
Abstract
The introduction of multi-agent chemotherapy for the treatment of Hodgkin lymphoma is one of the major breakthroughs in clinical oncology. Treatment with multi-agent chemotherapy and improved radiation methods has significantly improved the chance of curing these patients from less than 5% in 1963 to about 80% at present. However, there is still a substantial need to improve current treatment approaches particularly for elderly patients or those with relapsed and refractory disease. Cured patients unfortunately are at high risk for late side effects including second malignancies, cardiac toxicity, infertility, and fatigue. Thus, there is a clear need for new and safer drugs that are more selective in targeting the malignant Hodgkin and Reed-Sternberg (HRS) cells in this disease while sparing normal tissues.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Hodgkin lymphoma
- Autologous stem cell transplantation
- Anaplastic large cell lymphoma
- Brentuximab vedotin
- John Cunningham virus
1 Introduction
The introduction of multi-agent chemotherapy for the treatment of Hodgkin lymphoma is one of the major breakthroughs in clinical oncology. Chemotherapy and improved radiation methods have significantly improved the chance of curing these patients from less than 5% in 1963 to about 80% at present [1,2,3]. However, there is still a substantial need to improve current treatment approaches particularly for elderly patients or those with relapsed and refractory disease [4,5,6]. Cured patients unfortunately are at high risk for late side effects including second malignancies, cardiac toxicity, infertility, and fatigue [7,8,9]. Thus, there is a clear need for new and safer drugs that are more selective in targeting the malignant Hodgkin and Reed-Sternberg (HRS) cells in this disease while sparing normal tissues.
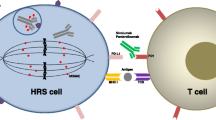
CD30 is a cell surface protein that is highly expressed on HRS cells (Figs. 22.1 and 22.2) and is rarely expressed by normal tissue, making it ideal for targeted therapy. In fact, soon after the identification and characterization of CD30, monoclonal antibodies against this protein were evaluated as potential therapeutics. Although several preclinical experiments established the proof of principle for this treatment strategy, early clinical trials with either naked monoclonal antibodies or a variety of immunoconjugates, including immunotoxins and radioimmunoconjugates against CD30, either did not demonstrate sufficient clinical activity or were too toxic [10,11,12,13,14,15]. The lack of meaningful clinical efficacy of naked anti-CD30 antibodies in patients with Hodgkin lymphoma (HL) remains poorly understood, but several hypotheses have been proposed: CD30 is internalized and, thus, does not allow sufficient time for engagement with effector cells. In addition, CD30 is shed in the serum in a soluble form, which may neutralize the efficacy of the antibodies; the early versions of anti-CD30 antibody were not ideal for binding CD30 or effector cells. More recently, advances in linker technology allowed the development of novel and potent antibody-drug conjugates (ADC), such as brentuximab vedotin. This overview will highlight pathophysiology and current clinical experience when targeting CD30 in patients with Hodgkin lymphoma.

cHL_NS (nodule of CHL, nodular sclerosing (NS) type). Courtesy from Pileri S

cHL_MC (classical Hodgkin lymphoma (CHL) of the mixed cellularity (MC) type). Courtesy from Pileri S
2 Structure and Function of CD30
In a landmark paper published in 1982, Stein and colleagues identified a new monoclonal antibody called Ki-1 that recognized a new antigen expressed on HRS cell, called CD30 [16]. Originally thought to be specific for HRS cells of Hodgkin lymphoma (HL), it was later found on small subsets of paracortical lymphocytes and a few other malignancies, including anaplastic large cell lymphoma (ALCL) [17,18,19]. The major limitation of the Ki-1 antibody was the need for fresh or frozen material, which allowed its application only in a limited number of reference centers. This was overcome by the generation of the Ber-H2 monoclonal antibody detecting an epitope of the molecule different from Ki-1 and applicable in routine formalin-fixed paraffin-embedded tissue samples.
Ten years after the identification of the CD30 antigen, the same group cloned the cDNAs coding for CD30 from expression libraries of the human HUT-102 cell line using the monoclonal antibodies Ki-1 and Ber-H2. The open reading frame of the cDNA predicted a 595-amino acid transmembrane protein. The extracellular domain contained six cysteine-rich motifs and shared sequence homology with members of the tumor necrosis factor (TNF) superfamily [20, 21]. The cytoplasmic tail contains several TNF receptor-associated factor (TRAF)-binding sequences that mediate activation of pleiotropic signals, including activation of nuclear factor kappa-B (NK-κB) [22, 23]. CD30 has a broad range of biologic effects depending on the cellular context, including regulation of cytokine secretion and inflammation, induction of apoptosis, and promotion of cell survival and proliferation [24]. The ligand for CD30 (CD30L, CD153) is a 26-kDa type II transmembrane protein that belongs to the TNF superfamily and maps to chromosome 9q33 [25]. CD30L is expressed in both resting and activated B cells, activated T lymphocytes, monocytes, granulocytes, and natural killer cells [26, 27].
