
Abstract
Accumulation of senescent cells has emerged as a major pathogenic factor in aging and multiple age-related diseases. Studies on the biology of senescent cells have identified vulnerabilities to eliminate them using a novel class of drugs called senolytics. These drugs kill senescent cells by blocking their resistance to apoptosis, by reactivating latent p53 or by increasing oxidative stress. Other compounds inhibit the senescence associated secretory phenotype or SASP. Senolytics and SASP modulators have been effective to improve natural aging and age-related diseases in mice models leading to ongoing clinical trials in humans.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Cellular senescence is a programmed response triggered by both physiological or pathological factors that results in a phenotype characterized by an inability to respond to proliferative signals, resistance to apoptosis and the secretion of a variety of proteins and lipids with potent proinflammatory activity (Ferbeyre 2018; Lopes-Paciencia et al. 2019). In vivo, senescent cells can be divided into three distinct categories: embryonic, acute and chronic. Embryonic senescent cells help to shape developing tissues in mammals, fish and amphibia (Davaapil et al. 2017; Yun et al. 2015; Storer et al. 2013; Munoz-Espin et al. 2013; Villiard et al. 2017) whereas acute senescent cells are a protective response to abrupt stress such as a wound or an oncogenic signal. Both acute and embryonic senescent cells are beneficial and are eliminated through the immune system. Chronic senescence, on the other hand, may result from slowly accumulating damage at the macromolecular level and is associated to aging, cancer and age-related diseases (Childs et al. 2015). Accumulation of DNA damage, including damage in telomeric regions, have been linked to aging (Sedelnikova et al. 2004; Liu et al. 2005; Lombard et al. 2005; Herbig et al. 2006; Sahin and Depinho 2010; Hewitt et al. 2012). DNA damage activates the DNA damage response, which is measurable using antibodies that recognize proteins phosphorylated by DNA damage-activated kinases and includes the histone variant γ-H2AX (Mallette et al. 2007; Mallette et al. 2007; Halazonetis et al. 2008; Di Micco et al. 2006). However, the precise triggers of DNA damage and senescence during aging are still unknown.
2 Evidence of Senescent Cell Accumulation in Vivo
A lot of research confirms that accumulation of senescent cells is a hallmark of aging (López-Otín et al. 2013; He and Sharpless 2017). This might be the consequence of an increased generation of senescent cells with aging and/or a decrease in senescent cell clearance as the immune system gets older (Childs et al. 2015). In fact, senescent cells have a longer half-life in old animals (Karin et al. 2019), suggesting that their clearance becomes less effective with aging. Accumulation of senescent cells has been demonstrated using several senescence biomarkers in old zebrafish (Kishi et al. 2008) and many mammals (Jeyapalan and Sedivy 2008).
A biomarker commonly used to detect senescent cells is the staining for the Senescence-Associated β-Galactosidase (SA-β-Gal), a lysosomal enzyme upregulated in senescent cells (Kurz et al. 2000; Bandyopadhyay et al. 2005). The other standard biomarker is the induction of the cyclin-dependent kinase inhibitor (CKI) p16Ink4a mRNA levels. Expression of this tumor suppressor is undetectable in young rodents, but it increases with age in older tissues (Krishnamurthy et al. 2006; Berkenkamp et al. 2014; Burd et al. 2013) including stem cells (Janzen et al. 2006; Molofsky et al. 2006). P16Ink4a is mechanistically connected to senescence since inhibition of its expression in stem cells reduces their aging phenotype and allows faster tissue repair (Janzen et al. 2006; Molofsky et al. 2006). Telomere shortening, another well-known cause of senescence, can be measured using in situ hybridization with telomeric probes and can be used as a biomarker in some tissues. Telomere length decreases with age in the gut and liver in mice (Hewitt et al. 2012) and primates (Jeyapalan et al. 2007). Given the lack of a universal marker for senescence, quantification of senescent cells should use methods combining several biomarkers. For example, by combining the SA-β-Gal assay, DNA damage response markers and the depletion of HMGB1 from the cell nucleus, Biran et al. found senescent cells to be 10–20 times more abundant in old than in young mice (Biran et al. 2017). Using a similar strategy based on several biomarkers, Herbig et al. showed that the percentage of senescent cells in baboons’ skin increased exponentially from 2% in young individuals to more than 15% in aged ones (Herbig et al. 2006). This was confirmed by another study and it was suggested that it would be the case for any mitotic tissue (Jeyapalan et al. 2007).
In humans, the levels of p16Ink4a and p27 (another CKI) are accurate biomarkers of aging in kidneys (Chkhotua et al. 2003; Melk et al. 2004), while the abundance of SA-β-Gal positive cells correlated with age in skin samples (Dimri et al. 1995). This has been confirmed using other senescence-associated markers in the skin (Wang and Dreesen 2018), bones, mesenchymal stem cells (Zhou et al. 2008; Farr and Khosla 2019) and human peripheral blood T lymphocytes (Liu et al. 2009). All of this research convincingly shows that senescent cells do accumulate with age in mammals. Importantly, senescent cells are also more readily detected in many ailing tissues and age-related conditions (Table 4.1) (Jaul and Barron 2017; Franceschi et al. 2018). This suggests that drugs acting on senescent cells will have a major impact in gerontology and healthy aging.
3 Elimination of Senescent Cells: Senolytics
Elimination of senescent cells using suicide genes in genetically modified mice or drugs that kill senescent cells, called senolytics, improve many age-related diseases (Table 4.2). Senolytics are thus posed to have broad medical applications. Here, we will discuss them according to their mechanism of action, illustrated in Fig. 4.1.

Schematic overview of the different strategies pursued to eliminate senescent cells or alleviate the detrimental effects of the SASP
3.1 Senolytics that Inhibit the Bcl2 Family
In multicellular organisms, cells can be eliminated by a process of programmed cell death called apoptosis. Apoptosis can be triggered in two ways. The extrinsic pathway involves a death receptor situated on the cytoplasmic membrane that can be activated by several death effector cytokines. The intrinsic pathway is triggered by endogenous damage that engages mitochondria to release pro-apoptotic factors such as cytochrome c. In both cases, Bcl-2 family proteins (Bcl-2, Bcl-xl, Bcl-w, Bfl-1 or Mcl-1) antagonize this process (Azmi et al. 2011).
Senescent cells are particularly resistant to apoptosis. For example, senescent human fibroblasts express high levels of Bcl-2 family members and can last as long as four weeks in media lacking serum without signs of apoptosis (Wang 1995). Senescent cells also secrete many cytokines and lipids, collectively known as the Senescence-Associated Secretory Phenotype (SASP), that may have anti-apoptotic functions. Together these Senescent Cell Anti-apoptotic Pathways protect senescent cells from cell death (Kirkland and Tchkonia 2017) and targeting them could be a promising way to selectively kill senescent cells.
