
Abstract
Several small molecule effectors of myosin function that target the motor domains of myosin classes I, II, V, and VI have been identified. Four distinct binding sites in the myosin motor domain have been reported with unique properties and mechanisms of action. This chapter describes the structural basis and activities of known small molecule effectors that allosterically target the myosin motor domain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Introduction
The superfamily of myosin motors has emerged as an attractive target for the modulation of their physiological function by small molecule effectors. To date, at least 14 types of allosteric small molecule effectors of the myosin heavy chain have been reported (Fig. 5.1). Small effector molecules that inhibit or activate the function of well-defined myosin isoforms do not only have great potential for the development of new therapeutics and the targeted modulation of dysfunctional myosins and myosin-related diseases, but have proven to be invaluable tools for the investigation of cytoskeletal processes in cellular systems.

Schematic overview of known allosteric small molecule effectors of myosin function. The small molecule effectors are grouped according to their specificities. Color code: blue = class II myosin inhibitors, red = class I myosin inhibitors, cyan = class V myosin inhibitors, purple = class VI myosin inhibitors, white = no particular specificities defined, however inhibition of myosin II is reported in the literature, green = class II myosin activators. Determined potencies of the inhibitors or activators are given as IC50 or AC50 values
A major challenge for a specific modulation of distinct myosin isoforms is the ubiquitous occurrence of these motor proteins in the human body and in important pathogens. Myosin isoforms can be grouped into at least 35 myosin classes (Foth et al. 2006; Odronitz and Kollmar 2007) with diverse and prominent roles for a wide range of intracellular processes. Myosins are involved in processes such as muscle contraction, intracellular transport, tethering, signaling and cell division, as well as organization of the cytoskeleton. The human genome contains more than 40 genes that encode myosin isoforms from 12 different classes – myosin classes I, II, III, V, VI, VII, IX, X, XV, XVI, XVIII, and XIX (Berg et al. 2001; Kollmar and Mühlhausen 2017). Structurally, myosins feature broad diversities in their tail regions, which are designed for cargo transport and interactions with other components of the cell, while the catalytically active motor domain is largely conserved across the myosinome. The approximately 80 kDa motor domain harbors the active site and the actin-binding region. Polar actin filaments (F-actin) serve as tracks for the directional movement of myosin motors. Several small molecule effectors have been shown to preferentially affect the generic myosin motor domain of specific isoforms. Compounds with well-defined specificity profiles serve currently as lead compounds for the development of treatments of myosin-associated diseases, in particular heart failure and hypertrophic cardiomyopathy (www.clinicaltrials.gov, identifiers NCT01300013, NCT01786512, NCT02929329, NCT02329184, NCT03470545, NCT0376855). Together, the ability of small effector molecules to exert their effect on certain myosin isoforms and the identification of at least four allosteric binding sites in the myosin motor domain illustrate the druggability of members of the myosin superfamily.
The specific modulation of individual myosin isoforms within the scope of the common “one-target-one-disease” paradigm, also known as Paul Ehrlich’s magic bullet (Strebhardt and Ullrich 2008), can provide innovative access to major human diseases that are currently difficult to treat. The major focus of ongoing pharmaceutical research programs lies on diseases affecting skeletal, smooth and cardiac muscle performance (Spudich 2014; Greenberg 2016; Varian and Tang 2017; Kaplinsky and Mallarkey 2018). However, strong links have also been established between major human pathologies and cytoskeletal myosin motors (see Chap. 12). Changes in the abundance, functional competence, and regulation of cytoskeletal myosin isoforms can play causal roles in the development of neurodegenerative disorders (Nadif Kasri and Van Aelst 2008; Hur et al. 2011; Ma and Adelstein 2014), diabetes (Charlton et al. 1997; Papadaki et al. 2018), various forms of cancer, with at least seven classes of myosins participating in tumorigenesis and cancer proliferation (Li et al. 2016b), viral, bacterial, and parasitic infections (Xiong et al. 2015; Cymerys et al. 2016; Cowman et al. 2017; Tan et al. 2019), and drug addiction (Young et al. 2015; Volkow and Morales 2015). The identification of specific small molecule effectors that change the function of the associated myosin isoforms in a well-defined manner can provide means to cure or at least treat the affected patients. These diseases are however complex, with diverse causative factors, versatile mechanisms to adapt to altered conditions and drug treatments, and several groups of myosin isoforms displaying functional redundancy. Therefore, analogous to the multitarget drug discovery concept (Roth et al. 2004; Morphy and Rankovic 2005; Keith et al. 2005; Ramsay et al. 2018), which yielded promising applications in various areas, such as e.g. multikinase inhibitors, accounting for the majority of the approved kinase inhibitor agents (Cohen and Alessi 2013), the development of multimyosin drugs has the potential to yield compounds with improved therapeutic potency and superior safety profiles.
This chapter will review the current status of small molecule effectors of myosin function, and focus on compounds that allosterically target the myosin motor domain.
Four classes of myosins, classes I, II, V, and VI, were shown to be affected by small effector molecules. The first part of this chapter will outline the structural basis underlying the modulating effect of the small molecules. How do the effector molecules bind to their target myosins; what factors determine their isoform specificity; and what is the structural mechanism behind their modulating effect? The second part will elaborate on the specificity of the small molecule effectors towards myosin isoforms or classes, in particular associated diseases, and their application in scientific studies of the function of myosins in the cell. Finally, allosteric modulation triggered by protein-protein interactions will be discussed in the third part. While the small molecule effectors in general either increase or reduce ATP turnover, motor function or force development of myosins, they can modulate the motor properties of myosins in an even more intricate manner by affecting specific substeps of the mechanochemical cycle. Myosins share a common motor cycle with actin-detached and -attached states that go along with large conformational changes in the myosin motor domain.
5.2 Binding Sites of Small Molecule Effectors in the Myosin Motor Domain
Structural studies have so far revealed four distinct allosteric binding pockets in the myosin motor domain for small molecule effectors (Allingham et al. 2005; Fedorov et al. 2009; Preller et al. 2011b; Sirigu et al. 2016; Planelles-Herrero et al. 2017). These binding pockets share a primarily hydrophobic nature but with both apolar and polar residues contributing to the interaction with the small molecule effectors. The binding pockets are found in regions that are known to be important for mechanochemical coupling between critical remote sites in the motor domain.
During myosin motor activity, ATP binding, hydrolysis, and release of hydrolysis products are coupled to large scale rearrangements of structural elements that affect actin binding and the position of the lever arm. An actomyosin ATPase cycle that associates chemical changes affecting the nucleotide with large-scale conformational changes was first proposed by Lymn and Taylor (Lymn and Taylor 1971) and further extended over time by the introduction of additional substrates, contributing to myosin function (Preller and Manstein 2017). Critical structural elements or regions of the myosin motor domain that determine the state and the properties of the myosin motor domain include the actin-binding cleft, the central seven-stranded β-sheet, also known as the transducer, the relay helix, the SH1/SH2-region, the converter domain together with the adjacent lever arm, and the active site elements switch-1, switch-2, and the P-loop. To briefly recapture the actomyosin ATPase cycle, starting with the nucleotide-free, strongly actin-bound rigor state of myosin, the motor domain features a closed actin-binding cleft, which is responsible for the favorable and strong interaction with the actin filament. The converter and lever arm in this state are in the down position, the relay helix is straight, and the transducer is twisted. Binding of ATP leads to small-scale rearrangements in the active site, the associated opening of the actin-binding cleft, and the dissociation of the myosin motor domain from the actin filament. This ATP-bound state is referred to as the post-rigor state with the converter and lever arm still in the down position. In addition to the coupling between the active site and the actin-binding cleft, conformational changes in the active site are coupled via the transducer, the relay helix, the SH1/SH2-region, and the converter domain to a swinging of the lever arm. Subsequent to ATP-induced actin detachment, the converter domain rotates from the down to the up position, a process also known as the recovery stroke of myosin, which primes the lever arm for force generation. The recovery stroke includes structural changes in the central β-sheet, which untwists and a bending of the long relay helix, which in turn is coupled to repositioning of the SH1/SH2-region to allow the converter being rotated. The recovery stroke is suggested to follow a stepwise seesaw mechanism (Fischer et al. 2005; Kintses et al. 2008) leading to the catalytically competent pre-power stroke state, in which ATP is hydrolyzed to ADP and Pi. Hydrolysis leads to a relaxation of binding constraints in the active site. As a consequence of conformational changes that are coupled to the subsequent opening of the active site, the myosin reattaches to the actin filament by closing of the actin-binding cleft, which together with the release of the inorganic phosphate initiates twisting of the central β-sheet, straightening of the relay helix, and finally the power stroke, which drives the converter and lever arm from the up position back to the down position. In the last substep of the cycle, ADP is released from myosin and drives the motor domain back to the rigor state. ATP binding starts a new round of the ATPase cycle (Geeves et al. 2005; Houdusse and Sweeney 2016).
The four identified allosteric binding sites for blebbistatin, the series of halogenated pseudilins, CK-571, and omecamtiv mecarbil involve structural elements that have been shown to play a crucial role in the coupling mechanisms within the myosin motor domain (Fig. 5.2). The binding site for blebbistatin directly involves active site switch-2 residues as well as residues of the relay helix and the actin-binding cleft, explaining the effect of blebbistatin on the actomyosin interaction and product release. Halogenated pseudilins bind approximately 7.5 Å apart from the blebbistatin site near actin-binding residues at the top of the cleft. Their binding involves residues of loop 2 and the strut loop of myosin, which were shown to support coupling between the active site state and the actin-binding region. CK-571 binding involves residues of the SH1/SH2-region and the relay helix, thereby inhibiting the coupling between ATP hydrolysis and converter rotation. Omecamtiv mecarbil involves extensive interactions with the N-terminal subdomain and the converter, in addition to contacts with the relay helix and the transducer. The clustering of allosteric binding pockets at critical intersections of the communication pathways in the motor domain brings up the question whether further effector molecules and binding pockets can be identified along the pathways that couple distant sites in the myosin motor domain and how differences between myosin isoforms can be exploited to support the development of potent and specific small molecule myosin effectors (Table 5.1).

