) The lysophosphatidic acid acyltransferases (acylglycerophosphate acyltransferases) family: one reaction, five enzymes, many roles. Curr Opin Lipidol 29(2):110–115. https://doi.org/10.1097/MOL.0000000000000492
Lu B, Jiang YJ, Zhou Y, Xu FY, Hatch GM, Choy PC (2005) Cloning and characterization of murine 1-acyl-sn-glycerol 3-phosphate acyltransferases and their regulation by PPARalpha in murine heart. Biochem J 385(Pt 2):469–477. https://doi.org/10.1042/BJ20041348
Yuki K, Shindou H, Hishikawa D, Shimizu T (2009) Characterization of mouse lysophosphatidic acid acyltransferase 3: an enzyme with dual functions in the testis. J Lipid Res 50(5):860–869. https://doi.org/10.1194/jlr.M800468-JLR200
Eto M, Shindou H, Shimizu T (2014) A novel lysophosphatidic acid acyltransferase enzyme (LPAAT4) with a possible role for incorporating docosahexaenoic acid into brain glycerophospholipids. Biochem Biophys Res Commun 443(2):718–724. https://doi.org/10.1016/j.bbrc.2013.12.043
Shindou H, Shimizu T (2009) Acyl-CoA:lysophospholipid acyltransferases. J Biol Chem 284(1):1–5. https://doi.org/10.1074/jbc.R800046200
Yamashita A, Hayashi Y, Matsumoto N, Nemoto-Sasaki Y, Oka S, Tanikawa T, Sugiura T (2014) Glycerophosphate/Acylglycerophosphate acyltransferases. Biology (Basel) 3(4):801–830. https://doi.org/10.3390/biology3040801
Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, Shimizu T (2008) Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc Natl Acad Sci U S A 105(8):2830–2835. https://doi.org/10.1073/pnas.0712245105
Shindou H, Eto M, Morimoto R, Shimizu T (2009) Identification of membrane O-acyltransferase family motifs. Biochem Biophys Res Commun 383(3):320–325. https://doi.org/10.1016/j.bbrc.2009.04.013
Korbes AP, Kulcheski FR, Margis R, Margis-Pinheiro M, Turchetto-Zolet AC (2016) Molecular evolution of the lysophosphatidic acid acyltransferase (LPAAT) gene family. Mol Phylogenet Evol 96:55–69. https://doi.org/10.1016/j.ympev.2015.12.001
Agarwal AK, Sukumaran S, Cortes VA, Tunison K, Mizrachi D, Sankella S, Gerard RD, Horton JD, Garg A (2011) Human 1-acylglycerol-3-phosphate O-acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2(−/−) gene lipodystrophic mice. J Biol Chem 286(43):37676–37691. https://doi.org/10.1074/jbc.M111.250449
Agarwal AK, Arioglu E, De Almeida S, Akkoc N, Taylor SI, Bowcock AM, Barnes RI, Garg A (2002) AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet 31(1):21–23. https://doi.org/10.1038/ng880
Garg A, Agarwal AK (2009) Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta 1791(6):507–513. https://doi.org/10.1016/j.bbalip.2008.12.014
Agarwal AK, Tunison K, Dalal JS, Nagamma SS, Hamra FK, Sankella S, Shao X, Auchus RJ, Garg A (2017) Metabolic, reproductive, and neurologic abnormalities in Agpat1-null mice. Endocrinology 158(11):3954–3973. https://doi.org/10.1210/en.2017-00511
Garg A, Wilson R, Barnes R, Arioglu E, Zaidi Z, Gurakan F, Kocak N, O’Rahilly S, Taylor SI, Patel SB, Bowcock AM (1999) A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab 84(9):3390–3394. https://doi.org/10.1210/jcem.84.9.6103
Broekema MF, Massink MPG, De Ligt J, Stigter ECA, Monajemi H, De Ridder J, Burgering BMT, van Haaften GW, Kalkhoven E (2018) A single complex Agpat2 allele in a patient with partial lipodystrophy. Front Physiol 9:1363. https://doi.org/10.3389/fphys.2018.01363
Cortes VA, Curtis DE, Sukumaran S, Shao X, Parameswara V, Rashid S, Smith AR, Ren J, Esser V, Hammer RE, Agarwal AK, Horton JD, Garg A (2009) Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab 9(2):165–176. https://doi.org/10.1016/j.cmet.2009.01.002
