
Abstract
The cell surface heparan sulfate proteoglycan Syndecan-1 acts as an important co-receptor for receptor tyrosine kinases and chemokine receptors, and as an adhesion receptor for structural glycoproteins of the extracellular matrix. It serves as a substrate for heparanase, an endo-β-glucuronidase that degrades specific domains of heparan sulfate carbohydrate chains and thereby alters the functional status of the proteoglycan and of Syndecan-1-bound ligands. Syndecan-1 and heparanase show multiple levels of functional interactions, resulting in mutual regulation of their expression, processing, and activity. These interactions are of particular relevance in the context of inflammation and malignant disease. Studies in animal models have revealed a mechanistic role of Syndecan-1 and heparanase in the regulation of contact allergies, kidney inflammation, multiple sclerosis, inflammatory bowel disease, and inflammation-associated tumorigenesis. Moreover, functional interactions between Syndecan-1 and heparanase modulate virtually all steps of tumor progression as defined in the Hallmarks of Cancer. Due to their prognostic value in cancer, and their mechanistic involvement in tumor progression, Syndecan-1 and heparanase have emerged as important drug targets. Data in preclinical models and preclinical phase I/II studies have already yielded promising results that provide a translational perspective.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
1.1 The Syndecan Family of Heparan Sulfate Proteoglycans
Glycosaminoglycans (GAG) are sulfated polysaccharides composed of unbranched chains of repetitive disaccharide units of uronic acid and glucosamine. One of the most studied GAGs is Heparan sulfate (HS), a complex GAG present in virtually all animal cells [1, 2] (Fig. 4.1). Via a linkage tetrasaccharide, HS can be O-glycosidically attached to serine or threonine residues of proteins in the cell membrane, or to proteins which are secreted into the extracellular matrix (ECM), thus forming Heparan Sulfate Proteoglycans (HSPG)[1, 2]. The HS chains present a vast structural complexity due to the possibility of inserting different disaccharide units during biosynthesis. The insertion of sulfate radicals residues largely contributes to its negative charge characteristic, and the degree of sulfation usually ranges between 0.6–1.5 sulfates per disaccharide [3]. The iduronic or hexuronic acid can be sulfated at the position 2, while the glucosamine can be N-sulfated, N-acetylated and/or O-sulfated in positions 6 and 3. Besides its sulfation pattern, the molecular weight can widely vary between HS chains, ranging frequently between 5–70 kDa [4]. Another important characteristic is the considerable amount of non-sulfated regions of glucuronic acid and N-acetyl glucosamine, allowing the HS chains to organize itself in N-acetylated and N-sulfated domains. Each domain has distinct structural characteristics that dictate the kinds of molecules that can interact with them [2]. The synthesis of HSPGs occurs in the Golgi apparatus and is dependent on many enzymes which catalyze the different steps of HS elongation and modification in a sequential manner. The majority of HS deregulation in disease occurs as a result of alterations in the expression of enzymes involved in its synthesis, however, in some cases, the deregulation is also due to alterations in the core protein [5].

Structure and specific protein and glycosidic domains of Syndecan-1 and Heparanase function. (A) The representative structure of the Syndecan-1 core protein. The different domains are represented on the right side and the GAG chains are represented in blue (Heparan Sulfate) and orange (Chondroitin Sulfate). The red arrows represent the various protein domains important for the interaction with other proteins. (B) Example of a Heparan Sulfate chain. The different monosaccharidic components are represented. The Heparanase cleavage site is represented in red
Syndecans (Sdc) are a family of transmembrane HSPGs expressed at the surface of several cell types, although they have also been found in the nucleus [6]. Moreover, their intact extracellular domains can be shed into the extracellular environment [7]. In mammals, this family is composed of four members that are expressed in a highly regulated cell-specific manner [8]. Among the syndecans, Sdc1 is the most highly expressed HSPG on the cell membrane of epithelial and plasma cells. Both the GAGs heparan sulfate and chondroitin sulfate (CS) are bound to its protein core [9]. Neuronal cells and cartilage express mainly Sdc3, while mesenchymal cells express Sdc2 and Sdc4, but have Sdc1 as well [10]. Sdc4 is expressed by most cell types [8]. All syndecans are composed of three different domains: an extracellular N-terminal domain where several glycosaminoglycan chains are covalently attached, a single transmembrane domain and a cytoplasmic C-terminal domain, which is subdivided into two constant domains separated by a variable region (Fig. 4.1). The extracellular domains of syndecans mediate various cell-cell and ECM-cell interactions dependent on their GAG composition. This domain, in turn, varies significantly between the members of the syndecan family, unlike the transmembrane and cytoplasmic domains, which are highly conserved [11]. Although most studies focus on describing the importance of the extracellular and intracellular domains of syndecans, the transmembrane region was described to be sufficient in reducing cell migration via alterations in focal adhesion dynamics [12]. The GAGs attached to syndecans have a structure capable of directly interacting with many growth factors , cytokines, chemokines, and other macromolecules and enzymes found in the ECM, leading to the presentation to their receptors on the cell membrane. These interactions mediated by the GAG chains imply several physiological activities attributed to syndecans, such as cell proliferation, migration, and invasion [13]. The cytoplasmic domains of syndecans are capable of interacting with various intracellular kinases and the cytoskeleton, promoting important intracellular signaling [11]. Syndecans can also interact with other family members as structural analysis has shown that Sdc1 is less likely to form homodimers than the other syndecans, and can form heterodimers with Sdc2 and Sdc3 but not with Sdc4 [14, 15]. Further studies are needed to better understand the implications of these interactions. These diverse intra- and extracellular interactions with different molecules characterize the syndecans as key molecules in various physiological and pathological cellular functions. In this review, we will mainly focus on the role of the prototype member of the family, Sdc1, which is best characterized for its functional interplay with heparanase in the context of inflammation and cancer.
The extracellular domain of Sdc 1 harbors five GAG-attachment sites. Three are close to the N-terminus and two are close to the cell membrane. While HS and CS can be located near the N-terminus, only CS is found near the plasma membrane. Cells can vary the number of HS and CS chains added to Sdc1, but it always has at least one HS chain at its N-terminus [10]. Studies have shown that the number of GAGs attached to Sdc1 also affects its function, such as reducing its ability to mediate cell-cell interaction and cell invasion through the ECM [16]. The extracellular domain of Sdc1 can be shed by many proteases, such as matrix metalloproteinases (MMPs) and A-disintegrin and matrix metalloproteinases (ADAMs) [17]. Currently, there are no reports of endogenous extracellular trimming of CS chains in proteoglycans of mammalian cells. The presence of extracellular CS chains close to the plasma membrane can alter its susceptibility to cleavage and shedding by proteases, as well as change its ability to associate with receptors and other syndecans. Injuries or bacterial infections may lead to upregulation of Sdc1, heparanase, and proteases that can release the ectodomain fragment of Sdc1 [18,19,20]. In vitro, Sdc1 ectodomain has been shown to decrease proliferation of tumor cells [21], whereas another study using tumor cells expressing uncleavable membrane-bound Sdc1 showed increased proliferation and decreased invasion [22]. These events are regulated by HS chains in Sdc1. The length and sulfation of these chains can vary between cell types due to the action of several enzymes that regulate glycan elongation and modification [23]. Of these enzymes, cleavage by heparanase is of particular importance.
1.2 Heparanase – A Key Enzyme in ECM Remodeling
Heparanase is an endo-β-glucuronidase that degrades specific domains of HS chains (Fig. 4.1). After being produced in the rough endoplasmic reticulum, it is sent to the Golgi apparatus and secreted to the ECM in its inactive, latent form [24]. Subsequently, it can interact with Sdc1 at the plasma membrane and be endocytosed to reach the lysosomes, where it can be proteolytically activated [25]. Following this processing step, heparanase can reach the ECM again, where it executes extracellular functions [25, 26], or act inside the cell and modulate intracellular processes [26]. Despite its enzymatic activity, studies using mutated inactive heparanase have demonstrated that it can also exert activities unrelated to HS degradation. Enzymatically inactive heparanase can facilitate adhesion and migration of endothelial cells [27], promote phosphorylation of signaling molecules such as Akt and Src [27,28,29], as well as activate receptor tyrosine kinases such as EGFR [30]. For example, in head and neck carcinoma, heparanase was also shown to facilitate the formation of lymphatic vessels and the migration of tumor cells toward these vessels [31].
Most of what is known about heparanase function are based on its activity in cancer, where heparanase RNA and protein levels are increased in many different forms of malignant diseases [32] (Table 4.1). By degrading HS chains, heparanase alters many regulatory paths, mainly by augmenting the bioavailability of growth factors previously bound to the HS. This diffusion of growth factors and cytokines affects many different physiological processes, such as cell migration, angiogenesis, inflammation, coagulation, autophagy, and exosome production [33, 34]. Most members of the syndecan family are involved in these activities. In angiogenesis, for instance, Sdc1, Sdc2, and Sdc3 have important effects related to their ability to bind and present pro- and anti-angiogenic factors [35,36,37].
2 The Heparanase-Syndecan Axis
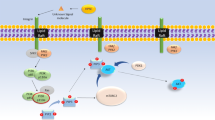
There is ample evidence that heparanase can affect cell behavior by regulating the structure and function of HSPGs. The Heparanase/Syndecan Axis (commonly referred as the Heparanase/Syndecan-1 axis due to most evidence showing a major role for Sdc1 on this process) has been shown to affect signaling cascades in healthy or malignant cells and biological systems (Fig. 4.2). The most studied growth factors that are strongly regulated by this axis are hepatocyte growth factor (HGF ) and vascular endothelial growth factor (VEGF) . Other than just stimulating syndecan shedding, the cleavage of heparan sulfate chains by heparanase also makes it easier for additional proteins to recognize and bind to syndecan [38]. One example is the binding of the protein lacritin, a prosecretory epithelial mitogen that is expressed in the tear ducts and can bind directly to syndecan-1 core protein only after the trimming of HS chains by heparanase [39]. The heparin-binding domains of heparanase also facilitate the clustering of Sdc1 and Sdc4 on the membrane of human glioma cells, initiating signaling cascades that lead to augmented cell adhesion and spreading. Notably, this activity does not require the HS degrading activity of heparanase [40].

The different functions of Syndecan-1 and how it can be affected by Heparanase. Syndecan-1 participates in many important physiological functions, such as (A) presenting growth factors to receptors on the cell surface or on the membrane of neighboring cells. (B) The trimming of HS chains mediated by heparanase exposes Sdc1 to the recognition and shedding by proteases, what can lead to (B1) binding and accumulation of factors in the extracellular matrix and presentation to receptors in distant cells, or (B2) binding to both integrins and directly to some receptors, activating them and leading to activation of intracellular pathways, what can be inhibited by Synstatins. (C) Some extracellular factors can trigger Sdc1 endocytosis, this endosome can be degraded or translocated into the cell nucleus and the factors can act as transcription factors. (D) Sdc1 can sequester factors from the extracellular environment, binding them to their GAG chains. Heparanase releases them into the extracellular matrix allowing them to act on their specific receptors. (E) The translocation of Sdc1 to the nucleus is highly dependent on tubulin, and it is also responsible for FGF-2 shuttling to the nucleus. (F) Sdc1 known nuclear actions include inhibiting DNA topoisomerase I activity and blocking the enzyme histone acetyltransferase (HAT) activity, controlling cell proliferation and differentiation. The trimming of the HS chains by heparanase decreases the level of nuclear Sdc1
2.1 Heparanase Mediated Sdc1 Shedding
The proteoglycans present in the cell membrane have many important roles in health and disease. One example is their strong interaction with the tumor stroma, allowing the activation of numerous pathways in cancer cells [7]. Several of these proteoglycans can also undergo an enzymatic shedding of their extracellular domains by proteases known as ‘sheddases’. In this process, the intact ectodomain of the proteoglycan is converted to a soluble molecule, allowing them to stay functional even after solubilization [7]. The shedding of membrane-bound proteoglycans is a controlled process that regulates many physiological and pathological conditions. With the exception of glycosylphosphatidyl-inositol (GPI)-anchored proteoglycans, such as the Glypican family, all membrane-bound proteoglycans are shed by a protease [1, 7].
The shedding of proteoglycans can be regulated at different levels. One of the mechanism is the regulation of the expression of sheddases and proteases inhibitors, such as tissue inhibitors of metalloproteinases (TIMPs), or the binding of these inhibitors to the GAG chains, either inhibiting or enhancing the proteoglycan activity, or even protecting them from degradation [41, 42]. The shedding occurs constitutively in cells, but it can also be enhanced by several stimuli, such as growth factor signaling, bacterial and viral stimuli, cell stress, among others [43, 44]. One of the most well-characterized actions of heparanase is its ability to facilitate Sdc1 shedding [45,46,47]. By trimming the HS chains on Sdc1, the core protein becomes more vulnerable to the actions of proteases [38]. All syndecans can be shed in vivo and in vitro by a process mediated by plasmin, thrombin, MMPs and/or ADAMs, and its proteolytic cleavage can be modulated by different signaling pathways such as protein kinase C (PKC), and the protein tyrosine kinase (PTK) MAP kinase [41]. After Sdc1 shedding, its remaining cytoplasmic C-terminal fragment (cCTF) undergoes proteolysis by γ-secretase and the proteasome. Nevertheless, this fragment has also been shown to inhibit lung cancer cell migration and invasion through the phosphorylation of Src, FAK, and Rho-GTPase, which blocks Sdc1-dependent migration and invasion, and also reduces lung tumor formation by these cells [48].
In tumors, shed Sdc1 plays multiple roles such as the delivery of growth factors to both tumor and host cells and triggering of signal transduction events at the cell surface [1, 41]. Most known functions of Sdc in the tumor context are mediated by the membrane-bound Sdc on cancer cells. However, tumor stroma syndecan has been shown to also have an important role in this context, either bound in the membrane of stromal cells or soluble ectodomains, which can be generated by cleavage from the tumor cells or other cell types such as cancer-associated fibroblasts, endothelial cells or leukocytes. In myeloma cells, the up-regulation of heparanase or the addition of recombinant heparanase to the media was shown to lead to enhanced Sdc1 expression and shedding [45, 46, 49]. This was mostly due to the heparanase mediated activation of ERK signaling, leading to enhanced expression of MMP-9. This proteinase cleaves Sdc1 in the juxtamembrane region, releasing an intact ectodomain to the extracellular matrix [50, 51]. This effect in myeloma cells is interesting because it is highly dependent on the HS degrading activity of heparanase, whereas in other cells types ERK signaling can be triggered by heparanase, independent of its enzymatic activity [52, 53]. In a lung inflammation model in mice, soluble Sdc1 has been shown to be required for the formation of chemotactic gradients regulating leukocyte-endothelial interactions and angiogenesis [54, 55]. Another interesting finding regarding the shed ectodomain of Sdc1 is the observation that it can be transported to the nucleus, where it can influence transcription by the inhibition of histone acetylation [6, 10, 56] (see also Purushothaman and Sanderson, Chap. 12 in this volume).
2.2 Heparanase and Sdc1 in the Nucleus
Sdc1 has been found in the nucleus of many cell types such including malignant mesothelioma, myeloma, neuroblastoma, lung adenocarcinoma, and breast carcinoma [57]. In addition, Sdc2 has been found in the nucleus of injured cerebral cortex neurons, astrocytes, and chondrosarcoma [58, 59]. Apart from localization to the cytoplasm and cell membrane, heparanase can also be found in its active form in the nucleus and this localization is correlated with cell differentiation [60]. From a mechanistic perspective, a lot remains to be uncovered about how HSPGs enter the nucleus and regulate nuclear processes. In the nucleus of mesothelioma cells, Sdc1 has been shown to co-localize with heparanase and fibroblast growth factor-2 (FGF-2) [61]. The amount of HSPGs in the nucleus is increased after inhibition of PKC and drastically diminished after its stimulation [62]. Apparently, FGF-2 seems to trigger its nuclear translocation by stimulating the dephosphorylation of the protein core [63].