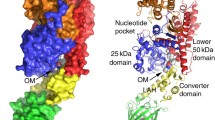
Overview of the four identified allosteric binding pockets in the myosin motor domain of small molecule effectors, mapped to the crystal structure of myosin II. The binding sites were determined by co-crystallization of myosin in complex with (−)-blebbistatin (pdb 1yv3), halogenated pseudilins (pdb 2jhr, 2xo8, 2xel), CK-571 (pdb 5 t45), and OM (pdb 5n69). Subdomains and critical structural elements of the motor domain are color coded and labeled in the diagram
5.2.1 Binding Site at the Apex of the Actin-Binding Cleft (Blebbistatin Site)
Blebbistatin was identified as a potent inhibitor of skeletal myosin II and nonmuscle myosin II isoforms in 2003 (Straight et al. 2003) and was the first myosin inhibitor whose structural mechanism was determined using X-ray crystallography (Allingham et al. 2005). The small molecule effector inhibits myosin II isoforms with half maximal inhibitory concentration (IC50) values between 0.5 and 80 μM, while myosins from classes I, V, and X have IC50 > 150 μM (Limouze et al. 2004). Blebbistatin is a 1-phenyl-2-pyrrolidinone derivative possessing a tricyclic core scaffold (A-, B-, and C-rings) and a D-ring phenyl substituent, as well as a chiral stereocenter bearing a hydroxy group. Blebbistatin was found to bind to a primarily hydrophobic pocket in the myosin motor domain, adjacent to the γ-phosphate sensor loops (switch-1, switch-2, P-loop) of the active site, at the apex of the actin-binding cleft (Allingham et al. 2005) (Fig. 5.3). In the crystal structure (pdb: 1yv3), numerous hydrophobic interactions of blebbistatin and Dictyostelium discoideum (Dd) myosin II are formed with residues of the U50 linker, a loop following β7-strand (Tyr261 and Leu262), residues of the relay helix (Phe466, Glu467, Ile471, and Thr474), and residues of the W-helix (Val630, Tyr634, Glu637, and Leu641), which contribute largely to the high binding affinity of blebbistatin. In addition, three direct hydrogen bonds stabilize the binding pose of blebbistatin in the allosteric myosin pocket: a hydrogen bond of the chiral hydroxy group with the main chain amide group of Gly240, a residue directly following switch-1, and a hydrogen bond with the main chain carbonyl oxygen of Leu262, as well as a hydrogen bond of the carbonyl oxygen of blebbistatin with switch-2 residue Ser456. The interactions of the chiral hydroxy group with both Leu262 and Gly240 are critical for the effective binding and inhibition of myosin by the S-(−)-enantiomer of blebbistatin. The R-(+)-enantiomer, which is not capable of forming hydrogen bonds with Leu262 and Gly240, has no detectable effect on myosin (Straight et al. 2003).

Binding site of (−)-blebbistatin in the motor domain of Dd myosin II in the pre-power stroke state at the apex of the actin-binding cleft (pdb 1yv3) and 2-dimensional interaction diagram, showing the favorable non-covalent interactions of blebbistatin with polar and hydrophobic residues of the myosin binding pocket. Color code: purple arrows = hydrogen bonds, blue amino acids = positively charged residues, red amino acids = negatively charged residues, green amino acids = hydrophobic residues, cyan amino acids = polar residues, white amino acid = glycine residues
As mentioned before, blebbistatin is reported to show a clear specificity for conventional class II myosins over unconventional myosins, particularly from classes I, V, and X (Limouze et al. 2004). The structural basis for this specificity was established around the interacting residues Ser456, Thr474, Tyr634, and Glu637, which were found to show the highest degree of variability among myosin isoforms (Allingham et al. 2005). A crucial role was demonstrated for residue Ser456. This amino acid is part of the γ-phosphate-sensing switch-2 with its consensus sequence DIXGFE, with X being an alanine or serine in conventional class II myosins but a bulky, aromatic tyrosine or phenylalanine in most unconventional myosins, including classes I, V, and X. Mutational studies, replacing the native alanine residue at this position in smooth muscle and nonmuscle myosin II by bulkier amino acids, such as phenylalanine, tyrosine, tryptophan, arginine, or glutamate, resulted in a loss of blebbistatin’s inhibitory potency against these mutated myosins (Zhang et al. 2017). Additionally, the adjacent residue Ile455 has been suggested to be responsible for the blebbistatin insensitivity of wildtype Drosophila melanogaster (Dm) nonmuscle myosin II, which possesses a methionine at that particular position. However, blebbistatin is able to inhibit mutant Dm nonmuscle myosin II, in which the methionine is replaced by an isoleucine, and loses its potency to inhibit mutant Hs nonmuscle myosin II, when isoleucine is exchange to methionine (Zhang et al. 2017).
Structural rearrangements of the side chains of Leu262 and Tyr634 by approximately 4.5 Å and 3 Å are required to accommodate blebbistatin in its binding pocket, suggesting an induced fit mechanism (Allingham et al. 2005). Through its direct interactions with the switch-2 motif and further structural elements of the actin-binding cleft, blebbistatin affects switch-1 and switch-2 opening-closing movements of the active site, and thus coupling with the actin-binding region. Blebbistatin traps the myosin motor domain in an actin-detached pre-power stroke state, impairing Pi release and actin-binding cleft closure, thereby the transition to the force-generating, strongly actin-bound states. The co-crystallized structure of blebbistatin bound to Dd myosin II in the presence of the non-hydrolyzable ATP transition state analogue ADP·VO4 highly resembles other Dd myosin II pre-power stroke state structures that were obtained in the presence of transition state ATP analogues. Interestingly, the structure of myosin in the presence of ADP and blebbistatin was reported to adopt a previously unobserved structural state, priming the converter and lever arm for the power stroke, while at the same time not affecting actin filament interaction (Takács et al. 2010). The low-resolution electron microscopy structure of this myosin II·ADP·blebbistatin complex was interpreted as the start-of-power stroke state, prior to force generation, similar to proposed structural models of the start-of-power stroke state (Preller and Holmes 2013), and showed that in the presence of ADP, blebbistatin blocks switch-2 opening, thereby priming the lever arm. However, switch-1 opening can occur, which is coupled to the closure of the actin-binding cleft and a concomitant increase in actin affinity. The binding site and pose of blebbistatin has been confirmed by co-crystallization of A-ring derivatives, where the position of the methyl group at the A-ring was modified, and which bind identically to the myosin motor domain (Lucas-Lopez et al. 2008).
5.2.2 Binding Site at the Top of the Actin-Binding Cleft (Pseudilin Site)
Halogenated pseudilins are a class of marine alkaloids with a 2-phenylpyrrole scaffold that was shown to inhibit different classes of myosins and myosin-dependent cellular processes with IC50 values in the low micromolar range (Fedorov et al. 2009; Chinthalapudi et al. 2011; Preller et al. 2011b; Martin et al. 2014). Structural analysis revealed that the halogenated pseudilins bind to an allosteric binding pocket in the myosin motor domain at the top of the actin-binding cleft, close to the actin-binding region, about 7.5 Å apart from the blebbistatin-binding site and 16 Å from the active site (Fedorov et al. 2009). The binding pocket is primarily formed by two helices of the U50 kDa domain, the strut loop, and loop 2 (Fig. 5.4). It is open to the actin-binding cleft as well as the protein surface, giving access for the halogenated pseudilins to enter the binding site.

Binding site of the halogenated pseudilin pentabromopseudilin in the motor domain of Dd myosin II in the pre-power stroke state at the top of the actin-binding cleft (pdb 2jhr) and 2-dimensional interaction diagram, showing the favorable interactions of pentabromopseudilin with the myosin residues. Color code: purple arrows = hydrogen bonds, orange arrows = halogen bonds, blue amino acids = positively charged residues, red amino acids = negatively charged residues, green amino acids = hydrophobic residues, cyan amino acids = polar residues, white amino acid = glycine residues
In the crystal structures of Dd myosin II co-crystallized with three different halogenated pseudilins (pdb 2jhr, 2xo8, 2xel), the small molecule inhibitors bind to the allosteric pocket through both hydrogen bonds and numerous hydrophobic interactions with residues, including Lys265, Ala420, Ala424, Arg428, Leu431, Asp590, Ile617, and Ala618. A particularly important role has been suggested for the interaction with Lys265, which is hydrogen bonded either to the hydroxy group or the amine group of the halogenated pseudilins. Upon binding of the small molecule inhibitors, the side chain of Lys265 reorients by up to 3.5 Å as compared to myosin structures in the same structural state and without bound pseudilin. This interaction leads to a cascade of small rearrangements of residue side chains along the so-called signal relay pathway, thereby affecting the active site and preventing the proper positioning of the lytic water molecule for an in-line attack of the ATP’s γ-phosphate (Chinthalapudi et al. 2011; Preller et al. 2011b). Mutational studies, replacing this lysine residue, confirmed the importance of this residue for myosin inhibition by halogenated pseudilins, since mutant Dd myosin II in which Lys265 is replaced by alanine becomes insensitive to pseudilins (Behrens et al. 2017). In addition, the mutational study revealed that this conserved Lys265 is a key residue in the coupling pathway between the active site and the actin-binding region (Behrens et al. 2017). Similar roles for the coupling between actin-binding region and active site have been earlier demonstrated for the strut loop and loop 2 (Geeves et al. 2005). Kinetic analysis of the inhibitory effect of halogenated pseudilins on myosin revealed that the pseudilins reduce the coupling between the active site and the actin-binding region, which can be rationalized by the direct interaction of the small molecule effectors with Lys265 as well as the strut loop and loop 2. Additionally, the pseudilins affect the active site, through the signal relay pathway, which correlates with the experimentally determined effect of halogenated pseudilins on ATP binding and hydrolysis, as well as ADP dissociation.
The three crystallized pseudilin derivatives were found to adopt slightly different binding poses within the allosteric binding pocket (Preller et al. 2011b). Depending on the halogen substituents, the pseudilins show specificities towards certain myosin classes. While highly chlorinated pseudilins exhibit their strongest effect on class I myosins, highly brominated pseudilins preferentially affect class V myosins. Pseudilins with mixed halogenations do not feature any preference for myosin classes. These specificity behaviors of the pseudilins are strongly correlated with differences in the polarities and physicochemical properties of the allosteric binding pocket in different myosin isoforms and classes (Preller et al. 2011b).
5.2.3 Binding Site Between the SH1/SH2 Region and the Relay Helix (CK-571 Site)
The third known allosteric binding site for a small molecule effector in the myosin motor domain is the binding pocket of the smooth muscle myosin II-specific inhibitor CK-2018571 (CK-571) (Sirigu et al. 2016). CK-571 inhibits smooth muscle myosin II with a high potency (IC50 ∼ 9 nM) and kinetically traps the myosin in an ATP-bound, actin-detached intermediate state of the recovery stroke. Hence, in contrast to the crystal structures of myosin II with blebbistatin or halogenated pseudilins, which both are crystallized in the pre-power stroke state, the smooth muscle myosin II·CK-571 structure (pdb 5t45) has similarities with post-rigor state structures, featuring an open actin-binding cleft and the converter and lever arm in the down position. However, other critical structural elements, such as the central β-sheet or switch-2, are found in intermediate conformations between the post-rigor and pre-power stroke states.