Wang H, Airola MV, Reue K (2017) How lipid droplets “TAG” along: glycerolipid synthetic enzymes and lipid storage. Biochim Biophys Acta Mol Cell Biol Lipids 1862(10 Pt B):1131–1145. https://doi.org/10.1016/j.bbalip.2017.06.010
Hishikawa D, Valentine WJ, Iizuka-Hishikawa Y, Shindou H, Shimizu T (2017) Metabolism and functions of docosahexaenoic acid-containing membrane glycerophospholipids. FEBS Lett 591(18):2730–2744. https://doi.org/10.1002/1873-3468.12825
Rawicz W, Olbrich KC, McIntosh T, Needham D, Evans E (2000) Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys J 79(1):328–339. https://doi.org/10.1016/S0006-3495(00)76295-3
Forman BM, Chen J, Evans RM (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A 94(9):4312–4317. https://doi.org/10.1073/pnas.94.9.4312
Serhan CN, Levy BD (2018) Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest 128(7):2657–2669. https://doi.org/10.1172/JCI97943
Koeberle A, Shindou H, Harayama T, Shimizu T (2010) Role of lysophosphatidic acid acyltransferase 3 for the supply of highly polyunsaturated fatty acids in TM4 Sertoli cells. FASEB J 24(12):4929–4938. https://doi.org/10.1096/fj.10-162818
Koeberle A, Shindou H, Harayama T, Yuki K, Shimizu T (2012) Polyunsaturated fatty acids are incorporated into maturating male mouse germ cells by lysophosphatidic acid acyltransferase 3. FASEB J 26(1):169–180. https://doi.org/10.1096/fj.11-184879
Schmidt JA, Brown WJ (2009) Lysophosphatidic acid acyltransferase 3 regulates Golgi complex structure and function. J Cell Biol 186(2):211–218. https://doi.org/10.1083/jcb.200904147
Iizuka-Hishikawa Y, Hishikawa D, Sasaki J, Takubo K, Goto M, Nagata K, Nakanishi H, Shindou H, Okamura T, Ito C, Toshimori K, Sasaki T, Shimizu T (2017) Lysophosphatidic acid acyltransferase 3 tunes the membrane status of germ cells by incorporating docosahexaenoic acid during spermatogenesis. J Biol Chem 292(29):12065–12076. https://doi.org/10.1074/jbc.M117.791277
Shindou H, Koso H, Sasaki J, Nakanishi H, Sagara H, Nakagawa KM, Takahashi Y, Hishikawa D, Iizuka-Hishikawa Y, Tokumasu F, Noguchi H, Watanabe S, Sasaki T, Shimizu T (2017) Docosahexaenoic acid preserves visual function by maintaining correct disc morphology in retinal photoreceptor cells. J Biol Chem 292(29):12054–12064. https://doi.org/10.1074/jbc.M117.790568
Valentine WJ, Tokuoka SM, Hishikawa D, Kita Y, Shindou H, Shimizu T (2018) LPAAT3 incorporates docosahexaenoic acid into skeletal muscle cell membranes and is upregulated by PPARdelta activation. J Lipid Res 59(2):184–194. https://doi.org/10.1194/jlr.M077321
Spector AA, Kim HY (2015) Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim Biophys Acta 1851(4):356–365. https://doi.org/10.1016/j.bbalip.2014.07.020
Koppers AJ, Garg ML, Aitken RJ (2010) Stimulation of mitochondrial reactive oxygen species production by unesterified, unsaturated fatty acids in defective human spermatozoa. Free Radic Biol Med 48(1):112–119. https://doi.org/10.1016/j.freeradbiomed.2009.10.033
Lenzi A, Gandini L, Maresca V, Rago R, Sgro P, Dondero F, Picardo M (2000) Fatty acid composition of spermatozoa and immature germ cells. Mol Hum Reprod 6(3):226–231. https://doi.org/10.1093/molehr/6.3.226
Gonzalez-Ravina C, Aguirre-Lipperheide M, Pinto F, Martin-Lozano D, Fernandez-Sanchez M, Blasco V, Santamaria-Lopez E, Candenas L (2018) Effect of dietary supplementation with a highly pure and concentrated docosahexaenoic acid (DHA) supplement on human sperm function. Reprod Biol 18(3):282–288. https://doi.org/10.1016/j.repbio.2018.06.002