The nuclear translocation of Sdc1 has been shown to be highly dependent on tubulin both in malignant and normal cells. Sdc1 co-localizes with tubulin at the mitotic spindle during all phases of mitosis and the interference with tubulin integrity blocks the transport of Sdc1 to the nucleus [6]. During mitosis in malignant mesothelioma cells, Sdc1 accumulates in the nucleus by associating with tubulin structures and inhibits cell cycle progression, proliferation, and migration [57]. Nuclear FGF-2 and HS also seem to regulate cell cycle in many cell types. The nuclear entry of FGF-2 happens in the G1 restriction point of the cell cycle and exogenous HS arrests cells in the G1 phase in a transient manner [64, 65]. A decrease in nuclear HS allows the regular cell cycle progression. The arrest of cells in the G2/M phase of the cell cycle efficiently blocks the nuclear translocation of Sdc1. It has been shown that the entire molecule is present in the nucleus, with both ecto- and endodomains and the HS chains [6]. The HS chains of HSPG were found to be important for the uptake and nuclear translocation of many different molecules, including heparanase [60]. The mechanism of FGF-2 shuttling into the nucleus is dependent on both the FGF receptor and HSPGs [66]. Sdc1 and FGF-2 share the same tubulin-mediated route to the nucleus, where they co-localize with heparanase. The minimal peptide sequence required for the tubulin-dependent nuclear translocation of Sdc1 is the juxtamembrane RMKKK motif that acts as a nuclear localization signal (NLS) [61]. Replacement of arginine (R) in the RMKKK motif dramatically reduces the amount of nuclear Sdc1, and complete deletion of this sequence abrogates the nuclear translocation of Sdc1 [67].
Many of the known nuclear functions of Sdc1 are associated with the HS interaction with various growth factors and nuclear structures. It is known that Sdc1 can be found in the nucleus as an intact HSPG, but it is still unclear if its actions are dependent only on the GAG chains or the entire molecule. The HS chains are important for nuclear translocation or degradation of Sdc1 and its negative charges facilitate their nuclear interactions. HS can compete with DNA for the binding of proteins such as transcription factors and enzymes. Sdc1 main known nuclear actions are to shuttle FGF-2 into the nucleus, inhibit DNA topoisomerase I activity and inhibit the activity of the enzyme histone acetyl transferase (HAT), controlling cell proliferation and differentiation [63, 64, 68]. Trimming of the HS chains by heparanase decreases the level of nuclear Sdc1, leading to an enhanced HAT activity and, consequently, augmented expression of genes that promote an aggressive phenotype in tumor cells such as MMP-9, VEGF, HGF and RANKL [69, 70]. It has been shown that HS competes with the DNA for topoisomerase I binding in the nucleus and is also able to dissociate the DNA-topoisomerase I complex, that is necessary for the unwinding of supercoiled DNA during transcription. This would suggest a negative effect of nuclear HS on gene transcription [68]. Activation of EGFR leads to nuclear translocation of heparanase and degradation of HS, increasing the activity of topoisomerase I and cell proliferation [71].
2.3 Effects on Exosome Formation and Function
Exosomes are secreted to the extracellular matrix by virtually all cell types [72]. They are very small vesicles ranging between 30–100 nm in diameter, but their classification comes from their endosomal origin rather than size. In mammalian cells, the biogenesis of exosomes begins with the invagination of the cell membrane forming the primary endocytic vesicles, which leads to the fusion of these vesicles and the formation of the early endosomal compartment [72]. During its maturation into late endosomes, it changes the protein composition of its membrane and receives many components derived from the trans-Golgi network. In the meantime, a second invagination occurs at the interior of the endosome forming the intraluminal vesicles (ILVs), that, consequently, have the same topology of the cell membrane (inside-in/outside-out) [73]. The late endosome containing many ILVs is called a multivesicular body (MVB). When it fuses with the plasma membrane, their ILVs are released to the extracellular environment and, upon release, the ILVs are called exosomes [72, 73]. Both MVB and ILV formation is dependent on the endosomal sorting complex required for transport (ESCRT), but also on specific membrane domains and oligomerization/clustering processes [74, 75]. ESCRT-0 leads the clustering of mono-ubiquitinated cargo proteins into the endosome. ESCRT-I and ESCRT-II are related to the membrane deformation and the intraluminal budding of these cargos. ESCRT-I recruits ESCRT-III via ESCRT-II or through the adaptor protein ALG-2 interacting protein X (ALIX), resulting in ESCRT-III stabilization. ESCRT-III induces vesicle scission, and the vacuolar protein sorting (Vps) 4 ATPase mediates the final step of dissociation and recycling of ESCRT-III leading to IVL formation [74].
The way by which Syndecans influence the biogenesis of exosomes is through the interaction with the syntenin/ALIX complex[76,77,78] (Fig. 4.3). Syntenin is a small cytosolic protein that contains two PSD95/Dlg/Zonula occludens 1 (PDZ) domains. Both PDZ domains are necessary for syntenin membrane localization and the high-affinity interaction with syndecans [79, 80]. Syntenin can interact with both Sdc core protein and ALIX, which leads ALIX to bind to ESCRT-III, the complex required for the IVL formation in multivesicular bodies. The HS chains in Sdc are essential for this activity [76]. In some human cancer cell types, when the cells are exposed to exogenous heparanase or the endogenous expression of heparanase is enhanced, exosome secretion increases dramatically. This process is dependent on the HS trimming activity of heparanase, as enzymatically inactive forms of the enzyme do not have the same effect [81]. Heparanase acts in this pathway by trimming the long HS chains in Sdc into shorter ones, which facilitates the clustering of syndecans [82, 83]. This clustering stimulates the binding of the cytoplasmic domains of syndecan to syntenin, driving ALIX-ESCRT-mediated sorting into exosomes [82,83,84]. This action of heparanase also facilitates the recruitment CD63 to exosomes, in a syntenin dependent fashion [82, 83] (Fig. 4.3). Consequently, heparanase inhibitors or syntenin inhibitors could be of particular interest for the treatment of cancer patients, as both exosome release and heparanase expression are frequently elevated in more aggressive subtypes [47, 81]. Heparanase has been shown to up-regulate the biogenesis of exosomes and affect its composition and function in myeloma cells [81]. Heparanase has also been found in exosomes isolated from ascites of ovarian cancer patients [85], and the levels of Sdc1, VEGF and HGF in exosomes derived from heparanase high expressing cells seem to be higher compared to heparanase low expressing cells [81]. Interestingly, exosomes secreted from heparanase high expressing cells were shown to better stimulate spreading of tumor cells on fibronectin and also stimulate invasion of endothelial cells compared to exosomes from heparanase low expression cells, suggesting a role in cancer angiogenesis and the spreading of cancer cells [81] (see also David and Zimmermann; Purushothaman and Sanderson, Chaps. 10 and 12 in this volume).

Syndecan-1 and Heparanase mediated exosome formation. (A) Heparanase acts by trimming the HS chains in Sdc1, which facilitates its clustering. This, stimulates the binding of the cytoplasmic domains of syndecan to syntenin and its internalization. (B) This cluster of Sdc1 is recognized by proteases and cleaved. (C-D) After cleaving of the core protein, syntenin interacts with both Sdc1 and ALIX, which leads ALIX to bind to ESCRT-III and (E) intraluminal vesicles formation in the multivesicular bodies. (F) After releasing to the extracellular environment, these vesicles are called exosomes and can carry many different factors to distant sites throughout the body
2.4 Effects on Growth Factor Signaling
The shedding of syndecans exposes domains in the core protein that can bind to different receptors. One well-described mechanism that affects signaling processes in the cell membrane is the one mediated by syndecan binding to integrin and to a tyrosine kinase receptor such as HER2, EGFR or IGF-1R. This tertiary complex activates various functions in the cells such as cancer progression and angiogenesis [86,87,88,89,90]. In these cases, the signaling mechanism can be disrupted by synthetic peptides called Synstatins (SSTNs), that mimic the binding sites of syndecan and compete with coupling to the tyrosine kinase receptors. Sdc1 shedding exposes a domain on the core protein that can bind to very late antigen 4 (VLA-4) integrin and vascular endothelial growth factor receptor 2 (VEGFR2). When Sdc1 binds and couples these receptors, VEGFR2 becomes activated and stimulates invasion in tumor cells [49]. The same mechanism potentiates endothelial tube formation and angiogenesis. Synstatin peptides based on either the VEGFR2 or VLA-4 binding site in Sdc1 can prevent invasion of tumor cells and endothelial tube formation [49]. Interestingly, the heparanase inhibitor Roneparstat, a chemically modified nonanticoagulant heparin, can decrease tumor invasion and angiogenesis by preventing Sdc1 shedding [49] (see also Noseda and Barbieri, Chap. 21 in this volume).
HSPGs are also involved in the uptake and nuclear translocation of growth factors and cytokines [91]. The vast majority of growth factors with nuclear translocation bind to HSPGs, which can efficiently deliver molecules to their intracellular targets [92, 93]. Apart from growth factors, a myriad of ligands, viruses, nucleic acids, peptides, lipoproteins, and exosomes enter the cells via HSPG-mediated endocytosis [94]. The juxtamembrane MKKK motif is the required peptide sequence for the efficient raft dependent endocytosis, and this sequence is also part of the same RMKKK motif necessary for Sdc1 nuclear translocation, being crucial for both endocytosis and nuclear transport of Sdc1 and any associated molecule [95].
Both heparanase and Sdc1 can regulate HGF function. In myeloma cells, heparanase enhances HGF expression [96]. This growth factor binds strongly to Sdc1 in the membrane, augmenting the interaction with the c-met receptor and facilitating tumor growth [97, 98]. Shed Sdc1 can also bind to HGF and some evidence suggests that c-met signaling in osteoblasts is stimulated by shed Sdc1/HGF complexes [99]. Heparanase also stimulates VEGF secretion in tumor cells [31, 50]. Secreted VEGF can form a complex with shed Sdc1 that positively regulates VEGF receptor by activating the extracellular signal-regulating kinase (ERK) signaling pathway, leading to augmented endothelial invasion and angiogenesis [50]. Immuno-depletion of the VEGF/Sdc1 complex or treatment with heparinase III, a HS degrading enzyme derived from bacteria, blocks the augmented phosphorylation of ERK. It is interesting to notice that Sdc1 also activates αvβ3 integrin in endothelial cells, which is a key regulator of endothelial activation and angiogenesis [50, 100, 101]. In that manner, Sdc1 promotes endothelial cell activation, angiogenesis, and tube formation. This is mediated not only by αvβ3 integrin activation but also by binding to VEGF presenting it to its high-affinity receptor as a tertiary complex described above. Heparanase plays a central role in this process by up-regulating Sdc1 shedding. Heparanase also inhibits FGF2 signaling in melanoma cells by degrading membrane-bound HS [102]. Modification of these chains is required for effective binding of FGF2 to the cell surface and subsequent stimulation of ERK and FAK phosphorylation [102]. FGF2 high-affinity binding requires HS chains of a minimum size and some specific structural features. Upon cleavage of HS by heparanase, specific sequences in the HS chains that bind to FGF2 could be either removed or revealed [103, 104]. In addition, interplay between heparanase and Sdc1 is required for renal tubular cells to undergo epithelial to mesenchymal transition induced by FGF2 [105] (see also Masola et al.; van der Vlag and Buijsers, Chap. 26 and 27 in this volume).
3 Functional Cooperation of Syndecan-1 and Heparanase in Inflammation
Inflammation is a complex process that involves interactions of various cell types, most notably leukocytes and endothelial cells, which exchange signals via cytokines and chemokines and their respective transmembrane receptors. Physical interactions between these cells are mediated through cell surface receptors of the selectin, integrin, and the immunoglobulin superfamily of cell adhesion molecules (CAMs) such as ICAM and VCAM [106,107,108]. In order to fight pathogens and toxins, leukocytes need to be recruited from the circulation to inflammatory sites via a controlled hierarchy of low- and high-affinity interactions with the endothelium, ultimately resulting in leukocyte transmigration, or diapedesis [108]. A role for HS in leukocyte recruitment has been documented in numerous studies and involves all key steps of this process (reviewed in [54, 108]). At early stages of leukocyte recruitment, HS promotes proinflammatory signaling processes by providing binding sites for growth factors and chemokines, resulting in the formation and stabilization of ternary complexes with their receptors [109, 110], and the establishment of chemokine gradients that are a crucial element in the leukocyte recruitment cascade [55, 111, 112]. Moreover, HS itself shows a distinct basolateral gradient pattern across blood vessels [113]. HS is also needed to ensure directional transendothelial chemokine transport, thus allowing for their presentation at the luminal surface endothelium [114]. Activated endothelial cells express adhesion molecules of the selectin family, which mediate low-affinity interactions with leukocytes that result in leukocyte rolling along the vessel wall [107]. Indeed, HS was shown to interact with L- and P-selectin, and L-selectin-HS complex formation appears to play an important role in enhancing leukocyte rolling mediated by P-selectin-PSGL-1 interactions [108, 114]. As will be pointed out in detail in the following section, Sdc1 plays a crucial role in modulating the next step of leukocyte adhesion, which is mediated by interactions of leukocyte β2 integrins with ICAM-1. Moreover, HPSE-mediated degradation of HS is a means of regulating intraluminal crawling of leukocytes within blood vessels, a process that involves interactions between the leukocyte β2 integrin Mac-1 and endothelial ICAM-1 [115]. Finally, HS modulates leukocyte diapedesis not only by facilitating chemokine gradient formation, but also via interactions with the leukocyte integrins LFA-1 and VLA4, and their binding partners ICAM-1 and VCAM-1 (reviewed in [55, 108]). Notably, several of the HS binding molecular and cellular mediators of inflammation are regulated by Sdc1 and heparanase, respectively, as will be detailed in the following section.
3.1 Lessons from Mouse Models
The relevance of Sdc1 and HPSE as regulators of inflammatory processes in vivo has been explored with the help of transgenic and knockout mouse models in a variety of experimental models of inflammation and repair [32, 54, 55, 108]. Early studies in Sdc1 knockout mice revealed that these mice show increased recruitment of leukocytes to the endothelium of the ocular vasculature [116]. Bone marrow transplantation experiments demonstrated that the increased adhesion was due to the lack of Sdc1 on leukocytes rather than the endothelium. Notably, intravital microscopy of TNFα-stimulated mesentery venules demonstrated that loss of Sdc1 was associated not only with a massively increased adhesion of leukocytes to blood vessels, but also with a substantial increase in leukocyte diapedesis [116]. Further mechanistic studies revealed that increased adhesion of Sdc1-deficient polymorphonuclear cells (PMNs) and monocytes to human umbilical vein endothelial cells (HUVEC) in vitro could be inhibited by heparin if the endothelium was not activated. The increased adhesion was not altered by heparin when Sdc1-deficient leukocytes were allowed to adhere to TNFα-stimulated HUVECs, suggesting that the Sdc1-dependent adhesion phenotype involves HS/heparin-sensitive and insensitive steps, that depend on the activation state of the endothelium [117]. Further studies employing Sdc1-deficient and WT leukocytes in vitro revealed that increased adhesion of Sdc1-deficient PMNs to ICAM-1 could be inhibited by heparin, suggesting a role for HS in this process [118]. Moreover, increased adhesion of Sdc1 KO PMNs to ICAM-1 could be efficiently blocked with antibodies directed against the leukocyte integrin CD18 [119]. In summary, these data suggest that the lack of Sdc1 on leukocytes may allow them to interact with non-activated endothelium, and enhances leukocyte-endothelial interactions in HS-dependent manner at the level of ICAM-1-CD18 interactions.
