
Abstract
Glypican-1 (GPC-1) is a cell surface heparan sulphate proteoglycan that is critical during normal development, but which is not required for normal homoeostasis in the adult. It is, however, overexpressed in a variety of solid tumours and is known to regulate tumour growth, invasion, metastasis and progression, through modulation of tumour cell biology as well as influence on the tumour microenvironment (TME). The role of GPC-1 in the TME and on the tumour cell is broad, as GPC-1 regulates signalling by several growth factors, including FGF, HGF, TGF-β, Wnt and Hedgehog (Hh). Signalling via these pathways promotes tumour growth and invasive and metastatic ability (drives epithelial-to-mesenchymal transition (EMT)) and influences angiogenesis, affecting both tumour and stromal cells. Broad modulation of the TME via inhibition of GPC-1 may represent a novel therapeutic strategy for inhibition of tumour progression. Here, we discuss the complex role of GPC-1 in tumour cells and the TME, with discussion of potential therapeutic targeting strategies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Glypican-1
- GPC-1
- Growth factor signalling
- Therapeutics
- Pancreatic cancer
- Tumour microenvironment
- Metastasis
- Angiogenesis
- Invasion
- Stroma
8.1 Introduction
The survival, growth and metastasis of a mass of tumour cells relies on its successful interaction with the collection of resident cells such as cancer-associated fibroblasts (CAFs) and infiltrating host cells such as immune cells including macrophages, cytokines, growth factors, blood vessels and extracellular matrix proteins. These are collectively known as the tumour microenvironment (TME). From a therapeutic perspective, tumour control may be achieved by manipulation of this interaction, by targeting elements of the TME crucial to tumour growth and/or survival. Understanding the complex interplay between tumour and TME will help in the identification of the most promising therapeutic targets.
Glypican-1 (GPC-1) is a cell surface heparan sulphate proteoglycan that is overexpressed in a variety of solid tumours, as well as during development, but whose expression is suppressed in most adult normal tissues [1, 2]. Glypican-1 is known to play a critical role in the biology of the tumour cell, being involved in cell cycle and proliferation, as well as invasive and metastatic capability [3, 4]. This review will focus on the complex role of GPC-1 in the solid tumour microenvironment and how its interaction with growth factors in the TME influences tumour growth, invasion and metastasis as well as CAF biology and angiogenesis. We will consider the role of GPC-1 in establishment and maintenance of cellular components of the stroma, with a special focus on the role of GPC-1 in pancreatic cancer. Finally, we will discuss potential therapeutic strategies for targeting of GPC-1 in the TME.
8.2 Glypican-1
Glypican-1 belongs to the family of glypicans (1–6), all of which are anchored to the cell surface by glycosylphosphatidylinositol. The heparan sulphate chains are covalently linked to the core protein, and these chains, when anchored to a cell, allow glypicans the ability to sequester and retain growth factors from the environment close to signalling molecules on the cell surface, facilitating the initiation and perpetuation of cell signalling [5]. In this respect, GPC-1 acts as a form of co-receptor for a range of signalling molecules, influencing signalling pathways including Wnt, Hedgehog (Hh), TGF-β and fibroblast growth factor (FGF) [6].
Glypican-1 is normally expressed during embryonic development in a temporally and spatially regulated manner. Animal studies have revealed a requirement for GPC-1 in normal brain development [7], and it is expressed in the developing skeletal system, bone marrow and kidneys; however, it is not required for normal homoeostasis [8]. Reports examining expression in normal adult tissue by immunohistochemistry have differed somewhat in reported expression, potentially attributable to the use of different antibodies; however, collectively, these studies show that GPC-1 expression is not observed in most normal adult tissues [1, 2].
Glypican-1 is known to promote tumour growth, metastasis and invasion, as detailed in this review and elsewhere [9]. In line with this, expression of GPC-1 has been described in a variety of solid tumours. Expression of GPC-1 was seen in 80% of prostate tumours, with no expression in benign prostate biopsy tissue [1]. Expression has been demonstrated in pancreatic cancer specimens but not normal pancreata [10]. Immunohistochemistry of normal pancreata (n = 169), pancreatic adenocarcinoma tissue (n = 186) and metastatic tumours in the liver (n = 4) revealed GPC-1 expression in tumour tissue but not in normal tissue [11]. Glypican-1 expression was seen in breast cancer by IHC and confirmed by in situ hybridisation, but not in normal tissue [4]. Significantly higher GPC-1 expression was observed in glioma specimens (astrocytomas n = 49 and oligodendrogliomas n = 7) than in non-malignant tissue [12]. Another study demonstrated expression in 27 of 53 surgically resected glioblastoma samples [13]. Expression in oesophageal squamous cell carcinoma (ESCC) was observed in 98.8% of specimens [14]. Expression of GPC-1 has been described in cervical cancer (adenocarcinoma and squamous cell carcinoma) [15] and in epithelioid mesothelioma in two studies [16, 17]. Interestingly, the study by Amatya and colleagues (2018) showed that expression of GPC-1 could differentiate epithelioid mesothelioma from lung adenocarcinoma with almost 100% sensitivity and 97% specificity, whilst the work described by Chiu et al. (2018) showed no differentiation between the two malignancies as most specimens for both indications stained positive for GPC-1.
Our understanding of the role of GPC-1 in tumour invasion and metastasis would predict the link between high tumour GPC-1 expression and poor clinical prognosis. For example, high GPC-1 expression in ESCC tumours, as measured by IHC, was associated with worse clinical outcomes than those tumours expressing low levels of GPC-1, potentially related to a relationship between GPC-1 expression and chemoresistance [14]. In glioblastoma, patients whose tumours stained positive for GPC-1 had a shorter overall survival than those whose tumours were negative [13]. In pancreatic ductal adenocarcinoma, higher GPC-1 levels in the tumour were associated with worse tumour biological features, including worse pathological differentiation and larger tumour sizes [11]. In line with these observations, patients whose tumours expressed GPC-1 had a shorter overall survival time.