The exact physiologic function of CD30/CD30L in healthy individuals remains poorly understood, as no human diseases have been associated with alterations in CD30 or CD30L genes. Furthermore, CD30 knockout mice experiments gave conflicting results regarding a possible role of CD30 in thymocyte negative selection [28, 29]. Other studies suggested that CD30-CD30L signaling may be involved in immunoregulation, such as class-switch DNA recombination and antibody production in B cells [30]. CD30 may also play a role in self-tolerance and pathogenesis of autoimmune disorders [31, 32], in addition to regulating Th1 and Th2 cell responses [33,34,35], CD4+ T-cell-mediated graft-versus-host disease [36], and CD30+ Treg cells [37].
3 Therapeutic Targeting of CD30
CD30 is an excellent target for monoclonal antibody therapy due to its restricted expression. A few years after the initial description of the first monoclonal antibody against CD30, Ki-1 [16], monoclonal antibodies such as Ki-4 and Ber-H2 were generated that had higher affinity for the CD30 antigen [30]. Subsequently, these antibodies were conjugated to ricin A chain to form specific immunoreagents. These so-called immunotoxins were extremely effective and specific in vitro and in different animal models [10, 11]. However, a subsequent clinical phase I/II trial using the ricin A-chain immunotoxin Ki-4.dgA targeting CD30 showed little clinical activity in a total of 18 patients with refractory HL. This immunotoxin was associated with vascular leak syndrome as dose-limiting toxicity [14]. In addition, most patients developed anti-ricin antibodies so that further clinical development of this immunotoxin in HL was abandoned. An alternate strategy used the murine anti-CD30 monoclonal antibody (Ber-H2) as carrier for a cytotoxic agent by covalently linking Ber-H2 to saporin (SO6), a type 1 ribosome-inactivating protein [12]. Four patients with advanced refractory HL were treated, and three patients had transient tumor reduction [13]. Human antibodies, however, developed against the murine antibody and the toxin in all patients preventing repeat dosing; thus, further development of this immunotoxin was also stopped.
4 Monoclonal Antibodies
Clinical results from first-generation naked monoclonal antibodies targeting CD30 were disappointing, possibly due to their poor antigen-binding properties, ineffective activation of effector cells, and neutralization by soluble CD30 [14, 38, 39]. MDX-060, a fully human anti-CD30 monoclonal antibody, was tested in a phase I/II study in patients with HL, ALCL, and CD30+ PTCL. This antibody had minimal toxicity, and the maximum tolerated dose (MTD) was not reached [15]. However, MDX-060 had minimal clinical activity with six responses in 72 patients and was subsequently abandoned. SGN-30, a CD30-specific chimeric antibody constructed from the variable regions of the anti-CD30 murine monoclonal AC10 and human gamma 1 heavy chain and kappa light chain constant regions, was also tested in phase I/II studies. A phase I study of SGN-30 in 24 HL or CD30+ non-Hodgkin lymphoma (NHL) patients demonstrated that SGN-30 was well tolerated, but only one patient with cutaneous ALCL achieved a complete response (CR) [38]. The phase II results of SGN-30 also showed only modest clinical activity with 9% overall response (2 CRs and 5 partial responses (PR) of 79 patients treated); all responses were limited to patients with ALCL [40]. Given preliminary evidence of selective efficacy of SGN-30 in cutaneous ALCL, SGN-30 was further tested in a phase II study of cutaneous diseases including cutaneous ALCL, lymphomatoid papulosis, and transformed mycosis fungoides; the response rate in this trial was 70% [41]. SGN-30 was subsequently combined with chemotherapy because preclinical data showed that SGN-30 sensitizes tumor cells to cytotoxic agents and single-agent phase I/II data demonstrated only modest efficacy [42]. In a Cancer and Leukemia Group B randomized phase II trial of SGN-30 with gemcitabine, vinorelbine, and pegylated liposomal doxorubicin (GVD) in relapsed HL patients, 30 patients were treated; however, five patients developed grade 3–5 pneumonitis, leading to premature closure of the trial [43]. The combination of SGN-30 and GVD was not only associated with significant toxicity but also was not associated with better outcomes compared to GVD alone. Given the disappointing results with first-generation naked monoclonal antibodies, a second-generation anti-CD30 humanized antibody, XmAb2513, with improved antigen-binding and enhanced Fcγ receptor IIIA affinity was developed demonstrating increased efficacy in vitro when compared to MDX-060 or SGN-30 [44]. Preliminary results of the phase 1 study of XmAb2513 found the drug to be well tolerated, but not associated with superior efficacy compared to first-generation monoclonal antibodies. Of 13 HL patients treated, tumor reduction was observed in three patients [45].
5 Bispecific Monoclonal Antibodies
A different approach to targeting CD30 was the development of bispecific monoclonal antibodies, engaging NK cells or neutrophils as effector cells [10, 46]. A construct based on the anti-CD30 monoclonal antibody Ki-4 and the human anti-CD64 monoclonal H22 showed very promising preclinical activity. In the phase I clinical trial, H22xKi-4 was very well tolerated; responses included one CR, four PRs, and four SDs in a total of ten patients treated [10]. More recently, a bispecific TandAb antibody, AFM13, was reported [47]. AFM13 targets both CD30 on HL tumor cells and CD16A on NK cells. Preclinical data demonstrated antitumor activity with engagement of NK immune effector cells. A phase 1 study of AFM13 in 28 HL patients found the drug safe and well tolerated, but with a modest activity. Overall, 3 of 28 patients achieved partial remissions [47].