In addition to Sdc1, the role of heparanase in leukocyte recruitment has been studied in vitro and in vivo. In vivo studies in rats demonstrated that intraperitoneal injection of heparanase resulted in increased recruitment of inflammatory cells to the peritoneal cavity, and an increase in leukocyte rolling and adhesion in postcapillary venules, as evidenced by intravital microscopy of mesentery microvessels [120]. Moreover, in vitro adhesion assays showed that heparanase treatment increased neutrophil and mononuclear cell adhesion to HUVEC cells [120]. Surprisingly, in contrast, a study utilizing heparanase knockout and overexpressing mice found that monocyte, but not neutrophil, recruitment into peritoneal cavities inflamed by zymosan treatment depended on heparanase [121]. Moreover, although heparanase was upregulated in effector T-cells, it was not required for extravasation inside inflamed lymph nodes or skin in adoptive transfer experiments. However, in an experimental mouse model of sepsis-associated acute lung injury , inhibition of heparanase prevented endotoxemia-associated glycocalyx loss and neutrophil adhesion [122]. Moreover, in an experimental model of inflammation that utilized heparanase overexpressing vs WT mice, heparanase was shown to interfere with the process of intraluminal crawling of leukocytes within blood vessels [115]. Cleavage of HS by heparanase perturbed an experimentally applied gradient of the chemokine CXCL2 in the cremaster muscle, resulting in a loss of directionality of intraluminal leukocyte crawling. Finally, a study on transendothelial migration of hepatocellular carcinoma cells revealed that cell lines expressing high levels of heparanase show a higher transendothelial migration rate compared to cells with lower expression and that heparanase inhibition or downregulation suppressed this process both in vitro and in vivo [123]. In conclusion, in spite of context- and cell-type-specific effects, the aforementioned studies suggest that heparanase has a similar effect on leukocyte recruitment as the knockout of Sdc1.
As mentioned above, increased recruitment of Sdc1-deficient leukocytes to sites of inflammation was observed in a variety of experimental models of inflammation [55]. Interestingly, a range of similar or even identical disease models has been used to study the function of heparanase in inflammation employing transgenic and knockout mice with altered heparanase expression. In the following section, we will discuss the effect of Sdc1 and heparanase on four selected experimental models of inflammation (contact allergy, colitis, kidney inflammation, and experimental autoimmune encephalomyelitis), as they will allow us to compare the individual functions of Sdc1 and heparanase in different inflammatory diseases.
3.1.1 Role of Sdc1 and Heparanase in Delayed-Type Hypersensitivity
Delayed-type hypersensitivity (DTH) is a mouse model for allergic contact dermatitis, which consists of a sensitization phase involving covalent modification of surface proteins with haptens such as oxazolone or TNCB (2,4,6-trinitro-1-chlorobenzene), which are subsequently taken up and processed by dendritic and Langerhans cells. These cells migrate to lymph nodes and prime hapten-specific T cell populations, which are recruited and activated during the elicitation phase, resulting in cytokine and chemokine release, mast cell degranulation and massive leukocyte infiltration of the skin [124, 125]. Consistent with the previously described increase in leukocyte recruitment in Sdc1-KO mice, elicitation of an oxazolone-mediated DTH response resulted in increased leukocyte recruitment, and increased and prolonged edema formation. Compared to wild-type mice, expression of ICAM-1, cytokines (i.e., TNFα and IL-6), and chemokines (i.e., CCL5/RANTES, CCL-3/MIP-1α) was increased in Sdc1-deficient animals [119]. Interestingly, loss of Sdc1 can compensate for the loss of another proteoglycan, decorin, in this model: decorin-deficient mice show reduced DTH responses, associated with attenuation of leukocyte infiltration, which is overcome in the absence of Sdc1 [125]. However, while the in vivo data suggest that loss of Sdc1 releases the block in diapedesis observed in decorin-deficient mice, the detailed mechanisms underlying this observation still need to be elucidated. In the TNBS model of DTH, Sdc1-deficient dendritic cells migrated at a higher rate and faster to draining lymph nodes, resulting in an increased DTH response both in Sdc1 KO mice and in WT mice subjected to adoptive transfer of Sdc1 KO dendritic cells [126]. Moreover, upregulation of CCL2, CCL3, VCAM1 and talin, and a prolonged presence of CCR7 at the cell surface was observed on Sdc1 KO vs WT cells during dendritic cell maturation. Notably, Sdc1-KO dendritic cells showed an increased migration towards CCL21 and CCL19 compared to WT cells [126]. In addition to Sdc1, the role of heparanase during DTH has been studied in vivo. While Sdc1 expression is downregulated prominently in the epithelium of inflamed skin during oxazolon-induced DTH response [119], heparanase is upregulated particularly by the endothelium at the site of DTH-induced inflammation [127]. Moreover, heparanase-overexpressing mice showed a substantially increased DTH response compared to WT animals, whereas heparanase inhibition in WT mice resulted in a reduced DTH response and less vascular leakage [127]. In summary, these data suggest that upregulation of heparanase and absence of Sdc1 generate a similar, pro-inflammatory phenotype during DTH in mice. However, while Sdc1 negatively regulates endothelial leukocyte recruitment and dendritic cell migration during DTH responses, heparanase appears to primarily act at the level of the endothelium, where it regulates vascular permeability .
3.1.2 Role of Sdc1 and Heparanase in Anti-Glomerular Basement Membrane Glomerulonephritis
Experimental anti-glomerular basement membrane (anti-GBM) nephritis is a model of inflammation that mimics aspects of the autoimmune disease Goodpasture syndrome [128]. This experimental animal model is based on the injection of antibodies directed against antigenic material from the GBM of mice into recipient mice. An initial heterologous phase, where leukocyte influx peaks within hours and albuminuria become apparent can be observed within 24 h. This phase is followed by an autologous phase during which endogenous anti-GBM IgG is produced, leading to persistent albuminiuria [128]. In Wild-type (WT) mice, induction of anti-GBM glomerulonephritis resulted in an upregulation of Sdc1, and Sdc4 protein 2 and 18 h after induction of the disease, which normalized after 4 days [129]. In contrast, heparanase is upregulated both in the heterologous and autologous phase of the disease [130]. 4 and 8 days after administration of rabbit anti-GBM IgG, glomerular deposition of mouse anti-rabbit IgG was higher in Sdc1 KO compared to WT mice [129]. Notably, the numbers of PMNs and macrophages were significantly higher in inflamed glomeruli of Sdc1 KO mice in the heterologous phase, whereas the numbers of CD4+ and CD8+ T-cells were higher in Sdc1 KO mice in the autologous phase compared to WT. As a result, Sdc1 KO mice developed more severe albuminuria and showed worsened kidney function compared to WT mice. These changes were accompanied by significant increases of ICAM-1, L-selectin, IL-1β, MCP-1, IL-6 and IL-10 expression in Sdc1 KO mice vs WT during the heterologous phase, and in numerous ECM proteins (fibronectin, collagen IV, collagen XVIII, laminins, MMP7 and MMP9) along with L-selectin and MCP-1 during the autologous phase of the disease. Overall, Sdc1 KO mice showed a shift of the Th1/Th2 balance towards a Th2 response [129]. Compared to WT mice, heparanase-deficient mice showed better kidney function in this experimental model, which was accompanied by a reduced influx of PMNs and macrophages, reduced glomerular damage, and a reduction of the expression of numerous inflammatory factors along with reduced Sdc1 levels [130]. Heparanase KO mice showed reduced expression of both Th1 and Th2 cytokines. Reduced degradation of basement membrane HS was identified as a mechanistic aspect of the beneficial effects of heparanase deficiency, as shown in vitro using heparanase-silenced mouse glomerular endothelial cells, which displayed a lower transendothelial albumin passage compared to controls. It appears that Sdc1 and heparanase may play distinct mechanistic roles in this experimental model, although it is conceivable that heparanase deficiency may result in reduced Sdc1 ectodomain shedding, which may dampen the inflammatory response by reducing the influx of leukocytes [129, 130].
3.1.3 Sdc1 and Heparanase in Experimental Autoimmune Encephalitis (EAE)
Experimental autoimmune encephalitis (EAE) is an experimental T-cell dependent in vivo model of multiple sclerosis. In EAE, antigen-specific CD4+ Th1 cells cause inflammatory damage in the central nervous system, thus mimicking the demyelination, axonal loss and progressive paralysis caused by autoreactive T-cells in human multiple sclerosis [131, 132]. In the EAE model, Sdc1-deficient mice showed a higher severity of the disease compared to their WT counterparts and recovered more slowly [133]. Mechanistically, Sdc1 is upregulated at the transcriptional level, but shed from epithelial cells of the choroid plexus to the cerebrospinal fluid in WT mice, resulting in a loss of cell-surface bound CCL20 chemokine, which showed a partial co-localization with Sdc1 in naïve WT mouse brain. Notably, in Sdc1 KO mice, early recruitment of leukocytes, and levels of IL-6 were enhanced, resulting in recruitment of Th17 cells and aggravation of the inflammatory reaction. Moreover, enhanced plasma cell levels and higher levels of myelin oligodendrocyte glycoprotein–specific antibodies – a driving factor of the disease – in Sdc1 KO mice may be the underlying cause for delayed recovery from EAE [133]. The role of heparanase in EAE was studied applying recombinant heparanase via daily intraperitoneal administration starting from the day of immunization with proteolipid protein until day +7 or day +17 [134]. In this study, heparanase ameliorated the clinical signs of the disease. Moreover, the formation of clusters of inflammatory cells, as seen in WT mice in the white matter zone of the spinal cord, was not observed in heparanase-treated mice. Mechanistic in vitro and in vivo experiments revealed that heparanase treatment caused a shift of the cytokine spectrum toward Th2 cytokines (IL-4, IL-6, IL-10), resulting in an inhibition of a mixed lymphocyte reaction and mitogen-induced splenocyte proliferation [134]. While these data clearly demonstrate a role for both Sdc1 and heparanase in EAE, both molecules acted via different mechanisms in the two studies presented, and the focus on different mechanistic aspects and application of different analytical assays impedes a direct comparison. Further investigations may help to clarify the possible interplay of Sdc1 and heparanase in EAE. For example, it could be envisaged that heparanase is involved in the shedding of Sdc1, however, it is not clear if it is expressed in the epithelium of the choroid plexus during the EAE reaction. Moreover, while upregulation of IL-6 in Sdc1 KO mice was seen as a factor promoting disease progression in the study by Zhang et al. [133], it was presented as part of an anti-inflammatory Th2 signature in the study by Bitan et al. [134]. Differences in the time-courses of IL-6 expression, and in the tissues the cytokine is derived from, may account for the deviating interpretations of the respective phenotype .
3.1.4 Role of Sdc1 and Heparanase in Inflammatory Bowel Disease and Colitis-Associated Colon Cancer
Inflammatory bowel diseases are complex diseases that constitute a major health burden, which is characterized by an aberrant immune response in the gastrointestinal tract [135]. An important experimental model of inflammatory bowel diseases like Crohn’s disease and ulcerative colitis is the dextran sodium sulfate (DSS) colitis murine model, which is based on DSS-induced epithelial cell injury, which is followed by the entry of luminal bacteria and associated antigens into the mucosa and a subsequent inflammatory reaction [136]. Of note, reduced expression of Sdc1 has been observed in patients with ulcerative colitis, which has been linked to disrupt healing of colonic ulcers [137].
When the mechanistic role of Sdc1 in colitis was studied by inducing the disease with 3% DSS in Sdc1 KO mice, a substantial increase in mortality was observed compared to WT animals (61% versus 5%) [118]. Sdc1 KO mice showed prolonged recruitment of leukocytes and impaired mucosal healing, which were accompanied by an upregulation of TNFα, CCL3/MCP1, and VCAM-1 in the inflamed tissue. Notably, treatment with enoxaparin improved mucosal wound repair and reduced lethality of Sdc1 KO mice, suggesting that heparin may be able to compensate for the loss of the heparan sulfate chains of Sdc1 and the associated poor outcome in this disease model [118]. Of particular clinical relevance, chronic inflammatory bowel disease increases the risk of colon cancer [138], a disease that is also associated with a downregulation of Sdc1 expression in the colonic epithelium [139, 140]. Colitis-associated cancer can be experimentally modeled in mice by application of the carcinogen azoxymethane (AOM) and subsequent induction of chronic colitis with DSS[141]. Sdc1 KO mice developed more severe inflammation during chronic DSS colitis, associated with increased recruitment of inflammatory cells, increased crypt damage, and increased weight loss compared to WT mice [142]. IL-6 expression and activation of STAT3 were increased in the inflamed colon tissue of Sdc1 KO vs. Wt mice. Notably, Sdc1 KO mice formed larger tumors than their WT controls in the AOM-DSS model, which was attributed to increased activation of STAT3, and an upregulation of cyclin D1, CCL2, and c-Myc in the tumor tissue [142]. Overall, these data suggest that the increased inflammation and tissue damage in the absence of Sdc1 drive colon cancer progression via enhanced signaling through the IL-6/STAT pathway. While Sdc1 is downregulated in IBD and colon cancer, heparanase is upregulated in the inflamed and tumor tissue in these diseases [143, 144], and in the experimental AOM / DSS and the acute and chronic DSS animal models [145]. Notably, when mice that transgenically overexpress heparanase were subjected to DSS colitis, high heparanase expression preserved the inflammatory conditions, along with increased expression of TNFa and Cyclin D1 (thus showing a resemblance to Sdc1 KO mice in this model) [142], and a substantially increased recruitment of TNFa-expressing macrophages [145]. Similar to Sdc1 KO mice, tumors were larger in heparanase transgenic mice [142, 145]. Moreover, the number of tumors was higher compared to WT mice, and tumor angiogenesis was enhanced. Overall, these data suggest that heparanase drives a vicious cycle that promotes colitis and chronic-inflammation-related tumorigenesis. The phenotype shows undeniable similarities to the Sdc1 KO mouse in this experimental model, however, the interrelation between these molecules still awaits experimental investigation. Table 4.2 summarizes the phenotypes and molecular mechanisms of the mouse models that were presented in this section, revealing similarities and disparities in the mechanisms by which Sdc1 and heparanase modulate inflammatory processes in vivo.