Recent publications have shown several compounds that could act as effective senolytics via inhibition of antiapoptotic pathways. ABT-263, also known as Navitoclax, preferentially induces apoptosis in senescent fibroblasts and vein epithelial cells by inhibiting Bcl-2, Bcl-xl and Bcl-w (Zhu et al. 2016). These results were also observed in vivo in different mice models (Chang et al. 2016, 2016; Pan et al. 2017). TW-37, an inhibitor of Bcl-2, Bcl-xl and Mcl-1, was less senolytic than ABT-263 (Zhu et al. 2016), suggesting that Bcl-w plays an important role in protecting senescent cells from apoptosis. ABT-263 was ineffective against human senescent primary preadipocytes (Zhu et al. 2016), demonstrating that senolytics act in a tissue specific manner, a factor that should be taken into account for their use. ABT-737, an inhibitor of Bcl-xl and Bcl-w, preferentially kills senescent cells induced by DNA damage in the lung and senescent cells induced by p14ARF expression in the epidermis (Yosef et al. 2016). Interestingly, the elimination of senescent cells in the epidermis led to an increase in hair follicle stem cell proliferation (Yosef et al. 2016). Of note, the anti-Bcl2 family of drugs cause neutropenia and thrombocytopenia (Roberts et al. 2012), side effects that could limit their application in healthy old individuals.
Other inhibitors of the Bcl2 family with senolytic activity include fisetin, a flavone molecule that induces apoptosis in senescent fibroblasts and endothelial cells but not in senescent preadipocytes (Zhu et al. 2017). In progeroid Ercc1−/Δ mice, fisetin killed senescent cells and reduced senescence biomarkers. In old naturally aged C57BL/6 mice, a 5-day diet of fisetin was able to significantly reduce the proportion of senescent cells in different tissues and extend median and maximal lifespan even when the treatment was initiated in old animals (Yousefzadeh et al. 2018). In cancer cells, fisetin can cause apoptosis by activating both the intrinsic and the extrinsic pathways and had beneficial effects to treat inflammation and metastasis (Kashyap et al. 2018). Fisetin is present in many fruits and vegetables, suggesting that it can be safely used as a senolytic and anti-aging agent in humans (Kashyap et al. 2018). Epigallocatechin gallate, a phytochemical found in green tea, inhibits both the anti-apoptotic Bcl-2 family and mTOR. The latter controls the SASP by regulating the translation of mRNAs coding for inflammatory cytokines (Herranz et al. 2015; Laberge et al. 2015). Epigallocatechin can thus act both as a SASP modulator by inhibiting mTOR and as a senolytic (Kumar et al. 2019).
Panobinostat is a deacetylase inhibitor used to treat multiple myeloma. Panobinostat is particularly potent against all deacetylases of class I, II and IV (Laubach et al. 2015). In non-small cell lung cancer and head and neck squamous cell carcinoma, senescent cells have altered H3 acetylation and Bcl-xl expression. Panobinostat inhibits Bcl-xl and kill senescent cells induced by chemotherapy (Samaraweera et al. 2017). However, this drug may cause a few adverse effects, including diarrhea, asthenia and a lower count of immune blood cells (Van Veggel et al. 2018; Hennika et al. 2017).
Finally, EF24, a natural compound found in turmeric (Curcuma longa) that is similar to curcumin, can kill senescent cells by downregulating Bcl-xl (Li et al. 2019). EF24 could be used in synergy with ABT-263 to kill senescent cells more effectively, and at the same time, prevent ABT-263’s cytotoxic effects (Li et al. 2019).
3.2 Proapoptotic Cocktail Dasatinib + Quercetin
Based on the fact that senescent cells are resistant to apoptosis induced by serum deprivation and other stresses, Zhu et al. hypothesized that it could be possible to kill them by inhibiting their antiapoptotic pathways. A screening using molecules that block these pathways showed that the drugs dasatinib and quercetin were particularly efficient to kill senescent cells and improves health span in progeroid mice (Zhu et al. 2015). The synergistic combination proved to be efficient to counteract the effect of irradiation in vivo, and to extend lifespan in naturally-aged mice (Xu et al. 2018). Dasatinib is a competitive inhibitor of tyrosine kinases and is currently used to treat leukemia (Zarbock 2012). The drug strongly suppresses the SASP and genes related to senescence in human subjects with systemic sclerosis (Martyanov et al. 2019). Quercetin is a flavonol found in some fruits and vegetables that has anti-inflammatory properties (Li et al. 2016). The cocktail with both drugs targets different anti-apoptotic pathways in senescent cells (Kirkland and Tchkonia 2017). It significantly lowered the number of senescent cells in human adipose tissues without killing macrophages. This subsequently had a positive impact on inflammation and frailty (Xu et al. 2018). However, senescent hepatocellular carcinoma cells resist the effect of the combination dasatinib/quercetin (Kovacovicova et al. 2018) indicating again the tissue specific mode of action of senolytics.
3.3 Senolytics that Activate P53: Nutlin and FOXO4DRI
Most types of stress that trigger senescence also activate the tumor suppressor p53 (Qian and Chen 2010). P53 acts as a transcription factor inducing the expression of genes such as promyelocytic leukemia (PML) (de Stanchina et al. 2004; Ablain et al. 2014), p21 (Fang et al. 1999), PAI1 (Kortlever et al. 2006) and E2F7 (Aksoy et al. 2012) that mediate the growth arrest phenotype of senescent cells. P53 can also trigger cell death but the key molecular switches that control cell fate downstream of p53 activation are not well understood (Macip et al. 2003). Reactive oxygen species (ROS) can convert a p53-dependent senescence response into apoptosis but the mechanism has not been totally elucidated (Macip et al. 2003). It has been proposed that p53 must overcome a concentration threshold to trigger apoptosis (Kracikova et al. 2013). Another pertinent characteristic of p53 in this regard is its ability to translocate to mitochondria (Mihara et al. 2003) and inhibit Bcl-2 and Bcl-xl, preventing their anti-apoptotic activities (Hagn et al. 2010). P53 translocation to mitochondria is inhibited by the nucleolar protein nucleophosmin (Dhar and St Clair 2009) and the E3 ubiquitin ligase TRAF6 (Zhang et al. 2016).