6 Radiolabeled Antibodies
Schnell et al. developed a radioimmunoconjugate consisting of the murine anti-CD30 monoclonal antibody Ki-4 labeled with iodine-131 (131I). Twenty-two HL patients were treated with 131I-Ki-4 to total body doses ranging from 0.035 to 0.99 Gy. Although there were six responses (one CR and five PRs), a significant rate of severe hematologic toxicity was observed with seven patients having grade four hematologic toxicity 4–8 weeks posttreatment, leading to the cessation of its further development [48].
7 Chimeric Antigen Receptor (CAR) T-Cell Therapy
First-generation anti-CD30 CAR T cells were developed in the 1990s, and preclinical studies demonstrated the ability of these cells to lyse CD30-expressing HL cell lines in vitro [49, 50]. Indeed, Epstein-Barr virus-specific cytotoxic T cells transduced with an anti-CD30 CAR have been shown to have activity against CD30+ cancer cell lines in vitro, as well as in vivo, in a mouse xenograft model [51, 52]. In a phase I clinical trial of anti-CD30 CAR T cells with a CD28 co-stimulatory domain including seven cHL and two ALCL patients, four patients had stable disease, one had a complete response, and one had a partial response, while three had disease progression [53, 54]. In another phase I trial, 18 patients (17 with HL and one with cutaneous ALCL) were treated with anti-CD30 CAR containing a 4-1BB co-stimulatory domain and seven patients had a PR, with a median PFS of 6 months [55]. A number of other clinical trials of anti-CD30 CAR-T-cell therapy are ongoing and will provide more information on the efficacy of this approach [56].
8 Antibody-Drug Conjugates
8.1 Single-Agent Experience with Brentuximab Vedotin
Brentuximab vedotin (BV) is an antibody-drug conjugate (ADC) consisting of the chimeric monoclonal antibody, cAC10, that was conjugated to monomethyl auristatin E (MMAE) [57, 58]. MMAE is a synthetic analog of the natural product dolastatin 10 and functions as a tubulin inhibitor. MMAE is covalently linked to cAC10 via a maleimidocaproyl-valyl-citrullinyl-p-aminobenzylcarbamate linker [59]. On average, four molecules of MMAE are conjugated to one cAC10. The mechanism of action of brentuximab vedotin is shown in Fig. 22.3 and involves the following steps: (1) binding of the anti-CD30 ADC via the antibody moiety to CD30 expressed on tumor cells in high density, (2) receptor-mediated endocytosis of brentuximab vedotin and intracellular internalization occurring via clathrin-mediated uptake, (3) uptake of the drug into lysosomal vesicles, (4) MMAE which is released from the antibody by reduction or acid hydrolysis within lysosomes, and (5) MMAE which is released into cytoplasm and inhibits microtubule polymerization leading to arrest of the G2/M phase of the cell cycle, thereby inducing cellular apoptosis [58]. In addition, there is also a small amount of MMAE released into the tumor microenvironment that may alter survival signaling to the HRS cell. Preclinical studies with cAC10-vcMMAE demonstrated stable linkage of the ADC in circulation and efficient release upon internalization into target cells. In addition, cAC10-vcMMAE was found to have significant antitumor activity in HL and ALCL cell lines with an IC50 of 10 ng/mL and antitumor activity in subcutaneous disease xenograft models [59].

Mechanism of action of brentuximab vedotin (SGN-35) (Figure was adapted from: Katz et al. [58])
The initial first-in-man, multicenter, dose-escalation phase I study enrolled 45 patients with relapsed or refractory CD30-positive hematologic cancers, including 42 HL and 3 ALCL patients. BV was administered intravenously every 3 weeks at doses ranging from 0.1 to 3.6 mg/kg. Dose-limiting toxicities were grade 4 thrombocytopenia, grade 3 hyperglycemia, and febrile neutropenia. Remarkably, tumor regression was seen in 86% of evaluable patients, and the MTD was defined at 1.8 mg/kg every 3 weeks. Eleven patients achieved complete responses and six achieved partial remissions. The median duration of response was at least 9.7 months. When the analysis was restricted to patients receiving the dose of 1.8 mg/kg or greater, six of twelve patients responded (50%), including four complete remissions [60].
A second phase I study evaluated the safety and efficacy of BV given on days 1, 8, and 15 in a 28-day cycle (3 weeks on, followed by 1 week of rest). This study demonstrated similar efficacy (ORR 59% and tumor regression in 85% of patients). Given the ease of administration of every 3-week dosing and similar response rates across the two dosing schedules, the 1.8 mg/kg every 3 weeks was selected for further development in phase II studies [61].