4 Syndecan-1 and Heparanase as Pathogenesis Factors and Therapeutic Targets in Malignant Disease
The investigation of biopsies and blood from cancer patients has revealed that Syndecan-1 and heparanase are mis-expressed in a large number of cancers, underscoring their clinicopathological relevance (see Table 4.1). As of August 2019, close to 1000 publications for each molecule have described a role for Syndecan-1 or heparanase in different types of cancer in the PubMed database , generating a need to present selected examples of their molecular functions in malignant disease. The molecular mechanisms that govern tumorigenesis and cancer progression have been conceptually summarized by Hanahan and Weinberg in their landmark article ‘The Hallmarks of Cancer’, which originally included sustained proliferation, evasion of growth suppression, death resistance, replicative immortality, induced angiogenesis, and initiation of invasion [146]. Additional hallmarks were defined 10 years later in an updated version of the article and included the aspect of chronic inflammation (see Sect. 4.3) and avoidance of immune destruction [147] (see reference [148] for a recent review on the role of cell surface proteoglycans in immunotherapy). Notably, proteoglycans such as Sdc1 and enzymes like heparanase, which utilize HS proteoglycans as substrates have been shown to modulate most, if not all of these Hallmarks [148, 149]. For example, regarding the cancer hallmark of sustained proliferation , Syndecan-1 acts as a co-receptor for receptor tyrosine kinases, thus contributing to proliferative signaling and tumor growth, as demonstrated for example in breast cancer, colon cancer, and multiple myeloma [22, 97, 108]. Notably, it has been shown that the soluble ectodomain of Syndecan-1 can competitively inhibit mitogenicity of the cytokine FGF2, whereas platelet heparanase is able to convert the HS chains of soluble Syndecan-1 from an inhibitor into heparin-like HS fragments that substantially activate FGF-2 mitogenicity [150]. Moreover, both latent heparanase and its mature active form promote signaling through multiple pathways with relevance to tumor progression, including the Src, MAPK, HGF-, IGF-, and EGF-receptor pathways [151]. The insensitivity to antigrowth signals has been defined as an additional hallmark of cancer. An important growth-inhibitory cytokine and mediator of epithelial-to-mesenchymal transition is TGFβ which has been shown to be negatively regulated by Syndecan-1 during liver fibrogenesis [152]. Interestingly, heparanase-mediated shedding of Syndecan-1 has been shown to result in an upregulation of TGFβ in hepatocellular carcinoma, providing a link between heparanase and Syndecan-1 in the regulation of growth-inhibitory signals [153]. The enabling of replicative immortality adds to the six original hallmarks of cancer [146]. Replicative immortality is a shared feature of cancer stem cells and is partially linked to the activity of telomerase, which prevents the shortening of chromosome ends [154]. Syndecan-1 has emerged as an important regulator of the cancer stem cell phenotype: The reduction of a wnt-responsive precursor cell population has been identified as a molecular mechanism underlying the resistance of juvenile Sdc1 KO mice to breast cancer and additional forms of experimentally induced cancer [154,155,156]. Moreover, siRNA mediated knockdown of Sdc1 in human breast cancer cell lines representative of different molecular classifications resulted in a reduction of stem cell properties, including the expression of typical cancer stem cell markers (CD44+/CD24low, side population, ALDH) as well as colony and mammosphere formation [157, 158]. Modulation of the stemness-related notch, wnt, and IL-6/STAT signaling pathways by Syndecan-1 was identified as the mechanistic basis for this finding. Notably siRNA knockdown of Syndecan-1 also resulted in an upregulation of heparanase expression in this model system [159]. Indeed, modulation of stem cell properties in the context of malignant disease has not only been ascribed to Syndecan-1, but also to heparanase, which acts in a context-dependent manner. In breast cancer, nuclear heparanase has been shown to induce tumor cell differentiation [160]. Moreover, heparanase-mediated modifications in the bone marrow microenvironment regulate the retention and proliferation of hematopoietic progenitor cells [161], modulate clonogenicity, proliferative potential and migration of mesenchymal stem cells in the bone marrow [162], and shift the differentiation potential of osteoblast progenitors within the myeloma bone microenvironment from osteoblastogenesis to adipogenesis [163]. Moreover, embryonic stem cells overexpressing heparanase proliferated faster than wild-type controls in culture and formed larger teratomas in vivo [164], indicating an important role for heparanase in (cancer) stem cells and the modulation of replicative immortality. Closely linked to the previous hallmarks of cancer is the resistance to cell death , as it allows for the proliferation of tumor cells which carrying mutations, ultimately leading to another enabling characteristic, genome instability. Several studies have documented a role for Syndecan-1 as a regulator of apoptosis in an oncological setting. For example, Syndecan-1 suppresses apoptosis in multiple myeloma by activating the IGF1 receptor [88], and in endometrial cancer by enhancing Erk and Akt activation [165]. In contrast, Syndecan-1-dependent MAPK-signaling was shown to be of importance for the pro-apoptotic effect of the n-3 polyunsaturated fatty acid docosahexaenoic acid in prostate and breast cancer cells. Likewise, heparanase modulates cancer cell apoptosis in a context-dependent manner [166, 167]. Early studies indicated a positive correlation between heparanase expression and spontaneous apoptosis in hepatocellular carcinoma [168]. Along these lines, an apoptosis-enhancing function of heparanase was revealed in growth-hormone secreting pituitary tumor cells [169]. In contrast, orthotopic xenograft experiments utilizing heparanase overexpressing breast cancer cells identified heparanase as a survival factor for breast cancer cells in vivo [170]. Similarly, overexpression of heparanase inhibited apoptosis in cervical cancer cells [171]. Since Syndecan-1 and heparanase modulate similar signal transduction pathways, it can be envisaged that they cooperate in the regulation of cell death, however, more studies are needed to confirm the mechanistic involvement in more detail. The induction of angiogenesis is another hallmark of cancer that is modulated both by Syndecan-1 and heparanase. The sprouting and growth of blood vessels from existing blood vessels is a carefully orchestrated physiological process, that is hijacked by tumor cells via secretion of angiogenic factors and by causing an imbalance between pro-and anti-angiogenic factors [89, 172]. Tumor angiogenesis is a prerequisite for supplying the tumor with nutrients and oxygen, and for promoting metastatic spread via the circulation. Of note, syndecan-1 expression has been identified as part of a molecular signature marking an angiogenic switch in early stages of breast cancer [173] and stromal syndecan-1 expression was shown to correlate with microvessel density and blood vessel area both in human breast cancer specimens and in xenograft models [174]. In vivo studies have demonstrated that absence and overexpression of syndecan-1 modulate angiogenesis via enhanced leukocyte recruitment and by promoting proteolysis, respectively [42, 116]. Moreover, syndecan-1 promotes angiogenesis as a classical co-receptor for angiogenic factors such as FGF-2, VEGF, and c-Met [97, 173, 175]. In addition, MMP9-induced syndecan-1 is part of a mechanism that is responsible for radiation-induced angiogenesis in medulloblastoma [176]. Moreover, syndecan-1 regulates tumor angiogenesis via lateral association of its extracellular core protein domain with proangiogenic integrins [86, 89]. Notably, in multiple myeloma, heparanase stimulates shedding of syndecan-1, which leads to a deposition of VEGF that is bound to the syndecan-1 HS chains in the ECM, where it stimulates endothelial invasion [50]. Heparanase-induced syndecan-1 shedding also promotes hepatocarcinoma lymphangiogenesis via the VEGF-C/ERK pathway [175]. However, heparanase promotes tumor angiogenesis not only via induction of syndecan-1 shedding but also via the release of proangiogenic factors from heparan sulfate on cell surfaces and the ECM, through Src-dependent upregulation of VEGF expression, enhancement of Akt signaling and stimulation of PI3K- and p38-dependent endothelial cell migration and invasion [177].
Finally, the activation of invasion and metastasis has been defined as a hallmark of cancer, which is of utmost importance as cancer-associated metastasis to vital organs is the leading cause of cancer-related mortality [178]. The misexpression of Syndecan-1 that is observed in numerous tumor entities (Table 4.1) contributes to metastatic behavior in several ways. While the membrane-bound form of Syndecan-1 had an invasion-inhibiting effect on the human breast cancer cell line MCF-7, overexpression of the soluble ectodomain and of the intact proteoglycan resulted in a substantial increase in Matrigel invasion chamber assays [22, 159]. The increase in invasiveness could be attributed to a downregulation of the MMP inhibitor TIMP-1 and upregulation of urokinase-type plasminogen activator receptor (uPAR) in cells overexpressing soluble Syndecan-1. Moreover, a downregulation of the anti-invasive homotypic cell adhesion molecule was observed both in MCF-7 breast cancer cells overexpressing soluble Syndecan-1, and in the highly invasive breast cancer cell line MDA-MB-231 upon Syndecan-1 depletion [22, 159]. While this observation may suggest a potential involvement of syndecan-1 in regulating the pro-metastatic process of epithelial-to-mesenchymal transition (EMT) in breast cancer, no consistent upregulation of mesenchymal markers was observed upon Syndecan-1 depletion in the same experimental system [157]. However, an indirect regulatory impact on EMT of prostate cancer cells was recently proposed, involving alterations in EMT-regulating microRNA processing that was modulated by syndecan-1-dependent changes in expression of the miRNA processing enzyme, Dicer [179]; Syndecan-1 promotes EMT in hepatocellular carcinoma by enhancing TGFbeta function [180]. In breast cancer, time-lapse microscopy analysis revealed that silencing of Syndecan-1 in human MDA-MB-231 breast cancer cells results in a substantial increase in cell motility [159] and migration through fibronectin-coated filter membranes [181]. Inhibitor studies and signal transduction analysis revealed that this increase could be attributed to increased activation of focal adhesion kinase and dysregulation of Rho-GTPases. In lung cancer, Syndecan-1 was shown to inhibit metastasis in vivo and invasiveness in vitro via a mechanism that relies on the cleavage of Syndecan-1 core protein by ADAM17 and gamma-secretase, and the generation of a cytoplasmic fragment with biological activity [48]. Moreover, similar to its function during inflammation, Syndecan-1 promotes chemotaxis of breast and lung cancer cells as a co-receptor for chemokines such as MIP1 [182]. Of note, heparanase-mediated processing of Syndecan-1 has an important role in the regulation of metastasis. Heparanase promotes shedding of syndecan-1 in myeloma and breast cancer cells, whereas syndecan-1 promotes processing of heparanase [45, 183]. Moreover, heparanase regulates the secretion of tumor exosomes, including their Syndecan-1 content, thus driving metastatic behaviour [81] (Fig. 4.3). Finally, Syndecan-1 can inhibit heparanase-mediated in vitro invasion of melanoma cells in an HS-dependent manner, indicating complex regulatory circuits between heparanase and Syndecan-1 [184]. Apart from the mechanisms involving Syndecan-1 processing, heparanase has been shown to promote invasive growth and metastasis of a wide range of tumor entities [185] (Table 4.1). Indeed, heparanase promotes metastasis via several mechanisms, as demonstrated in transgenic mouse models overexpressing heparanase and in tumor cell lines subjected to heparanase silencing of inhibition. These mechanisms including a degradation of basement membrane HS, thus removing a physical barrier of metastasis in epithelia and endothelia [186, 187], through the release of stored growth factors and chemokines from the ECM and cell surfaces, thus promoting chemotaxis and pro-invasive signaling [103, 188, 189], and through the impact on exosome secretion and function [190], as mentioned above (Fig. 4.3).
Given the importance of Syndecan-1 and heparanase as prognostic markers in various tumor entities, and as mechanistically relevant factors in driving cancer progression, it is no surprise that these molecules have been identified as therapeutic targets in malignant disease [188, 191]. Regarding a therapeutic targeting of Syndecan-1, it has to be considered that it is highly expressed on several healthy epithelia, including the gut, skin, and lung [8, 41], thus increasing the danger of an unfavorable side effects profile upon therapeutic targeting. Nevertheless, promising results have been achieved upon Syndecan-1-targeting in preclinical models. One approach includes the interference of Syndecan-1 integrin interactions with the Syndecan-1-derived peptide synstatin, which successfully disrupted tumor angiogenesis in vivo [86, 89]. Moreover, Syndecan-1-conjugates with cytotoxic drugs (i.e., indatuximab and ravtansine) have shown efficacy in preclinical in vivo models of triple-negative breast cancer [192]. In patients with relapsed or refractory multiple myeloma, this antibody-drug conjugate entered phase I/IIa clinical trials and showed efficiency with respect to clinical activity, although side effects on epithelial tissues were also observed [148, 193]. Finally, CAR-T cell therapy has been employed to target Syndecan-1, showing efficacy in a preclinical model of multiple myeloma, and promising results in patients, including stable disease for over 3 months, and a reduction of myeloma cells in the peripheral blood, respectively [148, 194, 195]. Compared to Syndecan-1, heparanase represents an even more attractive drug target, as it is only expressed in a few cell types in healthy adults (mainly leukocytes), thus limiting potential therapy-associated side effects [189]. A recent comprehensive review provided an overview of different heparanase inhibitors that have been explored in preclinical studies and partially also in clinical trials [196]. These inhibitors include heparin and its derivatives (e.g., roneparstat/SST0001 and mupofastat/PI-88), nucleic acid-based inhibitors (e.g. defibrotide), synthetic inhibitors (e.g., suramin) and heparanase-neutralizing antibodies. Apart from targeting tumor cell-derived heparanase, these inhibitors can also have an impact on the tumor stroma and inhibit processes that support tumor progression, e.g. tumor-promoting inflammation and angiogenesis. Indeed, several of these inhibitors have demonstrated anti-tumor, anti-angiogenic and anti-metastatic efficacy in preclinical animal models of the disease, including e.g. myeloma [51], lymphoma [197], and sarcoma models [198]. Moreover, heparanase inhibitors have shown to be largely well-tolerated in phase I and phase II clinical trials [196, 199,200,201], providing a positive outlook for progress in targeted cancer therapy in the near future (see Noseda and Barbieri; Hammond and Dredge; Chhabra and Ferro, Chaps. 19, 21 and 22 in this volume).
5 Concluding Remarks
Syndecan-1 and heparanase fulfill important and pleiotropic physiological functions during development and tissue homeostasis. Notably, several processes regulated by Syndecan-1 and heparanase are also relevant in the context of inflammatory and malignant disease. These processes include the regulation of cell proliferation and survival, cell motility and cell invasion, leukocyte recruitment and angiogenesis, which are achieved by modulation of growth factor-, chemokine- and morphogen-mediated signaling processes and via regulation of the composition and functional properties of the ECM. While Syndecan-1 and heparanase retain some autonomous functions, many of their properties are functionally linked. Syndecan-1 can act as a substrate for heparanase, and heparanase promotes Syndecan-1 shedding as an important mechanism of conversion of this membrane-bound molecule into a soluble paracrine effector. In turn, Syndecan-1-dependent signaling mechanisms regulate heparanase expression, and the cytoplasmic domain of Syndecan-1 plays an important role in heparanase processing. Moreover, ligands bound to the HS chains of Syndecan-1 can be released by the action of heparanase, resulting in an altered functionality. In contrast, the HS chains of Syndecan-1 can have an inhibitory impact on heparanase activity, depending on their fine structure. In most cases, synergistic effects of Syndecan-1 and heparanase are observed, which contribute to the pathogenesis of inflammatory diseases and tumor progression. Therefore, Syndecan-1 and heparanase have emerged as important targets for therapeutic approaches. Due to their pleiotropic functions and their mechanistic involvement in inflammatory and malignant diseases, their targeting represents a highly promising approach, as it can be expected that therapeutics simultaneously inhibit multiple processes related to disease progression. Substantial evidence from preclinical models and promising results from phase I/II clinical trials provide a promising perspective regarding the translation into a clinical setting.
References
Prydz, K., & Dalen, K. T. (2000). Synthesis and sorting of proteoglycans. Journal of Cell Science, 113, 193–205.
Lamanna, W. C., et al. (2007). The heparanome—The enigma of encoding and decoding heparan sulfate sulfation. Journal of Biotechnology, 129, 290–307.
Dreyfuss, J. L., et al. (2009). Heparan sulfate proteoglycans: Structure, protein interactions and cell signaling. Anais da Academia Brasileira de Ciências, 81, 409–429.
Turnbull, J. E. (2001). Analytical and preparative strong anion-exchange HPLC of heparan sulfate and heparin saccharides. Methods Mol Biol Clifton NJ, 171, 141–147.
Nagarajan, A., Malvi, P., & Wajapeyee, N. (2018). Heparan Sulfate and Heparan Sulfate proteoglycans in Cancer initiation and progression. Frontiers in Endocrinology, 9.