Nutlin (Nutley inhibitor) was designed by Vassilev and colleagues to prevent the binding of Mdm2 to p53. The nutlin binding site is situated in a deep hydrophobic pocket in the Mdm2 protein (Vassilev et al. 2004). Mdm2, an E3 ubiquitin ligase, inhibits p53 through three mechanisms: (1) degradation of p53 by poly-ubiquitylation and targeting it to the proteasome; (2) export of p53 out of the nucleus by mono-ubiquitylation and (3) direct binding to p53 preventing its activity as a transcription factor (Wu and Prives 2018). Since p53 is needed to express Mdm2, levels of p53 are autoregulated in a feedback loop (Lessel et al. 2017).
Nutlin is not genotoxic and does not promote p53 modifications associated to DNA damage, it only stabilizes p53 by protecting it from Mdm2. Although senescent cells activate p53, this activation is limited (Huang and Vassilev 2009) suggesting that p53 cannot attain the levels needed to trigger apoptosis. It is then quite possible that nutlin could stabilize latent p53 in senescent cells and trigger apoptosis. This has been shown in cultures of senescent chondrocytes obtained from patients with osteoarthrosis or in vivo in a mouse model of osteoarthrosis triggered by transection of the anterior cruciate ligament (Jeon et al. 2017). However, p53 reactivation could lead to toxic effects in normal cells, including death by apoptosis (Burgess et al. 2016), or could generate a selective pressure for mutations of p53 (Aziz et al. 2011). Also, nutlin could potentially bind to other protein pockets with similar shapes and physicochemical properties, leading to potential toxic effects (Nguyen et al. 2019).
Proteolysis Targeting Chimera (PROTAC) consist of two protein binding fragments, one capable of binding to a target protein and another that binds to an E3 ubiquitin ligase (Bondeson et al. 2018). PROTACs having an Mdm2-binding fragment such as nutlin could have a double effect. First, they can activate p53 by preventing its inhibition by Mdm2. Second, they could target another protein for degradation by the proteasome by promoting its interaction with Mdm2. Such a PROTAC targeting BRD4 was shown to be very effective against cancer cells with wild type p53 (Hines et al. 2019) but it could also be modified by coupling nutlin to anti-Bcl2 family compounds such as ABT-273 to better kill senescent cells.
FOXO4 is a member of the Forkhead box O (FOXO) family of transcription factors which are negatively regulated by insulin or IGF-1 via AKT-dependent phosphorylation and cytoplasmic retention (Martins et al. 2016). In the nucleus, FOXO factors can localize to PML nuclear bodies (Trotman et al. 2006) which are particularly induced in senescent cells (Bourdeau et al. 2009). FOXO factors are involved in resistance to stress, metabolism, cell cycle arrest and apoptosis (Martins et al. 2016).
In response to acute damage, FOXO4 was shown to favor senescence instead of apoptosis. During senescence, p53 is phosphorylated by ATM (Ataxia-Telangiectasia Mutated), which would prevent its inhibition by Mdm2 (Mallette et al. 2007). In this situation, p53 localizes to chromatin having persistent DNA damage (DNA-SCARS), next to PML bodies containing FOXO4. FOXO4 would then limit p53’s ability to promote apoptosis by sequestering p53 in PML bodies (Baar et al. 2017). Based on this, Baar and colleagues synthesized a FOXO4-derived peptide that would prevent the binding of FOXO4 to p53 (Baar et al. 2017). This peptide was designed as a D-retro inverse isoform (DRI) so that it would have a better potency than its natural L-isoform counterpart. The FOXO4-DRI peptide was effective in relocating p53 to mitochondria, promoting apoptosis selectively in senescent cells both in vitro and in vivo. This peptide had potent anti-aging effects both in progeroid and wild-type mice illustrating once again the causal relationship between senescent cells and aging (Baar et al. 2017). FOXO4-DRI showed a tenfold selectivity to senescent cells compared to normal cells. Although the peptide was safe in rodents upon repeated administration, long living humans may require even more injections. It would be preferable to optimize this peptide to achieve a higher degree of selectivity to avoid potential toxicities in humans (Baar et al. 2018).
3.4 Metabolic Inhibitors: Targeting Glycolysis and REDOX Metabolism
Senescent cells have a dramatic upregulation of glucose utilization in association to their mitochondrial dysfunction (Moiseeva et al. 2009). For instance, therapy-induced senescence (TIS) in lymphoma cells is accompanied by an increase in glucose utilization and autophagy that together support the ATP and metabolic demands of senescent cells. A combination of deoxyglucose with the autophagy inhibitor bafilomycin A1 selectively killed these senescent cells (Dörr et al. 2013). The glucose transport inhibitors phloretin and cytochalasin B or the lactate dehydrogenase inhibitor oxamate were also selectively toxic for TIS cells (Dörr et al. 2013). This was actually the first demonstration of a pharmacological approach to kill senescent cells.
The screening of a library of small molecules supposed to target important pathways for senescent cells showed that piperlongumine was a promising candidate as a senolytic (Wang et al. 2016). The drug induced apoptosis selectively in senescent fibroblasts compared to control cells. A significant synergy between piperlongumine and ABT-263 could allow for a lower dose of ABT-263. The latter causes thrombocytopenia and neutropenia because of inhibition of Bcl-xl in platelets (Wang et al. 2016). Although piperlongumine’s mode of action is not fully understood, the drug was shown to bind the protein Oxidation Resistance 1 (OXR1) leading to its degradation by the ubiquitin-proteasome system (Liu et al. 2018). Targeting OXR1 kills senescent cells by promoting oxidative stress (Yang et al. 2014; Zhang et al. 2018).
Senescent chondrocytes have been linked to osteoarthritis, a disease for which there is no cure. Nogueira-Recalde and colleagues found fenofibrate, an agonist of PPARα (peroxisome proliferator-activated receptor alpha) as a senolytic after interrogating the Prestwick chemical library for molecules that kill senescent cells. They showed that fenofibrate effectively and selectively killed senescent chondrocytes by promoting apoptosis in vitro. Since this drug was found by screening chemical compounds it is not yet clear how it selectively kills senescent cells. The authors correlated the effects of fenofibrate with inhibition of the mTOR effector S6 kinase (Nogueira-Recalde et al. 2019). Since mTOR is required for protein synthesis, this strategy may work by inhibiting the expression of anti-apoptotic proteins. In a retrospective study, human osteoarthritis patients taking fenofibrate reported a significant decrease in disability and pain leading to fewer joint surgeries (Nogueira-Recalde et al. 2019). The use of fenofibrate as a senolytic could be however limited by its hepatotoxicity (Hedrington and Davis 2018).