The pivotal phase 2 study that led to the FDA approval of BV was conducted in 102 patients with relapsed and refractory HL after receiving autologous stem cell transplantation (ASCT), to determine the efficacy and safety of brentuximab vedotin [62]. Patients received 1.8 mg/kg brentuximab vedotin every 3 weeks as a 30-min outpatient infusion (capped dose at 180 mg) for up to 16 cycles. There was no limit on the number of prior treatment regimens (median of 3.5, range 1–13 regimens). All patients had failed ASCT with a median time to relapse after ASCT of 6.7 months (range 0–131 months). Patients received a median of nine cycles of brentuximab vedotin, and the overall response rate was 75% (33% CRs). In a waterfall plot analysis (Fig. 22.4), 94% of patients had tumor regression. Responses were rapid, with a median time to treatment response of 5.7 weeks and the median time to achieving complete remission of 12 weeks. The median progression-free survival for all patients was 5.6 months. This study led to the FDA approval of brentuximab vedotin for the following indications: (1) Hodgkin lymphoma after failure of ASCT and (2) HL patients who are not ASCT candidates after failure of at least two prior therapies. The 5-year end-of-study results were reported and in 34 patients who achieved a CR the median PFS and OS were not reached and 13 patients remained in remission at time of study closure. This suggests that a proportion of patients who achieve a CR with single-agent brentuximab vedotin will have long-term disease control and may potentially be cured [63].

Maximum percent reduction in sum of the product of diameters in individual patients (n = 98) in the pivotal phase II trial of brentuximab vedotin for relapsed and refractory Hodgkin lymphoma (Figure was adapted from Younes et al. with permission [62])
9 Safety and Tolerability of Brentuximab Vedotin
In the two phase I studies of brentuximab vedotin, the dose-limiting toxicities included cytopenias, diarrhea, vomiting, and hyperglycemia [60, 61]. Data from phase I and II studies of brentuximab vedotin have characterized the adverse effects of the drug, including peripheral sensory neuropathy, nausea, fatigue, neutropenia, diarrhea, pyrexia, vomiting, arthralgia, pruritus, myalgia, peripheral motor neuropathy, and alopecia [62]. In phase II studies, approximately 55% of patients experienced adverse grade 3 and 4 events including peripheral sensory neuropathy (8–12%), neutropenia (20–21%), anemia (6–7%), and thrombocytopenia (8–14%). The associated peripheral neuropathy is typically cumulative and most commonly grade 1–2 characterized by numbness or tingling in the fingers and toes. In addition, 11–14% of patients had grade 3 peripheral neuropathy; no grade 4 was seen. Approximately 80% of patients with peripheral neuropathy experienced clinical improvement after dose reduction or cessation of drug, and 50% experienced complete resolution. As a result of these data, significant cytopenias or neuropathy should prompt consideration for dose modification, delay, or discontinuation. Overall, brentuximab vedotin is well tolerated with manageable side effects and few serious adverse events. Additional rare, but serious adverse events have been reported including pancreatitis and fatal progressive multifocal leukoencephalopathy associated with John Cunningham (JC) virus infection [64].
9.1 Brentuximab Vedotin in Frontline Setting for HL
Brentuximab vedotin was successfully combined with chemotherapy for the up-front treatment HL. BV has been evaluated for the frontline treatment of early-stage disease, advanced-stage disease, and for elderly patients. In a phase I study of brentuximab vedotin combined with ABVD (adriamycin, bleomycin, vinblastine, and dacarbazine) chemotherapy, significant pulmonary toxicity (40%) was described in conjunction with bleomycin. Therefore, the combination of BV with AVD chemotherapy (without bleomycin) has been established as a safe combination [65].
9.1.1 Early-Stage Disease
The aim of incorporating BV into early-stage treatment protocols has been to eliminate RT, particularly in early-stage patients with unfavorable features, such as disease bulk. In a pilot study in early-stage cHL patients with unfavorable risk disease, BV + AVD for four cycles followed by 30 Gy involved-site radiotherapy was found to be safe and well-tolerated without evidence of significant pulmonary toxicity. Among the 30 patients treated, 77% had disease bulk by Memorial Sloan Kettering Cancer Center criteria (>7 cm in transverse or coronal dimension) and reported outcomes were promising with high rates of PET negativity after chemotherapy and a 1-year progression-free survival of 93% [66, 67]. Subsequent cohorts of the study tested if consolidation after BV + AVD × 4 cycles can be decreased to minimize late toxicities associated with radiotherapy. In cohort 2, a lower dose of ISRT (20Gy) was applied post-chemotherapy. In cohort 3, a smaller radiation field to treat only the residual disease post chemotherapy was used and in cohort 4 had no radiation consolidation [67]. Other clinical trials also incorporate BV into early-stage programs with the aim of eliminating RT or enhancing efficacy of short-course chemotherapy.
9.1.2 Advanced-Stage Disease
The ECHELON-1 study was a large, international, multicenter, randomized control trial comparing the efficacy of standard ABVD × 6 cycles vs. BV + AVD × 6 cycles for the treatment of advanced-stage HL [68]. This study found a modest benefit in terms of modified 2-year progression-free survival in favor of BV + AVD vs. ABVD (82.1% vs. 77.2%). In certain subgroups of patients, there was a higher degree of benefit observed, including stage IV disease, males, and in patients with high-risk international prognostic scores. However, the BV + AVD treatment program was found to have increased toxicities including peripheral neuropathy and febrile neutropenia; the latter risk was attenuated when growth factor support was used. Importantly, this study established BV + AVD as an FDA-approved frontline regimen for advanced-stage HL. BV has also been incorporated into other ongoing frontline clinical trials, such as the modified BEACOPP regimen with inclusion of BV, called BrECAPP or BrECADD [69].
