
Abstract
The main sources of reactive oxygen species (ROS) in cells are mitochondria, whose components would be primary targets of ROS. Both facts are responsible for the key role of these organelles in aging according to the “mitochondrial theory of aging”. Oxidative damage to mitochondrial DNA (mtDNA) is especially important since it would have the longest-term consequences impairing mitochondrial function. This would lead to a decrease in ATP production, but also to an increased ROS generation. In turn, CoQ, which acts as an electron carrier in mitochondria, is an essential factor for cell bioenergetics and an equilibrated CoQ pool is expected to perform a better electron flow adaptation. Moreover, it is a lipid-soluble antioxidant and efficiently prevents oxidation of DNA along with other macromolecules. Other interesting attributed roles include interaction with cell signaling cascades, anti-inflammatory activities and interference with programmed cell death. Due to this pleiotropic effect, most of interventions with CoQ have been focused on multiple processes related to mitochondria. In this sense, its effects have been investigated in mitochondrial diseases and pathological conditions related with aging whose patients have shown a higher frequency of mtDNA alterations. In addition, dietary CoQ also has been tested in combination with different diets rich in particular type of fatty acids due to the role of these in biological membranes and oxidative stress, as well as aging. This chapter aims to review the effect of CoQ on aging and mitochondrial dysfunction, with especial interest in their actions on mtDNA or the consequences of mtDNA alterations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
There is a growing body of evidences indicating that mitochondria have a key role in aging phenomenon particularly in organs or tissues with an important dependence of aerobic metabolism. A major fact explaining this possible role is that mitochondria are the main source of reactive oxygen species (ROS) in most cells. An impairment of mitochondrial function led to lower production of ATP, but other important downstream consequences also should be considered. Among other, the increase in the generation of reactive oxygen species is usually considered as the most relevant.
In turn, oxidative stress also seems a major factor influencing mitochondria “health” since primary target of ROS would be mitochondrial components. Oxidative damage to mitochondrial DNA (mtDNA) would be especially important since it would have the longest-term consequences in mitochondrial function. For these reason, there are several theories of aging that have suggested a key role of mitochondria and in particular of mtDNA in this phenomenon.
This chapter aims to review the effect of coenzyme Q (CoQ) on aging and mitochondrial dysfunction, with especial interest in their actions on mtDNA or mtDNA mutations consequences. Additionally, to improve the understanding of implications in aging of oxidative damage to mtDNA, this chapter provides an approximation to mtDNA processes as well as an analysis of mechanisms that try to explaining their relationship with aging.
2 Mitochondrial DNA Features
Unlike the rest of organelles (except chloroplasts in vegetal cells), mitochondria had their own extrachromosomal DNA molecules. mtDNA presents structural and functional features very different respect than nuclear DNA (nDNA). For this reason, a preliminary approach to these unique features is needed to properly understand the relationship of mtDNA with mitochondrial functionality and aging.
MtDNA is a circular double-stranded molecule located in the mitochondrial matrix whose size ranged from 16,000 to 18,000 base pairs (bp) in vertebrates. Namely, human mtDNA size is 16569 bp and its sequence was determined in 1981 (Anderson et al. 1981). To differentiate the two mtDNA strands, it has been established the terms “heavy strand” (H-strand) and the “light strand” (L-strand) according to their GCT content. This is due to their behavior when strands are separated on denaturing cesium chloride gradients. MtDNA has no histones but rather packaged into nucleoids. These consist in stable protein-mtDNA macrocomplexes primarily associated to inner mitochondrial membrane (Wang and Bogenhagen 2006; Holt et al. 2007). Each nucleoid has an average diameter of 100 nm (Kukat et al. 2011) and they may be exchanged between mitochondria (Wang and Bogenhagen 2006; Holt et al. 2007).
Mitochondrial genome encodes 13 polypetides, two rRNAs and 22 tRNAs (Anderson et al. 1981). Encoded polypetides are subunits of mitochondrial electron transport chain (mtETC) complexes whereas rRNAs and tRNAs are required for the intramitochondrial translation of the protein-coding units. In contrast to nuclear, mitochondrial genome does not contain introns within mtDNA coding region and almost no noncoding nucleotides exist between genes. In vertebrates, an exception is a noncoding region closely associated to origin of H-strand DNA replication (OH) that contains the transcription promotors (Clayton 1982, 1991). In most of cases (some species present two), each strand contain only a promoter. The transcription processes initiated from each of them yield two large polycistronic transcripts that are processed later to generate mature tRNAs, rRNAs, and mRNA by precise endonucleolytic cleavages. In most cases, such cleavages occur, immediately before and after a tRNA sequence. Depending on which strand acts as the template for transcription, promoters are designated as light-strand promoter (LSP) or heavy-strand promoter (HSP) (Ojala et al. 1981; Attardi and Schatz 1988; Clayton 1992). In addition to 13 mtDNA encoded subunits, there are other approximately 70 components of mitochondrial respiratory chain and other proteins that participate in mitochondrial metabolism and maintenance, which are encoded in the nuclear genome and require specialized import systems to be imported to mitochondria (Mokranjac and Neupert 2005).
Several molecules (usually from two to ten) of mtDNA coexist in a mitochondrion and there are mitochondria in every cell. Therefore, a cell possesses hundreds of copies of mtDNA, a condition termed as polyplasmy. When all mtDNA in a cell present the same sequence, the condition is known as homoplasmy. In contrast, heteroplasmy occurs when two or more different molecules of mtDNA can be also present in a cell or organism.
Because of zygote does not receive mitochondria from sperm in mammals or possible transmitted mitochondria are selected against during replication, all mitochondria are inherited from the mother. Then, mitochondria divide and proliferate during development, but also in adult life increasing mitochondrial mass. During this process known as mitochondrial biogenesis synthesis of new mitochondrial proteins, but also mtDNA, is required. It is known that biogenesis is under the control of nuclear factors, although the exact mechanism has not been fully unraveled yet.
2.1 Replication of Mitochondrial DNA
It is assumed that mtDNA replication is not necessary linked to the cell cycle (Clayton 1982) and mtDNA is continuously turned over. Thus, mitochondrial and nuclear genomes would be independently replicated (Bogenhagen and Clayton 1977). For this reason, mtDNA is also replicated in postmitotic cells, which has been also evidenced (Pohjoismäki et al. 2009). Notwithstanding, there is a increasing number of publications reporting some relationship between mitochondrial function and cell cycle (Arakaki et al. 2006; Owusu-Ansah et al. 2008; Mitra et al. 2009), which suggests a possible connection between mtDNA replication and the cell cycle.
The exact mechanism of human mtDNA replication is not completely known yet. The two most important replication models proposed until now are: the strand-asynchronous method that is the most traditional and the leading-lagging strand model (Holt et al. 2000; Fish et al. 2004). In both, DNA replication is initiating at OH that is located downstream of the LSP in the D-loop region. In the first one, subsequent elongation of a nascent newly-synthesized H-strand leads to the parental H-strand displacement from the H-strand. Because of the origin of L-strand DNA replication (OL) is located approximately two thirds the genomic distance away from OH on the mtDNA molecule, L-strand synthesis, that proceeds in the opposite direction, will not be initiated until almost two-thirds of new H-strand have been synthesized and OL is exposed (Clayton 1992). Here, the adoption of a particular configuration by the H-strand allows a mitochondrial DNA primase initiate L-strand DNA synthesis (Wong and Clayton 1985a, b). In the second model, L-strand synthesis starts in a coordinately way shortly after replication in form of short Okazaki fragments that will be joined then (Yasukawa et al. 2006).
In any of the models, after DNA strand synthesis, the two daughter molecules will be separated, RNA primers removed, and remaining DNA gaps will be filled and ligated. An additional step introducing superhelical turns into the closed molecule also occurs (Shadel y Clayton 1997). The existence of a model does not necessarily exclude the other and their occurrence may depend on cell type. It has been suggested that cells requiring rapid mtDNA synthesis present a strand-displacement mechanism whereas the leading-lagging strand one would be more prevalent in cells which are in a steady-state (Holt et al. 2000; Fish et al. 2004; Jacobs et al. 2006; Tuppen et al. 2010).
Regardless of the model, various nuclear DNA (nDNA)-encoded proteins are needed to form the mitochondrial replisome and accomplish mtDNA replication. These include a 5′–3′ DNA helicase named Twinkle, some mitochondrial SSB proteins (mt-SBB) and the polymerase γ (Polγ) that contains two subunits, one catalytic with 5′-3′ exonuclease activity (PolγA) and other processivity (PolγB) (Korhonen et al. 2004).
2.2 Mutations and Mitochondrial DNA
Molecular defects in mtDNA have a significant role in human disease and aging and they have been found in each type of mitochondrial gene. MtDNA mutations range from single base changes in the genome (point mutations) up to large rearrangements (deletions and duplications). In turn, these changes in mtDNA sequence can be maternally inherited or somatic (i.e. created in situ). In general, deletions and duplications are most often sporadic or somatic whereas maternally inherited alterations are commonly point mutations (Leonard y Schapira 2000).
Alterations in mtDNA sequence have been strongly associated with deleterious effects on organisms. There are at least two reasons explaining this relationship. On the one hand mtDNA and has no introns, so that a random mutation will usually strike a coding DNA sequence. On the other hand, estimations indicate a 10–20 fold higher mutation frequency in human mtDNA than in nDNA (Brown et al. 1979). This higher rate could be due to the combination of two factors. On the one hand, maybe the number of systems of DNA repair is insufficient for all the damage that occurs, although there are a growing number of reports indicating that in mitochondria there are more enzyme activities for repair of damaged nucleotides of which it was believed at first. On the other hand, it seems that mtDNA has an increased susceptibility to mutation (Shadel and Clayton 1997). Several differential features are responsible for this susceptibility to mutation:
-
Mitochondrial oxidative environment, mainly generated by free radicals generated at the electron transport chain, although there are other sources.
-
The absence of protective histones, although mtDNA is packaged into protein-mtDNA aggregates termed nucleoids (Kukat et al. 2011) that are believed to protect mtDNA from chemical damage in some degree (Lagouge and Larsson 2013).
-
Failure of proof-reading by mtDNA polymerases during mtDNA replication that usually also occurs although cell does not divide.
-
Lack of recombination as consequence of maternal heritage that allows sequential accumulations of mutations through maternal lineages.
Mutations in mtDNA have received great interest because it is known that specific mtDNA mutations found in humans are likely causative in different diseases. Moreover, common neurodegenerative disorders and others disease often associated with aging also have been associated with mtDNA sequence alterations (Larsson and Clayton 1995). Because of cells present polyplasmy, normal and mutant mtDNA can coexist within the same cell. Actually, the existence of either completely normal or completely mutant mtDNA is rare. In this context, heteroplasmy is particularly important since it can allow an otherwise lethal mutation to persist. This is due to a certain minimal amount or threshold level is required to have deleterious effects in cell (Larsson 2010). In that sense, there are selection pressures at the molecular and cellular levels, as well as at the level of the organism itself. The proportion of mutant mitochondrial DNA required for the occurrence of a deleterious phenotype, known as the threshold effect, varies among persons, among organ systems, and within a given tissue.
After mtDNA sequence alterations are produced, several processes can modify their frequencies in the cell. In this sense, it has been suggested that changes in the frequencies of different mtDNA molecules follows principles of population genetics but rather Mendelian laws. Both, de novo and inherited mutations in mtDNA, if there are present in heteroplasmy, are subject to mitotic segregation. Consequently, frequency of different mtDNA molecules can shift in daughter cells since they are randomly segregated during mitosis. Thus, mutated mtDNA can increase with possible deleterious effects or decrease to disappear, particularly in fast-dividing tissues (Tuppen et al. 2010). Anyway, mechanisms for mitotic segregation need to be studied further. On the other hand, a mtDNA molecule may be replicated many times or not at all as a cell divide. If mtDNA molecules are selectively replicated, proportions of mutant and normal molecules in mother cells would be modified. In addition, it is important to note that replication of mtDNA also occurs in the absence of cell division. Thus, mtDNA is replicated also in postmitotic cells, so it can undergo similar types of segregation (Larsson 2010). It has been suggested that it is caused by random genetic drift, in conditions of relaxed mtDNA replication (Elson et al. 2001). Actually, expansion in postmitotic tissues, a preferential amplification of mtDNA mutations might occur termed clonal (Larsson et al. 1990; Weber et al. 1997).
In mammals, a rapid segregation in heteroplasmic mtDNA genotypes returning to homoplasmy in some descendants has been reported (Upholt and Dawid 1977; Olivo et al. 1983; Holt et al. 1989; Vilkki et al. 1990; Larsson et al. 1992; Blok et al. 1997; Brown 1997). The existence of a mtDNA bottleneck during development has been proposed to explain these observations (Tuppen et al. 2010). Different mechanisms by which this bottleneck is present have been hypothesized, but discussion about this topic remains. A relatively well-accepted hypothesis suggests that a marked reduction in mtDNA copy number would take place in the germ line leading to a genetic bottleneck during embryonic development (Jenuth et al. 1996; Cree et al. 2008). In contrast, other authors have suggested that, during oogenesis, there is a preferential replication of a particular mtDNA or a subgroup of them, but neither reduction of mtDNA copy number is produced in germ line (Cao et al. 2007). Other recently proposed explanation suggests that mtDNA subpopulation is selectively replicated during postnatal folliculogenesis, thus the mtDNA bottleneck would not occur during oogenesis. In single germ cells, mtDNA heteroplasmy and copy number vary throughout oogenesis (Wai et al. 2008), a finding that support that hypothesis. Anyway, more research is needed to clarify the mtDNA bottleneck exact nature (Tuppen et al. 2010).
