
Abstract
Conventional osteosarcoma (OS) is a high-grade intraosseous malignancy with production of osteoid matrix; however, a deeper dive into the underlying genetics reveals genomic complexity and instability that result in significant tumor heterogeneity. While early karyotyping studies demonstrated aneuploidy with chromosomal complexity and structural rearrangements, further investigations have identified few recurrent genetic alterations with the exception of the tumor suppressors TP53 and RB1. More recent studies utilizing next-generation sequencing (NGS; whole-exome sequencing, WES; and whole-genome sequencing, WGS) reveal a genomic landscape predominantly characterized by somatic copy number alterations rather than point/indel mutations. Despite its genomic complexity, OS has shown variable immune infiltrate and limited immunogenicity. In the current chapter, we review the hallmarks of OS genomics across recent NGS studies and the immune profile of OS including a large institutional cohort of OS patients with recurrent and metastatic disease. Understanding the genomic and immune landscape of OS may provide opportunities for translation in both molecularly targeted therapies and novel immuno-oncology approaches.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Osteosarcoma
- Genomics
- Next-generation sequencing (NGS)
- Chromothripsis
- Telomere lengthening
- Immune profiling
Introduction
Osteosarcoma (OS) is the most common primary malignancy of the bone predominantly occurring in adolescents with a second peak in incidence as secondary OS among older adults [44]. For patients presenting with localized disease at diagnosis, standard multi-agent chemotherapy combined with surgical resection yields long-term survival rates of ~70% [6, 44]. Metastatic disease either at diagnosis or at the time of recurrence portends a poor prognosis with survival of 20–30% [28, 42]. Thus, there has been a long-standing interest in understanding the underlying biology of OS tumorigenesis, evolution, and metastasis in order to identify novel treatment strategies and improve survival outcomes.
Recent progress made in next-generation sequencing (NGS) and molecular genetic studies of osteosarcoma has broadened our view of the genetic hallmarks of the disease and potential therapeutic approaches for patients. The point mutation burden of OS is around 1.1~1.5 per Mb [13, 49, 71], making it the highest mutation burden among pediatric solid tumors but intermediate overall and significantly lower than melanoma or non-small cell lung cancer (Fig. 2.1). The OS genome is characterized by genomic complexity and instability, enriched with rearrangements, and somatic copy number alterations. Figure 2.2 shows the MD Anderson Cancer Center osteosarcoma (MDACC OS) cohort has a higher level of rearrangements than most of other tumor types. This suggests that rearrangements and copy number alterations are major driving forces contributing to OS oncogenesis. In addition, it has become clear that genome instability has a significant impact on the interaction between the tumor cells and immune system [47]. In this chapter, we review the molecular genetics of OS, which are associated with genome instability and immune landscape, based on the findings of recent whole-genome/whole-exome sequencing (WGS/WES) studies (Table 2.1).

Somatic point mutation burden in osteosarcoma as compared to other cancer types within the TCGA [61]

Frequency of structural variants in OS (bone osteosarcoma) and other human cancer types within the ICGC [37]
Genomic Landscape of Osteosarcoma
Key Altered Genes and Pathways Associated with OS Genome Instability and Oncogenesis
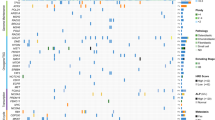
OS is characterized by complex genome instability and high level of genetic heterogeneity [4, 13]. The majority of the resultant genetic alterations are associated with copy number changes and genome rearrangement. Genome instability can lead to changes in both the cancer genome and the tumor microenvironment. Elucidating the mechanisms of genome instability in OS would thus aid in our understanding of tumorigenesis, evolution, progression, and metastasis in order to develop new therapeutic approaches [76]. This section reviews key altered genes and pathways associated with OS genome instability and oncogenesis identified in recent WGS/WES studies . The most frequently altered genes and their associated cancer signaling pathways of our MDACC OS cohort of recurrent and metastatic OS patients (MDACC OS ) are shown in Fig. 2.3. Importantly, the majority of these pathways are associated with the underlying genome instability that is a hallmark of OS.

The mutation landscape of the MDACC OS cohort. Genomic alteration identified by WGS for selected genes and key pathways
TP53
TP53 , a tumor suppressor gene, codes for a protein that can respond to diverse cellular stresses and thereby induce cell cycle arrest, apoptosis, senescence, and DNA repair. Somatic mutations in TP53 are one of the most frequent alterations in human cancers, in which the majority of genetic alterations across cancer types are missense substitutions [50]. With WES and targeted sequencing, it had previously been estimated that only 20%~50% of osteosarcomas carry mutations in the p53 pathway, and other portion of the tumors were identified as so-called TP53 wild type [33, 79]). However, in a WGS study of 34 pediatric OS samples, Chen et al. [13] discovered new insights into alterations in TP53 to promote the OS oncogenesis. In their cohort, they identified 55% of TP53 mutations were caused by structural variations, whose breakpoints were mostly confined to the first intron of the gene [13], thereby inactivating TP53. In the 36 samples of our MDACC OS cohort, we also identified 9 samples (25%) as having TP53 structural variations. Therefore, it now is suspected that up to 75–90% of OS patients harbor various types of TP53 genetic alterations [13, 49, 71], which is the most prevalent genetic alteration in OS.