9.1.3 Elderly Patients
Older patients with cHL have poor outcomes due to more aggressive biologic features and poor tolerance of standard chemotherapy such as ABVD. Incorporating BV into treatment regimens for elderly patients can increase efficacy of therapy and decrease toxicity. A study in cHL aged 60 years or older and stage IIB, III, and IV disease with initial BV × 2 cycles then AVD × 6 cycles followed by BV × 4 cycles demonstrated 2-year PFS and OS of 84% and 93%, respectively [70]. These are promising results compared to historically reported outcomes in this patient population. BV has also been used as a single agent and combined with bendamustine and dacarbazine in the older HL population [71, 72].
9.2 Brentuximab Vedotin Pre-ASCT
Brentuximab vedotin has been studied in relapsed/refractory HL as a second-line salvage prior to high-dose therapy and autologous stem cell transplant (HDCT-ASCT). In one study, patients were treated with single-agent brentuximab vedotin for two cycles (1.2 mg/kg IV weekly, 3 weeks on and 1 week off), followed by response assessment using PET imaging. Patients who achieved a complete remission with a negative PET (Deauville 1, 2) were allowed to proceed to stem cell collection followed by ASCT, thus avoiding chemotherapy. Patients with PET-positive scans after two cycles of brentuximab vedotin were treated with augmented ICE chemotherapy, followed by ASCT. Using this PET-adapted strategy, approximately 30% of patients achieved CR after two cycles of brentuximab vedotin, avoiding ICE-based therapy [73]. Other dosing schedules of BV pre-ASCT have also been published (1.8 mg/kg IV every 3 weeks for 2–4 cycles) [74]. In addition, BV has been combined with other chemotherapy regimens including bendamustine, ICE, DHAP, and ESHAP [75,76,77,78,79]. Another promising chemotherapy-free salvage treatment program combines BV and nivolumab, a checkpoint inhibitor, for 2–4 cycles pre-ASCT [80]. The combination was well-tolerated and was associated with a complete response rate of 61% (61/62) with an objective response rate of 82%.
9.3 Brentuximab Vedotin Maintenance Post Autologous Stem Cell Transplant
To study the role of adjuvant brentuximab vedotin after autologous stem cell transplant, the randomized phase III ATHERA study includes an investigational arm of brentuximab vedotin 1.8 mg/kg administered every 3 weeks for approximately 1 year (a maximum of 16 doses) vs. placebo after ASCT in high-risk HL patients [81]. High-risk features included presence of extranodal disease, B-symptoms, relapse within 1 year of initial treatment, primary refractory disease, less than CR to salvage therapy, or requiring ≥2 salvage therapies before ASCT. The median PFS in the BV group was superior to placebo (42.9 vs. 24.1 months, P = 0.0013) and this led to FDA approval for BV maintenance post-ASCT. All patients who were enrolled in ATHERA were BV naïve, so the applicability of these findings may be limited in the modern era when most patients will receive BV in the first- or second-line setting.
9.4 Brentuximab Vedotin-Based Combinations in Posttransplant Settings
Although brentuximab vedotin produces a high overall response rate in patients with relapsed HL, most responses are partial and of short duration. Therefore, there is a need to combine brentuximab vedotin with other active agents to increase the proportion of complete remissions and to prolong the duration of response. Based on preclinical data to suggest synergy between brentuximab vedotin and other agents, BV is being combined with other agents such as bendamustine, temsirolimus, HDAC inhibitors, and PD1/PDL1 monoclonal antibodies.
10 Conclusions
With the identification of the CD30 antigen on Hodgkin and Sternberg-Reed cells, different constructs such as the naked monoclonal antibodies Ki-1 and Ber-H2 as well as the ligand for CD30 were initially assessed for therapeutically targeting of Hodgkin lymphoma cells via CD30. Since these constructs had little clinical efficacy against Hodgkin lymphoma, other immunoreagents include immunotoxins, bispecifics, as well as humanized anti-CD30 antibodies such as MDX-060 or SGN-30. The latter was subsequently linked to MMAE, a potent anti-tubulin agent. This construct, SGN-35, was later termed brentuximab vedotin. The efficacy, tolerability, and broad applicability of brentuximab vedotin have dramatically changed treatment paradigms in cHL, improving outcomes for patients at every phase of the disease. In the future, we anticipate there will be advances in risk stratification of cHL patients, allowing for a more individualized approach to treatment and identification of which patients will benefit the most from novel therapies, including BV.
