
Abstract
Secondary hyperparathyroidism (SHPT) is an integral component of chronic kidney disease-mineral and bone disorder (CKD-MBD). Many factors have been associated with its development and progression, but the presence of skeletal or calcemic resistance to the action of PTH in CKD has often gone unnoticed. The term hyporesponsiveness to PTH is currently preferred and, in this chapter, we will review not only the scientific history but also the factors linked to this impaired response to PTH in CKD. Decreased calcitriol levels, phosphate retention, down-regulation of PTH receptors, uremia per se, abnormal metabolism of PTH and PTH fragments, downstream competing signals, local factors (inflammation, cytokines, oxidative stress, ion concentration etc.), and systemic factors (age, race, diabetes, fibroblast growth factor-23, klotho, Wnt antagonists such as sclerostin, etc.) have all been implicated. Additionally, important clinical implications are reviewed since hyporesponsiveness to PTH explains not only progressive PTH hypersecretion and parathyroid hyperplasia but also the increasing prevalence of adynamic bone disease in the CKD population. Therefore, hyporesponsiveness to PTH should be taken into account when treating SHPT in CKD patients, and its significance underlines the importance of avoiding the normalization of PTH levels at any CKD stage. On the other hand, progressively increasing PTH levels may indicate a change from an adaptive to a maladaptive clinical situation requiring therapeutic decisions. Finally, resistance to the biological action of several other hormones is a well-known feature of CKD (uremia as a “receptor disease”), and consequently future additional studies at cellular and molecular levels may be of value beyond their significance for hyporesponsiveness to PTH.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- PTH
- Skeletal resistance
- Calcemic response
- Hyporesponsiveness
- Secondary hyperparathyroidism
- Adynamic bone disease
- Phosphate
- Calcitriol
- Down-regulation
Introduction
Chronic kidney disease (CKD) is an important global health problem, involving about 10% of the population worldwide and entailing high cardiovascular and mortality risks [1, 2]. CKD-mineral and bone disorder (CKD-MBD) is one of the many complications associated with CKD and represents a systemic disorder manifested by either one or a combination of: (a) abnormalities of calcium (Ca), phosphate (P), parathyroid hormone (PTH ), or vitamin D metabolism; (b) abnormalities in bone turnover, mineralization, volume, linear growth, or strength (formerly known as renal osteodystrophy); and (c) vascular or other soft tissue calcifications [3,4,5]. Consequently, renal osteodystrophy is currently considered one measure of the skeletal component of CKD-MBD, which is quantifiable by bone histomorphometry, and recent knowledge indicates bone to be an endocrine organ at the heart of metabolic and cardiovascular complications of CKD [6].
Increased PTH levels [classically referred to as secondary hyperparathyroidism (SHPT)] are an integral component of the CKD-MBD syndrome and if they remain uncontrolled, they will worsen the laboratory abnormalities of CKD-MBD (e.g., calcium and phosphate levels), the bone structure (high-turnover bone disease), and cardiovascular parameters (e.g., vascular calcification) [7,8,9]. Moreover, PTH has also classically been considered a “uremic toxin” [10] since the effect of PTH appears not to be limited to bone [11, 12]. Indeed SHPT has been claimed to contribute to many “off-target” extraskeletal clinical manifestations frequently present in CKD [11, 12]. For instance, PTH produces an increase in cytosolic “intracellular calcium” in many different cell types [13, 14] and this may be a common pathway that explains the relationship of PTH with many other systemic effects such as cognitive decline, cachexia, peripheral neuropathy, increasing osmotic fragility of erythrocytes, abnormalities of the immune system, muscular dysfunction and wasting, decreased testosterone and increased prolactin serum levels, carbohydrate intolerance and lipid abnormalities, renin-angiotensin-aldosterone system stimulation, myocardial dysfunction, and cardiac hypertrophy and fibrosis [11, 12, 15,16,17]. The improvement of some of these deleterious outcomes (e.g., left ventricular hypertrophy) following parathyroidectomy (PTX) supports the hypothesis of a causal link with PTH, although evidence of a direct role is still lacking [12, 18]. The above-mentioned toxic effects of PTH may explain the association of SHPT with CKD progression and atheromatous and non-atheromatous cardiovascular disease, as well as with all-cause and/or cardiovascular morbidity and mortality [19,20,21,22,23].
SHPT is classically considered a common, serious, and progressive complication of CKD. In the United States, the estimated prevalence of SHPT in CKD patients ranges from 2 to nearly 5 million individuals, with 30–50% of dialysis patients being affected with SHPT [24]. However, very low or relatively low PTH levels (which are associated with adynamic bone disease ) are also considered an important contributory factor to the high morbidity and mortality in CKD patients [20, 25]. An increased risk of fractures and cardiovascular calcifications have been related to low PTH levels and is attributable to the inability of bone to buffer an excess of Ca and P loading [26]. Therefore, it is not surprising that associations between mortality and PTH are recognized to be complex, except at both extremes (high and low) of serum PTH levels [27]. Similarly, cohort studies have clearly indicated that only extreme levels are able to predict with acceptable sensitivity/specificity the bone turnover status [from adynamic bone disease (low-turnover) to osteitis fibrosa (high-turnover bone disease) [4, 28]. Although PTH is a crucial determinant of bone remodeling [12], in uremic conditions it does not offer information on bone properties and strength, similarly to most bone markers [12, 29, 30]. Beyond the CKD population, investigations of cohorts of primarily cardiology patients have confirmed the independent association between high PTH levels, cardiovascular events, and mortality (including sudden cardiac death) [31,32,33].
Pathophysiology of Secondary Hyperparathyroidism
It is well known that progressive loss of kidney function leads to an increased P load (retention) [34, 35], increased fibroblast growth factor 23 (FGF23) and decreased α-klotho [36,37,38], and decreased synthesis and increased catabolism of calcitriol (1,25-dihydroxyvitamin D; active vitamin D), among many other metabolic derangements [15, 39]. All these factors, extensively reviewed elsewhere [15, 40, 41], involve various closely interacting mechanisms, including the down-regulation of Ca-sensing, vitamin D, and FGF23 receptors, and contribute in increasing the synthesis and secretion of PTH by the parathyroid glands as well as the proliferation (polyclonal and/or monoclonal) of parathyroid cells. It is well known that hyperplasia of the parathyroid glands starts to develop during the very early stages of CKD [11, 15, 42, 43].
Although it is currently known that P can both directly and indirectly induce SHPT in patients with CKD [44,45,46], it is widely accepted that Ca is the most important regulator of parathyroid gland function [12]. In order to correct hypocalcemia, rapid release of stored PTH is necessary, with increased synthesis and, eventually, parathyroid cell proliferation if required to produce the necessary amount of PTH to correct Ca. Thus, most of the previously mentioned mechanisms (e.g., P retention, decreased calcitriol production) share in common the capability to lead to secondary multifactorial hypocalcemia and hence induce SHPT. In the parathyroid cell, the transmembrane Ca-sensing receptor (CaSR), the complex FGFR1-klotho receptor complex, and the nuclear parathyroid vitamin D receptor (VDR) are down-regulated, resulting in increased PTH synthesis and secretion as well as parathyroid cell proliferation [47, 48]. It is beyond the scope of this review to analyze the complex interrelationships among all these pathophysiologic factors leading to SHPT . Rather, the focus will be on analysis of the importance of skeletal resistance to the action of PTH in the pathogenesis of SHPT and, on the other hand, the development of adynamic bone disease , since this remains greatly underappreciated by the nephrology community.
Skeletal Resistance to PTH Action
“Skeletal” resistance to the action of PTH in CKD (also known as calcemic resistance to PTH or PTH resistance) is an old concept [49], recently recognized as “hyporesponsiveness ” to PTH [27, 50]. In fact, both bone and renal responses to the action of PTH are progressively impaired in CKD [27] and the term “hyporesponsiveness” may be more appropriate because the response to PTH is blunted but not completely absent [27].
A decreased calcemic response to PTH was first described by J. M. Evanson in 1966 [49] when he reported the calcemic response to an infusion of crude parathyroid extract (1 U/kg/h over 10 h) to be significantly lower in 12 hypocalcemic patients with CKD than in normal subjects or patients with primary hyperparathyroidism [49]. He postulated that vitamin D is necessary for the action of PTH on bone, since a decreased calcemic response was also observed in hypocalcemic patients with steatorrhea or rickets [49]. Subsequently, Massry et al. observed that the calcemic response to a parathyroid extract (2 IU/kg over 8 h) in thyroparathyroidectomized (T-PTX) dogs, before and after the induction of uremia by bilateral ureteral ligation (BUL) or by bilateral nephrectomy, was markedly impaired, but that the impairment was more severe one day after nephrectomy [51]. They also observed that the calcemic response after 2 or 3 days of BUL was similar to that seen at one day after nephrectomy and that calcitriol partially restored the calcemic response to PTH [51]. The authors concluded that a deficiency of calcitriol was at least partly responsible for the skeletal resistance to the calcemic action of PTH in the presence of uremia and that uremia, per se, could also have contributed to this phenomenon. In addition, it was observed that the calcemic response to PTH was lower in patients with moderate and advanced CKD (including hemodialysis and renal transplant patients) than in normal healthy subjects [52]. This reduced calcemic response was not related to the initial levels of serum Ca, P, or PTH and was not reversed by hemodialysis [52].
Llach et al. also noted a decreased calcemic response to endogenous PTH and an altered divalent ion metabolism in patients with early CKD [53, 54]. For instance, they described delayed recovery from ethylene diamine tetra-acetic acid (EDTA)-induced hypocalcemia in these patients as compared with normal subjects, despite higher serum PTH levels in the CKD group [53]. Such observations indicated that the impaired calcemic response to PTH appears early in the course of CKD and that a direct consequence is a continuous requirement for a greater concentration of circulating PTH in order to maintain a normal Ca homeostasis in affected patients. Ever since the study by Albright et al. [55] it has been hypothesized that P retention and reciprocal blood Ca lowering in CKD patients might cause parathyroid hyperplasia and renal osteitis fibrosa. With the formulation of the “trade-off” hypothesis by Bricker and Slatopolsky [34, 56,57,58], the reduction in the level of serum Ca in CKD was considered to be the main driver of increased synthesis and secretion of PTH, as well as parathyroid hyperplasia. However, the presence of skeletal resistance to PTH in CKD provides a usually forgotten additional mechanism in the pathogenesis of hypocalcemia and SHPT in CKD [15, 59, 60]. The appearance of skeletal resistance to the action of PTH early in the course of renal failure would induce hypocalcemia, which in turn would stimulate the parathyroid glands, resulting in continuously increased secretion of the hormone and hyperplasia. At the other extreme, the late appearance of detectable hypocalcemia in advanced stages of CKD [61] clearly represents the final failure of PTH to restore Ca levels to normal, and the maximal clinical expression of hyporesponsiveness to PTH.