Ebert S, Weigelt K, Walczak Y, Drobnik W, Mauerer R, Hume DA, Weber BH, Langmann T (2009) Docosahexaenoic acid attenuates microglial activation and delays early retinal degeneration. J Neurochem 110(6):1863–1875. https://doi.org/10.1111/j.1471-4159.2009.06286.x
Querques G, Forte R, Souied EH (2011) Retina and omega-3. J Nutr Metab 2011:748361. https://doi.org/10.1155/2011/748361
Senoo N, Miyoshi N, Goto-Inoue N, Minami K, Yoshimura R, Morita A, Sawada N, Matsuda J, Ogawa Y, Setou M, Kamei Y, Miura S (2015) PGC-1alpha-mediated changes in phospholipid profiles of exercise-trained skeletal muscle. J Lipid Res 56(12):2286–2296. https://doi.org/10.1194/jlr.M060533
Goto-Inoue N, Yamada K, Inagaki A, Furuichi Y, Ogino S, Manabe Y, Setou M, Fujii NL (2013) Lipidomics analysis revealed the phospholipid compositional changes in muscle by chronic exercise and high-fat diet. Sci Rep 3:3267. https://doi.org/10.1038/srep03267
Helge JW, Wu BJ, Willer M, Daugaard JR, Storlien LH, Kiens B (2001) Training affects muscle phospholipid fatty acid composition in humans. J Appl Physiol (1985) 90(2):670–677. https://doi.org/10.1152/jappl.2001.90.2.670
Andersson A, Sjodin A, Hedman A, Olsson R, Vessby B (2000) Fatty acid profile of skeletal muscle phospholipids in trained and untrained young men. Am J Physiol Endocrinol Metab 279(4):E744–E751. https://doi.org/10.1152/ajpendo.2000.279.4.E744
Mitchell TW, Turner N, Hulbert AJ, Else PL, Hawley JA, Lee JS, Bruce CR, Blanksby SJ (2004) Exercise alters the profile of phospholipid molecular species in rat skeletal muscle. J Appl Physiol (1985) 97(5):1823–1829. https://doi.org/10.1152/japplphysiol.00344.2004
Fan W, Evans RM (2017) Exercise mimetics: impact on health and performance. Cell Metab 25(2):242–247. https://doi.org/10.1016/j.cmet.2016.10.022
Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM (2004) Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol 2(10):e294. https://doi.org/10.1371/journal.pbio.0020294
Manio MC, Inoue K, Fujitani M, Matsumura S, Fushiki T (2016) Combined pharmacological activation of AMPK and PPARdelta potentiates the effects of exercise in trained mice. Physiol Rep 4(5). https://doi.org/10.14814/phy2.12625
Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM (2008) AMPK and PPARdelta agonists are exercise mimetics. Cell 134(3):405–415. https://doi.org/10.1016/j.cell.2008.06.051
Batenburg JJ (1992) Surfactant phospholipids: synthesis and storage. Am J Phys 262(4 Pt 1):L367–L385. https://doi.org/10.1152/ajplung.1992.262.4.L367
Veldhuizen EJ, Haagsman HP (2000) Role of pulmonary surfactant components in surface film formation and dynamics. Biochim Biophys Acta 1467(2):255–270. https://doi.org/10.1016/s0005-2736(00)00256-x
Creuwels LA, van Golde LM, Haagsman HP (1997) The pulmonary surfactant system: biochemical and clinical aspects. Lung 175(1):1–39
Whitsett JA, Wert SE, Weaver TE (2010) Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med 61:105–119. https://doi.org/10.1146/annurev.med.60.041807.123500
Matthay MA, Zemans RL (2011) The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol 6:147–163. https://doi.org/10.1146/annurev-pathol-011110-130158
Post M, Schuurmans EA, Batenburg JJ, Van Golde LM (1983) Mechanisms involved in the synthesis of disaturated phosphatidylcholine by alveolar type II cells isolated from adult rat lung. Biochim Biophys Acta 750(1):68–77. https://doi.org/10.1016/0005-2760(83)90205-9