Brockstedt, U., Dobra, K., Nurminen, M., & Hjerpe, A. (2002). Immunoreactivity to cell surface Syndecans in cytoplasm and nucleus: Tubulin-dependent rearrangements. Experimental Cell Research, 274, 235–245.
Piperigkou, Z., Mohr, B., Karamanos, N., & Götte, M. (2016). Shed proteoglycans in tumor stroma. Cell and Tissue Research, 365, 643–655.
Kim, C. W., Goldberger, O. A., Gallo, R. L., & Bernfield, M. (1994). Members of the syndecan family of heparan sulfate proteoglycans are expressed in distinct cell-, tissue-, and development-specific patterns. Molecular Biology of the Cell, 5, 797–805.
Sarrazin, S., Lamanna, W. C., & Esko, J. D. (2011). Heparan sulfate proteoglycans. Cold Spring Harbor Perspectives in Biology, 3.
Stepp, M. A., Pal-Ghosh, S., Tadvalkar, G., & Pajoohesh-Ganji, A. (2015). Syndecan-1 and its expanding list of contacts. Advances in Wound Care, 4, 235–249.
Afratis, N. A., et al. (2017). Syndecans - key regulators of cell signaling and biological functions. The FEBS Journal, 284, 27–41.
Altemeier, W. A., et al. (2012). Transmembrane and extracellular domains of Syndecan-1 have distinct functions in regulating lung epithelial migration and adhesion. The Journal of Biological Chemistry, 287, 34927–34935.
Bishop, J. R., Schuksz, M., & Esko, J. D. (2007). Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature, 446, 1030–1037.
Chakravarti, R., Sapountzi, V., & Adams, J. C. (2005). Functional role of Syndecan-1 cytoplasmic V region in Lamellipodial spreading, actin bundling, and cell migration. Molecular Biology of the Cell, 16, 3678–3691.
Dews, I. C., & MacKenzie, K. R. (2007). Transmembrane domains of the syndecan family of growth factor coreceptors display a hierarchy of homotypic and heterotypic interactions. Proceedings of the National Academy of Sciences, 104, 20782–20787.
Langford, J. K., Stanley, M. J., Cao, D., & Sanderson, R. D. (1998). Multiple Heparan Sulfate chains are required for optimal Syndecan-1 function. The Journal of Biological Chemistry, 273, 29965–29971.
Manon-Jensen, T., Multhaupt, H. A. B., & Couchman, J. R. (2013). Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. The FEBS Journal, 280, 2320–2331.
Teng, Y. H.-F., Aquino, R. S., & Park, P. W. (2012). Molecular functions of syndecan-1 in disease. Matrix biology : Journal of the International Society for Matrix Biology, 31, 3–16.
Alexopoulou, A. N., Multhaupt, H. A. B., & Couchman, J. R. (2007). Syndecans in wound healing, inflammation and vascular biology. The International Journal of Biochemistry & Cell Biology, 39, 505–528.
Haynes, A., et al. (2005). Syndecan 1 shedding contributes to Pseudomonas aeruginosa sepsis. Infection and Immunity, 73, 7914–7921.
Mali, M., Andtfolk, H., Miettinen, H. M., & Jalkanen, M. (1994). Suppression of tumor cell growth by syndecan-1 ectodomain. The Journal of Biological Chemistry, 269, 27795–27798.
Nikolova, V., et al. (2009). Differential roles for membrane-bound and soluble syndecan-1 (CD138) in breast cancer progression. Carcinogenesis, 30, 397–407.
Zhang, L. (2010). Glycosaminoglycan (GAG) biosynthesis and GAG-binding proteins. Progress in Molecular Biology and Translational Science, 93, 1–17.
Fairbanks, M. B., et al. (1999). Processing of the human Heparanase precursor and evidence that the active enzyme is a heterodimer. The Journal of Biological Chemistry, 274, 29587–29590.
Abboud-Jarrous, G., et al. (2005). Site-directed mutagenesis, Proteolytic cleavage, and activation of human Proheparanase. The Journal of Biological Chemistry, 280, 13568–13575.
Levy-Adam, F., Miao, H.-Q., Heinrikson, R. L., Vlodavsky, I., & Ilan, N. (2003). Heterodimer formation is essential for heparanase enzymatic activity. Biochemical and Biophysical Research Communications, 308, 885–891.
Gingis-Velitski, S., Zetser, A., Flugelman, M. Y., Vlodavsky, I., & Ilan, N. (2004). Heparanase induces endothelial cell migration via protein kinase B/Akt activation. The Journal of Biological Chemistry, 279, 23536–23541.
Doviner, V., et al. (2006). Spatial and temporal heparanase expression in colon mucosa throughout the adenoma-carcinoma sequence. Modern Pathology, 19, 878–888.
Zetser, A., Bashenko, Y., Miao, H.-Q., Vlodavsky, I., & Ilan, N. (2003). Heparanase affects adhesive and tumorigenic potential of human Glioma cells. Cancer Research, 63, 7733–7741.
Cohen-Kaplan, V., Doweck, I., Naroditsky, I., Vlodavsky, I., & Ilan, N. (2008). Heparanase augments epidermal growth factor receptor phosphorylation: Correlation with head and neck tumor progression. Cancer Research, 68, 10077–10085.
Cohen-Kaplan, V., et al. (2008). Heparanase induces VEGF C and facilitates tumor lymphangiogenesis. International Journal of Cancer, 123, 2566–2573.
Sanderson, R. D., Elkin, M., Rapraeger, A. C., Ilan, N., & Vlodavsky, I. (2017). Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy. The FEBS Journal, 284, 42–55.
Jin, H., & Zhou, S. (2017). The functions of Heparanase in human diseases. Mini Reviews in Medicinal Chemistry, 17, 541–548.
Masola, V., Bellin, G., Gambaro, G., & Onisto, M. (2018). Heparanase: A multitasking protein involved in Extracellular Matrix (ECM) Remodeling and intracellular events. Cell, 7, 236.
Lamorte, S., et al. (2012). Syndecan-1 promotes the angiogenic phenotype of multiple myeloma endothelial cells. Leukemia, 26, 1081–1090.
Noguer, O., Villena, J., Lorita, J., Vilaró, S., & Reina, M. (2009). Syndecan-2 downregulation impairs angiogenesis in human microvascular endothelial cells. Experimental Cell Research, 315, 795–808.
De Rossi, G., & Whiteford, J. R. (2013). A novel role for syndecan-3 in angiogenesis. F1000Research, 2, 270.
Ramani, V. C., Pruett, P. S., Thompson, C. A., DeLucas, L. D., & Sanderson, R. D. (2012). Heparan Sulfate chains of Syndecan-1 regulate Ectodomain shedding. The Journal of Biological Chemistry, 287, 9952–9961.
Ma, P., et al. (2006). Heparanase deglycanation of syndecan-1 is required for binding of the epithelial-restricted prosecretory mitogen lacritin. The Journal of Cell Biology, 174, 1097–1106.
Levy-Adam, F., Feld, S., Suss-Toby, E., Vlodavsky, I., & Ilan, N. (2008). Heparanase facilitates cell adhesion and spreading by clustering of cell surface Heparan Sulfate proteoglycans. PLoS One, 3, e2319.
Bernfield, M., et al. (1999). Functions of cell surface Heparan Sulfate proteoglycans. Annual Review of Biochemistry, 68, 729–777.
Elenius, V., Götte, M., Reizes, O., Elenius, K., & Bernfield, M. (2004). Inhibition by the soluble Syndecan-1 Ectodomains delays wound repair in mice overexpressing Syndecan-1. The Journal of Biological Chemistry, 279, 41928–41935.
Manon-Jensen, T., Itoh, Y., & Couchman, J. R. (2010). Proteoglycans in health and disease: The multiple roles of syndecan shedding. The FEBS Journal, 277, 3876–3889.
Nam, E. J., & Park, P. W. (2012). Shedding of cell membrane-bound Proteoglycans. In F. Rédini (Ed.), Proteoglycans: Methods and protocols (pp. 291–305). Humana Press. https://doi.org/10.1007/978-1-61779-498-8_19.
Yang, Y., et al. (2007). Heparanase enhances syndecan-1 shedding: A novel mechanism for stimulation of tumor growth and metastasis. The Journal of Biological Chemistry, 282, 13326–13333.
Mahtouk, K., et al. (2007). Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood, 109, 4914–4923.
Ramani, V. C., et al. (2013). The heparanase/syndecan-1 axis in cancer: Mechanisms and therapies. The FEBS Journal, 280, 2294–2306.
Pasqualon, T., et al. (2015). A cytoplasmic C-terminal fragment of syndecan-1 is generated by sequential proteolysis and antagonizes syndecan-1 dependent lung tumor cell migration. Oncotarget, 6, 31295–31312.
Jung, O., et al. (2016). Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: Prevention by novel synstatins. Oncogene, 5, e202.
Purushothaman, A., et al. (2010). Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood, 115, 2449–2457.
Ritchie, J. P., et al. (2011). SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the Heparanase/Syndecan-1 Axis. Clinical Cancer Research, 17, 1382–1393.
Purushothaman, A., Chen, L., Yang, Y., & Sanderson, R. D. (2008). Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. The Journal of Biological Chemistry, 283, 32628–32636.
Sotnikov, I., et al. (2004). Enzymatically quiescent Heparanase augments T cell interactions with VCAM-1 and extracellular matrix components under versatile dynamic contexts. Journal of Immunology, 172, 5185–5193.
Götte, M. (2003). Syndecans in inflammation. The FASEB Journal, 17, 575–591.
Götte, M., & Echtermeyer, F. (2003). Syndecan-1 as a regulator of chemokine function. The Scientific World Journal. https://doi.org/10.1100/tsw.2003.118.
Stewart, M. D., Ramani, V. C., & Sanderson, R. D. (2015). Shed Syndecan-1 Translocates to the nucleus of cells delivering growth factors and inhibiting histone acetylation a novel mechanism of tumor-host cross-talk. The Journal of Biological Chemistry, 290, 941–949.
Kovalszky, I., Hjerpe, A., & Dobra, K. (2014). Nuclear translocation of heparan sulfate proteoglycans and their functional significance. Biochimica et Biophysica Acta (BBA) – General Subjects, 1840, 2491–2497.
Leadbeater, W. E., et al. (2006). Intracellular trafficking in neurones and glia of fibroblast growth factor-2, fibroblast growth factor receptor 1 and heparan sulphate proteoglycans in the injured adult rat cerebral cortex. Journal of Neurochemistry, 96, 1189–1200.
Schrage, Y. M., et al. (2009). Aberrant Heparan Sulfate proteoglycan localization, despite Normal Exostosin, in central Chondrosarcoma. The American Journal of Pathology, 174, 979–988.
Schubert, S. Y., et al. (2004). Human heparanase nuclear localization and enzymatic activity. Laboratory Investigation, 84, 535–544.
Zong, F., et al. (2009). Syndecan-1 and FGF-2, but not FGF Receptor-1, share a common transport route and co-localize with Heparanase in the nuclei of Mesenchymal tumor cells. PLoS One, 4, e7346.
Richardson, T. P., Trinkaus-Randall, V., & Nugent, M. A. (2001). Regulation of heparan sulfate proteoglycan nuclear localization by fibronectin. Journal of Cell Science, 114, 1613–1623.
Hsia, E., Richardson, T. P., & Nugent, M. A. (2003). Nuclear localization of basic fibroblast growth factor is mediated by heparan sulfate proteoglycans through protein kinase C signaling. Journal of Cellular Biochemistry, 88, 1214–1225.
Fedarko, N. S., Ishihara, M., & Conrad, H. E. (1989). Control of cell division in hepatoma cells by exogenous heparan sulfate proteoglycan. Journal of Cellular Physiology, 139, 287–294.
Baldin, V., Roman, A. M., Bosc-Bierne, I., Amalric, F., & Bouche, G. (1990). Translocation of bFGF to the nucleus is G1 phase cell cycle specific in bovine aortic endothelial cells. The EMBO Journal, 9, 1511–1517.
Roghani, M., & Moscatelli, D. (1992). Basic fibroblast growth factor is internalized through both receptor-mediated and heparan sulfate-mediated mechanisms. The Journal of Biological Chemistry, 267, 22156–22162.
Zong, F., et al. (2011). Specific Syndecan-1 domains regulate Mesenchymal tumor cell adhesion, motility and migration. PLoS One, 6, e14816.
Kovalszky, I., et al. (1998). Inhibition of DNA topoisomerase I activity by heparan sulfate and modulation by basic fibroblast growth factor. Molecular and Cellular Biochemistry, 183, 11–23.
Chen, L., & Sanderson, R. D. (2009). Heparanase regulates levels of Syndecan-1 in the nucleus. PLoS One, 4, e4947.
Purushothaman, A., et al. (2011). Heparanase-mediated loss of nuclear Syndecan-1 enhances histone Acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. The Journal of Biological Chemistry, 286, 30377–30383.
Zhang, L., Sullivan, P., Suyama, J., & Marchetti, D. (2010). Epidermal growth factor-induced heparanase nucleolar localization augments DNA topoisomerase I activity in brain metastatic breast cancer. Molecular Cancer Research, 8, 278–290.
Gould, G. W., & Lippincott-Schwartz, J. (2009). New roles for endosomes: From vesicular carriers to multi-purpose platforms. Nature Reviews. Molecular Cell Biology, 10, 287–292.
Klumperman, J., & Raposo, G. (2014). The complex ultrastructure of the Endolysosomal system. Cold Spring Harbor Perspectives in Biology, 6, a016857.
Henne, W. M., Buchkovich, N. J., & Emr, S. D. (2011). The ESCRT pathway. Developmental Cell, 21, 77–91.
Hanson, P. I., & Cashikar, A. (2012). Multivesicular body morphogenesis. Annual Review of Cell and Developmental Biology, 28, 337–362.
Baietti, M. F., et al. (2012). Syndecan–syntenin–ALIX regulates the biogenesis of exosomes. Nature Cell Biology, 14, 677–685.
Peinado, H., Lavotshkin, S., & Lyden, D. (2011). The secreted factors responsible for pre-metastatic niche formation: Old sayings and new thoughts. Seminars in Cancer Biology, 21, 139–146.
Ostrowski, M., et al. (2010). Rab27a and Rab27b control different steps of the exosome secretion pathway. Nature Cell Biology, 12, 19–30.
Grootjans, J. J., Reekmans, G., Ceulemans, H., & David, G. (2000). Syntenin-Syndecan binding requires Syndecan-Synteny and the co-operation of both PDZ domains of Syntenin. The Journal of Biological Chemistry, 275, 19933–19941.
Zimmermann, P., et al. (2001). Characterization of Syntenin, a Syndecan-binding PDZ protein, as a component of cell adhesion sites and microfilaments. Molecular Biology of the Cell, 12, 339–350.
Thompson, C. A., Purushothaman, A., Ramani, V. C., Vlodavsky, I., & Sanderson, R. D. (2013). Heparanase regulates secretion, composition, and function of tumor cell-derived Exosomes. The Journal of Biological Chemistry, 288, 10093–10099.
Roucourt, B., Meeussen, S., Bao, J., Zimmermann, P., & David, G. (2015). Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Research, 25, 412–428.
David, G., & Zimmermann, P. (2016). Heparanase tailors syndecan for exosome production. Molecular & Cellular Oncology, 3, e1047556.
Friand, V., David, G., & Zimmermann, P. (2015). Syntenin and syndecan in the biogenesis of exosomes. Biology of the Cell, 107, 331–341.
Keller, S., et al. (2009). Systemic presence and tumor-growth promoting effect of ovarian carcinoma released exosomes. Cancer Letters, 278, 73–81.