3.5 HSP90 Inhibitors
Robbins and colleagues screened a library of autophagic regulators for compounds that killed senescent ercc1 null murine embryonic fibroblasts. They identified the HSP90 inhibitors geldanamycin, 17-AAG (tenespimycin) and 17-DMAG (alvespimycin) as potent senolytics that triggered apoptosis in senescent cells (Fuhrmann-Stroissnigg et al. 2017). The mechanism of senolytic activity of HSP90 inhibitors included the destabilization of phospho-AKT (Fuhrmann-Stroissnigg et al. 2017). Of note, treating progeroid ercc1−/Δ mice with 17-DMAG reduced the expression of senescence biomarkers in the kidneys and delayed the onset of age-related phenotypes (Fuhrmann-Stroissnigg et al. 2017).
3.6 Sodium/Potassium ATPase Inhibitors
Produced naturally by many plants, cardiac glycosides have been recently found to be senolytic agents. They prevent cytoplasmic transmembrane Na+/K+ pumps to maintain the resting potential across the membrane by binding to their alpha 1 subunit (Langford and Boor 1996). Using a high throughput screening method on compounds found in the Prestwick library, Triana-Martinez et al. identified 9 cardiac glycosides as senolytics. Among them, digoxin had the highest senolytic index (Triana-Martinez et al. 2019). Cardiac glycosides selectively trigger apoptosis of senescent cells by increasing intracellular concentration of Na+ ions. This in turn would inhibit Na+/Ca2+ and Na+/H+ exchangers, leading to increasing concentrations of Ca2+ and H+. Since senescent cells already have a lower cytosolic pH than normal cells, digoxin could activate both the intrinsic and the extrinsic apoptosis pathways in these cells only (Majdi et al. 2016). In a similar fashion, ouabain, another cardiac glycoside, was also found to have senolytic properties (Guerrero et al. 2019). Interestingly, in this study ouabain was shown to induce the expression of the proapoptotic protein Noxa, suggesting that changes in gene expression underpin the senolytic activity of cardiac glycosides.
3.7 Immunotherapy
Human senescent fibroblasts express higher levels of the cell surface marker dipeptidyl peptidase 4 (DPP4) (Kim et al. 2017). An antibody-dependent cell-mediated cytotoxicity (ADCC) assay showed that NK cells can recognize and selectively kill these DPP4 positive senescent fibroblasts (Kim et al. 2017). As senescence can also occur in immune cells, the immune system can become impaired, leading to accumulation of senescent cells. Thus, targeting specifically immune senescent cells could have an indirect senolytic effect on senescent cells from other tissues. In mice, removal of senescent hematopoietic stem cells had a rejuvenating effect on aged tissues (Chang et al. 2016). Also, clearance of senescent cells in irradiated mouse spleen restored the functions of T cells and macrophages (Palacio et al. 2019). It seems also possible to engineer immune cells against senescent cells expressing IL-6, and then promote their death via cell fusion (Qudrat et al. 2017).
3.8 Drug Delivery System Targeting Senescent Cells
Another interesting way of killing senescent cells is to encapsulate a cytotoxic compound in a shell that would preferentially target these cells. Since they have a high level of lysosomal β-galactosidase compared to normal cells, Muñoz-Espín et al. used nanoparticles covered with galacto-oligosaccharides on a silica scaffold. These beads are integrated by most cells via endocytosis and then quickly released via exocytosis. However, in the case of senescent cells, β-galactosidase will digest the polysaccharide allowing the release of drugs inside the nanoparticles before exocytosis. Fluorophores were used to show that these nanoparticles identified senescent cells in vivo and when loaded with doxorubicin they selectively killed senescent cells (Muñoz-Espín et al. 2018).
3.9 SASP Modulation
Another way to fight the detrimental effects of senescent cells is to attenuate their inflammatory secretions. The hundreds of cytokines, chemokines, growth factors and metalloproteases come mainly from two distinct pathways: NF-κB and C/EBPβ (Paez-Ribes et al. 2019). Targeting these pathways or their upstream regulators could help reduce inflammation linked to aging. Rapamycin decreases secretion of interleukin-6 (IL-6) and other inflammatory cytokines by inhibiting their translation (Herranz et al. 2015; Laberge et al. 2015). MAPK pathway inhibitors, such as ginsenosides, were able to suppress the SASP in senescent astrocytes (Hou et al. 2018) or hematopoietic stem cells (Tang et al. 2015). The NF-κB pathway could also be inhibited by the antidiabetic drug metformin, preventing the expression of several SASP cytokines (Moiseeva et al. 2013; Oubaha et al. 2016). Resveratrol, a polyphenol, was able to inhibit the SASP through SIRT1/NF-κB pathway on melanoma cells (Menicacci et al. 2017) or in the gut of the fish N. guentheri (Liu et al. 2018). Administration of a JAK inhibitor, ruxolitinib, reduced inflammation in aged mice by down-regulating the C/EBPβ pathway (Xu et al. 2015). Glucocorticoids such as corticosterone or cortisol were shown to suppress IL-6 as well as several other SASP components. They inhibit IL-1α signaling upstream of NF-κB, which in turn, stimulates the expression of IL-1α, in a positive feedback loop (Laberge et al. 2012).
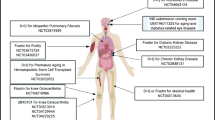
4 Clinical Trials and Future Directions
Since aging is still not recognized as a disease, very few of the aforementioned senolytics have made their ways to clinical studies. Unity biotechnology is currently testing the effect of a compound called UBX0101 on osteoarthritis. After a successful phase I (NCT03513016) they are recruiting patients for a phase II trial (NCT04129944). UBX0101 is supposed to eliminate senescent cells that accumulate in joints, which should decrease local inflammation and alleviate the pain. The Mayo clinic is testing the cocktail D + Q in 2 different clinical trials. One is aimed at chronic kidney diseases (NCT02848131) and the other one is targeted against Alzheimer’s disease (NCT04063124). Mayo clinic is also investigating the senolytic effect of fisetin on frail elderly syndrome (NCT03430037). They are currently recruiting for phase II on these 3 studies. A preliminary report of the phase I trial with D + Q in 9 patients with diabetic kidney disease claims a reduction in adipose tissue senescent cells burden 11 days after completion of a 3-days senolytic treatment (Hickson et al. 2019). If confirmed in a large number of patients, this study suggests that all the beneficial effects observed in mice treated with senolytics will be also attained in humans.