Vereyken JM, Montfoort A, van Golde LM (1972) Some studies on the biosynthesis of the molecular species of phosphatidylcholine from rat lung and phosphatidylcholine and phosphatidylethanolamine from rat liver. Biochim Biophys Acta 260(1):70–81. https://doi.org/10.1016/0005-2760(72)90075-6
Nakanishi H, Shindou H, Hishikawa D, Harayama T, Ogasawara R, Suwabe A, Taguchi R, Shimizu T (2006) Cloning and characterization of mouse lung-type acyl-CoA:lysophosphatidylcholine acyltransferase 1 (LPCAT1). Expression in alveolar type II cells and possible involvement in surfactant production. J Biol Chem 281(29):20140–20147. https://doi.org/10.1074/jbc.M600225200
Chen X, Hyatt BA, Mucenski ML, Mason RJ, Shannon JM (2006) Identification and characterization of a lysophosphatidylcholine acyltransferase in alveolar type II cells. Proc Natl Acad Sci U S A 103(31):11724–11729. https://doi.org/10.1073/pnas.0604946103
Harayama T, Shindou H, Shimizu T (2009) Biosynthesis of phosphatidylcholine by human lysophosphatidylcholine acyltransferase 1. J Lipid Res 50(9):1824–1831. https://doi.org/10.1194/jlr.M800500-JLR200
Heath RJ, Rock CO (1998) A conserved histidine is essential for glycerolipid acyltransferase catalysis. J Bacteriol 180(6):1425–1430
Bridges JP, Ikegami M, Brilli LL, Chen X, Mason RJ, Shannon JM (2010) LPCAT1 regulates surfactant phospholipid synthesis and is required for transitioning to air breathing in mice. J Clin Invest 120(5):1736–1748. https://doi.org/10.1172/JCI38061
Harayama T, Eto M, Shindou H, Kita Y, Otsubo E, Hishikawa D, Ishii S, Sakimura K, Mishina M, Shimizu T (2014) Lysophospholipid acyltransferases mediate phosphatidylcholine diversification to achieve the physical properties required in vivo. Cell Metab 20(2):295–305. https://doi.org/10.1016/j.cmet.2014.05.019
Friedman JS, Chang B, Krauth DS et al (2010) Loss of lysophosphatidylcholine acyltransferase 1 leads to photoreceptor degeneration in rd11 mice. Proc Natl Acad Sci U S A 107(35):15523–15528. https://doi.org/10.1073/pnas.1002897107
Akagi S, Kono N, Ariyama H, Shindou H, Shimizu T, Arai H (2016) Lysophosphatidylcholine acyltransferase 1 protects against cytotoxicity induced by polyunsaturated fatty acids. FASEB J 30(5):2027–2039. https://doi.org/10.1096/fj.201500149
Wei C, Dong X, Lu H, Tong F, Chen L, Zhang R, Dong J, Hu Y, Wu G, Dong X (2019) LPCAT1 promotes brain metastasis of lung adenocarcinoma by up-regulating PI3K/AKT/MYC pathway. J Exp Clin Cancer Res 38(1):95. https://doi.org/10.1186/s13046-019-1092-4
Bi J, Ichu TA, Zanca C et al (2019) Oncogene amplification in growth factor signaling pathways renders cancers dependent on membrane lipid remodeling. Cell Metab 30(3):525–538 e528. https://doi.org/10.1016/j.cmet.2019.06.014
Wang B, Tontonoz P (2019) Phospholipid remodeling in physiology and disease. Annu Rev Physiol 81:165–188. https://doi.org/10.1146/annurev-physiol-020518-114444
Shindou H, Hishikawa D, Nakanishi H, Harayama T, Ishii S, Taguchi R, Shimizu T (2007) A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:LYSO-PAF acetyltransferase. J Biol Chem 282(9):6532–6539. https://doi.org/10.1074/jbc.M609641200
Benveniste J, Henson PM, Cochrane CG (1972) Leukocyte-dependent histamine release from rabbit platelets. The role of IgE, basophils, and a platelet-activating factor. J Exp Med 136(6):1356–1377. https://doi.org/10.1084/jem.136.6.1356
Morimoto R, Shindou H, Tarui M, Shimizu T (2014) Rapid production of platelet-activating factor is induced by protein kinase Calpha-mediated phosphorylation of lysophosphatidylcholine acyltransferase 2 protein. J Biol Chem 289(22):15566–15576. https://doi.org/10.1074/jbc.M114.558874