2.3 Mitochondrial DNA Repair Systems
Although its importance is currently discussed (Richter et al. 1988; Hegler et al. 1993), oxidative damage occurs normally and can be elevated in cells and tissues (LeDoux et al. 1992; Mecocci et al. 1993, 1994; Driggers et al. 1993; Shigenaga et al. 1994). However, mitochondria have their own repair systems for damaged mtDNA that help to maintain mtDNA integrity, although their number seems to be more limited than in nucleus.
Base excision repair (BER) is one of the most studied mitochondrial mechanisms for mtDNA repair. In fact, intially it was though that short-patch BER was the unique pathway to repair mtDNA damage, especially oxidative damage (Stierum et al. 1999). This mechanism represents the main pathway for repairing oxidized modifications (Slupphaug et al. 2003), but it is also a primary pathway for alkylation and deamination-derived modifications repair (Dianov et al. 2001; Chan et al. 2006). First step in BER is the cleaving the N-glycosidic bond leading to an abasic site. This reaction is catalyzed by different DNA glycosylases that are responsible to recognize modified bases and also present AP lyase activity to cleavage DNA backbone (Robertson et al. 2009). Among other, these include the uracil DNA glycosylase (UNG), the endonuclease III homolog (NTH1), and the 8-oxoguanine DNA glycosylase-1 (OGG1). OGG1 is particularly interesting since it is required for the recognition and cleavage of 8-oxoguanine (8-oxoG) from double-stranded DNA (Kuznetsov et al. 2005). In this step also participates the AP endonuclease (APE1) that cleaves on the immediate 5´ side of the apurinic/apyrimidinic (AP) site, leaving a 3´ hydroxyl and 5´-deoxyribose-5-phosphate (5´-dRP) residue (Masuda et al. 1998). Then, the resultant gap is filled with the correct nucleotide by the mitochondrial DNA polymerase -i.e. Polγ- (Ropp and Copeland 1996).
Here, it is possible to distinguish two BER pathways according to the number of nucleotides incorporated to the gap. When a single nucleotide is incorporated, the mechanism termed short-patch BER is relatively simple. In contrast, long-patch BER, which involves the incorporation of multiple nucleotides (commonly ranged from 2 to 7) is more complex and additional enzymatic activities are required (Robertson et al. 2009). Such enzymatic activities would deal with the exposure of the original DNA strand as a single-stranded overhang or a flap structure that is the main difficulty generated by the incorporation of several nucleotides (Xu et al. 2008). Finally, the nick generated is sealed by the mitochondrial DNA ligase, ligase III (Lakshmipathy and Campbell 1999a).
Other known nuclear DNA repair mechanisms has been proposed to exist in a mitochondrial version. It seem that the most clear additional mechanism is homologous recombination (LeDoux et al. 1992; Ling et al. 1995; Sage et al. 2010) that is the primary pathway to repair double-strand breaks. That plays a critical role in facilitating the progression of replication when advancing polymerase complex progress is blocked by the presence of a DNA lesion. There are also evidences for existence of mismatch repair (Mason et al. 2003) and non-homologous end-joining activities (Lakshmipathy and Campbell 1999b) at mitochondria but more research is needed to confirm them. Other hypothesized mechanisms especially useful to repair 8-oxoG have been nucleotide excision repair (Stevnsner et al. 2002) and translesion synthesis (Pinz et al. 1995; Graziewicz et al. 2004, 2007), although none enzyme activity related to them has been reported in mitochondria up to date.
3 Aging and Mitochondrial DNA
3.1 Aging and Mitochondrial DNA Mutations Relationship: A Conceptual Framework
Overall, different studies have indicated mutated mtDNA molecules accumulate with aging since elderly people has shown higher levels of somatic point mutations or deletions in mtDNA from different tissue types (Cortopassi and Arnheim 1990; Simonetti et al. 1992; Laderman et al. 1996: Melov et al. 1999; Berneburg et al. 2004; Bender et al. 2006; Marín-García et al. 2006; Krishnan et al. 2008). This association between aging and mtDNA alterations also has been in found studies in rodents (Pikó et al. 1988; Quiles et al. 2006, 2010; Ochoa et al. 2011). However, discussion exists about what is the magnitude of such accumulation and its importance in aging. Still, rather low, the differences between young and old individuals in frequency of mtDNA mutations are statistically significant (Pikó et al. 1988). Additionally, an accumulation of multiple mtDNA deletions has also been reported in individuals with neurodegenerative diseases, such as Alzheimer and Parkinson’s disease (Cortopassi et al. 1992; Coskun et al. 2004; Bender et al. 2006; Kraytsberg et al. 2006 Krishnan et al. 2008). Similarly, overall mtDNA heteroplasmy seem to increase with aging indicating that additional somatic mutations are continuously appearing during adult life (Pliss et al. 2011; Sondheimer et al. 2011; Diot et al. 2016). Most of these observations support the idea of mtDNA alterations and their subsequent accumulation during life are responsible or at least contribute to the senescent phenotype. Several proposed theories that related mitochondria and aging providing an explanation for this phenomenon.
As indicated above, mitochondrion is a major site of ROS production in the cell (especially at mtETC) which would makes mitochondria the prime targets for oxidative damage (Harman and others 1955; Miquel et al. 1980). This fact was taken into account by Harman and other authors (Harman and others 1955; Miquel et al. 1980) to considered mtDNA mutations to be the initiating, primary event in the aging process in their mitochondrial free radical theory of aging. According to that, a vicious cycle would be established whereby oxidative damage to mtDNA and other mitochondrial components leads to respiratory chain dysfunction, which in turn leads to increased generation of ROS , further facilitating respiratory chain components damage and thus creating a self-amplifying deterioration. The mitochondrial free radical theory of aging, thus, suggests the existence of a vicious cycle that results in an exponential increase in mtDNA mutations with time. Interestingly, Greaves et al. (2014), using next-generation sequencing, has reported that mtDNA mutation rate does not seem to increase with age (Greaves et al. 2014). This fact would be contradictory with the exponential increase in mtDNA mutations proposed by the mitochondrial free radical theory of aging.
More recently, new evidences have emerged that give more subtle roles beyond those as damaging agent to ROS. Actually, despite lifespan and ROS production is correlated, it has been shown that ROS are not directly responsible for aging (Sanz et al. 2010). However, it has been progressively appreciated that ROS also can function as signaling molecules, facilitating adaptation to stress in a wide variety of physiological situations (Sena et al. 2008). In this context, Hekimi and colleagues (2011) proposed the gradual ROS response hypothesis that suggests that “ROS generation is not a cause of aging, but rather represents a stress signal in response to age-dependent damage”. Concerning mitochondria, when respiratory chain dysfunction coupled with moderate increases in ROS levels, these act as stress signal that activates protective quality control pathways improving mitochondria quality. These finding also have resulted in the mitohormesis hypothesis (Tapia 2006; Ristow and Zarse 2010). Despite of the existence of protective quality control pathways, a continuous or strong dysfunction of the mitochondrial respiratory chain would lead to a substantial ROS accumulation. Consequently, protective and defense mechanisms against oxidative stress would be overwhelmed (Tapia 2006; Zelenka et al. 2015). Indeed, evidences suggest that mtDNA controls longevity (Sanz et al. 2010) which is consistent with this theory.
Lastly, possible link between mitochondria (and mtDNA) and other important event in aging process have been also proposed. In this sense, Ahmed et al. (2008) have suggested that telomerase protects mitochondria from mild oxidative stress. Other possible causes would include the repression of PGC-1 promoter by p53 activated as consequence of telomere dysfunction or TERT activity effect on mtDNA repair (Monickaraj et al 2012; Tyrka et al. 2015).
3.2 Generation and Accumulation of Mitochondrial DNA Mutations
Different mechanisms have been proposed to explain the accumulation of mtDNA mutations with aging. Oxidative damage to mtDNA is often assumed as main responsible for age-associated somatic mtDNA mutations generation, although there are other agents able to produce DNA lesions that also could accumulate that might be important under some conditions. If the amount of mtDNA (oxidative) damage overwhelm the mtDNA repair mechanism, a progressively accumulation of mtDNA alterations or mutations would occur with aging. The involvement of ROS in the creation of mtDNA mutations is central to the mitochondrial free radical theory of aging and it is supported by correlative data showing higher levels of somatic mtDNA mutations in older than in younger mammals including humans (Larsson 2010). It is known that DNA bases can suffer until 24 oxidative lesions different (Evans et al. 2004). There are also 13 additional major products of oxidative damage to the sugar moiety (Evans et al. 2004). In spite of this number, most of investigations has been focused on the guanine adduct 7,8-dihydro-8-oxo-deoxyguanosine (8-oxodG) (Evans et al. 2004) that is considered one of the most abundant oxidative lesions that accumulate in mtDNA over time. One consequence of 8-oxodG presence in DNA is the transversion with adenine during replication due to the mispairing of 8-oxoG, but the biological significance for the majority of the lesions remains unknow (Larsson 2010). It has been reported that accumulation of 8-oxoG in mtDNA occurs with age (Szczesny et al. 2003). Initially, it was thought that in vivo levels of 8-oxodG were very high (Richter et al. 1988), although then this finding was attributed to overestimation by methodological problems (Hamilton et al. 2001). More recently a sequencing study in mouse showed that mtDNA transversion mutations not increased with age, so oxidative damage may not be a major source of formation of mtDNA mutations (Ameur et al. 2011).
Although the exact steady state level of oxidative damage in mtDNA is variable among tissues and the importance is discussed in the literature (Richter et al. 1988; Hegler et al. 1993; Shadel and Clayton 1997), such damage occurs normally and can be elevated in cells and tissues under certain conditions. These include exposure to certain chemical agents (Driggers et al. 1993) and antiviral drugs (Lewis and Dalakas 1995), UV radiations (Berneburg et al. 2004; Krishnan et al. 2008; Birket y Birch-Machin 2007) or pathologies (Mecocci et al. 1994). In this context, dietary conditions could results especially interesting for mtDNA mutation implications in aging since certain nutritional conditions may be maintained over life. Recent experimental studies indicate that reduction in the degree of unsaturation of fatty acids in the diet induces less oxidative damage and alterations in mitochondrial DNA (mtDNA) in different tissues including liver (Quiles et al. 2006), brain (Ochoa et al. 2011) and heart (Quiles et al. 2010).
An alternative source of mtDNA mutations is pol γ that would produce somatic mtDNA mutations by slipped mispairing during mtDNA replication. Namely, these replication errors have suggested being an important mechanism for formation of mtDNA deletions (Madsen et al. 1993). In human mtDNA deletions that mainly occur between OH and OL, are typically flanked by short direct repeated sequences (Mita et al. 1990; Samuels et al. 2004; Bua et al. 2006), which supports this hypothesis. Moreover, an in vitro analysis of the mutations generated by wild-type Pol γ showed a good concordance with those observed in vivo in human, including a paucity of G:C to T:A transversions (Zheng et al. 2006). This hypothesis is also supported by mathematical modeling (Cortopassi and Arnheim 1990). Still, the fact of true turnover rate of mtDNA in mammalian tissues is largely unknown complicates studies in this area (Larsson 2010).
However, most important evidences in favor of this mechanism come from studies using a well-established knock-in murine model (Trifunovic et al. 2004; Kujoth et al. 2005). This has homozygous genotype for a mutated version of PolγA with increased proofreading activity, so it provides a critical test of the replication error hypothesis. Expression of the proof-reading deficient PolγA leads to a rapid accumulation of mtDNA point mutations and deletion during embryogenesis, which are clearly present in midgestation (Trifunovic et al. 2004). Moreover, in adult life, accumulation of mtDNA mutations goes on in a linear manner leading to the progressive and random accumulation of mtDNA point mutations during mitochondrial biogenesis (Trifunovic et al. 2004). Because of this amount of mtDNA mutations, it has been generally named as Polγ mutator mouse . Most of the mutations generated in mtDNA mutator mice are transitions (Trifunovic et al. 2004) and their pattern after germ line transmission resembles the mutation spectra found in natural populations of mice and humans ( Stewart et al. 2008a, b). Because of mitochondrial free radical theory of aging predicts an exponential increase in the mutation burden throughout life; findings from these models are in contradiction with it. Interestingly, accumulation of mtDNA mutations has no major increase in oxidative damage in many different tissues in adults (Trifunovic et al. 2005).
Most researchers consider replication to be the most likely mechanism of deletion formation (Lloret et al. 2009; Lagouge and Larsson 2013), but Krishnan et al. (2008) proposed that “mtDNA deletions arise during the repair of damaged mtDNA”. Although they remain unclear, some mechanisms have been proposed to explain the impairment or restriction of repair machinery efficiency with aging. A possibility would be the age-associated decline in import capacity of the mitochondria. Accumulation into the mitochondrial intermembrane space and importation failure inside the mitochondrial matrix of an unprocessed form of DNA glycosylase OGG1 that is involved in BER of 8-oxoG has been proposed to occur with aging (Szczesny et al. 2003). This would explain why 8-oxoG is so abundant among oxidative lesions that accumulate in mtDNA. In fact, mice lacking this enzyme have increased levels of 8-oxodG in mtDNA (de Souza-Pinto et al. 2001). In turn, mispairing of 8-oxoG during replications would extend the mutation
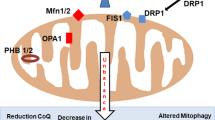
Several processes cooperate to maintain mitochondrial quality, among which highlights mitophagy that is the only mechanism known to turn over whole mitochondrial genomes (Kim et al. 2012; Diot et al. 2016). As it is expected, to keep the pool of mitochondria healthy, replacement by biogenesis is needed that must be adequately coordinated with mitophagy. Although mtDNA turnover in differentiated tissues is not well defined, if this results affected by aging, accumulation of mutant mtDNA can occur. In this sense, it has been reported a decline of mitophagy with aging (Diot et al. 2015) thus disadvantages both the turnover of dysfunctional mitochondria and the production of fresh mitochondria. Interestingly, Greaves et al. (2014), using next-generation sequencing, have shown that mtDNA mutation rate could not increase with age, which enhances the importance of autophagy decline in mutations accumulation.