References
Ablain J, Rice K, Soilihi H, de Reynies A, Minucci S, de The H (2014) Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat Med 20(2):167–174
Aguayo-Mazzucato C, Andle J, Lee TB, Jr., Midha A, Talemal L, Chipashvili V, et al (2019) Acceleration of beta cell aging determines diabetes and senolysis improves disease outcomes. Cell Metabolism 30(1):129–42 e4
Aguayo-Mazzucato C, Andle J, Lee TB, Midha A, Talemal L, Chipashvili V, et al (2019) Acceleration of β cell aging determines diabetes and senolysis improves disease outcomes. Cell Metabolism 30(1):129–42 e4
Aksoy O, Chicas A, Zeng T, Zhao Z, McCurrach M, Wang X et al (2012) The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev 26(14):1546–1557
Aziz MH, Shen H, Maki CG (2011) Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 30(46):4678–4686
Azmi AS, Wang Z, Philip PA, Mohammad RM, Sarkar FH (2011) Emerging Bcl-2 inhibitors for the treatment of cancer. Expert Opin Emerg Drugs 16(1):59–70
Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM et al (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to Chemotoxicity and aging. Cell 169(1):132–47.e16
Baar MP, Perdiguero E, Munoz-Canoves P, de Keizer PL (2018) Musculoskeletal senescence: a moving target ready to be eliminated. Curr Opin Pharmacol 40:147–155
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J et al (2016) Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530(7589):184–189
Balint B, Yin H, Nong Z, Arpino J-M, O’Neil C, Rogers SR et al (2019) Seno-destructive smooth muscle cells in the ascending aorta of patients with bicuspid aortic valve disease. EBioMedicine 43:54–66
Bandyopadhyay D, Gatza C, Donehower LA, Medrano EE (2005) Analysis of cellular senescence in culture in vivo: the senescence-associated beta-galactosidase assay. Current protocols in cell biology/editorial board, Juan S Bonifacino [et al. Chapter 18: Unit 18 9
Barnes PJ, Baker J, Donnelly LE (2019) Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med
Berkenkamp B, Susnik N, Baisantry A, Kuznetsova I, Jacobi C, Sörensen-Zender I et al (2014) In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS ONE 9(2):e88071
Bestilny LJ, Gill MJ, Mody CH, Riabowol KT (2000) Accelerated replicative senescence of the peripheral immune system induced by HIV infection. AIDS. 14(7):771–780
Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU et al (2012) Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 7(9):e45069
Biran A, Zada L, Karam PA, Vadai E, Roitman L, Ovadya Y et al (2017) Quantitative identification of senescent cells in aging and disease. Aging Cell 16(4):661–671
Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S et al (2018) Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem Biol 25(1):78–87.e5
Bourdeau V, Baudry D, Ferbeyre G (2009) PML links aberrant cytokine signaling and oncogenic stress to cellular senescence. Front Biosci 14:475–485
Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM et al (2013a) Monitoring tumorigenesis and senescence in vivo with a p16INK4a-luciferase model. Cell 152:340–351
Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM et al (2013b) Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 152(1–2):340–351
Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E (2016) Clinical overview of MDM2/X-Targeted therapies. Front Oncol 6:7
Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ (2018) Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562(7728):578–582
Castro P, Giri D, Lamb D, Ittmann M (2003) Cellular senescence in the pathogenesis of benign prostatic hyperplasia. Prostate 55(1):30–38
Castro P, Xia C, Gomez L, Lamb DJ, Ittmann M (2004) Interleukin-8 expression is increased in senescent prostatic epithelial cells and promotes the development of benign prostatic hyperplasia. Prostate 60(2):153–159
Chalan P, van den Berg A, Kroesen B-J, Brouwer L, Boots A (2015) Rheumatoid arthritis, immunosenescence and the hallmarks of aging. Curr Aging Sci 8(2):131–146
Chang J, Wang Y, Shao L, Laberge R-M, Demaria M, Campisi J et al (2016) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22(1):78–83
Childs BG, Durik M, Baker DJ, van Deursen JM (2015) Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21(12):1424–1435
Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM (2016) Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (New York, NY) 354(6311):472–477
Chinta SJ, Lieu CA, Demaria M, Laberge RM, Campisi J, Andersen JK (2013) Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson’s disease? J Intern Med 273(5):429–436
Chkhotua AB, Gabusi E, Altimari A, D’Errico A, Yakubovich M, Vienken J et al (2003) Increased expression of p16(INK4a) and p27(Kip1) cyclin-dependent kinase inhibitor genes in aging human kidney and chronic allograft nephropathy. Am J Kidney Dis 41(6):1303–1313
Choi J, Shendrik I, Peacocke M, Peehl D, Buttyan R, Ikeguchi EF et al (2000) Expression of senescence-associated beta-galactosidase in enlarged prostates from men with benign prostatic hyperplasia [In Process Citation]. Urology 56(1):160–166
Davaapil H, Brockes JP, Yun MH (2017) Conserved and novel functions of programmed cellular senescence during vertebrate development. Development (Cambridge, England). 144(1):106–114
de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G et al (2004) PML is a direct p53 target that modulates p53 effector functions. Mol Cell 13(4):523–535
Deschenes-Simard X, Gaumont-Leclerc MF, Bourdeau V, Lessard F, Moiseeva O, Forest V et al (2013) Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev 27(8):900–915
Deschenes-Simard X, Parisotto M, Rowell MC, Le Calve B, Igelmann S, Moineau-Vallee K et al (2019) Circumventing senescence is associated with stem cell properties and metformin sensitivity. Aging Cell 18(2):e12889
Dhar SK, St Clair DK (2009) Nucleophosmin blocks mitochondrial localization of p53 and apoptosis. J Biol Chem 284(24):16409–16418
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C et al (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444(7119):638–642
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C et al (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92(20):9363–9367
Dörr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Däbritz JHM et al (2013) Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501:421
Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, Aaronson SA (1999) p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene 18(18):2789–2797
Farr JN, Khosla S (2019) Cellular senescence in bone. Bone 121:121–133
Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL et al (2017) Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 23(9):1072–1079
Ferbeyre G (2018) Aberrant signaling and senescence associated protein degradation. Exp Gerontol 107:50–54
Fessler J, Husic R, Schwetz V, Lerchbaum E, Aberer F, Fasching P et al (2018) Senescent T-cells promote bone loss in rheumatoid arthritis. Front Immunol 9:95
Franceschi C, Garagnani P, Morsiani C, Conte M, Santoro A, Grignolio A et al (2018) The continuum of aging and age-related diseases: common mechanisms but different rates. Front Med (Lausanne) 5:61
Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW et al (2017) Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun 8(1):422
Fülöp T, Herbein G, Cossarizza A, Witkowski JM, Frost E, Dupuis G et al (2017) Cellular senescence, Immunosenescence and HIV. Interdiscip Top Gerontol Geriatr 42:28–46
Garrido AM, Bennett M (2016) Assessment and consequences of cell senescence in atherosclerosis. Curr Opin Lipidol 27(5):431–438
Garwood CJ, Simpson JE, Al Mashhadi S, Axe C, Wilson S, Heath PR et al (2014) DNA damage response and senescence in endothelial cells of human cerebral cortex and relation to Alzheimer’s neuropathology progression: a population-based study in the Medical Research Council Cognitive Function and Ageing Study (MRC-CFAS) cohort. Neuropathol Appl Neurobiol 40(7):802–814
Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R et al (2019) Cardiac glycosides are broad-spectrum senolytics. Nature Metabolism 1(11):1074–1088
Gutierrez-Reyes G, del Carmen Garcia de Leon M, Varela-Fascinetto G, Valencia P, Perez Tamayo R, Rosado CG, et al (2010) Cellular senescence in livers from children with end stage liver disease. PloS One 5(4):e10231
Guzik TJ, Touyz RM (2017) Oxidative Stress, inflammation, and vascular aging in hypertension. Hypertension 70(4):660–667
Hagn F, Klein C, Demmer O, Marchenko N, Vaseva A, Moll UM et al (2010) BclxL changes conformation upon binding to wild-type but not mutant p53 DNA binding domain. J Biol Chem 285(5):3439–3450
Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene-induced DNA damage model for cancer development. Science 319(5868):1352–1355
He S, Sharpless NE (2017) Senescence in health and disease. Cell 169(6):1000–1011
Hedrington MS, Davis SN (2018) Peroxisome proliferator-activated receptor alpha-mediated drug toxicity in the liver. Expert Opin Drug Metab Toxicol. 14(7):671–677
Hennika T, Hu G, Olaciregui NG, Barton KL, Ehteda A, Chitranjan A et al (2017) Pre-clinical study of Panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS ONE 12(1):e0169485
Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM (2006a) Cellular senescence in aging primates. Science 311(5765):1257
Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM (2006b) Cellular senescence in aging primates. Science (New York, NY). 311(5765):1257
Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ et al (2015) mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17(9):1205–1217
Hewitt G, Jurk D, Marques FDM, Correia-Melo C, Hardy T, Gackowska A et al (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nature Commun 3:708
Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK et al (2019) Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47:446–456
Hines J, Lartigue S, Dong H, Qian Y, Crews CM (2019) MDM2-Recruiting PROTAC offers superior, synergistic Antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Can Res 79(1):251–262
Hou J, Cui C, Kim S, Sung C, Choi C (2018) Ginsenoside F1 suppresses astrocytic senescence-associated secretory phenotype. Chem Biol Interact 283:75–83
Huang B, Vassilev LT (2009) Reduced transcriptional activity in the p53 pathway of senescent cells revealed by the MDM2 antagonist nutlin-3. Aging (Albany NY) 1(10):845–854
Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM et al (2006) Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443(7110):421–426
Jaul E, Barron J (2017) Age-related diseases and clinical and public health implications for the 85 years old and over population. Front Public Health 5:335
Jeon OH, Kim C, Laberge R-M, Demaria M, Rathod S, Vasserot AP et al (2017) Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23(6):775
Jeyapalan JC, Sedivy JM (2008) Cellular senescence and organismal aging. Mech Ageing Dev 129(7–8):467–474
Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U (2007) Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev 128(1):36–44
Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK et al (2019) Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40:554–563
Karin O, Agrawal A, Porat Z, Krizhanovsky V, Alon U (2019) Senescent cells and the dynamics of aging. bioRxiv, 470500
Kashyap D, Sharma A, Sak K, Tuli HS, Buttar HS, Bishayee A (2018) Fisetin: a bioactive phytochemical with potential for cancer prevention and pharmacotherapy. Life Sci 194:75–87
Kim KM, Noh JH, Bodogai M, Martindale JL, Yang X, Indig FE et al (2017) Identification of senescent cell surface targetable protein DPP4. Genes Dev 31(15):1529–1534
Kirkland JL, Tchkonia T (2017) Cellular senescence: a translational perspective. EBioMedic 21:21–28
Kishi S, Bayliss PE, Uchiyama J, Koshimizu E, Qi J, Nanjappa P et al (2008) The identification of zebrafish mutants showing alterations in senescence-associated biomarkers. PLoS Genet 4(8):e1000152
Kortlever RM, Higgins PJ, Bernards R (2006) Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol 8(8):877–884
Kovacovicova K, Skolnaja M, Heinmaa M, Mistrik M, Pata P, Pata I, et al (2018) Senolytic Cocktail Dasatinib + Quercetin (D + Q) does not enhance the efficacy of senescence-inducing chemotherapy in liver cancer. Front Oncol 8:459
Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA (2013) A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ 20(4):576–588
Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S et al (2006) p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443(7110):453–457
Kumar R, Sharma A, Kumari A, Gulati A, Padwad Y, Sharma R (2019) Epigallocatechin gallate suppresses premature senescence of preadipocytes by inhibition of PI3K/Akt/mTOR pathway and induces senescent cell death by regulation of Bax/Bcl-2 pathway. Biogerontology 20(2):171–189
Kurz DJ, Decary S, Hong Y, Erusalimsky JD (2000) Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 113(Pt 20):3613–3622
Laberge RM, Zhou L, Sarantos MR, Rodier F, Freund A, de Keizer PL et al (2012) Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 11(4):569–578
Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L et al (2015) MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17(8):1049–1061
Langford SD, Boor PJ (1996) Oleander toxicity: an examination of human and animal toxic exposures. Toxicology 109(1):1–13
Lanna A, Henson SM, Escors D, Akbar AN (2014) The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat Immunol 15(10):965–972
Laubach JP, Moreau P, San-Miguel JF, Richardson PG (2015) Panobinostat for the treatment of multiple Myeloma. Clin Cancer Res 21(21):4767–4773
Lessel D, Wu D, Trujillo C, Ramezani T, Lessel I, Alwasiyah MK et al (2017) Dysfunction of the MDM2/p53 axis is linked to premature aging. J Clin Investig 127(10):3598–3608
Li Y, Yao J, Han C, Yang J, Chaudhry MT, Wang S, et al (2016) Quercetin, Inflammation and Immunity. Nutrients 8(3):167
Li W, He Y, Zhang R, Zheng G, Zhou D (2019) The curcumin analog EF24 is a novel senolytic agent. Aging (Albany NY) 11(2):771–782
Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X et al (2005) Genomic instability in laminopathy-based premature aging. Nat Med 11(7):780–785
Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG et al (2009) Expression of p16INK4a in peripheral blood T-cells is a biomarker of human aging. Aging Cell 8(4):439–448