Morimoto R, Shindou H, Oda Y, Shimizu T (2010) Phosphorylation of lysophosphatidylcholine acyltransferase 2 at Ser34 enhances platelet-activating factor production in endotoxin-stimulated macrophages. J Biol Chem 285(39):29857–29862. https://doi.org/10.1074/jbc.M110.147025
Cheng L, Han X, Shi Y (2009) A regulatory role of LPCAT1 in the synthesis of inflammatory lipids, PAF and LPC, in the retina of diabetic mice. Am J Physiol Endocrinol Metab 297(6):E1276–E1282. https://doi.org/10.1152/ajpendo.00475.2009
Liang L, Willis-Owen SAG, Laprise C et al (2015) An epigenome-wide association study of total serum immunoglobulin E concentration. Nature 520(7549):670–674. https://doi.org/10.1038/nature14125
Cotte AK, Aires V, Fredon M, Limagne E, Derangere V, Thibaudin M, Humblin E, Scagliarini A, de Barros JP, Hillon P, Ghiringhelli F, Delmas D (2018) Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat Commun 9(1):322. https://doi.org/10.1038/s41467-017-02732-5
Hall Z, Bond NJ, Ashmore T, Sanders F, Ament Z, Wang X, Murray AJ, Bellafante E, Virtue S, Vidal-Puig A, Allison M, Davies SE, Koulman A, Vacca M, Griffin JL (2017) Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 65(4):1165–1180. https://doi.org/10.1002/hep.28953
Kihara Y, Yanagida K, Masago K, Kita Y, Hishikawa D, Shindou H, Ishii S, Shimizu T (2008) Platelet-activating factor production in the spinal cord of experimental allergic encephalomyelitis mice via the group IVA cytosolic phospholipase A2-lyso-PAFAT axis. J Immunol 181(7):5008–5014. https://doi.org/10.4049/jimmunol.181.7.5008
Shindou H, Shiraishi S, Tokuoka SM, Takahashi Y, Harayama T, Abe T, Bando K, Miyano K, Kita Y, Uezono Y, Shimizu T (2017) Relief from neuropathic pain by blocking of the platelet-activating factor-pain loop. FASEB J 31(7):2973–2980. https://doi.org/10.1096/fj.201601183R
van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N (2014) Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 155(4):654–662. https://doi.org/10.1016/j.pain.2013.11.013
Woolf CJ, Salter MW (2000) Neuronal plasticity: increasing the gain in pain. Science 288(5472):1765–1769. https://doi.org/10.1126/science.288.5472.1765
Braz J, Solorzano C, Wang X, Basbaum AI (2014) Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron 82(3):522–536. https://doi.org/10.1016/j.neuron.2014.01.018
Bonnet J, Loiseau AM, Orvoen M, Bessin P (1981) Platelet-activating factor acether (PAF-acether) involvement in acute inflammatory and pain processes. Agents Actions 11(6–7):559–562. https://doi.org/10.1007/bf01978740
Dallob A, Guindon Y, Goldenberg MM (1987) Pharmacological evidence for a role of lipoxygenase products in platelet-activating factor (PAF)-induced hyperalgesia. Biochem Pharmacol 36(19):3201–3204. https://doi.org/10.1016/0006-2952(87)90633-2
Basran GS, Page CP, Paul W, Morley J (1984) Platelet-activating factor: a possible mediator of the dual response to allergen? Clin Allergy 14(1):75–79
Morita K, Morioka N, Abdin J, Kitayama S, Nakata Y, Dohi T (2004) Development of tactile allodynia and thermal hyperalgesia by intrathecally administered platelet-activating factor in mice. Pain 111(3):351–359. https://doi.org/10.1016/j.pain.2004.07.016
Hasegawa S, Kohro Y, Shiratori M, Ishii S, Shimizu T, Tsuda M, Inoue K (2010) Role of PAF receptor in proinflammatory cytokine expression in the dorsal root ganglion and tactile allodynia in a rodent model of neuropathic pain. PLoS One 5(5):e10467. https://doi.org/10.1371/journal.pone.0010467