Factors Linked to Impaired Calcemic Response to PTH (See Table 1)
Decreased Levels of Calcitriol
Since the earliest work in this field it was considered that vitamin D appears to be necessary for the proper action of PTH on bone [49]. Later, it was demonstrated that the administration of calcitriol restored, at least partially, the blunted calcemic response to PTH in experimental animals (rats and dogs) [51, 62, 63] leading to the conclusion that this impaired calcemic response to PTH is related to the decreased calcitriol levels observed in early CKD. Also, in patients with early CKD, daily administration of 0.5 µg of calcitriol for 6 weeks improved the calcemic response to PTH [54]. Finally, studies in experimental animals also exhibited complete correction of the calcemic response to PTH after the administration of calcitriol [1,25(OH)2D3] together with 24,25(OH)2D3 [64]. Consequently, although an adequate mechanistic explanation for these observations is still lacking, it seems clear that reduced serum vitamin D levels play a role in the impaired calcemic response to PTH, and that vitamin D could enhance the action of PTH on bone. However, other investigators did not confirm this beneficial effect of calcitriol on the calcemic response to PTH [62, 65]. There is also no scientific evidence demonstrating that the correction of native vitamin D deficiency (25OHD < 15 ng/ml), which is often the case in CKD, by restoring serum 25OHD levels, would improve the calcemic response to PTH.
Phosphate Retention
It is also well known that P retention significantly decreases the calcemic response to PTH in CKD [63, 66]. Somerville and Kaye reinfused urine into T-PTX rats, inducing acute uremia in the presence of normal kidneys, and observed that the calcemic response to PTH after 5 h of PTH extract infusion was clearly blunted. Removal of P by treating urine with zirconium oxide completely restored the calcemic response to PTH [66]. Moreover, other groups of T-PTX rats given PTH extract were infused with an electrolyte solution containing varying amounts of P up to a maximum similar to the amount that a urine-infused rat would receive. A highly significant inverse relationship was found between the dose of P infused with the electrolyte solution and the measured calcemic response to PTH [66]. Since P and calcitriol are also closely interrelated (P inhibits the synthesis of calcitriol), it could not be excluded that at least some of these observations might be mediated indirectly by inhibition of calcitriol. Thus, using a different model, Rodriguez et al. [63] demonstrated that rats with CKD fed with a low-P diet exhibited an improvement in the calcemic response during a constant 48-h infusion of 1–34 rat PTH with an ALZET® pump; moreover, rats with either moderate or advanced CKD had an impaired calcemic response to PTH and low levels of calcitriol . The low-P diet enhanced the calcemic response to PTH in both groups of rats, but only those with moderate CKD had a significant increment in calcitriol levels. Thus, with moderate CKD, P restriction improved the calcemic response to PTH, and this effect could be due to higher levels of calcitriol. However, in advanced CKD, the calcemic response to PTH improved with P restriction independently of calcitriol . As a matter of fact, we observed that the negative effect of P retention on the calcemic response to PTH may be far superior to the effect of calcitriol deficiency in rats with CKD [67, 68] (See Fig. 1).
Improvement of the calcemic response to a standardized infusion of PTH after P restriction has also been demonstrated in patients with mild CKD [69]. Interestingly, in rats with normal renal function that received a high-P diet, the calcemic response to PTH was reduced in the absence of any change in serum P levels; this indicates that the diet itself was responsible for the reduction in response. This is an issue that should be taken into consideration currently, given the lack of a clear recommendation on whether dietary P restriction should be applied in CKD stages 2–3. The intrinsic mechanism that leads to a P-induced decrease in the calcemic response to PTH is not completely known but, in addition to the effect of P on calcitriol levels, the ambient P concentration in bone may affect the amount of exchangeable Ca that can be mobilized by PTH [62, 70].
All of the above experiments were done before the discovery of FGF23, and to date the possibility that high FGF23 (which may act on osteoblasts) may reduce the calcemic effect of PTH on bone has not been excluded. It could be that high FGF23 either directly or indirectly (via calcitriol suppression, Dkk1 stimulation or sclerostin) interferes with PTH-mediated Ca efflux from bone. Moreover, multiple binding sites for Ca2+ and PO43− ions have been described at the CaSR [71]. While Ca2+ ions stabilize its active state, PO43− ions reinforce the inactive conformation of the CaSR, thus contributing to increased PTH secretion [71]. A similar inactivating effect of P on the bone CaSR could also be involved in such reduced calcemic response to PTH in CKD.
Although both P restriction and calcitriol supplementation improve the calcemic response to PTH, they do not seem to completely restore it, either alone or together. Conversely, in PTX dogs or rats [63, 65, 72], the removal of circulating PTH corrects the calcemic response to PTH despite the presence of hyperphosphatemia and low calcitriol levels.
Down-Regulation of PTH Receptors
The aforementioned findings suggested that high levels of endogenous PTH levels in PTX uremic animals could have desensitized the skeleton to the administration of exogenous PTH [65, 73]. The role of down-regulation of PTH bone receptors by high PTH levels was established after the observation that the removal of PTH seemed to restore the responsiveness to its receptors. Likewise, it was described in isolated perfused bones from uremic dogs with acute or chronic renal failure that the release of cyclic adenosine monophosphate (cAMP) was blunted in response to PTH, whereas it was restored by T-PTX [74, 75]. Although PTX corrected the calcemic response to PTH in experimental uremia, we noted that holding PTH levels in the normal range in rats with CKD fed with a low-P diet did not correct the calcemic response to PTH [72]. Thus, uremic rats with normal PTH levels, achieved with partial PTX, still showed a 50% decrease in the calcemic response to PTH as compared with normal rats [72]. This is consistent with clinical studies showing that subtotal PTX resulted in almost normal PTH levels but did not enhance the calcemic response to PTH [52]. Similarly, PTX improved the calcemic response to PTH not only in animals with CKD but also in sham-operated rats [72]. Therefore, PTH-induced down-regulation of PTH receptors appeared not to be the sole explanation for the decrease in the calcemic response to PTH in CKD. It was hypothesized that the significant improvement in the calcemic response to PTH after PTX could be due to a phenomenon of hypersensitization following hormone depletion, as described for other hormonal systems, among other potential explanations affecting the exchangeable Ca pool in bone described elsewhere [72, 76].
All of these findings do not exclude down-regulation of PTH receptors as a potential cause of the decreased calcemic response to PTH [77]. Thus, the cloning of a ubiquitous PTH receptor (PTHR) gene—a common receptor for both PTH and PTH-related peptide (PTHrP) [78, 79]—has allowed demonstration that the PTH1R (PTHR-1) is not only widely distributed in tissues [80] but also down-regulated in uremic kidneys, epiphyseal cartilage growth plates, and osteoblasts, at least at the transcription level [81,82,83,84]. Human data are less consistent, with some investigators demonstrating decreased expression [85] while others have recently reported increased expression [86]. Methodological issues and variation in the characteristics of the studied populations may explain this apparent discrepancy [27].
The finding of down-regulation of the PTH1R mRNA in bone tissue and not in the liver or heart suggested that PTH1R expression is regulated in a cell-specific manner regardless of the uremic state [87]. It is likely, however, that factors other than the increased PTH levels down-regulate this receptor [82, 87]. Thus, Ureña et al. showed the expression of PTH1R mRNA to be decreased in kidneys from both rats with CKD and rats with CKD that were subjected to PTX [87]. Since PTX did not restore the expression of PTH1R mRNA, elevation of PTH levels would not be necessary to induce its down-regulation in CKD. Their data also demonstrated that neither an increase in plasma PTH and P nor a decrease in plasma Ca is important in renal PTH1R down-regulation during CKD, and that it is also unlikely that an increase in the locally produced renal PTHrP could down-regulate its own receptor. However, other authors observed increased expression of PTH1R mRNA after PTX, but controls were not available [83].
Although the mechanisms responsible for the putative desensitization or down-regulation of PTH1R in CKD remain very poorly defined, several studies have implicated several uremic factors and C-terminal PTH fragments (see below). The available information on PTH1R regulation is very limited and even contradictory, and beyond the scope of this review [88,89,90,91].
Uremia
Importantly, in experimental rats with CKD we observed a significant decrease in the calcemic response to a 48-h rat 1–34 PTH infusion despite the presence of normal serum Ca, P, calcitriol , and PTH levels. This finding suggests that factors intrinsic to uremia may, per se, impair the calcemic response to PTH [72]. These data were subsequently confirmed in a different model when Berdud et al. [92] observed that maintenance of normal PTH levels in uremic PTX rats by constant infusion of PTH did not correct the impaired calcemic response to PTH.
The presence of unknown uremic factors, beyond P, that are potentially responsible for the decreased calcemic response to PTH in CKD had already been postulated previously [52]. Similarly, Wills and Jenkins had also shown that serum from uremic patients inhibited PTH-induced bone resorption in an in vitro model, whereas serum obtained after dialysis did not [93]. Low molecular weight inhibitors of osteoblast mitogenesis in uremic plasma were described [94], and subsequent experimental studies pointed towards different uremic toxins [95] triggering oxidative stress, such as indoxyl sulfate (IS) and p-cresyl sulfate (PCS) [96, 97] and/or inflammatory bioactive oxidized low-density lipoproteins [98]. Increased oxidative stress and low-degree inflammation are common conditions in CKD, and thus they may be not only causal but also common pathways (e.g., P retention or calcitriol deficiency is also associated with oxidative stress and inflammation) linking CKD and PTH hyporesponsiveness [27]. In vitro studies [99] have shown that the uremic toxin IS inhibits not only osteoblast but also osteoclast differentiation and function, and that these effects are enhanced in the presence of high P concentrations. In vivo studies [100] have also shown that the administration of the oral charcoal adsorbent AST-120 prevented low-turnover bone in uremic rats. Nii-Kono et al. [96] further showed that IS induces a state of PTH resistance, consisting in a reduction of PTH-induced cAMP and PTHR gene expression, and decreases the viability of osteoblasts maintained in culture. These authors also demonstrated that free radical production in osteoblasts increases in relation to the concentration of IS added [96]. Furthermore, their results suggested that IS is taken up by osteoblasts via the organic anion transporter-3, augmenting oxidative stress to impair osteoblast function and down-regulate PTH1R expression [96]. On the other hand, contradictory results have recently been reported by Barreto et al. [101], who showed a positive association of serum IS with osteoblast surface and bone formation rate in 49 CKD stage 2–3 patients (36 ± 17 ml/min/1.73 m2); however, this study may have been largely underpowered to address this question. Although the temporal sequence is unknown, it is possible that early accumulation of uremic toxins induces skeletal PTH hyporesponsiveness and therefore contributes to the initial adaptive increase in PTH (initial biochemical SHPT ) in order to normalize the important serum Ca (and P) levels. At a certain point, the progressive increase in serum PTH levels could then override the described direct inhibitory effects of IS on bone turnover [39]. Finally, uremic toxins may also stimulate PTH secretion indirectly by decreasing calcitriol synthesis and binding to DNA vitamin D response elements [102, 103], inducing resistance to the inhibitory action of calcitriol.