Liu X, Wang Y, Zhang X, Gao Z, Zhang S, Shi P et al (2018a) Senolytic activity of piperlongumine analogues: Synthesis and biological evaluation. Bioorg Med Chem 26(14):3925–3938
Liu S, Zheng Z, Ji S, Liu T, Hou Y, Li S et al (2018b) Resveratrol reduces senescence-associated secretory phenotype by SIRT1/NF-kappaB pathway in gut of the annual fish Nothobranchius guentheri. Fish Shellfish Immunol 80:473–479
Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW (2005) DNA repair, genome stability, and aging. Cell 120(4):497–512
Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y et al (2017) Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov 7(5):522–538
Lopes-Paciencia S, Saint-Germain E, Rowell MC, Ruiz AF, Kalegari P, Ferbeyre G (2019) The senescence-associated secretory phenotype and its regulation. Cytokine 117:15–22
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153(6):1194–1217
Macip S, Igarashi M, Berggren P, Yu J, Lee SW, Aaronson SA (2003) Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol Cell Biol 23(23):8576–8585
Majdi A, Mahmoudi J, Sadigh-Eteghad S, Golzari SEJ, Sabermarouf B, Reyhani-Rad S (2016) Permissive role of cytosolic pH acidification in neurodegeneration: A closer look at its causes and consequences. J Neurosci Res 94(10):879–887
Maldonado JL, Timmerman L, Fridlyand J, Bastian BC (2004) Mechanisms of cell-cycle arrest in Spitz nevi with constitutive activation of the MAP-kinase pathway. Am J Pathol 164(5):1783–1787
Mallette FA, Ferbeyre G (2007) The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell cycle (Georgetown, Tex 6(15):1831–6
Mallette FA, Gaumont-Leclerc MF, Ferbeyre G (2007) The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev 21(1):43–48
Martins R, Lithgow GJ, Link W (2016) Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15(2):196–207
Martyanov V, Whitfield ML, Varga J (2019) Senescence signature in skin biopsies from systemic sclerosis patients treated with senolytic therapy: potential predictor of clinical response? Arthritis Rheumatol
Melk A, Schmidt BMW, Takeuchi O, Sawitzki B, Rayner DC, Halloran PF (2004) Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int 65(2):510–520
Mendelsohn AR, Larrick JW (2018) Cellular senescence as the key intermediate in tau-mediated neurodegeneration. Rejuvenation Res 21(6):572–579
Menicacci B, Laurenzana A, Chilla A, Margheri F, Peppicelli S, Tanganelli E et al (2017) Chronic Resveratrol treatment inhibits MRC5 Fibroblast SASP-related protumoral effects on melanoma cells. J Gerontol A Biol Sci Med Sci 72(9):1187–1195
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM et al (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436(7051):720–724
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P et al (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11(3):577–590
Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I (2002) Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105(13):1541–1544
Moiseeva O, Bourdeau V, Roux A, Deschenes-Simard X, Ferbeyre G (2009) Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol 29(16):4495–4507
Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE et al (2013) Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 12(3):489–498
Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J et al (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443(7110):448–452
Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S et al (2013) Programmed cell senescence during mammalian embryonic development. Cell 155(5):1104–1118
Muñoz-Espín D, Rovira M, Galiana I, Giménez C, Lozano-Torres B, Paez-Ribes M et al (2018) A versatile drug delivery system targeting senescent cells. EMBO Molecul Medic 10(9):e9355
Nakamura S, Nishioka K (2003) Enhanced expression of p16 in seborrhoeic keratosis; a lesion of accumulated senescent epidermal cells in G1 arrest. BC J Dermatol 149(3):560–565
Nguyen MN, Sen N, Lin M, Joseph TL, Vaz C, Tanavde V et al (2019) Discovering putative protein targets of small molecules: a study of the p53 activator nutlin. J Chem Inf Model 59(4):1529–1546
Nogueira-Recalde U, Lorenzo-Gomez I, Blanco FJ, Loza MI, Grassi D, Shirinsky V et al (2019) Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 45:588–605
Noureddine H, Gary-Bobo G, Alifano M, Marcos E, Saker M, Vienney N et al (2011) Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ Res 109(5):543–553
Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al (2017) Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 8:15691
Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, Germain M-A, et al (2016) Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Medic 8(362):362ra144
Paez-Ribes M, Gonzalez-Gualda E, Doherty GJ, Munoz-Espin D (2019) Targeting senescent cells in translational medicine. EMBO Mol Med, e10234
Palacio L, Goyer ML, Maggiorani D, Espinosa A, Villeneuve N, Bourbonnais S et al (2019) Restored immune cell functions upon clearance of senescence in the irradiated splenic environment. Aging Cell 18(4):e12971
Palmer AK, Tchkonia T, LeBrasseur NK, Chini EN, Xu M, Kirkland JL (2015) Cellular senescence in type 2 diabetes: a therapeutic opportunity. Diabetes 64(7):2289–2298
Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM et al (2019) Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18(3):e12950
Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M et al (2017) Inhibition of Bcl-2/xl With ABT-263 selectively kills senescent Type II Pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Phys 99(2):353–361
Papatheodoridi A-M, Chrysavgis L, Koutsilieris M, Chatzigeorgiou A (2019) The role of senescence in the development of non-alcoholic fatty liver disease and progression to non-alcoholic steatohepatitis. Hepatology (Baltimore, Md)
Paradis V, Youssef N, Dargere D, Ba N, Bonvoust F, Deschatrette J et al (2001) Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum Pathol 32(3):327–332
Patil P, Dong Q, Wang D, Chang J, Wiley C, Demaria M et al (2019) Systemic clearance of p16(INK4a) -positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell 18(3):e12927
Petersen LE, Schuch JB, de Azeredo LA, Baptista TSA, Motta JG, do Prado AD, et al (2019) Characterization of senescence biomarkers in rheumatoid arthritis: relevance to disease progression. Clin Rheumatol
Qian Y, Chen X (2010) Tumor suppression by p53: making cells senescent. Histol Histopathol 25(4):515–526
Qudrat A, Wong J, Truong K (2017) Engineering mammalian cells to seek senescence-associated secretory phenotypes. J Cell Sci 130(18):3116–3123
Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL et al (2012) Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol 30(5):488–496
Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM et al (2016) Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15(5):973–977
Rotter Sopasakis V, Sandstedt J, Johansson M, Lundqvist A, Bergström G, Jeppsson A, et al (2019) Toll-like receptor-mediated inflammation markers are strongly induced in heart tissue in patients with cardiac disease under both ischemic and non-ischemic conditions. Int J Cardiol