Honda Z, Nakamura M, Miki I, Minami M, Watanabe T, Seyama Y, Okado H, Toh H, Ito K, Miyamoto T et al (1991) Cloning by functional expression of platelet-activating factor receptor from Guinea-pig lung. Nature 349(6307):342–346. https://doi.org/10.1038/349342a0
Okubo M, Yamanaka H, Kobayashi K, Kanda H, Dai Y, Noguchi K (2012) Up-regulation of platelet-activating factor synthases and its receptor in spinal cord contribute to development of neuropathic pain following peripheral nerve injury. Mol Pain 8:8. https://doi.org/10.1186/1744-8069-8-8
Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K (2003) P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424(6950):778–783. https://doi.org/10.1038/nature01786
Tozaki-Saitoh H, Tsuda M, Miyata H, Ueda K, Kohsaka S, Inoue K (2008) P2Y12 receptors in spinal microglia are required for neuropathic pain after peripheral nerve injury. J Neurosci 28(19):4949–4956. https://doi.org/10.1523/JNEUROSCI.0323-08.2008
Kobayashi K, Takahashi E, Miyagawa Y, Yamanaka H, Noguchi K (2011) Induction of the P2X7 receptor in spinal microglia in a neuropathic pain model. Neurosci Lett 504(1):57–61. https://doi.org/10.1016/j.neulet.2011.08.058
Masuda T, Ozono Y, Mikuriya S, Kohro Y, Tozaki-Saitoh H, Iwatsuki K, Uneyama H, Ichikawa R, Salter MW, Tsuda M, Inoue K (2016) Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat Commun 7:12529. https://doi.org/10.1038/ncomms12529
Jin SX, Zhuang ZY, Woolf CJ, Ji RR (2003) p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci 23(10):4017–4022
Tanga FY, Nutile-McMenemy N, DeLeo JA (2005) The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A 102(16):5856–5861. https://doi.org/10.1073/pnas.0501634102
Kasperska-Zajac A, Brzoza Z, Rogala B (2008) Platelet-activating factor (PAF): a review of its role in asthma and clinical efficacy of PAF antagonists in the disease therapy. Recent Patents Inflamm Allergy Drug Discov 2(1):72–76
Tarui M, Shindou H, Kumagai K, Morimoto R, Harayama T, Hashidate T, Kojima H, Okabe T, Nagano T, Nagase T, Shimizu T (2014) Selective inhibitors of a PAF biosynthetic enzyme lysophosphatidylcholine acyltransferase 2. J Lipid Res 55(7):1386–1396. https://doi.org/10.1194/jlr.M049205
Zhao Y, Chen YQ, Bonacci TM, Bredt DS, Li S, Bensch WR, Moller DE, Kowala M, Konrad RJ, Cao G (2008) Identification and characterization of a major liver lysophosphatidylcholine acyltransferase. J Biol Chem 283(13):8258–8265. https://doi.org/10.1074/jbc.M710422200
Gijon MA, Riekhof WR, Zarini S, Murphy RC, Voelker DR (2008) Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J Biol Chem 283(44):30235–30245. https://doi.org/10.1074/jbc.M806194200
Matsuda S, Inoue T, Lee HC, Kono N, Tanaka F, Gengyo-Ando K, Mitani S, Arai H (2008) Member of the membrane-bound O-acyltransferase (MBOAT) family encodes a lysophospholipid acyltransferase with broad substrate specificity. Genes Cells 13(8):879–888. https://doi.org/10.1111/j.1365-2443.2008.01212.x
Kita Y, Shindou H, Shimizu T (2019) Cytosolic phospholipase A2 and lysophospholipid acyltransferases. Biochim Biophys Acta Mol Cell Biol Lipids 1864(6):838–845. https://doi.org/10.1016/j.bbalip.2018.08.006
Rong X, Wang B, Dunham MM, Hedde PN, Wong JS, Gratton E, Young SG, Ford DA, Tontonoz P (2015) Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. elife 4. https://doi.org/10.7554/eLife.06557
Hashidate-Yoshida T, Harayama T, Hishikawa D et al (2015) Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. elife 4. https://doi.org/10.7554/eLife.06328