Beauvais, D. M., Ell, B. J., McWhorter, A. R., & Rapraeger, A. C. (2009). Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. The Journal of Experimental Medicine, 206, 691–705.
Beauvais, D. M., & Rapraeger, A. C. (2010). Syndecan-1 couples the insulin-like growth factor-1 receptor to inside-out integrin activation. Journal of Cell Science, 123, 3796–3807.
Beauvais, D. M., Jung, O., Yang, Y., Sanderson, R. D., & Rapraeger, A. C. (2016). Syndecan-1 (CD138) suppresses apoptosis in multiple myeloma by activating IGF1 receptor: Prevention by SynstatinIGF1R inhibits tumor growth. Cancer Research, 76, 4981–4993.
Rapraeger, A. C. (2013). Synstatin: A selective inhibitor of the syndecan-1-coupled IGF1R–αvβ3 integrin complex in tumorigenesis and angiogenesis. The FEBS Journal, 280, 2207–2215.
Wang, H., Jin, H., & Rapraeger, A. C. (2015). Syndecan-1 and Syndecan-4 Capture epidermal growth factor receptor family members and the α3β1 integrin via binding sites in their Ectodomains novel synstatins prevent kinase capture and inhibit α6β4-integrin-dependent epithelial cell motility. The Journal of Biological Chemistry, 290, 26103–26113.
Belting, M. (2003). Heparan sulfate proteoglycan as a plasma membrane carrier. Trends in Biochemical Sciences, 28, 145–151.
Olsnes, S., Klingenberg, O., & Wiedłocha, A. (2003). Transport of Exogenous Growth Factors and Cytokines to the Cytosol and to the Nucleus. Physiological Reviews, 83, 163–182.
Keresztes, M., & Boonstra, J. (1999). Import(ance) of growth factors in(to) the nucleus. The Journal of Cell Biology, 145, 421–424.
Belting, M., Sandgren, S., & Wittrup, A. (2005). Nuclear delivery of macromolecules: Barriers and carriers. Advanced Drug Delivery Reviews, 57, 505–527.
Chen, K., & Williams, K. J. (2013). Molecular mediators for raft-dependent endocytosis of Syndecan-1, a highly conserved, multifunctional receptor. Journal of Biological Chemistry, 288, 13988–13999.
Ramani, V. C., Yang, Y., Ren, Y., Nan, L., & Sanderson, R. D. (2011). Heparanase plays a dual role in driving Hepatocyte Growth Factor (HGF) signaling by enhancing HGF expression and activity. The Journal of Biological Chemistry, 286, 6490–6499.
Derksen, P. W. B., et al. (2002). Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes met signaling in multiple myeloma. Blood, 99, 1405–1410.
Deakin, J. A., & Lyon, M. (1999). Differential regulation of hepatocyte growth factor/scatter factor by cell surface proteoglycans and free glycosaminoglycan chains. Journal of Cell Science, 112, 1999–2009.
Seidel, C., et al. (2000). High levels of soluble syndecan-1 in myeloma-derived bone marrow: Modulation of hepatocyte growth factor activity. Blood, 96, 3139–3146.
Brooks, P. C., Clark, R. A., & Cheresh, D. A. (1994). Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science, 264, 569–571.
Mahabeleshwar, G. H., Feng, W., Reddy, K., Plow, E. F., & Byzova, T. V. (2007). Mechanisms of integrin–vascular endothelial growth factor receptor cross-activation in angiogenesis. Circulation Research, 101, 570–580.
Reiland, J., Kempf, D., Roy, M., Denkins, Y., & Marchetti, D. (2006). FGF2 binding, Signaling, and angiogenesis are modulated by Heparanase in metastatic melanoma cells. Neoplasia N. Y. N, 8, 596–606.
Kato, M., et al. (1998). Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nature Medicine, 4, 691–697.
Liu, D., Shriver, Z., Venkataraman, G., Shabrawi, Y. E., & Sasisekharan, R. (2002). Tumor cell surface heparan sulfate as cryptic promoters or inhibitors of tumor growth and metastasis. Proceedings of the National Academy of Sciences, 99, 568–573.
Masola, V., et al. (2012). Heparanase and Syndecan-1 interplay orchestrates fibroblast growth Factor-2-induced epithelial-Mesenchymal transition in renal tubular cells. The Journal of Biological Chemistry, 287, 1478–1488.
Margraf, A., Ley, K., & Zarbock, A. (2019 July). Neutrophil recruitment: From model systems to tissue-specific patterns. Trends in Immunology, 40(7), 613–634.
Borsig, L. (2018, September 1). Selectins in cancer immunity. Glycobiology, 28(9), 648–655.
Kumar, A. V., Katakam, S. K., Urbanowitz, A. K., & Gotte, M. (2015). Heparan sulphate as a regulator of leukocyte recruitment in inflammation. Current Protein & Peptide Science, 16(1), 77–86.
Schlessinger, J., Plotnikov, A. N., Ibrahimi, O. A., Eliseenkova, A. V., Yeh, B. K., Yayon, A., Linhardt, R. J., & Mohammadi, M. (2000). Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Molecular Cell, 6, 743–750.
Lortat-Jacob, H. (2009). The molecular basis and functional implications of chemokine interactions with heparan sulfate. Current Opinion in Structural Biology, 19, 543–548.
Hayashida, K., Parks, W. C., & Park, P. W. (2009). Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered CXC chemokines. Blood, 114, 3033–3043.
Hoogewerf, A. J., Kuschert, G. S., Proudfoot, A. E., Borlat, F., Clark-Lewis, I., Power, C. A., & Wells, T. N. (1997). Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry, 36, 13570–13578.
Stoler-Barak, L., Moussion, C., Shezen, E., Hatzav, M., Sixt, M., & Alon, R. (2014, January 22). Blood vessels pattern heparan sulfate gradients between their apical and basolateral aspects. PLoS One, 9(1), e85699.
Wang, L., Fuster, M., Sriramarao, P., & Esko, J. D. (2005). Endothelial heparan sulfate deficiency impairs L-selectin- and chemokinemediated neutrophil trafficking during inflammatory responses. Nature Immunology, 6, 902–910.
Massena, S., Christoffersson, G., Hjertstrom, E., Zcharia, E., Vlodavsky, I., Ausmees, N., Rolny, C., Li, J. P., & Phillipson, M. (2010). A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood, 116, 1924–1931.
Götte, M., Joussen, A. M., Klein, C., Andre, P., Wagner, D. D., Hinkes, M. T., Kirchhof, B., Adamis, A. P., & Bernfield, M. (2002, April). Role of syndecan-1 in leukocyte-endothelial interactions in the ocular vasculature. Investigative Ophthalmology & Visual Science, 43(4), 1135–1141.
Götte, M., Bernfield, M., & Joussen, A. M. (2005, June). Increased leukocyte-endothelial interactions in syndecan-1-deficient mice involve heparan sulfate-dependent and -independent steps. Current Eye Research, 30(6), 417–422.
Floer, M., Götte, M., Wild, M. K., Heidemann, J., Gassar, E. S., Domschke, W., Kiesel, L., Luegering, A., & Kucharzik, T. (2010, January). Enoxaparin improves the course of dextran sodium sulfate-induced colitis in syndecan-1-deficient mice. The American Journal of Pathology, 176(1), 146–157.
Kharabi Masouleh, B., Ten Dam, G. B., Wild, M. K., Seelige, R., van der Vlag, J., Rops, A. L., Echtermeyer, F. G., Vestweber, D., van Kuppevelt, T. H., Kiesel, L., & Götte, M. (2009, April 15). Role of the heparan sulfate proteoglycan syndecan-1 (CD138) in delayed-type hypersensitivity. Journal of Immunology, 182(8), 4985–4993.
Lever, R., Rose, M. J., McKenzie, E. A., & Page, C. P. (2014, June 15). Heparanase induces inflammatory cell recruitment in vivo by promoting adhesion to vascular endothelium. American Journal of Physiology. Cell Physiology, 306(12), C1184–C1190.
Stoler-Barak, L., Petrovich, E., Aychek, T., Gurevich, I., Tal, O., Hatzav, M., Ilan, N., Feigelson, S. W., Shakhar, G., Vlodavsky, I., & Alon, R. (2015). Heparanase of murine effector lymphocytes and neutrophils is not required for their diapedesis into sites of inflammation. FASEB Journal, 29, 2010–2021.
Schmidt, E. P., Yang, Y., Janssen, W. J., Gandjeva, A., Perez, M. J., Barthel, L., Zemans, R. L., Bowman, J. C., Koyanagi, D. E., Yunt, Z. X., Smith, L. P., Cheng, S. S., Overdier, K. H., Thompson, K. R., Geraci, M. W., Douglas, I. S., Pearse, D. B., & Tuder, R. M. (2012). The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nature Medicine, 18, 1217–1223.
Chen, X., Jiang, W., Yue, C., Zhang, W., Tong, C., Dai, D., Cheng, B., Huang, C., & Lu, L. (2017, September 16). Heparanase contributes to trans-endothelial migration of hepatocellular carcinoma cells. Journal of Cancer, 8(16), 3309–3317.
Grabbe, S., & Schwarz, T. (1998). Immunoregulatory mechanisms involved in elicitation of allergic contact hypersensitivity. Immunology Today, 19, 37–44.
Seidler, D. G., Mohamed, N. A., Bocian, C., Stadtmann, A., Hermann, S., Schäfers, K., Schäfers, M., Iozzo, R. V., Zarbock, A., & Götte, M. (2011, December 1). The role for decorin in delayed-type hypersensitivity. Journal of Immunology, 187(11), 6108–6119.
Averbeck, M., Kuhn, S., Bühligen, J., Götte, M., Simon, J. C., & Polte, T. (2017, November). Syndecan-1 regulates dendritic cell migration in cutaneous hypersensitivity to haptens. Experimental Dermatology, 26(11), 1060–1067.
Edovitsky, E., Lerner, I., Zcharia, E., Peretz, T., Vlodavsky, I., & Elkin, M. (2006, May 1). Role of endothelial heparanase in delayed-type hypersensitivity. Blood, 107(9), 3609–3616.
Reynolds, J. (2011, June). Strain differences and the genetic basis of experimental autoimmune anti-glomerular basement membrane glomerulonephritis. International Journal of Experimental Pathology, 92(3), 211–217.
Rops, A. L., Götte, M., Baselmans, M. H., van den Hoven, M. J., Steenbergen, E. J., Lensen, J. F., Wijnhoven, T. J., Cevikbas, F., van den Heuvel, L. P., van Kuppevelt, T. H., Berden, J. H., & van der Vlag, J. (2007, November). Syndecan-1 deficiency aggravates anti-glomerular basement membrane nephritis. Kidney International, 72(10), 1204–1215.
Garsen, M., Benner, M., Dijkman, H. B., van Kuppevelt, T. H., Li, J. P., Rabelink, T. J., Vlodavsky, I., Berden, J. H., Rops, A. L., Elkin, M., & van der Vlag, J. (2016, April). Heparanase is essential for the development of acute experimental glomerulonephritis. The American Journal of Pathology, 186(4), 805–815.
Begolka, W. S., Vanderlugt, C. L., Rahbe, S. M., & Miller, S. D. (1998). Differential expression of inflammatory cytokines parallels progression of central nervous system pathology in two clinically distinct models of multiple sclerosis. Journal of Immunology, 161, 4437–4446.
Hemmer, B., Archelos, J. J., & Hartung, H. P. (2002). New concepts in the immunopathogenesis of multiple sclerosis. Nature Reviews. Neuroscience, 3, 291–301.
Zhang, X., Wu, C., Song, J., Götte, M., & Sorokin, L. (2013, November 1). Syndecan-1, a cell surface proteoglycan, negatively regulates initial leukocyte recruitment to the brain across the choroid plexus in murine experimental autoimmune encephalomyelitis. Journal of Immunology, 191(9), 4551–4561.
Bitan, M., Weiss, L., Reibstein, I., Zeira, M., Fellig, Y., Slavin, S., Zcharia, E., Nagler, A., & Vlodavsky, I. (2010, June). Heparanase upregulates Th2 cytokines, ameliorating experimental autoimmune encephalitis. Molecular Immunology, 47(10), 1890–1898.
Blander, J. M. (2016, July). Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. The FEBS Journal, 283(14), 2720–2730.
Eichele, D. D., & Kharbanda, K. K. (2017, September 7). Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World Journal of Gastroenterology, 23(33), 6016–6029.
Day, R., Ilyas, M., Daszak, P., Talbot, I., & Forbes, A. (1999, December). Expression of syndecan-1 in inflammatory bowel disease and a possible mechanism of heparin therapy. Digestive Diseases and Sciences, 44(12), 2508–2515.
Lopez, A., Pouillon, L., Beaugerie, L., Danese, S., & Peyrin-Biroulet, L. (2018 February – April). Colorectal cancer prevention in patients with ulcerative colitis. Best Practice & Research Clinical Gastroenterology, 32–33, 103–109.
Pap, Z., Pávai, Z., Dénes, L., Kovalszky, I., & Jung, J. (2009, December). An immunohistochemical study of colon adenomas and carcinomas: E-cadherin, Syndecan-1, Ets-1. Pathology Oncology Research, 15(4), 579–587.
Peretti, T., Waisberg, J., Mader, A. M., de Matos, L. L., da Costa, R. B., Conceição, G. M., Lopes, A. C., Nader, H. B., & Pinhal, M. A. (2008, August). Heparanase-2, syndecan-1, and extracellular matrix remodeling in colorectal carcinoma. European Journal of Gastroenterology & Hepatology, 20(8), 756–765.
Snider, A. J., Bialkowska, A. B., Ghaleb, A. M., Yang, V. W., Obeid, L. M., & Hannun, Y. A. (2016). Murine model for colitis-associated Cancer of the Colon. Methods in Molecular Biology, 1438, 245–254.
Binder Gallimidi, A., Nussbaum, G., Hermano, E., Weizman, B., Meirovitz, A., Vlodavsky, I., Götte, M., Elkin, M. (2017, March 28). Syndecan-1 deficiency promotes tumor growth in a murine model of colitis-induced colon carcinoma. PLoS One; 12(3):e0174343. https://doi.org/10.1371/journal.pone.0174343. eCollection 2017.
Waterman, M., Ben-Izhak, O., Eliakim, R., Groisman, G., Vlodavsky, I., & Ilan, N. (2007, January). Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Modern Pathology, 20(1), 8–14.
Friedmann, Y., Vlodavsky, I., Aingorn, H., Aviv, A., Peretz, T., Pecker, I., & Pappo, O. (2000, October). Expression of heparanase in normal, dysplastic, and neoplastic human colonic mucosa and stroma. Evidence for its role in colonic tumorigenesis. The American Journal of Pathology, 157(4), 1167–1175.
Lerner, I., Hermano, E., Zcharia, E., Rodkin, D., Bulvik, R., Doviner, V., Rubinstein, A. M., Ishai-Michaeli, R., Atzmon, R., Sherman, Y., Meirovitz, A., Peretz, T., Vlodavsky, I., & Elkin, M. (2011, May). Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. The Journal of Clinical Investigation, 121(5), 1709–1721.
Hanahan, D., & Weinberg, R. A. (2000, January 7). The hallmarks of cancer. Cell, 100(1), 57–70.
Hanahan, D., & Weinberg, R. A. (2011, March 4). Hallmarks of cancer: The next generation. Cell, 144(5), 646–674.
Espinoza-Sánchez, N. A., & Götte, M. (2019, July 20) Role of cell surface proteoglycans in cancer immunotherapy. Seminars in Cancer Biology. pii: S1044-579X(19)30041–0. https://doi.org/10.1016/j.semcancer.2019.07.012. [Epub ahead of print] Review.PMID:31336150.