Along with the aforementioned mechanisms, changes in mitochondrial dynamics also are very important, as well as affecting fusion and fission of membranes, modulate mitochondrial turnover. Fission disrupted mitochondria segregation (Katajisto et al. 2015) whereas fusion would mix content of different mitochondria including mtDNA molecules (Chan 2012; Tam et al. 2013). When the frequency of fusion/fission cycles is reduced, mtDNA mutations tend to accumulate and there is less mtDNA mixing (Diot et al. 2016). In support of these mechanisms importance, a mathematical model by Tam et al. (2014) suggests that a combination of rapid mitochondrial fission, fusion and mitophagy can extend lifespan because mitochondrial function maintenance would be achieved.
3.3 The Impact of Mitochondrial DNA Alterations on Mitochondrial Function and Aging
Independently of the actual cause of a given mutation, it is possible to suppose at least some of the consequences of changes in mitochondrial DNA sequence. Most mtDNA sequence alterations are neutral polymorphisms (Ingman et al. 2000), but when this not occurs, their magnitude could be different depending on gene affected. Point mutations can affect to protein, tRNA, or rRNA genes within mtDNA. Phenotypical consequence of mutations in a protein-coding gene would be a functional alteration of a particular complex of mitochondrial respiratory chain to which the corresponding protein belongs (Tuppen et al. 2010). In turn, mutations in genes encoding for mt-tRNAs might impair overall translation of mtDNA by reducing functional mt-tRNAs availability (Tuppen et al. 2010). Regarding mtDNA rearrangements, it has been reported that most of them are large-scale deletions ranged from 1.3 to 8 kb that span several genes (Schon et al. 1989).
It is expected that accumulation of somatic mtDNA mutations would lead to mitochondria with respiratory chain deficiencies. Notwithstanding, an important feature of mtDNA further must be considered to understand the consequences of mtDNA alterations in cell and/or tissue. As mentioned, cells are polyplasmic for mtDNA, which implies that de novo somatic mutations in mtDNA would be in heteroplasmy, at least at beginning. Moreover, mutated mtDNA frequencies can vary dramatically between tissues (Shoffner et al. 1990; Goto et al. 1990). Actually, even in mitochondrial disorder patients, there is considerable clinical heterogeneity with mostly mtDNA mutations in heteroplasmy that are also considered highly recessive (Tuppen et al. 2010). This is particularly important for mutations causing lethal impairments that would be viable only in heteroplasmy. When heteroplasmy is present, there is a minimum critical frequency of mutated mtDNAs necessary to biochemical defects and tissue dysfunction become apparent. In humans, it has been reported that pathogenic mtDNA mutations only cause respiratory chain dysfunction when they are present above a certain threshold level, which is 60% for single large mtDNA deletions (Hayashi et al. 1991) and 90% for certain point mutations in tRNA genes (Chomyn et al. 1992). Therefore, threshold value varies for each mutation but it also differs amongst tissues according to the dependence on the oxidative metabolism presented by the tissue. It would be higher in tissues that need to obtain most energy from oxidative phosphorylation than in those that can rely on anaerobic glycolysis (Schultz and Harrington 2003). However, it is important to note that mutations in nuclear genes and in mitochondrial genes other than those in the respiratory chain also can lead to mitochondrial dysfunction. These are mainly nuclear genes encoding for respiratory chain subunits, as well as those controlling mtDNA structure and function (Leonard y Schapira 2000).
Overall, a moderate decline of respiratory chain function with age has been widely reported (Trounce et al. 1989). However, in many cases, respiratory chain deficient cells by accumulation of mtDNA alterations would represent only a part of the cells present in a tissue or organ. In addition, age-associated somatic mtDNA mutations tend to undergo clonal expansion and thereby cause focal respiratory chain deficiency. Focal respiratory chain deficiency is a ubiquitous phenomenon in human aging tissues (Müller-Höcker 1989, 1990; Trifunovic and Larsson 2008) supporting that mitochondrial dysfunction is important in human aging. Mosaic respiratory chain deficiency has been also found in many different types of aged tissues in humans including heart (Müller-Höcker 1989), skeletal muscle (Fayet et al. 2002; Bua et al. 2006; Park et al. 2009), hippocampal neurons (Cottrell et al. 2001), choroid plexus (Cottrell et al. 2001), midbrain dopaminergic neurons (Bender et al. 2006), and colon (Taylor et al. 2003). However, the responsibility of age-associated accumulation mtDNA alterations for oxidative phosphorylation impairment has been discussed since low levels of mutated mtDNA has been found in aged humans. Still, there are evidences in favor of mtDNA effects on mitochondria function. Initially, clonal accumulation of deleted mtDNA was associated with focal respiratory chain deficiency in skeletal muscle fiber segments (Fayet et al. 2002). Similarly, it has been found that mtDNA deletions are common (Bender et al. 2006) in respiratory chain-deficient dopaminergic neurons (Reeve et al. 2009). Studies in rats, rhesus monkeys, and humans have shown that accumulation of deleted mtDNA colocalizes with respiratory chain deficiency (Wanagat et al. 2001; Bua et al. 2006). A severe respiratory chain dysfunction has been also found in cardiomyocytes from Tfam homozygous knockout mice that present an associated mtDNA depletion (Wang et al. 2001).
In humans, most published cases until now show that clonally expanded mutations are single large mtDNA deletions. The deletions differ in various respiratory chain-deficient cells of the same tissue, which is in agreement with their somatic nature (Larsson 2010). Along with deletions, clonally expanded point mutations also have been found in other tissues from aging subjects. For instance, in colonic crypts from elderly humans which show focal respiratory chain deficiency in more than 15% of all colonic crypts (Taylor et al. 2003). It has been suggested that they would be originated in the crypts stem cells and they clonally expand with division. It has been suggested that accumulated mutation type by a particular tissues depend on its mitotic activity.
Various experimental models have improved the understanding of the functional consequences of mtDNA mutations and their molecular mechanisms. Biochemical effects of mtDNA mutations have been well described in all experimental systems and are invariably characterized by lower mitochondrial respiration, compromised mtETC complex activity, and reduced ATP synthesis. However, mitochondria also play important roles in different cellular pathways beyond ATP production. These include apoptosis and nucleotide synthesis, calcium regulation (Smeitink et al. 2006; Diot et al. 2016). Therefore alterations in mitochondrial function might affect to different processes that can modulate aging at distinct levels. Oxidative stress has been studied in various animal models that accumulate mtDNA mutations with associated mitochondrial dysfunction. In Tfam homozygous knockout mice there are an initial increased ROS production as consequence of mtDNA depletion and respiratory chain dysfunction (Wang et al. 2001). In contrast, mouse strains knockout for complex I Ndufs4 protein (Kruse et al. 2008) and apoptosis inducer factor (AIF) (Pospisilik et al. 2007) do not have substantially increased ROS production or oxidative damage. Evidence of oxidative stress was almost also absent in Polγ “mutator” (Kujoth et al. 2005; Trifunovic et al. 2005; Niu et al. 2007), although there are rare exceptions (Geromel et al. 2001). In contrast, Tfam heterozygous knockout mice, which also undergo mild mtDNA depletion exhibit increased oxidative mtDNA damage susceptibility (Woo et al. 2012). In addition, apoptotic cell loss can be a common feature in respiratory chain deficiency (Trifunovic and Larsson 2008). These mutator mice displayed a massive increase in apoptosis (Kujoth et al. 2005; Trifunovic et al. 2005; Niu et al. 2007) that has been also observed in mice conditionally knockout for Tfam that led to abolished mtDNA expression (Wang et al. 2001).
The accumulation of deficient mitochondria in a cell can lead to compensatory increase in mitochondria number. This would be mediated by the increase in mitochondrial biogenesis, which in turn leads to an increased number of mtDNA copy number in the cell. In skeletal muscle, it has been reported that ragged-red fibers, which are featured by an extraordinary accumulation of mitochondria, have very high proportions of mutated mtDNA in comparison with adjacent normal-appearing fibers (Moraes et al. 1992). It has been suggested that so massive amount of mitochondria is due to the activated mitochondrial biogenesis indeed, which is a futile response in this case. This is expected since new mtDNA molecules also harbor the same mutations, thus, many additional mitochondria remain dysfunctional. Moreover, this compensatory increase in copy number could lead to mutated mtDNA accumulation (Elson et al. 2001). Still, it has been reported that a higher number of mitochondria compensates for a decrease of oxidative phosphorylation capacity in associated to a mitochondrial myopathy in mouse skeletal muscle (Wredenberg et al. 2002; Wenz et al. 2008).
More recently, some investigations have been directed particularly to stem cells. Neural stem cells from mtDNA mutator mouse showed decreased renewal in vitro and quiescent pools of neural stem cells were decreased, whereas the haematopoietic stem cells showed a skewed lineage differentiation leading to anaemia and lymphopenia (Ahlqvist et al. 2012). In contrast, other model known as mtDNA “deletor” mice (Tyynismaa et al. 2005) that accumulate large-scale mtDNA deletions in postmitotic tissues and exhibited a similar late-onset respiratory chain deficiency not present any signs of premature aging as the mtDNA mutator mouse . This last probably is correlated with the fact that they have no similar somatic stem cell phenotypes (Ahlqvist et al. 2012). Therefore, consequences of mtDNA mutations in stem cells may explain at least in part the aging phenotypes.
Consequences of impaired mitochondrial respiratory chain function derived from accumulation of mtDNA mutations, combined or separately, would contribute to age-associated organ dysfunction and disease onset. Studies have suggested an association of deleted mtDNA with areas of fiber atrophy and splitting, thus, that mitochondrial dysfunction have a role in age-associated sarcopenia (Pak et al. 2003). Similarly, frequency of mtDNA mutation is higher in patients with parkinson´s disease that in age-matched controls (Bender et al. 2006). Concerning cancer, colon (Polyak et al. 1998) and prostate (Chinnery et al. 2002) cancer cases has been also associated to mtDNA mutations. Curiously, different evidences suggest that the most important factors in determining clinical symptoms are not the size and location of the deletions but tissue distribution (Zeviani et al. 1988; Moraes et al. 1995; Vielhaber et al. 2000).
The Polγ mutator mouse also results useful in this aspect since certainly shows that high levels of mtDNA mutations cause a phenotype that included shortened life-span, weight loss, osteoporosis, kyphosis, reduced subcutaneous fat, alopecia, reduced fertility, and cardiac hypertrophy (Trifunovic et al. 2004). This suggests a link between mtDNA mutations and aging phenotypes in mammals. However, the existence of a premature aging syndrome does not necessarily implies that mtDNA mutation levels found in normal aging are high enough to cause aging-related pathology. Additional experiments to test whether a decrease in somatic mtDNA mutations extends lifespan need to be done to confirm that. In contrast, the mtDNA mutator mice show no signs of premature aging (Tyynismaa et al. 2005) in spite of similarities in mutations accumulations and mitochondrial dysfunctions. Because of between featured stem cells mentioned above (Ahlqvist et al. 2012), it has been suggested that somatic stem cell dysfunction has a crucial role of in generating the progeroid phenotype seen in mtDNA mutator mice.
In some occasions, the consequences in mitochondria and cell physiology of mtDNA mutation accumulation led enhanced aging alterations or age-associated diseases progression. Actually, abnormalities of mtDNA have been described in several diseases and high levels of deletions and point mutations cause human mitochondrial disease or syndromes. It has been characterised up to 250 pathogenic mtDNA mutations (point mutations and rearrangements) (Schaefer et al. 2008) causing a wide variety of diseases with a heterogeneity of phenotypes and a variable age of onset (McFarland et al. 2007). Although many mutations are heteroplasmic, there are also an increasing number of pathogenic homoplasmic mutations, often affecting just a single tissue and characterized by incomplete penetrance (McFarland et al. 2002, 2004, 2007; Temperley et al. 2003; Taylor et al. 2003; Yang et al. 2009). In concordance with previous observation, mitochondrial disorders share common cellular consequences including a decreased ATP production, an increased reliance on alternative anaerobic energy sources, and an increased production of reactive oxygen species. Regardless alteration responsible for these, studies in patients with mitochondrial disease were thus able to establish a clear cause and-effect relationship between mtDNA mutations and respiratory chain dysfunction. In addition, an increased ROS production has been described in different models with mutations associated to any of these diseases (Wong et al. 2002; Baracca et al. 2007; Li et al. 2008).
4 Therapies Based on Coenzyme Q Against Diseases Associated with mtDNA Alterations
Despite the role of the alterations in mtDNA in aging have not clarified yet, different treatments have been tested to retard aging or to attenuate aging consequences, which, among other possible effects, can prevent mtDNA alterations. CoQ, usually CoQ10, has been used with this aim in humans and in different experimental models with this aim. Traditionally, the interest in this molecule usually comes from two main roles or activities. On the one hand, CoQ is an essential factor for cell bioenergetics as consequence of its activity as electron carrier in mitochondria. Actually, it has been proposed that an equilibrated CoQ pool may perform a better electron flow adaptation than a higher or lower CoQ pool by keeping a better mitochondrial homeostasis control (López-Lluch et al. 2010). In addition, it also seems to affect protein complex activity and structure (López-Lluch et al. 2010). On the other hand, CoQ also is considered as an endogenously synthetized lipid-soluble antioxidant in biological membranes and it has been shown to efficiently prevent oxidation of DNA along with other macromolecules (Ernster and Forsmark-Andrée 1993). However, other roles that also result interesting for aging have been reported. These include interaction with cell signaling cascades, certain anti-inflammatory activities and even the prevention of events leading to programmed cell death. As consequence of the possible pleiotropic effect of CoQ on cell, many interventions did not aimed specifically to attenuate accumulation of mtDNA mutations. Instead, most of studies have focused on different processes related to mitochondria that have been associated with the generalized role of this organelle in aging. However, the present section of the chapter will be mainly devoted to those studies that evaluated the effect of CoQ on mtDNA.