Li Z, Jiang H, Ding T, Lou C, Bui HH, Kuo MS, Jiang XC (2015) Deficiency in lysophosphatidylcholine acyltransferase 3 reduces plasma levels of lipids by reducing lipid absorption in mice. Gastroenterology 149(6):1519–1529. https://doi.org/10.1053/j.gastro.2015.07.012
Kabir I, Li Z, Bui HH, Kuo MS, Gao G, Jiang XC (2016) Small intestine but not liver Lysophosphatidylcholine acyltransferase 3 (Lpcat3) deficiency has a dominant effect on plasma lipid metabolism. J Biol Chem 291(14):7651–7660. https://doi.org/10.1074/jbc.M115.697011
Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martin MG, Alrefai WA, Ford DA, Tontonoz P (2018) Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 22(2):206–220. e204. https://doi.org/10.1016/j.stem.2017.12.017
Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P (2007) Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc Natl Acad Sci U S A 104(25):10601–10606. https://doi.org/10.1073/pnas.0701096104
Demeure O, Lecerf F, Duby C, Desert C, Ducheix S, Guillou H, Lagarrigue S (2011) Regulation of LPCAT3 by LXR. Gene 470(1–2):7–11. https://doi.org/10.1016/j.gene.2010.09.002
Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, Jung ME, Ford DA, Tontonoz P (2013) LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab 18(5):685–697. https://doi.org/10.1016/j.cmet.2013.10.002
Ishibashi M, Varin A, Filomenko R, Lopez T, Athias A, Gambert P, Blache D, Thomas C, Gautier T, Lagrost L, Masson D (2013) Liver x receptor regulates arachidonic acid distribution and eicosanoid release in human macrophages: a key role for lysophosphatidylcholine acyltransferase 3. Arterioscler Thromb Vasc Biol 33(6):1171–1179. https://doi.org/10.1161/ATVBAHA.112.300812
Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14(22):2819–2830. https://doi.org/10.1101/gad.844900
Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B (2000) Role of LXRs in control of lipogenesis. Genes Dev 14(22):2831–2838. https://doi.org/10.1101/gad.850400
Li Z, Ding T, Pan X, Li Y, Li R, Sanders PE, Kuo MS, Hussain MM, Cao G, Jiang XC (2012) Lysophosphatidylcholine acyltransferase 3 knockdown-mediated liver lysophosphatidylcholine accumulation promotes very low density lipoprotein production by enhancing microsomal triglyceride transfer protein expression. J Biol Chem 287(24):20122–20131. https://doi.org/10.1074/jbc.M111.334664
Cash JG, Hui DY (2016) Liver-specific overexpression of LPCAT3 reduces postprandial hyperglycemia and improves lipoprotein metabolic profile in mice. Nutr Diabetes 6:e206. https://doi.org/10.1038/nutd.2016.12
Rong X, Wang B, Palladino EN, de Aguiar Vallim TQ, Ford DA, Tontonoz P (2017) ER phospholipid composition modulates lipogenesis during feeding and in obesity. J Clin Invest 127(10):3640–3651. https://doi.org/10.1172/JCI93616
Tanaka H, Zaima N, Sasaki T, Yamamoto N, Inuzuka K, Yata T, Iwaki T, Umemura K, Sano H, Suzuki Y, Urano T, Setou M, Unno N (2017) Lysophosphatidylcholine Acyltransferase-3 expression is associated with atherosclerosis progression. J Vasc Res 54(4):200–208. https://doi.org/10.1159/000473879
Thomas C, Jalil A, Magnani C, Ishibashi M, Quere R, Bourgeois T, Bergas V, Menegaut L, Patoli D, Le Guern N, Labbe J, Gautier T, de Barros JPP, Lagrost L, Masson D (2018) LPCAT3 deficiency in hematopoietic cells alters cholesterol and phospholipid homeostasis and promotes atherosclerosis. Atherosclerosis 275:409–418. https://doi.org/10.1016/j.atherosclerosis.2018.05.023
Taniguchi K, Hikiji H, Okinaga T, Hashidate-Yoshida T, Shindou H, Ariyoshi W, Shimizu T, Tominaga K, Nishihara T (2015) Essential role of Lysophosphatidylcholine acyltransferase 3 in the induction of macrophage polarization in PMA-treated U937 cells. J Cell Biochem 116(12):2840–2848. https://doi.org/10.1002/jcb.25230