References
Bonadonna G, Santoro A (1982) ABVD chemotherapy in the treatment of Hodgkin’s disease. Cancer Treat Rev 9:21–35
De Vita VT, Serpick A (1967) Combination chemotherapy in the treatment of advanced Hodgkin’s disease. Proc Am Assoc Cancer Res 8:13
De Vita VT (1981) The consequences of the chemotherapy of Hodgkin’s disease: the 10th David a. Karnofsky memorial lecture. Cancer 47:1–13
Josting A, Muller H, Borchmann P et al (2010) Dose intensity of chemotherapy in patients with relapsed Hodgkin’s lymphoma. J Clin Oncol 28:5074–5080
Kuruvilla J, Keating A, Crump M (2011) How I treat relapsed and refractory Hodgkin lymphoma. Blood 117:4208–4217
Moskowitz AJ, Perales MA, Kewalramani T et al (2009) Outcomes for patients who fail high dose chemoradiotherapy and autologous stem cell rescue for relapsed and primary refractory Hodgkin lymphoma. Br J Haematol 146:158–163
Ng AK, Bernardo MP, Weller E et al (2002) Long-term survival and competing causes of death in patients with early-stage Hodgkin’s disease treated at age 50 or younger. J Clin Oncol 20:2101–2108
Specht L (2003) Very long-term follow-up of the Danish National Hodgkin Study Group’s randomized trial of radiotherapy (RT) alone vs. combined modality treatment (CMT) for early stage Hodgkin lymphoma, with special reference to second tumors and overall survival. Blood 102:637A
van Leeuwen FE, Klokman WJ, Veer MB et al (2000) Long-term risk of second malignancy in survivors of Hodgkin’s disease treated during adolescence or young adulthood. J Clin Oncol 18:487–497
Engert A, Burrows F, Jung W et al (1990) Evaluation of ricin a chain-containing immunotoxins directed against the CD30 antigen as potential reagents for the treatment of Hodgkin’s disease. Cancer Res 50:84–88
Engert A, Martin G, Pfreundschuh M et al (1990) Antitumor effects of ricin a chain immunotoxins prepared from intact antibodies and fab’ fragments on solid human Hodgkin’s disease tumors in mice. Cancer Res 50:2929–2935
Falini B, Flenghi L, Fedeli L et al (1992) In vivo targeting of Hodgkin and reed-Sternberg cells of Hodgkin’s disease with monoclonal antibody Ber-H2 (CD30): immunohistological evidence. Br J Haematol 82:38–45
Falini B, Bolognesi A, Flenghi L et al (1992) Response of refractory Hodgkin’s disease to monoclonal anti-CD30 immunotoxin. Lancet 339:1195–1196
Schnell R, Staak O, Borchmann P et al (2002) A phase I study with an anti-CD30 ricin A-chain immunotoxin (Ki-4.dgA) in patients with refractory CD30+ Hodgkin’s and non-Hodgkin’s lymphoma. Clin Cancer Res 8:1779–1786
Borchmann P, Schnell R, Fuss I et al (2002) Phase 1 trial of the novel bispecific molecule H22xKi-4 in patients with refractory Hodgkin lymphoma. Blood 100:3101–3107
Stein H, Mason DY, Gerdes J et al (1985) The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 66:848–858
Schwab U, Stein H, Gerdes J et al (1982) Production of a monoclonal antibody specific for Hodgkin and Sternberg-reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature 299:65–67
Hecht TT, Longo DL, Cossman J et al (1985) Production and characterization of a monoclonal antibody that binds reed-Sternberg cells. J Immunol 134:4231–4236
Schwarting R, Gerdes J, Durkop H, Falini B, Pileri S, Stein H (1989) BER-H2: a new anti-Ki-1 (CD30) monoclonal antibody directed at a formol-resistant epitope. Blood 74:1678–1689
Durkop H, Latza U, Hummel M, Eitelbach F, Seed B, Stein H (1992) Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell 68:421–427
Fonatsch C, Latza U, Durkop H, Rieder H, Stein H (1992) Assignment of the human CD30 (Ki-1) gene to 1p36. Genomics 14:825–826
Duckett CS, Gedrich RW, Gilfillan MC, Thompson CB (1997) Induction of nuclear factor kappaB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol Cell Biol 17:1535–1542
Duckett CS, Thompson CB (1997) CD30-dependent degradation of TRAF2: implications for negative regulation of TRAF signaling and the control of cell survival. Genes Dev 11:2810–2821
Mir SS, Richter BW, Duckett CS (2000) Differential effects of CD30 activation in anaplastic large cell lymphoma and Hodgkin disease cells. Blood 96:4307–4312
Smith CA, Gruss HJ, Davis T et al (1993) CD30 antigen, a marker for Hodgkin’s lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell 73:1349–1360
Younes A, Consoli U, Zhao S et al (1996) CD30 ligand is expressed on resting normal and malignant human B lymphocytes. Br J Haematol 93:569–571
Gruss HJ, Boiani N, Williams DE, Armitage RJ, Smith CA, Goodwin RG (1994) Pleiotropic effects of the CD30 ligand on CD30-expressing cells and lymphoma cell lines. Blood 83:2045–2056
Amakawa R, Hakem A, Kundig TM et al (1996) Impaired negative selection of T cells in Hodgkin’s disease antigen CD30-deficient mice. Cell 84:551–562
DeYoung AL, Duramad O, Winoto A (2000) The TNF receptor family member CD30 is not essential for negative selection. J Immunol 165:6170–6173
Bowen MA, Lee RK, Miragliotta G, Nam SY, Podack ER (1996) Structure and expression of murine CD30 and its role in cytokine production. J Immunol 156:442–449