The simplest approach has been the administration of CoQ to subjects with mitochondrial diseases or syndromes due to one or more specific mtDNA mutations (both, point mutations and deletions). The objective of this treatment was to stop the progression or to reduce some of the symptoms of these diseases. There are two features that make CoQ10 particularly popular in the management of patients with these disorders: its already mentioned rol as component of mtETC and its action as antioxidant , together with its well-documented safety, even at very high doses. Different trials have been carried out in this sense (Bresolin et al. 1990; Chan et al. 1998; Abe et al. 1999; Glover et al. 2010). In most of cases syndromes considered were mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), but also other rare myopathies and/or encephalopathies. In general, main parameters tested in related to mitochondrial function has been post-exercise serum (Bresolin et al. 1990; Abe et al. 1999; Glover et al. 2010) or platelets (Bresolin et al. 1990) lactate levels, which were reduced by CoQ10 supplementation (Bresolin et al. 1990; Abe et al. 1999; Glover et al. 2010), although not in all studies (Bresolin et al. 1990). Similarly, a study showed a reduction in lactate/pyruvate ratio that also proved to be the clinically most useful parameter in the evaluation and monitoring of mitochondrial function (Chan et al. 1998). Differences in CoQ dosage and treatment duration explain contradictory results in most cases, but in a study (Bresolin et al. 1990) observed also inter-individual differences for the same treatment that need to be clarified. It has been reported that responsiveness to treatment was apparently not related to CoQ10 levels in serum and platelets or to the presence or absence of mtDNA deletions (Bresolin et al. 1990). Furthermore, when they have been evaluated, it did not affect other clinically relevant variables such as strength or resting lactate (Glover et al. 2010). Therefore, these trials suggest that CoQ10 supplementation in relative high amount offers some improve of mitochondrial function, but the relevance for overall health is not confirmed. Likewise, most studies had a very small sample size and a rigorous placebo-controlled trial is still lacking. In addition to the above mentioned studies, there are also a set of studies using CoQ10 usually as a component of a “cocktail” that also includes L-carnitine, vitamin B complex, vitamin C and vitamin K1 (Marriage et al. 2004).
To understand possible mechanisms under the effect of CoQ treatment on mitochondrial function more research is still needed. Nevertheless, an in vitro assay suggested that reduction of oxidative stress and inhibition of apoptosis signaling cascades could be implicated. With more detail, it has been reported that preincubation with CoQ10 reduced both, ROS production and activation of caspase 3, after induction by ultraviolet light in cybrids carrying mtDNA with a large-scale deletions associated to chronic progressive external ophthalmoplegia (i.e., 4366-bp and 4977-bp large-scale deletions) (Lee et al. 2005). A particular studied mitochondrial disorder is maternally inherited diabetes mellitus and deafness (MIDD) that is featured by progressive insulin secretory defect and neurosensory deafness. In a randomized-controlled trial, daily oral administration of 150 mg of CoQ10 for 3 years led to higher insulin secretory response than in the control group. Likewise, it improved lactate levels and prevented progressive hearing loss, although other diabetic complications and clinical symptoms remained unchanged (Suzuki et al. 1998).
In addition, dietary CoQ has shown to enhance electron transfer and ATP synthesis in some pathological situations related with aging such as cardiac failure (Rosenfeldt et al. 2005; Molyneux et al. 2009), Parkinson’s disease (Beal 1999; Shults 2003; Young et al. 2007; Thomas and Beal 2010), Alzheimer’s disease (Dumont et al. 2010; Yang et al. 2010; Dumont and Beal 2011) and Friedreich’s ataxia (Hart et al. 2005). Although they are not specifically mitochondrial disorders, patients affected with most of these pathologies have shown a higher frequency of mtDNA alterations. In relation to these diseases, it has been reported that presence of CoQ10 restored the activity of impaired respiratory chain complexes I and IV in cultured fibroblasts from Parkinson´s patients. Some beneficial CoQ effects have been also observed in patients affected by HIV. This pathology is associated with alterations in the amount of mtDNA, as well as with presence of lipodystrophy and peripheral neuropathy with mitochondrial toxicity induced by reverse-transcriptase inhibitors. The administration of 100 mg of CoQ twice a day for 3 months improved the general condition and well-being in asymptomatic HIV-infected patients. However, the treatment aggravated pain in patients with peripheral neuropathy and it did not change mtDNA levels in fat and peripheral blood mononuclear cells (Rabing Christensen et al. 2004).
5 Coenzyme Q, Dietary Fat and Aging in Relation to mtDNA Alterations
Another approach to the study of CoQ in relation to mtDNA alterations has been the life-long dietary administration of low dosages of the molecule to rodents, in order to investigate some aspects of the interaction between nutrition and aging, mainly in relation to dietary fat. Dietary fat has been shown to be particularly interesting because of the importance of phospholipid acyl chain of mitochondrial membrane in their susceptibility to oxidative damage as well as in membrane function and structure. This is due to the fatty acids that form them present different chemical reactivity (Pamplona 2008). Unsaturated fatty acids are more susceptible to damage from ROS molecules owing to the high presence of unstable electrons near their double bonds, and also because its sensitivity to lipid peroxidation is greater as molecules have more double bond (Bielski et al. 1983; Holman 1954). Further, they also can participate in free radical chain reactions and lipid peroxidation product would produce covalent modifications of other macromolecules as proteins and DNA. Thus, a low degree of unsaturation in the fatty acids of biological membranes would decrease their sensitivity to lipid peroxidation, which, in turn, can protect damage other lipooxidation-derivative molecules (Mataix et al. 1998). In fact, some studies in mammals have shown that fatty acids unsaturation degree in biological membranes of various tissues is negatively correlated with longevity (Pamplona 2008; Pamplona et al. 2000).
There are enough evidences indicating that fatty acids present in the diet modify the lipid profile of biological membranes, including mitochondrial membranes (Huertas et al. 1991, 1999; Ochoa et al. 2001; Quiles et al. 1999). Thus, dietary fat affects the structure and mitochondrial function, as well as its susceptibility to oxidative stress. In this sense, if we could build "customized" biological membranes depending on the type of dietary fat, maybe we could positively change the way in which the organs age. This working hypothesis represented a new approach to the study of aging from the point of view of nutrition, and had important implications for aging phenomenon study (González-Alonso et al. 2015a). This was the basis for the work of our research group in a series of experiments performed on a rat model of aging for the last 20 years. In these studies, male Wistar rats were life-long maintained on different diets with different fat sources (virgin olive oil, sunflower oil or fish oil) which notably varying in their unsaturated fatty acids profiles to evaluate how this component of the diet affected to aging of different tissues and organs. Because of mitochondria role in aging and oxidative stress, evaluations were focused on mitochondrial aspects as ultrastructural alterations, mtDNA and/or respiratory chain functionality, as well as oxidative stress (including oxidative damage and antioxidant defense components) (González-Alonso et al. 2015a). As consequence of CoQ importance in mitochondria and oxidative stress, most of the experiments were carried out by using these dietary fats without or with a supplement of CoQ10.
In early interventions, rats were fed diets based on AIN-93 (Reeves 1997; Reeves et al. 1993) criteria but with different dietary fat source virgin olive oil (rich in MUFA) or sunflower oil (rich in n-6 PUFA). Animals were sacrificed at different ages to study how dietary fat and CoQ modulated aging in different tissues (Quiles et al. 2002, 2004a, b, 2005, 2006; Ochoa et al. 2003, 2011). In this context, mitochondria isolated from three different tissues, liver, heart and skeletal muscle, were compared at 6, 12, 18, and 24 months of age. Lipid peroxidation markers used (i.e. hydroperoxides levels) indicated that, in general, postmitotic tissues (i.e. heart as skeletal muscle) were more prone to suffer oxidation, but n-6 PUFA-rich diets led to a higher degree of membrane polyunsaturation and peroxidation. In addition, the degree of polyunsaturation in mitochondria was found to correlate with those in diet , confirming a very good degree of membrane adaption to diet (Ochoa et al. 2003). Similar experiments also showed a worsening of aging effects by n-6 PUFA on different tissue and/or markers. These included total antioxidant capacity and DNA double-strand breaks which, respectively, decreased and increased in all animals as they age. In spite of all this effects of n-6 PUFA can affect to onset of some diseases, particularly those associated to aging, no changes in mean or maximal lifespan were observed (Quiles et al. 2004b).
In liver, ROS-mediated damage products (Hydroperoxides and TBARS) relative amounts were higher in 24 months old animals than in those aged 6 months but only in those receiving n-6 PUFA-rich diets, whereas rats fed virgin olive oil showed the lowest values at both ages. In most of case these levels correlates with activities of antioxidant enzymes (SOD, catalase and GPX) and concentrations of lipophilic antioxidant (α-tocopherol and CoQ). This suggests that this tissue as it ages triggers protection mechanism against oxidative stress probably as response to higher levels ROS or ROS-mediated damage products (Quiles et al. 2006). Interestingly, this study were even more focused on mitochondria an effects of diet and aging on mitochondrial ultrastructure and mtDNA were also evaluated. Namely, possible effects on mtDNA were evaluated using a particular deletion in the region encoded for mtETC complex I components (Nd4 gene) since it has been suggested that is one of the complexes most affected by aging (Sanz et al. 2006). An age-related increase in mtDNA deletion frequency was observed in all animals but this was higher in rats fed sunflower oil. Likewise, old animals fed on n-6 PUFA rich diet displayed a lower crests number and higher circularity, factors that have been linked to a reduced functionality of mitochondria (Quiles et al. 2006). These findings, thus, revealed a relationship among ROS production and alterations of ultrastructure and mtDNA with aging at liver mitochondria in rats. But the most interesting was to see how these aspects (including accumulation of mtDNA deletion), which could be defining the appearance of aging phenotype, could be modulated through diet by choosing more or less unsaturated fat source and, which gives rise to the possibility modular aging through diet .
Based on negative consequences of n-6 PUFA intake found in above mentioned investigations, other studies were carried out where two experimental groups received similar sunflower oil-based diets, but with or without a supplementation on CoQ10 to reach a daily dosage of 0.7 g/kg (Ochoa et al. 2005; Quiles et al. 2004a, 2005). In heart, long-term supplementation with CoQ10, led to lower hydroperoxide levels, higher content of lipophilic antioxidants (α-tocopherol and coenzyme Q), and a higher catalase activity. Also, a slightly lower decrease in certain key activities for mitochondrial function when animals with age of 6, 12, or 24 months were compared (Ochoa et al. 2005). At the systemic level, an age-associated increase in nDNA strand breaks in peripheral blood lymphocytes was observed. This increase associated to age was lower in animals supplemented on CoQ10. If it is assumed that main cause of such breaks is oxidative damage, this suggests that CoQ10 by means of both, reactive species scavenging and antioxidant recycling, protects DNA against oxidative damage. It could be possible that also can occur in mitochondria at least under certain conditions. However, this finding could also result indirectly from of CoQ10 effects on mitochondria or other organelles that would reduce ROS levels and consequent attacks to nDNA (Quiles et al. 2005). In liver, similar effects on DNA double-strand breaks, CoQ levels at mitochondrial membrane have been reported. Lastly, it has also been noted that CoQ10-supplemented animals reached a significantly higher mean life span and a significantly higher maximum life span (Quiles et al. 2004a). This emphasized the importance of oxidative stress, DNA damage and mitochondria in aging since CoQ has shown effect on all them (Ochoa et al. 2005; Quiles et al. 2005).
According to previous finding, it seems that life-long supplementation with CoQ10 of n-6 PUFA-rich diet resulted interesting to attenuate aging consequences, but it was necessary to check if CoQ10 led even to better results than virgin olive oil. For this reason, additional experiments similar to the previous one but including also a group fed on a virgin olive oil-based diet were carried out (Ochoa et al. 2011; Quiles et al. 2010). Thus, three diets rich in MUFA, n-6 PUFA and CoQ10-supplemented n-6 PUFA were compared. Because of previous results and their importance in ROS generation and aging, mitochondria received a greater attention and mtDNA and ultrastructure were also analyzed in most of cases along with ROS and antioxidants levels. Again, the frequency of a specific deletion in mtDNA corresponding to the mETC complex I of the was used as marker of mtDNA alterations, This experimental design was used to evaluate diet and aging interaction in two tissues, both postmitotic, brain and heart.
In heart, animals fed virgin olive oil showed a lower increase in the frequency of studied mtDNA deletion than those fed sunflower oil. However, the addition of CoQ10 to the n-6 PUFA-rich fat source (i.e. sunflower oil) reduced the difference between young and old animals although the lowest values were present by MUFA-fed animals (Quiles et al. 2010). Concerning mitochondrial ultrastructure, dietary fat used had similar effects (Quiles et al. 2010) to those achieved in liver tissue in absence of CoQ10 (Quiles et al. 2006), whereas CoQ10 treatment led to lower mitochondrial perimeter in this case (Quiles et al. 2010). CoQ10 also prevented the decrease in cytochrome C oxidase activity and mtETC complex I levels suggested for old subjects fed on the same dietary fat. Therefore, it would prevent mitochondrial respiratory chain dysfunction in some degree. Aged animals receiving CoQ also showed lower hydroperoxide levels than those fed on sunflower or virgin olive oil not supplemented (Quiles et al. 2010). This would suggest that CoQ contributes to decrease oxidative stress, although there are several possible mechanisms. In any case, the effect found for dietary CoQ10 on either ultrastructure, mtDNA and some respiratory chain components would alleviate ROS production associated to age.