Ibrahim, S. A., Hassan, H., & Götte, M. (2014, November). MicroRNA regulation of proteoglycan function in cancer. The FEBS Journal, 281(22), 5009–5022.
Kato, M., Wang, H., Kainulainen, V., Fitzgerald, M. L., Ledbetter, S., Ornitz, D. M., & Bernfield, M. (1998, June). Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nature Medicine, 4(6), 691–697.
Vlodavsky, I., Gross-Cohen, M., Weissmann, M., Ilan, N., & Sanderson, R. D. (2018, January). Opposing functions of Heparanase-1 and Heparanase-2 in Cancer progression. Trends in Biochemical Sciences, 43(1), 18–31.
Regős, E., Abdelfattah, H. H., Reszegi, A., Szilák, L., Werling, K., Szabó, G., Kiss, A., Schaff, Z., Kovalszky, I., & Baghy, K. (2018, August). Syndecan-1 inhibits early stages of liver fibrogenesis by interfering with TGFβ1 action and upregulating MMP14. Matrix Biology, 68–69, 474–489.
Zeng, Y., Yao, X., Chen, L., Yan, Z., Liu, J., Zhang, Y., Feng, T., Wu, J., & Liu, X. (2016, September 27). Sphingosine-1-phosphate induced epithelial-mesenchymal transition of hepatocellular carcinoma via an MMP-7/ syndecan-1/TGF-β autocrine loop. Oncotarget, 7(39), 63324–63337.
Vitale, D., Kumar Katakam, S., Greve, B., Jang, B., Oh, E. S., Alaniz, L., & Götte, M. (2019, August). Proteoglycans and glycosaminoglycans as regulators of cancer stem cell function and therapeutic resistance. The FEBS Journal, 286(15), 2870–2882.
McDermott, S. P., Ranheim, E. A., Leatherberry, V. S., Khwaja, S. S., Klos, K. S., & Alexander, C. M. (2007, March 1). Juvenile syndecan-1 null mice are protected from carcinogen-induced tumor development. Oncogene, 26(10), 1407–1416.
Liu, B. Y., McDermott, S. P., Khwaja, S. S., & Alexander, C. M. (2004, March 23). The transforming activity of Wnt effectors correlates with their ability to induce the accumulation of mammary progenitor cells. Proceedings of the National Academy of Sciences of the United States of America, 101(12), 4158–4163.
Ibrahim, S. A., Hassan, H., Vilardo, L., Kumar, S. K., Kumar, A. V., Kelsch, R., Schneider, C., Kiesel, L., Eich, H. T., Zucchi, I., Reinbold, R., Greve, B., & Götte, M. (2013, December 31). Syndecan-1 (CD138) modulates triple-negative breast cancer stem cell properties via regulation of LRP-6 and IL-6-mediated STAT3 signaling. PLoS One, 8(12), e85737.
Ibrahim, S. A., Gadalla, R., El-Ghonaimy, E. A., Samir, O., Mohamed, H. T., Hassan, H., Greve, B., El-Shinawi, M., Mohamed, M. M., & Götte, M. (2017, March 7). Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, notch and EGFR signaling pathways. Molecular Cancer, 16(1), 57.
Ibrahim, S. A., Yip, G. W., Stock, C., Pan, J. W., Neubauer, C., Poeter, M., Pupjalis, D., Koo, C. Y., Kelsch, R., Schüle, R., Rescher, U., Kiesel, L., & Götte, M. (2012, September 15). Targeting of syndecan-1 by microRNA miR-10b promotes breast cancer cell motility and invasiveness via a rho-GTPase- and E-cadherin-dependent mechanism. International Journal of Cancer, 131(6), E884–E896.
Nobuhisa, T., Naomoto, Y., Takaoka, M., Tabuchi, Y., Ookawa, K., Kitamoto, D., Gunduz, E., Gunduz, M., Nagatsuka, H., Haisa, M., Matsuoka, J., Nakajima, M., & Tanaka, N. (2005, May 27). Emergence of nuclear heparanase induces differentiation of human mammary cancer cells. Biochemical and Biophysical Research Communications, 331(1), 175–180.
Spiegel, A., Zcharia, E., Vagima, Y., Itkin, T., Kalinkovich, A., Dar, A., Kollet, O., Netzer, N., Golan, K., Shafat, I., Ilan, N., Nagler, A., Vlodavsky, I., & Lapidot, T. (2008, May 15). Heparanase regulates retention and proliferation of primitive Sca-1+/c-kit+/Lin- cells via modulation of the bone marrow microenvironment. Blood, 111(10):4934–4943. https://doi.org/10.1182/blood-2007-10-116145. Epub 2008 Mar 11.
Cheng, C. C., Lee, Y. H., Lin, S. P., Huangfu, W. C., & Liu, I. H. (2014, March 13). Cell-autonomous heparanase modulates self-renewal and migration in bone marrow-derived mesenchymal stem cells. Journal of Biomedical Science, 21, 21.
Ruan, J., Trotter, T. N., Nan, L., Luo, R., Javed, A., Sanderson, R. D., Suva, L. J., & Yang, Y. (2013, November). Heparanase inhibits osteoblastogenesis and shifts bone marrow progenitor cell fate in myeloma bone disease. Bone, 57(1):10–17. https://doi.org/10.1016/j.bone.2013.07.024. Epub 2013 Jul 27.
Xiong, A., Kundu, S., Forsberg, M., Xiong, Y., Bergström, T., Paavilainen, T., Kjellén, L., Li, J. P., & Forsberg-Nilsson, K. (2017, October). Heparanase confers a growth advantage to differentiating murine embryonic stem cells, and enhances oligodendrocyte formation. Matrix Biology, 62, 92–104.
Choi, D. S., Kim, J. H., Ryu, H. S., Kim, H. C., Han, J. H., Lee, J. S., & Min, C. K. (2007, August 15). Syndecan-1, a key regulator of cell viability in endometrial cancer. International Journal of Cancer, 121(4), 741–750.
Sun, H., Hu, Y., Gu, Z., Owens, R. T., Chen, Y. Q., & Edwards, I. J. (2011, October). Omega-3 fatty acids induce apoptosis in human breast cancer cells and mouse mammary tissue through syndecan-1 inhibition of the MEK-Erk pathway. Carcinogenesis, 32(10), 1518–1524.
Hu, Y., Sun, H., O'Flaherty, J. T., & Edwards, I. J. (2013, January). 15-Lipoxygenase-1-mediated metabolism of docosahexaenoic acid is required for syndecan-1 signaling and apoptosis in prostate cancer cells. Carcinogenesis, 34(1), 176–182.
Ikeguchi, M., Hirooka, Y., & Kaibara, N. (2003, January). Heparanase gene expression and its correlation with spontaneous apoptosis in hepatocytes of cirrhotic liver and carcinoma. European Journal of Cancer, 39(1), 86–90.
Rubinfeld, H., Cohen-Kaplan, V., Nass, D., Ilan, N., Meisel, S., Cohen, Z. R., Hadani, M., Vlodavsky, I., & Shimon, I. (2011, December). Heparanase is highly expressed and regulates proliferation in GH-secreting pituitary tumor cells. Endocrinology, 152(12), 4562–4570.
Cohen, I., Pappo, O., Elkin, M., San, T., Bar-Shavit, R., Hazan, R., Peretz, T., Vlodavsky, I., & Abramovitch, R. (2006, April 1). Heparanase promotes growth, angiogenesis and survival of primary breast tumors. International Journal of Cancer, 118(7), 1609–1617.
Zeng, C., Ke, Z. F., Luo, W. R., Yao, Y. H., Hu, X. R., Jie, W., Yin, J. B., & Sun, S. J. (2013, March). Heparanase overexpression participates in tumor growth of cervical cancer in vitro and in vivo. Medical Oncology, 30(1), 403.
Folkman, J. (2007, April). Angiogenesis: An organizing principle for drug discovery? Nature Reviews. Drug Discovery, 6(4), 273–286.
Götte, M., Kersting, C., Radke, I., Kiesel, L., & Wülfing, P. (2007). An expression signature of syndecan-1 (CD138), E-cadherin and c-met is associated with factors of angiogenesis and lymphangiogenesis in ductal breast carcinoma in situ. Breast Cancer Research, 9(1), R8.
Maeda, T., Desouky, J., & Friedl, A. (2006, March 2). Syndecan-1 expression by stromal fibroblasts promotes breast carcinoma growth in vivo and stimulates tumor angiogenesis. Oncogene, 25(9), 1408–1412.
Yu, S., Lv, H., Zhang, H., Jiang, Y., Hong, Y., Xia, R., Zhang, Q., Ju, W., Jiang, L., Ou, G., Zhang, J., Wang, S., & Zhang, J. (2017, April 1). Heparanase-1-induced shedding of heparan sulfate from syndecan-1 in hepatocarcinoma cell facilitates lymphatic endothelial cell proliferation via VEGF-C/ERK pathway. Biochemical and Biophysical Research Communications, 485(2), 432–439.
Asuthkar, S., Velpula, K. K., Nalla, A. K., Gogineni, V. R., Gondi, C. S., & Rao, J. S. (2014, April 10). Irradiation-induced angiogenesis is associated with an MMP-9-miR-494-syndecan-1 regulatory loop in medulloblastoma cells. Oncogene, 33(15), 1922–1933.
Ilan, N., Elkin, M., & Vlodavsky, I. (2006). Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. The International Journal of Biochemistry & Cell Biology, 38(12), 2018–2039.
Jiang, W. G., Sanders, A. J., Katoh, M., Ungefroren, H., Gieseler, F., Prince, M., Thompson, S. K., Zollo, M., Spano, D., Dhawan, P., Sliva, D., Subbarayan, P. R., Sarkar, M., Honoki, K., Fujii, H., Georgakilas, A. G., Amedei, A., Niccolai, E., Amin, A., Ashraf, S. S., Ye, L., Helferich, W. G., Yang, X., Boosani, C. S., Guha, G., Ciriolo, M. R., Aquilano, K., Chen, S., Azmi, A. S., Keith, W. N., Bilsland, A., Bhakta, D., Halicka, D., Nowsheen, S., Pantano, F., & Santini, D. (2015, December). Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Seminars in Cancer Biology, 35(Suppl), S244–S275.
Fujii, T., Shimada, K., Tatsumi, Y., Tanaka, N., Fujimoto, K., & Konishi, N. (2016, September). Syndecan-1 up-regulates microRNA-331-3p and mediates epithelial-to-mesenchymal transition in prostate cancer. Molecular Carcinogenesis, 55(9), 1378–1386.
Hassan, H., Greve, B., Pavao, M. S., Kiesel, L., Ibrahim, S. A., & Götte, M. (2013, May). Syndecan-1 modulates β-integrin-dependent and interleukin-6-dependent functions in breast cancer cell adhesion, migration, and resistance to irradiation. The FEBS Journal, 280(10), 2216–2227.
Pasqualon, T., Lue, H., Groening, S., Pruessmeyer, J., Jahr, H., Denecke, B., Bernhagen, J., & Ludwig, A. (2016, April). Cell surface syndecan-1 contributes to binding and function of macrophage migration inhibitory factor (MIF) on epithelial tumor cells. Biochimica et Biophysica Acta, 1863(4), 717–726.
Shteingauz, A., Ilan, N., & Vlodavsky, I. (2014, November). Processing of heparanase is mediated by syndecan-1 cytoplasmic domain and involves syntenin and α-actinin. Cellular and Molecular Life Sciences, 71(22), 4457–4470.
Reiland, J., Sanderson, R. D., Waguespack, M., Barker, S. A., Long, R., Carson, D. D., & Marchetti, D. (2004, February 27). Heparanase degrades syndecan-1 and perlecan heparan sulfate: Functional implications for tumor cell invasion. The Journal of Biological Chemistry, 279(9):8047–8055. Epub 2003 Nov 20.
Barash, U., Cohen-Kaplan, V., Dowek, I., Sanderson, R. D., Ilan, N., & Vlodavsky, I. (2010, October). Proteoglycans in health and disease: New concepts for heparanase function in tumor progression and metastasis. The FEBS Journal, 277(19), 3890–3903.
Yang, Y., Macleod, V., Bendre, M., Huang, Y., Theus, A. M., Miao, H. Q., Kussie, P., Yaccoby, S., Epstein, J., Suva, L. J., Kelly, T., & Sanderson, R. D. (2005, February 1). Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood, 105(3), 1303–1309.
Edovitsky, E., Elkin, M., Zcharia, E., Peretz, T., & Vlodavsky, I. (2004, August 18). Heparanase gene silencing, tumor invasiveness, angiogenesis, and metastasis. Journal of the National Cancer Institute, 96(16), 1219–1230.
Roy, M., & Marchetti, D. (2009, February 1). Cell surface heparan sulfate released by heparanase promotes melanoma cell migration and angiogenesis. Journal of Cellular Biochemistry, 106(2), 200–209.
Götte, M., & Yip, G. W. (2006, November 1). Heparanase, hyaluronan, and CD44 in cancers: A breast carcinoma perspective. Cancer Research, 66(21), 10233–10237.
Bandari, S. K., Purushothaman, A., Ramani, V. C., Brinkley, G. J., Chandrashekar, D. S., Varambally, S., Mobley, J. A., Zhang, Y., Brown, E. E., Vlodavsky, I., & Sanderson, R. D. (2018, January). Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biology, 65, 104–118.
Yip, G. W., Smollich, M., & Götte, M. (2006, September). Therapeutic value of glycosaminoglycans in cancer. Molecular Cancer Therapeutics, 5(9), 2139–2148.
Schönfeld, K., Herbener, P., Zuber, C., Häder, T., Bernöster, K., Uherek, C., & Schüttrumpf, J. (2018, April 17). Activity of Indatuximab Ravtansine against triple-negative breast Cancer in preclinical tumor models. Pharmaceutical Research, 35(6), 118.
Jagannath, S., Chanan-Khan, A., Heffner, L. T., Avigan, D., Zimmerman, T. M., Lonial, S., Lutz, R. J., Engling, A., Uherek, C., Osterroth, F., Ruehle, M., Beelitz, M. A., Niemann, G., Wartenberg-Demand, A., Haeder, T., Anderson, K. C., & Munshi, N. C. (2011). BT062, An antibody-drug conjugate directed against CD138, shows clinical activity in patients with relapsed or relapsed/refractory multiple Myeloma. Blood, 118(21), 305–305.
Zhao, S., Han, Z., Ji, C., An, G., Meng, H., & Yang, L. (2018). The research significance of concomitant use of CAR-CD138-NK and CAR-CD19-NK to target multiple myelomas. European Journal of Inflammation, 16, 2058739218788968.
Guo, B., Chen, M., Han, Q., Hui, F., Dai, H., Zhang, W., Zhang, Y., Wang, Y., Zhu, H., & Han, W. (2016). CD138-directed adoptive immunotherapy of Chimeric Antigen Receptor (CAR)-modified T cells for multiple myeloma. Journal of Cellular Immunotherapy, 2(1), 28–35.
Mohan, C. D., Hari, S., Preetham, H. D., Rangappa, S., Barash, U., Ilan, N., Nayak, S. C., Gupta, V. K., Basappa, V. I., & Rangappa, K. S. (2019, May 31). Targeting heparanase in cancer: Inhibition by synthetic, chemically modified, and natural compounds. iScience, 15, 360–390.
Weissmann, M., Arvatz, G., Horowitz, N., Feld, S., Naroditsky, I., Zhang, Y., Ng, M., Hammond, E., Nevo, E., Vlodavsky, I., & Ilan, N. (2016, January 19). Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America, 113(3), 704–709.