The identified allosteric binding pocket for CK-571 is located between the SH1-helix and the relay helix in the myosin motor domain, 22 Å apart from the active site (Fig. 5.5). Interestingly, this pocket is not accessible either in the post-rigor or the pre-power stroke state of myosin, and is suggested to only open during the recovery stroke (Sirigu et al. 2016). Binding of CK-571 is primarily mediated by hydrophobic interactions with residues of the N-terminal domain (Asp88, Met89, Ala90, Leu95, Val100, Ser117, Gly118, Leu119, and Phe120), relay helix (Phe493, Asn494, Met497, and Glu501), SH1-helix (Gly709, Val710, Glu712, Gly713, Ile716, and Cys717), and converter domain (Pro722 and Arg724). In the crystal structure with smooth muscle myosin II, no attractive hydrogens bonds were detected between CK-571 and myosin. The structural basis for the inhibition of smooth muscle myosin II by CK-571 has been attributed to the trapping of the myosin motor domain in an intermediate state, resembling the post-rigor state, by interfering with the required conformational changes of the relay helix and the SH1-helix, which in turn prevents myosin from transitioning through the recovery stroke and adopting the catalytically active pre-power stroke state. This effect is similar to the reported consequences of cold-sensitive single-point mutations G680A and G680 V in the SH1-helix, which were shown via kinetic and structural analyses to freeze the mutated myosin in a state similar to the post-rigor state, impairing myosin motor function (Preller et al. 2011a). Small-angle X-ray scattering experiments confirmed that CK-571-bound myosin preferentially adopts such an intermediate post-rigor-like structure. Particularly switch-2 appears affected by CK-571. Marked differences between the CK-571 intermediate state and the wild type post-rigor state have been suggested for the catalytically important switch-2, which is no longer able to close around the ATP γ-phosphate to position the lytic water for the hydrolysis reaction (Sirigu et al. 2016).

Binding site of CK-571 in the motor domain of smooth muscle myosin II in an intermediate state between post-rigor and pre-power stroke states, between the SH1/SH2-region and the relay helix (pdb 5t45) and 2-dimensional interaction diagram, showing the favorable interactions of CK-571 with the myosin residues. Color code: purple arrow = hydrogen bonds, blue amino acids = positively charged residues, red amino acids = negatively charged residues, green amino acids = hydrophobic residues, cyan amino acids = polar residues, white amino acid = glycine residues
All residues of the binding pocket, which are involved in interaction with CK-571, are highly conserved within the class II myosins, preventing establishment of the structural basis for the specificity behavior of CK-571 towards smooth muscle myosin II over other members of the myosin class II.
5.2.4 Binding Site at the L50 kDa – Converter Interface (OM Site)
Omecamtiv mecarbil (OM), earlier denoted as CK-1827452, is a diaryl-urea-based small molecule activator of cardiac myosin II, which is currently in phase 3 clinical trials as a treatment of systolic heart failure. OM was shown to specifically bind to the cardiac myosin motor domain with an affinity of 1.3 μM, and to accelerate the transition rate of the β-cardiac myosin II motor domain into actin-bound, force-generating states (Malik et al. 2011). The co-crystallized high-resolution structure of cardiac myosin II in complex with OM has been solved in two different structural states, with controversial results and binding positions (Winkelmann et al. 2015; Planelles-Herrero et al. 2017). However, the determined kinetic mechanism of action of OM on cardiac myosin II (Malik et al. 2011; Liu et al. 2015; Swenson et al. 2017), as well as binding studies using isothermal calorimetry, and low-resolution solution structural analysis by small angle X-ray scattering (Sirigu et al. 2016) indicate a preference of OM for cardiac myosin II states directly prior to the power stroke and at the end of the recovery stroke, with the converter and lever arm in the up position, i.d. the pre-power stroke state.
The high-resolution structure of cardiac myosin II in complex with OM, crystallized in the pre-power stroke state (pdb 5n69), reveals a binding pocket for OM that is located approximately 31 Å apart from the active site, at a coupling hot spot in the motor domain, critical for regulation of the converter domain and the lever arm swing, and hence for force production (Sirigu et al. 2016) (Fig. 5.6). All structural elements that are directly involved in binding OM play a role in communication between the active site and the force-generating lever arm swing. OM binds to a pocket of predominantly hydrophobic nature. One side of the pocket is built by the converter domain, and only existent if the converter domain together with the lever arm is in the up position, as in the pre-power stroke. Indeed, comparison of crystal structures in the same state, however in the presence and absence of OM, showed, that OM is required to fully close the binding pocket by pulling the converter closer to itself. Hence, OM seems to bind through an induced fit mechanism to the cardiac myosin II motor domain, and stabilize states with the lever arm in the up position, primed for the power stroke. Local rearrangements were found surrounding the bound OM, mostly involving residues of the converter domain. Hydrophobic interactions stabilize OM in the pocket and are formed with residues of the N-terminal domain (Lys146, Arg147, Asn160, Gln163, Tyr164, Thr167, and Asp168), the relay helix (His492), the third β-strand of the central transducer (His666), and the converter domain (Pro710, Ile713, Arg721, Tyr726, Phe765, and Leu770). In addition, polar interactions are found with Asp168, Asn711, Arg712, and the urea moiety of OM, as well as potentially Tyr164 and Glu774, and the piperazine ring of OM. The OM-mediated formation of this tight interaction network leads to a stabilization of motor domain conformations with the converter and lever arm in the up position. This leads to an associated shift of the ensemble of conformers to states where most motor domains are primed for rebinding to actin and the force-generating power stroke (Sirigu et al. 2016). Earlier studies using a photo-reactive benzophenone derivative of OM suggested already this critical hot spot and particularly interactions with the N-terminal domain, as the photo-reactive OM derivative was found to crosslink to Ser148 in the N-terminal domain (Malik et al. 2011). Myosin isoform-specific differences in the interacting residues have been attributed to the high specificity of OM within class II towards cardiac myosin II, including Tyr164, one of the hydrogen bonded residues, which is a serine in smooth muscle myosin II and a phenylalanine in skeletal myosin II isoforms, His666, which is replaced by a threonine residue in smooth muscle myosin II, Asn711, which is a serine in skeletal muscle myosin, and Ile713, which is replaced by a valine residue in skeletal muscle myosin. Other residues of the binding pocket that show variations between the different myosin isoforms include Asp168, His492, and Arg712.

Binding site of OM in the motor domain of cardiac myosin II in the pre-power stroke state at the L50 kDa – converter interface (pdb 5n69) and 2-dimensional interaction diagram, showing the favorable interactions of OM with the myosin residues. Color code: purple arrow = hydrogen bonds, green line = π-π-stacking, blue amino acids = positively charged residues, red amino acids = negatively charged residues, green amino acids = hydrophobic residues, cyan amino acids = polar residues, white amino acid = glycine residues
As mentioned before, several reported experiments indicate that OM preferentially acts on force-generating myosin states with the lever arm in the up position: (1) according to kinetic studies, OM has an impact on the equilibrium of ATP hydrolysis and lever arm priming, shifting it towards intermediate states with bound ADP·Pi prior to power stroke (Liu et al. 2015), (2) measurements of the binding affinity of OM to different myosin states revealed a 20-fold higher affinity towards the pre-power stroke state (in the presence of the non-hydrolysable nucleotide analogue ADP·VO4 that traps myosin in the intermediate state) as compared to post-rigor state (in the presence of ADP) (Sirigu et al. 2016), (3) in solution, cardiac myosin II in the presence of OM preferentially adopts a conformation that closely resembles the pre-power stroke as determined by small-angle X-ray scattering (Sirigu et al. 2016).
The structural mechanism for OM activation of cardiac myosin function seems to be a stabilization of the pre-power stroke state with OM supporting the interaction of the relay helix, the N-terminal domain, and the converter domain, and thereby the priming of the lever arm in the up position. This increased population of catalytically competent states leads to an elevated number of myosin motor domain with bound ADP·Pi, which possess a higher affinity for actin filament. The rebinding of actin filaments rapidly releases Pi and allows force production. This postulated mechanism is compatible with the findings that OM reduces the ATP activity in the absence of actin (basal ATPase activity) (Malik et al. 2011; Liu et al. 2015; Swenson et al. 2017), due to the increased population of ADP·Pi states.
5.3 Myosin Classes Targeted by Small Molecule Effectors
5.3.1 Preference Towards Conventional Myosins (Class II)
5.3.1.1 Small Molecule Inhibitors of Myosin II
One of the first identified myosin II inhibitor was 2,3-butanedione monoxime (BDM) (Higuchi and Takemori 1989; Mckillop et al. 1994). BDM binds with a low affinity of Ki ≈ 5 mM to skeletal muscle myosin II, inhibiting myosin ATPase activity in a noncompetitive and reversible manner (Herrmann et al. 1992). Despite the high concentrations required to inhibit myosin II, BDM was used widely in studies of myosin II function in cellular systems. However, the applicability of BDM as a general myosin II inhibitor is limited due to controversially discussed uncertainties about the specificity of BDM towards skeletal muscle myosin II and possible inhibitory effects on other myosin classes and isoforms, such as nonmuscle myosin II, class I, V, and VI myosins (Cramer and Mitchison 1995; Ostap 2002). In addition, BDM was found to affect many other non-myosin proteins (Sellin and McArdle 1994), including acetylcholinesterase (Wilson and Ginsburg 1955), potassium channels (Schlichter et al. 1992; Lopatin and Nichols 1993), L-type calcium channels (Ferreira et al. 1997), serine and threonine phosphorylation (Stapleton et al. 1998), and myosin II light-chain kinase (Siegman et al. 1994).
Targeted screening of a chemical library led to the identification of N-benzyl-p-toluene sulfonamide (BTS) as a reversible small molecule inhibitor of fast muscle myosin II with IC50 values of ∼5 μM for fast skeletal muscle myosin II (Cheung et al. 2002). According to nucleotide competition experiments, it was suggested that BTS binds noncompetitively to the myosin motor domain. BTS was shown to inhibit contraction and force production in skeletal muscle fibers, and possesses 100-fold specificity towards fast skeletal muscle myosin II over other class II isoforms, including slow skeletal muscle myosin II, cardiac myosin II, platelet myosin II, and nonmuscle myosin II (Cheung et al. 2002).