PTH Abnormal Metabolism and Fragments in CKD
An increased rate of PTH secretion is primarily responsible for high plasma levels of PTH in CKD. However, evidence demonstrates that the kidneys play an important role in the degradation of PTH and that, in patients with CKD, the metabolic clearance of PTH, like that of other peptide hormones, is reduced.
PTH is a hormone actively secreted mainly by chief cells of the parathyroid gland as a single-chain polypeptide containing 84 amino acids (1–84 PTH) [104, 105]. The physiological role of the increased number of parathyroid oxyphil cells in CKD or the chief-to-oxyphil transdifferentiation is still largely unknown [106, 107]. As mentioned previously, PTH increases serum Ca by activation of the PTH1R, which is mainly present in bone and kidney but is also found in a variety of tissues not regarded as classic PTH targets. This explains the widespread effects of PTH and illustrates why PTH is considered to be a clinically relevant uremic toxin, at least in patients with advanced CKD [11].
The amino-terminal extreme of PTH is required for activation of adenyl-cyclase. The PTH1R, coupled to G-proteins, is activated equivalently by 1–84 PTH, amino-terminal PTH, and PTHrP.
PTH is physiologically released in episodic secretory bursts that are superimposed on a basal, tonic mode of secretion [108], and this pulsatile secretion may determine the balance between its catabolic and anabolic effects on the skeleton [109, 110]. Such pulsatility has been detected in both normal subjects and patients with CKD [108, 111]. Despite the pulsatile character of PTH release, intact PTH levels were found to be continuously maintained within the normal range in a control group and in CKD patients with normal parathyroid function whereas in CKD patients with SHPT , PTH changes were restricted within a level of hyperparathyroidism [111].
Secreted 1–84 PTH is degraded rapidly (half-life of approximately 2–4 min) to amino-terminal and carboxy-terminal fragments [112]. The amino-terminal residues bind to the PTH1R, activate cellular responses, and mimic all the Ca-regulating actions of PTH in animals [113]. The carboxy-terminal fragment of PTH seems to be essential for hormone processing (efficient transport across the endoplasmic reticulum) and secretion [114]. Although 1–84 PTH is the main source of secreted PTH, it is also known that the gland can secrete carboxy-terminal fragments and that the relative secretion of these fragments increases or decreases in the presence of hypercalcemia or hypocalcemia, respectively [115, 116]. Studies have shown that preferential secretion of carboxy-terminal fragments may not occur in primary hyperparathyroidism as it does in other hypercalcemic states [117], and that some subpopulations of parathyroid cells from hyperplastic or adenomatous glands can secrete in vitro more amino-terminal fragments than 1–84 PTH [118].
Because of the shorter half-lives of both 1–84 PTH and amino-terminal fragments, the carboxy-terminal fragments become the predominating PTH peptide in the circulation. The parent peptide is rapidly degraded in the peripheral tissues, particularly in the kidneys and the liver [11, 112, 119]. The liver has great capacity to degrade the peptide, but it may not play an important role in the degradation of fragments [120]. By contrast, the kidney can extract and degrade the complete molecule and its fragments [121], at least partially via the action of cathepsin D [122]. Thus, impairment of renal function causes accumulation of carboxy-terminal fragments, and although amino-terminal fragments are also produced by cleavage of 1–84 PTH (i.e., by Kupffer cells), they are rapidly degraded, unlike the corresponding carboxy-terminal fragments [11].
As mentioned above, carboxy-terminal fragments are mainly catabolized in the kidney, and the degradation process involves solely glomerular filtration and tubular reabsorption, whereas the amino-terminal fragment undergoes both tubular reabsorption and peritubular uptake, like 1–84 PTH [112]. These pathways of PTH metabolism are altered in the presence of CKD, and renal excretion of PTH and its fragments (mainly carboxy-terminal) is decreased [112, 123]. Therefore, such alterations in PTH metabolism also partially account for the elevated PTH levels observed in CKD.
Although PTH measurement with the current immunoradiometric (IRMA) and immunochemiluminescence assays directed to the so-called intact PTH (the most widely implemented worldwide) has significantly improved clinical management, several fragments still affect the measurement and interpretation of these second-generation intact PTH assays. Thus, it is now well known that there are non-(1–84)-PTH truncated fragments in the circulation (such as 7–84 PTH) which, in addition to 1–84 PTH , are measured by most IRMA intact PTH assays, giving erroneously high PTH values [27, 124, 125]. This fact is especially important now that many different (automated and non-automated) non-standardized intact PTH kits are available on the market, using different antibodies and without a common calibrator [11, 27, 126,127,128,129]. Consequently, there is wide variability among commercially available intact PTH assays [128]. Third-generation assays (measuring the so-called “whole” or “bioactive” PTH) seem to detect only the biologically active 1–84 PTH molecule because the detection antibody is more specific for the first four amino acids at the amino-terminal end [130, 131]. While these new-generation “whole PTH” assays do not interact with 7–84 PTH fragments, they seem to measure, in addition to 1–84 PTH, a post-translational form called amino-PTH [132]. In any case, “intact” and “whole” PTH assays appear to be of similar clinical value for the diagnosis of SHPT and the follow-up of CKD-MBD, and “whole”/“bio-intact” PTH measurement is not yet fully recommended in any guideline [4].
The recent description of non-active oxidized-PTH adds complexity to the clinical interpretation of PTH values [112, 133,134,135,136], and it has recently been debated whether the increased mortality risk associated with PTH might actually reflect an oxidative stress-related mortality [19, 112, 135]. Whereas it is widely accepted that PTH measurement (especially trends) is an appropriate marker of parathyroid activity, PTH is only indirectly and weakly associated with bone dynamics [137]. Therefore, over the last decade there has been increased controversy over the validity of PTH as a surrogate marker of CKD-MBD and/or bone turnover [137, 138], as well as for the definition of optimal PTH targets in both non-dialysis and dialysis CKD patients [4, 139].
Furthermore, regarding the importance of these abnormalities in the metabolism of PTH for the calcemic response to PTH , it has been reported that, in PTX rats fed a calcium-deficient diet, 7–84 PTH was not only biologically inactive but also had antagonistic effects on the PTHR in kidney and bone [130]. In these animals, plasma Ca was increased 2 h after 1–84 PTH treatment, while 7–84 PTH had no effect. However, when 1–84 PTH and 7–84 PTH were given simultaneously in a 1:1 molar ratio, the calcemic response to 1–84 PTH was decreased by 94%. Moreover, the administration of 1–84 PTH increased the renal fractional excretion of P in normal rats. However, when 1–84 PTH and 7–84 PTH were given simultaneously, the 7–84 PTH decreased the phosphaturic response by 50.2%. Finally, in surgically excised parathyroid glands from six uremic patients, the authors found that 44.1% of the total intracellular PTH was the non-PTH (1–84), most likely PTH 7–84. They concluded that in patients with CKD, the presence of high circulating levels of non-1–84 PTH fragments detected by the “intact” assay and the antagonistic effects of 7–84 PTH on the biological activity of 1–84 PTH explain the need for higher levels of “intact” PTH to prevent adynamic bone disease and provide a novel mechanism for skeletal resistance to PTH in uremia [130].
In addition to the “classic” PTH1R, it is currently accepted that a carboxy-terminal PTHR (PTH4R or PTHR-C) may mediate these different actions [140,141,142]. Increasing evidence indicates that the C-terminal PTH fragments, by binding and competing with 1–84 PTH for the PTH1R or by binding to the PTHR-C, exert these different/opposite biological effects as compared to 1–84 PTH [11, 27, 130, 141, 143,144,145]. Actually, it has been shown that the effects of 7–84 PTH on 1–84 PTH secretion and on plasma Ca levels are not associated with changes in PTH, PTH1R, CaSR, and PTHrP gene expression in rat parathyroid glands [145], and it has been hypothesized that PTH 7–84 regulates PTH secretion via an autocrine/paracrine regulatory mechanism [145]. The biological significance of this system may relate to the fact that during hypercalcemia the PTHR-C may help to maintain normal bone formation when the carboxy-terminal fragments exceed those of 1–84 PTH. Therefore, it has been postulated that the ratio of 1–84 PTH (or amino-terminal PTH ) to carboxy-terminal PTH, which would be equivalent to the opposite effects of PTH1R/PTHR-C receptor activation, may be important in understanding not only the changes in the parathyroid gland but also the bone activity observed in CKD patients [11]. Moreover, the increased production of large carboxy-terminal PTH fragments by parathyroid glands during hypercalcemia, mentioned earlier, may help to restore Ca by inhibition of bone resorption [11, 115]. This relative increase in C-terminal fragments has been demonstrated in hemodialysis patients exposed to low and high Ca concentrations in the dialysis bath [116].
Other PTH receptors (PTH2R and PTH3R) have also been described but their effects in humans and in CKD are largely unknown [12, 146, 147]. PTH2R is not expressed in renal tubules and bone and is not activated by PTHrP. Like PTH1R, PTH2R responds to PTH with generation of cAMP and an increase in intracellular calcium.
Additional information on PTH metabolism and signaling, both in health and in CKD and including classic and non-classic target organs for PTH, may be found in recent review papers [11, 27].
Downstream Competing Signals, Local or Systemic Factors
It is possible that the downstream effects of PTH may be offset by associated competing and/or inhibitory endocrine or paracrine signals, mediated by, for example, FGF23, klotho, calcitonin, osteoprotegerin, bone morphogenetic proteins, Wnt antagonists, and insulin-like growth factors, in addition to local environmental factors (i.e., inflammation, cytokines, oxidative stress, acid-base disturbances, and Ca, P, magnesium, and aluminum concentrations) [27, 39].
Thus, a recent study demonstrated that a recombinant human klotho protein interacted with human PTH1R to inhibit the binding of human PTH in renal tubular cells. It also inhibited the PTH-induced 1-α-hydroxylase expression by tubular cells both in vitro and in vivo [148]. These results suggest that free klotho mediates the FGF23-induced inhibition of calcitriol synthesis [148], and it has been hypothesized that as long as PTH underlies basal production of calcitriol, free klotho mediates, at least in part, the decrease in calcitriol levels in response to FGF23 by impairing PTH signaling [148].
The role of endogenous calcitonin production by thyroid C cells in the pathogenesis of SHPT —in general, in the protection against hypercalcemia, and in the decreased calcemic response to PTH in CKD—has also been analyzed [149,150,151,152,153,154]. It has been shown that in the absence of calcitonin, the calcemic response to PTH increased in rats regardless of whether they had CKD or not. In the presence of SHPT and hypercalcemia, calcitonin was an important modifier of the calcemic response to PTH, especially in animals with CKD [149, 151].
There is scarce information about a potential role of osteoprotegerin (OPG, a potent osteoclast activation inhibitor) in the CKD-MBD complex. Since skeletal resistance to PTH appears as an anti-calcemic effect against exogenous PTH load in the physiological aspect, and as the discrepancy between serum PTH level and bone turnover in the morphological aspect, OPG has been postulated to be a common pathogenic mediator of both high- and low-bone turnover diseases [155, 156]. Thus, the high circulating OPG levels found in CKD [156] may promote skeletal resistance to PTH through suppression of osteoclastogenesis [155, 157].