Preclinical studies provide significant insight into the critical role of GPC-1 in tumour cell growth, invasion and metastasis in a variety of solid tumours. Indeed, GPC-1 is physiologically necessary for signalling via some mitogenic pathways that are required for tumour cell proliferation, characterised for breast and pancreatic cancers [4, 18]. For example, reduction of GPC-1 expression by transfection of Colo-357 pancreatic cancer cells with an anti-sense construct reduced anchorage-dependent and anchorage-independent cell growth [19]. PANC-1 cells in which GPC-1 was knocked down showed inhibited cell growth in vitro (longer doubling times and reduced anchorage-independent cell growth) and were less able to form tumours in vivo in immune-deficient mice, additionally demonstrating reduced angiogenesis and metastasis associated with GPC-1 knockdown [3]. Interesting studies into the role of glycosaminoglycans (GAGs) as mechanosensors for interstitial flow on cancer cells have helped elucidate the role of GPC-1 in metastasis, as knockdown of GPC-1 in a metastatic renal carcinoma cell line SN12L1 completely inhibited migration, with the GPC-1 core protein acting as a link between the GAG and cell, promoting metastasis [20]. Glypican-1 knockdown inhibited tumour growth in a KrasG12D-driven genetic pancreatic cancer model, affecting angiogenesis and metastasis [21]. In ESCC, blocking GPC-1 with an antibody inhibits tumour growth in a mouse PDX model [2].
Importantly, GPC-1 expression is not just required in the tumour cell for effective tumour growth, as GPC-1null mice exhibited defects in tumour angiogenesis and metastasis in an orthotopic xenograft model of pancreatic cancer, where the tumour cells were wild type for GPC-1 (control tumour cells) [3]. This points to a critical role for GPC-1 not only in the tumour cell but also in the TME, in both angiogenesis and the modulation of resident (host) cell function.
Here, we examine how GPC-1 interacts with and influences elements of the TME to control tumour cell survival, proliferation, invasion and metastasis.
8.3 Glypican-1 in the Tumour Microenvironment
Growth factors are critical components of the complex network of chemical mediators that may be derived from host or tumour cells and act in a paracrine or autocrine manner to activate signalling pathways that influence tumour growth, proliferation, invasion and metastasis in both tumour cells and host cells of the TME. Here we discuss evidence for the interaction of GPC-1 with factors, such as Wnts, fibroblast growth factor (FGF), hepatocyte growth factor (HGF), TGF-β and VEGF-A, and the influence of this interaction on cellular elements of the stroma, angiogenesis and tumour cell growth, proliferation, invasion and metastasis. Growth factors influence stromal cells (including CAFs which in turn provide growth factors to the TME), which then influence tumour drug resistance, metastasis, and proliferation and affect angiogenesis. Thus, GPC-1 exerts control over various pathways associated with tumour progression.
8.4 GPC-1 Enhances Growth Factor Signalling to Promote Proliferation, Invasion and Metastatic Potential
The Wnt family of proteins trigger canonical Wnt signalling by binding to FRIZZLED receptors on the cell surface, which inhibits degradation of β-catenin, triggering the translocation of β-catenin to the nucleus and accumulation there, where it regulates the expression of Wnt target genes. Wnt signalling in homoeostasis is involved in cell differentiation, self-renewal and cell migration but is also known to be involved in the establishment and progression of solid tumours (reviewed in [22], including colorectal [23], prostate [24], breast cancer [25, 26] and melanoma [27]).
The glypicans are known to interact with Wnts [28, 29]. Glypican-3 has been shown to promote canonical Wnt signalling and, hence, hepatocellular carcinoma growth, by two mechanisms: firstly, by binding Wnt and promoting the accumulation of Wnts at the cell membrane, in contact with FRIZZLED, and, secondly, by interacting with FRIZZLED itself, to promote the formation of signalling complexes of Wnt and FRIZZLED [30]. Studies of the interaction between GPC-1 and Wnts are limited, with just one study examining the relationship between Wnt signalling and GPC-1 expression in the forming of trigeminal ganglions of chickens, showing that overexpression of GPC-1 actually phenocopies inhibition of Wnt signalling, thus suggesting a negative regulatory role for GPC-1 (although this is in normal development) [31]. Further investigations into the role of GPC-1 in Wnt signalling in the TME are required to understand this relationship and its implication in tumour biology. Indeed, it is foreseeable that GPC-1 would be involved in Wnt signalling in tumour cells, as GPC-1 is overexpressed in cancers where Wnt signalling is also implicated in tumour establishment/progression, for example, in prostate and breast cancers.
The family of fibroblast growth factors (FGFs) consist of secreted signalling proteins that are expressed in almost all tissues and serve a critical role in embryonic development, organogenesis, and, in the adult, maintenance of homoeostasis with roles in metabolism and tissue maintenance and repair. Signalling involves binding of FGF to FGFR and a heparin sulphate [32]. In the TME, FGFs have broad biological function, influencing angiogenesis, tumour cell migration and invasion and cross-talk between epithelial and stromal cells (reviewed in [32]).
Glypican-1 is known to act as a co-receptor for FGF-2 and heparin binding epidermal growth factor (HB-EGF) [7, 33], influencing the mitogenic response of tumour cells and thus tumour growth. It is thought that the heparan sulphate chains interact with FGF to stabilise the FGF-FGFR complex on the cell surface and/or retain FGF in close proximity to FGFR to encourage signalling [34, 35]. Cleavage of the heparin sulphate chains of GPC-1 in two breast cancer cell lines MDA-MB-231 and MDA-MB-468 arrested the mitogenic response to FGF2 and HB-EGF [4]. Moreover, reducing GPC-1 protein expression by transfection with a GPC-1 anti-sense construct reduced the mitogenic response to both growth factors. Reduction of GPC-1 expression either by enzymatic cleavage of the cell surface expressed GPC-1 or by transfection with an anti-sense construct, in two pancreatic cell lines, resulted in suppression of mitogenic responses to FGF2 and HB-EGF [18]. In ESCC cells, phosphorylation of EGFR was reduced in cells transfected with GPC-1 siRNA following stimulation with HB-EGF [2]. In vivo, treatment of NOD/SCID mice (lacking functional NK cells and CDC) with an antibody that recognises GPC-1 inhibited the growth of an ESCC PDX , in a complement-dependent cytotoxicity (CDC)-independent and antibody-dependent cellular cytotoxicity (ADCC)-independent manner, potentially attributable to inhibition of mitogenic response, although this mechanism was not investigated [2].