The antipsychotic drug trifluoperazine (TFP) was reported to inhibit the myosin II ATPase activity and motility at concentrations of 100–200 μM (Sellers et al. 2003). TFP was known earlier as an efficient calmodulin antagonist that leads to removal of the regulatory light chains of smooth muscle myosins (Trybus et al. 1994; Yang and Sweeney 1995) and scallop striated muscle myosins (Patel et al. 2000) at concentrations in the millimolar range.
3-[4-(3-pheny-2-pyrazolin-1-yl) benzene-1-sulfonylamido]-phenylboronic acid (PPBA) is a fluorescent dye that was initially surmised to competitively inhibit skeletal myosin II function with a submicromolar potency (Ki ≈ 0.8 μM) (Hiratsuka 1994). However, this was later revised and PPBA was suggested to bind to the nucleotide-bound myosin motor domain and act through an allosteric mechanism (Hiratsuka 2006). On the basis of manual fitting the structure of PPBA to the crystal structure of Dd myosin II, a hydrophobic pocket distant from the active site was speculated to be the binding site of PPBA, with direct interactions of the small molecule inhibitor and Phe472.
In contrast to the small molecule inhibitors described above, blebbistatin and blebbistatin derivatives are well characterized small molecule myosin inhibitors that exhibit marked specificities towards class II myosins with no or weak activities on class I, V, and X myosins (Limouze et al. 2004). The highest IC50 of 0.5 μM was determined for skeletal muscle myosin II, while cardiac myosin II and nonmuscle myosin II isoforms are affected in the range of 1–10 μM. Larger variations of the potencies of blebbistatin for smooth muscle myosin II are reported ranging from 3 to 80 μM. The myosin II-specific small molecule inhibitors have been applied in numerous studies to reveal the physiological and pathological role of myosin II isoforms in biological processes, including the spatiotemporal control of cytokinesis (Straight et al. 2003), control of cell migration and antigen capture in dendritic cells (Chabaud et al. 2015), regulation of detrusor contractility in partial bladder outlet obstruction (Zhang et al. 2011), and in optical mapping of the heart (Swift et al. 2012). Hence, blebbistatin and its derivatives have advanced to front line research tools for studying myosin-dependent processes. The mechanism as well as the structural basis of myosin inhibition by blebbistatin has been determined (Ramamurthy et al. 2004; Kovács et al. 2004; Allingham et al. 2005). However, blebbistatin has several drawbacks that interfere with its general use, such as photo-toxicity and -instability, high fluorescence, cytotoxic effects, and poor solubility. Therefore, comprehensive optimizations of blebbistatin were carried out, particularly centering on derivatizations of the A- and D-ring of blebbistatin to improve the physicochemical properties and the inhibitory potency of the small molecules (Képiró et al. 2014; Várkuti et al. 2016; Verhasselt et al. 2017a, b, c; Roman et al. 2018; Rauscher et al. 2018). As a reminder, blebbistatin has a tricyclic tetrahydropyrroloquinolinone scaffold forming the A-, B-, and C-rings, and a phenyl ring, the D-ring, linked to the pyrrole (C-ring) nitrogen. A-ring modifications had minor or negative impacts on the properties and potencies of the small molecule blebbistatin derivatives (Lucas-Lopez et al. 2008; Verhasselt et al. 2017c). However, superior physicochemical properties or potencies were observed for modifications of the D-ring at the meta- or para-position of the phenyl ring (Rauscher et al. 2018). Addition of small polar functional groups, particularly para-nitro and para-amino, as well as meta-hydroxy or meta-nitro groups improved the photosensitivity, hence the structural stability of the small molecules, and the solubility, while at the same time leading to a reduced phototoxicity and fluorescence (Képiró et al. 2014; Várkuti et al. 2016; Verhasselt et al. 2017b). Such optimized scaffolds potentially open the way for safer use of these compounds in scientific studies of myosin-dependent processes and for directed targeting of myosin II-associated diseases.
CK-2018571 (CK-571) is a small molecule inhibitor that was identified by high-throughput screenings and chemical modification to target smooth muscle myosin II and muscle contraction for potential use in associated diseases, including asthma and chronic obstructive pulmonary disease (Sirigu et al. 2016). Co-crystallization of CK-571 with smooth muscle myosin II confirmed the allosteric mechanism of CK-571. Considerable specificities within myosin class II were observed for CK-571 with high potency towards smooth muscle myosin II (IC50 = 9 nM), and 280-fold and 1255-fold lower potencies against cardiac myosin II and skeletal muscle myosin II, as well as 8.5-fold lower potency for the closely related nonmuscle myosin II. The small molecule inhibitor interferes with ATP hydrolysis and actin reattachment by trapping the myosin in an ATP-bound intermediate state of the recovery stroke (Sirigu et al. 2016). In vivo experiments verify the relaxing effect of CK-571 on different smooth muscle tissues and decreased force production in stimulated skinned artery rings. Concentrations in the range of 1 μM were required to relax skinned and intact tissue. In addition, CK-571 was shown to inhibit methacholine-induced bronchoconstriction in naïve dogs with comparable effect as obtained with albuterol, as the clinically-relevant β2-adrenergic agonist, highlighting the therapeutic potential of CK-571 (Sirigu et al. 2016).
The recently identified small molecule inhibitor of cardiac myosin II mavacamten (MYK-461) shows potent inhibitory effects on the ATPase function of cardiac myosin II (IC50 = 0.3–0.7 μM) and the weak-to-strong transition of myosin (Green et al. 2016; Kawas et al. 2017). Mavacamten was developed specifically to target mutated cardiac myosins, a prime cause for hypertrophic cardiomyopathy (HCM) and is currently tested in phase 3 clinical trials for treatment of HCM. The cardiac myosin inhibitor reduces phosphate release as well as the myosin duty ratio – the fraction of the time myosin stays strongly bound to the actin filament (Kawas et al. 2017). Mavacamten has additionally been suggested to stabilize an autoinhibited super-relaxed state with markedly reduced ATPase activity, which might be common for a fraction of healthy cardiac myosins, and which is prevented by HCM mutations in the cardiac myosin heavy chain (Rohde et al. 2018; Anderson et al. 2018). In transgenic mice harboring heterozygous mutations in β-cardiac myosin, mavacamten improved cardiac function, as evidenced from reduced fractional shortening and left ventricular hypertrophy, myocyte disarray and fibrosis (Green et al. 2016). Additionally, a clinical feline model of HCM treated with mavacamten displayed an improved sarcomere contractility and relieved left ventricular outflow tract obstruction (Stern et al. 2016).
5.3.1.2 Pharmacological Chaperones
Myosin family members are among the most abundant proteins in our bodies. All of our movement, each step and every heartbeat, requires the force and tension-generating action of myosins. These activities subject myosins to physical stress that, together with the extremes of pH and temperature found in the cell, can lead to the formation of roadblock-like, strongly-bound complexes of “dead” myosin motors with actin filament tracks (Murphy and Spudich 1999). Even a slight increase in the occurrence of such roadblocks can trigger catastrophic consequences in actomyosin-rich organs. Here, the restoration of normal myosin homeostasis by pharmacological chaperones promises to provide an efficient means of interfering with the progression of an acute crisis and to support long-term stabilization of patients (Radke et al. 2014; Wustman et al. 2014).
The thiadiazenone EMD57033 is a cardiotonic agent that enhances both systolic and diastolic function in failing hearts at minimal energetic cost (Solaro et al. 1993; Senzaki et al. 2000). The well-known effects of EMD57033 as an inotropic Ca2+ sensitizer and activator of cardiac actomyosin (Ferroni et al. 1991; Beier et al. 1991; Gambassi et al. 1993) are fully explained by an EMD57033-mediated shift in the duty-ratio and their consequences on thin-filament regulation (Radke et al. 2014). EMD57033 binds with an affinity of 7.3 μM to an allosteric pocket in the β-cardiac myosin II motor domain and protects myosin against heat stress and thermal denaturation (Radke et al. 2014). Addition of the compound to heat-inactivated β-cardiac myosin results in refolding and reactivation of ATPase and motile activities. Moreover, in heat-stressed cardiomyocytes expression of the stress-marker atrial natriuretic peptide is suppressed by EMD 57033. Thus, EMD 57033 displays a much wider spectrum of activities than those previously associated with small, drug-like compounds (Radke et al. 2014). Current efforts by our groups have led to the discovery of further thiadiazenones that act as pharmacological chaperones for β-cardiac myosin. We see the greatest potential for the therapeutic use of pharmacological chaperones in the area of cardiac and cytoskeletal myosin II isoforms.
5.3.2 Preference Towards Unconventional Class I Myosins
Two small molecule inhibitors of class I myosins have been described in the literature with clear isoform or even species specificities. The fully chlorinated marine antibiotic pentachloropseudilin (PClP) inhibits the enzymatic and motor function of myosin I in a reversible manner with IC50 values in the range of 1–5 μM for mammalian class I myosins (Chinthalapudi et al. 2011). PClP shows largely effects on most tested myosin I isoforms in the low micromolar range, and 20- to 90-fold higher potencies as compared to class II und V myosins. In addition, even at 100 μM concentrations, PClP does not inhibit human myosin VI or myosin VII isoforms. Comparative structural studies attributed the specificity of highly chlorinated halogenated pseudilins, such as PClP, to the isoform-specific differences in polarity of the allosteric binding pocket in the myosin motor domain (Preller et al. 2011b). PClP is active in cells and tissue and was shown to induce comparable effects in HeLa cells at low micromolar concentrations as observed with myosin IC knockdown cells, resulting in abnormalities in lysosome morphology and distribution (Chinthalapudi et al. 2011). Above concentrations of 25 μM, PClP showed cytotoxic effects on the HeLa cells. Hence, PClP was used as a tool compound for studying myosin I functions in cellular systems. Selective inhibition of myosin I by PClP demonstrated the role of class I myosins in autophagosome-lysosome fusion (Brandstaetter et al. 2014), as well as a critical function of myosin I in the maintenance and formation of blastodisc morphology, cell-division and dynamics as well as distribution of lipid droplets within the blastodisc in early zebrafish embryos (Gupta et al. 2017). Additionally, both PClP and the related myosin class V-specific pentabromopseudilin were used to elucidate that changes in cell shape, apical contraction, and cell intercalation highly depend on myosin II function, whereas myosin I and myosin V are involved in the assembly of supercellular cable-like myosin II structures at cell junctions that mediate primitive streak formation (Rozbicki et al. 2015).