Importantly, Wnt antagonists such as sclerostin, the product of the SOST gene and mainly produced by osteocytes, was originally believed to be a non-classic BMP antagonist [158]. Sclerostin has now been identified as a soluble inhibitor of the Wnt signaling pathway via binding to LRP5/6 receptors [159, 160], and hence it may lead to decreased bone formation by inhibiting osteoblastogenesis (in contrast to OPG). Sclerostin may also play a role in the mediation of systemic and local factors such as calcitriol , PTH , glucocorticoids, and tumor necrosis factor-α [161]. Circulating sclerostin levels increase with age and with declining renal function [161,162,163] and are also increased in diabetic patients independently of gender and age [164]. Levels decrease rapidly after renal transplantation [165, 166]. Nevertheless, it remains a matter of debate to what extent circulating sclerostin levels reflect bone expression and affect local signaling, since discrepant findings have been described [167]. Increased osteocytic sclerostin expression has indeed been observed in early-stage CKD despite still normal serum PTH levels [168], and the increase persists in dialysis patients, although to a lesser extent, despite elevated PTH levels [169].
Sclerostin and the related Dickkopf-1 (Dkk1) or secreted frizzled-related protein 4 (sFRP4) have been postulated to be potentially important mediators of the development of adynamic bone disease [12, 39, 97, 170] and/or skeletal resistance to the action of PTH [39, 97, 170]. A new paradigm is evolving and it has been proposed that early inhibition of the osteocyte Wnt pathway with an increase in the expression of sclerostin and other inhibitors of the Wnt/β catenin signaling pathway may be the initial stage of renal osteodystrophy and may explain observations of a relatively high and increasing prevalence of adynamic bone disease [101, 171,172,173]. It has even been hypothesized that in early CKD stages, low bone turnover prevails, with adynamic bone disease being the predominant form [39, 97]. With the progression of CKD to more advanced stages and increasing circulating levels of PTH, the steadily increasing activation of the PTH1R eventually overcomes the skeletal resistance to PTH and osteitis fibrosa ensues, if left untreated [12, 39]. Whether FGF23 and α-klotho play a direct role in this postulated transition from low- to high-turnover bone disease or participate only indirectly via regulating PTH secretion remains to be seen [39], but osteocyte dysfunction has been shown to be altered early in the course of CKD [39]. Of note, the use of an anti-sclerostin antibody in a CKD rat model of progressive renal osteodystrophy was shown to increase trabecular bone volume/total volume and trabecular mineralization surface in animals with low, but not high, PTH [174]. Similarly, bone properties (bone volume, cortical geometry, and biomechanical properties) improved only when PTH levels were low [174]. Whether high sclerostin levels are the cause or the consequence of PTH hyporesponsiveness in CKD remains to be clarified [166].
It also has to be taken into account that other factors such as racial and sex differences [175,176,177], the higher age and increased prevalence of diabetes among the CKD population, and overzealous PTH control (e.g., normalization of PTH in non-dialysis CKD patients) may influence the evaluation of PTH hyporesponsiveness [144].
Clinical Implications of Skeletal Resistance to the Action of PTH
Skeletal resistance to PTH was initially suggested as a mechanism of PTH hypersecretion in CKD. Interest in this background abnormality has been resuscitated by the effective, potentially excessive suppression of PTH by different therapies (i.e., Ca-based P binders, vitamin D, calcimimetics) and the increasing prevalence of adynamic bone disease and its associated risks (including vascular calcification and fractures) [9, 27, 60, 97, 170, 178]. As mentioned previously, skeletal resistance to PTH is currently recognized as “hyporesponsiveness ” to PTH or “relative hypoparathyroidism” in terms of its relation to bone turnover [27, 39, 60, 97]. London et al., analyzing the presence of a bone–vascular axis and/or bone–vascular cross-talk [179], had already shown an inverse association between vascular calcification and lower serum PTH , low osteoclast numbers and smaller osteoblastic surfaces, and smaller or absent double tetracycline labeling surfaces, although also with high percentages of aluminum-stained surfaces [180]. In a recent cross-sectional study, these authors also found peripheral artery disease to be associated with significant reductions in the skeletal anabolic response to PTH, as demonstrated by weaker correlation coefficients (slopes) between serum PTH and double-labelled surfaces or osteoblast surfaces in patients with peripheral artery disease [181].
Additional evidence that PTH hyporesponsiveness is an important factor in the development of SHPT and/or adynamic bone disease derives from clinical studies demonstrating that a high level of circulating PTH is necessary to maintain normal bone remodeling [182,183,184]. Consequently, for instance in dialysis patients, current guidelines [4, 185] suggest that treatment should be modified to keep PTH levels higher than twice the upper limit of normal (better in conjunction with evaluation of bone-specific alkaline phosphatase) to avoid a low bone turnover. Also, “predialysis patients” with CKD need higher levels of PTH to maintain a normal osteoblastic surface [183], a fact that suggests that maintenance dialysis may enhance the skeletal response to PTH. However, reversibility by dialysis is not a uniform observation [52]. Finally, the presence of this multifactorial complex hyporesponsiveness to PTH may also explain the absence of clear associations between circulating PTH levels and outcomes in CKD patients (usually U-, J-, or inverted J-shaped, and overall rather weak) as opposed to the linear associations observed in primary hyperparathyroidism [27].
Conclusion
According to the previously described experimental observations demonstrating the presence of a decreased calcemic response using a PTH infusion, either with extracts in CKD patients or with synthetic PTH in different experimental models, hyporesponsiveness to PTH is just as much an integral component of CKD-MBD as are elevated circulating PTH levels [27]. Clear differences in the calcemic response to PTH among CKD and normal subjects or animals cannot just be explained by the presence of distinct inactive or antagonistic PTH fragments, since all individuals and experimental animals received the same PTH compound (usually from the same batch) at a constant rate [67, 72]. Phosphate retention, calcitriol deficiency, sclerostin, and other uremic factors may play a role by desensitizing and/or down-regulating the PTHR or downstream signaling.
Although skeletal resistance to PTH was initially suggested as a mechanism of PTH hypersecretion in CKD, “hyporesponsiveness ” to PTH has also been associated with the increasing prevalence of low-turnover bone disease, which is explained by, among other factors, an increasing number of elderly and diabetic patients with CKD and treatment overshooting. Therefore, hyporesponsiveness to PTH should be taken into account when treating SHPT in CKD patients and it is important to avoid the normalization of PTH levels in these patients [139, 185]; on the other hand, progressively increasing PTH levels may indicate a change from an adaptive to a maladaptive clinical situation that requires therapeutic decisions (See Fig. 2).
Defining an optimal PTH target may be challenging but accomplishable at the population level, but it may be very difficult at the individual patient level [27]. Whether it is best to wait for onset of severe SHPT before starting antiparathyroid treatment, as suggested by the recent KDIGO guidelines [4], or to avoid complete normalization of PTH levels, as suggested by others [139], remains to be determined. Probably one single PTH recommendation does not fit all CKD patients, and nephrologists will be drawn towards a more personalized and individual management by the need to take into account other factors such as age, diabetic status, presence of metabolic syndrome, fracture risk, vascular calcification, and other biochemical markers, as well as recently identified factors that require further investigation (uremic toxins, FGF23/klotho, Wnt/β-catenin, type 2 activin A receptor pathways, etc.). Interestingly, recent evidence has indicated that osteocytes are crucial cellular targets of PTH , and the concept of “osteocytic osteolysis” has been proposed as a mechanism through which PTH rapidly increases blood calcium levels [186]. One attractive mechanism through which PTH signaling in osteocytes influences skeletal remodeling is by coordinated transcriptional regulation of paracrine mediators, including SOST [myocyte enhancer factors (MEF2)] and receptor activator of NF-κB ligand (RANKL) [186]. Beyond SOST and RANKL, PTH/PTHrP signaling in osteocytes may directly influence the way osteocytes remodel their perilacunar environment to influence bone homeostasis in a cell-autonomous manner [186].
In the meantime, despite its limitations [187], no other biomarker or therapeutic strategy has been proven to be superior to PTH , and efforts seem mandatory to improve diagnosis. More frequent monitoring, enabling PTH trends to be captured, seems the appropriate way to proceed [27] until better new molecular targets and treatments become available that demonstrate proven efficacy in clinical practice [137, 188].
Finally, resistance to the biological action of several other hormones, such as insulin, calcitriol , growth hormone, and FGF23, is also a well-known feature of CKD [189,190,191,192], as is decreased expression of several other receptors (i.e. VDR, CaSR, FGFR/klotho) [15, 72, 193,194,195,196,197,198,199,200,201]. As a matter of fact, uremia may thus be considered a disease which extensively affects different types of receptor (uremia as a “receptor disease”) [137]. Additional studies at cellular and molecular levels are needed to establish preventive and therapeutic modalities which may be of value beyond their significance for hyporesponsiveness to PTH .

Adapted from Ref. [68]
Calcemic response after a 48 h PTH infusion in rats, according to different degrees of renal function (normal, moderate and advanced renal failure) and dietary phosphate (HPD: high phosphorus diet; MPD: moderate phosphorus diet; LPD: low phosphorus diet). During the PTH infusion, rats received a calcium-free-low phosphorus diet. The magnitude of the calcemic response to PTH inversely depends on the degree of renal failure and the content of phosphorus in the diet. The term “calcemic response” to PTH, “skeletal response” to PTH or “end-organ resistance” to PTH evolved to “hyporesponsiveness ” to PTH (see text)

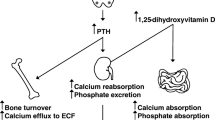
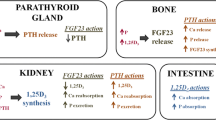
Causes and consequences of parathyroid hormone (PTH) hyporesponsiveness in chronic kidney disease (CKD). P = Phosphate ; FGF-23 = Fibroblast Growth Factor 23; IS = indoxyl-sulfate; PTHR = PTH receptor; PT = parathyroid; CTR = calcitriol ; OF = Osteitis fibrosa; ABD = Adynamic bone disease
References
Martínez-Castelao A, Górriz JL, Segura-de la Morena J, et al. Consensus document for the detection and management of chronic kidney disease. Nefrologia. 2014;34:243–62.
Covic A, Vervloet M, Massy ZA, et al. Bone and mineral disorders in chronic kidney disease: implications for cardiovascular health and ageing in the general population. Lancet Diabetes Endocrinol. 2018;6:319–31.
Moe SM, Drueke T, Lameire N, Eknoyan G. Chronic kidney disease-mineral bone disorder: a new paradigm. Adv Chronic Kidney Dis. 2007;14:3–12.
Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017 clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Suppl. 2017;7:1–59.
Cozzolino M, Urena-Torres P, Vervloet MG, Brandenburg V, Bover J, Goldsmith D, Larsson TE, Massy ZA, Mazzaferro S. Is chronic kidney disease-mineral bone disorder (CKD-MBD) really a syndrome? Nephrol Dial Transpl. 2014;29:1815–20.