Cassinelli, G., Favini, E., Dal Bo, L., Tortoreto, M., De Maglie, M., Dagrada, G., Pilotti, S., Zunino, F., Zaffaroni, N., & Lanzi, C. (2016, July 26). Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases. Oncotarget, 7(30), 47848–47863.
Liu, C. J., Lee, P. H., Lin, D. Y., Wu, C. C., Jeng, L. B., Lin, P. W., Mok, K. T., Lee, W. C., Yeh, H. Z., Ho, M. C., Yang, S. S., Lee, C. C., Yu, M. C., Hu, R. H., Peng, C. Y., Lai, K. L., Chang, S. S., & Chen, P. J. (2009, May). Heparanase inhibitor PI-88 as adjuvant therapy for hepatocellular carcinoma after curative resection: A randomized phase II trial for safety and optimal dosage. Journal of Hepatology, 50(5), 958–968.
Liu, C. J., Chang, J., Lee, P. H., Lin, D. Y., Wu, C. C., Jeng, L. B., Lin, Y. J., Mok, K. T., Lee, W. C., Yeh, H. Z., Ho, M. C., Yang, S. S., Yang, M. D., Yu, M. C., Hu, R. H., Peng, C. Y., Lai, K. L., Chang, S. S., & Chen, P. J. (2014, August 28). Adjuvant heparanase inhibitor PI-88 therapy for hepatocellular carcinoma recurrence. World Journal of Gastroenterology, 20(32), 11384–11393.
Galli, M., Chatterjee, M., Grasso, M., Specchia, G., Magen, H., Einsele, H., Celeghini, I., Barbieri, P., Paoletti, D., Pace, S., Sanderson, R. D., Rambaldi, A., & Nagler, A. (2018, October). Phase I study of the heparanase inhibitor roneparstat: An innovative approach for ultiple myeloma therapy. Haematologica, 103(10), e469–e472.
Kim, J. H., & Park, J. (2014, September). Prognostic significance of heme oxygenase-1, S100 calcium-binding protein A4, and syndecan-1 expression in primary non-muscle-invasive bladder cancer. Human Pathology, 45(9), 1830–1838.
Szarvas, T., Reis, H., Kramer, G., Shariat, S. F., Vom Dorp, F., Tschirdewahn, S., et al. (2014, April). Enhanced stromal syndecan-1 expression is an independent risk factor for poor survival in bladder cancer. Human Pathology, 45(4), 674–682.
Gohji, K., Okamoto, M., Kitazawa, S., Toyoshima, M., Dong, J., Katsuoka, Y., et al. (2001, October). Heparanase protein and gene expression in bladder cancer. The Journal of Urology, 166(4), 1286–1290.
Shafat, I., Pode, D., Peretz, T., Ilan, N., Vlodavsky, I., & Nisman, B. (2008, February). Clinical significance of urine heparanase in bladder cancer progression. Neoplasia N Y N., 10(2), 125–130.
Loussouarn, D., Campion, L., Sagan, C., Frenel, J.-S., Dravet, F., Classe, J.-M., et al. (2008, June 17). Prognostic impact of syndecan-1 expression in invasive ductal breast carcinomas. British Journal of Cancer, 98(12), 1993–1998.
Nguyen, T. L., Grizzle, W. E., Zhang, K., Hameed, O., Siegal, G. P., & Wei, S. (2013, October). Syndecan-1 overexpression is associated with nonluminal subtypes and poor prognosis in advanced breast cancer. American Journal of Clinical Pathology, 140(4), 468–474.
Leivonen, M., Lundin, J., Nordling, S., von Boguslawski, K., & Haglund, C. (2004). Prognostic value of syndecan-1 expression in breast cancer. Oncology, 67(1), 11–18.
Barbareschi, M., Maisonneuve, P., Aldovini, D., Cangi, M. G., Pecciarini, L., Angelo Mauri, F., Veronese, S., Caffo, O., Lucenti, A., Palma, P. D., Galligioni, E., & Doglioni, C. (2003, August 1). High syndecan-1 expression in breast carcinoma is related to an aggressive phenotype and to poorer prognosis. Cancer, 98(3), 474–483.
Baba, F., Swartz, K., van Buren, R., Eickhoff, J., Zhang, Y., Wolberg, W., et al. (2006, July). Syndecan-1 and syndecan-4 are overexpressed in an estrogen receptor-negative, highly proliferative breast carcinoma subtype. Breast Cancer Research and Treatment, 98(1), 91–98.
Vornicova, O., Naroditsky, I., Boyango, I., Shachar, S. S., Mashiach, T., Ilan, N., et al. (2017, December 21). Prognostic significance of heparanase expression in primary and metastatic breast carcinoma. Oncotarget, 9(5), 6238–6244.
Sun, X., Zhang, G., Nian, J., Yu, M., Chen, S., Zhang, Y., et al. (2017, June 27). Elevated heparanase expression is associated with poor prognosis in breast cancer: A study based on systematic review and TCGA data. Oncotarget, 8(26), 43521–43535.
Hashimoto, Y., Skacel, M., & Adams, J. C. (2008, June 30). Association of loss of epithelial syndecan-1 with stage and local metastasis of colorectal adenocarcinomas: An immunohistochemical study of clinically annotated tumors. BMC Cancer, 8, 185.
Kim, S. Y., Choi, E. J., Yun, J. A., Jung, E. S., Oh, S. T., Kim, J. G., et al. (2015). Syndecan-1 expression is associated with tumor size and EGFR expression in colorectal carcinoma: A clinicopathological study of 230 cases. International Journal of Medical Sciences, 12(2), 92–99.
Lundin, M., Nordling, S., Lundin, J., Isola, J., Wiksten, J.-P., & Haglund, C. (2005). Epithelial syndecan-1 expression is associated with stage and grade in colorectal cancer. Oncology, 68(4–6), 306–313.
Nobuhisa, T., Naomoto, Y., Ohkawa, T., Takaoka, M., Ono, R., Murata, T., et al. (2005, April). Heparanase expression correlates with malignant potential in human colon cancer. Journal of Cancer Research and Clinical Oncology, 131(4), 229–237.
Roh, Y. H., Kim, Y. H., Choi, H. J., Lee, K. E., & Roh, M. S. (2008). Syndecan-1 expression in gallbladder cancer and its prognostic significance. European Surgical Research, 41(2), 245–250.
Wu, W., Pan, C., Yu, H., Gong, H., & Wang, Y. (2008, March). Heparanase expression in gallbladder carcinoma and its correlation to prognosis. Journal of Gastroenterology and Hepatology, 23(3), 491–497.
Wiksten, J.-P., Lundin, J., Nordling, S., Kokkola, A., & Haglund, C. (2008, August). Comparison of the prognostic value of a panel of tissue tumor markers and established clinicopathological factors in patients with gastric cancer. Anticancer Research, 28(4C), 2279–2287.
Wiksten, J. P., Lundin, J., Nordling, S., Lundin, M., Kokkola, A., von Boguslawski, K., et al. (2001, January 20). Epithelial and stromal syndecan-1 expression as predictor of outcome in patients with gastric cancer. International Journal of Cancer, 95(1), 1–6.
Takaoka, M., Naomoto, Y., Ohkawa, T., Uetsuka, H., Shirakawa, Y., Uno, F., et al. (2003, May). Heparanase expression correlates with invasion and poor prognosis in gastric cancers. Laboratory Investigation; A Journal of Technical Methods and Pathology, 83(5), 613–622.
Inki, P., Joensuu, H., Grénman, R., Klemi, P., & Jalkanen, M. (1994, August). Association between syndecan-1 expression and clinical outcome in squamous cell carcinoma of the head and neck. British Journal of Cancer, 70(2), 319–323.
Anttonen, A., Kajanti, M., Heikkilä, P., Jalkanen, M., & Joensuu, H. (1999, February). Syndecan-1 expression has prognostic significance in head and neck carcinoma. British Journal of Cancer, 79(3–4), 558–564.
Mukunyadzi, P., Liu, K., Hanna, E. Y., Suen, J. Y., & Fan, C.-Y. (2003, August). Induced expression of syndecan-1 in the stroma of head and neck squamous cell carcinoma. Modern Pathology, 16(8), 796–801.
Beckhove, P., Helmke, B. M., Ziouta, Y., Bucur, M., Dörner, W., Mogler, C., et al. (2005, April 15). Heparanase expression at the invasion front of human head and neck cancers and correlation with poor prognosis. Clinical Cancer Research, 11(8), 2899–2906.
Gökden, N., Greene, G. F., Bayer-Garner, I. B., Spencer, H. J., Sanderson, R. D., & Gökden, M. (2006, June). Expression of CD138 (Syndecan-1) in renal cell carcinoma is reduced with increasing nuclear grade. Applied Immunohistochemistry & Molecular Morphology, 14(2), 173–177.
Mikami, S., Oya, M., Shimoda, M., Mizuno, R., Ishida, M., Kosaka, T., et al. (2008, October 1). Expression of heparanase in renal cell carcinomas: Implications for tumor invasion and prognosis. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 14(19), 6055–6061.
Ren, J., Liu, H., Yan, L., Tian, S., Li, D., & Xu, Z. (2011, December 2). Microvessel density and heparanase over-expression in clear cell renal cell cancer: Correlations and prognostic significances. World Journal of Surgical Oncology, 9, 158.
Harada, K., Masuda, S., Hirano, M., & Nakanuma, Y. (2003, September). Reduced expression of syndecan-1 correlates with histologic dedifferentiation, lymph node metastasis, and poor prognosis in intrahepatic cholangiocarcinoma. Human Pathology, 34(9), 857–863.
Nault, J.-C., Guyot, E., Laguillier, C., Chevret, S., Ganne-Carrie, N., N’Kontchou, G., et al. (2013, August). Serum proteoglycans as prognostic biomarkers of hepatocellular carcinoma in patients with alcoholic cirrhosis. Cancer Epidemiology, Biomarkers & Prevention : A Publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 22(8), 1343–1352.
El-Assal, O. N., Yamanoi, A., Ono, T., Kohno, H., & Nagasue, N. (2001, May). The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 7(5), 1299–1305.
Anttonen, A., Heikkilä, P., Kajanti, M., Jalkanen, M., & Joensuu, H. (2001, June). High syndecan-1 expression is associated with favourable outcome in squamous cell lung carcinoma treated with radical surgery. Lung Cancer, 32(3), 297–305.
Joensuu, H., Anttonen, A., Eriksson, M., Mäkitaro, R., Alfthan, H., Kinnula, V., et al. (2002, September 15). Soluble syndecan-1 and serum basic fibroblast growth factor are new prognostic factors in lung cancer. Cancer Research, 62(18), 5210–5217.
Anttonen, A., Leppä, S., Ruotsalainen, T., Alfthan, H., Mattson, K., & Joensuu, H. (2003, August). Pretreatment serum syndecan-1 levels and outcome in small cell lung cancer patients treated with platinum-based chemotherapy. Lung Cancer, 41(2), 171–177.
Cohen, E., Doweck, I., Naroditsky, I., Ben-Izhak, O., Kremer, R., Best, L. A., et al. (2008, September 1). Heparanase is overexpressed in lung cancer and correlates inversely with patient survival. Cancer, 113(5), 1004–1011.
Fernandes dos Santos, T. C., Gomes, A. M., Paschoal, M. E. M., Stelling, M. P., Rumjanek, V. M. B. D., Junior A do R, et al. (2014, August). Heparanase expression and localization in different types of human lung cancer. Biochimica et Biophysica Acta, 1840(8), 2599–2608.
Neagu, M., Constantin, C., & Zurac, S. (2013). Immune parameters in the prognosis and therapy monitoring of cutaneous melanoma patients: Experience, role, and limitations. BioMed Research International, 2013, 107940.
Wang, X., Wen, W., Wu, H., Chen, Y., Ren, G., & Guo, W. (2013, June 21). Heparanase expression correlates with poor survival in oral mucosal melanoma. Medical Oncology, 30(3), 633.
Xu, S., Yang, Z., Zhang, J., Jiang, Y., Chen, Y., Li, H., et al. (2014, October 24). Increased levels of β-catenin, LEF-1, and HPA-1 correlate with poor prognosis for Acral melanoma with negative BRAF and NRAS mutation in BRAF exons 11 and 15 and NRAS exons 1 and 2. DNA and Cell Biology, 34(1), 69–77.
Vornicova, O., Boyango, I., Feld, S., Naroditsky, I., Kazarin, O., Zohar, Y., et al. (2016, October 6). The prognostic significance of heparanase expression in metastatic melanoma. Oncotarget, 7(46), 74678–74685.
Kurokawa, H., Zhang, M., Matsumoto, S., Yamashita, Y., Tanaka, T., Takamori, K., et al. (2006, May). Reduced syndecan-1 expression is correlated with the histological grade of malignancy at the deep invasive front in oral squamous cell carcinoma. Journal of Oral Pathology and Medicine, 35(5), 301–306.
Máthé, M., Suba, Z., Németh, Z., Tátrai, P., Füle, T., Borgulya, G., et al. (2006, May). Stromal syndecan-1 expression is an adverse prognostic factor in oral carcinomas. Oral Oncology, 42(5), 493–500.
Leiser, Y., Abu-El-Naaj, I., Sabo, E., Akrish, S., Ilan, N., Ben-Izhak, O., et al. (2011, June). Prognostic value of heparanase expression and cellular localization in oral cancer. Head & Neck, 33(6), 871–877.
Kusumoto, T., Kodama, J., Seki, N., Nakamura, K., Hongo, A., & Hiramatsu, Y. (2010, April). Clinical significance of syndecan-1 and versican expression in human epithelial ovarian cancer. Oncology Reports, 23(4), 917–925.
Davies, E. J., Blackhall, F. H., Shanks, J. H., David, G., McGown, A. T., Swindell, R., et al. (2004, August 1). Distribution and clinical significance of heparan sulfate proteoglycans in ovarian cancer. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 10(15), 5178–5186.
Davidson, B., Shafat, I., Risberg, B., & Ilan, N. (2007, February). Trope’ CG, Vlodavsky I, et al. Heparanase expression correlates with poor survival in metastatic ovarian carcinoma. Gynecologic Oncology, 104(2), 311–319.
Juuti, A., Nordling, S., Lundin, J., Louhimo, J., & Haglund, C. (2005). Syndecan-1 expression—a novel prognostic marker in pancreatic cancer. Oncology, 68(2–3), 97–106.
Rohloff, J., Zinke, J., Schoppmeyer, K., Tannapfel, A., Witzigmann, H., Mössner, J., et al. (2002, April 22). Heparanase expression is a prognostic indicator for postoperative survival in pancreatic adenocarcinoma. British Journal of Cancer, 86(8), 1270–1275.
Quiros, R. M., Rao, G., Plate, J., Harris, J. E., Brunn, G. J., Platt, J. L., Gattuso, P., Prinz, R. A., Xu, X. (2006). Elevated serum heparanase-1 levels in patients with pancreatic carcinoma are associated with poor survival. Cancer, 106(3), 532–40.
Acknowledgement
Work in the authors’ laboratory has been supported by EU H2020 RISE MSCA-2014 project grant number 645756 (GLYCANC) (to MG) and CNPq Sandwich Doctorate (SWE) grant number 290231/2017-5 – SWE (to FCOBT).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Teixeira, F.C.O.B., Götte, M. (2020). Involvement of Syndecan-1 and Heparanase in Cancer and Inflammation. In: Vlodavsky, I., Sanderson, R., Ilan, N. (eds) Heparanase. Advances in Experimental Medicine and Biology, vol 1221. Springer, Cham. https://doi.org/10.1007/978-3-030-34521-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-34521-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-34520-4
Online ISBN: 978-3-030-34521-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)