Loss of the TP53 pathways that disable the cell’s ability to respond to DNA damage mediates genome instability and triggers OS oncogenesis [40]. Several TP53-deficient cell lines and genetically engineered mouse models also have been developed to model OS oncogenesis and indicated the causal relation between TP53 alterations and OS initiation/genome instability [23, 66]. Taken together, these mechanistic studies and associations observed from sequenced patient samples identify TP53 alterations as having the strongest association with genome instability and oncogenesis in OS .
RB1 and Other DNA Damage Repair Pathways
Retinoblastoma transcriptional corepressor 1 (RB1) , a key regulator of cell cycle progression by controlling the G1/S phase transition, is another prevalent genetic alteration in OS. Alterations in RB pathway can prevent cell cycle arrest in response to DNA damage to induce genome instability and promote oncogenesis [65]. Alterations in RB1 have been identified in 50%–78% of OS across NGS studies [13, 33, 49, 71]. Unlike TP53, the depletion of RB alone was not sufficient to induce OS formation in mouse models, and studies speculated that RB alterations may synergize with TP53 inactivation during OS oncogenesis [52].
Breast cancer susceptibility genes (BRCA1/2) encode nuclear phosphoproteins that are involved in molecular signaling in transcription, DNA repair of double-stranded breaks, and recombination, thereby playing a role in maintaining genomic stability and acting as a tumor suppressor [73]. Alterations in these genes are known to be responsible for inherited breast and ovarian cancers. In OS, Kovac et al. [33] identified BRCA1/2 inactivation in 112 (91%) and 96 (78%) of their 123 samples, primarily caused by copy number alterations. They also showed that BRCA alterations in OS cell lines are associated with sensitivity to PARP inhibition, a strategy that was shown to induce cell cycle arrest and apoptosis in BRCA1-, BRCA2-, and PALB2-deficient breast cancers [62]. We also identified alterations (mostly copy number LOH) in BRCA in 89% of our 36 MDACC OS samples [71]. By analyzing the mutation spectrum of their sample cohort, Kovac [33] also identified COSMIC signature 3 and signature 5 in their WES data. Signature 5 is associated with an age-related mutational process, whereas signature 3 is characterized by a pattern enriching of C > G substitutions that is strongly associated with BRCA1 and BRCA2 mutations in breast, pancreatic, and ovarian cancers [1]. However, the mutation spectrum of the MDACC OS WGS sample cohort is dominated by C > T substitutions, C > A, and T > C substitutions. We identified two prevalent mutation signatures: COSMIC signature 5 and signature 8. Behjati et al. [4] also identified signature 5 and 8 are the most prevalent mutation signatures in their OS WGS cohort. We found that signature 8 is significantly associated with worse prognosis, but its etiology is still unknown. The difference of the mutation signature analysis results may be related to the lower mutation burden observed in OS WES data, compared to OS WGS data. In addition, some studies recently proposed that the genetic association of mutation signatures would be tissue-specific or cancer type-specific [7, 26]. Therefore, the association between signature 3 and BRCA in OS warrants further validation in other OS cohorts.
Telomere Lengthening Pathways
Telomeres can protect chromosomes of a cell from DNA damage but become shorter with each cell division, eventually leading to senescence or apoptosis [40]. During oncogenesis, cancer cells frequently activate either telomerase-dependent or telomerase-independent elongation mechanisms in order to protect against telomere shortening in accelerated cell division cycle and maintain unlimited growth and clonal evolution of genomically unstable cells [40]. Through the process of telomere lengthening, cancer cells can accumulate large amounts of genome alterations.
Promoter mutations of TERT , an active component of telomerase, were previously identified in 1 of 23 (4.3%) OS patient samples [31]. No TERT mutation was found in current OS NGS studies [4, 13, 33, 49] except our MDACC OS cohort. We identified TERT promoter mutations (chr5: 1295228 C > T) in two patient samples, but we found the mutations and expression of TERT are not significantly associated with longer telomeres. Therefore, an association between TERT mutations with telomere lengthening in OS has not been well-established.
A telomerase-independent mechanism termed alternative lengthening of telomeres (ALT) also has been frequently identified in several cancer types [20]. Cancers utilizing ALT often have lost function of ATRX, a chromatin remodeling protein, and/or DAXX, a death domain associated protein, through DNA mutation or deletion. Chen’s study [13] identified five samples with point mutations in ATRX and five with focal deletions or structural variation affecting the coding region of the gene. They also found samples with ATRX alterations tend to have a greater number of telomeric reads estimated from the sequencing data. Our MDACC OS cohort also identified seven patients with deleterious alterations in ATRX (7/36, 20%) as well as one patient with copy number loss in DAXX who all had telomere lengths greater than the cohort median. Lower expression levels of ATRX were also significantly correlated with longer telomere lengths. We also found that patient samples with the longest telomere length carried alterations in both TP53 and ATRX, supporting the permissive context in which TP53 alterations can allow for activation of ALT in OS. In addition, our MDACC dataset also showed that the expression levels of known telomere maintenance genes, including HNRNPA2B1, WRN, and HUS1, were also significantly correlated with telomere length [14]. However, the exact mechanisms surrounding telomere maintenance in the ALT pathway are unclear, and the effects of the telomere-related mutations on ALT are still needed to be explored. Based upon these findings, there is a growing interest in investigating ATR inhibitors or other agents that target DNA damage response in OS .