The second class I-specific myosin inhibitor is the cyanoacrylate-based compound phenamacril, a well-known fungicide, whose direct inhibitory effect on Fusarium graminearum myosin I has recently been described and characterized (Zhang et al. 2015; Wollenberg et al. 2019). Phenamacril reversibly inhibits the ATPase activity and in vitro motility of Fusarium graminearum and Fusarium avenaceum myosin I with potencies up to IC50 = 0.36 μM but does not affect the related Fusarium solani myosin I, nor human myosin IC or class I and II isoforms of the slime mold D. discoideum. Phenamacril is an environmentally-friendly fungicide that is widely used in regulation of cereal infections by fungi of the Fusarium family in China (Li et al. 2008). Docking experiments predicted the binding site of phenamacril to be in the actin-binding cleft of myosin, in proximity of the binding pockets of halogenated pseudilins or blebbistatin, which correlates with identified mutations in the myosin motor domain that are implicated in resistance development of the fungi against phenamacril (Li et al. 2016a; Hou et al. 2018).
5.3.3 Preference Towards Unconventional Class V Myosins
Based on the concept of privileged chemical scaffolds that were derived from known bioactive compounds, particularly kinase inhibitors (Peters et al. 2006), a search for class V-specific myosin inhibitors was performed and yielded the small molecule inhibitor of myosin V, based on a pyrazolopyrimidine scaffold, Myo-Vin1 (Islam et al. 2010). Myo-Vin1 demonstrates a preference for class V myosins, which are inhibited in the low micromolar range (IC50 ≈ 6 μM), over skeletal muscle and nonmuscle myosin II isoforms, which are not affected at concentrations up to 50 μM. In addition, no inhibition of many representative kinases was observed at 100 μM compound concentrations.
Highly brominated members of the halogenated pseudilin family of small molecule myosin inhibitors, such as pentabromopseudilin (PBP), were identified as potent and reversible inhibitors with specificities towards class V myosins over class I and II myosins (Fedorov et al. 2009). Halogenated pseudilins were initially reported as marine alkaloids with antitumor and phytotoxic activities, and effects on 12- and 15-human lipoxygenases (Ohri et al. 2005). In vitro, PBP potently affects the enzymatic ATPase activity and the motor function of myosin V with IC50 values of 1.2 μM for chicken myosin VA, as well as isometric tension development and unloaded shortening velocity in muscle. Cellular studies have shown that the phenotypic effect of PBP on mitochondrial fragmentation closely resembles the phenotype observed with mutant yeast, leading to an impaired expression of myosin V (Fedorov et al. 2009).
5.3.4 Preference Towards Unconventional Class VI Myosins
Myosin VI has been shown to play a critical role in migration, metastasis, and tumorigenesis of cancer cells (Yoshida et al. 2004; Dunn et al. 2006). In addition, strong links have been made between mutations in myosin VI and diseases such as hypertrophic cardiomyopathy (Mohiddin et al. 2004), Snell’s Waltzer deafness and nonsyndromic hearing loss (Melchionda et al. 2001). There is only a single small molecule effector of class VI myosins reported to date: 2,4,6-triiodophenol inhibits human myosin VI actin-activated ATPase activity following a biphasic behavior with a Ki,1 of 0.8 μM, contributing 37% to the inhibition, and a second Ki,2 of 37 μM, contributing 63% to the inhibition (Heissler et al. 2012). This biphasic behavior, observed in the inhibition assays, was interpreted as the binding to two independent binding sites in the myosin VI motor domain. However, no structural data are currently available to support the existence of two binding sites for 2,4,6-triiodophenol. No effect of 2,4,6-triiodophenol on the ATPase activity of Dd myosin ID, human nonmuscle myosin IIC, or porcine β-cardiac myosin II were observed up to concentrations of 50 μM of the small molecule effector, hence 2,4,6-triiodophenol seems to show a preferential binding and inhibition of class VI myosins (Heissler et al. 2012). 2,4,6-Triiodophenol was earlier shown to possess nonsteroid anti-inflammatory effects by inhibiting leukotriene B4 synthesis (Trocóniz et al. 2006). Accompanying live-cell studies confirmed the inhibitory effect of 2,4,6-triiodophenol on the physiological function of myosin VI during the final stages of the secretory pathway, with a comparable efficiency as myosin VI knockdown, indicating an IC50 of 1.6 μM (Heissler et al. 2012).
5.4 Allosteric Modulation by Protein-Protein Interactions
Myosins are multifunctional enzymes that typically function within the framework of supramolecular complexes whose function is regulated and modulated by multiple allosteric trigger events. In addition to the binding of small effector molecules, allosteric trigger events include post-translational modifications, protein-protein interactions, and disease-causing mutations. Thus, allosteric trigger events are contributed by more than 100 gene products encoding actin- and myosin-binding proteins (Dos Remedios et al. 2003; Winder and Ayscough 2005), hundreds of post-translational modifications for which actin and actin-binding proteins are major targets (Li et al. 2015; Terman and Kashina 2013), and well over 100 disease causing mutations affecting core components of actin-dependent contractile complexes (Richard et al. 2003; Goebel and Laing 2009; Ampe and Van Troys 2017; Marian and Braunwald 2017). This brings up the question of how we can address the resulting functional and regulatory complexity. The answer is to address one complex at a time in a stringently defined state, to be complete in the analysis of the individual components, to report the results using the correct nomenclature, and to develop and refine structure-based models that explain the changes brought about by individual allosteric perturbations. The availability of highly sensitive, rapid, and quantitative analytical methods, high-resolution structures of complexes composed of F-actin-Tpm (tropomyosin) cofilaments and myosin motor domains (A-Tpm-M), the stringent use of an unambiguous systematic protein nomenclature, and the increasing performance of computational approaches will help to unravel the complexity and to move programs for the identification of small allosteric effectors of myosin function to a new level. The following paragraph describes the need for and the advantages of such an approach using thin filament regulation by Tpm as an example.
Tpm form a large family of double-stranded alpha-helical coiled-coil actin-binding proteins that play a key role in regulating the interaction of actin filaments with both sarcomeric myosin and cytoskeletal myosin isoforms (Schaub and Ermini 1969; Galińska-Rakoczy et al. 2008; Meiring et al. 2018; Manstein et al. 2019). Over 40 Tpm isoforms are produced by alternate promoter selection and splicing of four different genes – TPM1–4. The resulting types of A-Tpm co-polymers show remarkably little functional redundancy (Bryce et al. 2003; Tojkander et al. 2011; Gateva et al. 2017). Modelling, which is based on structural and kinetic analysis of A-M-Tpm-complexes and their individual components (Behrmann et al. 2012; Münnich et al. 2014a, b; von der Ecken et al. 2015; Hundt et al. 2016; Ecken et al. 2016; Chinthalapudi et al. 2017; Pathan-Chhatbar et al. 2018), predicts that the mechanism by which Tpm isoforms support cytoskeletal functions differs fundamentally from the regulatory role described for sarcomeric Tpm isoforms. In the cytoskeletal environment, the higher duty-ratio of the interacting myosin isoforms and the absence of a mechanism for the stringent synchronization of different types and classes of myosin motors are predicted to keep Tpm permanently in an “open” state on F-actin. This means that myosins can always interact with A-Tpm cofilaments. In the absence of a “blocked” state, the contribution of Tpm to the stereospecific interaction surface with myosin has three distinct consequences. A widely accepted consequence is the function of a gatekeeper that favours the interaction between certain combinations of myosin and Tpm isoforms while inhibiting others (Coulton et al. 2010; Gunning et al. 2015; Moore et al. 2016; Manstein and Mulvihill 2016; Gateva et al. 2017). The second, equally well-accepted consequence is the cooperative behaviour of actomyosin in the presence of Tpm. Here, it is well known that in the presence of bare F-actin the activation of ATP turnover by myosin subfragment-1 (S1) is linear as a function of added S1, yet becomes sigmoidal when Tpm is present (Lehrer and Morris 1982; Moraczewska et al. 1999; Tobacman 2008). Tpm inhibits the actomyosin ATPase at low S1:F-actin ratios and activates the ATPase at moderate to high S1:F-actin ratios more strongly than bare actin filaments. Inhibition and activation by Tpm are greatest when troponin and Ca2+ are present, but strong cooperative behaviour is also observed with the cytoskeletal isoforms in the absence of troponin (Lehrer and Morris 1982, 1984). The third consequence is a Tpm isoform-dependent change in key enzymatic parameters of the myosin motor. The Tpm isoform defines the rate of ATP turnover, the duty-ratio, thermodynamic coupling, maximal velocity, and strain-sensitivity of product release steps (Hundt et al. 2016; Gateva et al. 2017; Pathan-Chhatbar et al. 2018). Accordingly, motor activity is geared towards slower or faster movement, tension holding, or active constriction. The elucidation of the exact role of individual Tpm isoforms, the mechanisms that are responsible for the sorting of the different isoforms, and the temporal and spatial control of complex formation and turnover are arguably amongst the most important tasks in cell biology. The profound and diverse Tpm-mediated changes in myosin motor activity challenge current concepts and call for their revision by a classification system in which myosin motor function is not only classified on the basis of the interaction with bare F-actin. In the context of small allosteric effectors of myosin motor activity, the terms activator and inhibitor should be best avoided or at least more systematically defined. An appropriate definition needs to reflect protein context, physiological function, changes in apparent actin affinity in the presence of ATP, maximal ATP-turnover in the absence and presence of F-actin, maximal velocity, cooperativity, duty ratio, and stall force.