There is extensive evidence for the role of both Wnt and FGF signalling in the promotion of cancer EMT, invasion and metastasis [36,37,38]. High levels of Wnt signalling are associated with a more aggressive phenotype of cancer, for example, in breast cancer [25]. In young prostate cancer patients (<50 years of age), an age group associated with more aggressive cancer, Wnt signalling is higher, and co-expression of β-catenin and androgen receptor (AR) correlates with higher Gleason scores [39]. In metastatic castration-resistant prostate cancer (mCRPC), mutations in the Wnt pathway are more common [40], and those men who have Wnt pathway mutations are less responsive to abiraterone/enzalutamide therapy [41]. Indeed, prostate cancer cells themselves secrete Wnts which act in an autocrine manner to promote tumour progression, but also act on the epithelium [24]. In line with the role of GPC-1 in Wnt and FGF signalling pathways, there is both experimental and clinical evidence for a role of GPC-1 in tumour invasiveness, as high GPC-1 expression in pancreatic cancer is associated with perineural invasion [10]. Loss of GPC-1 in a GPC-1nullmouse model of KrasG12D-driven pancreatic cancer inhibited spontaneous pancreatic tumour invasiveness into surrounding tissue [21]. Pancreatic cancer cells isolated from the tumours of GPC-1nullKrasG12D mice were less invasive when stimulated with FGF-2 ex vivo, as compared to tumours from GPC-1+/+ mice, and when these cancer cells were then grafted back into mice as tumours, they were less metastatic than tumours from GPC-1+/+ mice.
TGF-β binds to cell surface kinase receptors that phosphorylate cytoplasmic Smad proteins, which in turn interact with Smad4 proteins, then translocating to the nucleus to drive gene expression. The role of TGF-β signalling in cancer is an interesting one, as TGF-β is not only known to inhibit tumour growth but also to promote tumour invasion in later-stage disease [42]. Interestingly, once established, tumour cells lose the ability to be growth inhibited by TGF-β, but retention of semi-functional TGF-β signalling favours a more aggressive phenotype [42]. Indeed, a loss of TGF-β signalling is thought to be critical to the pathogenesis of pancreatic cancer [43]. On the other hand, TGF-β ligands, which are commonly seen to be overexpressed in pancreatic cancer, can drive EMT and an invasive phenotype of tumour cell [44, 45]. Studies in a cell line of pancreatic cancer have demonstrated that GPC-1 is involved in TGF-β signalling, as knockdown of GPC-1 resulted in an insensitivity to TGF-β-mediated growth inhibition [19], suggesting a role for GPC-1 in tumour progression through modulation of TGF-β signalling.
It is thought that the Wnt and TGF-β signalling pathways act synergistically to regulate gene transcription involved in tumourigenesis [46]. Interestingly, gene expression of GPC-1 is upregulated when tumour cells are stimulated with Wnt and TGF-β in combination, but not when cells are stimulated with either ligand alone, implying the involvement of GPC-1 in the Wnt/TGF-β collaboration [46].
Cancer stem cells (CSCs) are a subpopulation of tumour cells that are “stem-like” in phenotype, with capacity for self-renewal and differentiation, and that are resistant to chemotherapy and radiation, acting as a pool of cancer cells that can drive recurrence after therapy [47]. This population is thought to be involved in establishment of metastases [48]. Cancer stem cells are described in several solid tumours, including pancreatic cancer, where they are controlled by TGF-β, signalling via which is required for self-renewal and EMT, influencing subsequent invasion. Incubation of pancreatic CSCs with TGF-β drives a mesenchymal phenotype (in morphology and gene expression), and exposed cells display increased invasion in a Matrigel assay [49]. The expression of GPC-1 in CSCs is not yet described and is an area worthy of further research.
8.5 GPC-1 in Angiogenesis
Promotion of growth factor signalling by GPC-1 influences angiogenesis, a process necessary for tumour growth and metastasis. FGF-2 acts as a pro-angiogenic signal to endothelial cells, binding FGFR1 [50]. Glypican-1 is postulated to act as a co-receptor for FGF, encouraging interaction with FGFR1. GPC-1 also interacts with VEGF-A (a potent mitogen for endothelial cells), enhancing signalling through the VEGF receptor. Indeed, expression of GPC-1 has been demonstrated in endothelial cells from glioma samples, but not endothelial cells from normal brain [51]. Wnt signalling, known to be regulated by GPC-1, is critically involved in angiogenesis, through regulation of VEGF transcription [52].
Functionally, knockdown of GPC-1 inhibits angiogenesis in vivo in mice in a human pancreatic cancer xenograft model [3]. In a GPC-1null mouse model of KrasG12D-driven pancreatic cancer, angiogenesis was inhibited and this was associated with smaller tumours [21]. This suppression in angiogenesis was associated with a reduction in mRNA expression of various pro-angiogenic factors, including VEGF, in the tumour. Moreover, tumours from GPC-1null mice expressed less CD34, a marker of angiogenesis. Endothelial cells isolated from GPC-1null mice were non-migratory in response to VEGF-A, evidence for a role for GPC-1 in establishment of angiogenesis. In a mouse model, treatment of mice bearing ESCC PDXs with an anti-GPC-1antibody (reactive to mouse GPC-1) inhibited tumour growth, and this inhibition was independent of antibody effector functions (ADCC and CDC) [2]. Instead, the authors described expression of GPC-1 in the vascular endothelium of the tumours and postulated that the reduction in tumour growth associated with anti-GPC-1 antibody treatment may be attributed to inhibition of angiogenesis. Indeed, treatment with the anti-GPC-1 antibody, as compared to an isotype control antibody, was associated with a decrease in the concentration of blood vessels (identified by CD31 positivity) in the tumour [2].
8.5.1 The Influence of GPC-1 on Stromal Cells: Highlight on Pancreatic Cancer
Pancreatic cancer is a highly lethal malignancy. Survival rates are dismal with the 5-year survival rate at just 17%. Normally diagnosed at late stage, the standard of care is gemcitabine after surgical resection which provides only a marginal survival benefit [53]. The complex interaction between the stroma and the tumour cells themselves must be understood to develop novel therapeutic approaches.