IGF Signaling/PI3K-mTOR
The insulin-like growth factor (IGF) signaling includes three ligands (INS, IGF1, and IGF2), three receptors (IR, IGF1R, and IGF2R), as well as six IGF-binding proteins (IGFBPs), which provide a potent proliferative signaling system that can block apoptosis and stimulate growth and differentiation in many cell types. Numerous studies have demonstrated the role of IGF signaling in the development and progression of various cancer types as well as its role in resistance to chemotherapeutic agents [38]. Given the association between IGF signaling and bone growth [72], disorders of IGF signaling are thought to be implicated in OS pathogenesis. Recently, Behjati et al. [4] identified recurrent alterations in IGF signaling as a potential therapeutic target in OS treatments. They found alterations of IGF signaling in 8 of 112 (7%) WES and WGS samples and validated the observation with IGF1R amplifications observed in 14% of 87 OS samples using fluorescence in situ hybridization (FISH). We also identified alterations of IGF signaling in 7 of the 36 MDACC OS samples (20%) (Fig. 2.3). Interestingly, some studies have shown that alteration in TP53 and DNA repair defects in tumor cells may activate IGF1R signaling [3, 69]; however, additional studies are still needed to explore this cause-effect relation. In addition, Perry et al. [49] also found recurrent mutations in the downstream signaling pathways of IGF signaling, the PI3K/mTOR pathway, in 14 of the 59 OS samples, a similar rate to the Behjati cohort which identified downstream pathway alterations including PI3K or MAPK signaling in 27% of tumors. Perry et al. [49] also demonstrated OS cell lines are responsive to pharmacologic and genetic inhibition of the PI3K/mTOR pathway both in vitro and in vivo and proposed this pathway as a therapeutic target for the treatment of OS. Our MDACC cohort similarly identified a high frequency of alterations in the PI3K/mTOR pathway in 28 of the 36 (78%) OS samples (Fig. 2.3). Across these cohort studies, these findings support further investigation of IGF1R, PI3K, or mTOR inhibition in patients with OS which may have greater activity or relevance in a biomarker selected patient population .
Other Genomic Events Associated with OS Genome Instability
While several of the aforementioned pathways can mediate genomic complexity and instability in OS, other genetic or epigenetic alterations and events have been implicated in increasing genome instability in OS. This section will review several genomic events associated with genomic complexity and instability of OS which have been further elucidated by current WGS/WES studies.
Genome Doubling
During clonal evolution in oncogenesis, genomically unstable cells continually lose and gain whole and/or parts of chromosomes to provide potent selective pressure for clonal expansion. However, genome instability beyond a certain threshold is likely to cause cancer cells with unviable karyotypes [76]. Therefore, OS cells need to maintain viability of their TP53/RB1 mutation-induced unstable genomes through multiple mechanisms. Whole-genome doubling (WGD) is one mechanism that can increase viability of cancer cells with significant chromosomal instability [18, 80, 81]). By applying the allele-specific copy number profiles inferred from WGS data and the algorithm modified from the previously published studies [11, 18], we identified WGD in 58% (22/38) of samples in the MDACC OS cohort, a comparable frequency to what has been observed in colorectal and breast cancer [11]. In addition, we also found that OS samples with WGD tend to have a higher number of rearrangements and copy number alterations than those without WGD. Furthermore, 50% (18/36) of patients had losses of heterozygosity (LOH) in TP53 and/or RB1 along with WGD. Given the inherent lower likelihood of losing two copies after WGD, these findings support that TP53 and RB1 aberrations likely occurred prior to WGD [11].
Chromothripsis
Chromothripsis is the genomic process by which massive genomic rearrangements are acquired in a single catastrophic event [21]. Chromothripsis may generate genetic drivers in oncogenesis through DNA copy number gain and loss as well as rearrangements, such as translocations. Chromothripsis is associated with both somatic and germ line TP53 mutations in pediatric medulloblastoma and acute myeloid leukemia [51]. In addition, chromothripsis can be associated with telomere crisis induced by telomere shortening in accelerated cancer cell cycle division [39]. In this study, the authors showed that telomere loss promotes end-to-end chromosome fusions and dicentric chromosomes during mitosis and undergoes breakage-fusion-bridge cycles, eventually resulting in hundreds of DNA breaks [39]. However, these associations warrant further investigation and validation, particularly in relation to OS pathogenesis. Chromothripsis has been observed at varying frequencies (20–89%) in OS patient samples of all ages across OS studies [4, 13, 71]. To date, the etiological factors and mechanisms underlying OS chromothripsis are still unknown, and no specific genomic regions and genes were found to be significantly associated with chromothripsis in OS samples. Our MDACC OS studies recently found that there was a trend for younger patients to have rearrangements that are clustered and associated with chromothripsis as compared with older patients. This result was also observed in the TARGET OS cohort dataset. This suggests that oncogenesis may be more driven by catastrophic chromothripsis events in young OS patients as compared to older adults.
Kataegis
Kataegis is a pattern of localized hypermutation (enriched of C > T and C > G changes at TpC dinucleotides) associated with the APOBEC deaminases [55]. Chen et al. [13] and Perry et al. [49] found approximately 50–85% of their OS patient samples showed kataegis patterns. Approximately 60% of the MDACC OS samples also showed the kataegis patterns. No important cancer genes were found to be recurrently located in the kataegis regions identified in OS samples. However, most of these kataegis patterns occurred in no-coding regions, some of which may include cis- and trans-regulatory elements or may be transcribed into functional noncoding RNA molecules, such as transfer RNA, ribosomal RNA, and regulatory RNAs. More investigations on the association of these kataegis regions with OS oncogenesis are necessary in future studies.