Very similar aspects were also evaluated in brain, but in this case in the experiment performed an additional group consisted in a virgin olive oil-based diet supplemented with CoQ10. In this, mtDNA deletion was higher in old groups fed on n-6 PUFA-rich diets but no age-associated differences were found for animals fed virgin olive oil. However, in this case CoQ10 did not show effects on mtDNA deletions in animals fed on sunflower oil-based diet at 24 months. In relation to oxidative stress markers, CoQ led to lower values of lipid peroxidation (hydroperoxides) at 24 months, although the lowest values were found in the two virgin olive oil fed groups (Ochoa et al. 2011).
These organs (heart and brain) are clear affected by aging and their alteration lead to overall health impairment reducing longevity. Moreover mitochondrial alterations and oxidative stress are key aspects in aging of these organs as it has been previously reported. Altogether, these findings revealed that CoQ, at least under certain conditions, can modulate aging effects on different tissues affecting to mtDNA, but also to mitochondrial ultrastructure and ROS production. Again, a key finding is the possibility of modulating mtDNA mutations associated to age through diet .
In more recent experiments, a third diet type has started to be compared with diets similar to previously described studies. So, as a new fat source namely fish oil, very rich in n-3 PUFA, was used. In addition, fat content was the half of the amount used in previous experiments (4% versus 8% w/w) according to more recent actualization of AIN93 criteria (Reeves 1997) until that moment. As previously, some additional experiments were carried out to test the effects of these diets under CoQ10 supplementation. Moreover, new organ/tissues, not previously studied, like pancreas and periodontum, were included in the experiments. In pancreas, it has been reported that dietary fat affected to endocrine and exocrine pancreas in a different way (Roche et al. 2014). In 24-months-old animals, n-6 PUFA rich-diets consumption was associated with a greater number of β-cells that correlated with an increase in insulin content and hyperleptinemia (Roche et al. 2014), signs that have been described in obesity, glucose intolerance, insulin resistance, disruption of adipoinsular axis or prediabetes (Sattar et al. 2008). Concerning exocrine compartment, old rats fed with n-3 PUFA-rich diets (Roche et al. 2014) led to histological features resembling those observed in pancreatic fibrosis in elderly people (Klöppel et al. 2004). In other experiments, it was observed that dietary CoQ10 improved endocrine pancreas structure and in particular β-cell mass from rat fed on n-6 PUFA resembling positive effects of virgin olive oil (González-Alonso et al. 2015b). Because of importance of mitochondria in this organ, CoQ10 effect could be mediated by effect on mtDNA previously reported (Quiles et al. 2010). However, oxidative damage or alterations of mtDNA sequence have not been directly analyzed yet. In a study focused on the pancreas of 24 months old rat fed on these diet , the profile of serum fatty acids confirmed, that animals an adaptation to the diet at 6 months of age since they resembled lipid profile of the diets. The percentages of circulating MUFA were significantly higher in rats fed virgin olive oil; the highest levels of n-6 PUFA were achieved in rats fed with sunflower oil, and the highest levels of n-3 PUFA were found in those rats fed fish oil (Roche et al. 2014; González-Alonso et al. 2015b). Moreover, the effect of this fat sources and CoQ on some bone metabolism markers at serum and age-associated alveolar bone have been also studied (Bullon et al. 2013). Alveolar bone loss is a major clinical outcome of periodontitis (Page and Kornman 1997), a disease with high prevalence in elderly people that in the last years has been associated with systemic diseases such as atherosclerosis and metabolic syndrome that would have oxidative stress as potential link (Bullon et al. 2009, 2011). Again, feeding on an n-6 PUFA-rich diet led to worse consequences in health since it was associated to the highest age-associated alveolar bone loss (Bullon et al. 2013). Although mtDNA alterations were not directly measured, expression of genes LC3 and ATG5 that are implicated in autophagy and the biogenesis markers Tfam and PGC-1α suggests that both processes increase with aging in gingival tissue, but not in animals fed n-6 PUFA. The combination of both processes would reduce or prevent accumulation of damage in mtDNA and its possible consequences. Moreover, this effect also was associated affecting to some mtETC components and antioxidant enzymes expression. In other study, CoQ supplementation eliminated differences in age-associated alveolar bone loss among dietary groups (Varela-Lopez et al. 2015). CoQ10 had no effect on age-associated changes in expression of genes of autophagy markers in rats fed on n-6 PUFA-rich diet (Varela-Lopez et al. 2015). An increase in the expression of the biogenesis marker Tfam was observed in n-6 PUFA fed animals indicating that there was an increase in mitochondria and probably in mtDNA copies. Although this mechanism possibly does not reduce the accumulation of altered or mutated mtDNA molecules, it seems that it can compensate, at least in part, the associated loss of function, as it has been reported for skeletal muscle. Sumarizing all these experiments on aged rats, it could be concluded that the basis for a putative beneficial effect of CoQ on mtDNA disturbances could be the enhancement of the cellular antioxidant protection systems in cell membranes where CoQ preventing lipid peroxidation and consequently reduced oxidative stress and mtDNA damage by ROS .
Finally, in healthy humans, comparisons between CoQ10 supplementation to diet have been also established following a cross-over design, although only for a short period of time (4 weeks). In this regard, elderly subjects following a Mediterranean diet (rich in MUFA) supplemented and not with CoQ10 or a Western diet rich in SFA were studied (Yubero-Serrano et al. 2010, 2012; Gutierrez-Mariscal et al. 2011, 2014; González-Guardia et al. 2015). Some postprandial oxidative stress marker levels were reduced by CoQ10 addition to the MUFA-rich diet (Yubero-Serrano et al. 2010, 2012). Interestingly, dietary CoQ also improved DNA repair systems (Gutierrez-Mariscal et al. 2011; Yubero-Serrano et al. 2012). These results suggest that CoQ may also protect mtDNA against the accumulation of mutations by this mechanism in addition to the prevention of ROS-mediated damage discussed for the rat models.
References
Abe K, Matsuo Y, Kadekawa J, Inoue S, Yanagihara T (1999) Effect of coenzyme Q10 in patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): evaluation by noninvasive tissue oximetry. J Neurol Sci 162:65–68
Ahlqvist KJ, Hämäläinen RH, Yatsuga S, Uutela M, Terzioglu M, Götz A, Forsström S, Salven P, Angers-Loustau A, Kopra OH, Tyynismaa H, Larsson N-G, Wartiovaara K, Prolla T, Trifunovic A, Suomalainen A (2012) Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab 15:100–109. https://doi.org/10.1016/j.cmet.2011.11.012
Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G (2008) Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci 121 (7):1046–1053
Ameur A, Stewart JB, Freyer C, Hagström E, Ingman M, Larsson N-G, Gyllensten U (2011) Ultra-deep sequencing of mouse mitochondrial DNA: mutational patterns and their origins. PLoS Genet 7:e1002028. https://doi.org/10.1371/journal.pgen.1002028
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465
Arakaki N, Nishihama T, Owaki H, Kuramoto Y, Suenaga M, Miyoshi E, Emoto Y, Shibata H, Shono M, Higuti T (2006) Dynamics of mitochondria during the cell cycle. Biol Pharm Bull 29:1962–1965
Attardi G, Schatz G (1988) Biogenesis of mitochondria. Annu Rev Cell Biol 4:289–333. https://doi.org/10.1146/annurev.cb.04.110188.001445
Baracca A, Sgarbi G, Mattiazzi M, Casalena G, Pagnotta E, Valentino ML, Moggio M, Lenaz G, Carelli V, Solaini G (2007) Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993. Biochim Biophys Acta BBA Bioenerg 1767:913–919. https://doi.org/10.1016/j.bbabio.2007.05.005
Beal MF (1999) Coenzyme Q10 administration and its potential for treatment of neurodegenerative diseases. Biofactors 9:261–266
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517. https://doi.org/10.1038/ng1769
Berneburg M, Plettenberg H, Medve-König K, Pfahlberg A, Gers-Barlag H, Gefeller O, Krutmann J (2004) Induction of the photoaging-associated mitochondrial common deletion in vivo in normal human skin. J Invest Dermatol 122:1277–1283. https://doi.org/10.1111/j.0022-202X.2004.22502.x
Bielski BH, Arudi RL, Sutherland MW (1983) A study of the reactivity of HO2/O2- with unsaturated fatty acids. J Biol Chem 258:4759–4761
Birket MJ, Birch-Machin MA, (2007) Ultraviolet radiation exposure accelerates the accumulation of the aging-dependent T414G mitochondrial DNA mutation in human skin. Aging Cell 6(4):557–564
Blok RB, Gook DA, Thorburn DR, Dahl H-HM (1997) Skewed segregation of the mtDNA nt 8993 (TrG) mutation in human oocytes. Am J Hum Genet 60:1495–1501. https://doi.org/10.1086/515453
Bogenhagen D, Clayton DA (1977) Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell 11:719–727. https://doi.org/10.1016/0092-8674(77)90286-0
Bresolin N, Doriguzzi C, Ponzetto C, Angelini C, Moroni I, Castelli E, Cossutta E, Binda A, Gallanti A, Gabellini S (1990) Ubidecarenone in the treatment of mitochondrial myopathies: a multi-center double-blind trial. J Neurol Sci 100:70–78
Brown GK (1997) Bottlenecks and beyond: mitochondrial DNA segregation in health and disease. J Inherit Metab Dis 20:2–8
Brown WM, George M, Wilson AC (1979) Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA 76:1967–1971
Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, Aiken JM (2006) Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet 79:469–480. https://doi.org/10.1086/507132
Bullon P, Morillo JM, Ramírez-Tortosa MC, Quiles JL, Newman HN, Battino M (2009) Metabolic syndrome and periodontitis: is oxidative stress a common link? J Dent Res 88:503–518. https://doi.org/10.1177/0022034509337479
Bullon P, Cordero MD, Quiles JL, Morillo JM, Ramírez-Tortosa MC, Battino M (2011) Mitochondrial dysfunction promoted by Porphyromonas gingivalis lipopolysaccharide as a possible link between cardiovascular disease and periodontitis. Free Radic Biol Med 50:1336–1343. https://doi.org/10.1016/j.freeradbiomed.2011.02.018
Bullon P, Battino M, Varela-Lopez A, Perez-Lopez P, Granados-Principal S, Ramirez-Tortosa MC, Ochoa JJ, Cordero MD, Gonzalez-Alonso A, Ramirez-Tortosa CL, Rubini C, Zizzi A, Quiles JL (2013) Diets based on virgin olive oil or fish oil but not on sunflower oil prevent age-related alveolar bone resorption by mitochondrial-related mechanisms. PLoS One 8:e74234. https://doi.org/10.1371/journal.pone.0074234. eCollection 2013
Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, Hayashi J-I, Yonekawa H (2007) The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet 39:386–390. https://doi.org/10.1038/ng1970
Chan DC (2012) Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46:265–287. https://doi.org/10.1146/annurev-genet-110410-132529
Chan A, Reichmann H, Kögel A, Beck A, Gold R (1998) Metabolic changes in patients with mitochondrial myopathies and effects of coenzyme Q10 therapy. J Neurol 245:681–685
Chan KKL, Zhang Q-M, Dianov GL (2006) Base excision repair fidelity in normal and cancer cells. Mutagenesis 21:173–178. https://doi.org/10.1093/mutage/gel020
Chinnery PF, Samuels DC, Elson J, Turnbull DM (2002) Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet Lond Engl 360:1323–1325. https://doi.org/10.1016/S0140-6736(02)11310-9
Chomyn A, Martinuzzi A, Yoneda M, Daga A, Hurko O, Johns D, Lai ST, Nonaka I, Angelini C, Attardi G (1992) MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc Natl Acad Sci USA 89:4221–4225
Clayton DA (1982) Replication of animal mitochondrial DNA. Cell 28:693–705
Clayton DA (1991) Replication and transcription of vertebrate mitochondrial DNA. Annu Rev Cell Biol 7:453–478. https://doi.org/10.1146/annurev.cb.07.110191.002321
Clayton DA (1992) Structure and function of the mitochondrial genome. J Inherit Metab Dis 15:439–447
Cortopassi GA, Arnheim N (1990) Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res 18:6927–6933
Cortopassi GA, Shibata D, Soong NW, Arnheim N (1992) A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Natl Acad Sci U S A 89:7370–7374
Coskun PE, Beal MF, Wallace DC (2004) Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A 101:10726–10731. https://doi.org/10.1073/pnas.0403649101