Vervloet MG, Massy ZA, Brandenburg VM, Mazzaferro S, Cozzolino M, Ureña-Torres P, Bover J, Goldsmith D. Bone: a new endocrine organ at the heart of chronic kidney disease and mineral and bone disorders. Lancet Diabetes Endocrinol. 2014;2:427–36.
Rodriguez M, Salmeron MD, Martin-Malo A, Barbieri C, Mari F, Molina RI, Costa P, Aljama P. A new data analysis system to quantify associations between biochemical parameters of chronic kidney disease-mineral bone disease. PLoS ONE. 2016;11:e0146801.
Behets GJ, Spasovski G, Sterling LR, Goodman WG, Spiegel DM, De Broe ME, D’Haese PC. Bone histomorphometry before and after long-term treatment with cinacalcet in dialysis patients with secondary hyperparathyroidism. Kidney Int. 2015;87:846–56.
Noordzij M, Cranenburg EM, Engelsman LF, et al. Progression of aortic calcification is associated with disorders of mineral metabolism and mortality in chronic dialysis patients. Nephrol Dial Transpl. 2011;26:1662–9.
Massry SG. Is parathyroid hormone a uremic toxin? Nephron. 1977;19:125–30.
Rodriguez M, Lorenzo V. Parathyroid hormone, a uremic toxin. Semin Dial. 2009;22:363–8.
Ureña-Torres PA, Vervloet M, Mazzaferro S, Oury F, Brandenburg V, Bover J et al. Novel insights into parathyroid hormone: report of The Parathyroid Day in chronic kidney disease. Clin Kidney J. 2018. https://doi.org/10.1093/ckj/sfy061 (Epub ahead online: https://academic.oup.com/ckj/advance-article/doi/10.1093/ckj/sfy061/5056879).
Slatopolsky E, Martin K, Hruska K. Parathyroid hormone metabolism and its potential as a uremic toxin. Am J Physiol Physiol. 1980;239:F1–12.
Morii H, Nishizawa Y, Smogorzewski M, Inaba M, Massry SG. New actions of parathyroid hormone. Introduction. Miner Electrolyte Metab. 1995;21:7–8.
Llach F, Bover J. Renal Osteodystrophies. In: Brenner BM, editor. The kidney, 6th ed. Philadelphia: W.B. Saunders Company;2000. p. 2103–2186.
Vaidya A, Brown JM, Williams JS. The renin–angiotensin–aldosterone system and calcium-regulatory hormones. J Hum Hypertens. 2015;29:515–21.
Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, Hodin RA, Spiegelman BM. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab. 2016;23:315–23.
McMahon DJ, Carrelli A, Palmeri N, Zhang C, DiTullio M, Silverberg SJ, Walker MD. Effect of parathyroidectomy upon left ventricular mass in primary hyperparathyroidism: a meta-analysis. J Clin Endocrinol Metab. 2015;100:4399–407.
Seiler-Mussler S, Limbach AS, Emrich IE, Pickering JW, Roth HJ, Fliser D, Heine GH. Association of nonoxidized parathyroid hormone with cardiovascular and kidney disease outcomes in chronic kidney disease. Clin J Am Soc Nephrol. 2018;13:569–76.
Floege J, Kim J, Ireland E, et al. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis population. Nephrol Dial Transpl. 2011;26:1948–55.
Young EW, Akiba T, Albert JM, McCarthy JT, Kerr PG, Mendelssohn DC, Jadoul M. Magnitude and impact of abnormal mineral metabolism in hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2004;44:34–8.
Ureña Torres PA, De Broe M. Calcium-sensing receptor, calcimimetics, and cardiovascular calcifications in chronic kidney disease. Kidney Int. 2012;82:19–25.
Wheeler DC, London GM, Parfrey PS, Block GA, Correa-Rotter R, Dehmel B, Drüeke TB, Floege J, Kubo Y, Mahaffey KW, Goodman WG, Moe SM, Trotman ML, Abdalla S, Chertow GM, Herzog CA. Effects of cinacalcet on atherosclerotic and nonatherosclerotic cardiovascular events in patients receiving hemodialysis: the EValuation Of Cinacalcet HCl Therapy to Lower CardioVascular Events (EVOLVE) trial. J Am Heart Assoc. 2012;4(1):e000570.
Joy MS, Karagiannis PC, Peyerl FW. Outcomes of secondary hyperparathyroidism in chronic kidney disease and the direct costs of treatment. J Manag Care Pharm. 2007;13:397–411.
Fernández-Martín JL, Martínez-Camblor P, Dionisi MP, Floege J, Ketteler M, London G, Locatelli F, Gorriz JL, Rutkowski B, Ferreira A, Bos WJ, Covic A, Rodríguez-García M, Sánchez JE, Rodríguez-Puyol D, Cannata-Andia JB, COSMOS group. Improvement of mineral and bone metabolism markers is associated with better survival in haemodialysis patients: the COSMOS study. Nephrol Dial Transplant. 2015;30(9):1542–51.
Kurz P, Monier-Faugere MC, Bognar B, Werner E, Roth P, Vlachojannis J, Malluche HH. Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int. 1994;46:855–61.
Evenepoel P, Bover J, Ureña Torres P. Parathyroid hormone metabolism and signaling in health and chronic kidney disease. Kidney Int. 2016;90:1184–90.
Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl. 2009;76:S1–130.
Kazama JJ, Iwasaki Y, Fukagawa M. Uremic osteoporosis. Kidney Int Suppl. 2013;3:446–50.
Kazama JJ, Matsuo K, Iwasaki Y, Fukagawa M. Chronic kidney disease and bone metabolism. J Bone Miner Metab. 2015;33:245–52.
Pilz S, Tomaschitz A, Drechsler C, Ritz E, Boehm BO, Grammer TB, Marz W. Parathyroid hormone level is associated with mortality and cardiovascular events in patients undergoing coronary angiography. Eur Heart J. 2010;31:1591–8.
London G. Cardiovascular disease in end-stage renal failure: role of calcium-phosphate disturbances and hyperparathyroidism. J Nephrol. 2002;15:209–10.
London GM, De Vernejoul MC, Fabiani F, Marchais SJ, Guerin AP, Metivier F, London AM, Llach F. Secondary hyperparathyroidism and cardiac hypertrophy in hemodialysis patients. Kidney Int. 1987;32:900–7.
Slatopolsky E, Caglar S, Pennell JP, Taggart DD, Canterbury JM, Reiss E, Bricker NS. On the pathogenesis of hyperparathyroidism in chronic experimental renal insufficiency in the dog. J Clin Invest. 1971;50:492–9.
Slatopolsky E, Bricker NS. The role of phosphorus restriction in the prevention of secondary hyperparathyroidism in chronic renal disease. Kidney Int. 1973;4:141–5.
Kuro-o M. Klotho, phosphate and FGF-23 in ageing and disturbed mineral metabolism. Nat Rev Nephrol. 2013;9:650–60.
Kuro-o M, Moe OW. FGF23-αKlotho as a paradigm for a kidney-bone network. Bone. 2017;100:4–18.
Wolf M. Forging forward with 10 burning questions on FGF23 in kidney disease. J Am Soc Nephrol. 2010;21:1427–35.
Drüeke TB, Massy ZA. Changing bone patterns with progression of chronic kidney disease. Kidney Int. 2016;89:289–302.
Hruska KA, Sugatani T, Agapova O, Fang Y. The chronic kidney disease—mineral bone disorder (CKD-MBD): advances in pathophysiology. Bone. 2017;100:80–6.
Cunningham J, Locatelli F, Rodriguez M. Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. Clin J Am Soc Nephrol. 2011;6:913–21.
Rodriguez M, Nemeth E, Martin D. The calcium-sensing receptor: a key factor in the pathogenesis of secondary hyperparathyroidism. Am J Physiol Renal Physiol. 2005;288:F253–64.
Tominaga Y, Takagi H. Molecular genetics of hyperparathyroid disease. Curr Opin Nephrol Hypertens. 1996;5:336–41.
Almaden Y, Canalejo A, Hernandez A, Ballesteros E, Garcia-Navarro S, Torres A, Rodriguez M, Rodriguez M. Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J Bone Miner Res. 1996;11:970–6.
Almaden Y, Hernandez A, Torregrosa V, Canalejo A, Sabate L, Fernandez Cruz L, Campistol JM, Torres A, Rodriguez M. High phosphate level directly stimulates parathyroid hormone secretion and synthesis by human parathyroid tissue in vitro. J Am Soc Nephrol. 1998;9:1845–52.
Slatopolsky E, Finch J, Denda M, Ritter C, Zhong M, Dusso A, MacDonald PN, Brown AJ. Phosphorus restriction prevents parathyroid gland growth. High phosphorus directly stimulates PTH secretion in vitro. J Clin Invest. 1996;97:2534–40.
Rodriguez-Ortiz ME, Lopez I, Muñoz-Castañeda JR, et al. Calcium deficiency reduces circulating levels of FGF23. J Am Soc Nephrol. 2012;23:1190–7.
Mace ML, Gravesen E, Nordholm A, Olgaard K, Lewin E. Fibroblast Growth Factor (FGF) 23 regulates the plasma levels of parathyroid hormone in vivo through the FGF receptor in normocalcemia, but not in hypocalcemia. Calcif Tissue Int. 2018;102:85–92.
Evanson JM. The response to the infusion of parathyroid extract in hypocalcaemic states. Clin Sci. 1966;31:63–75.
Kruse K, Kracht U, Wohlfart K, Kruse U. Biochemical markers of bone turnover, intact serum parathyroid hormone and renal calcium excretion in patients with pseudohypoparathyroidism and hypoparathyroidism before and during vitamin D treatment. Eur J Pediatr. 1989;148:535–9.
Massry SG, Stein R, Garty J, Arieff AI, Coburn JW, Norman AW, Friedler RM. Skeletal resistance to the calcemic action of parathyroid hormone in uremia: role of 1,25 (OH)2 D3. Kidney Int. 1976;9:467–74.
Massry SG, Coburn JW, Lee DB, Jowsey J, Kleeman CR. Skeletal resistance to parathyroid hormone in renal failure. Studies in 105 human subjects. Ann Intern Med. 1973;78:357–64.
Llach F, Massry SG, Singer FR, Kurokawa K, Kaye JH, Coburn JW. Skeletal resistance to endogenous parathyroid hormone in patients with early renal failure. A possible cause for secondary hyperparathyroidism. J Clin Endocrinol Metab. 1975;41:339–45.
Wilson L, Felsenfeld A, Drezner MK, Llach F. Altered divalent ion metabolism in early renal failure: role of 1,25(OH)2D. Kidney Int. 1985;27:565–73.
Albright F, Drake TG, Sulkowitch HW. Renal osteitis fibrosa cystica. Bull Johns Hopkins Hosp. 1937;60:377–99.