The Osteosarcoma Immune Landscape and Immunogenomic Interplay
Interactions between the immune system and tumor play an important role in effective tumor control. Aberrations in this interaction can lead to ineffective tumor surveillance, enhance tumor growth, and enable metastatic disease progression. For this reason, there has been a long-standing interest in targeting this interaction and modulating the host’s immune response as a strategy to eliminate cancer. Targeting immune checkpoints , such as cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death 1 (PD-1)/ligand 1 (PD-L1), has been an overwhelmingly successful step forward for immunotherapy in the treatment of cancer. Immune cells, including CD8+ tumor-infiltrating lymphocytes , are initially attracted to tumor cells by the presence of tumor-specific antigens which are encoded by somatic alterations in cancer cells. Tumors can escape immune-surveillance by modulating antigen expression and upregulating inhibitory immune checkpoints to lead to immune cell apoptosis, anergy, and tolerance. Immune checkpoint inhibitors (ICI) block such signals in order to activate an antitumor immune response. The success of ICIs in the clinic, yielding durable responses in a subset of patients with previously incurable metastatic disease, such as melanoma and lung cancer, has revived enthusiasm for immunotherapy and established a new paradigm for cancer treatment [16]. The Food and Drug Administration (FDA) has approved a number of ICIs including anti-PD-1, anti-PD-L1, and anti-CTLA4 antibodies among others for the treatment of a wide range of malignancies. Despite their remarkable success, only a subset of cancer patients benefit from these therapies, and responses are varied across patients and cancer types. Therefore, there is a growing need to understand mechanisms of the resistance to ICI and identify predictive biomarkers for personalized immunotherapy approaches.
Osteosarcoma demonstrates significant genetic complexity and genome instability with resultant high levels of genomic rearrangements and the highest point mutation burden as compared to other pediatric cancers, suggesting that these genomics factors may yield neoantigens capable of eliciting an immune response. However, despite this rationale, recent clinical trials using immune checkpoint inhibitors in OS have been disappointing. Therefore, this section will outline the OS immune landscape and genomic features that may contribute to resistance to ICI and other immunotherapy agents in OS.
OS Immune Landscape
Transcriptome profiles derived from bulk RNAseq and other methods have been used to study features of the tumor and the microenvironment that are associated with tumor response or resistance to ICI in several cancer types including OS. Most often, these studies compare the transcriptome of responders vs. nonresponders to identify key differentially expressed genes that may account for response or resistance. While several studies have explored the transcriptional signatures linked to ICI responses across various clinical patient cohorts [25], the small sample size of most of these studies limits their generalizability [43]. This is particularly true for rare cancers such as OS. One particular challenge in OS is the overall lack of responders to ICI and the concurrent lack of high-quality patient samples to undertake such a study. Given the limited number of responses on clinical trials to date such as the SARC028 study of pembrolizumab in soft tissue and bone sarcoma [59], such a transcriptome analysis of responders vs. nonresponders has not been feasible. Therefore, we focused on a larger cohort of OS patients with poor risk – those with recurrent metastatic disease – as this cohort is thought to represent OS patients who would be considered for treatment with ICI. The MDACC OS cohort also includes four pretreatment specimens from patients treated with anti-PD-L1 in combination with anti-CTLA4 therapy, all of whom did not respond to treatment.
In the ongoing search for biomarkers, immune infiltration levels (and, in particular, tumor-infiltrating CD8+ T cells) and PD-1/PD-L1 expressions have been shown to be associated with response to ICIs. However, expression is variable, and the significance across studies and across tumor types remains controverted as a considerable number of patients with high levels of immune infiltration and high PD-1/PD-L1 expression have poor responses or fail to respond to treatment [56]. These points aside, several studies have recently been conducted in OS evaluating the prevalence and prognostic significance of immune infiltrate and PD-1/PD-L1 expression. Shen et al. [57] first measured RNA expression levels for PD-L1 in 38 OS samples using quantitative real-time RT-PCR and found that high levels of PD-L1 are expressed in a subset of OS and that PD-L1 expression is positively correlated with tumor-infiltrating lymphocytes. Koirala et al. [32] and Palmerini et al. [48] also identified the similar results in much larger OS cohorts. Further, they identified an association between CD8+ infiltrate and superior overall survival, whereas infiltration with dendritic cells and macrophages as well as PD-L1 expression was associated with a poor prognosis. Within the MDACC OS cohort, we did not observe an association between immune infiltrate, CD8+ TIL, or PD-L1 expression , and overall survival. This may be due in part to the poor risk nature of the patients we included for study. Further translational studies are needed to determine if either immune infiltrate or PD-L1 expression correlates with clinical benefit from ICIs in the treatment of OS.