Cottrell DA, Blakely EL, Johnson MA, Ince PG, Turnbull DM (2001) Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology 57:260–264
Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, Dahl H-HM, Chinnery PF (2008) A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet 40:249–254. https://doi.org/10.1038/ng.2007.63
de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA (2001) Repair of 8-oxodeoxyguanosine lesions in mitochondrial Dna depends on the Oxoguanine Dna Glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial Dna of OGG1-defective mice. Cancer Res 61:5378–5381
Dianov GL, Souza-Pinto N, Nyaga SG, Thybo T, Stevnsner T, Bohr VA (2001) Base excision repair in nuclear and mitochondrial DNA. Prog Nucleic Acid Res Mol Biol 68:285–297
Diot A, Hinks-Roberts A, Lodge T, Liao C, Dombi E, Morten K, Brady S, Fratter C, Carver J, Muir R, Davis R, Green CJ, Johnston I, Hilton-Jones D, Sue C, Mortiboys H, Poulton J (2015) A novel quantitative assay of mitophagy: combining high content fluorescence microscopy and mitochondrial DNA load to quantify mitophagy and identify novel pharmacological tools against pathogenic heteroplasmic mtDNA. Pharmacol Res 100:24–35. https://doi.org/10.1016/j.phrs.2015.07.014
Diot A, Morten K, Poulton J (2016) Mitophagy plays a central role in mitochondrial ageing. Mamm Genome 27(7–8):381–395
Driggers WJ, LeDoux SP, Wilson GL (1993) Repair of oxidative damage within the mitochondrial DNA of RINr 38 cells. J Biol Chem 268:22042–22045
Dumont M, Beal MF (2011) Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic Biol Med 51:1014–1026. https://doi.org/10.1016/j.freeradbiomed.2010.11.026
Dumont M, Lin MT, Beal MF (2010) Mitochondria and antioxidant targeted therapeutic strategies for Alzheimer’s disease. J Alzheimers Dis 20(Suppl 2):S633–S643. https://doi.org/10.3233/JAD-2010-100507
Elson JL, Samuels DC, Turnbull DM, Chinnery PF (2001) Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet 68:802–806. https://doi.org/10.1086/318801
Ernster L, Forsmark-Andrée P (1993) Ubiquinol: an endogenous antioxidant in aerobic organisms. Clin Investig 71(S8)
Evans MD, Dizdaroglu M, Cooke MS (2004) Oxidative DNA damage and disease: induction, repair and significance. Mutat Res 567:1–61. https://doi.org/10.1016/j.mrrev.2003.11.001
Fayet G, Jansson M, Sternberg D, Moslemi AR, Blondy P, Lombès A, Fardeau M, Oldfors A (2002) Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord NMD 12:484–493
Fish J, Raule N, Attardi G (2004) Discovery of a major D-loop replication origin reveals two modes of human mtDNA synthesis. Science 306:2098–2101. https://doi.org/10.1126/science.1102077
Geromel V, Kadhom N, Cebalos-Picot I, Ouari O, Polidori A, Munnich A, Rötig A, Rustin P (2001) Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the Neurogenic Ataxia Retinitis Pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA. Hum Mol Genet 10:1221–1228
Glover EI, Martin J, Maher A, Thornhill RE, Moran GR, Tarnopolsky MA (2010) A randomized trial of coenzyme Q10 in mitochondrial disorders. Muscle Nerve 42:739–748. https://doi.org/10.1002/mus.21758
González-Alonso A, Pérez-López P, Varela-López A, Ramírez-Tortosa MC, Battino M, Quiles JL (2015a) Experimental evidence on the role of different types unsaturated fats in the diet on ageing. Rev Esp Geriatría Gerontol 50:285–288. https://doi.org/10.1016/j.regg.2015.05.003
González-Alonso A, Ramírez-Tortosa CL, Varela-López A, Roche E, Arribas MI, Ramírez-Tortosa MC, Giampieri F, Ochoa JJ, Quiles JL (2015b) Sunflower oil but not fish oil resembles positive effects of virgin olive oil on aged pancreas after life-long coenzyme Q addition. Int J Mol Sci 16:23425–23445. https://doi.org/10.3390/ijms161023425
González-Guardia L, Yubero-Serrano EM, Delgado-Lista J, Perez-Martinez P, Garcia-Rios A, Marin C, Camargo A, Delgado-Casado N, Roche HM, Perez-Jimenez F, Brennan L, López-Miranda J (2015) Effects of the Mediterranean diet supplemented with coenzyme Q10 on metabolomic profiles in elderly men and women. J Gerontol Biol Sci Med Sci 70:78–84. https://doi.org/10.1093/gerona/glu098
Goto YI, Nonaka I, Horai S (1990) A mutation in the tRNA Leu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348:651–653. https://doi.org/10.1038/348651a0
Graziewicz MA, Sayer JM, Jerina DM, Copeland WC (2004) Nucleotide incorporation by human DNA polymerase gamma opposite benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyguanosine and deoxyadenosine. Nucleic Acids Res 32:397–405. https://doi.org/10.1093/nar/gkh213
Graziewicz MA, Bienstock RJ, Copeland WC (2007) The DNA polymerase gamma Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7,8-dihydro-8-oxo-2’-deoxyguanosine. Hum Mol Genet 16:2729–2739. https://doi.org/10.1093/hmg/ddm227
Greaves LC, Nooteboom M, Elson JL, Tuppen HAL, Taylor GA, Commane DM, Arasaradnam RP, Khrapko K, Taylor RW, Kirkwood TBL, Mathers JC, Turnbull DM (2014) Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet 10:e1004620. https://doi.org/10.1371/journal.pgen.1004620
Gutierrez-Mariscal FM, Perez-Martinez P, Delgado-Lista J, Yubero-Serrano EM, Camargo A, Delgado-Casado N, Cruz-Teno C, Santos-Gonzalez M, Rodriguez-Cantalejo F, Castaño JP, Villalba-Montoro JM, Fuentes F, Perez-Jimenez F, Lopez-Miranda J (2011) Mediterranean diet supplemented with coenzyme Q10 induces postprandial changes in p53 in response to oxidative DNA damage in elderly subjects. Age Drodr 34:389–403. https://doi.org/10.1007/s11357-011-9229-1
Gutierrez-Mariscal FM, Yubero-Serrano EM, Rangel-Zúñiga OA, Marín C, García-Rios A, Perez-Martinez P, Delgado-Lista J, Malagón MM, Tinahones FJ, Pérez-Jimenez F, López-Miranda J (2014) Postprandial activation of P53-dependent DNA repair is Modified by Mediterranean diet supplemented with coenzyme Q10 in elderly subjects. J Gerontol Biol Sci Med Sci 69:886–893. https://doi.org/10.1093/gerona/glt174
Hamilton ML, Guo Z, Fuller CD, Van Remmen H, Ward WF, Austad SN, Troyer DA, Thompson I, Richardson A (2001) A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA. Nucleic Acids Res 29:2117–2126
Harman D, others (1955) Aging: a theory based on free radical and radiation chemistry
Hart PE, Lodi R, Rajagopalan B, Bradley JL, Crilley JG, Turner C, Blamire AM, Manners D, Styles P, Schapira AHV, Cooper JM (2005) Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol 62:621–626. https://doi.org/10.1001/archneur.62.4.621
Hayashi J-I, Ohta S, Kikuchi A, Takemitsu M, Goto Y-I, Nonaka I (1991) Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc Natl Acad Sci USA 88:10614–10618
Hegler J, Bittner D, Boiteux S, Epe B (1993) Quantification of oxidative DNA modifications in mitochondria. Carcinogenesis 14:2309–2312
Hekimi S, Lapointe J, Wen Y (2011) Taking a “good” look at free radicals in the aging process. Trends Cell Biol 21:569–576. https://doi.org/10.1016/j.tcb.2011.06.008
Holman RT (1954) Autoxidation of fats and related substances. In: Holman RT, Lundberg WO, Malkin T (eds) Progress in chemistry of fats and other lipids. Pergamon Press, London, pp 51–98
Holt IJ, Miller DH, Harding AE (1989) Genetic heterogeneity and mitochondrial DNA heteroplasmy in Leber’s hereditary optic neuropathy. J Med Genet 26:739–743. https://doi.org/10.1136/jmg.26.12.739
Holt IJ, Lorimer HE, Jacobs HT (2000) Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell 100:515–524. https://doi.org/10.1016/S0092-8674(00)80688-1
Holt IJ, He J, Mao CC, Boyd-Kirkup JD, Martinsson P, Sembongi H, Reyes A, Spelbrink JN (2007) Mammalian mitochondrial nucleoids: organizing an independently minded genome. Mitochondrion 7:311–321. https://doi.org/10.1016/j.mito.2007.06.004
Huertas JR, Battino M, Lenaz G, Mataix FJ (1991) Changes in mitochondrial and microsomal rat liver coenzyme Q9 and Q10 content induced by dietary fat and endogenous lipid peroxidation. FEBS Lett 287:89–92. https://doi.org/10.1016/0014-5793(91)80022-U
Huertas JR, Martínez-Velasco E, Ibáñez S, López-Frías M, Ochoa JJ, Quiles JL, Parenti-Castelli G, Mataix J, Lenaz G (1999) Virgin olive oil protect heart mitochondria from peroxidative damage during aging. BioFactors 9:337–343. https://doi.org/10.1002/biof.5520090233
Ingman M, Kaessmann H, Pääbo S, Gyllensten U (2000) Mitochondrial genome variation and the origin of modern humans. Nature 408:708–713. https://doi.org/10.1038/35047064
Jacobs LJAM, Wert G de, JPM G, Coo IFM de, HJM S (2006) The transmission of OXPHOS disease and methods to prevent this. Hum Reprod Update 12:119–136. https://doi.org/10.1093/humupd/dmi042
Jenuth JP, Peterson AC, Fu K, Shoubridge EA (1996) Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet 14:146–151. https://doi.org/10.1038/ng1096-146
Katajisto P, Döhla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, Sabatini DM (2015) Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348:340–343. https://doi.org/10.1126/science.1260384
Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, Zhang J, Zong NC, Lam MP, Ping P (2012) Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol Cell Proteomics 11(12):1586–1594. https://doi.org/10.1074/mcp.M112.021162
Klöppel G, Detlefsen S, Feyerabend B (2004) Fibrosis of the pancreas: The initial tissue damage and the resulting pattern. Virchows Arch 445:1–8. https://doi.org/10.1007/s00428-004-1021-5
Korhonen JA, Pham XH, Pellegrini M, Falkenberg M (2004) Reconstitution of a minimal mtDNA replisome in vitro. EMBO J 23:2423–2429. https://doi.org/10.1038/sj.emboj.7600257
Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K (2006) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 38:518–520. https://doi.org/10.1038/ng1778
Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM (2008) What causes mitochondrial DNA deletions in human cells? Nat Genet 40:275–279. https://doi.org/10.1038/ng.f.94
Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD (2008) Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab 7:312–320. https://doi.org/10.1016/j.cmet.2008.02.004
Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481–484. https://doi.org/10.1126/science.1112125
Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson N-G, Jakobs S (2011) Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A 108:13534–13539. https://doi.org/10.1073/pnas.1109263108
Kuznetsov NA, Koval VV, Zharkov DO, Nevinsky GA, Douglas KT, Fedorova OS (2005) Kinetics of substrate recognition and cleavage by human 8-oxoguanine-DNA glycosylase. Nucleic Acids Res 33:3919–3931. https://doi.org/10.1093/nar/gki694
Laderman KA, Penny JR, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G (1996) Aging-dependent functional alterations of mitochondrial DNA (mtDNA) from human fibroblasts transferred into mtDNA-less cells. J Biol Chem 271:15891–15897
Lagouge M, Larsson N-G (2013) The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med 273:529–543. https://doi.org/10.1111/joim.12055
Lakshmipathy U, Campbell C (1999a) The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol Cell Biol 19:3869–3876
Lakshmipathy U, Campbell C (1999b) Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res 27:1198–1204
Larsson N-G (2010) Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 79:683–706. https://doi.org/10.1146/annurev-biochem-060408-093701
Larsson NG, Clayton DA (1995) Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet 29:151–178. https://doi.org/10.1146/annurev.ge.29.120195.001055
Larsson N, Holme E, Kristiansson B, Oldfors A, Tulinius M (1990) Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediatr Res 28:131–136. https://doi.org/10.1203/00006450-199008000-00011
Larsson NG, Tulinius MH, Holme E, Oldfors A, Andersen O, Wahlström J, Aasly J (1992) Segregation and manifestations of the mtDNA tRNA(Lys) A-->G(8344) mutation of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. Am J Hum Genet 51:1201–1212
LeDoux SP, Wilson GL, Beecham EJ, Stevnsner T, Wassermann K, Bohr VA (1992) Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 13:1967–1973
Lee C-F, Liu C-Y, Chen S-M, Sikorska M, Lin C-Y, Chen T-L, Wei Y-H (2005) Attenuation of UV-Induced apoptosis by Coenzyme Q in human cells harboring large-scale deletion of mitochondrial DNA. Ann N Y Acad Sci 1042(1):429–438
Leonard JV, Schapira AHV (2000) Mitochondrial respiratory chain disorders I: mitochondrial DNA defects. Lancet 355(9200):299–304
Lewis W, Dalakas MC (1995) Mitochondrial toxicity of antiviral drugs. Nat Med 1:417–422
Li J, Zhou K, Meng X, Wu Q, Li S, Liu Y, Wang J (2008) Increased ROS generation and SOD activity in heteroplasmic tissues of transmitochondrial mice with A3243G mitochondrial DNA mutation. Genet Mol Res GMR 7:1054–1062
Ling F, Makishima F, Morishima N, Shibata T (1995) A nuclear mutation defective in mitochondrial recombination in yeast. EMBO J 14:4090–4101