Eto M, Shindou H, Koeberle A, Harayama T, Yanagida K, Shimizu T (2012) Lysophosphatidylcholine acyltransferase 3 is the key enzyme for incorporating arachidonic acid into glycerophospholipids during adipocyte differentiation. Int J Mol Sci 13(12):16267–16280. https://doi.org/10.3390/ijms131216267
Lee HC, Inoue T, Imae R, Kono N, Shirae S, Matsuda S, Gengyo-Ando K, Mitani S, Arai H (2008) Caenorhabditis elegans mboa-7, a member of the MBOAT family, is required for selective incorporation of polyunsaturated fatty acids into phosphatidylinositol. Mol Biol Cell 19(3):1174–1184. https://doi.org/10.1091/mbc.e07-09-0893
Lee HC, Inoue T, Sasaki J, Kubo T, Matsuda S, Nakasaki Y, Hattori M, Tanaka F, Udagawa O, Kono N, Itoh T, Ogiso H, Taguchi R, Arita M, Sasaki T, Arai H (2012) LPIAT1 regulates arachidonic acid content in phosphatidylinositol and is required for cortical lamination in mice. Mol Biol Cell 23(24):4689–4700. https://doi.org/10.1091/mbc.E12-09-0673
Anderson KE, Kielkowska A, Durrant TN, Juvin V, Clark J, Stephens LR, Hawkins PT (2013) Lysophosphatidylinositol-acyltransferase-1 (LPIAT1) is required to maintain physiological levels of PtdIns and PtdInsP(2) in the mouse. PLoS One 8(3):e58425. https://doi.org/10.1371/journal.pone.0058425
Johansen A, Rosti RO, Musaev D et al (2016) Mutations in MBOAT7, encoding Lysophosphatidylinositol acyltransferase I, Lead to intellectual disability accompanied by epilepsy and autistic features. Am J Hum Genet 99(4):912–916. https://doi.org/10.1016/j.ajhg.2016.07.019
Buch S, Stickel F, Trepo E et al (2015) A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet 47(12):1443–1448. https://doi.org/10.1038/ng.3417
Mancina RM, Dongiovanni P, Petta S et al (2016) The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 150(5):1219–1230 e1216. https://doi.org/10.1053/j.gastro.2016.01.032
Thabet K, Asimakopoulos A, Shojaei M et al (2016) MBOAT7 rs641738 increases risk of liver inflammation and transition to fibrosis in chronic hepatitis C. Nat Commun 7:12757. https://doi.org/10.1038/ncomms12757
Sookoian S, Flichman D, Garaycoechea ME, Gazzi C, Martino JS, Castano GO, Pirola CJ (2018) Lack of evidence supporting a role of TMC4-rs641738 missense variant-MBOAT7- intergenic downstream variant-in the susceptibility to nonalcoholic fatty liver disease. Sci Rep 8(1):5097. https://doi.org/10.1038/s41598-018-23453-9
Acknowledgements
The Lipid Signaling Project, National Center for Global Health and Medicine receives funding from ONO Pharmaceuticals. This work was supported by AMED-CREST 19gm0910011 (to HS), AMED-P-CREATE 19 cm0106116 (to HS), AMED Program for Basic and Clinical Research on Hepatitis JP19fk0210041 (to HS).
Conflicts of Interest
The authors have no conflicts of interest affecting the objectivity of this review.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Valentine, W.J., Hashidate–Yoshida, T., Yamamoto, S., Shindou, H. (2020). Biosynthetic Enzymes of Membrane Glycerophospholipid Diversity as Therapeutic Targets for Drug Development. In: Kihara, Y. (eds) Druggable Lipid Signaling Pathways. Advances in Experimental Medicine and Biology, vol 1274. Springer, Cham. https://doi.org/10.1007/978-3-030-50621-6_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-50621-6_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-50620-9
Online ISBN: 978-3-030-50621-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)