Saeed H, Abdallah BM, Ditzel N, Catala-Lehnen P, Qiu W, Amling M et al (2011) Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. J Bone Miner Res 26(7):1494–1505
Sahin E, Depinho RA (2010) Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464(7288):520–528
Samaraweera L, Adomako A, Rodriguez-Gabin A, McDaid HM (2017) A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Scie Reports 7(1):1900
Sasaki Y, Ikeda Y, Miyauchi T, Uchikado Y, Akasaki Y, Ohishi M (2019) Estrogen-SIRT1 axis plays a pivotal role in protecting arteries against menopause-induced senescence and atherosclerosis. J Atheroscler Thromb
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ et al (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8:14532
Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC (2004) Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol 6(2):168–170
Shimizu I, Minamino T (2019) Cellular senescence in cardiac diseases. J Cardiol
Snijders T, Parise G (2017) Role of muscle stem cells in sarcopenia. Curr Opin Clin Nutrition Metabolic Care 20(3):186–190
Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V et al (2014a) Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506(7488):316–321
Sousa-Victor P, Perdiguero E, Munoz-Canoves P (2014) Geroconversion of aged muscle stem cells under regenerative pressure. Cell cycle (Georgetown, Tex. 13(20):3183–90
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V et al (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155(5):1119–1130
Tang YL, Zhou Y, Wang YP, Wang JW, Ding JC (2015) SIRT6/NF-κB signaling axis in ginsenoside Rg1-delayed hematopoietic stem/progenitor cell senescence. Int J Clin Exp Pathol 8(5):5591–5596
Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H et al (2010) Fat tissue, aging, and cellular senescence. Aging Cell 9(5):667–684
Thorin E, Thorin-Trescases N (2009) Vascular endothelial ageing, heartbeat after heartbeat. Cardiovasc Res 84(1):24–32
Triana-Martinez F, Picallos-Rabina P, Da Silva-Alvarez S, Pietrocola F, Llanos S, Rodilla V et al (2019) Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat Commun 10(1):4731
Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP (2006) Identification of a tumour suppressor network opposing nuclear Akt function. Nature 441(7092):523–7
Valentijn FA, Falke LL, Nguyen TQ, Goldschmeding R (2018) Cellular senescence in the aging and diseased kidney. J Cell Commun Signal 12(1):69–82
Van Veggel M, Westerman E, Hamberg P (2018) Clinical pharmacokinetics and pharmacodynamics of Panobinostat. Clin Pharmacokinet 57(1):21–29
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science (New York, NY) 303(5659):844–848
Vernier M, Bourdeau V, Gaumont-Leclerc MF, Moiseeva O, Begin V, Saad F et al (2011) Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev 25(1):41–50
Villiard E, Denis JF, Hashemi FS, Igelmann S, Ferbeyre G, Roy S (2017) Senescence gives insights into the morphogenetic evolution of anamniotes. Biol Open 6(6):891–896
Voghel G, Thorin-Trescases N, Mamarbachi AM, Villeneuve L, Mallette FA, Ferbeyre G et al (2010) Endogenous oxidative stress prevents telomerase-dependent immortalization of human endothelial cells. Mech Ageing Dev 131(5):354–363
Walaszczyk A, Dookun E, Redgrave R, Tual-Chalot S, Victorelli S, Spyridopoulos I et al (2019) Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 18(3):e12945
Wang E (1995) Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 55(11):2284–2292
Wang AS, Dreesen O (2018) Biomarkers of cellular senescence and skin aging. Front Genet 9:247
Wang Y, Chang J, Liu X, Zhang X, Zhang S, Zhang X et al (2016) Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY) 8(11):2915–2926
Watson N, Ding B, Zhu X, Frisina RD (2017) Chronic inflammation—inflammaging—in the ageing cochlea: a novel target for future presbycusis therapy. Ageing Res Rev 40:142–148
Wiemann SU, Satyanarayana A, Tsahuridu M, Tillmann HL, Zender L, Klempnauer J et al (2002) Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J 16(9):935–942
Wu D, Prives C (2018) Relevance of the p53–MDM2 axis to aging. Cell Death Differ 25(1):169–179
Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T et al (2015) JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci 112(46):E6301–E6310
Xu M, Bradley EW, Weivoda MM, Hwang SM, Pirtskhalava T, Decklever T et al (2016) Transplanted senescent cells induce an osteoarthritis-like condition in mice. J Gerontol A Biol Sci Med Sci 72(6):780–785
Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM et al (2018) Senolytics improve physical function and increase lifespan in old age. Nat Med 24(8):1246–1256
Yang M, Luna L, Sorbo JG, Alseth I, Johansen RF, Backe PH et al (2014) Human OXR1 maintains mitochondrial DNA integrity and counteracts hydrogen peroxide-induced oxidative stress by regulating antioxidant pathways involving p21. Free Radic Biol Med 77:41–48
Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S et al (2016) Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7:11190
Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S et al (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499(7456):97–101
Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M et al (2018) Fisetin is a senotherapeutic that extends health and lifespan. EBio Medic 36:18–28
Yun MH, Davaapil H, Brockes JP (2015) Recurrent turnover of senescent cells during regeneration of a complex structure. eLife; 4
Zarbock A (2012) The shady side of dasatinib. Blood 119(21):4817–4818
Zhang X, Li CF, Zhang L, Wu CY, Han L, Jin G et al (2016) TRAF6 Restricts p53 mitochondrial translocation, apoptosis, and tumor suppression. Mol Cell 64(4):803–814
Zhang X, Zhang S, Liu X, Wang Y, Chang J, Zhang X, et al (2018) Oxidation resistance 1 is a novel senolytic target. Aging Cell, e12780
Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S et al (2019) Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci 22(5):719–728
Zhou S, Greenberger JS, Epperly MW, Goff JP, Adler C, LeBoff MS et al (2008) Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell 7(3):335–343
Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N et al (2015) The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14(4):644–658
Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB et al (2016) Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15(3):428–435
Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H et al (2017) New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY) 9(3):955–963
Acknowledgements
We thank members of the Ferbeyre laboratory for their critical insights reading this manuscript. G.F. is supported by the CIBC chair for breast cancer research at the CRCHUM.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Buffard, T., Ferbeyre, G. (2020). Senolytics Target Senescent Cells and Improve Aging and Age-Related Diseases. In: Muñoz-Espin, D., Demaria, M. (eds) Senolytics in Disease, Ageing and Longevity. Healthy Ageing and Longevity, vol 11. Springer, Cham. https://doi.org/10.1007/978-3-030-44903-2_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-44903-2_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-44902-5
Online ISBN: 978-3-030-44903-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)