A role for GPC-1 in pancreatic cancer is well established, and clinical evidence demonstrates that high expression is associated with perineural invasion and associated poor prognosis [10, 18]. Two pertinent studies examined carefully the role of GPC-1 in pancreatic cancer initiation and progression, establishing a critical role for GPC-1 in both the pancreatic tumour cell and the host TME. The study by Aikawa and colleagues (2008) [3] showed that knockdown of GPC-1 in the human pancreatic cell lines PANC-1 and T3M4 using anti-sense constructs inhibited tumour growth, metastasis and angiogenesis when engrafted in mice in an orthotopic xenograft. Importantly, the same study demonstrated a requirement for GPC-1 in the host TME for efficient tumour progression, as reduced tumour angiogenesis and metastasis was observed after xeno-engraftment of human pancreatic cell lines into athymic nude mice on a GPC-1null background. The other pertinent study on the role of GPC-1 in pancreatic cancer progression was that by Whipple et al. (2012) [21]. In this study, a genetic mouse model of pancreatic ductal adenocarcinoma (PDAC) was established by pancreas-specific activation of the oncogene KrasG12Dand loss of pancreatic INK4A expression. This background was then combined with wild-type GPC-1 or GPC-1null. Only 1/10 GPC-1null mice developed tumours, as compared to 7/10 GPC-1+/+ mice, and at later-stage disease, tumours from GPC-1null mice were less invasive, as all GPC-1+/+ mice (14/14) had large, invasive tumours, whilst just 4/20 GPC-1null mice did. In line with these findings, levels of Ki67, a marker of proliferation, and CD34, a marker of angiogenesis, were reduced in the tumours of GPC-1null mice as compared to those of GPC-1+/+ mice. Tumours cultured ex vivo from GPC-1+/+ and GPC-1nullmice grew more slowly in vitro, were less invasive in response to FGF stimulation and, when tumour fragments were engrafted orthotopically into athymic nude mice, were unable to metastasise. These tumour fragments consisted tumour cells as well as endothelial cells and CAFs, which are involved in metastasis, pointing to a role for GPC-1 in controlling PDAC progression through modulation of both tumour cell and stroma. Here, we discuss the influence of GPC-1 on CAF biology, an important component of the stroma in PDAC.
In pancreatic cancer, CAFs largely arise from pancreatic stellate cells (PSCs) and mesenchymal stem cells (MSCs). The complex interplay between CAFs and tumour cells promotes growth, proliferation and invasion of the cancer cells, whilst CAFs also modify the stromal composition to ensure tumour survival and facilitate metastasis. CAFs secrete a variety of growth factors into the TME, including cytokines, growth factors and exosomes containing miRNAs, whose paracrine actions influence tumour cell function, including driving EMT and invasive ability [54]. This role of CAFs in promoting tumour progression is not limited to pancreatic cancer, but it has also been shown in breast, colorectal and bladder cancers [55,56,57,58]. One of the major molecules secreted by CAFs that drives invasion and EMT in tumour cells is TGF-β, a molecule that plays a complex role in the pathogenesis of pancreatic cancer, suppressing early-stage disease, whilst driving invasion, promoting metastasis and angiogenesis and contributing to immune evasion in later stages [59,60,61]. Glypican-1 is known to modulate the TGF-β signalling pathway in pancreatic tumour cells, potentially playing a role in tumour cell proliferation [19, 62]. Importantly, signalling via TGF-β (that is derived from the tumour cell) activates normal fibroblasts, driving them to a CAF phenotype (Fig. 8.1). Given that GPC-1 is expressed on fibroblasts found adjacent to pancreatic tumour cells in biopsy tissue [18], we postulate that GPC-1 likely modulates fibroblast activation in PDAC. In support of this idea, stromal cells from GPC-1null Kras-driven pancreatic tumour-bearing mice express less Ki67 (a marker of proliferation) than those from GPC-1-expressing tumour-bearing mice, suggesting GPC-1 controls growth signalling in these cells [21].

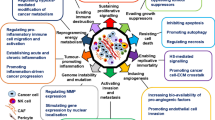
The role of GPC-1 in cell signalling – modulation of factors critical to tumour progression Glypican-1 acts as a co-receptor for TGF-β; signalling via TGFR1 and TGFR11 promotes epithelial-to-mesenchymal transition (EMT) and invasion in tumour cells. TGF-β activates CAFs which influence the tumour and TME. Glypican-1 sequesters FGF in proximity to FGFR driving signalling via the FGF pathway which promotes angiogenesis, drives migration and invasion and is mitogenic in tumour cells. Delivering Wnt to FRIZZLED, GPC-1 influences the β-catenin signalling pathway which drives metastasis and invasion. Hedgehog signalling is influenced by GPC-1 and promotes the activation of TME cells, e.g. stromal cells. TGF-β transforming growth factor-β, EMT epithelial-to-mesenchymal transition, CAFs cancer-associated fibroblasts, Hh Hedgehog, FGF fibroblast growth factor
In turn, activated CAFs secrete HGF and FGF which drive cancer cell proliferation, migration, invasion and metastasis. Moreover, these signalling molecules can act in an autocrine manner, driving CAF proliferation [63]. Indeed, fibroblasts isolated from GPC-1null mice were unresponsive to stimulation with FGF in a cell migration assay [21]. Glypican-1 may influence growth factor signalling in CAFs, with immediate effects on CAF biology, but also with knock-on effects for tumour cell signalling via inhibition of CAF growth factor release.
The Hedgehog (Hh) signalling pathway plays a crucial role in normal development. In pancreatic cancer, it is known to drive the formation of the stroma by activating PSCs [64], and inhibition of Hh signalling reduces the establishment of the stroma and allows exposure of the tumour to chemotherapy [65, 66]. The Hh ligands, derived from the tumour cell, act in a paracrine manner on the PSCs, driving proliferation [67]. Glypican-1 is known to regulate Hh signalling, acting as a co-receptor for Sonic Hedgehog (Shh) and regulating Hh signalling in normal cells, for example, cholangiocytes and neurons [68, 69]. We postulate that GPC-1 may play a role in controlling Hh signalling in PSCs, thus influencing establishment of the stromal compartment in pancreatic cancer (Fig. 8.2).
8.6 Perspective: Glypican-1 as a Therapeutic Target
The critical role of GPC-1 in modulating tumour biology and the TME, as well as its overexpression in a variety of solid tumours and link to poor clinical prognosis, suggests its potential as a therapeutic target. Indeed, several studies have demonstrated the efficacy of targeting GPC-1 in different cancers. Knockdown of GPC-1 reduces the proliferation, migration and invasion of breast and pancreatic cancers and inhibits tumour growth in vivo [3, 4, 18]. Targeting of GPC-1 with an antibody inhibits tumour growth through the direct action of the antibody (as opposed to CDC or ADCC). The safety of targeting GPC-1 is evidenced by several studies. Protein expression of GPC-1 in normal adult tissue is limited [1, 2]. Moreover, safety studies in mice using a high dose of anti-GPC-1 antibody (50 mg/kg) that recognises mouse GPC-1 showed no adverse effects associated with targeting of the antigen [2, 15].