Bricker NS, Slatopolsky E, Reiss E, Avioli LV. Calcium, phosphorus, and bone in renal disease and transplantation. Arch Intern Med. 1969;123:543–53.
Slatopolsky E, Caglar S, Gradowska L, Canterbury J, Reiss E, Bricker NS. On the prevention of secondary hyperparathyroidism in experimental chronic renal disease using proportional reduction of dietary phosphorus intake. Kidney Int. 1972;2:147–51.
Deykin D, Balko C, Bricker NS. On the pathogenesis of the uremic state. N Engl J Med. 1972;286:1093–9.
Llach F. Secondary hyperparathyroidism in renal failure: the trade-off hypothesis revisited. Am J Kidney Dis. 1995;25:663–79.
Fukagawa M, Kazama JJ, Shigematsu T. Skeletal resistance to PTH as a basic abnormality underlying uremic bone diseases. Am J Kidney Dis. 2001;38:S152–5.
Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA, Andress DL. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71:31–8.
Somerville PJ, Kaye M. Resistance to parathyroid hormone in renal failure: role of vitamin D metabolites. Kidney Int. 1978;14:245–54.
Rodriguez M, Felsenfeld AJ, Llach F. Calcemic response to parathyroid hormone in renal failure: role of calcitriol and the effect of parathyroidectomy. Kidney Int. 1991;40:1063–8.
Massry SG, Tuma S, Dua S, Goldstein DA. Reversal of skeletal resistance to parathyroid hormone in uremia by vitamin D metabolites: evidence for the requirement of 1,25(OH)2D3 and 24,25(OH)2D3. J Lab Clin Med. 1979;94:152–7.
Galceran T, Martin KJ, Morrissey JJ, Slatopolsky E. Role of 1,25-dihydroxyvitamin D on the skeletal resistance to parathyroid hormone. Kidney Int. 1987;32:801–7.
Somerville PJ, Kaye M. Evidence that resistance to the calcemic action of parathyroid hormone in rats with acute uremia is caused by phosphate retention. Kidney Int. 1979;16:552–60.
Bover J, Rodriguez M, Trinidad P, Jara A, Martinez ME, Machado L, Llach F, Felsenfeld AJ. Factors in the development of secondary hyperparathyroidism during graded renal failure in the rat. Kidney Int. 1994;45:953–61.
Bover J, Jara A, Trinidad P, Rodriguez M, Felsenfeld AJ. Dynamics of skeletal resistance to parathyroid hormone in the rat: effect of renal failure and dietary phosphorus. Bone. 1999;25:279–85.
Llach F, Massry SG. On the mechanism of secondary hyperparathyroidism in moderate renal insufficiency. J Clin Endocrinol Metab. 1985;61:601–6.
Yates AJ, Oreffo RO, Mayor K, Mundy GR. Inhibition of bone resorption by inorganic phosphate is mediated by both reduced osteoclast formation and decreased activity of mature osteoclasts. J Bone Miner Res. 1991;6:473–8.
Geng Y, Mosyak L, Kurinov I, et al. Structural mechanism of ligand activation in human calcium-sensing receptor. eLife. 2016;5;pii:e13662.
Bover J, Jara A, Trinidad P, Rodriguez M, Martin-Malo A, Felsenfeld AJ. The calcemic response to PTH in the rat: effect of elevated PTH levels and uremia. Kidney Int. 1994;46:310–7.
Fujimori A, Miyauchi A, Hruska KA, Martin KJ, Avioli LV, Civitelli R. Desensitization of calcium messenger system in parathyroid hormone-stimulated opossum kidney cells. Am J Physiol. 1993;264:E918–24.
Olgaard K, Arbelaez M, Schwartz J, Klahr S, Slatopolsky E. Abnormal skeletal response to parathyroid hormone in dogs with chronic uremia. Calcif Tissue Int. 1982;34:403–7.
Olgaard K, Schwartz J, Finco D, Arbelaez M, Korkor A, Martin K, Klahr S, Slatopolsky E. Extraction of parathyroid hormone and release of adenosine 3’,5’-monophosphate by isolated perfused bones obtained from dogs with acute uremia. Endocrinology. 1982;111:1678–82.
Roth J, Grunfeld C. Endocrine systems: mechanisms of disease, target cells, and receptors. In: RH Williams, editor. Textbook of endocrinology, 6th ed. Philadelphia: W.B. Saunders Comp., Williams & Wilkins;1981. p. 41–3.
Drüeke TB. Abnormal skeletal response to parathyroid hormone and the expression of its receptor in chronic uremia. Pediatr Nephrol. 1996;10:348–50.
Jüppner H, Abou-Samra AB, Freeman M, Kong XF, Schipani E, Richards J, Kolakowski LF, Hock J, Potts JT, Kronenberg HM. A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science. 1991;254:1024–6.
Abou-Samra AB, Jüppner H, Force T, Freeman MW, Kong XF, Schipani E, Urena P, Richards J, Bonventre JV, Potts JT. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc Natl Acad Sci U S A. 1992;89:2732–6.
Ureña P, Kong XF, Abou-Samra AB, Jüppner H, Kronenberg HM, Potts JT, Segre GV. Parathyroid hormone (PTH)/PTH-related peptide receptor messenger ribonucleic acids are widely distributed in rat tissues. Endocrinology. 1993;133:617–23.
Iwasaki-Ishizuka Y, Yamato H, Nii-Kono T, Kurokawa K, Fukagawa M. Downregulation of parathyroid hormone receptor gene expression and osteoblastic dysfunction associated with skeletal resistance to parathyroid hormone in a rat model of renal failure with low turnover bone. Nephrol Dial Transplant. 2005;20:1904–11.
Ureña P, Kubrusly M, Mannstadt M, Hruby M, Trinh MM, Silve C, Lacour B, Abou-Samra AB, Segre GV, Drüeke T. The renal PTH/PTHrP receptor is down-regulated in rats with chronic renal failure. Kidney Int. 1994;45:605–11.
Tian J, Smogorzewski M, Kedes L, Massry SG. PTH-PTHrP receptor mRNA is downregulated in chronic renal failure. Am J Nephrol. 1994;14:41–6.
Ureña P, Ferreira A, Morieux C, Drüeke T, de Vernejoul MC. PTH/PTHrP receptor mRNA is down-regulated in epiphyseal cartilage growth plate of uraemic rats. Nephrol Dial Transpl. 1996;11:2008–16.
Picton ML, Moore PR, Mawer EB, Houghton D, Freemont AJ, Hutchison AJ, Gokal R, Hoyland JA. Down-regulation of human osteoblast PTH/PTHrP receptor mRNA in end-stage renal failure. Kidney Int. 2000;58:1440–9.
Pereira RC, Delany AM, Khouzam NM, Bowen RE, Freymiller EG, Salusky IB, Wesseling-Perry K. Primary osteoblast-like cells from patients with end-stage kidney disease reflect gene expression, proliferation, and mineralization characteristics ex vivo. Kidney Int. 2015;87:593–601.
Ureña P, Mannstadt M, Hruby M, Ferreira A, Schmitt F, Silve C, Ardaillou R, Lacour B, Abou-Samra AB, Segre GV. Parathyroidectomy does not prevent the renal PTH/PTHrP receptor down-regulation in uremic rats. Kidney Int. 1995;47:1797–805.
Sanchez C, Salusky I, Willsey P et al. Calcitriol upregulates PTH/PTHrP receptor mRNA in rat growth plate cartilage. J Am Soc Nephrol. 1995;6:970 (Abstract).
Gonzalez EA MK. Calcitriol decreases PTH/PTHrp receptor gene expression in UMR 106-01 cells. J Am Soc Nephrol. 1994);5:880 (Abstract).
Zoccali C, Mallamaci F LD et al. Autoregulation of PTH secretion. J Am Soc Nephrol. 1994;5:892 (Abstract).
Suarez-Bregua P, Cal L, Cañestro C, Rotllant J. PTH reloaded: a new evolutionary perspective. Front Physiol. 2017;8:776.
Berdud I, Martin-Malo A, Almaden Y, Tallon S, Concepcion MT, Torres A, Felsenfeld A, Aljama P, Rodriguez M. Abnormal calcaemic response to PTH in the uraemic rat without secondary hyperparathyroidism. Nephrol Dial Transpl. 1996;11:1292–8.
Wills MR, Jenkins MV. The effect of uraemic metabolites on parathyroid extract-induced bone resorption in vitro. Clin Chim Acta. 1976;73:121–5.
Andress DL, Howard GA, Birnbaum RS. Identification of a low molecular weight inhibitor of osteoblast mitogenesis in uremic plasma. Kidney Int. 1991;39:942–5.
Disthabanchong S, Hassan H, McConkey CL, Martin KJ, Gonzalez EA. Regulation of PTH1 receptor expression by uremic ultrafiltrate in UMR 106–01 osteoblast-like cells. Kidney Int. 2004;65:897–903.
Nii-Kono T, Iwasaki Y, Uchida M, Fujieda A, Hosokawa A, Motojima M, Yamato H, Kurokawa K, Fukagawa M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007;71:738–43.
Massy Z, Drueke T. Adynamic bone disease is a predominant bone pattern in early stages of chronic kidney disease. J Nephrol. 2017;30:629–34.
Sage AP, Lu J, Atti E, Tetradis S, Ascenzi M-G, Adams DJ, Demer LL, Tintut Y. Hyperlipidemia induces resistance to PTH bone anabolism in mice via oxidized lipids. J Bone Miner Res. 2011;26:1197–206.
Mozar A, Louvet L, Godin C, Mentaverri R, Brazier M, Kamel S, Massy ZA. Indoxyl sulphate inhibits osteoclast differentiation and function. Nephrol Dial Transpl. 2012;27:2176–81.
Iwasaki Y, Yamato H, Nii-Kono T, Fujieda A, Uchida M, Hosokawa A, Motojima M, Fukagawa M. Administration of oral charcoal adsorbent (AST-120) suppresses low-turnover bone progression in uraemic rats. Nephrol Dial Transpl. 2006;21:2768–74.
Barreto FC, Barreto DV, Canziani MEF, Tomiyama C, Higa A, Mozar A, Glorieux G, Vanholder R, Massy Z, Carvalho AB. Association between indoxyl sulfate and bone histomorphometry in pre-dialysis chronic kidney disease patients. J Bras Nefrol. 2014;36:289–96.
Hsu CH, Patel S. Uremic plasma contains factors inhibiting 1 alpha-hydroxylase activity. J Am Soc Nephrol. 1992;3:947–52.
Patel SR, Ke HQ, Vanholder R, Koenig RJ, Hsu CH. Inhibition of calcitriol receptor binding to vitamin D response elements by uremic toxins. J Clin Invest. 1995;96:50–9.
Habener JF, Potts JT. Biosynthesis of parathyroid hormone. N Engl J Med. 1978;299:635–44.
Kakuta T, Sawada K. New developments in CKD-MBD. Cell biology of parathyroid in CKD. Clin Calcium. 2014;24:1801–8.
Basile C, Lomonte C. The function of the parathyroid oxyphil cells in uremia: still a mystery? Kidney Int. 2017;92:1046–8.