To understand the immune infiltrate level of OS in a broader context, we compared the immune infiltration score [74] derived from the bulk RNAseq data of our cohort against other tumor types profiled in the Cancer Genome Atlas (TCGA) , the 87 TARET OS samples, the 7 OS samples from the Behjati’s study [4], as well as 4 patients with metastatic OS who were treated with ICIs but exhibited no objective responses [71]. All four OS cohorts have comparable median immune score (Fig. 2.4). We observed that these four OS cohorts have intermediate median immune scores that are lower than many of the cancer types that have shown clinical benefit and treatment responses to ICIs with high immune infiltrate levels such as melanoma (TCGA-SKCM) and lung cancer (TCGA-LUAD and TCGA-LUSC) but are higher than those that have shown minimal activity with current immunotherapy approaches such as uveal melanoma (TCGA-UVM) (Fig. 2.4). When compared to other sarcoma subtypes, the median immune scores of dedifferentiated liposarcoma (TCGA-DDLPS) and undifferentiated pleomorphic sarcoma (TCGA-UPS) – two soft tissue sarcoma subtypes where ICI are active – are higher than OS samples. We also found less than 10% of OS samples whose immune infiltration levels were among the highest quartile across tumor types. These results suggest that while most OS specimens may have insufficient immune infiltrate to elicit meaningful responses to ICI, there is a subset of patients who would be predicted to benefit. However, immune infiltrate alone is inadequate to predict treatment response.

Immune scores in TCGA tumor types and the four OS cohorts. ESTIMATE scores derived from RNAseq show intermediate immune score for OS across cohorts as compared to the TCGA
Bulk RNAseq data were also used to infer profiles of different infiltrated immune cells [12, 36, 63]. We characterized the composition of various infiltrated immune cells across our MDACC OS samples [71] and identified three clusters of our samples: immune-high, intermediate, and low (Fig. 2.5). The immune-low and the immune-high samples, respectively, have the lowest and highest levels of all immune cell types including CD8+ lymphocytes. Several known tumor-intrinsic immunosuppressive pathways were found to be deregulated between the immune-high and low cluster samples, such as PD-1 signaling, CTLA4 pathway, and IFNG signaling, suggesting that the immune-high tumors may upregulate immune-suppressive signals that inhibit T-cell activation.

Immune cell profiling of the MDACC OS cohort. RNAseq identifies three unique clusters with low (C1), intermediate (C2), and high (C3) immune infiltrate
OS Genomic Features Associated with the Immune Response
Immune modulation in cancer cells is recognized as a hallmark of cancer initiation and progression, implying that tumor cell-intrinsic factors are associated with tumor response/resistance to ICIs [56]. Therefore, this section reviews several OS genomic features revealed by WES/WGS/RNA-seq studies that may be associated with OS response/resistance to ICIs. These factors may explain in part why the majority of OS patients have failed to benefit from treatment with ICIs and also present opportunities for novel therapeutic approaches.
Neoantigens
During oncogenesis , tumors accumulate thousands of genetic alterations, including point mutations, indels, and rearrangements. Some of them alter the amino acid sequence of the encoded proteins, called neoantigens, which do not present in normal cells [30]. The immune system can discriminate self from these non-self-antigens expressed by cancer cells and activate immune response to kill cancer cells. ICI can enhance and strengthen the immune response to non-self-antigens and promote antitumor activity .
Correlations between tumor-specific antigen and tumor response to checkpoint blockade therapies have largely been focused on tumor mutation burden, specifically point mutations. Several studies showed that nonsynonymous point mutation burden is one of the most reliable predictive biomarkers associated with responsiveness to ICIs in both melanoma and lung cancer [77, 78]. Recently, Turajlic et al. [64] investigated whether the frameshift nature of indel mutations can create novel open reading frames and a large quantity of neoantigens, which might contribute to the immunogenic response. They found renal cell carcinomas, one of the cancer types that have clinical benefit and response to immune checkpoint blockade related to high immune infiltrate levels, have the highest proportion and number of indel mutations, compared to other cancer types in TCGA [64]. Their analysis also showed that frameshift indel count is significantly associated with response to ICIs.
Although OS has a much lower point mutation burden compared to those of melanoma and lung cancer (Fig. 2.3), OS demonstrates significant genomic complexity with the high levels of genomic rearrangements that could potentially generate high-level neoantigens. However, we found that most of mutations detected by WGS (i.e., whole genome DNA-seq) were not detected by RNAseq in our MDACC OS cohort samples [71]. Unexpressed mutations tended to occur in genes that have low expression or whose variant allele frequencies were low. The limited overlap of point mutations identified in both WGS and RNAseq also has been observed in non-small cell lung cancer and glioblastoma [15, 45]. In addition, we observed that very few predicted rearrangements involving coding regions were expressed, suggesting that most of the rearrangements are truncated or harbor premature termination codons [71]. Therefore, the nonsense-mediated mRNA decay (NMD) pathway, which selectively degrades mRNAs harboring premature termination codons [9], may contribute to the low level of neoantigens generated from large amount of rearrangements in OS. In the MDACC OS cohort, we observed a positive association between NMD factors and the number of gene-containing rearrangements as well as immune infiltration, indicating that there may be substantial transcript suppression in rearranged OS genomes [71]. These indicate that highly mutated and rearranged OS genome may not generate sufficient neoantigens to elicit an immune response. Strategies that enhance neoantigen expression in combination with immune checkpoint inhibition warrant further evaluation in OS .