Lloret A, Badía M-C, Mora NJ, Pallardó FV, Alonso M-D, Viña J (2009) Vitamin E paradox in Alzheimer’s disease: it does not prevent loss of cognition and may even be detrimental. J Alzheimers Dis JAD 17:143–149. https://doi.org/10.3233/JAD-2009-1033
López-Lluch G, Rodríguez-Aguilera JC, Santos-Ocaña C, Navas P (2010) Is coenzyme Q a key factor in aging? Mech Ageing Dev 131:225–235. https://doi.org/10.1016/j.mad.2010.02.003
Madsen CS, Ghivizzani SC, Hauswirth WW (1993) In vivo and in vitro evidence for slipped mispairing in mammalian mitochondria. Proc Natl Acad Sci U S A 90:7671–7675
Marín-García J, Pi Y, Goldenthal MJ (2006) Mitochondrial-nuclear cross-talk in the aging and failing heart. Cardiovasc Drugs Ther Spons Int Soc Cardiovasc Pharmacother 20:477–491. https://doi.org/10.1007/s10557-006-0584-6
Marriage BJ, Clandinin MT, Macdonald IM, Glerum DM (2004) Cofactor treatment improves ATP synthetic capacity in patients with oxidative phosphorylation disorders. Mol Genet Metab 81:263–272. https://doi.org/10.1016/j.ymgme.2003.12.008
Mason PA, Matheson EC, Hall AG, Lightowlers RN (2003) Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res 31:1052–1058
Masuda Y, Bennett RA, Demple B (1998) Dynamics of the interaction of human apurinic endonuclease (Ape1) with its substrate and product. J Biol Chem 273:30352–30359
Mataix J, Quiles JL, Huertas JR, Battino M, Mañas M (1998) Tissue specific interactions of exercise, dietary fatty acids, and vitamin E in lipid peroxidation. Free Radic Biol Med 24:511–521. https://doi.org/10.1016/S0891-5849(97)00288-8
McFarland R, Clark KM, Morris AAM, Taylor RW, Macphail S, Lightowlers RN, Turnbull DM (2002) Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet 30:145–146. https://doi.org/10.1038/ng819
McFarland R, Schaefer AM, Gardner JL, Lynn S, Hayes CM, Barron MJ, Walker M, Chinnery PF, Taylor RW, Turnbull DM (2004) Familial myopathy: new insights into the T14709C mitochondrial tRNA mutation. Ann Neurol 55:478–484. https://doi.org/10.1002/ana.20004
McFarland R, Chinnery PF, Blakely EL, Schaefer AM, Morris AAM, Foster SM, Tuppen HAL, Ramesh V, Dorman PJ, Turnbull DM, Taylor RW (2007) Homoplasmy, heteroplasmy, and mitochondrial dystonia. Neurology 69:911–916. https://doi.org/10.1212/01.wnl.0000267843.10977.4a
Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF (1993) Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol 34:609–616. https://doi.org/10.1002/ana.410340416
Mecocci P, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol 36:747–751. https://doi.org/10.1002/ana.410360510
Melov S, Schneider JA, Coskun PE, Bennett DA, Wallace DC (1999) Mitochondrial DNA rearrangements in aging human brain and in situ PCR of mtDNA. Neurobiol Aging 20:565–571
Miquel J, Economos AC, Fleming J, Johnson JE (1980) Mitochondrial role in cell aging. Exp Gerontol 15:575–591
Mita S, Rizzuto R, Moraes CT, Shanske S, Arnaudo E, Fabrizi GM, Koga Y, DiMauro S, Schon EA (1990) Recombination via flanking direct repeats is a major cause of large-scale deletions of human mitochondrial DNA. Nucleic Acids Res 18:561–567
Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J (2009) A hyperfused mitochondrial state achieved at G1–S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci 106:11960–11965. https://doi.org/10.1073/pnas.0904875106
Mokranjac D, Neupert W (2005) Protein import into mitochondria. Biochem Soc Trans 33:1019–1023. https://doi.org/10.1042/BST20051019
Molyneux SL, Florkowski CM, Richards AM, Lever M, Young JM, George PM (2009) Coenzyme Q10; an adjunctive therapy for congestive heart failure? N Z Med J 122:74–79
Moraes CT, Ricci E, Petruzzella V, Shanske S, DiMauro S, Schon EA, Bonilla E (1992) Molecular analysis of the muscle pathology associated with mitochondrial DNA deletions. Nat Genet 1:359–367. https://doi.org/10.1038/ng0892-359
Moraes CT, Sciacco M, Ricci E, Tengan CH, Hao H, Bonilla E, Schon EA, DiMauro S (1995) Phenotype-genotype correlations in skeletal muscle of patients with mtDNA deletions. Muscle Nerve Suppl 3:S150–S153
Müller-Höcker J (1989) Cytochrome-c-oxidase deficient cardiomyocytes in the human heart−an age-related phenomenon. A histochemical ultracytochemical study. Am J Pathol 134:1167–1173
Müller-Höcker J (1990) Cytochrome c oxidase deficient fibres in the limb muscle and diaphragm of man without muscular disease: an age-related alteration. J Neurol Sci 100:14–21
Niu X, Trifunovic A, Larsson N-G, Canlon B (2007) Somatic mtDNA mutations cause progressive hearing loss in the mouse. Exp Cell Res 313:3924–3934. https://doi.org/10.1016/j.yexcr.2007.05.029
Ochoa JJ, Huertas JR, Quiles JL, Mataix J (2001) Dietary oils high in oleic acid, but with different non-glyceride contents, have different effects on lipid profiles and peroxidation in rabbit hepatic mitochondria. J Nutr Biochem 12:357–364. https://doi.org/10.1016/S0955-2863(01)00150-4
Ochoa JJ, Quiles JL, Ibáñez S, Martínez E, López-Frías M, Huertas JR, Mataix J (2003) Aging-related oxidative stress depends on dietary lipid source in rat postmitotic tissues. J Bioenerg Biomembr 35:267–275
Ochoa JJ, Quiles JL, Huertas JR, Mataix J (2005) Coenzyme Q10 protects from aging-related oxidative stress and improves mitochondrial function in heart of rats fed a polyunsaturated fatty acid (PUFA)-rich diet. J Gerontol Biol Sci Med Sci 60:970–975
Ochoa JJ, Pamplona R, Ramirez-Tortosa MC, Granados-Principal S, Perez-Lopez P, Naudí A, Portero-Otin M, López-Frías M, Battino M, Quiles JL (2011) Age-related changes in brain mitochondrial DNA deletion and oxidative stress are differentially modulated by dietary fat type and coenzyme Q10. Free Radic Biol Med 50:1053–1064. https://doi.org/10.1016/j.freeradbiomed.2011.02.004
Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290:470–474
Olivo PD, Van de Walle MJ, Laipis PJ, Hauswirth WW (1983) Nucleotide sequence evidence for rapid genotypic shifts in the bovine mitochondrial DNA D-loop. Nature 306:400–402. https://doi.org/10.1038/306400a0
Owusu-Ansah E, Yavari A, Mandal S, Banerjee U (2008) Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet 40:356–361. https://doi.org/10.1038/ng.2007.50
Page RC, Kornman KS (1997) The pathogenesis of human periodontitis: an introduction. Periodontology 2000 14:9–11. https://doi.org/10.1111/j.1600-0757.1997.tb00189.x
Pak JW, Herbst A, Bua E, Gokey N, McKenzie D, Aiken JM (2003) Mitochondrial DNA mutations as a fundamental mechanism in physiological declines associated with aging. Aging Cell 2:1–7
Pamplona R (2008) Membrane phospholipids, lipoxidative damage and molecu-lar integrity: a causal role in aging and longevity. Biochim Biophys Acta 1777:1249–1262. https://doi.org/10.1016/j.bbabio.2008.07.003
Pamplona R, Portero-Otín M, Ruiz C, Gredilla R, Herrero A, Barja G (2000) Double bond content of phospholipids and lipid peroxidation negatively correlate with maximum longevity in the heart of mammals. Mech Ageing Dev 112:169–183. https://doi.org/10.1016/S0047-6374(99)00045-7
Park KS, Lim JW, Kim H (2009) Inhibitory mechanism of omega-3 fatty acids in pancreatic inflammation and apoptosis. Ann N Y Acad Sci 1171:421–427. https://doi.org/10.1111/j.1749-6632.2009.04887.x
Pikó L, Hougham AJ, Bulpitt KJ (1988) Studies of sequence heterogeneity of mitochondrial DNA from rat and mouse tissues: evidence for an increased frequency of deletions/additions with aging. Mech Ageing Dev 43:279–293
Pinz KG, Shibutani S, Bogenhagen DF (1995) Action of mitochondrial DNA polymerase gamma at sites of base loss or oxidative damage. J Biol Chem 270:9202–9206
Pliss L, Brakmanis A, Ranka R, Elferts D, Krumina A, Baumanis V (2011) The link between mitochondrial DNA hypervariable segment I heteroplasmy and ageing among genetically unrelated Latvians. Exp Gerontol 46:560–568. https://doi.org/10.1016/j.exger.2011.02.016
Pohjoismäki JLO, Goffart S, Tyynismaa H, Willcox S, Ide T, Kang D, Suomalainen A, Karhunen PJ, Griffith JD, Holt IJ, Jacobs HT (2009) Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J Biol Chem 284:21446–21457. https://doi.org/10.1074/jbc.M109.016600
Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B (1998) Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet 20:291–293. https://doi.org/10.1038/3108
Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM (2007) Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131:476–491. https://doi.org/10.1016/j.cell.2007.08.047
Quiles JL, Huertas JR, Mañas M, Battino M, Mataix J (1999) Physical exercise affects the lipid profile of mitochondrial membranes in rats fed with virgin olive oil or sunflower oil. Br J Nutr 81:21–24
Quiles JL, Martínez E, Ibáñez S, Ochoa JJ, Martín Y, López-Frías M, Huertas JR, Mataix J (2002) Ageing-related tissue-specific alterations in mitocondrial composition and function are modulated by dietary fat type in the rat. J Bioenerg Biomembr 34:517–524
Quiles JL, Ochoa JJ, Huertas JR, Mataix J (2004a) Coenzyme Q supplementation protects from age-related DNA double-strand breaks and increases lifespan in rats fed on a PUFA-rich diet. Exp Gerontol 39:189–194. https://doi.org/10.1016/j.exger.2003.10.002
Quiles JL, Ochoa JJ, Ramirez-Tortosa C, Battino M, Huertas JR, Martín Y, Mataix J (2004b) Dietary fat type (virgin olive vs. sunflower oils) affects age-related changes in DNA double-strand-breaks, antioxidant capacity and blood lipids in rats. Exp Gerontol 39:1189–1198. https://doi.org/10.1016/j.exger.2004.05.002
Quiles JL, Ochoa JJ, Battino M, Gutierrez-Rios P, Nepomuceno EA, Frías ML, Huertas JR, Mataix J (2005) Life-long supplementation with a low dosage of coenzyme Q10 in the rat: effects on antioxidant status and DNA damage. Biofactors 25:73–86
Quiles JL, Ochoa JJ, Ramírez-Tortosa MC, Huertas JR, Mataix J (2006) Age-related mitocondrial DNA deletion in rat liver depends on dietary fat unsaturation. J Gerontol A Biol Sci Med Sci 61:107–114
Quiles JL, Pamplona R, Ramirez-Tortosa MC, Naudí A, Portero-Otin M, Araujo-Nepomuceno E, López-Frías M, Battino M, Ochoa JJ (2010) Coenzyme Q addition to an n-6 PUFA-rich diet resembles benefits on age-related mitochondrial DNA deletion and oxidative stress of a MUFA-rich diet in rat heart. Mech Ageing Dev 131:38–47. https://doi.org/10.1016/j.mad.2009.11.004
Rabing Christensen E, Stegger M, Jensen-Fangel S, Laursen AL, Ostergaard L (2004) Mitochondrial DNA levels in fat and blood cells from patients with lipodystrophy or peripheral neuropathy and the effect of 90 days of high-dose coenzyme Q treatment: a randomized, double-blind, placebo-controlled pilot study. Clin Infect Dis 39:1371–1379. https://doi.org/10.1086/424881
Reeves PG (1997) Components of the AIN-93 diets as improvements in the AIN-76A diet. J Nut 127:838S–841S
Reeves PG, Nielsen FH, Fahey GC (1993) AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr 123:1939–1951
Reeve AK, Krishnan KJ, Taylor G, Elson JL, Bender A, Taylor RW, Morris CM, Turnbull DM (2009) The low abundance of clonally expanded mitochondrial DNA point mutations in aged substantia nigra neurons. Aging Cell 8(4):496–498
Richter C, Park JW, Ames BN (1988) Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A 85:6465–6467
Ristow M, Zarse K (2010) How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp Gerontol 45:410–418. https://doi.org/10.1016/j.exger.2010.03.014
Robertson AB, Klungland A, Rognes T, Leiros I (2009) DNA repair in mammalian cells: Base excision repair: the long and short of it. Cell Mol Life Sci CMLS 66:981–993. https://doi.org/10.1007/s00018-009-8736-z
Roche E, Ramírez-Tortosa CL, Arribas MI, Ochoa JJ, Sirvent-Belando JE, Battino M, Ramírez-Tortosa MC, González-Alonso A, Pérez-López MP, Quiles JL (2014) Comparative analysis of pancreatic changes in aged rats fed life long with sunflower, fish, or olive oils. J Gerontol A Biol Sci Med Sci 69:934–944. https://doi.org/10.1093/gerona/glt157
Ropp PA, Copeland WC (1996) Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics 36:449–458. https://doi.org/10.1006/geno.1996.0490
Rosenfeldt F, Marasco S, Lyon W, Wowk M, Sheeran F, Bailey M, Esmore D, Davis B, Pick A, Rabinov M, Smith J, Nagley P, Pepe S (2005) Coenzyme Q10 therapy before cardiac surgery improves mitochondrial function and in vitro contractility of myocardial tissue. J Thorac Cardiovasc Surg 129:25–32. https://doi.org/10.1016/j.jtcvs.2004.03.034
Sage JM, Gildemeister OS, Knight KL (2010) Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J Biol Chem 285:18984–18990. https://doi.org/10.1074/jbc.M109.099846
Samuels DC, Schon EA, Chinnery PF (2004) Two direct repeats cause most human mtDNA deletions. Trends Genet TIG 20:393–398. https://doi.org/10.1016/j.tig.2004.07.003
Sanz A, Caro P, Sanchez JG, Barja G (2006) Effect of lipid restriction on mitochondrial free radical production and oxidative DNA damage. Ann N Acad Sci 1067:200–209. https://doi.org/10.1196/annals.1354.024