Ritter C, Miller B, Coyne DW, Gupta D, Zheng S, Brown AJ, Slatopolsky E. Paricalcitol and cinacalcet have disparate actions on parathyroid oxyphil cell content in patients with chronic kidney disease. Kidney Int. 2017;92:1217–22.
Kitamura N, Shigeno C, Shiomi K, et al. Episodic fluctuation in serum intact parathyroid hormone concentration in men. J Clin Endocrinol Metab. 1990;70:252–63.
Tam CS, Heersche JNM, Murray TM, Parsons JA. Parathyroid hormone stimulates the bone apposition rate independently of its resorptive action: differential effects of intermittent and continuous administration. Endocrinology. 1982;110:506–12.
Hock JM, Gera I. Effects of continuous and intermittent administration and inhibition of resorption on the anabolic response of bone to parathyroid hormone. J Bone Miner Res. 1992;7:65–72.
de Francisco AL, Amado JA, Cotorruelo JG, González M, de Castro SS, Canga E, de Bonis E, Ruiz JC, Arias M, Gonzalez-Macías J. Pulsatile-secretion of parathyroid hormone in patients with chronic renal failure. Clin Nephrol. 1993;39:224–8.
Hocher B, Zeng S. Clear the fog around parathyroid hormone assays: what do iPTH assays really measure? Clin J Am Soc Nephrol. 2018;13:524–6.
Tregear GW, Van Rietschoten J, Greene E, Keutmann HT, Niall HD, Reit B, Parsons JA, Potts JT. Bovine parathyroid hormone: minimum chain length of synthetic peptide required for biological activity. Endocrinology. 1973;93:1349–53.
Lim SK, Gardella TJ, Baba H, Nussbaum SR, Kronenberg HM. The carboxy-terminus of parathyroid hormone is essential for hormone processing and secretion. Endocrinology. 1992;131:2325–30.
Maye GP, Keaton JA, Hurst JG, Habener JF. Effects of plasma calcium concentration on the relative proportion of hormone and carboxyl fragments in parathyroid venous blood. Endocrinology. 1979;104:1778–84.
Santamaria R, Almaden Y, Felsenfeld A, Martin-Malo A, Gao P, Cantor T, Aljama P, Rodriguez M. Dynamics of PTH secretion in hemodialysis patients as determined by the intact and whole PTH assays. Kidney Int. 2003;64:1867–73.
Brossard JH, Whittom S, Lepage R, D’Amour P. Carboxyl-terminal fragments of parathyroid hormone are not secreted preferentially in primary hyperparathyroidism as they are in other hypercalcemic conditions. J Clin Endocrinol Metab. 1993;77:413–9.
el-Hajj Fuleihan G, Chen CJ, Rivkees SA, Marynick SP, Stock J, Pallotta JA, Brown EM. Calcium-dependent release of N-terminal fragments and intact immunoreactive parathyroid hormone by human pathological parathyroid tissue in vitro. J Clin Endocrinol Metab. 1989;69:860–7.
Catherwood BD, Friedler RM, Singer FR. Sites of clearance of endogenous parathyroid hormone in the vitamin D-deficient dog. Endocrinology. 1976;98:228–36.
Martin K, Hruska K, Greenwalt A, Klahr S, Slatopolsky E. Selective uptake of intact parathyroid hormone by the liver: differences between hepatic and renal uptake. J Clin Invest. 1976;58:781–8.
Martin KJ, Hruska KA, Lewis J, Anderson C, Slatopolsky E. The renal handling of parathyroid hormone. Role of peritubular uptake and glomerular filtration. J Clin Invest. 1977;60:808–14.
Zull JE, Chuang J. Characterization of parathyroid hormone fragments produced by cathepsin D. J Biol Chem. 1985;260:1608–13.
Freitag J, Martin KJ, Hruska KA, Anderson C, Conrades M, Ladenson J, Klahr S, Slatopolsky E. Impaired parathyroid hormone metabolism in patients with chronic renal failure. N Engl J Med. 1978;298:29–32.
Brossard JH, Cloutier M, Roy L, Lepage R, Gascon-Barré M, D’Amour P. Accumulation of a non-(1-84) molecular form of parathyroid hormone (PTH) detected by intact PTH assay in renal failure: importance in the interpretation of PTH values. J Clin Endocrinol Metab. 1996;81:3923–9.
Lepage R, Roy L, Brossard JH, Rousseau L, Dorais C, Lazure C, D’Amour P. A non-(1-84) circulating parathyroid hormone (PTH) fragment interferes significantly with intact PTH commercial assay measurements in uremic samples. Clin Chem. 1998;44:805–9.
Cavalier E, Delanaye P, Vranken L, Bekaert AC, Carlisi A, Chapelle JP, Souberbielle JC. Interpretation of serum PTH concentrations with different kits in dialysis patients according to the KDIGO guidelines: importance of the reference (normal) values. Nephrol Dial Transpl. 2012;27:1950–6.
Souberbielle JC, Friedlander G, Cormier C. Practical considerations in PTH testing. Clin Chim Acta. 2006;366:81–9.
Souberbielle JCP, Roth H, Fouque DP. Parathyroid hormone measurement in CKD. Kidney Int. 2010;77:93–100.
Souberbielle JC, Boutten A, Carlier MC, et al. Inter-method variability in PTH measurement: implication for the care of CKD patients. Kidney Int. 2006;70:345–50.
Slatopolsky E, Finch J, Clay P, Martin D, Sicard G, Singer G, Gao P, Cantor T, Dusso A. A novel mechanism for skeletal resistance in uremia. Kidney Int. 2000;58:753–61.
Malluche HH, Mawad H, Trueba D, Monier-Faugere MC. Parathyroid hormone assays–evolution and revolutions in the care of dialysis patients. Clin Nephrol. 2003;59:313–8.
González-Casaus ML, González-Parra E, Sánchez-González C, Albalate M, de la Piedra-Gordo C, Fernández E, Torregrosa V, Rodríguez M, Lorenzo V. A lower proportion of circulating active parathyroid hormone in peritoneal dialysis does not allow the pth inter-method adjustment proposed for haemodialysis. Nefrologia. 2014;34:330–40.
Hocher B, Armbruster FP, Stoeva S, Reichetzeder C, Grön HJ, Lieker I, Khadzhynov D, Slowinski T, Roth HJ. Measuring Parathyroid Hormone (PTH) in patients with oxidative stress—do we need a fourth generation parathyroid hormone assay? PLoS ONE. 2012;7:e40242.
Hocher B, Oberthür D, Slowinski T, et al. Modeling of oxidized PTH (oxPTH) and non-oxidized PTH (n-oxPTH) receptor binding and relationship of oxidized to non-oxidized PTH in children with chronic renal failure, adult patients on hemodialysis and kidney transplant recipients. Kidney Blood Press Res. 2013;37:240–51.
Tepel M, Armbruster FP, Grön HJ, Scholze A, Reichetzeder C, Roth HJ, Hocher B. Nonoxidized, biologically active parathyroid hormone determines mortality in hemodialysis patients. J Clin Endocrinol Metab. 2013;98:4744–51.
Souberbielle JC, Massart C, Brailly-Tabard S, Cormier C, Cavalier E, Delanaye P, Chanson P. Serum PTH reference values established by an automated third-generation assay in vitamin D-replete subjects with normal renal function: consequences of diagnosing primary hyperparathyroidism and the classification of dialysis patients. Eur J Endocrinol. 2016;174:315–23.
Bover J, Ureña P, Aguilar A, Mazzaferro S, Benito S, López-Báez V, Ramos A, daSilva I, Cozzolino M. Alkaline phosphatases in the complex chronic kidney disease-mineral and bone disorders. Calcif Tissue Int. 2018;103:111–24.
Tolouian RGA. The need for a reliable bone biomarker to better assess chronic kidney disease mineral and bone disorder. J Parathyr Dis. 2018;6:36–8.
Torregrosa V, Bover J, Cannata J et al. Spanish Society of Nephrology recommendations for controlling mineral and bone disorder in chronic kidney disease patients (S.E.N.-M.B.D.). Nefrologia. 2019 (submitted).
Divieti P, John MR, Jüppner H, Bringhurst FR. Human PTH-(7-84) inhibits bone resorption in vitro via actions independent of the type 1 PTH/PTHrP receptor. Endocrinology. 2002;143:171–6.
Nguyen M, He B, Karaplis A. Nuclear forms of parathyroid hormone-related peptide are translated from non-AUG start sites downstream from the initiator methionine. Endocrinology. 2001;142:694–703.
Murray TM, Rao LG, Divieti P, Bringhurst FR. Parathyroid hormone secretion and action: evidence for discrete receptors for the carboxyl-terminal region and related biological actions of carboxyl-terminal ligands. Endocr Rev. 2005;26:78–113.
Inomata N, Akiyama M, Kubota N, Jüppner H. Characterization of a novel parathyroid hormone (PTH) receptor with specificity for the carboxyl-terminal region of PTH-(1-84). Endocrinology. 1995;136:4732–40.
Wesseling-Perry K, Harkins GC, Wang H, Elashoff R, Gales B, Horwitz MJ, Stewart AF, Jüppner H, Salusky IB. The calcemic response to continuous parathyroid hormone (PTH)(1-34) infusion in end-stage kidney disease varies according to bone turnover: a potential role for PTH(7-84). J Clin Endocrinol Metab. 2010;95:2772–80.
Huan J, Olgaard K, Nielsen LB, Lewin E. Parathyroid hormone 7-84 induces hypocalcemia and inhibits the parathyroid hormone 1-84 secretory response to hypocalcemia in rats with intact parathyroid glands. J Am Soc Nephrol. 2006;17:1923–30.
Behar V, Pines M, Nakamoto C, et al. The human PTH2 receptor: binding and signal transduction properties of the stably expressed recombinant receptor. Endocrinology. 1996;137:2748–57.
Ureña P. The PTH/PTHrP receptor: biological implications. Nefrologia. 2003;23(Suppl 2):12–7.
Takenaka T, Inoue T, Miyazaki T, Hayashi M, Suzuki H. Xeno-klotho inhibits parathyroid hormone signaling. J Bone Miner Res. 2016;31:455–62.
Torres A, Rodriguez M, Felsenfeld A, Martin-Malo A, Llach F. Sigmoidal relationship between calcitonin and calcium: studies in normal, parathyroidectomized, and azotemic rats. Kidney Int. 1991;40:700–4.
Felsenfeld AJ, Machado L, Rodriguez M. The relationship between serum calcitonin and calcium in the hemodialysis patient. Am J Kidney Dis. 1993;21:292–9.
Rodriguez M, Felsenfeld AJ, Torres A, Pederson L, Llach F. Calcitonin, an important factor in the calcemic response to parathyroid hormone in the rat. Kidney Int. 1991;40:219–25.
Quesada JM, Rodriguez M, Calderon de la Barca JM, Alvarez-Lara A, Martín-Malo A, Mateo A, Martinez ME, Aljama P. Effect of calcitriol replacement on serum calcitonin and parathyroid hormone levels in CAPD patients. Nephrol Dial Transpl. 1995;10:70–4.