Aneuploidy
In addition to increasing the mutational burden , genome instability in cancer cells also leads to chromosome copy number alterations that are categorized in two major classes: whole or arm level copy number changes known as aneuploidy or focal copy number changes [5]. While focal copy number changes that involve tumor suppressor genes or oncogenes are often considered actionable targets in cancer therapy, the functional relationship between aneuploidy and oncogenesis is not well understood. Several recent studies have shown that aneuploidy is associated with the immune suppression across multiple cancer types [17, 47, 53, 60]. Of interest, Davoli et al. [17] found that chromosome and arm level of copy number alterations have a greater contribution to immune suppression than focal level of copy number alterations. They hypothesize that chromosome and arm level of copy number alterations can impact gene expression of a large number of genes and may thus impair or deregulate cellular signaling needs for cytotoxic immune cell infiltration. However, their studies did not specifically explore the impact of copy number gain and/or loss on immune infiltration. In melanoma, Roh et al. [53] identified a higher burden of copy number loss in nonresponders to CTLA-4 and PD-1 blockade as compared to responders and found that these copy number losses were associated with decreased expression of genes in immune-related pathways. However, the same association was not identified between copy number gain and immune response. Although frequent, significantly less is known about aneuploidy and copy number changes as they relate to immune suppression in OS. In the MDACC OS cohort, we similarly found that copy number loss has a significant negative correlation with the immune scores such that OS samples with high levels of copy number loss had significantly less immune cell infiltrate. Similarly, such a correlation was not observed between copy number gains and the immune scores in OS. We hypothesized that copy number loss may impact gene expression balance more than copy number gain because copy loss may lead to permanent loss of many genes and eventually impact immune response .
Genetic Alterations and Pathways
A range of genetic/epigenetic aberrations in tumor cells can influence various aspects of the immune landscape, such as activation of immunosuppressive immune cells, regulation of dendritic cell activation and T-cell priming, instigation of tumor resistance to T-cell attack, and deregulation of immune checkpoint molecule expression [30, 43, 54, 68]. Alterations and deregulation of multiple oncogenic pathways such as MAPK/PTEN/PI3K, WNT/beta-catenin, and JAK/STAT (termed genetic T-cell exclusion) have specifically been shown to be associated with resistance to ICIs. Aberrations in antigen processing/presentation pathway or interferon-gamma signaling have also been implicated in primary resistance to immunotherapy [2, 56, 58, 75].
In our MDACC OS cohort , we also found many of these tumor-intrinsic immunosuppressive pathways such as IFNG, MAPK/PI3K/mTOR, and JAK/STAT and antigen presenting pathways are dysregulated between the immune-high and low samples that were identified by RNAseq (sect. 3.1). Some of these pathways were also found to be deregulated between OS and normal samples through the analysis of RNAseq data [81]). These pathways are currently being explored as potential combination strategies that could extend the benefits of ICIs for OS treatments.
In our MDACC OS cohort, we also integrated genomic aberrations and transcriptomic analyses to identify genes whose aberrations and gene expression are significantly associated with immune infiltration, such as TP53 and PARP2. Among other findings, we identified a negative association between PARP2, a druggable target involved in the DNA damage response, amplification (35% samples), and gene expression with the immune infiltration score (such that OS samples with high PARP2 expression had significantly lower immune infiltrate). While prior studies have demonstrated the sensitivity of OS cell lines to PARP inhibitors [19, 33], these studies largely focused on the role of BRCA2 deletion in DNA damage response and its synergy with PARP inhibitions. However, PARP2 amplification was found in the samples of these OS studies [4, 33], and the association of PARP2 amplification with immune response in OS was not previously identified. PARP appears to play an important role in modulating the immune response. PARP inhibition can increase intratumoral CD8+ T cells and drive production of IFNg and TNFa in murine ovarian tumors [24] and can upregulate of PD-L1 expression, providing further rationale for its combination of immune checkpoint blockade [27]. Several trials are currently underway evaluating PARP inhibitors in combination with immune checkpoint inhibitors across a range of solid tumors but have not yet been undertaken in OS [10].
Translational Applications
Across NGS studies, few recurrent potentially actionable alterations have been characterized including PI3K/AKT/mTOR pathway, IGF signaling, and the potential role of PARP inhibitors among a subset of OS with BRCA signatures. However, many of these targets also contribute to immune modulation and therefore may be relevant in combination with immunotherapy for the treatment of OS. For example, mutations in PI3K or AKT can lead in constitutive PD-L1 expression [33], whereas mTOR inhibition can enhance the production of CD8+ memory T cells [35]. PARP inhibition has been shown to increase intratumoral CD8+ T cells, drive production of IFNγ and TNFα [24], and upregulate PD-L1 expression independent of its role in the DNA damage response [27] . In the MDACC OS cohort, PARP2 was functionally associated with the MHC class I antigen presentation pathway further supporting the rationale for exploring PARP + immunotherapy combinations in osteosarcoma. Data to support combining IGF signaling inhibitors with immunotherapies are limited but may warrant further exploration [67]. A practical challenge facing targeted therapy + immunotherapy combinations in the treatment of osteosarcoma is the lack of single agent efficacy data for either immune checkpoint inhibitors or many of the available targeted therapies in unselected patient populations. Immunotherapy combinations with VEGFR and/or mTOR inhibitors may be an appropriate starting point for clinical trials given their activity in metastatic osteosarcoma and early promising combination data in selected soft tissue sarcoma subtypes [70].
Conclusion
NGS studies in OS have yielded additional insight into its genomic complexity and heterogeneity. Predominant genomic features such as aneuploidy and pathway alterations as well as limited neoantigen expression influence the immune landscape of OS and result in a similarly diverse and heterogeneous immune spectrum of tumors. However, these immunosuppressive mechanisms in OS may themselves present opportunities for novel therapeutic combinations.