References
Allingham JS, Smith R, Rayment I (2005) The structural basis of blebbistatin inhibition and specificity for myosin II. Nat Struct Mol Biol 12:378–379. https://doi.org/10.1038/nsmb908
Ampe C, Van Troys M (2017) Mammalian actins: isoform-specific functions and diseases. Handb Exp Pharmacol 235:1–37. https://doi.org/10.1007/164_2016_43
Anderson RL, Trivedi DV, Sarkar SS et al (2018) Deciphering the super relaxed state of human β-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc Natl Acad Sci 115:E8143–E8152. https://doi.org/10.1073/PNAS.1809540115
Behrens VA, Münnich S, Adler-Gunzelmann G et al (2017) The conserved lysine-265 allosterically modulates nucleotide- and actin-binding site coupling in myosin-2. Sci Rep 7:7650. https://doi.org/10.1038/s41598-017-07933-y
Behrmann E, Müller M, Penczek PA et al (2012) Structure of the rigor actin-tropomyosin-myosin complex. Cell 150:327–338. https://doi.org/10.1016/J.CELL.2012.05.037
Beier N, Harting J, Jonas R et al (1991) The novel cardiotonic agent EMD 53 998 is a potent “calcium sensitizer”. J Cardiovasc Pharmacol 18:17–27
Berg JS, Powell BC, Cheney RE (2001) A millennial myosin census. Mol Biol Cell 12:780–794. https://doi.org/10.1091/mbc.12.4.780
Brandstaetter H, Kishi-Itakura C, Tumbarello DA et al (2014) Loss of functional MYO1C/myosin 1c, a motor protein involved in lipid raft trafficking, disrupts autophagosome-lysosome fusion. Autophagy 10:2310–2323. https://doi.org/10.4161/15548627.2014.984272
Bryce NS, Schevzov G, Ferguson V et al (2003) Specification of actin filament function and molecular composition by tropomyosin isoforms. Mol Biol Cell 14:1002–1016. https://doi.org/10.1091/mbc.e02-04-0244
Chabaud M, Heuzé ML, Bretou M et al (2015) Cell migration and antigen capture are antagonistic processes coupled by myosin II in dendritic cells. Nat Commun 6:7526. https://doi.org/10.1038/ncomms8526
Charlton MR, Balagopal P, Nair KS (1997) Skeletal muscle myosin heavy chain synthesis in type 1 diabetes. Diabetes 46:1336–1340. https://doi.org/10.2337/diab.46.8.1336
Cheung A, Dantzig JA, Hollingworth S et al (2002) A small-molecule inhibitor of skeletal muscle myosin II. Nat Cell Biol 4:83–88. https://doi.org/10.1038/ncb734
Chinthalapudi K, Taft MH, Martin R et al (2011) Mechanism and specificity of pentachloropseudilin-mediated inhibition of myosin motor activity. J Biol Chem 286:29700–29708. https://doi.org/10.1074/jbc.M111.239210
Chinthalapudi K, Heissler SM, Preller M et al (2017) Mechanistic insights into the active site and allosteric communication pathways in human nonmuscle myosin-2C. Elife 6:1–24. https://doi.org/10.7554/eLife.32742
Cohen P, Alessi DR (2013) Kinase drug discovery – what’s next in the field? ACS Chem Biol 8:96–104. https://doi.org/10.1021/cb300610s
Coulton AT, East DA, Galinska-Rakoczy A et al (2010) The recruitment of acetylated and unacetylated tropomyosin to distinct actin polymers permits the discrete regulation of specific myosins in fission yeast. J Cell Sci 123:3235–3243. https://doi.org/10.1242/JCS.069971
Cowman AF, Tonkin CJ, Tham WH, Duraisingh MT (2017) The molecular basis of erythrocyte invasion by malaria parasites. Cell Host Microbe 22:232–245. https://doi.org/10.1016/j.chom.2017.07.003
Cramer LP, Mitchison TJ (1995) Myosin is involved in postmitotic cell spreading. J Cell Biol 131:179–189. https://doi.org/10.1083/jcb.131.1.179
Cymerys J, SŁonska A, Skwarska J, Banbura MW (2016) Function of myosin during entry and egress of equid herpesvirus type 1 in primary murine neurons. Acta Virol 60:410–416. https://doi.org/10.4149/av_2016_04_410
Dos Remedios CG, Chhabra D, Kekic M et al (2003) Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev 83:433–473. https://doi.org/10.1152/physrev.00026.2002
Dunn TA, Chen S, Faith DA et al (2006) A novel role of myosin VI in human prostate cancer. Am J Pathol 169:1843–1854. https://doi.org/10.2353/AJPATH.2006.060316
Fedorov R, Böhl M, Tsiavaliaris G et al (2009) The mechanism of pentabromopseudilin inhibition of myosin motor activity. Nat Struct Mol Biol 16:80–88. https://doi.org/10.1038/nsmb.1542
Ferreira G, Artigas P, Pizarro Gustavo Brum G (1997) Butanedione Monoxime promotes voltage-dependent inactivation of L-type calcium channels in heart. Effects on gating currents. J Mol Cell Cardiol 29:777–787. https://doi.org/10.1006/JMCC.1996.0321
Ferroni C, Hano O, Ventura C et al (1991) A novel positive inotropic substance enhances contractility without increasing the Ca2+ transient in rat myocardium. J Mol Cell Cardiol 23:325–331. https://doi.org/10.1016/0022-2828(91)90068-W
Fischer S, Windshügel B, Horak D et al (2005) Structural mechanism of the recovery stroke in the myosin molecular motor. Proc Natl Acad Sci U S A 102:6873–6878. https://doi.org/10.1073/pnas.0408784102
Foth BJ, Goedecke MC, Soldati D (2006) New insights into myosin evolution and classification. Proc Natl Acad Sci 103:3681–3686. https://doi.org/10.1073/pnas.0506307103
Galińska-Rakoczy A, Engel P, Xu C et al (2008) Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J Mol Biol 379:929–935. https://doi.org/10.1016/J.JMB.2008.04.062
Gambassi G, Capogrossi MC, Klockow M, Lakatta EG (1993) Enantiomeric dissection of the effects of the inotropic agent, EMD 53998, in single cardiac myocytes. Am J Physiol 264:H728–H738. https://doi.org/10.1152/ajpheart.1993.264.3.H728
Gateva G, Kremneva E, Reindl T et al (2017) Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Curr Biol 27:705–713. https://doi.org/10.1016/J.CUB.2017.01.018
Geeves MA, Fedorov R, Manstein DJ (2005) Molecular mechanism of actomyosin-based motility. Cell Mol Life Sci 62:1462–1477. https://doi.org/10.1007/s00018-005-5015-5
Goebel HH, Laing NG (2009) Actinopathies and myosinopathies: Mini-symposium: Protein aggregate myopathies. Brain Pathol 19:516–522. https://doi.org/10.1111/j.1750-3639.2009.00287.x
Green EM, Wakimoto H, Anderson RL et al (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351:617–621. https://doi.org/10.1126/science.aad3456
Greenberg B (2016) Novel therapies for heart failure – where do they stand? Circ J 80:1882–1891. https://doi.org/10.1253/circj.CJ-16-0742
Gunning PW, Hardeman EC, Lappalainen P, Mulvihill DP (2015) Tropomyosin – master regulator of actin filament function in the cytoskeleton. J Cell Sci 128:2965–2974. https://doi.org/10.1242/jcs.172502
Gupta P, Martin R, Knölker H-J et al (2017) Myosin-1 inhibition by PClP affects membrane shape, cortical actin distribution and lipid droplet dynamics in early Zebrafish embryos. PLoS One 12:e0180301. https://doi.org/10.1371/journal.pone.0180301
Heissler SM, Selvadurai J, Bond LM et al (2012) Kinetic properties and small-molecule inhibition of human myosin-6. FEBS Lett 586:3208–3214. https://doi.org/10.1016/j.febslet.2012.07.014
Herrmann C, Wray J, Travers F, Barman T (1992) Effect of 2,3-butanedione monoxime on myosin and myofibrillar ATPases. An example of an uncompetitive inhibitor. Biochemistry 31:12227–12232. https://doi.org/10.1021/bi00163a036
Higuchi H, Takemori S (1989) Butanedione monoxime suppresses contraction and ATPase activity of rabbit skeletal muscle. J Biochem 105:638–643. https://doi.org/10.1093/oxfordjournals.jbchem.a122717
Hiratsuk T (1994) Nucleotide-induced closure of the ATP-binding pocket in myosin subfragment-1. J Biol Chem 269:27251–27257
Hiratsuka T (2006) The interaction of Phe472 with a fluorescent inhibitor bound to the complex of myosin subfragment-1 with nucleotide. Biochemistry 45:1234–1241. https://doi.org/10.1021/bi051373l
Hou Y-P, Qu X-P, Mao X-W et al (2018) Resistance mechanism of Fusarium fujikuroi to phenamacril in the field. Pest Manag Sci 74:607–616. https://doi.org/10.1002/ps.4742
Houdusse A, Sweeney HL (2016) How myosin generates force on actin filaments. Trends Biochem Sci 41:989–997
Hundt N, Steffen W, Pathan-Chhatbar S et al (2016) Load-dependent modulation of non-muscle myosin-2A function by tropomyosin 4.2. Sci Rep 6:20554. https://doi.org/10.1038/srep20554
Hur E-M, Yang IH, Kim D-H et al (2011) Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc Natl Acad Sci U S A 108:5057–5062. https://doi.org/10.1073/pnas.1011258108
Islam K, Chin HF, Olivares AO et al (2010) A myosin V inhibitor based on privileged chemical scaffolds. Angew Chem Int Ed 49:8484–8488. https://doi.org/10.1002/anie.201004026
Kaplinsky E, Mallarkey G (2018) Cardiac myosin activators for heart failure therapy: focus on omecamtiv mecarbil. Drugs Context 7:1–10. https://doi.org/10.7573/dic.212518
Kawas RF, Anderson RL, Bartholomew Ingle SR et al (2017) A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J Biol Chem 292:16571–16577. https://doi.org/10.1074/jbc.M117.776815
Keith CT, Borisy AA, Stockwell BR (2005) Multicomponent therapeutics for networked systems. Nat Rev Drug Discov 4:71–78. https://doi.org/10.1038/nrd1609
Képiró M, Várkuti BH, Végner L et al (2014) para-nitroblebbistatin, the non-cytotoxic and photostable myosin II inhibitor. Angew Chem Int Ed 53:8211–8215. https://doi.org/10.1002/anie.201403540
Kintses B, Yang Z, Málnási-Csizmadia A (2008) Experimental investigation of the seesaw mechanism of the relay region that moves the myosin lever arm. J Biol Chem 283:34121–34128. https://doi.org/10.1074/jbc.M805848200
Kollmar M, Mühlhausen S (2017) Myosin repertoire expansion coincides with eukaryotic diversification in the Mesoproterozoic era. BMC Evol Biol 17:211. https://doi.org/10.1186/s12862-017-1056-2