Kurts C, Carbone FR, Krummel MF, Koch KM, Miller JF, Heath WR (1999) Signalling through CD30 protects against autoimmune diabetes mediated by CD8 T cells. Nature 398:341–344
Gaspal FM, Kim MY, McConnell FM, Raykundalia C, Bekiaris V, Lane PJ (2005) Mice deficient in OX40 and CD30 signals lack memory antibody responses because of deficient CD4 T cell memory. J Immunol 174:3891–3896
Gerli R, Lunardi C, Vinante F, Bistoni O, Pizzolo G, Pitzalis C (2001) Role of CD30+ T cells in rheumatoid arthritis: a counter-regulatory paradigm for Th1-driven diseases. Trends Immunol 22:72–77
Sun X, Somada S, Shibata K et al (2008) A critical role of CD30 ligand/CD30 in controlling inflammatory bowel diseases in mice. Gastroenterology 134:447–458
Sun X, Yamada H, Shibata K et al (2010) CD30 ligand is a target for a novel biological therapy against colitis associated with Th17 responses. J Immunol 185:7671–7680
Blazar BR, Levy RB, Mak TW et al (2004) CD30/CD30 ligand (CD153) interaction regulates CD4+ T cell-mediated graft-versus-host disease. J Immunol 173:2933–2941
Dai Z, Li Q, Wang Y et al (2004) CD4+CD25+ regulatory T cells suppress allograft rejection mediated by memory CD8+ T cells via a CD30-dependent mechanism. J Clin Invest 113:310–317
Bartlett NL, Younes A, Carabasi MH et al (2008) A phase 1 multidose study of SGN-30 immunotherapy in patients with refractory or recurrent CD30+ hematologic malignancies. Blood 111:1848–1854
Ansell SM, Horwitz SM, Engert A et al (2007) Phase I/II study of an anti-CD30 monoclonal antibody (MDX-060) in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. J Clin Oncol 25:2764–2769
Forero-Torres A, Leonard JP, Younes A et al (2009) A phase II study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br J Haematol 146:171–179
Duvic M, Reddy SA, Pinter-Brown L et al (2009) A phase II study of SGN-30 in cutaneous anaplastic large cell lymphoma and related lymphoproliferative disorders. Clin Cancer Res 15:6217–6224
Cerveny CG, Law CL, McCormick RS et al (2005) Signaling via the anti-CD30 mAb SGN-30 sensitizes Hodgkin’s disease cells to conventional chemotherapeutics. Leukemia 19:1648–1655
Blum KA, Jung SH, Johnson JL et al (2010) Serious pulmonary toxicity in patients with Hodgkin’s lymphoma with SGN-30, gemcitabine, vinorelbine, and liposomal doxorubicin is associated with an FcgammaRIIIa-158 V/F polymorphism. Ann Oncol 21:2246–2254
Lawrence CE, Hammond P, Zalevsky J et al (2007) XmAbTM2513, an fc engineered humanized anti-CD30 monoclonal antibody, has potent in vitro and in vivo activities, and has the potential for treating hematologic malignancies. Blood (ASH Annual Meeting Abstracts) 110:2340
Blum KA, Smith M, Fung H et al (2009) Phase I study of an anti-CD30 fc engineered humanized monoclonal antibody in Hodgkin lymphoma (HL) or anaplastic large cell lymphoma (ALCL) patients: safety, pharmacokinetics (PK), immunogenicity, and efficacy. ASCO Annu Meet (Abstr) 27:8531
Hartmann F, Renner C, Jung W et al (2001) Anti-CD16/CD30 bispecific antibody treatment for Hodgkin’s disease: role of infusion schedule and costimulation with cytokines. Clin Cancer Res 7:1873–1881
Zhukovsky E, Achim R, von Tesckow B et al (2013) A phase I study of an anti-CD30 x anti-CD16A bispecific Tandab antibody, AFM13, in patients with relapsed or refractory Hodgkin lymphoma. Blood (ASH Annual Meeting Abstracts) 122:5116
Schnell R, Dietlein M, Staak JO et al (2005) Treatment of refractory Hodgkin’s lymphoma patients with an iodine-131-labeled murine anti-CD30 monoclonal antibody. J Clin Oncol 23:4669–4678
Hombach A et al (1998) An anti-CD30 chimeric receptor that mediates CD3-zeta-independent T-cell activation against Hodgkin’s lymphoma cells in the presence of soluble CD30. Cancer Res 58:1116–1119
Hombach A et al (1999) Characterization of a chimeric T-cell receptor with specificity for the Hodgkin’s lymphoma-associated CD30 antigen. J Immunother 22:473–480, 473,475,477,479
Savoldo B et al (2007) Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30ζ artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood 110:2620–2630
Di Stasi A et al (2009) T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 113:6392–6402
Ramos CA, Heslop HE, Brenner MK (2016) CAR-T cell therapy for lymphoma. Annu Rev Med 67:165–183
Ramos CA et al (2015) Chimeric T cells for therapy of CD30+ Hodgkin and non-Hodgkin lymphomas. Blood 126:185
Wang C et al (2017) Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin’s lymphoma: an open-label phase I trial. Clin Cancer Res 23:1156–1166
Brudno JN, Kochenderfer JF (2018) Chimeric antigen receptor T-cell therapies for lymphoma. Nat Rev Clin Oncol 15:31–46
Oki Y, Younes A (2012) Brentuximab vedotin in systemic T-cell lymphoma. Expert Opin Biol Ther 12:623–632
Katz J, Janik JE, Younes A (2011) Brentuximab vedotin (SGN-35). Clin Cancer Res 17:6428–6436