Given the crucial role for growth factor signalling pathways in tumour progression, many therapies have been designed to modulate individual signalling pathways. Inhibition of FGFR1 and FGFR2 in pancreatic stellate cells using a silencing RNA or chemical inhibitor has been shown to reduce proliferation and, in organoid cultures, prevents invasion in both pancreatic stellate cells and pancreatic tumour cells [63]. Complete ablation of FGFR in stellate cells completely inhibited invasive ability of the cancer. Inhibition of HGF using a neutralising antibody (AMG102) hindered tumour growth in vivo in an orthotopic model of pancreatic cancer to the same degree as gemcitabine, but had a more profound effect on angiogenesis and metastasis than the chemotherapeutic treatment [70]. Interestingly, this inhibition of metastasis was lost when the HGF inhibitor therapy was combined with gemcitabine. The authors postulated that this was likely due to selection by gemcitabine for a stem-like population more inclined to EMT. Therapeutic targeting of TGF-β signalling using blocking antibodies or anti-sense oligonucleotides (for blocking of ligand and/or receptor) has delivered complex experimental results. In pancreatic cancer, the TGFβR1 small molecule inhibitor galunisertib inhibited proliferation to some degree in cell line studies, but its real effect was in vivo inhibition of invasion [71, 72]. However, the pitfalls of targeting a single pathway therapeutically were highlighted by in vitro studies in 3D tumour-stroma cultures of pancreatic stellate and tumour cells. In fibroblasts, cancer cell-derived TGF-β inhibits HGF secretion; thus, “therapeutic” inhibition of TGF-β signalling led to an increase in fibroblast HGF secretion and, consequently, increased tumour cell invasion, reminding us of the complex interplay between tumour cells and the TME [73].
Despite promising preclinical data, therapies aimed at inhibiting growth factor signalling have had varied clinical success. In pancreatic cancer, inhibition of the Hh pathway was predicted to inhibit PSC growth, thus making the tumour more accessible to gemcitabine. Efficacy was expected based on promising preclinical studies [66]; however, when tested in phase II clinical trials in combination with gemcitabine, the Hh inhibitor saridegib did not show benefit beyond gemcitabine alone (NCT01130142; [74]). Similarly, sulindac, an approved nonsteroidal anti-inflammatory drug (NSAID) known to target the accumulation of β-catenin in the nucleus and influence Wnt signalling, failed to show clinical benefit in lung cancer in phase II clinical trials [75]. These clinical results may not be wholly unexpected, given the complex interplay of tumour and TME and the many signalling pathways involved. Indeed, it is now known that some signalling pathways collaborate to drive tumour progression, which is a difficult phenomenon to target therapeutically using single agents, in part not only because of the complex interplay between pathways but also because our understanding of these complexities is limited. Glypican-1 broadly modulates multiple signalling pathways critical to tumour growth and progression, in both the tumour cells and the TME, whilst not being required for normal homoeostasis, making targeting GPC-1 an attractive therapeutic approach. A blocking antibody or GPC-1 binding peptide may be an appropriate therapeutic agent. Indeed, there is preclinical evidence for the therapeutic potential of blocking glypican family members. Blocking of GPC-3 with the humanised monoclonal antibody HS20 inhibits Wnt signalling and prevents liver tumour growth in vitro and in vivo in nude mice [76].
Targeting of GPC-1 may inhibit fibroblast activation and CAF establishment of the stroma resulting in a more vulnerable tumour, through modulation of various signalling pathways including FGF and Hh (which may be a superior approach to therapeutic targeting of a single pathway). Moreover, inhibition of CAF activation inhibits the release of TGF-β, critical for maintaining immune suppression in the tumour, as well as driving tumour cell EMT. TGF-β signalling in CAFs is thought to promote the production of extracellular matrix (ECM), establishing fibrosis. Indeed, TGFβR1 inhibition with kinase inhibitor SD-208 in an orthotopic PANC-1 xenograft reduced tumour growth and fibrosis [77]. Moreover, Hh signalling, another pathway mediated by GPC-1, drives fibrosis [64]. Thus, targeting of GPC-1 may be a means to inhibit fibrosis. Reduction in CAF-released FGF and HGF inhibits tumour cell proliferation, migration, invasion and metastasis and autocrine CAF activation. Inhibition of GPC-1 signalling (FGF, VEGF and Wnt) inhibits angiogenesis, crucial to tumour survival, by modulating the function of endothelial cells. Finally, targeting of GPC-1 signalling inhibits tumour cell growth factor signalling required for growth, invasion and metastasis, including signalling via FGF, HGF, TGF-β and Wnt, and may modify the response to collaborative pathways such as Wnt/TGF-β. The broad inhibition achieved by blocking of GPC-1 would likely inhibit tumour growth, invasion and metastasis, in line with experimental work demonstrating the therapeutic effects of GPC-1 knockdown or blocking in vitro and in mouse studies and clinical evidence demonstrating a role for GPC-1 in tumour invasion. Work performed by our collaborators has demonstrated that knockdown of GPC-1 by siRNA inhibits proliferation, migration and invasion of a variety of prostate cancer cell lines in vitro (manuscript in preparation). Importantly, in vivo, knockdown not only inhibits the growth of subcutaneous prostate cancer tumours but also inhibits metastasis in a PC3 intracardiac metastatic model (manuscript in preparation).