Kovács M, Tóth J, Hetényi C et al (2004) Mechanism of blebbistatin inhibition of myosin II. J Biol Chem 279:35557–35563. https://doi.org/10.1074/jbc.M405319200
Lehrer SS, Morris EP (1982) Dual effects of tropomyosin and troponin-tropomyosin on actomyosin subfragment 1 ATPase. J Biol Chem 257:8073–8080
Lehrer SS, Morris EP (1984) Comparison of the effects of smooth and skeletal tropomyosin on skeletal actomyosin subfragment 1 ATPase. J Biol Chem 259:2070–2072
Li H, Diao Y, Wang J et al (2008) JS399-19, a new fungicide against wheat scab. Crop Prot 27:90–95. https://doi.org/10.1016/J.CROPRO.2007.04.010
Li M, Ogilvie H, Ochala J, Artemenko K, Iwamoto H, Yagi N, Bergquist J, Larsson L (2015) Aberrant post-translational modifications compromise human myosin motor function in old age. Aging Cell 14:228–235. https://doi.org/10.1111/acel.12307
Li B, Zheng Z, Liu X et al (2016a) Genotypes and characteristics of phenamacril-resistant mutants in Fusarium asiaticum. Plant Dis 100:1754–1761. https://doi.org/10.1094/PDIS-02-16-0169-RE
Li Y-R, Yang W-X, Li Y-R, Yang W-X (2016b) Myosins as fundamental components during tumorigenesis: diverse and indispensable. Oncotarget 7:46785–46812. https://doi.org/10.18632/oncotarget.8800
Limouze J, Straight AF, Mitchison T, Sellers JR (2004) Specificity of blebbistatin, an inhibitor of myosin II. J Muscle Res Cell Motil 25:337–341. https://doi.org/10.1007/s10974-004-6060-7
Liu Y, White HD, Belknap B et al (2015) Omecamtiv mecarbil modulates the kinetic and motile properties of porcine β-cardiac myosin. Biochemistry 54:1963–1975. https://doi.org/10.1021/bi5015166
Lopatin AN, Nichols CG (1993) Block of delayed rectifier (DRK1) K+ channels by internal 2,3-butanedione monoxime in Xenopus oocytes. Receptors Channels 1:279–286
Lucas-Lopez C, Allingham JS, Lebl T et al (2008) The small molecule tool (S)-(−)-blebbistatin: novel insights of relevance to myosin inhibitor design. Org Biomol Chem 6:2076. https://doi.org/10.1039/b801223g
Lymn RW, Taylor EW (1971) Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10:4617–4624. https://doi.org/10.1021/bi00801a004
Ma X, Adelstein RS (2014) The role of vertebrate nonmuscle myosin II in development and human disease. BioArchitecture 4:88–102. https://doi.org/10.4161/bioa.29766
Malik FI, Hartman JJ, Elias KA et al (2011) Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science 331:1439–1443. https://doi.org/10.1126/science.1200113
Manstein DJ, Mulvihill DP (2016) Tropomyosin-mediated regulation of cytoplasmic myosins. Traffic 17:872–877. https://doi.org/10.1111/tra.12399
Manstein DJ, Meiring JCM, Hardeman EC, Gunning PW (2019) Actin–tropomyosin distribution in non-muscle cells. J Muscle Res Cell Motil:1–12. https://doi.org/10.1007/s10974-019-09514-0
Marian AJ, Braunwald E (2017) Hypertrophic cardiomyopathy. Circ Res 121:749–770. https://doi.org/10.1161/CIRCRESAHA.117.311059
Martin R, Risacher C, Barthel A et al (2014) Silver(I)-catalyzed route to pyrroles: synthesis of halogenated pseudilins as allosteric inhibitors for myosin atpase and x-ray crystal structures of the protein-inhibitor complexes. European J Org Chem 2014:4487–4505. https://doi.org/10.1002/ejoc.201402177
Mckillop DFA, Fortune NS, Ranatunga KW, Geeves MA (1994) The influence of 2,3-butanedione 2-monoxime (BDM) on the interaction between actin and myosin in solution and in skinned muscle fibres. J Muscle Res Cell Motil 15:309–318. https://doi.org/10.1007/BF00123483
Meiring JCM, Bryce NS, Wang Y et al (2018) Co-polymers of actin and tropomyosin account for a major fraction of the human actin cytoskeleton. Curr Biol 28:2331–2337.e5. https://doi.org/10.1016/j.cub.2018.05.053
Melchionda S, Ahituv N, Bisceglia L et al (2001) MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am J Hum Genet 69:635–640. https://doi.org/10.1086/323156
Mohiddin SA, Ahmed ZM, Griffith AJ et al (2004) Novel association of hypertrophic cardiomyopathy, sensorineural deafness, and a mutation in unconventional myosin VI (MYO6). J Med Genet 41:309–314. https://doi.org/10.1136/jmg.2003.011973
Moore JR, Campbell SG, Lehman W (2016) Structural determinants of muscle thin filament cooperativity. Arch Biochem Biophys 594:8–17. https://doi.org/10.1016/j.abb.2016.02.016
Moraczewska J, Nicholson-Flynn K, Hitchcock-DeGregori SE (1999) The ends of tropomyosin are major determinants of actin affinity and myosin subfragment 1-induced binding to F-actin in the open state. Biochemistry 38:15885–15892. https://doi.org/10.1021/bi991816j
Morgan BP, Muci A, Lu PP et al (2010) Discovery of omecamtiv mecarbil the first, selective, small molecule activator of cardiac myosin. ACS Med Chem Lett 1:472–477. https://doi.org/10.1021/ml100138q
Morphy R, Rankovic Z (2005) Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem 48:6523–6543. https://doi.org/10.1021/JM058225D
Münnich S, Pathan-Chhatbar S, Manstein DJ (2014a) Crystal structure of the rigor-like human non-muscle myosin-2 motor domain. FEBS Lett 588:4754–4760. https://doi.org/10.1016/j.febslet.2014.11.007
Münnich S, Taft MH, Manstein DJ (2014b) Crystal structure of human myosin 1c—the motor in GLUT4 exocytosis: implications for Ca2+ regulation and 14-3-3 binding. J Mol Biol 426:2070–2081. https://doi.org/10.1016/j.jmb.2014.03.004
Murphy CT, Spudich JA (1999) The sequence of the myosin 50−20K loop affects myosin’s affinity for actin throughout the actin−myosin ATPase cycle and its maximum ATPase activity. Biochemistry 38:3785–3792. https://doi.org/10.1021/BI9826815
Nadif Kasri N, Van Aelst L (2008) Rho-linked genes and neurological disorders. Pflügers Arch – Eur J Physiol 455:787–797. https://doi.org/10.1007/s00424-007-0385-1
Odronitz F, Kollmar M (2007) Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol 8:R196. https://doi.org/10.1186/gb-2007-8-9-r196
Ohri RV, Radosevich AT, James Hrovat K et al (2005) A Re(V)-catalyzed C−N bond-forming route to human lipoxygenase inhibitors. Org Lett 7:2501. https://doi.org/10.1021/OL050897A
Ostap EM (2002) 2,3-Butanedione monoxime (BDM) as a myosin inhibitor. J Muscle Res Cell Motil 23:305–308. https://doi.org/10.1023/A:1022047102064
Papadaki M, Holewinski RJ, Previs SB et al (2018) Diabetes with heart failure increases methylglyoxal modifications in the sarcomere, which inhibit function. JCI Insight 3. https://doi.org/10.1172/JCI.INSIGHT.121264
Patel H, Margossian SS, Chantler PD (2000) Locking regulatory myosin in the off-state with trifluoperazine. J Biol Chem 275:4880–4888. https://doi.org/10.1074/jbc.275.7.4880
Pathan-Chhatbar S, Taft MH, Reindl T et al (2018) Three mammalian tropomyosin isoforms have different regulatory effects on nonmuscle myosin-2B and filamentous β-actin in vitro. J Biol Chem 293:863–875. https://doi.org/10.1074/jbc.M117.806521
Peters U, Cherian J, Kim JH et al (2006) Probing cell-division phenotype space and Polo-like kinase function using small molecules. Nat Chem Biol 2:618–626. https://doi.org/10.1038/nchembio826
Planelles-Herrero VJ, Hartman JJ, Robert-Paganin J et al (2017) Mechanistic and structural basis for activation of cardiac myosin force production by omecamtiv mecarbil. Nat Commun 8:190. https://doi.org/10.1038/s41467-017-00176-5
Preller M, Holmes KC (2013) The myosin start-of-power stroke state and how actin binding drives the power stroke. Cytoskeleton 70:651–660. https://doi.org/10.1002/cm.21125
Preller M, Manstein DJ (2017) Myosin motors: structural aspects and functionality. In: Reference module in life sciences
Preller M, Bauer S, Adamek N et al (2011a) Structural basis for the allosteric interference of myosin function by reactive thiol region mutations G680A and G680V. J Biol Chem 286:35051–35060. https://doi.org/10.1074/jbc.M111.265298
Preller M, Chinthalapudi K, Martin R et al (2011b) Inhibition of myosin ATPase activity by halogenated pseudilins: A structure-activity study. J Med Chem 54:3675–3685. https://doi.org/10.1021/jm200259f
Radke MB, Taft MH, Stapel B et al (2014) Small molecule-mediated refolding and activation of myosin motor function. Elife 2014:1–19. https://doi.org/10.7554/eLife.01603
Ramamurthy B, Yengo CM, Straight AF et al (2004) Kinetic mechanism of blebbistatin inhibition of nonmuscle myosin IIB. Biochemistry 43:14832–14839. https://doi.org/10.1021/BI0490284
Ramsay RR, Popovic-Nikolic MR, Nikolic K et al (2018) A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med 7:3. https://doi.org/10.1186/s40169-017-0181-2
Rauscher AÁ, Gyimesi M, Kovács M, Málnási-Csizmadia A (2018) Targeting myosin by blebbistatin derivatives: optimization and pharmacological potential. Trends Biochem Sci 43:700–713. https://doi.org/10.1016/J.TIBS.2018.06.006
Richard P, Charron P, Carrier L et al (2003) Hypertrophic cardiomyopathy. Circulation 107:2227–2232. https://doi.org/10.1161/01.CIR.0000066323.15244.54
Rohde JA, Roopnarine O, Thomas DD, Muretta JM (2018) Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proc Natl Acad Sci U S A 115:E7486–E7494. https://doi.org/10.1073/pnas.1720342115
Roman BI, Verhasselt S, Stevens CV (2018) Medicinal chemistry and use of myosin II inhibitor (S)-blebbistatin and its derivatives. J Med Chem 61:9410–9428. https://doi.org/10.1021/acs.jmedchem.8b00503
Roth BL, Sheffler DJ, Kroeze WK (2004) Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov 3:353–359. https://doi.org/10.1038/nrd1346