Francisco JA, Cerveny CG, Meyer DL et al (2003) cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 102:1458–1465
Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL et al (2010) Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 363(19):1812–1821
Fanale MA, Forero-Torres A, Rosenblatt JD, Advani RH, Franklin AR, Kennedy DA et al (2012) A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30-positive hematologic malignancies. Clin Cancer Res 18(1):248–255
Younes A, Gopal AK, Smith SE et al (2012) Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol 30:2183–2189
Chen R et al (2016) Five-year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood 128(12):1562–1566
von Geldern G, Pardo CA, Calabresi PA, Newsome SD (2012) PML-IRIS in a patient treated with brentuximab. Neurology 79:2075–2077
Younes A, Connors JM, Park SI et al (2013) Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncol 14:1348–1356
Kumar A, Burger IA, Zhang Z et al (2016) Definition of bulky disease in early stage Hodgkin lymphoma in computed tomography era: prognostic significance of measurements in the coronal and transverse planes. Haematologica 101(10):1237–1243
Kumar A, Casulo C, Yahalom J et al (2016) Brentuximab vedotin and AVD followed by involved-site radiotherapy in early stage, unfavorable risk Hodgkin lymphoma. Blood 128(11):1458–1464
Connors JM, Jurczak W, Straus DJ et al (2018) ECHELON-1 study group. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin’s lymphoma. N Engl J Med 378(4):331–344
Eichenauer DA, Plutschow A, Kreissl S et al (2017) Incorporation of brentuximab vedotin into first-line treatment of advanced classical Hodgkin’s lymphoma: final analysis of a phase 2 randomised trial by the German Hodgkin study group. Lancet Oncol 18(12):1680–1687
Evens AM, Advani RH, Helenowski IB et al (2018) Multicenter phase II study of sequential brentuximab vedotin and doxorubicin, vinblastine, and dacarbazine chemotherapy for older patients with untreated classical Hodgkin lymphoma. J Clin Oncol 36:3015
Forero-Torres A, Holkova B, Goldschmidt J et al (2015) Phase 2 study of frontline brentuximab vedotin monotherapy in Hodgkin lymphoma patients aged 60 years and older. Blood 126(26):2798–2804
Friedberg JW, Forero-Torres A, Bordoni RE et al (2017) Frontline brentuximab vedotin in combination with dacarbazine or bendamustine in patients aged ≥60 years with HL. Blood 130(26):2829–2837
Moskowitz AJ, Schöder H, Yahalom J et al (2015) PET-adapted sequential salvage therapy with brentuximab vedotin followed by augmented ifosamide, carboplatin, and etoposide for patients with relapsed and refractory Hodgkin’s lymphoma: a non-randomised, open-label, single-Centre, phase 2 study. Lancet Oncol 16(3):284–292
Chen R, Palmer JM, Martin P et al (2015) Results of a multicenter phase II trial of brentuximab vedotin as second-line therapy before autologous transplantation in relapsed/refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 21(12):2136–2140
Cassaday RD, Fromm J, Cowan AJ et al (2016) Safety and activity of brentuximab vedotin (BV) plus ifosfamide, carboplatin, and etoposide (ICE) for relapsed/refractory (Rel/ref) classical Hodgkin lymphoma (cHL): initial results of a phase I/II trial. Blood 128(22):1834
Hagenbeek A, Zijlstra J, Lugtenburg P et al (2016) Transplant BRaVE: combining brentuximab vedotin with DHAP as salvage treatment in relapsed/refractory Hodgkin’s lymphoma. A phase 1 dose-escalation study. Haematologica 101(s5):44
Garcia-Sanz R, Sureda A, Gonzalez AP et al (2016) Brentuximab vedotin plus ESHAP (BRESHAP) is a highly effective combination for inducing remission in refractory and relapsed Hodgkin lymphoma patients prior to autologous stem cell transplant: a trial of the Spanish Group of Lymphoma and Bone Marrow Transplantation (GELTAMO). Blood 128(22):1109
LaCasce AS, Bociek RG, Sawas A et al (2018) Brentuximab vedotin plus bendamustine: a highly active first salvage regimen for relapsed or refractory Hodgkin lymphoma. Blood 132(1):40–48
O’Connor OA, Lue JK, Sawas A et al (2018) Brentuximab vedotin plus bendamustine in relapsed or refractory Hodgkin’s lymphoma: an international, multicentre, single-arm, phase 1-2 trial. Lancet Oncol 19(2):257–266
Herrera AF, Moskowitz AJ, Bartlett NL et al (2018) Interim results of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 131(11):1183–1194
Moskowitz CM, Nademanee A, Masszi T et al (2015) Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Lancet 385(9980):1853–1862
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kumar, A., Pileri, S., Younes, A., Engert, A. (2020). Targeting CD30 in Patients with Hodgkin Lymphoma. In: Engert, A., Younes, A. (eds) Hodgkin Lymphoma. Hematologic Malignancies. Springer, Cham. https://doi.org/10.1007/978-3-030-32482-7_22
Download citation
DOI: https://doi.org/10.1007/978-3-030-32482-7_22
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-32481-0
Online ISBN: 978-3-030-32482-7
eBook Packages: MedicineMedicine (R0)