8.7 GPC-1 and Cancer Stem Cells
Cancer stem cells are a subpopulation of cancer cells that are critical to tumour recurrence and to the establishment of metastases. Signalling via TGF-β modulates the function of CSCs, including driving EMT. For example, in glioblastoma, the stem-like population glioma-initiating cells (GICs) rely on TGF-β for self-renewal, through activation of the JAK-STAT pathway [78]. Thus, targeting of GPC-1 may inhibit the function of CSCs via modulation of TGF-β signalling. For some indications, the CSC population plays a more prominent role in pathogenesis, for example, in triple-negative breast cancer (TNBC). This phenotype of breast cancer has a worse prognosis than the ER- and HER2-positive tumour phenotypes, and this is thought to be related to the presence of virulent CSCs [79]. There is precedence for targeting of the TGF-β pathway in this indication, as in vitro work using genetic or pharmacological inhibitors of TGF-β prevented the expansion of CSCs [80]. The expression of GPC-1 in triple-negative breast cancer has not yet been described; however, this is a subject of investigation in our lab. If expression is high, then a GPC-1 blocking strategy may be therapeutically appropriate for this indication.
Glypican-1 blocking therapies may have potential in combination with standard-of-care therapies. Anti-oestrogens (e.g. tamoxifen) are used as an adjuvant treatment for oestrogen receptor-positive (ER+) breast cancers; however, resistance is commonplace, developing in one-third of patients and close to all patients with metastatic disease [81]. It has been postulated that signalling via FGF promotes resistance to anti-oestrogen therapy, through upregulation of cyclin D and downregulation of the pro-apoptotic factor Bim [82]. Blockade of FGFR or FGFs reversed drug resistance in murine PDX models of drug-resistant breast cancer. Given the critical role of GPC-1 in promoting FGF signalling, there is a clear rationale for a combination of a GPC-1 targeting therapeutic with hormone therapy in hormone-responsive breast cancers [83].
In summary, GPC-1 plays a critical role in the tumour and TME, modulating tumour growth factor signalling, influencing angiogenesis and controlling activity of components of the stroma, including CAFs. The end point of these complex interactions is the promotion of tumour growth, invasion and metastasis. Importantly, in adults, modulation by GPC-1 is largely restricted to tumour sites, so therapeutic targeting of GPC-1 would not affect normal homoeostasis (an idea supported by preclinical safety studies). Thus, targeting of GPC-1 may provide an effective, broad-ranging therapeutic strategy to address multiple underlying issues in the tumour and TME, including oncogenic growth factor signalling, establishment of angiogenesis and therapy-resistant CSCs that contribute to disease recurrence.

The role of GPC-1 in cell signalling in the tumour and its microenvironment. Glypican-1 is expressed on the surface of endothelial cells, on fibroblasts, on the tumour cell itself and, putatively, on cancer stem cells. Glypican-1 acts as a co-receptor for various signalling molecules, influencing signalling via Wnts, VEGF, FGF and Hedgehogs (Hhs), molecules which may act in autocrine or paracrine manner. In endothelial cells, GPC-1 enhances FGF and VEGF signalling and modulates Wnt signalling which in turn influences VEGF gene expression levels. Fibroblasts differentiate into CAFs in response to tumour cell-derived TGF-β, signalling via which in fibroblasts is controlled by GPC-1. The activation of CAFs leads to the release of HGF, FGF and TGF-β, all of which act in an autocrine manner (regulated by GPC-1 as a co-receptor) or act on tumour cells to drive tumour cell migration, invasion and metastasis. Cancer-associated fibroblast (CAF)-derived VEGF also acts on endothelial cells to drive angiogenesis. Hedgehogs interact with pancreatic stellate cells (PSCs) to drive proliferation. CAF cancer-associated fibroblast, GPC-1 glypican-1, Hh Hedgehog, PSCs pancreatic stellate cells
References
Russell PJ et al (2004) Immunohistochemical characterisation of the monoclonal antibody BLCA-38 for the detection of prostate cancer. Cancer Immunol Immunother 53:995–1004
Harada E et al (2017) Glypican-1 targeted antibody-based therapy induces preclinical antitumor activity against esophageal squamous cell carcinoma. Oncotarget 8:24741–24752
Aikawa T et al (2008) Glypican-1 modulates the angiogenic and metastatic potential of human and mouse cancer cells. J Clin Invest 118:89–99
Matsuda K et al (2001) Glypican-1 is overexpressed in human breast cancer and modulates the mitogenic effects of multiple heparin-binding growth factors in breast cancer cells. Cancer Res 61:5562–5569
Häcker U, Nybakken K, Perrimon N (2005) Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol 6:530. https://doi.org/10.1038/nrm1681
Lin X (2004) Functions of heparan sulfate proteoglycans in cell signaling during development. Development 131:6009. https://doi.org/10.1242/dev.01522
Jen YHL, Musacchio M, Lander AD (2009) Glypican-1 controls brain size through regulation of fibroblast growth factor signaling in early neurogenesis. Neural Dev 4:1–19
Litwack ED et al (1998) Expression of the heparan sulfate proteoglyean glypican-1 in the developing rodent. Dev Dyn 211:72. https://doi.org/10.1002/(SICI)1097-0177(199801)211:1<72::AID-AJA7>3.0.CO;2-4
Kaur SP, Cummings BS (2019) Role of glypicans in regulation of the tumor microenvironment and cancer progression. Biochem Pharmacol 168:108–118
Duan L, Hu XQ, Feng DY, Lei SY, Hu GH (2013) GPC-1 may serve as a predictor of perineural invasion and a prognosticator of survival in pancreatic cancer. Asian J Surg 36:7–12
Lu H et al (2017) Elevated glypican-1 expression is associated with an unfavorable prognosis in pancreatic ductal adenocarcinoma. Cancer Med 6:1181–1191
Su G et al (2006) Glypican-1 is frequently overexpressed in human gliomas and enhances FGF-2 signaling in glioma cells. Am J Pathol 168:2014–2026
Saito T et al (2017) High expression of glypican-1 predicts dissemination and poor prognosis in glioblastomas. World Neurosurg 105:282–288