Arenas MD, de la Fuente V, Delgado P, Gil MT, Gutiérrez P, Ribero J, Rodríguez M, Almadén Y. Pharmacodynamics of cinacalcet over 48 hours in patients with controlled secondary hyperparathyroidism: useful data in clinical practice. J Clin Endocrinol Metab. 2013;98:1718–25.
Felsenfeld A, Rodriguez M, Levine B. New insights in regulation of calcium homeostasis. Curr Opin Nephrol Hypertens. 2013;22:371–6.
Kazama JJ. The skeletal resistance to PTH and osteoprotegerin. Clin Calcium. 2002;12:764–7.
Kim CS, Bae EH, Ma SK, et al. Association of Serum Osteoprotegerin Levels with bone loss in chronic kidney disease: insights from the KNOW-CKD study. PLoS ONE. 2016;11:e0166792.
Kazama JJ, Shigematsu T, Yano K, Tsuda E, Miura M, Iwasaki Y, Kawaguchi Y, Gejyo F, Kurokawa K, Fukagawa M. Increased circulating levels of osteoclastogenesis inhibitory factor (osteoprotegerin) in patients with chronic renal failure. Am J Kidney Dis. 2002;39:525–32.
Winkler DG, Sutherland MK, Geoghegan JC, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–76.
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical wnt signaling. J Biol Chem. 2005;280:19883–7.
Ellies DL, Viviano B, McCarthy J, Rey J-P, Itasaki N, Saunders S, Krumlauf R. Bone density ligand, sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006;21:1738–49.
Hay E, Bouaziz W, Funck-Brentano T, Cohen-Solal M. Sclerostin and bone aging: a mini-review. Gerontology. 2016;62:618–23.
Fang Y, Ginsberg C, Seifert M, Agapova O, Sugatani T, Register TC, Freedman BI, Monier-Faugere M-C, Malluche H, Hruska KA. CKD-induced wingless/integration1 inhibitors and phosphorus cause the CKD-mineral and bone disorder. J Am Soc Nephrol. 2014;25:1760–73.
Kanbay M, Siriopol D, Saglam M, et al. Serum sclerostin and adverse outcomes in nondialyzed chronic kidney disease patients. J Clin Endocrinol Metab. 2014;99:E1854–61.
García-Martín A, Rozas-Moreno P, Reyes-García R, Morales-Santana S, García-Fontana B, García-Salcedo JA, Muñoz-Torres M. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97:234–41.
Bonani M, Rodriguez D, Fehr T, Mohebbi N, Brockmann J, Blum M, Graf N, Frey D, Wüthrich RP. Sclerostin blood levels before and after kidney transplantation. Kidney Blood Press Res. 2014;39:230–9.
Evenepoel P, Claes K, Viaene L, Bammens B, Meijers B, Naesens M, Sprangers B, Kuypers D. Decreased circulating sclerostin levels in renal transplant recipients with persistent hyperparathyroidism. Transplantation. 2016;100:2188–93.
Roforth MM, Fujita K, McGregor UI, Kirmani S, McCready LK, Peterson JM, Drake MT, Monroe DG, Khosla S. Effects of age on bone mRNA levels of sclerostin and other genes relevant to bone metabolism in humans. Bone. 2014;59:1–6.
Sabbagh Y, Graciolli FG, O’Brien S, et al. Repression of osteocyte Wnt/β-catenin signaling is an early event in the progression of renal osteodystrophy. J Bone Miner Res. 2012;27:1757–72.
Cejka D, Herberth J, Branscum AJ, Fardo DW, Monier-Faugere M-C, Diarra D, Haas M, Malluche HH. Sclerostin and Dickkopf-1 in renal osteodystrophy. Clin J Am Soc Nephrol. 2011;6:877–82.
Bover J, Ureña P, Brandenburg V, et al. Adynamic bone disease: from bone to vessels in chronic kidney disease. Semin Nephrol. 2014;34:626–40.
Moe S, Drüeke T, Cunningham J, Goodman W, Martin K, Olgaard K, Ott S, Sprague S, Lameire N, Eknoyan G. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006;69:1945–53.
Coen G, Mazzaferro S, Ballanti P, Sardella D, Chicca S, Manni M, Bonucci E, Taggi F. Renal bone disease in 76 patients with varying degrees of predialysis chronic renal failure: a cross-sectional study. Nephrol Dial Transpl. 1996;11:813–9.
Graciolli FG, Neves KR, Barreto F, et al. The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017;91:1436–46.
Moe SM, Chen NX, Newman CL, Organ JM, Kneissel M, Kramer I, Gattone VH, Allen MR. Anti-sclerostin antibody treatment in a rat model of progressive renal osteodystrophy. J Bone Miner Res. 2015;30:499–509.
Gupta A, Kallenbach LR, Zasuwa G, Divine GW. Race is a major determinant of secondary hyperparathyroidism in uremic patients. J Am Soc Nephrol. 2000;11:330–4.
Malluche HH, Mawad HW, Monier-Faugere M-C. Renal osteodystrophy in the first decade of the new millennium: analysis of 630 bone biopsies in black and white patients. J Bone Miner Res. 2011;26:1368–76.
Cosman F, Morgan DC, Nieves JW, Shen V, Luckey MM, Dempster DW, Lindsay R, Parisien M. Resistance to bone resorbing effects of PTH in black women. J Bone Miner Res. 1997;12:958–66.
Fishbane S, Hazzan AD, Jhaveri KD, Ma L, Lacson E. Bone parameters and risk of hip and femur fractures in patients on hemodialysis. Clin J Am Soc Nephrol. 2016;11:1063–72.
London GM. Bone-vascular cross-talk. J Nephrol. 2012;25:619–25.
London GM, Marty C, Marchais SJ, Guerin AP, Metivier F, de Vernejoul M-C. Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol. 2004;15:1943–51.
London GM, Marchais SJ, Guerin AP, de Vernejoul MC. Ankle-brachial index and bone turnover in patients on dialysis. J Am Soc Nephrol. 2015;26:476–83.
Quarles LD, Lobaugh B, Murphy G. Intact parathyroid hormone overestimates the presence and severity of parathyroid-mediated osseous abnormalities in uremia. J Clin Endocrinol Metab. 1992;75:145–50.
Torres A, Lorenzo V, Hernández D, Rodríguez JC, Concepción MT, Rodríguez AP, Hernández A, de Bonis E, Darias E, González-Posada JM. Bone disease in predialysis, hemodialysis, and CAPD patients: evidence of a better bone response to PTH. Kidney Int. 1995;47:1434–42.
Hercz G, Pei Y, Greenwood C, Manuel A, Saiphoo C, Goodman WG, Segre GV, Fenton S, Sherrard DJ. Aplastic osteodystrophy without aluminum: the role of suppressed parathyroid function. Kidney Int. 1993;44:860–6.
Torregrosa JV, Bover J, Cannata Andia J, et al. Spanish Society of Nephrology recommendations for controlling mineral and bone disorder in chronic kidney disease patients (S.E.N.-M.B.D.). Nefrologia. 2011;31(Suppl 1):3–32.
Wein MN. Parathyroid hormone signaling in osteocytes. J Bone Miner Res Plus. 2018;2:22–30.
Garrett G, Sardiwal S, Lamb EJ, Goldsmith DJA. PTH—a particularly tricky hormone: why measure it at all in kidney patients? Clin J Am Soc Nephrol. 2013;8:299–312.
Yilmaz MI, Siriopol D, Saglam M, et al. Osteoprotegerin in chronic kidney disease: associations with vascular damage and cardiovascular events. Calcif Tissue Int. 2016;99:121–30.
DeFronzo RA, Alvestrand A, Smith D, Hendler R, Hendler E, Wahren J. Insulin resistance in uremia. J Clin Invest. 1981;67:563–8.
Blum WF, Ranke MB, Kietzmann K, Tönshoff B, Mehls O. Growth hormone resistance and inhibition of somatomedin activity by excess of insulin-like growth factor binding protein in uraemia. Pediatr Nephrol. 1991;5:539–44.
Koizumi M, Komaba H, Fukagawa M. Parathyroid function in chronic kidney disease: role of FGF23-Klotho axis. Contrib Nephrol. 2013;180:110–23.
Evenepoel P, Rodriguez M, Ketteler M. Laboratory abnormalities in CKD-MBD: markers, predictors, or mediators of disease? Semin Nephrol. 2014;34:151–63.
Román-García P, Carrillo-López N, Naves-Díaz M, Rodríguez I, Ortiz A, Cannata-Andía JB. Dual-specificity phosphatases are implicated in severe hyperplasia and lack of response to FGF23 of uremic parathyroid glands from rats. Endocrinology. 2012;153:1627–37.
Galitzer H, Ben-Dov IZ, Silver J, Naveh-Many T. Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease. Kidney Int. 2010;77:211–8.
Komaba H, Goto S, Fujii H, et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 2010;77:232–8.
Brown AJ, Ritter CS, Finch JL, Slatopolsky EA. Decreased calcium-sensing receptor expression in hyperplastic parathyroid glands of uremic rats: role of dietary phosphate. Kidney Int. 1999;55:1284–92.
Brown AJ, Dusso A, Lopez-Hilker S, Lewis-Finch J, Grooms P, Slatopolsky E. 1,25-(OH)2D receptors are decreased in parathyroid glands from chronically uremic dogs. Kidney Int. 1989;35:19–23.
Mithal A, Kifor O, Kifor I, Vassilev P, Butters R, Krapcho K, Simin R, Fuller F, Hebert SC, Brown EM. The reduced responsiveness of cultured bovine parathyroid cells to extracellular Ca2+ is associated with marked reduction in the expression of extracellular Ca(2+)-sensing receptor messenger ribonucleic acid and protein. Endocrinology. 1995;136:3087–92.
Ritter CS, Finch JL, Slatopolsky EA, Brown AJ. Parathyroid hyperplasia in uremic rats precedes down-regulation of the calcium receptor. Kidney Int. 2001;60:1737–44.
Fukuda N, Tanaka H, Tominaga Y, Fukagawa M, Kurokawa K, Seino Y. Decreased 1,25-dihydroxyvitamin D3 receptor density is associated with a more severe form of parathyroid hyperplasia in chronic uremic patients. J Clin Invest. 1993;92:1436–43.
Silver J, Kilav R, Naveh-Many T. Mechanisms of secondary hyperparathyroidism. Am J Physiol Physiol. 2002;283:F367–76.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bover, J., Ureña-Torres, P.A., Evenepoel, P., Lloret, M.J., Guirado, L., Rodríguez, M. (2020). PTH Receptors and Skeletal Resistance to PTH Action. In: Covic, A., Goldsmith, D., Ureña Torres, P. (eds) Parathyroid Glands in Chronic Kidney Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-43769-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-43769-5_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-43768-8
Online ISBN: 978-3-030-43769-5
eBook Packages: MedicineMedicine (R0)