Sanz A, Fernández-Ayala DJM, Stefanatos RK, Jacobs HT (2010) Mitochondrial ROS production correlates with, but does not directly regulate lifespan in Drosophila. Aging 2:200–223. https://doi.org/10.18632/aging.100137
Sattar N, Wannamethee SG, Forouhi NG (2008) Novel biochemical risk factors for type 2 diabetes: pathogenic insights or prediction possibilities? Diabetologia 51:926–940. https://doi.org/10.1007/s00125-008-0954-7
Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, Chinnery PF, Turnbull DM (2008) Prevalence of mitochondrial DNA disease in adults. Ann Neurol 63:35–39. https://doi.org/10.1002/ana.21217
Schon EA, Rizzuto R, Moraes CT, Nakase H, Zeviani M, DiMauro S (1989) A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science 244:346–349
Schultz DR, Harrington WJ Jr (2003) Apoptosis: programmed cell death at a molecular level. Semin Arthritis Rheum 32:345–369. https://doi.org/10.1053/sarh.2003.50005
Sena CM, Nunes E, Gomes A, Santos MS, Proença T, Martins MI, Seiça RM (2008) Supplementation of coenzyme Q10 and alpha-tocopherol lowers glycated hemoglobin level and lipid peroxidation in pancreas of diabetic rats. Nutr Res 28:113–121. https://doi.org/10.1016/j.nutres.2007.12.005
Shadel GS, Clayton DA (1997) Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem 66:409–435. https://doi.org/10.1146/annurev.biochem.66.1.409
Shigenaga MK, Hagen TM, Ames BN (1994) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A 91:10771–10778
Shoffner JM, Lott MT, Lezza AMS, Seibel P, Ballinger SW, Wallace DC (1990) Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 61:931–937. https://doi.org/10.1002/(SICI)1097-4598(199703)20:3<271::AID-MUS2>3.0.CO;2-8
Shults CW (2003) Coenzyme Q10 in neurodegenerative diseases. Curr Med Chem 10:1917–1921
Simonetti S, Chen X, DiMauro S, Schon EA (1992) Accumulation of deletions in human mitochondrial DNA during normal aging: analysis by quantitative PCR. Biochim Biophys Acta 1180:113–122
Slupphaug G, Kavli B, Krokan HE (2003) The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res Mol Mech Mutagen 531:231–251. https://doi.org/10.1016/j.mrfmmm.2003.06.002
Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT (2006) Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab 3:9–13. https://doi.org/10.1016/j.cmet.2005.12.001
Sondheimer N, Glatz CE, Tirone JE, Deardorff MA, Krieger AM, Hakonarson H (2011) Neutral mitochondrial heteroplasmy and the influence of aging. Hum Mol Genet 20:1653–1659. https://doi.org/10.1093/hmg/ddr043
Stevnsner T, Thorslund T, de Souza-Pinto NC, Bohr VA (2002) Mitochondrial repair of 8-oxoguanine and changes with aging. Exp Gerontol 37:1189–1196. https://doi.org/10.1016/S0531-5565(02)00142-0
Stewart JB, Freyer C, Elson JL, Larsson N-G (2008a) Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat Rev Genet 9:657–662. https://doi.org/10.1038/nrg2396
Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson N-G (2008b) Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol 6:e10. https://doi.org/10.1371/journal.pbio.0060010
Stierum RH, Dianov GL, Bohr VA (1999) Single-nucleotide patch base excision repair of uracil in DNA by mitochondrial protein extracts. Nucleic Acids Res 27:3712–3719
Suzuki S, Hinokio Y, Ohtomo M, Hirai M, Hirai A, Chiba M, Kasuga S, Satoh Y, Akai H, Toyota T (1998) The effects of coenzyme Q10 treatment on maternally inherited diabetes mellitus and deafness, and mitochondrial DNA 3243 (A to G) mutation. Diabetologia 41:584–588. https://doi.org/10.1007/s001250050950
Szczesny B, Hazra TK, Papaconstantinou J, Mitra S, Boldogh I (2003) Age-dependent deficiency in import of mitochondrial DNA glycosylases required for repair of oxidatively damaged bases. Proc Natl Acad Sci U S A 100:10670–10675. https://doi.org/10.1073/pnas.1932854100
Tam ZY, Gruber J, Halliwell B, Gunawan R (2013) Mathematical modeling of the role of mitochondrial fusion and fission in mitochondrial DNA maintenance. PloS One 8:e76230. https://doi.org/10.1371/journal.pone.0076230
Tam ZY, Gruber J, Ng LF, Halliwell B, Gunawan R (2014) Effects of lithium on age-related decline in mitochondrial turnover and function in Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci 69:810–820. https://doi.org/10.1093/gerona/glt210
Tapia PC (2006) Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Med Hypotheses 66:832–843. https://doi.org/10.1016/j.mehy.2005.09.009
Taylor RW, Barron MJ, Borthwick GM, Gospel A, Chinnery PF, Samuels DC, Taylor GA, Plusa SM, Needham SJ, Greaves LC, Kirkwood TBL, Turnbull DM (2003) Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest 112:1351–1360. https://doi.org/10.1172/JCI19435
Temperley RJ, Seneca SH, Tonska K, Bartnik E, Bindoff LA, Lightowlers RN, Chrzanowska-Lightowlers ZMA (2003) Investigation of a pathogenic mtDNA microdeletion reveals a translation-dependent deadenylation decay pathway in human mitochondria. Hum Mol Genet 12:2341–2348. https://doi.org/10.1093/hmg/ddg238
Thomas B, Beal MF (2010) Mitochondrial therapies for Parkinson’s disease. Mov Disord 25(Suppl 1):S155–S160. https://doi.org/10.1002/mds.22781
Trifunovic A, Larsson N-G (2008) Mitochondrial dysfunction as a cause of ageing. J Intern Med 263:167–178. https://doi.org/10.1111/j.1365-2796.2007.01905.x
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson N-G (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417–423. https://doi.org/10.1038/nature02517
Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson N-G (2005) Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A 102:17993–17998. https://doi.org/10.1073/pnas.0508886102
Trounce I, Byrne E, Marzuki S (1989) Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet Lond Engl 1:637–639
Tuppen HAL, Blakely EL, Turnbull DM, Taylor RW (2010) Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 1797:113–128. https://doi.org/10.1016/j.bbabio.2009.09.005
Tyrka AR, Carpenter LL, Kao H-T, Porton B, Philip NS, Ridout SJ, Ridout KK, Price LH (2015) Association of telomere length and mitochondrial DNA copy number in a community sample of healthy adults. Exp Gerontol 66:17–20
Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, Spelbrink JN, Paetau A, Suomalainen A (2005) Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A 102:17687–17692. https://doi.org/10.1073/pnas.0505551102
Upholt WB, Dawid IB (1977) Mapping of mitochondrial DNA of individual sheep and goats: rapid evolution in the D loop region. Cell 11:571–583. https://doi.org/10.1016/0092-8674(77)90075-7
Varela-Lopez A, Bullon P, Battino M, Ramirez-Tortosa MC, Ochoa JJ, Cordero MD, Ramirez-Tortosa CL, Rubini C, Zizzi A, Quiles JL (2015) Coenzyme Q protects against age-related alveolar bone loss associated to n-6 PUFA rich-diets by modulating mitochondrial mechanisms. J Gerontol A Biol Sci Med Sci 71:593–600
Vielhaber S, Kunz D, Winkler K, Wiedemann FR, Kirches E, Feistner H, Heinze HJ, Elger CE, Schubert W, Kunz WS (2000) Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain J Neurol 123(Pt 7):1339–1348
Vilkki J, Savontaus ML, Nikoskelainen EK (1990) Segregation of mitochondrial genomes in a heteroplasmic lineage with Leber hereditary optic neuroretinopathy. Am J Hum Genet 47:95–100
Wai T, Teoli D, Shoubridge EA (2008) The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat Genet 40:1484–1488. https://doi.org/10.1038/ng.258
Wanagat J, Cao Z, Pathare P, Aiken JM (2001) Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J Off Publ Fed Am Soc Exp Biol 15:322–332. https://doi.org/10.1096/fj.00-0320com
Wang Y, Bogenhagen DF (2006) Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J Biol Chem 281:25791–25802. https://doi.org/10.1074/jbc.M604501200
Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG (2001) Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci U S A 98:4038–4043. https://doi.org/10.1073/pnas.061038798
Weber K, Wilson JN, Taylor L, Brierley E, Johnson MA, Turnbull DM, Bindoff LA (1997) A new mtDNA mutation showing accumulation with time and restriction to skeletal muscle. Am J Hum Genet 60:373–380
Wenz T, Diaz F, Speigelman BM, Moraes CT (2008) Activation of the PPAR/PGC-1alpha pathway pre- vents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab 8:249–256. https://doi.org/10.1016/j.cmet.2008.07.006
Wong TW, Clayton DA (1985a) Isolation and characterization of a DNA primase from human mitochondria. J Biol Chem 260:11530–11535
Wong TW, Clayton DA (1985b) In vitro replication of human mitochondrial DNA: accurate initiation at the origin of light-strand synthesis. Cell 42:951–958
Wong A, Cavelier L, Collins-Schramm HE, Seldin MF, McGrogan M, Savontaus M-L, Cortopassi GA (2002) Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Hum Mol Genet 11:431–438. https://doi.org/10.1093/hmg/11.4.431
Woo DK, Green PD, Santos JH, D’Souza AD, Walther Z, Martin WD, Christian BE, Chandel NS, Shadel GS (2012) Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am J Pathol 180:24–31. https://doi.org/10.1016/j.ajpath.2011.10.003
Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG (2002) Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci USA 99:15066–15071. https://doi.org/10.1073/pnas.232591499
Xu G, Herzig M, Rotrekl V, Walter CA (2008) Base excision repair, aging and health span. Mech Ageing Dev 129:366–382. https://doi.org/10.1016/j.mad.2008.03.001
Yang J, Zhu Y, Tong Y, Chen L, Liu L, Zhang Z, Wang X, Huang D, Qiu W, Zhuang S, Ma X (2009) Confirmation of the mitochondrial ND1 gene mutation G3635A as a primary LHON mutation. Biochem Biophys Res Commun 386:50–54. https://doi.org/10.1016/j.bbrc.2009.05.127
Yang X, Dai G, Li G, Yang ES (2010) Coenzyme Q10 reduces beta-amyloid plaque in an APP/PS1 transgenic mouse model of Alzheimer’s disease. J Mol Neurosci 41:110–113. https://doi.org/10.1007/s12031-009-9297-1
Yasukawa T, Reyes A, Cluett TJ, Yang M-Y, Bowmaker M, Jacobs HT, Holt IJ (2006) Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J 25:5358–5371. https://doi.org/10.1038/sj.emboj.7601392
Young AJ, Johnson S, Steffens DC, Doraiswamy PM (2007) Coenzyme Q10: a review of its promise as a neuroprotectant. CNS Spectr 12:62–68
Yubero-Serrano EM, Delgado-Casado N, Delgado-Lista J, Perez-Martinez P, Tasset-Cuevas I, Santos-Gonzalez M, Caballero J, Garcia-Rios A, Marin C, Gutierrez-Mariscal FM, Fuentes F, Villalba JM, Tunez I, Perez-Jimenez F, Lopez-Miranda J (2010) Postprandial antioxidant effect of the Mediterranean diet supplemented with coenzyme Q10 in elderly men and women. Age Drodr 33:579–590. https://doi.org/10.1007/s11357-010-9199-8
Yubero-Serrano EM, Gonzalez-Guardia L, Rangel-Zuñiga O, Delgado-Lista J, Gutierrez-Mariscal FM, Perez-Martinez P, Delgado-Casado N, Cruz-Teno C, Tinahones FJ, Villalba JM, Perez-Jimenez F, Lopez-Miranda J (2012) Mediterranean diet supplemented with coenzyme Q10 modifies the expression of proinflammatory and endoplasmic reticulum stress–related genes in elderly men and women. J Gerontol Biol Sci Med Sci 67A:3–10. https://doi.org/10.1093/gerona/glr167
Zelenka J, Dvořák A, Alán L (2015) L-lactate protects skin fibroblasts against aging-associated mitochondrial dysfunction via mitohormesis. Oxid Med Cell Longev 2015:351698. https://doi.org/10.1155/2015/351698
Zeviani M, Moraes CT, DiMauro S, Nakase H, Bonilla E, Schon EA, Rowland LP (1988) Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 38:1339–1346
Zheng W, Khrapko K, Coller HA, Thilly WG, Copeland WC (2006) Origins of human mitochondrial point mutations as DNA polymerase gamma-mediated errors. Mutat Res 599:11–20. https://doi.org/10.1016/j.mrfmmm.2005.12.012
Acknowledgments
Dr. Alfonso Varela-López was recipient of a “Contrato Puente” from the University of Granada during 2017. At present is recipient of a research grant at universities or centers abroad from the Alfonso Martin Escudero Foundation. María Dolores Navarro Hortal has been awarded a Scholarship of Initiation to Research for undergraduate students of the Own Plan of the University of Granada during the academic year 2016–2017 and a Scholarship for Student Collaboration in University Departments for the academic year 2017–2018 in Research in University Departments of the Ministry of Education, Culture and Sports 2017/2018 call. Authors acknowledge to the University of Granada and the Autonomous Government of Andalusia for partial support of the research team.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Quiles, J.L., Varela-López, A., Navarro-Hortal, M.D., Battino, M. (2020). Coenzyme Q, mtDNA and Mitochondrial Dysfunction During Aging. In: López Lluch, G. (eds) Coenzyme Q in Aging. Springer, Cham. https://doi.org/10.1007/978-3-030-45642-9_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-45642-9_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-45641-2
Online ISBN: 978-3-030-45642-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)