Hara H et al (2016) Overexpression of glypican-1 implicates poor prognosis and their chemoresistance in oesophageal squamous cell carcinoma. Br J Cancer 115:66–75
Matsuzaki S et al (2018) Anti-glypican-1 antibody-drug conjugate exhibits potent preclinical antitumor activity against glypican-1 positive uterine cervical cancer. Int J Cancer 142:1056–1066
Amatya VJ et al (2018) Glypican-1 immunohistochemistry is a novel marker to differentiate epithelioid mesothelioma from lung adenocarcinoma. Mod Pathol 31:809–815
Chiu K, Lee L, Cheung S, Churg AM (2018) Glypican-1 immunohistochemistry does not separate mesothelioma from pulmonary adenocarcinoma. Mod Pathol 31:1400–1403
Kleeff J et al (1998) The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J Clin Invest 102:1662–1673
Li J et al (2004) Glypican-1 antisense transfection modulates TGF-β-dependent signaling in Colo-357 pancreatic cancer cells. Biochem Biophys Res Commun 320:1148–1155
Moran H et al (2019) The cancer cell glycocalyx proteoglycan glypican-1 mediates interstitial flow mechanotransduction to enhance cell migration and metastasis. Biorheology 56:151. https://doi.org/10.3233/BIR-180203
Whipple CA, Yung AL, Korc M (2012) A Kras^G12D-driven genetic mouse model of pancreatic cancer requires glypican-1 for efficient proliferation and angiogenesis. Oncogene 31:2535–2544
Zhan T, Rindtorff N, Boutros M (2017) Wnt signaling in cancer. Oncogene 36:1461. https://doi.org/10.1038/onc.2016.304
Novellasdemunt L, Antas P, Li VSW (2015) Targeting Wnt signaling in colorectal cancer. A review in the theme: cell signaling: proteins, pathways and mechanisms. Am J Physiol Cell Physiol 309:C511. https://doi.org/10.1152/ajpcell.00117.2015
Murillo-Garzón V, Kypta R (2017) WNT signalling in prostate cancer. Nat Rev Urol 14:683. https://doi.org/10.1038/nrurol.2017.144
Lin S-Y et al (2000) Beta -catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci 97:4262. https://doi.org/10.1073/pnas.060025397
Howe LR, Brown AMC (2004) Wht signaling and breast cancer. Cancer Biol Ther 3:36. https://doi.org/10.4161/cbt.3.1.561
Webster MR, Weeraratna AT (2013) A Wnt-er migration: the confusing role of β-catenin in melanoma metastasis. Sci Signal 6:pe11. https://doi.org/10.1126/scisignal.2004114
Ohkawara B (2003) Role of glypican 4 in the regulation of convergent extension movements during gastrulation in Xenopus laevis. Development 130:2129. https://doi.org/10.1242/dev.00435
De Cat B, David G (2001) Developmental roles of the glypicans. Semin Cell Dev Biol 12:117. https://doi.org/10.1006/scdb.2000.0240
Capurro M, Martin T, Shi W, Filmus J (2014) Glypican-3 binds to Frizzled and plays a direct role in the stimulation of canonical Wnt signaling. J Cell Sci 127:1565. https://doi.org/10.1242/jcs.140871
Shiau CE, Hu N, Bronner-Fraser M (2010) Altering Glypican-1 levels modulates canonical Wnt signaling during trigeminal placode development. Dev Biol 348:107. https://doi.org/10.1016/j.ydbio.2010.09.017
Turner N, Grose R (2010) Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 10:116. https://doi.org/10.1038/nrc2780
Zhang Z, Coomans C, David G (2001) Membrane heparan sulfate proteoglycan-supported FGF2-FGFR1 signaling: evidence in support of the ‘cooperative end structures’ model. J Biol Chem 276:41921. https://doi.org/10.1074/jbc.M106608200
Pellegrini L (2001) Role of heparan sulfate in fibroblast growth factor signalling: a structural view. Curr Opin Struct Biol 11:629. https://doi.org/10.1016/S0959-440X(00)00258-X
Ornitz DM (2000) FGFs, heparan sulfate and FGFRs: complex interactions essential for development. BioEssays 22:108. https://doi.org/10.1002/(SICI)1521-1878(200002)22:2<108::AID-BIES2>3.0.CO;2-M
Gonzalez DM, Medici D (2014) Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal 7:re8. https://doi.org/10.1126/scisignal.2005189
Wu Y et al (2012) Expression of Wnt3 activates Wnt/ -catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res 10:1597. https://doi.org/10.1158/1541-7786.MCR-12-0155-T
Zhao JH, Luo Y, Jiang YG, He DL, Wu CT (2011) Knockdown of β-catenin through shRNA cause a reversal of EMT and metastatic phenotypes induced by HIF-1α. Cancer Investig 29:377. https://doi.org/10.3109/07357907.2010.512595
Jung SJ et al (2013) Clinical significance of Wnt/β-catenin signalling and androgen receptor expression in prostate cancer. World J Men’s Health 31:36. https://doi.org/10.5534/wjmh.2013.31.1.36
Grasso CS et al (2012) The mutational landscape of lethal castration-resistant prostate cancer. Nature 487:239. https://doi.org/10.1038/nature11125
Isaacsson Velho P et al (2019) Wnt-pathway activating mutations are associated with resistance to first-line abiraterone and enzalutamide in castration-resistant prostate cancer. Eur Urol 77:1–8. https://doi.org/10.1016/j.eururo.2019.05.032
Akhurst RJ, Derynck R (2001) TGF-β signaling in cancer – a double-edged sword. Trends Cell Biol 11:S44–S51. https://doi.org/10.1016/S0962-8924(01)02130-4
Shen W et al (2017) TGF-β in pancreatic cancer initiation and progression: two sides of the same coin. Cell Biosci 7:39. https://doi.org/10.1186/s13578-017-0168-0
Horiguchi K et al (2009) Role of Ras signaling in the induction of snail by transforming growth factor-β. J Biol Chem 284:245. https://doi.org/10.1074/jbc.M804777200
Nolan-Stevaux O et al (2009) GLI1 is regulated through smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev 23:24. https://doi.org/10.1101/gad.1753809
Labbé E et al (2007) Transcriptional cooperation between the transforming growth factor-β and Wnt pathways in mammary and intestinal tumorigenesis. Cancer Res 67:75. https://doi.org/10.1158/0008-5472.CAN-06-2559
Kuşoğlu A, Biray Avcı Ç (2019) Cancer stem cells: a brief review of the current status. Gene 681:80. https://doi.org/10.1016/j.gene.2018.09.052
Nandy SB, Lakshmanaswamy R (2017) Cancer stem cells and metastasis. Prog Mol Biol Transl Sci 151:137–176. https://doi.org/10.1016/bs.pmbts.2017.07.007
Kabashima A et al (2009) Side population of pancreatic cancer cells predominates in TGF-β-mediated epithelial to mesenchymal transition and invasion. Int J Cancer 124:2771. https://doi.org/10.1002/ijc.24349
Presta M et al (2005) Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 16:159. https://doi.org/10.1016/j.cytogfr.2005.01.004