Rozbicki E, Chuai M, Karjalainen AI et al (2015) Myosin-II-mediated cell shape changes and cell intercalation contribute to primitive streak formation. Nat Cell Biol 17:397–408. https://doi.org/10.1038/ncb3138
Schaub MC, Ermini M (1969) Effect of bivalent cations on the adenosine triphosphatase of actomyosin and its modification by tropomyosin and troponin. Biochem J 111:777–783. https://doi.org/10.1042/bj1110777
Schlichter LC, Pahapill PA, Chung I (1992) Dual action of 2,3-butanedione monoxime (BDM) on K+ current in human T lymphocytes. J Pharmacol Exp Ther 261
Sellers JR, Wang F, Chantler PD (2003) Trifluoperazine inhibits the MgATPase activity and in vitro motility of conventional and unconventional myosins. J Muscle Res Cell Motil 24:579–585. https://doi.org/10.1023/B:JURE.0000009969.04562.58
Sellin LC, McArdle JJ (1994) Multiple effects of 2,3-butanedione monoxime. Pharmacol Toxicol 74:305–313. https://doi.org/10.1111/j.1600-0773.1994.tb01365.x
Senzaki H, Isoda T, Paolocci N et al (2000) Improved mechanoenergetics and cardiac rest and reserve function of in vivo failing heart by calcium sensitizer EMD-57033. Circulation 101:1040–1048. https://doi.org/10.1161/01.CIR.101.9.1040
Shaw MA, Ostap EM, Goldman YE (2003) Mechanism of inhibition of skeletal muscle actomyosin by N-benzyl-p-toluenesulfonamide. Biochemistry 42:6128–6135. https://doi.org/10.1021/BI026964F
Siegman MJ, Mooers SU, Warren TB et al (1994) Comparison of the effects of 2,3-butanedione monoxime on force production, myosin light chain phosphorylation and chemical energy usage in intact and permeabilized smooth and skeletal muscles. J Muscle Res Cell Motil 15:457–472. https://doi.org/10.1007/BF00122119
Sirigu S, Hartman JJ, Planelles-Herrero VJ et al (2016) Highly selective inhibition of myosin motors provides the basis of potential therapeutic application. Proc Natl Acad Sci 201609342. https://doi.org/10.1073/pnas.1609342113
Solaro RJ, Gambassi G, Warshaw DM et al (1993) Stereoselective actions of thiadiazinones on canine cardiac myocytes and myofilaments. Circ Res 73:981–990. https://doi.org/10.1161/01.RES.73.6.981
Spudich JA (2014) Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J 106:1236–1249. https://doi.org/10.1016/J.BPJ.2014.02.011
Stapleton MT, Fuchsbauer CM, Allshire AP (1998) BDM drives protein dephosphorylation and inhibits adenine nucleotide exchange in cardiomyocytes. Am J Physiol Circ Physiol 275:H1260–H1266. https://doi.org/10.1152/ajpheart.1998.275.4.H1260
Stern JA, Markova S, Ueda Y et al (2016) A small molecule inhibitor of sarcomere contractility acutely relieves left ventricular outflow tract obstruction in feline hypertrophic cardiomyopathy. PLoS One 11:e0168407. https://doi.org/10.1371/journal.pone.0168407
Straight AF, Cheung A, Limouze J et al (2003) Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science 299:1743–1747. https://doi.org/10.1126/science.1081412
Strebhardt K, Ullrich A (2008) Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer 8:473–480. https://doi.org/10.1038/nrc2394
Swenson AM, Tang W, Blair CA et al (2017) Omecamtiv mecarbil enhances the duty ratio of human β-cardiac myosin resulting in increased calcium sensitivity and slowed force development in cardiac muscle. J Biol Chem 292:3768–3778. https://doi.org/10.1074/jbc.M116.748780
Swift LM, Asfour H, Posnack NG et al (2012) Properties of blebbistatin for cardiac optical mapping and other imaging applications. Pflügers Arch – Eur J Physiol 464:503–512. https://doi.org/10.1007/s00424-012-1147-2
Takács B, Billington N, Gyimesi M et al (2010) Myosin complexed with ADP and blebbistatin reversibly adopts a conformation resembling the start point of the working stroke. Proc Natl Acad Sci U S A 107:6799–6804. https://doi.org/10.1073/pnas.0907585107
Tan L, Yuan X, Liu Y et al (2019) Non-muscle myosin II: role in microbial infection and its potential as a therapeutic target. Front Microbiol 10:401. https://doi.org/10.3389/fmicb.2019.00401
Terman JR, Kashina A (2013) Post-translational modification and regulation of actin. Curr Opin Cell Biol 25:30–38. https://doi.org/10.1016/J.CEB.2012.10.009
Tobacman LS (2008) Cooperative binding of tropomyosin to actin. Springer, New York, pp 85–94
Tojkander S, Gateva G, Schevzov G et al (2011) A molecular pathway for myosin II recruitment to stress fibers. Curr Biol 21:539–550. https://doi.org/10.1016/j.cub.2011.03.007
Trocóniz IF, Zsolt I, Garrido MJ et al (2006) Dealing with time-dependent pharmacokinetics during the early clinical development of a new leukotriene B4 synthesis inhibitor. Pharm Res 23:1533–1542. https://doi.org/10.1007/s11095-006-0254-1
Trybus KM, Waller GS, Chatman TA (1994) Coupling of ATPase activity and motility in smooth muscle myosin is mediated by the regulatory light chain. J Cell Biol 124:963–969. https://doi.org/10.1083/jcb.124.6.963
Varian K, Tang WHW (2017) Therapeutic strategies targeting inherited cardiomyopathies. Curr Heart Fail Rep 14:321–330. https://doi.org/10.1007/s11897-017-0346-8
Várkuti BH, Képiró M, Horváth IÁ et al (2016) A highly soluble, non-phototoxic, non-fluorescent blebbistatin derivative. Sci Rep 6:26141. https://doi.org/10.1038/srep26141
Verhasselt S, Roman BI, Bracke ME, Stevens CV (2017a) Improved synthesis and comparative analysis of the tool properties of new and existing D-ring modified (S)-blebbistatin analogs. Eur J Med Chem 136:85–103. https://doi.org/10.1016/J.EJMECH.2017.04.072
Verhasselt S, Roman BI, De Wever O et al (2017b) Discovery of (S)-3′-hydroxyblebbistatin and (S)-3′-aminoblebbistatin: polar myosin II inhibitors with superior research tool properties. Org Biomol Chem 15:2104–2118. https://doi.org/10.1039/C7OB00006E
Verhasselt S, Stevens CV, Van den broecke T et al (2017c) Insights into the myosin II inhibitory potency of A-ring-modified (S)-blebbistatin analogs. Bioorg Med Chem Lett 27:2986–2989. https://doi.org/10.1016/J.BMCL.2017.05.008
Volkow ND, Morales M (2015) The brain on drugs: from reward to addiction. Cell 162:712–725. https://doi.org/10.1016/J.CELL.2015.07.046
von der Ecken J, Müller M, Lehman W et al (2015) Structure of the F-actin–tropomyosin complex. Nature 519:114–117. https://doi.org/10.1038/nature14033
von der Ecken J, Heissler SM, Pathan-Chhatbar S et al (2016) Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 534:724–728. https://doi.org/10.1038/nature18295
Wilson IB, Ginsburg S (1955) A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. Biochim Biophys Acta 18:168–170. https://doi.org/10.1016/0006-3002(55)90040-8
Winder SJ, Ayscough KR (2005) Actin-binding proteins. J Cell Sci 118:651–654. https://doi.org/10.1242/jcs.01670
Winkelmann DA, Forgacs E, Miller MT, Stock AM (2015) Structural basis for drug-induced allosteric changes to human β-cardiac myosin motor activity. Nat Commun 6:7974. https://doi.org/10.1038/ncomms8974
Wollenberg RD, Taft MH, Giese S et al (2019) Phenamacril is a reversible and noncompetitive inhibitor of Fusarium class I myosin. J Biol Chem 294:1328–1337. https://doi.org/10.1074/jbc.RA118.005408
Wustman BA, Steele JW, Sjoberg ER, Stevens AC (2014) Bringing dead proteins back to life. Elife 3:e02189. https://doi.org/10.7554/eLife.02189
Xiong D, Du Y, Wang H-B et al (2015) Nonmuscle myosin heavy chain IIA mediates Epstein-Barr virus infection of nasopharyngeal epithelial cells. Proc Natl Acad Sci U S A 112:11036–11041. https://doi.org/10.1073/pnas.1513359112
Yang Z, Sweeney HL (1995) Restoration of phosphorylation-dependent regulation to the skeletal muscle myosin regulatory light chain. J Biol Chem 270:24646–24649. https://doi.org/10.1074/jbc.270.42.24646
Yoshida H, Cheng W, Hung J et al (2004) Lessons from border cell migration in the Drosophila ovary: a role for myosin VI in dissemination of human ovarian cancer. Proc Natl Acad Sci U S A 101:8144–8149. https://doi.org/10.1073/pnas.0400400101
Young E, Briggs S, Miller C (2015) The actin cytoskeleton as a therapeutic target for the prevention of relapse to methamphetamine use. CNS Neurol Disord Drug Targets 14:731–737. https://doi.org/10.2174/1871527314666150529145531
Zhang X, Seftel A, DiSanto ME (2011) Blebbistain, a myosin II inhibitor, as a novel strategy to regulate detrusor contractility in a rat model of partial bladder outlet obstruction. PLoS One 6:e25958. https://doi.org/10.1371/journal.pone.0025958
Zhang C, Chen Y, Yin Y et al (2015) A small molecule species specifically inhibits Fusarium myosin I. Environ Microbiol 17:2735–2746. https://doi.org/10.1111/1462-2920.12711
Zhang H-M, Ji H-H, Ni T et al (2017) Characterization of blebbistatin inhibition of smooth muscle myosin and nonmuscle myosin-2. Biochemistry 56:4235–4243. https://doi.org/10.1021/acs.biochem.7b00311
Acknowledgments
This work was supported by grants from Deutsche Forschungsgemeinschaft to D.J.M (MA1081-22/1) and M.P (PR1478-2/1).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Manstein, D.J., Preller, M. (2020). Small Molecule Effectors of Myosin Function. In: Coluccio, L. (eds) Myosins. Advances in Experimental Medicine and Biology, vol 1239. Springer, Cham. https://doi.org/10.1007/978-3-030-38062-5_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-38062-5_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-38061-8
Online ISBN: 978-3-030-38062-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)