References
Alexandrov LB, Nik-Zainal S, Wedge DC et al (2013) Signatures of mutational processes in human cancer. Nature 500(7463):415–421
Bai J, Gao Z, Li X et al (2017) Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget 8(66):110693–110707
Bayram F, Bitgen N, Donmez-Altuntas H et al (2014) Increased genome instability and oxidative DNA damage and their association with IGF-1 levels in patients with active acromegaly. Growth Hormon IGF Res 24(1):29–34
Behjati S, Tarpey PS, Haase K et al (2017) Recurrent mutation of IGF signaling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat Commun 8:ncomms15936
Beroukhim R, Mermel CH, Porter D et al (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463(7283):899–905
Bielack SS, Kempf-Bielack B, Delling GN et al (2002) Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 20:776–790
Blokzijl F, de Ligt J, Jager M et al (2016) Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538(7624):260–264
Bousquet M, Noirot C, Accadbled F et al (2016) Whole-exome sequencing in osteosarcoma reveals important heterogeneity of genetic alterations. Ann Oncol 27:738–744
Brogna S, Wen J (2009) Nonsense-mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol 16(2):107–113
Burgess MA, Bolejack V, Tine BAV et al (2017) Multicenter phase II study of pembrolizumab (P) in advanced soft tissue (STS) and bone sarcomas (BS): final results of SARC028 and biomarker analyses. J Clin Oncol 35:11008–11008
Carter SL, Cibulskis K, Helman E et al (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30(5):413–421
Charoentong P, Finotello F, Angelova M et al (2017) Pan-cancer Immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 18(1):248–262
Chen X, Bahrami A, Pappo A et al (2014) Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7:104–112
Chen Y-J, Hakin-Smith V, Teo M et al (2006) Association of mutant TP53 with alternative lengthening of telomeres and favorable prognosis in glioma. Cancer Res 66:6473–6476
Coudray A, Battenhouse AM, Bucher P et al (2018) Detection and benchmarking of somatic mutations in cancer genomes using RNA-seq data. bioRxiv 2018:249219
Couzin-Frankel J (2013) Breakthrough of the year 2013. Cancer immunotherapy. Science 342(6165):1432–1433
Davoli T, Uno H, Wooten EC et al (2017) Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355(6322):pii:eaaf8399
Dewhurst SM, McGranahan N, Burrell RA et al (2014) Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov 4(2):175–185
Engert F, Kovac M, Baumhoer D et al (2017) Osteosarcoma cells with genetic signatures of BRCAness are susceptible to the PARP inhibitor talazoparib alone or in combination with chemotherapeutics. Oncotarget 8(30):48794–48806
Flynn RL, Cox KE, Jeitany M et al (2015) Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347:273–277
Forment JV, Kaidi A, Jackson SP (2012) Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer 12(10):663–670
Gianferante DM, Mirabello L, Savage SA (2017) Germline and somatic genetics of osteosarcoma – connecting aetiology, biology and therapy. Nat Rev Endocrinol 13(8):480–491
He Y, de Castro LF, Shin MH et al (2015) p53 loss increases the osteogenic differentiation of bone marrow stromal cells. Stem Cells 33(4):1304–1319
Huang J, Wang L, Cong Z et al (2015) The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(−/−) murine model of ovarian cancer. Biochem Biophys Res Commun 463(4):551–556
Hugo W, Zaretsky JM, Sun L et al (2016) Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165(1):35–44
Jager M, Blokzijl F, Kuijk E et al (2019) Deficiency of global genome nucleotide excision repair explains mutational signature observed in cancer. Genome Res 29(7):1067–1077
Jiao S, Xia W, Yamaguchi H et al (2017) PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res 23:3711–3720
Kager L, Zoubek A, Pötschger U et al (2003) Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol 21:2011–2018
Kansara M, Teng MW, Smyth MJ et al (2014) Translational biology of osteosarcoma. Nat Rev Cancer 14(11):722–735
Keenan TE, Burke KP, Van Allen EM (2019) Genomic correlates of response to immune checkpoint blockade. Nat Med 25(3):389–402
Killela PJ, Reitman ZJ, Jiao Y et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A 110(15):6021–6026
Koirala P, Roth ME, Gill J et al (2016) Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci Rep 6:30093
Kovac M, Blattmann C, Ribi S et al (2015) Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun 6:8940
Lastwika KJ, Wilson W 3rd, Li QK et al (2015) Control of PD-L1 expression by oncogenic activation of the AKT/mTOR pathway in non-small cell lung cancer. Cancer Res 76(2):227–238
Li Q, Rao RR, Araki K et al (2011) A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity 34(4):541–553
Li B, Severson E, Pignon JC et al (2016) Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol 17:174
Li Y, Roberts ND, Weischenfeldt J et al (2017) Patterns of structural variation in human cancer. bioRxiv. https://doi.org/10.1101/181339v1
Li YS, Liu Q, He HB et al (2019) The possible role of insulin-like growth factor-1 in osteosarcoma. Curr Probl Cancer 43(3):228–235
Maciejowski J, Li Y, Bosco N et al (2015) Chromothripsis and kataegis induced by telomere crisis. Cell 163:1641–1654
Maciejowski J, de Lange T (2017) Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18(3):175–186
McEachron TA, Triche TJ, Sorenson L et al (2018) Profiling targetable immune checkpoints in osteosarcoma. Onco Targets Ther 7(12):e1475873