Qiao D, Meyer K, Mundhenke C, Drew SA, Friedl A (2003) Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 signaling in brain endothelial cells: specific role for glypican-1 in glioma angiogenesis. J Biol Chem 278:16045. https://doi.org/10.1074/jbc.M211259200
Easwaran V et al (2003) β-catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res 63:3145–3153
Neesse A et al (2019) Stromal biology and therapy in pancreatic cancer: ready for clinical translation? In: Gut, vol 68, p 159. https://doi.org/10.1136/gutjnl-2018-316451
Qu C, Wang Q, Meng Z, Wang P (2018) Cancer-associated fibroblasts in pancreatic cancer: should they be deleted or reeducated? Integr Cancer Ther 17:1016. https://doi.org/10.1177/1534735418794884
Yu Y et al (2014) Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer 110:724. https://doi.org/10.1038/bjc.2013.768
Fuyuhiro Y et al (2012) Cancer-associated orthotopic myofibroblasts stimulates the motility of gastric carcinoma cells. Cancer Sci 103:797. https://doi.org/10.1111/j.1349-7006.2012.02209.x
Zhuang J et al (2015) TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci Rep 5. https://doi.org/10.1038/srep11924
Shimao Y, Nabeshima K, Inoue T, Koono M (1999) Role of fibroblasts in HGF/SF-induced cohort migration of human colorectal carcinoma cells: fibroblasts stimulate migration associated with increased fibronectin production via upregulated TGF-β1. Int J Cancer 82:449. https://doi.org/10.1002/(SICI)1097-0215(19990730)82:3<449::AID-IJC20>3.0.CO;2-H
Teraoka H et al (2001) TGF-beta1 promotes liver metastasis of pancreatic cancer by modulating the capacity of cellular invasion. Int J Oncol 19:709–715
Culhaci N et al (2005) Expression of transforming growth factor-beta-1 and p27Kip1 in pancreatic adenocarcinomas: relation with cell-cycle-associated proteins and clinicopathologic characteristics. BMC Cancer 5:98. https://doi.org/10.1186/1471-2407-5-98
Wagner M, Kleeff J, Friess H, Büchler MW, Korc M (1999) Enhanced expression of the type II transforming growth factor-β receptor is associated with decreased survival in human pancreatic cancer. Pancreas 19:370. https://doi.org/10.1097/00006676-199911000-00008
Kayed H et al (2006) Correlation of glypican-1 expression with TGF-β, BMP, and activin receptors in pancreatic ductal adenocarcinoma. Int J Oncol 29:1139–1148
Coleman SJ et al (2014) Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol Med 6:467. https://doi.org/10.1002/emmm.201302698
Bai Y et al (2016) Hedgehog signaling in pancreatic fibrosis and cancer. Medicine (United States) 95:e2996. https://doi.org/10.1097/MD.0000000000002996
Bailey JM et al (2008) Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res 14:5995. https://doi.org/10.1158/1078-0432.CCR-08-0291
Olive KP et al (2009) Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (80-. ) 324:1457. https://doi.org/10.1126/science.1171362
Li X et al (2014) Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin Cancer Res 20:4326. https://doi.org/10.1158/1078-0432.CCR-13-3426
Wilson NH, Stoeckli ET (2013) Sonic Hedgehog regulates its own receptor on postcrossing commissural axons in a glypican1-dependent manner. Neuron 79:478–491
Cui S et al (2013) Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology 144:1107. https://doi.org/10.1053/j.gastro.2013.01.022
Pothula SP et al (2016) Hepatocyte growth factor inhibition: a novel therapeutic approach in pancreatic cancer. Br J Cancer 114:269. https://doi.org/10.1038/bjc.2015.478
Dituri F et al (2013) Differential inhibition of the TGF-β signaling pathway in HCC cells using the small molecule inhibitor LY2157299 and the D10 monoclonal antibody against TGF-β receptor type II. PLoS One 8:e67109. https://doi.org/10.1371/journal.pone.0067109
Serova M et al (2015) Effects of TGF-beta signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget 6:21614. https://doi.org/10.18632/oncotarget.4308
Oyanagi J et al (2014) Inhibition of transforming growth factor-β signaling potentiates tumor cell invasion into collagen matrix induced by fibroblast-derived hepatocyte growth factor. Exp Cell Res 326:267. https://doi.org/10.1016/j.yexcr.2014.04.009
Madden JI (2012) Infinity reports update from phase 2 study of saridegib plus gemcitabine in patients with metastatic pancreatic cancer. Infinity Pharmaceuticals 2012 [online], http://phx.corporate-ir.net/phoenix.zhtml?c=121941&p=irol-newsArticle&ID=1653550&highlight.
Limburg PJ et al (2013) Randomized phase II trial of sulindac for lung cancer chemoprevention. Lung Cancer 79:254. https://doi.org/10.1016/j.lungcan.2012.11.011
Gao W et al (2014) Inactivation of Wnt signaling by a human antibody that recognizes the heparan sulfate chains of glypican-3 for liver cancer therapy. Hepatology 60:576. https://doi.org/10.1002/hep.26996
Medicherla S et al (2007) Antitumor activity of TGF-β inhibitor is dependent on the microenvironment. Anticancer Res 27:4149–4157
Peñuelas S et al (2009) TGF-β increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 15:315. https://doi.org/10.1016/j.ccr.2009.02.011
Li X et al (2008) Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 100:672. https://doi.org/10.1093/jnci/djn123
Bhola NE et al (2013) TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest 123:1348. https://doi.org/10.1172/JCI65416
Abe O et al (2011) Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 378:771. https://doi.org/10.1016/S0140-6736(11)60993-8
Shee K et al (2018) Therapeutically targeting tumor microenvironment–mediated drug resistance in estrogen receptor–positive breast cancer. J Exp Med 215:895. https://doi.org/10.1084/jem.20171818
Wesche J, Haglund K, Haugsten EM (2011) Fibroblast growth factors and their receptors in cancer. Biochem J 437:199. https://doi.org/10.1042/BJ20101603
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lund, M.E., Campbell, D.H., Walsh, B.J. (2020). The Role of Glypican-1 in the Tumour Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1245. Springer, Cham. https://doi.org/10.1007/978-3-030-40146-7_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-40146-7_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-40145-0
Online ISBN: 978-3-030-40146-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)