Meyers PA, Heller G, Healey JH et al (1993) Osteogenic sarcoma with clinically detectable metastasis at initial presentation. J Clin Oncol 11:449–453
Miao D, Margolis CA, Vokes NI et al (2018) Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet 50(9):1271–1281
Mirabello L, Troisi RJ, Savage SA (2009) Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer 115:1531–1543
Mouw KW, Goldberg MS, Konstantinopoulos PA et al (2017) DNA damage and repair biomarkers of immunotherapy response. Cancer Discov 7(7):675–693
O’Brien TD, Jia P, Xia J et al (2015) Inconsistency and features of single nucleotide variants detected in whole exome sequencing versus transcriptome sequencing: a case study in lung cancer. Methods 83:118–127
Ock CY, Hwang JE, Keam B et al (2017) Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat Commun 8(1):1050
Palmerini E, Agostinelli C, Picci P et al (2017) Tumoral immune-infiltrate (IF), PD-L1 expression and role of CD8/TIA-1 lymphocytes in localized osteosarcoma patients treated within protocol ISG-OS1. Oncotarget 8(67):111836–111846
Perry JA, Kiezun A, Tonzi P et al (2014) Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci 111:E5564–E5573
Petitjean A, Achatz MI, Borresen-Dale AL et al (2007) TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 26(15):2157–2165
Rausch T, Jones David TW, Zapatka M et al (2012) Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148:59–71
Rickel K, Fang F, Tao J (2017) Molecular genetics of osteosarcoma. Bone 102:69–79
Roh W, Chen PL, Reuben A et al (2017) Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med 9(379):pii: eaah3560
Sade-Feldman M, Jiao YJ, Chen JH et al (2017) Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8(1):1136
Seplyarskiy VB, Soldatov RA, Popadin KY et al (2016) APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res 26(2):174–182
Sharma P, Hu-Lieskovan S, Wargo JA et al (2017) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168(4):707–723
Shen JK, Cote GM, Choy E et al (2014) Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol Res 2(7):690–698
Sun C, Mezzadra R, Schumacher TN (2018) Regulation and function of the PD-L1 checkpoint. Immunity 48(3):434–452
Tawbi HA, Burgess M, Bolejack V et al (2017) Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 18(11):1493–1501
Taylor AM, Shih J, Ha G et al (2018) Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 33(4):676–689
The Cancer Genome Atlas (2013) The cancer genome atlas pan-cancer analysis project. Nat Genet 45(10):1113–1120
Thompson LH (2012) Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res 751:158–246
Thorsson V, Gibbs DL, Brown SD et al (2018) The immune landscape of cancer. Immunity 48(4):812–830
Turajlic S, Litchfield K, Xu H et al (2017) Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 18(8):1009–1021
van Harn T, Foijer F, van Vugt M et al (2010) Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev 24(13):1377–1388
Walkley CR, Qudsi R, Sankaran VG et al (2008) Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev 22(12):1662–1676
Wargo JA, Cooper ZA, Flaherty KT (2014) Universes collide: combining immunotherapy with targeted therapy for cancer. Cancer Discov 4(12):1377–1386
Wellenstein MD, de Visser KE (2018) Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 48(3):399–416
Werner H, Sarfstein R, LeRoith D et al (2016) Insulin-like growth factor 1 signaling axis meets p53 genome protection pathways. Front Oncol 6:159
Wilky BA, Trucco MM, Subhawong TK et al (2019) Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-centre, single-arm, phase 2 trial. Lancet Oncol 20(6):837–848
Wu CC, Beird HC, Livingston JA et al (2019) Immuno-genomic landscape of osteosarcoma. Nat Commun 11(1):1008
Yakar S, Rosen CJ, Beamer WG et al (2002) Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest 110(6):771–781
Yoshida K, Miki Y (2004) Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci 95(11):866–871
Yoshihara K, Shahmoradgoli M, Martinez E et al (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4:2612
Zhao X, Subramanian S (2017) Oncogenic pathways that affect antitumor immune response and immune checkpoint blockade therapy. Pharmacol Ther 181:76–84
Sansregret L, Vanhaesebroeck B, Swanton C (2018) Determinants and clinical implications of chromosomal instability in cancer. Nature Reviews Clinical Oncology 15 (3):139–150
Brown SD, Warren RL, Gibb EA et al (2014) Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Research 24 (5):743–750
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348 (6230):69–74
Wunder JS, Gokgoz N, Parkes R et al(2005) TP53 mutations and outcome in osteosarcoma: a prospective, multicenter study. Journal of Clinical Oncology 23 (7):1483–1490
Holland AJ, Cleveland DW (2009) Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nature Reviews Molecular Cell Biology 10 (7):478–487
Yang Y, Zhang Y, Qu X et al (2016) Identification of differentially expressed genes in the development of osteosarcoma using RNA-seq. Oncotarget 7 (52)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Wu, CC., Livingston, J.A. (2020). Genomics and the Immune Landscape of Osteosarcoma. In: Kleinerman, E., Gorlick, R. (eds) Current Advances in the Science of Osteosarcoma. Advances in Experimental Medicine and Biology, vol 1258. Springer, Cham. https://doi.org/10.1007/978-3-030-43085-6_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-43085-6_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-43084-9
Online ISBN: 978-3-030-43085-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)